

Abstract
Background/Aims
IGF signaling has a relevant role in a variety of human malignancies. We analyzed the underlying molecular mechanisms of IGF signaling activation in early hepatocellular carcinoma (HCC; BCLC class 0 or A) and assessed novel targeted therapies blocking this pathway
Methods
An integrative molecular dissection of the axis was conducted in a cohort of 104 HCCs analyzing gene and miRNA expression, structural aberrations and protein activation. The therapeutic potential of a selective IGF-1R inhibitor, the monoclonal antibody A12, was assessed in vitro and in a xenograft model of HCC
Results
Activation of the IGF axis was observed in 21% of early HCCs. Several molecular aberrations were identified, such as overexpression of IGF2 –resulting from reactivation of fetal promoters P3 and P4-, IGFBP3 downregulation and allelic losses of IGF2R 25% of cases). A gene signature defining IGF-1R activation was developed. Overall, activation of IGF signaling in HCC was significantly associated with mTOR signaling (p=0.035) and was clearly enriched in the Proliferation subclass of the molecular classification of HCC (p=0.001). We also found an inverse correlation between IGF activation and miR-100/miR-216 levels (FDR<0.05). In vitro studies showed that A12-induced abrogation of IGF-1R activation and downstream signaling significantly decreased cell viability and proliferation. In vivoA12 delayed tumor growth and prolonged survival, reducing proliferation rates and inducing apoptosis
Conclusions
Integrative genomic analysis showed enrichment of activation of IGF signaling in the Proliferation subclass of HCC. Effective blockage of IGF signaling with A12 provides the rationale for testing this therapy in clinical trials.
Keywords: hepatocellular carcinoma, IGF signaling, molecular therapy, A12, miR-100
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common neoplasms in the world, representing the third cause of cancer-related mortality(1). Only 30%-40% of patients can benefit from curative treatments. The encouraging results derived from the SHARP phase III clinical trial with the multi-kinase inhibitor sorafenib has opened a new era in pre-clinical and clinical research in molecular targeted therapies in HCC(2). Growing understanding of the molecular mechanisms underlying HCC has evidenced the heterogenicity of HCC which involves chromosomal aberrations, gene mutations, epigenetic alterations and activation of complex signaling pathways(3).
The insulin-like growth factor (IGF) signaling system is an essential regulator for growth and development. Data derived from epidemiological studies and experimental models have unmasked the implication of the IGF system as an important player in the pathogenesis of several cancers(4). In response to the stimulatory ligands IGF-1 and IGF-2, activation of IGF-1R results in both proliferative and anti-apoptotic signals(5). Moreover, IGF signaling appears to confer resistance to anti-tumoral therapies(6).
Several observations support the role for the IGF system in HCC(7–12). Increased levels of the IGF-2 ligand have been reported in human HCC(7; 8), allelic losses and inactivating mutations of M6P/IGF2R9; 10and reduced expression of IGFBP-3(11). Unlike other human neoplasms, overexpression and activation of IGF-1R has not been properly assessed in HCC, although the hepatitis B viral X protein has been implicated in inducing IGF1R gene expression(12).
A12 (ImClone Systems Inc, NY) is a fully human monoclonal IgG1 antibody that binds IGF-1R with high specificity and affinity, promoting its internalization and degradation, thus preventing ligand-dependent activation of the pathway(13). Pre-clinical studies 5 showed that A12 has anti-tumoral activity against a wide range of malignancies(14). Several phase II clinical trials are currently ongoing in patients with advanced solid tumors. Preliminary data suggest evidence of clinical activity and good tolerance(15).
In the present study we investigated in a single comprehensive approach the main genomic alterations involved in the activation of IGF signaling pathway in a large cohort of human HCCs, integrating the results in our molecular classification of HCC(16). In addition, we provide pre-clinical evidence of non-toxic anti-tumoral efficacy of A12, a monoclonal antibody against IGF-1R.
