

Abstract
The new dialkyl(aryl) lithium zincates [(THF)2Li(C6H4−OMe)MeZnMe] (4), [(TMEDA)Li(C6H4−OMe)MeZnMe] (6), [(THF)3Li(C6H4−OMe)tBuZntBu] (7), and [(PMDETA)Li(C6H4−OMe)tBuZntBu] (8) have been prepared by co-complexation reactions of lithiated anisole with the relevant dialkylzinc compound and the relevant Lewis base. These new heterobimetallic compounds have been characterized in solution using 1H, 13C{H}, and 7Li NMR spectroscopy, and the molecular structures of 6 and 8 have been elucidated by X-ray crystallography. In 6 the distinct metals are connected through the anisole ligand which binds in an ambidentate fashion (through carbon−zinc and oxygen−lithium contacts) and also through one of the methyl groups, to close a [LiOCCZnC] six-membered ring; whereas 8 displays an open structure where anisole connects the two metals (in the same mode as in 6) but with the tert-butyl groups exclusively bonded terminally to zinc. Reactivity studies of zincates 4 and 7 with the amine TMP(H) supply experimental evidence that these heterobimetallic compounds are intermediates in the two-step deprotonation reaction of anisole by TMP−dialkyl zincates and show the relevance of the alkyl groups in the efficiency of TMP−dialkyl zincate bases. In addition, important solvent effects have also been evaluated. When hexane is added to THF solutions of compounds 4 or 7, the homoleptic tetraorganozincate [(THF)2Li2Zn(C6H4−OMe)4] (5) is obtained as the result of a disproportionation process. This lithium-rich zincate has also been spectroscopically and crystallographically characterized.
Introduction
Since first reported by Kondo and Uchiyama in 1999, di-tert-butyl-TMP zincate LiZn(TMP)tBu2 (TMP = 2,2,6,6-tetramethylpiperidide)1 has proven to be a powerful and effective regioselective base for directed ortho-metalation (DoM) of various arenes. Its great versatility allows the direct zincation of a broad range of substituted aromatic molecules with a rich variety of functional groups such as esters, amides, ethers, or cyanides.2 In addition to this high functional group tolerance, reactions can be carried out under much milder conditions (generally at room temperature) than when traditional organolithium reagents such as tBuLi or LiTMP are employed, where other undesirable reactions such as nucleophilic addition can compete with the desired metalation.3 The structural elucidation of the THF-supported mixed-metal base [(THF)Li(TMP)(tBu)Zn(tBu)] (1)4 and of some key zincated intermediates of its reaction with DoM substrates such as the tertiary aromatic amide N,N-diisopropyl benzamide,5 anisole (Scheme 1)6 or phenyl dialkyl carbamates7 have unequivocally established that these metalations are genuine direct zincations, where the position previously filled by a hydrogen atom in the aromatic molecule is now occupied by zinc. This reactivity is in stark contrast to the low kinetic basicity found for conventional homometallic zinc reagents.8 Branded as alkali-metal-mediated zincation (AMMZ),2 this synthetic methodology represents an excellent alternative to the conventional synthesis of aryl zinc reagents, usually prepared by treating the relevant aryllithium or arylmagnesium compound with ZnCl2.9 Arylzinc species are key intermediates in synthesis, being involved in many important organic transformations such as cross-coupling Negishi reactions with aryl chlorides, which represent one of the most efficient methods of preparing biaryl compounds.10
Scheme 1. Selective Lithium-Mediated Zincation of Anisole with TMP-Zincate 1.
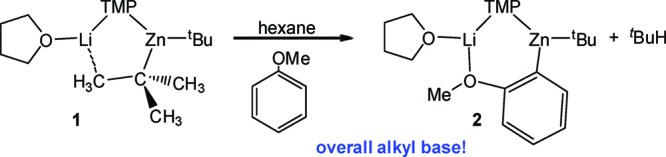
Recently we reported a sodium modification of 1, [(TMEDA)Na(TMP)(tBu)Zn(tBu)] (1-Na) (TMEDA= N,N,N′,N′-tetramethylethylenediamine)11 which exhibits an enhanced metalating power, successfully accomplishing not only monometalation but also regioselective dimetalation of nonactivated arenes such as benzene12 or naphthalene.13 The heteroleptic nature of the TMP-zincates 1 and [(TMEDA)Na(TMP)(tBu)Zn(tBu)] (1-Na) makes it possible that they can perform as either alkyl or amido bases. Definitive structural elucidations of metalated intermediates intimate that for the vast majority of the aromatic substrates studied both zincates react overall as alkyl bases, as illustrated in Scheme 1 for the AMMZ of anisole by 1 where the tert-butyl bridge of 1 is ultimately replaced by an ortho-deprotonated anisole fragment with isobutane as the coproduct of the reaction.6 As informative as they are, these X-ray crystallographically established results can only reveal part of the story, as any kinetic process taking place along the reaction coordinate between the reactants and the isolated product stages will be invisible to them. Important contributions toward filling this gap in our knowledge have come from theoretical (DFT) studies of several DoM subtrates (anisole,4a benzonitrile,14 methyl benzoate,14N,N-diisopropyl benzamide15) by Uchiyama, Nobuto, and Morokuma, which indicate that kinetically, amido basicity is preferred to the experimentally (X-ray) observed alkyl basicity due to significantly lower activation energies for the cleavage of the Zn−N bonds in comparison to Zn−C bonds. To explain the structures found experimentally for the isolated intermediates they propose that a two-step mechanism could be taking place where the arene is initially deprotonated by the amido ligand (due to the greater kinetic lability of the Zn−N bonds) affording a reaction intermediate [(S)M(areneide)(tBu)Zn(tBu)] (S = THF, M = Li for 1; S = TMEDA, M = Na for 1-Na) which could then react with concomitantly generated TMP(H) giving rise to the relevant [(S)M(TMP)(areneide)Zn(tBu)] (isolated) compound and tBuH, in agreement with the aforementioned experimentally established overall alkyl basicity.16 Although these studies provide good theoretical evidence for a two-step mechanism, one of their limitations is the simplified theoretical models employed for zincates 1 and 1-Na, [(Me2O)M(NMe2)(Me)Zn(Me)] (M = Li, Na) which undervalue the steric influence and the carboanionic character that bulky substituents tBu and TMP play in these metalations. In addition, our attempts to detect experimentally the proposed intermediates [(S)M(arenide)(tBu)Zn(tBu)] have been unsuccessful even when the metalations are carried out at low temperatures; this seems to indicate that if this two-step mechanism is taking place the second step must be extremely fast.
In order to shed new light upon the mechanisms involved in the reactions of these TMP-zincates with DoM substrates, herein we investigate in detail the reaction of 1 with anisole as a case study. Pioneering studies from Wittig and Gilman in the ortho-lithiation of anisole constituted a milestone in the development of directed ortho-metalation.17 The lithiation of this classical molecule has been extensively studied using NMR spectroscopy,18 semiempirical calculations,19 kinetic isotope effects,20 and X-ray crystallography.21 In 2006 we reported the first example of lithium-mediated zincation of anisole by mixed-metal base 1 in neat hexane at room temperature (Scheme 1). The metalated intermediate [(THF)Li(C6H4−OMe)(TMP)Zn(tBu)] (2) established that zincate 1 has behaved as an overall alkyl base.6 The same year Uchiyama and Morokuma published a theoretical study on the metalation of anisole by the related simple model zincate [(Me2O)Li(NMe2)(Me)Zn(Me)] which concluded that amido basicity is kinetically preferred over alkyl basicity, to generate a metalated intermediate [(Me2O)Li(C6H4−OMe)(Me)Zn(Me)(NHMe2)].4a
Herein we report the synthesis and structural elucidation of the putative intermediates [(S)xLi(C6H4−OMe)(R)Zn(R)] (S = THF, TMEDA; PMDETA; R = Me, tBu) suggested by the theoretical studies for the AMMZ of anisole. These are prepared through an indirect route in which anisole is first metalated by tBuLi and then co-complexed with R2Zn (R = Me, tBu). Mimicking the theoretical study, we also probe their reactivity toward TMP(H) and the influence that solvents with different donor abilities such as benzene and THF exert in these reactions.