MATERIALS AND METHODS
Human tissue samples
Human samples were collected upon Institutional Review Board approval and patient written informed consent. A total of 104 HCV-related HCC human samples from patients treated with liver resection or transplantation and 10 normal liver samples obtained from the healthy liver of patients undergoing resection for hepatic hemangioma (3), focal nodular hyperplasia (3), adenoma/cystadenoma (2), neuroendocrine tumor (1), and living donor liver transplantation (1) were collected in 3 centers of the HCC Genomic Consortium [Hospital Clínic-Barcelona, Mount Sinai-New York and National Cancer Institute-Milan]. For clinical correlations, we selected those patients who underwent liver resection (n=82). Overall, more than 80% of the tumors are clinically early HCC (Barcelona Clinic Liver Cancer-BCLC class 0 or A)(1). For the purpose of subgroup analysis, we divided these clinically early tumors, into 2 groups: histologically-early (single, well-moderately differentiated, and absence of vascular invasion), as opposed to histologically-advanced HCC (multinodular or >3cm with vascular invasion or poorly differentiated). See Supplementary Table 1
Reagents and Cell lines
Liver cancer cell lines Huh7, HepG2, Hep3B and BCLC-9(17) were cultured in DMEM (Gibco, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS). The monoclonal antibody A12 was provided by ImClone Systems, Inc (New York, NY)
RT-PCR and Quantitative Real-Time PCR
RNA extraction and quantitative Real-Time PCR was done as previously described(18). miR-100 was isolated with mirVana™ miRNA Isolation Kit (Ambion, Austin, TX) and converted to cDNA using a TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Levels of mRNA and miR-100 were measured with TaqMan® Gene Expression Assays. Ribosomal RNA (18S) and RNU6B were chosen for mRNA and miRNA normalization, respectively. TaqMan® probes are listed inSupplementary Table 2. Specific IGF2 transcripts derived from adult P1 promoter and fetal P3, P4 promoters were amplified following a modified protocol from a previous report(19) (Supplementary Table 3)
SNP array, oligonucleotide microarray and expression analysis
Copy number alterations at 238,000 loci were measured with the StyI chip of the 500K Human Mapping Array set (Affymetrix, Santa Clara, CA) as previously described(16). Gene expression was measured with Affymetrix U133Plus 2.0 arrays as previously described(16). Gene set enrichment analysis (GSEA)(20) was used to identify gene signatures significantly associated to the expression pattern found in p-IGF-1R positive samples. The association between expression levels of 358 microRNAs and activation of IGF-1R was assessed in 89 human HCCs. miRNA expression levels were assessed using a bead-based flow cytometry method, as previously reported(21)
Cell viability, cell proliferation, cell death assays and miRNA transfection
Cells were starved for 24 hours (DMEM + 0.5% FBS) and cultured with increasing concentrations of A12 [0.1–200 nM] or control human IgG for 24–72 hours. Cell viability was measured using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI). For proliferation assays, cells were incubated for 6 additional hours with 0.25 µCi of [3H]thymidine and DNA was recovered in 0.25M NaOH/0.25% SDS and 1N HCl. Thymidine incorporation was measured in a scintillation counter. Cell death was evaluated by determining the extent of hypodiploidy (G0–G1 population) through propidium iodide staining on a FACSCalibur flow cytometer (Becton Dickinson Instrument, San Jose, CA). Experiments were done in physiological concentrations of recombinant human IGF-1 (rh-IGF-1) [50 ng/ml] (PeproTech, Rocky Hill, NJ) to induce specifically IGF-1R activation(13;22), IGF-2 was not used since it can co-activate insulin receptor(4). For miRNA functional assays, an anti-miR-100 and a FAM-labeled negative control (Ambion) were transfected using the TransIT-siQuest transfection reagent (Supplementary Information). Each assay condition was measured in triplicates and results were averaged from at least 3 independent experiments. Mycoplasma was screened using the EZ-PCR Mycoplasma Test Kit (Biological industriesIsrael)
Western blotting
Serum starved cells were treated with A12 [50 nM] for 2 hours in serum-free medium and stimulated with rh-IGF-1 [50 ng/ml] for 15 minutes. Protein extracts were collected in lysis buffer (Supplementary Information). Equal amounts of total protein were resolved in polyacrylamide gels and transferred to nitrocellulose membranes (Pierce, Rockford, IL). Membranes were BSA-blocked and hybridized overnight at 4°C with primary antibodies (Supplementary Table 4) and later at room temperature with HRPconjugated secondary antibodies. Signals were visualized with ECL and SuperSignal® West Pico Substrates (Pierce Biotecnology, Rockford, IL) using a LAS-3000 imaging system (Fujifilm, Tokyo, Japan)
Human HCC xenograft model
All experimental procedures were performed after institutional ethical committee approval. 5×106 Huh7 cells were subcutaneously injected in Balb/C nu/nu athymic mice. Tumor development was monitored three times per week using a hand caliper (tumor volume= 0.5 × (width)2 × length). Once tumor volumes reached 100–200 mm3, mice were randomized in two treatment groups; control (PBS, n=16) or A12 (40 mg/kg, n=22). A12 and PBS were injected intraperitoneally three times per week until tumors reached 10% body weight (~2000 mm3), when mice were sacrificed according to Institutional ethical guidelines. We define survival as the time comprised between randomization and sacrifice. Collected tumors were fixed and apoptosis and proliferation were evaluated using the DeadEnd™ Colorimetric TUNEL System (Promega, Wisconsin, WI) and Ki-67 immunostaining (DakoCytomation, Glostrup, Denmark). We assessed glycemia and glycosylated hemoglobin levels to discard potential induction of glucose intolerance (Supplementary Information)
Immunohistochemistry of Tumor Sections
Paraffin-embedded sections were incubated at 4°C overnight with primary antibodies (Supplementary Table 4). Phosphorylation of IGF-1R was assessed using a very selective anti-pIGF-1R broadly reported in literature(23), provided by Dr. Rubini. DAB was used as a detection system (EnVision+ System-HRP, Dako). Immunoreactivity was independently graded by three liver pathologists (MS, ST, and SM) (Supplementary Information)
Statistical analysis
Statistical significance when comparing groups was evaluated with Fisher exact test and T-test or Mann-Whitney for categorical and continuous variables. Correlations were calculated either with the Pearson’s coefficient or the non-parametric Spearman’s coefficient. SNP array data and GSEA were processed as previously described(16). p-IGF-1R differential expressed genes were determined using the Significance analysis of Microarrays Package(24). Survival and recurrence data were plotted with Kaplan-Meier curves and significance was calculated with the Log rank test. All calculations were done by SPSS 14.0 (SPSS Inc, Chicago, IL).
RESULTS
Deregulated genes of the IGF system in HCC
IGF2 was overexpressed more than 10 fold in 12 HCC cases compared to healthy liver samples (Figure 1-A), three of the tumors showed an increase higher than 1000 fold. By contrast, IGF1 mRNA expression levels were overall significantly decreased among HCC samples (p<0.001). As previously reported, we observed reduced expression levels of IGFBP3 p<0.001)(25). No difference was observed in IGF1RIGF2RIRS1 and IRS2 expression in tumors compared to control samples. However, among tumors we detected a subgroup of samples with significant decrease of IGF2R expression (Supplementary Figure 1)
Figure 1.
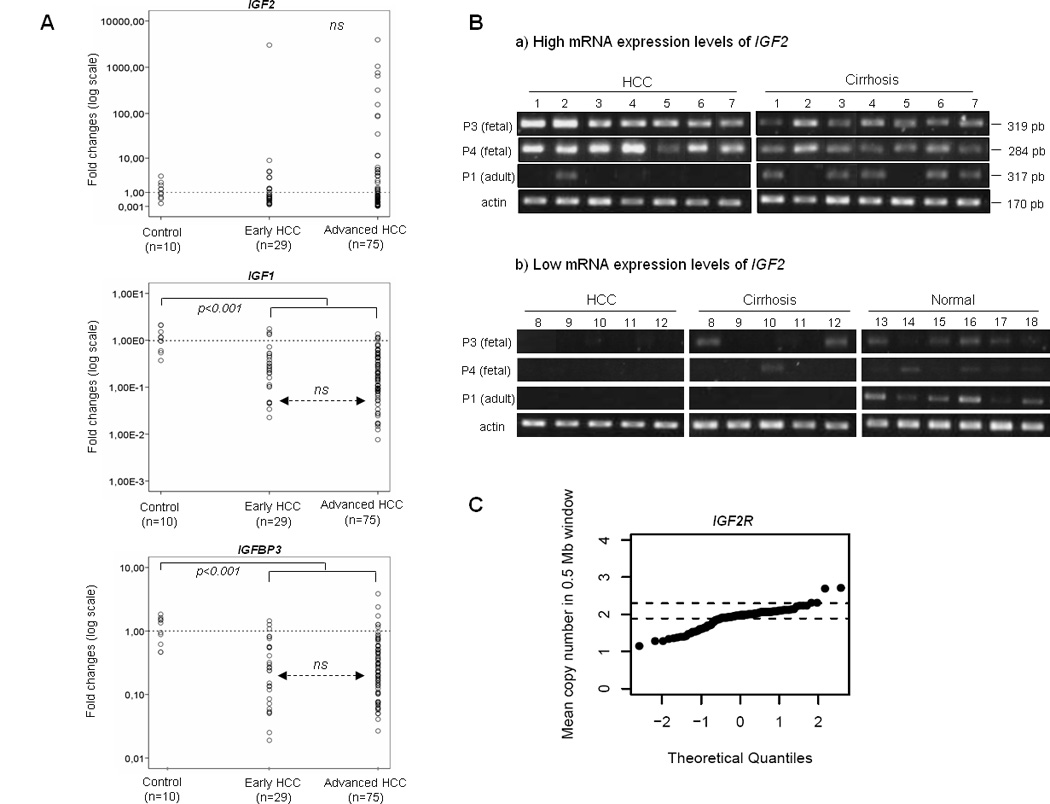
Deregulation of the IGF axis in clinically early HCV-related HCC samples, including both histologically-early and histologically-advanced tumors. (A) Quantitative Real-Time PCR analysis of IGF2 IGF1 and IGFBP3. Dots represent the mean expression value of each individual sample displayed as fold-changes normalized to 1 (mean expression value in normal liver). (B) Reactivation of IGF2 fetal promoters was analyzed by RT-PCR in HCC, cirrhosis and normal control tissue using specific primers corresponding to P1, P3 and P4 promoter derived transcripts. (C) Allelic losses of IGF2R. Each dot represents the mean copy number in a 0.5Mb window around IGF2R locus for each HCC sample. Horizontal dashed lines indicate the copy number range found among cirrhotic counterparts. Cut-off value <1.8 was established on the lower limit of inferred copy number in non-tumoral cirrhotic samples.