Results and Discussion
Co-complexation Synthesis of [(THF)xLi(C6H4−OMe)(R)Zn(R)] (R = Me, tBu)
Following literature procedures anisole can be easily ortho-lithiated by reaction with tert-butyl lithium in THF at 0 °C and on the addition of hexane colorless crystals of [Li4(C6H4−OMe)4(THF)2] (3) could be obtained which were characterized by 1H, 13C, and 7Li NMR spectroscopy (see Experimental Section and Table 1). One molar equivalent of dimethylzinc was added to a solution of 3 in THF which was prepared in situ and the mixture was allowed to stir at room temperature for 30 min to afford the mixed-metal zincate [(THF)2Li(C6H4−OMe)(Me)Zn(Me)] (4) as a pale yellow oil (Scheme 2).
Table 1. Selected Chemical Shifts in the 1H and 7Li NMR Spectra in C6D6 Solution.
| compound | δ(1H)Zn-R | δ(1H)ArH | δ(1H)OMe | δ(7Li) |
|---|---|---|---|---|
| [(THF)Li(C6H4−OMe)(TMP)Zn(tBu)](2) | 1.61 | 7.94, 7.22, 7.16, 6.56 | 3.31 | 3.40 |
| [Li4(C6H4−OMe)4(THF)2](3) | 8.15, 7.42, 7.25, 6.78 | 3.41 | 3.28 | |
| [(THF)2Li(C6H4−OMe)(Me)Zn(Me)](4) | −0.23 | 8.15, 7.20, 7.11, 6.74 | 3.39 | 0.72 |
| [(THF)2Li2Zn(C6H4−OMe)4](5) | 8.45, 7.23, 7.09, 6.72 | 3.24 | 0.97 | |
| [(TMEDA)Li(C6H4−OMe)(Me)Zn(Me)](6) | −0.14 | 8.27, 7.22, 6.77 | 3.42 | 0.67 |
| [(THF)3Li(C6H4−OMe)(tBu)Zn(tBu)](7) | 1.58 | 7.26, 7.14, 6.80, 6.74 | 3.35 | −0.28 |
| [(PMDETA)Li(C6H4−OMe)(tBu)Zn(tBu)](8) | 1.66 | 7.98, 7.13, 7.03, 6.21 | 3.80 | 0.09 |
| [(THF)Li(TMP)(Me)Zn(Me)](9) | −0.31 | 1.51 | ||
| Me2Zn | −0.52 | |||
| tBu2Zn | 1.02 | |||
| anisole | 7.12, 6.84, 6.81 | 3.31 |
Scheme 2. Co-complexation Reactions to Synthesize Compounds 4 and 7.

NMR characterization of this oil confirmed the formation of the mixed-metal complex. Its 1H NMR spectrum in deuterated benzene solution revealed a singlet at −0.23 ppm corresponding to the methyl groups, which is less upfield than that of pure (not co-complexed) dimethylzinc in the same solvent (−0.52 ppm, Table 1). This slight but significant difference in the chemical shifts indicates that, although part of a lithium−zinc compound, the methyl groups retain much of their original “zinc-character”.22 In addition the 7Li NMR spectrum of 4 showed a sharp resonance at 0.72 ppm which differs substantially to that found for the homometallic lithium compound 3. Remarkably the chemical shifts of the aromatic signals in the 1H and 13C NMR spectra of 4 show little variation in comparison with those found for 3, where the major difference found is for the metalated ortho-carbon in the 13C NMR spectrum which appears slightly more shielded in 4 (154.8 ppm) than in 3 (159.2 ppm).
Attempts to grow crystals of 4 from neat THF or toluene solution were unsuccessful due to the high solubility of 4 in these solvents, even at low temperatures. When a less polar solvent such as hexane was employed, a white solid precipitated which could be recrystallized by addition of toluene and was found to be the tetraorgano-dilithium-zincate [(THF)2Li2Zn(C6H4−OMe)4] (5). This unexpected product formed as result of a disproportionation process (vide infra). In a deliberate attempt to afford a crystalline derivative of 4 the reaction was repeated employing a stoichiometric amount of the chelating diamine TMEDA as mimic for two THF ligands. Thus, using the same approach as for 3, anisole was first ortho-lithiated with an equimolar mixture of tBuLi/TMEDA in hexane solution at 0 °C and then Me2Zn was introduced. The TMEDA-solvated zincate [(TMEDA)Li(C6H4−OMe)MeZnMe] (6) was obtained as colorless crystals (isolated yield, 62%). 1H, 13C, and 7Li NMR spectroscopy established the co-complexed nature of 6 (see Experimental Section and Table 1). This last point was also confirmed by the determination of the molecular structure of 6 by X-ray crystallography. Its molecular structure can be considered a contacted ion pair and can be viewed as a six-membered [LiOCCZnC] ring system where both metals are connected through a shared methyl and an ortho-deprotonated anisole ligand set (full details are provided in the Supporting Information).
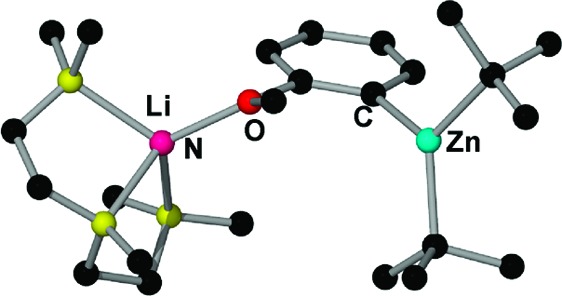
We next endeavored to synthesize a tert-butyl derivative of 4, using the same synthetic methodology. Anisole was first ortho-lithiated by tBuLi in THF at 0 °C followed by the addition of a solution of tBu2Zn in THF, which afforded [(THF)3Li(C6H4−OMe)(tBu)Zn(tBu)] (7) as a yellow oil (Scheme 2). This was characterized by 1H, 13C, and 7Li NMR spectroscopy. The 7Li NMR spectrum showed a sharp singlet at −0.28 ppm indicative of a single species. 1H NMR spectra in deuterated benzene solution revealed the presence of ortho-metalated anisole, tert-butyl, and THF ligands in a 1:2:3 ratio. The singlet for the tert-butyl groups appeared at a similar chemical shift (1.58 ppm) to that found for the related species [(THF)Li(C6H4−OMe)(TMP)Zn(tBu)] (2) (1.61 ppm) and further downfield to that for tBu2Zn in the same deuterated solvent (1.02 ppm) (Table 1). Unlike 4, the chemical shifts of the aromatic signals in the 1H and 13C NMR spectra are remarkably different from those of the lithiated anisole in compound 3, in particular for the meta-H adjacent to the metalated carbon in the 1H NMR spectrum (7.26 ppm in 7, 8.15 ppm in 3) and the ortho-carbon that has been metalated in the 13C NMR spectrum (150.38 ppm in 7, 159.2 ppm in 3). In addition, contrasting with the methyl derivative 4, surprisingly 7 contains three molecules of THF. This apparent contradiction that replacing small methyl ligands by bulkier tert-butyl ones leads to a higher degree of coligand (THF) support could be explained by 7 adopting an open structure with no Li···tBu contact as shown in Scheme 2, where lithium attains tetracoordination by bonding to three THF molecules and the oxygen of the anisole. Unfortunately attempts to grow crystals from THF solutions of 7 were unsuccessful due to the high solubility of this compound in this solvent and addition of hexane led to the disproportionation product 5 being obtained (vide infra). In the light of the successful isolation of compound 6 when the bidentate ligand TMEDA is employed, we next repeated the reaction that led to the formation of 7 in the absence of THF and using 1 molar equiv of the tridentate amine PMDETA (N,N,N′,N′′,N′′-pentamethyldiethylenetriamine) as a mimic for three THF ligands. Colorless crystals of [(PMDETA)Li(C6H4−OMe)(tBu)Zn(tBu)] (8) were obtained which were characterized in solution by NMR spectroscopy (see Experimental Section and Table 1) and in the solid state by X-ray crystallography. Its molecular structure (Figure 1) can be described as a contacted ion-pair. The anionic part contains a distorted trigonal planar zinc center (sum of the angles around Zn: 358.2°) bonded to three carbon atoms, two from the terminal tert-butyl groups, and one from the ortho-metalated anisole fragment. Contact to the cationic part of the molecule is established through the oxygen atom of the deprotonated anisole molecule which binds terminally to the lithium center, which is also bonded to the three nitrogen atoms of the PMDETA ligand, exhibiting a distorted tetrahedral geometry (average angle around Li: 108.2°). The open structure of 8 contrasts with the closed structure found for the methyl relative 6, where, in addition to the dual anisole bridge present in 8, the metals lithium and zinc are connected by one of the methyl groups. In 8 there is no contact between the lithium atom and any carbon atoms of the tert-butyl groups as shown by the elongated Li···C separation distances [Li···C5 = 5.611(11) Å; Li···C7 = 4.869(12) Å]. A similar open structural motif has been found for the lithium−aluminum product [(THF)3Li{O(=C)NiPr2(C6H4)}Al(iBu)3] of the direct alumination of N,N-diisopropylbenzamide.23 As previously found for 6, the chelating coordination of PMDETA must provide extra stabilization to this intermediate, precluding its disproportionation in nonpolar solvents such as hexane as previously observed for 7.