Reactivation of IGF2 fetal promoters in HCC
We further explored whether reactivation of fetal promoters P3 and P4 could be responsible for the marked increase of IGF2 expression observed. We could screen 7 out of the 12 samples showing IGF2 overexpression. Results showed that IGF2 transcripts derived from P3 and P4 fetal promoters were abundant in all analyzed HCCs exhibiting high mRNA levels (>10 fold-change; n=7) and to a lesser extent in their adjacent cirrhotic tissue, suggesting a robust fetal promoter reactivation in these tumors (Figure 1-B). By contrast, fetal promoter activity was reduced or absent in HCCs expressing normal to low levels of IGF2 n=10), in their adjacent cirrhotic tissue and in normal control samples (n=6). Finally, the adult P1 promoter was active in all normal liver samples and most of the cirrhotic paired samples
Alterations in gene copy number
We observed allelic losses in the IGF2R locus in 24.5% [25/104] of the HCCs (cut-off <1.8) (Figure 1-C). These losses correlated with a significant decrease in IGF2R expression (p=0.0001) (Supplementary Figure 1). We did not find amplifications or deletions of any other analyzed genes of the system (IGF1, IGF2, IGF1R, IRS1, IRS2, IGFBP1, IGFBP2, IGFBP3 and IGFBP4)
The IGF signaling pathway is activated in a subgroup of HCCs
We afterwards assessed IGF activation, defined by positive p-IGF-1R staining in paraffin-embedded human samples. IGF-1R was activated in 21.4% (18/84) of HCC samples, but in none of adjacent cirrhotic tissues (Figure 2-A). In addition, p-Akt staining was positive in 31.2% (29/92) of cases, p-ERK in 11.8% (10/85) and p-RPS6 in 47.1% (41/87) of tumors(25). Activation of IGF-1R was significantly associated with activation of RPS6 (p-RPS6 staining) (11/16 versus 25/63; p=0.035) and with IGF2 mRNA levels (>20 fold-change, p=0.035). These results observed in tissue samples suggest that IGF-2 might be responsible for IGF-1R activation, and that this might induce downstream activation of mTOR cascade. Our observation is consistent with other previously reported in HCC cell lines(26). These authors showed that in human HCC cells IGF-1R but not insulin receptor is involved in the oncogenic activity of IGF-2
Figure 2.
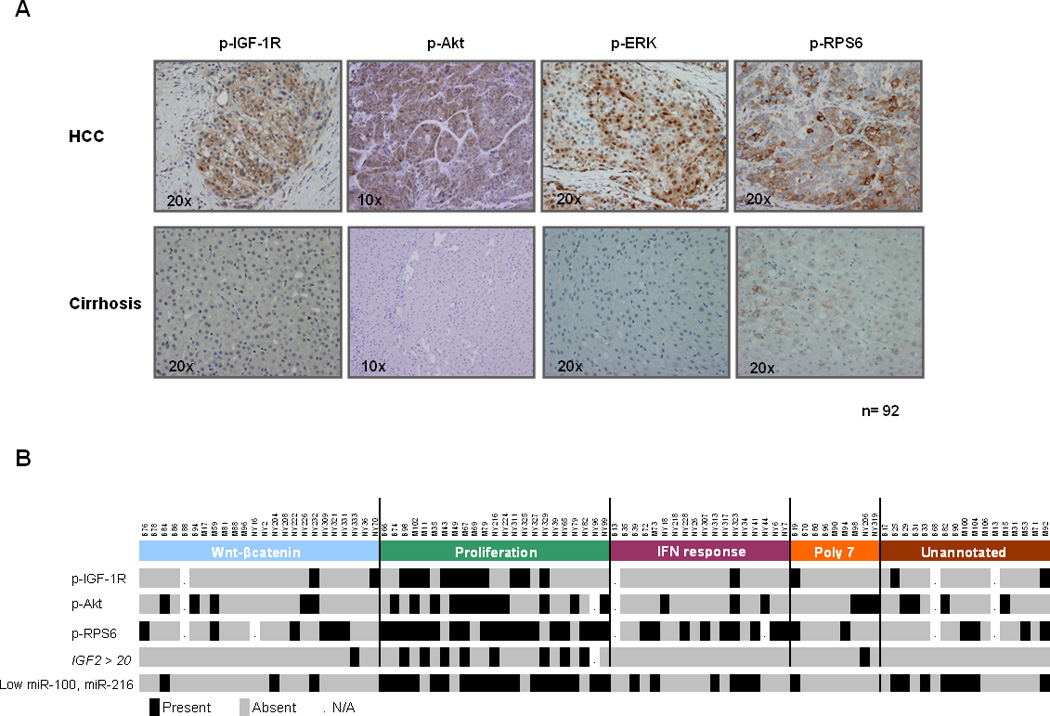
Activation of IGF axis and integration within the molecular classification of HCC. (A) Activation status of IGF-1R, Akt, ERK and RPS6 by immunohistochemistry human HCC samples and its adjacent cirrhotic tissue. (B) HCC samples showing IGF deregulated features are significantly enriched in the Proliferation subgroup of the molecular classification of 91 HCCs. Results for immunohistochemical characterization of p-IGF-1R, p-Akt and p-RPS6, IGF2 mRNA expression levels higher than 20 fold increase (IGF2 > 20) and low expression of both miR-100 and miR-216 are overlapped. Figure adapted from Chiang et al(16).