Figure 1.

Molecular structure of 8 with 30% probability displacement ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and bond angles (deg): Zn−C1 2.044(9), Zn−C5 1.975(8), Zn−C9 2.050(3), Li−N1 2.118(6), Li−N2 2.129(6), Li−N3 2.086(7), Li−O 1.929(4); C1−Zn−C5 124.2(5), C1−Zn−C9 117.2(3), C5−Zn−C9 116.8(3), O−Li−N1 117.8(2), O−Li−N2 123.3(2), O−Li−N3 116.2(2), N1−Li−N2 86.8(2), N1−Li−N3 119.2(2), N2−Li−N3 85.9(2).
Reactivity of [(THF)xLi(C6H4−OMe)(R)Zn(R)] (R = Me, tBu) with TMPH
According to theoretical studies, compounds 4 and 7 could be putative intermediates in the first step of the metalation of anisole by TMP-zincates [(THF)Li(TMP)(R)Zn(R)]. Thus, we next endeavored to study their reactivity toward TMP(H) and specifically whether they would yield the relevant [(THF)Li(C6H4−OMe)(TMP)Zn(R)] product. Since the reactivity of these mixed-metal reagents appears to be influenced by the coordinating ability of the solvent employed,24 these studies have been carried out in parallel using deuterated benzene or the more polar solvent THF. First the reactivity of 4 with 1 molar equiv of the amine TMP(H) in deuterated benzene was investigated and monitored by 1H and 7Li NMR spectroscopy. The 1H NMR spectrum provided evidence of the metalation of TMP(H), as shown by the inequivalence of the α-Me groups of the (deprotonated) TMP ligand (at 1.45 and 1.15 ppm).6 However, surprisingly, this spectrum also revealed the formation of free anisole (multiplets at 7.12, 6.84, 6.81 ppm) which indicates that 4 has reacted with TMP(H) in an unexpected way and, instead of forming [(THF)Li(TMP)(C6H4−OMe)Zn(Me)] and liberating methane, the TMP-zincate [(THF)Li(TMP)(Me)Zn(Me)] (9) and anisole were obtained (Scheme 3).25 Moreover, this reaction showed no dependence on the identity (coordination ability) of the solvent, thus the experiment repeated in the much more polar solvent THF gave the same results.
Scheme 3. Reactivity of Compound 4 with TMP(H).
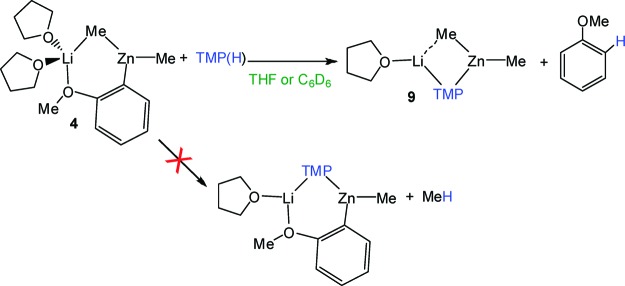
These results fill an important gap in our understanding of the reactivity of TMP−dialkyl zincates [(THF)Li(TMP)(R)Zn(R)]. In theory, if they behaved as amido bases it might be expected that the R groups would play a secondary spectating role in the basicity of these heteroleptic compounds, and that both zincates 1 (R = tBu) and 9 (R = Me) would exhibit similar reactivities. However, we have now established that whereas 1 ortho-deprotonates anisole smoothly at room temperature in hexane to yield 2 in an almost quantitative yield, its methyl congener 9 failed to behave likewise even under harsher refluxing conditions or with longer reactions times (up to 72 h).26 These differences in reactivity and the isolation of 2 as the product of direct zincation of anisole advocate for an alkyl basicity preference. Nevertheless the results from the reaction of 4 with TMP(H) provide a new answer to this apparent lack of reactivity of TMP−dimethyl zincate 9. If 9 behaved as an amido base in the reaction with anisole, it would first generate compound 4 as the metalated intermediate and TMP(H) as a coproduct, then the reverse reaction could take place to regenerate the starting materials and therefore no net reaction would be observed.27
Turning to the reactivity of the tert-butyl variant 7, we first explored its reaction with TMP(H) in deuterated benzene solution. The 1H NMR spectrum of the reaction solution revealed that the amine has been metalated (inequivalent α-Me resonances at 1.21 and 1.42 ppm, Figure 2). In this case, in contrast to the reactivity observed for 4, anisole remains metalated as shown by the presence of four multiplets at 7.86, 7.18, 7.09, and 6.57 ppm, which are modestly more deshielded than those found for starting material 7. In addition, a distinct doublet at 0.87 ppm and a multiplet at 1.56 ppm are observed (Figure 2) which confirmed the formation of isobutane (tBuH). These data establish that one of the tert-butyl groups of compound 7 has reacted with TMP(H) to afford the heteroleptic zincate 2 and isobutane as coproduct (Scheme 4). Since 2 is the product obtained from the direct zincation of anisole by 1, these results constitute the first experimental evidence of the possibility of a two-step mechanism suggested by previous theoretical studies where zincate 1 acts first as an amido base metalating the aromatic substrate (giving 7) and liberating TMP(H) which then can react in a second stage to afford a metalated product [(THF)Li(areneide)(TMP)Zn(tBu)] and concomitant gaseous isobutane (the loss of which means that the reaction cannot go backwards), showing an overall alkyl-basicity when kinetically it has been the amido group TMP which has selectively removed the hydrogen from the aromatic ring. This would be in agreement with the lack of basicity observed for trialkyl and tetraalkyl zincates, which is generally rationalized in terms of the low kinetic reactivity of the Zn−C bonds.28 Thus, whereas alkyl-amido 1 ortho-deprotonates anisole almost instantaneously at room temperature in hexane, we found that all-alkyl LiZntBu329 is inert toward anisole even under forcing reflux conditions. On the other hand, the formation of 2 and isobutane as the products of the reaction of 7 with TMP(H) illustrates again the important contributing role that the alkyl groups play in the reactivities of TMP−dialkyl zincates [(THF)Li(TMP)(R)Zn(R)]. The increase in the basicity of the R group in 7 (tBu) compared to 4 (Me) allows TMP(H) to be deprotonated by the alkyl group and not by the ortho-metalated anisole group.
Figure 2.
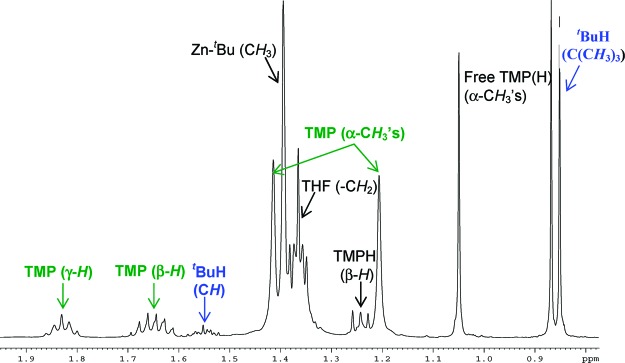
Aliphatic region of the 1H NMR spectrum of [(THF)3Li(C6H4−OMe)(tBu)Zn(tBu)] (7) in deuterated benzene solution on the addition of 1 equiv of TMP(H) after 2 h.
Scheme 4. Reactivity of Compound 7 with TMP(H).
In order to determine the influence of the donor quality of the solvent in these experiments, we repeated the reaction of 7 with TMP(H) using this time deuterated THF as solvent. The 1H NMR spectrum of 7 in deuterated THF is more complicated than in deuterated benzene: while a singlet at 0.94 is observed for the tert-butyl ligands, in the aromatic region two sets of ortho-metalated anisole fragments are observed, one of them with much broader resonances (at 7.50, 6.85, 6.71, and 6.59 ppm) than the other (at 7.31, 6.68, 6.53, and 6.43 ppm) (Figure 3a). A probable explanation is that in a donor solvent such as THF there is an equilibrium in solution between the contacted ion-pair structure (shown in Scheme 4) and solvent-separated [{Li(THF)x+{Zn(C6H4−OMe)tBu2}−].30 When 1 molar equiv of TMP(H) was introduced into the NMR tube and the reaction with 7 was monitored by 1H and 7Li NMR spectroscopy, metalation of the amine was definitively observed (as indicated by the inequivalent α-Me resonances at 1.21 and 0.97 ppm and at 1.06 and 0.92 ppm for two different metalated TMP(H) fragments). In addition, the aromatic region of the spectrum shows the presence of two ortho-metallated anisole fragments with resonances at different chemical shifts than those observed for 7. One of these sets of signals is, as previously observed for 7, significantly broader (at 7.35, 6.65, 6.52, and 6.44 ppm) than the other (7.48, 7.07, 6.86, and 6.74 ppm). Furthermore, this spectrum also showed the formation of free anisole (at 7.21 and 6.84 ppm) (see Figure 3b).31 A comparison of this spectrum with that obtained by dissolving crystals of 2 in deuterated THF (Figure 3c) shows that one of the products of the reaction of 7 with TMP(H) is the zincate 2, as a result of the reaction between some amine and one tert-butyl ligand.32 In addition, the formation of a substantial amount of free anisole also indicates that some more of the amine must react with 7 in a different way targeting the ortho-metalated anisole anion, to generate zincate 1 and free anisole (Scheme 4). These results suggest that, if AMMZ of anisole occurs via the intermediate 7, the yields of metalated anisole must be lower when THF is employed in comparison with a nondonor solvent such as hexane or benzene since, using the former, part of the base and anisole are regenerated. To confirm this, we carried out the reaction of zincate 1 with anisole in deuterated THF as a solvent and we found that after 2 h at room temperature the conversion of anisole to 2(33) was only 62% complete (Figure 3d), whereas in hexane solution under the same reaction conditions 2 was obtained in an almost quantitative yield. Thus, in the light of the results obtained from these reactivity studies, the AMMZ of anisole by zincate 1 seems to occur through the two-step mechanism previously predicted by Uchiyama et al. from computational studies, and therefore compound 7 is the intermediate of the first step of the reaction, where the amide first acts as a base deprotonating anisole. The coproduct of this metalation is the amine TMP(H), which can react with 7 through two different routes, (i) with the tert-butyl group to yield the final metallated product 2 and isobutane or (ii) with the metallated anisole to regenerate the base 1 and free anisole. When a nonpolar solvent such as hexane is employed (i) is the only significant reaction that takes place; however, in a Lewis donor base solvent such as THF both reaction pathways are feasible, which accordingly diminishes the final yield observed for the anisole metalation.
Figure 3.

Aromatic region of the 1H NMR spectra in deuterated THF of (a) [(THF)3Li(C6H4−OMe)(tBu)Zn(tBu)] (7), (b) [(THF)3Li(C6H4−OMe)(tBu)Zn(tBu)] (7) + TMP(H), 2 h, (c) [(THF)Li(C6H4−OMe)(TMP)Zn(tBu)] (2), and (d) [(THF)Li(TMP)(tBu)Zn(tBu)] (1) + 1 equiv of anisole, 2 h.
These reactivity studies provide experimental evidence that a two-step mechanism (as predicted by Uchiyama in his theoretical studies) can be taking place for the deprotonation of anisole by the TMP−dialkyl zincate 1.
If the amide TMP is the basic group that carries out the deprotonation in the first place by removing the ortho-hydrogen of anisole to form TMP(H), then it could be possible to carry out these metalations using trialkyl zincates in the presence of catalytic amounts of the amine TMP(H).34 However, we found that no net metalation of anisole is observed when a substoichiometric (0.1 equiv) or even stoichiometric amount of TMP(H) was injected into an equimolar solution of anisole and LiZntBu329 in deuterated benzene solution. Moreover, surprisingly LiZntBu329 failed to deprotonate TMP(H) in the same solvent and the 1H NMR spectrum of the reaction mixture showed only the presence of the unreacted starting materials. Recent reports have established that trialkylzincate LiZntBu3 (previously reported as an efficient reagent in metal−halogen exchange reactions of organic halides)35 disproportionates to tBu2Zn and Li2ZntBu4,36 implying that it is the latter mixed-metal compound that is the active species in these reactions. This tetraorganozincate is an excellent chemoselective reagent for zinc−halogen exchange reactions of aromatic halides, showing an unprecedented compatibility with unprotected acidic protons such as amide N−H or phenolic O−H. It can be also used in the anionic polymerization of N-isopropylacrylamide/styrene using protic solvents such as methanol or even water.36 Therefore, it is not surprising that in our case no metalation of the bulky secondary amine TMP(H) was observed.
Disproportionation of Compounds 4 and 7
As previously alluded to, when hexane is added to a THF solution of compound 4 or 7 a white solid precipitates. This could be recrystallized to produce colorless crystals characterized by 1H and 7Li NMR spectroscopy and by X-ray crystallography as the unprecedented homoleptic tetraorganozincate 5. Its molecular structure (Figure 4) can be viewed as a contacted ion-pair zincate in which distorted tetrahedral zinc is bonded to four ortho-metalated molecules of anisole by short (strong) covalent bonds [average Zn−C distance: 2.111 Å]. These Zn−C bonds are slightly more elongated than those found in triorganozincates 2 [2.0937(16) Å]6 or 6 [2.045(2) Å], probably as a consequence of the higher coordination number of zinc in 5. Each lithium in 5 exhibits a distorted tetrahedral geometry comprising three oxygen atoms (two from two anisole molecules and one from THF) and one ortho-carbon of one of the anisole molecules. These Li−C bonds [Li1−C22 = 2.308(6) Å; Li2−C1 = 2.373(6) Å] lie in the middle of the range found for the Li−C bond distances in tetrameric ortho-lithiated anisole [{Li(C6H4−OMe)}4]21 [2.18(1)−2.51(1) Å]. In forming these bonds, each lithium lies as near perpendicular to an anisole molecule as the molecular geometry allows [Li−C−C−O torsion angles are 65.9(3)° and 62.0(3)°], giving rise to a cation−π interaction with the aromatic ring which contrasts with the σ-nature of the Zn−C bonds, where zinc lies almost coplanar with the anisole rings [corresponding Zn−C−C−O torsion angles are −15.2(4)° and −17.6(4)°]. This σ/π bonding distinction has become a signature feature for mixed-metal ‘ate compounds.2a Only two of the four metalated anisole rings (one for each lithium) engage in these π-contacts with lithium (shown with thin lines in Figure 4), as the remaining two Li···Cortho separation distances [Li1···C8 = 3.013(5) Å, Li2···C15 = 3.018(6) Å] appear too long to be indicative of any significant interaction.37 These differences in bonding of the anisole molecules to lithium have an influence on the lengths of the Zn−Canisole bonds. Thus, the molecules that are not interacting with the lithium centers through the carbon bind more strongly to zinc [Zn1−C15 = 2.075(3) Å, Zn1−C8 = 2.072(3) Å] than those which form the Li−C π-contacts [Zn1−C1 = 2.148(3) Å, Zn1−C22 = 2.150(3) Å]. In addition, these Li−C π-interactions must contribute significantly to the marked nonlinearity of the Li···Zn···Li vector [91.09(15)°] which contrasts with the tendency of other compounds with this Li···Zn···Li motif to adopt near-linear arrangements, as for example in [Li2Zn{C6H4CH2NMe2}4] [Li···Zn···Li = 177.83°]38 and [(TMEDA)2Li2Zn(C≡CPh)4] [Li···Zn···Li = 146.70°].39
Figure 4.