Integration of the IGF deregulation analysis within the molecular classification
We recently described a molecular classification of HCC obtained through whole genome gene expression analysis identifying 5 different subclasses: Wnt-CTNNB1 30%), Proliferation 30%), IFN-response 18%), polysomy of chromosome 7 (10%), and unannotated 12%)(16). The molecular classification was established with the same cohort of samples used herein, enabling us to integrate the results obtained. There was a significant enrichment of p-IGF-1R positive samples in the Proliferation subclass, defined by proliferation-related genes (11/23 vs 7/61, p=0.001). In addition, samples with high levels of IGF2 mRNA were also significantly enriched in this molecular subclass (Figure 2-B, Table 1)
Gene signatures and miRNA expression associated with IGF-1R activation
Supervised analysis of oligonucleotide microarray data revealed differential gene expression associated with IGF-1R activation (p-IGF-1R positive staining) involving 296 up-regulated genes and 114 down-regulated genes (Supplementary Figure 2-A and Supplementary Tables 5 and 6). By means of GSEA we identified a significant correlation between the p-IGF-1R related expression pattern and a phosphatidylinositol gene signature (FDR= 0.16) (Supplementary Figure 2-B).
Low expression levels of both miR-100 and miR-216, were significantly associated to IGF-1R phosphorylation (FDR<0.05) and the Proliferation subclass (Figure 2-B and Figure 3-A). miR-100 could potentially target members of mTOR and IGF-1R pathways, as predicted by TargetScan software. We observed inverse correlation between the expression of miR-100 and genes involved in PI3K/Akt/mTOR signaling, as BUB1 (p<0.001), CDS1 (p=0.001) and AURKB (p<0.001). Functional assays showed increased phosphorylation levels of IGF-1R in HCC cells transfected with an anti-miR-100 (Figure 3 B–C)
Figure 3.
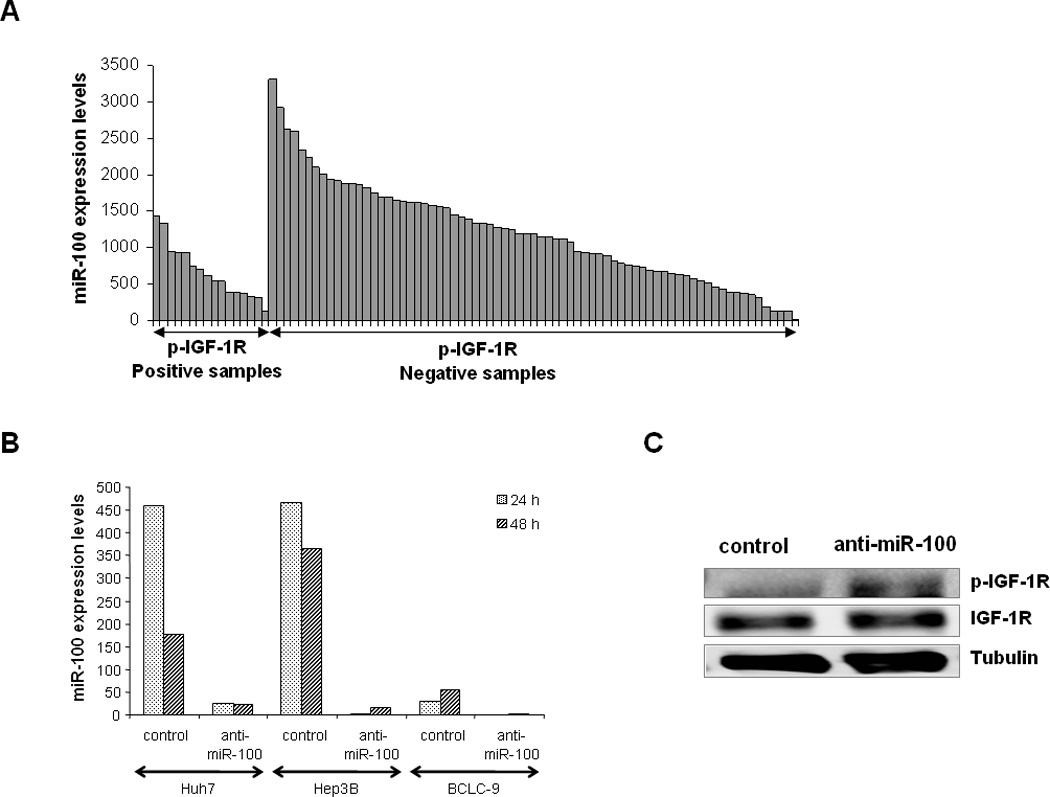
miR-100 is involved in aberrant IGF signaling pathway activation. (A) miR-100 expression among 89 HCCs is overall decreased in samples exhibiting phosphorylation of IGF-1R. (B) miR-100 expression decrease using an anti-mR-100 inhibitor in HCC cell lines. (C) Inhibition of miR-100 expression in Hep3B cells enhanced IGF-1R activation 24 hours post-transfection.