Molecular structure of 5 with 30% probability displacement ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (deg): Zn−C1 2.148(3), Zn−C8 2.072(3), Zn−C15 2.075(3), Zn−C22 2.150(3), Li1−O1 1.950(6), Li1−O2 1.973(5), Li1−O5 1.982(5), Li1−C22 2.308(6), Li2−O3 1.982(6), Li2−O4 1.950(5), Li2−O6 1.938(6), Li2−C1 2.373(6); Li1···Zn···Li2 91.09(15), C1−Zn−C8 111.64(10), C1−Zn−C15 103.12(11), C1−Zn−C22 113.34(10), C8−Zn−C15 112.95(11), C8−Zn−C22 103.12(10), C15−Zn−C22 113.01(10).
Turning to the possible reaction involved in its formation, 5 precipitates readily from hexane solutions of 4. On this evidence, a dismutation process must be taking place. A probable disproportionation pathway is shown in Scheme 5, which is supported by 1H NMR analysis of the filtrate, which revealed a singlet at −0.29 ppm for the methyl groups and two multiplets at 1.23 and 3.23 ppm for the coordinated THF. This chemical shift found for the methyl groups is less upfield than those found for methyl lithium (−1.42 ppm) and dimethyl zinc (−0.52 ppm) in the same deuterated benzene solution.40 In addition the 7Li NMR spectrum displayed a single resonance at 0.76 ppm.41 The presence of only one resonance for the methyl groups in the 1H spectrum must be the result of a rapid exchange process between the species present in solution.42
Scheme 5. Proposed Disproportionation Pathway of 4 for the Formation of 5.
Interestingly, this dismutation process appears to be reversible. To elaborate, when hexane is added to a solution of 4 in THF, compound 5 precipitates as a white solid. However, if at this stage the solvent is removed under vacuum and the oily solid residue (including 5) is dissolved in THF solution, compound 4 is the only species present in solution. The stability of 4 toward disproportionation was studied for other solvents and monitored by 1H and 7Li NMR spectroscopy. In deuterated THF and benzene solutions, no conversion to 5 was observed after 4 days at room temperature. However, after a week in deuterated benzene, some small crystals were formed in the NMR tube that were identified as 5, which shows that although much slower than in hexane, where 5 is formed almost instantaneously, compound 4, at least partially, also undergoes disproportionation to 5 in arene solutions. These disproportionation processes must be significantly slower than the reactions of the intermediates 4 and 7 with TMP(H) since 5 has not been detected in the metalation reaction of 1 with anisole in hexane or in the reactivity studies of 4 and 7 with TMP(H) when hexane is employed as solvent.
Conclusions
The new dialkyl(aryl) lithium zincates 4 and 7 have been prepared using a co-complexation approach by reaction of lithiated anisole 3 with the relevant dialkylzinc compound and characterized in solution by NMR spectroscopy. Compounds 4 and 7 are stable in THF solutions but in a less polar solvent such as hexane they disproportionate to yield the tetraorganozincate 5 whose structure has been determined by X-ray crystallography. The molecular structures of the TMEDA solvate of 4 and the PMDETA solvate for 7 have also been elucidated and they exhibit different structural motifs. Thus, in 6 the distinct metals are connected through the anisole ligand which binds in an ambidentate fashion (through the carbon−zinc and the oxygen−lithium contacts) and also through one of the methyl groups, forming a six-membered ring, whereas 8 displays an open structure where anisole connects the two metals (in the same mode as aforementioned) and the tert-butyl groups are solely bonded terminally to zinc.
Reactivity studies of compounds 4 and 7 with TMP(H) provide us with the first experimental evidence of the possibility of a two-step mechanism for the AMMZ of anisole using zincate 1, as previously predicted by theoretical calculations, whereby the arene is first ortho-metalated by the amide TMP to yield a putative intermediate 7 and concomitant TMP(H) which could then react in a second step giving rise to 2 and isobutane. In addition, the importance of the Lewis donor ability of the solvent employed in the reaction is also assessed showing that the use of a polar solvent such as THF diminishes the yield of 2 since in the second step of the reaction the amine TMP(H) can also react with the ortho-metalated anisole ligand in 7 to regenerate the starting materials 1 and anisole. This side reaction can be avoided when a nonpolar solvent such as hexane or benzene is employed. These reactivity studies also revealed the importance of the alkyl groups in the efficiency of dialkyl(TMP)zincates [(THF)Li(TMP)(R)Zn(R)], which far from being just mere spectators, play a crucial role for the final success of the metalation by reacting with the concomitant TMP(H) released in the first step of the reaction and therefore precluding the reversibility of the metalation. In addition, the lack of kinetic basicity of the Zn−C bonds prevents anisole for being metalated by the triorganozincate LiZntBu3 using catalytic (or even stoichiometric) amounts of the amine TMP(H).
Experimental Section
General
All reactions were performed under a protective argon atmosphere using standard Schlenk techniques. Hexane, THF, and toluene were dried by heating to reflux over sodium benzophenone ketyl and distilled under nitrogen prior to use. ZnMe2 was purchased from Aldrich Chemicals as a 2 M solution in toluene. tBu2Zn11 was prepared according to literature methods. NMR spectra were recorded on a Bruker DPX 400 MHz spectrometer, operating at 400.13 MHz for 1H, 150.32 MHz for 7Li, and 100.62 MHz for 13C{1H}.
X-ray Crystallography
Single-crystal diffraction data were recorded at 150 K on Nonius KappaCCD (5) and Oxford Diffraction Gemini A Ultra (6 and 8) diffractometers using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å).43 The structures were solved by direct methods and refined by full-matrix least-squares against F2 using SHELXTL.44 A summary of crystallographic data is given in Table 2. Two-fold disorder in some ligands was resolved satisfactorily; all non-hydrogen atoms were refined anisotropically, and H atoms were constrained with a riding model.
Table 2. Crystallographic Data.
| 5 | 6 | 8 | |
|---|---|---|---|
| formula | C36H44Li2O6Zn | C15H29LiN2OZn | C24H48LiN3OZn |
| Mr | 652.0 | 325.7 | 467.0 |
| cryst size (mm3) | 0.58 × 0.28 × 0.12 | 0.18 × 0.14 × 0.04 | 0.43 × 0.39 × 0.26 |
| cryst syst | monoclinic | orthorhombic | orthorhombic |
| space group | P21/c | P212121 | P212121 |
| a, Å | 12.030(5) | 8.3053(3) | 12.0463(3) |
| b, Å | 17.467(5) | 13.0406(3) | 14.9178(5) |
| c, Å | 17.5333(14) | 16.4544(5) | 15.5281(4) |
| β, deg | 107.689(11) | ||
| V, Å3 | 3510.2(17) | 1782.11(9) | 2790.47(14) |
| Z | 4 | 4 | 4 |
| Dcalcd, g cm−3 | 1.234 | 1.214 | 1.112 |
| μ, mm−1 | 0.74 | 1.38 | 0.90 |
| <121>max, deg | 27.5 | 29.5 | 29.4 |
| reflns measured | 26 446 | 5932 | 12 878 |
| unique reflns | 7763 | 3047 | 6292 |
| Rint (on F2) | 0.057 | 0.028 | 0.027 |
| no. of params | 484 | 189 | 456 |
| Ra [F2 > 2σ(F2)] | 0.048 | 0.029 | 0.038 |
| Rwb (all data) | 0.101 | 0.043 | 0.078 |
| GOFc (S) | 1.04 | 0.89 | 0.93 |
| max, min diff map, e Å−3 | 0.64, −0.37 | 0.31, −0.32 | 0.49, −0.39 |
R = Σ|Fo| − |Fc||/Σ|Fo|.