Clinical correlations and outcome involving IGF axis
Both activation of IGF-1R and high levels of IGF2 mRNA (>20 fold-change) were associated with high levels of α-fetoprotein (p=0.028 and p=0.035 respectively). Decreased IGFBP3 mRNA levels (5 fold-change) correlated with smaller tumor size (p=0.013), less vascular invasion (p=0.007), and lower incidence of early recurrence (p=0.006) (Table 1). This association disappeared in the multivariate analysis, when clinical-pathological variables were included
Effects of IGF-1R blockage on cell viability and proliferation
The four liver cancer cell lines showed variable expression of IGF-1R being BCLC-9 the one expressing greater levels of IGF1R as detected by quantitative Real-Time PCR and western blot (data not shown). A12 [10 nM] at 48 hours caused 30% reduction in Hep3B cell viability (69.5 ± 7.3, p<0.0001) (Figure 4-A). However, after 72 hours of incubation, viable cells showed a trend to increase up to 80%. Huh7 cell viability decreased by 20% at 48 hours (79.8 ± 8.7, p<0.02). Similar results were observed in HepG2 and BCLC-9 cells (data not shown). Regarding the inhibitory effects of A12 on cell proliferation, HepG2 cells exhibited the greatest degree of dosedependent inhibition, up to 36% at 48 hours [200 nM] (64 ±1.37, p<0.01) (Figure 4-B). By contrast, after 24 and 48 hours of treatment no evidence of induction of apoptosis or cell cycle arrest was observed
Figure 4.
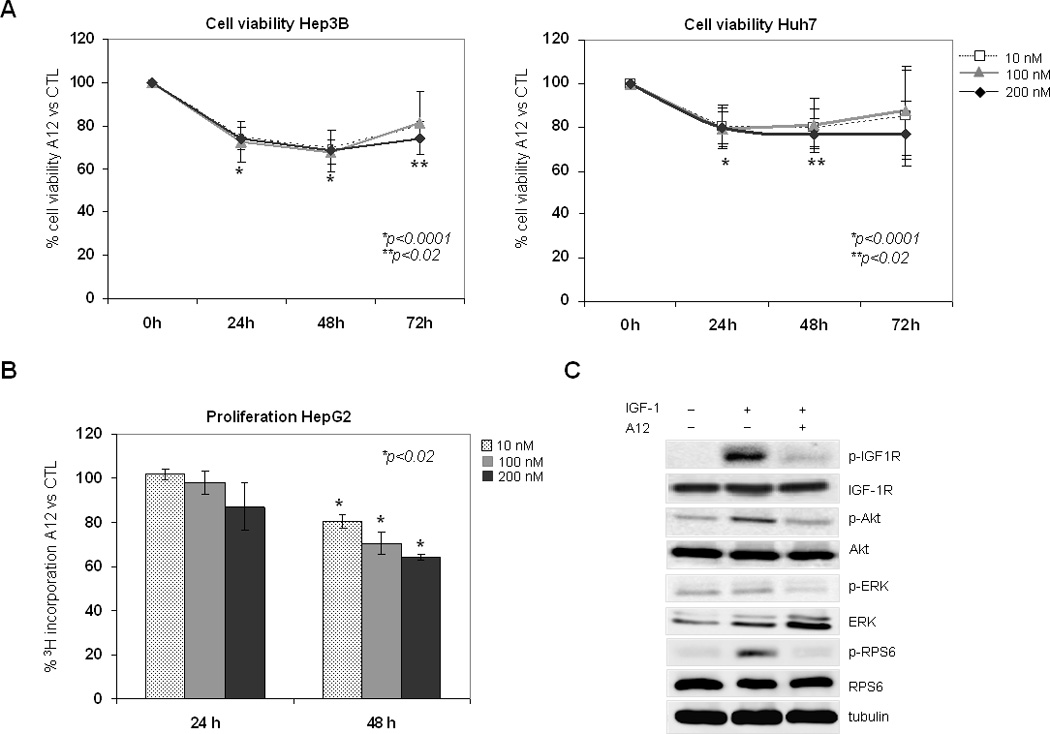
Inhibition of IGF-1R in liver cancer cell lines with the monoclonal antibody A12. (A) Cell viability was measured by MTT and the ratio of each experimental condition versus control is represented. (B) A12 effects on cell proliferation were assessed by [3H]thymidine incorporation assay. (C) Western blot from BCLC-9 lysates was performed to detect activation of IGF-1R and downstream molecules after treatment with A12 [50 nM] for 2 hours. Asterisks (*) indicate statistical significance at A12 experimental conditions compared to controls.