Rw = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2.
GOF = [Σw(Fo2 − Fc2)2/(no. of unique reflections − no. of parameters)]1/2.
Synthesis of [Li4(C6H4−OMe)4(THF)2](3)
To an oven-dried Schlenk tube were added dry THF (10 mL) and anisole (0.54 mL, 5 mmol), the solution then being cooled to 0 °C in an ice bath. tBuLi (1.7 M in pentane, 2.9 mL, 5 mmol) was then added to give a yellow solution, which was stirred at 0 °C for 25 min. The solvent was removed in vacuo to yield a yellow oil, which was dissolved in hexane (10 mL). This solution was concentrated by removing some solvent under vacuum and placed in the freezer (−20 °C). A crop of colorless crystals was deposited overnight (isolated yield 0.16 g, 22%; NMR analysis of the filtrate indicated the metalation is almost quantitative). 1H NMR (400.13 MHz, 298 K, C6D6) δ 8.18 (1H, d, Hmeta), 7.39 (1H, t, Hmeta*), 7.26 (1H, t, Hpara), 6.78 (1H, d, Hortho*), 3.40 (3H, s, −OCH3), 3.10 (2H, s, −OCH2, THF), 1.17 (2H, s, −CH2, THF). 13C {1H} NMR (100.62 MHz, 298 K, C6D6) δ 169.9 (Cispo), 159.2 (Cortho), 142.3 (Cmeta), 127.2 (Cmeta*), 121.8 (Cpara), 106.4 (Cortho*), 67.2 (−OCH3), 54.3 (−OCH2, THF), 25.2 (−CH2, THF). 7Li (298K, C6D6, reference LiCl in D2O at 0.00 ppm): δ 3.28.
Synthesis of [(THF)2Li(C6H4−OMe)MeZnMe] (4)
To an oven-dried Schlenk tube were added dry THF (10 mL) and anisole (0.54 mL, 5 mmol), the solution then being cooled to 0 °C in an ice bath. tBuLi (1.7 M in pentane, 2.9 mL, 5 mmol) was then added to give a yellow solution, which was stirred at 0 °C for 25 min. Me2Zn (2 M in toluene, 2.5 mL, 5 mmol) was added, and the solution slowly warmed to room temperature to give a very pale yellow solution, which was stirred for 15 min. The solvent was removed in vacuo to yield a yellow oil (4), which was analyzed by 1H, 7Li, and 13C NMR spectroscopy. 1H NMR (400.13 MHz, 298 K, C6D6) δ 8.15 (1H, d, Hmeta), 7.20 (1H, t, Hmeta*), 7.11 (1H, t, Hpara), 6.74 (1H, d, Hortho*), 3.40 (3H, s, OCH3), 3.24 (8H, s, OCH2, THF), 1.21 (8H, s, CH2, THF), −0.23 (6H, s, Zn(CH3)2). 13C{1H} NMR (100.62 MHz, 298 K, C6D6) δ 165.9 (Cispo), 154.8 (Zn−Cortho), 142.8 (Cmeta), 126.7 (Cmeta*), 122.8 (Cpara), 110.0 (Cortho*), 67.8 (OCH2, THF), 56.0 (OCH3), 24.8 (CH2, THF), −8.60 (Zn(CH3)2). 7Li NMR (298 K, C6D6, reference LiCl in D2O at 0.00 ppm): δ 0.72.
Synthesis of [(THF)2Li2Zn(C6H4−OMe)4] (5)
To an oven-dried Schlenk tube were added dry THF (10 mL) and anisole (0.54 mL, 5 mmol), the solution then being cooled to 0 °C in an ice bath. tBuLi (1.7 M in pentane, 2.9 mL, 5 mmol) was then added to give a yellow solution, which was stirred at 0 °C for 25 min. Me2Zn (2 M in toluene, 5 mL, 5 mmol) was added, and the solution slowly warmed to room temperature to give a colorless solution, which was stirred for 15 min. The solvent was removed in vacuo, to give a yellow oil (4). Hexane (5 mL) was then introduced, to give an oily white solid. The addition of toluene (5 mL) gave a white precipitate, and gently heating for a few minutes resulted in the formation of an yellow solution. The Schlenk tube was then placed in a Dewar of hot water, and after standing overnight yielded a white needle-like crystalline solid (5). Typical yield = 0.28 g (9%) (Maximum possible yield = 25%). 1H NMR (400.13 MHz, 298 K, C6D6) δ 8.35 (3H, d, Hmeta), 7.20 (3H, t, Hmeta*), 7.10 (3H, t, Hpara), 6.73 (3H, d, Hortho*), 3.24 (3H, s, OCH3), 3.16 (12H, t, OCH2, THF), 1.23 (11H, m, CH2, THF). 7Li NMR (298 K, C6D6, reference LiCl in D2O at 0.00 ppm): δ 0.96.
Synthesis of [(TMEDA)Li(C6H4−OMe)MeZnMe] (6)
To an oven-dried Schlenk tube were added dry hexane (10 mL), TMEDA (0.75 mL, 5 mmol), and anisole (0.54 mL, 5 mmol), the solution then being cooled to 0 °C in an ice bath. tBuLi (1.7 M in pentane, 2.9 mL, 5 mmol) was then added to give a pale yellow solution, which was stirred at 0 °C for 25 min. Then Me2Zn (2 M in toluene, 2.5 mL, 5 mmol) was added to afford a white precipitate. Toluene was introduced at this stage (2 mL), and the mixture was gently heated until all the solid had dissolved, affording a pale yellow solution. Allowing this solution to cool slowly to room temperature afforded a crop of colorless crystals of 4 (1.03 g, 62%) 1H NMR (400.13 MHz, 298 K, C6D6) δ 8.27 (1H, d, Hmeta), 7.22 (2H, m, Hmeta* and Hpara), 6.77 (1H, d, Hortho*), 3.42 (3H, s, OCH3), 1.65 (12H, s, CH3, TMEDA), 1.62 (2H, s, CH2, TMEDA), −0.14(6H, s, Zn(CH3)2). 13C{1H} NMR (100.62 MHz, 298 K, C6D6) δ 166.0 (Cispo), 155.1 (Zn-Cortho), 142.9 (Cmeta), 127.2 (Cmeta*), 124.1 (Cpara), 113.9 (Cortho*), 59.5 (OCH3), 56.0 (CH2, TMEDA), 45.0 (CH3, TMEDA), −9.2 (Zn(CH3)2). 7Li NMR (298 K, C6D6, reference LiCl in D2O at 0.00 ppm): δ 0.67.