A12 inhibited IGF-1 ligand-dependent activation of the pathway
Strong induction of IGF-1R activation and downstream effector proteins Akt, RPS6, and to a lesser extend ERK1/2, was observed when serum starved cells were stimulated with IGF-1. After two hours of incubation, A12 almost completely blocked IGF-1 induced activation of IGF-1R in HCC tumor cells (Figure 4-C). In addition, A12 efficiently suppressed Akt, ERK1/2 and RPS6 phosphorylation
A12 delays tumor growth and improves survival in an HCC xenograft model
After 10 days of treatment we observed a significant decrease in tumor growth between the two groups (A12 mean (n=22): 609.9 ± 443.6 mm3 versus control mean (n=12): 1048 ± 650 mm3, p=0.024) (Figure 5-A). Furthermore, A12 treated mice exhibited a significant increase in survival, being A12 median survival 21 days [IC95%: 17.4, 24.7] versus control median 16 days [IC95%: 13.5, 18.9] (p=0.039) (Figure 5-B). All mice were sacrificed following institutional ethical guidelines, either due to tumor progression (35/38) or weight loss greater than 15% body weight (3/38, all from A12 arm). Thus, A12 treatment was overall well tolerated and mice did not display significant toxicity. Additionally, there were no significant differences in blood glucose or glycosylated hemoglobin levels between control and A12 treated mice (Supplementary Figure 3).
Figure 5.
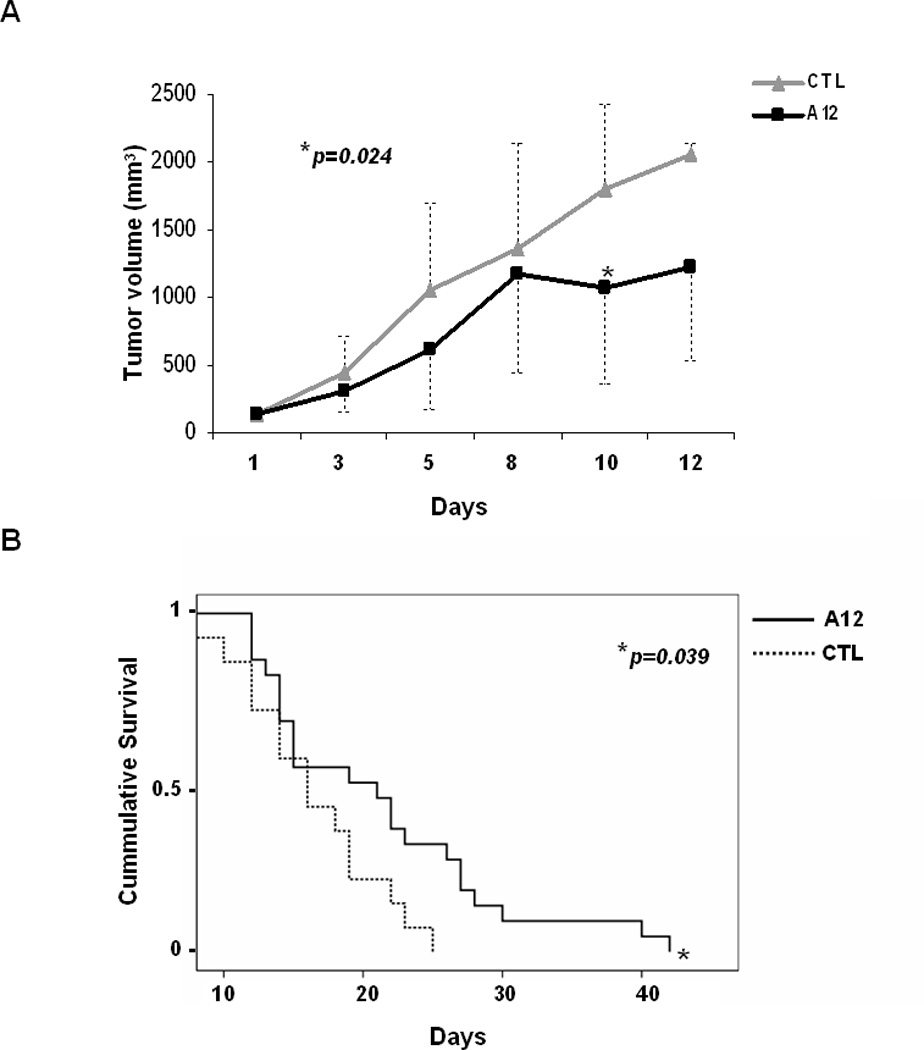
Effects of A12 on tumor growth and survival in xenograft tumors. (A) Tumor progression in mice treated with A12 (n=22) versus PBS (n=12). Plotted data represents mean values of all tumor volumes in the two groups in mm3, bars are standard deviation. (B) Kaplan-Meier curves represent the median survival in A12 treated versus control mice.