Synthesis of [(THF)3Li(C6H4−OMe)tBuZntBu] (7)
To an oven-dried Schlenk tube were added dry THF (4 mL) and anisole (0.22 mL, 2 mmol), the solution then being cooled to 0 °C in an ice bath. tBuLi (1.7 M in pentane, 1.2 mL, 2 mmol) was then added to give a yellow solution, which was stirred at 0 °C for 50 min. A solution of tBu2Zn in THF (2 mmol) was added via canula, and the solution was slowly warmed to room temperature and allowed to stir for 30 min. The solution was then placed in the freezer overnight. The solvent was removed in vacuo to yield a yellow oil (7), which was analyzed by 1H, 7Li, and 13C NMR spectroscopy. 1H NMR (400.13 MHz, 298 K, C6D6) δ 7.26 (1H, d, Hmeta), 7.16 (1H, t, Hmeta*), 6.80 (1H, t, Hpara), 6.74 (1H, d, Hortho*), 3.43 (3H, s, OCH3), 3.12 (12H, t, OCH2, THF), 1.58 (18H, s, CH3, tBu), 1.39 (12H, m, CH2, THF). 13C{1H} NMR (100.62 MHz, 298 K, C6D6)δ 167.0 (Cipso), 147.8 (Zn−Cortho), 140.9 (Cmeta), 129.7 (Cmeta*), 122.5 (Cpara), 110.6 (Cortho*), 68.3 (OCH2, THF), 56.3 (OCH3), 36.6 (CH3, tBu), 25.4 (CH2, THF), 24.2 (Zn−C, tBu). 7Li NMR (298 K, C6D6, reference LiCl in D2O at 0.00 ppm): δ −0.28.
Synthesis of [(PMDETA)Li(C6H4−OMe)tBuZntBu] (8)
To an oven-dried Schlenk tube were added dry hexane (10 mL), PMDETA (0.42 mL, 2mmol), and anisole (0.54 mL, 5 mmol), the solution then being cooled to 0 °C in an ice bath. tBuLi (1.7 M in pentane, 2.9 mL, 5 mmol) was then added to give a pale yellow solution, which was stirred at 0 °C for 25 min. A solution of tBu2Zn in hexane (0.36 g, 2mmol) was added, to afford two layers, a yellow oil, and a colorless solution. The reaction mixture was warmed slowly to room temperature and stirred for 1 h at room temperature. The solvent was then removed in vacuo, and toluene (3 mL) was added. Gentle heating afforded an orange solution, which was left in a Dewar filled with hot water overnight, resulting in the formation of small crystals (0.29 g, 31%). NMR analysis of the filtrate showed the formation of 8 is almost quantitative and the low yield of the isolated crystals is probably due to the high solubility of 8 in toluene solution. 1H NMR (400.13 MHz, 298 K, C6D6) δ 8.18 (1H, d, Hmeta), 7.22 (1H, t, Hmeta*), 7.08 (1H, t, Hpara), 6.23 (1H, d, Hortho*), 3.85 (3H, s, OCH3), 1.87 (7H, bs, CH3 and CH2, PMDETA), 1.83 (4H, m, CH2, PMDETA), 1.76 (12H, s, CH3, PMDETA), 1.66 (18H, s, CH3, tBu). 13C{1H} NMR (100.62 MHz, 298 K, C6D6) δ 163.4 (Cipso), 158.3 (Zn−Cortho), 141.2 (Cmeta), 123.9 (Cmeta*), 122.8 (Cpara), 112.5 (Cortho*), 56.4 (OCH3), 56.8 (CH2, PMDETA), 53.1 (CH2, PMDETA), 45.0 (CH3, PMDETA), 44.3 (CH3, PMDETA), 36.2 (CH3, tBu), 22.7 (Zn−C, tBu). 7Li NMR (298 K, C6D6, reference LiCl in D2O at 0.00 ppm): δ 0.25.
Synthesis of [(THF)Li(TMP)(Me)Zn(Me)] (9)
Me2Zn (2 M in toluene, 2 mL, 4 mmol) was added to a solution of LiTMP [prepared in situ by reaction of BuLi (2.50 mL of a 1.6 solution in hexane, 4 mmol) and TMP(H) (0.68 mL, 4 mmol)] to afford a white precipitate. After stirring for a further 15 min, THF (0.33 mL, 4 mmol) was added to give a yellow solution. This solution was concentrated by removing some solvent under vacuum and placed in the freezer (−20 °C). A crop of colorless crystals was deposited overnight (0.12 g, 10%). NMR analysis of the filtrate showed the formation of 9 is almost quantitative and the low yield of the isolated crystals is probably due to the high solubility of 9 in hexane solution at room temperature. 1H NMR (400.13 MHz, 298 K, C6D6) δ 3.26 (4H, t, −OCH2, THF), 1.91 (2H, bs, Hγ, TMP), 1.66 (4H, bs, Hβ, TMP), 1.42 (7H, bs, α-CH3, TMP), 1.21 (5H, m, −CH2, THF), 1.08 (6H, bs, α-CH3, TMP), −0.36 (6H, s, −Zn(CH3)2). 13C{1H} NMR (100.62 MHz, 298 K, C6D6) δ 68.78 (−OCH2, THF), 55.12 (Cα, TMP), 41.44 (Cβ, TMP), 36.25, 31.12 (CH3, TMP), 25.27 (−CH2, THF), 20.01 (Cγ, TMP), −6.88 (−Zn(CH3)2). 7Li (298 K, C6D6, reference LiCl in D2O at 0.00 ppm): δ 1.53.
Acknowledgments
We thank the Royal Society (University Research Fellowship to E.H.), the Faculty of Science, University of Strathclyde (starter grant to E.H.), and EPSRC (equipment grant to W.C.) for their generous sponsorship of this research.
Supporting Information Available
CIF files giving crystal data, detailed discussion on the molecular structure of 6, and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Kondo Y.; Shilai M.; Uchiyama M.; Sakamoto T. J. Am. Chem. Soc. 1999, 121, 3539. [Google Scholar]
- a Mulvey R. E. Organometallics 2006, 25, 1060. [Google Scholar]; b Mulvey R. E.; Mongin F.; Uchiyama M.; Kondo Y. Angew. Chem., Int. Ed. 2007, 46, 3802. [DOI] [PubMed] [Google Scholar]
- Clayden J.Organolithiums: Selectivity for Synthesis; Pergamon, Elsevier Science Ltd.: Oxford,2002. [Google Scholar]
- a Uchiyama M.; Matsumoto Y.; Nobuto D.; Furuyama T.; Yamaguchi K.; Morokuma K. J. Am. Chem. Soc. 2006, 128, 8748. [DOI] [PubMed] [Google Scholar]; b Clegg W.; Dale S. H.; Hevia E.; Honeyman G. W.; Mulvey R. E. Angew. Chem., Int. Ed. 2006, 45, 2370. [DOI] [PubMed] [Google Scholar]
- Clegg W.; Dale S. H.; Harrington R. W.; Hevia E.; Honeyman G. W.; Mulvey R. E. Angew. Chem., Int. Ed. 2006, 45, 2374. [DOI] [PubMed] [Google Scholar]
- Clegg W.; Dale S. H.; Drummond A. M.; Hevia E.; Honeyman G. W.; Mulvey R. E. J. Am. Chem. Soc. 2006, 128, 7434. [DOI] [PubMed] [Google Scholar]
- Garcia F.; McPartlin M.; Morey J. V.; Nobuto D.; Kondo Y.; Naka H.; Uchiyama M.; Wheatley A. E. H. Eur. J. Org. Chem. 2008, 644. [Google Scholar]
- Knochel P., Jones P. In Organozinc Reagents: A Practical Approach; Harwood L. H., Moody C. J., Ed.; Oxford University Press: Oxford, 1999. [Google Scholar]
- Erdik E. In Organozinc Reagents in Organic Synthesis; CRC Press: New York, 1996. [Google Scholar]
- Negishi E. I.Metal-Catalyzed Cross-Coupling Reactions; Diederich F., Stang P. J., Eds.; Wiley-VCH: New York, 1998, Chapter 1. [Google Scholar]
- Andrikopoulos P. C.; Armstrong D. R.; Barley H. R. L.; Clegg W.; Dale S. H.; Hevia E.; Honeyman G. W.; Kennedy A. R.; Mulvey R. E. J. Am. Chem. Soc. 2005, 127, 6184. [DOI] [PubMed] [Google Scholar]
- Armstrong D. R.; Clegg W.; Dale S. H.; Graham D. V.; Hevia E.; Hogg L. M.; Honeyman G. W.; Kennedy A. R.; Mulvey R. E. Chem. Commun. 2007, 598. [DOI] [PubMed] [Google Scholar]
- Clegg W.; Dale S. H.; Hevia E.; Hogg L. M.; Honeyman G. W.; Mulvey R. E.; O’Hara C. T. Angew. Chem., Int. Ed. 2006, 45, 6548. [DOI] [PubMed] [Google Scholar]
- Uchiyama M.; Matsumoto Y.; Usui S.; Hashimoto Y.; Morokuma K. Angew. Chem., Int. Ed. 2007, 46, 926. [DOI] [PubMed] [Google Scholar]
- Kondo Y.; Morey J. V.; Morgan J. C.; Naka H.; Nobuto D.; Raithby P. R.; Uchiyama M.; Wheatley A. E. H. J. Am. Chem. Soc. 2007, 129, 13360. [DOI] [PubMed] [Google Scholar]
- Nobuto D.; Uchiyama M. J. Org. Chem. 2008, 73, 1117. [DOI] [PubMed] [Google Scholar]
- a Gilman H.; Bebb R. L. J. Am. Chem. Soc. 1939, 61, 109. [Google Scholar]; b Wittig G.; Fuhrman G. Chem. Ber. 1940, 73, 1197. [Google Scholar]
- Bauer W.; Schleyer P. v. R. J. Am. Chem. Soc. 1989, 111, 7191. [Google Scholar]
- Saà J. M.; Deyà P. M.; Suner G. A.; Frontera A. J. Am. Chem. Soc. 1992, 114, 9093. [Google Scholar]
- Stratakis M. J. Org. Chem. 1997, 62, 3024. [DOI] [PubMed] [Google Scholar]
- Harder S.; Boersma J.; Brandsma L.; Kanters J. A. J. Organomet. Chem. 1988, 339, 7. [Google Scholar]
- This trend has been previously found for other alkyl zincates, see for example:; Barley H. R. L.; Clegg W.; Dale S. H.; Hevia E.; Honeyman G. W.; Kennedy A. R.; Mulvey R. E. Angew. Chem., Int. Ed. 2005, 44, 6018. [DOI] [PubMed] [Google Scholar]
- Conway B.; Hevia E.; Garcia-Alvarez J.; Graham D. V.; Kennedy A. R.; Mulvey R. E. Chem. Commun. 2007, 5241. [DOI] [PubMed] [Google Scholar]
- See for example:; Seggio A.; Lannou M. I.; Chevallier F.; Nobuto D.; Uchiyama M.; Golhen S.; Roisnel T.; Mongin F. Chem. Eur. J. 2007, 13, 9982. [DOI] [PubMed] [Google Scholar]
- The formation of 9 was confirmed by synthesising this compound using an alternative approach by the reaction of LiTMP, Me2Zn, and THF in hexane (see Experimental Section)
- For previous work where the importance of the alkyl groups in TMP-zincates is discussed in the context of the generation of functionalised benzynes see; a Uchiyama M.; Miyoshi T.; Kajihara Y.; Sakamoto T.; Otani Y.; Ohwada T.; Kondo Y. J. Am. Chem. Soc. 2002, 124, 8514. [DOI] [PubMed] [Google Scholar]; b Uchiyama M.; Kobayashi Y.; Furuyama T.; Nakamura S.; Kajihara Y.; Miyoshi T.; Sakamoto T.; Kondo Y.; Morokuma K. J. Am. Chem. Soc. 2008, 130, 472. [DOI] [PubMed] [Google Scholar]
- The same reactivity pattern was observed when TMEDA-solvated compound 6 was reacted with TMP(H) in deuterated benzene and in THF
- Uchiyama M.; Furuyama T.; Kobayashi M.; Matsumodo Y.; Tanaka K. J. Am. Chem. Soc. 2006, 128, 8404. [DOI] [PubMed] [Google Scholar]
- This compound was prepared in situ by combination of 1 molar equiv of tBuLi and one of tBu2Zn in THF. Recently Uchiyama has reported that in this solvent “LiZntBu3” disproportionates to ZntBu2 and Li2ZntBu4, see; Furuyama T.; Yonehara M.; Arimoto S.; Kobayashi M.; Matsumoto Y.; Uchiyama M. Chem. Eur. J. 2008, 11, 10348. [DOI] [PubMed] [Google Scholar]
- For both structures the resonances of the tert-butyl groups would be at similar chemical shifts since in each case these are terminal Zn−tBu bonds, whereas for the resonances of the ortho-metalated anisole a marked change in the chemical shifts could be expected since in the solvent-separated structure it bonds only to zinc whereas in the contacted ion-pair it also interacts with the polar lithium center.
- Note that a small amount of anisole is always found to be present in deuterated THF solutions of 7. This is probably due to unavoidable partial hydrolysis when preparing the sample. However, it must be noted that this small amount of anisole remains constant, even after 48 h. On the other hand, when TMP(H) is introduced to this solution of 7 in deuterated THF, the amount of anisole increases substantially (Figure 3b) which is consistent with the formation of anisole and zincate 1 as depicted in Scheme 4.
- In contrast to the reaction of 7 with TMP(H) in deuterated benzene solution, isobutane formation could not be detected in this reaction, probably due to the lower solubility of this nonpolar gas in the more polar solvent THF.
- These results contrast with a partial 13C NMR spectrum of the product of the reaction of LiZn(TMP)tBu2 with anisole in THF reported by Uchiyama (ref (4a)) where the authors show that the same 13C NMR spectrum was obtained when anisole was first treated with tBuLi and then tBu2Zn. In our hands we find that the product of the reaction of 1 with anisole in THF is the zincate 2 for which the 1H NMR spectrum clearly shows the presence of metallated TMP, see Supporting Information.
- For examples of the use of dialkylzinc reagents (ZnR2) as bases in the presence of catalytic amounts of amines see:; Hlavinka M. L.; Greco J. F.; Hagadorn J. R. Chem. Commun. 2005, 5304. [DOI] [PubMed] [Google Scholar]
- a Kondo Y.; Takazawa N.; Yamazaki C.; Sakamoto T. J. Org. Chem. 1994, 59, 4717. [Google Scholar]; b Kondo Y.; Fujinami M.; Uchiyama M.; Sakamoto T. J. Chem. Soc., Perkin Trans. 1 1997, 799. [Google Scholar]
- See reference in ref (29).
- 1H NMR spectrum of 5 in deuterated benzene solution exhibit a single type of anisole molecule which suggests that in solution the Li−C π-interactions must be negligible or alternatively and most likely that a dynamic process is taking place where the anisole molecules are rapidly exchanging.
- Rjinberg E.; Jastrzebski J. T. B. H.; Boersma J.; Koojiman H.; Veldman N.; Spek A. L.; van Koten G. Organometallics 1997, 16, 2239. [Google Scholar]
- Edwards A. J.; Fallaize A.; Raithby P. R.; Rennie M. A.; Steiner A.; Verhorevoort K. L.; Wright D. S. Dalton Trans. 1996, 133. [Google Scholar]
- This chemical shift (−0.29 ppm) can be also compared to those found for LiZnMe3 (−0.41 ppm) and Li2ZnMe4 (−0.45 ppm) in the same deuterated solvent. In addition, the 1H NMR spectrum of the crude product of the reaction of 2 equiv of MeLi with 3 equiv of Me2Zn in THF showed a single resonance for the methyl groups at −0.38 ppm.
- Compound 5 was also observed when 7 was dissolved in hexane; however, NMR analysis from the filtrates was not as informative as that obtained from the dismutation of compound 4 and showed also some degradation compounds. These solutions deposited a black solid after 24 h at room temperature. This is probably due to the poorer stability of the tert-butyl derivative compounds (in comparison with their methyl analogues) that must form from the disproportionation of 7.
- This has been previously observed in related compounds such as Li2ZnMe4, see:; Mobley T. A.; Berger S. Angew. Chem., Int. Ed. 1999, 38, 3070. [PubMed] [Google Scholar]
- COLLECT: Nonius BV, Delft, The Netherlands, 2000; CrysAlisPro: Oxford Diffraction Ltd., Abingdon, UK, 2008.
- Sheldrick G. M. Acta Crystallogr., Sect. A 2008, 64, 112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.