TUNEL staining revealed an increment of apoptotic nuclei in tumors of treated mice (A12 mean: 2.8 ± 1.6 versus control mean: 1.2 ± 0.86, p=0.008). A significant increase in the extension of necrotic areas was also observed in treated mice. Modest, yet significant reduction of Ki-67 staining was observed in A12 mice. Cell proliferation decreased by 16.4% according to stained nuclei per high power field (A12 mean: 134.8 ± 23.21 versus control mean: 161.3 ± 9.57, p=0.028) (Figure 6-B). Finally, the evaluation of the molecular response to IGF signaling blockage evidenced that A12 could down-regulate IGF-1 induced phosphorylation of both IGF-1R and RPS6 (Figure 6-A) showing the capacity of A12 to impair tumor growth and proliferation in vivo by abrogating IGF signaling pathway.
Figure 6.
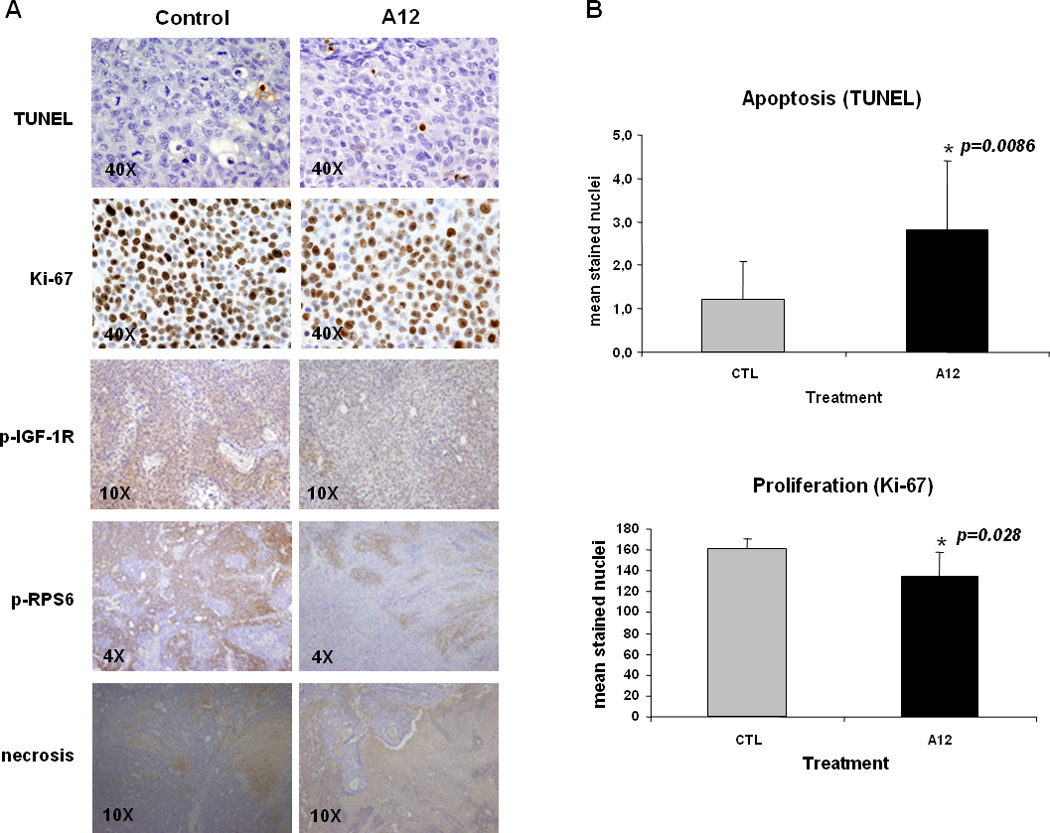
Molecular assessment of the antitumoral efficacy of A12 by immunohistochemistry in tumor sections. (A)Tissue sections were stained for Ki-67, TUNEL, p-IGF-1R and p-RPS6 staining. Extension of necrotic areas was also evaluated. (B) Apoptotic and proliferation indices are expressed as the mean number of positive stained nuclei in 10 high power fields (x40), bars are standard deviation.
DISCUSSION
The IGF signaling pathway has emerged as one of the most relevant molecular alterations in carcinogenesis(4). Herein we report that around 20% of patients with early HCC have altered the IGF system due to aberrant signaling through its receptor. IGF-1R activation results from ligand interaction. It has been suggested that some tumors may be dependent on the host for the ligand and later acquire the capacity to produce the ligand in an autocrine manner(4). In our cohort of 104 HCV-related HCCs, 12 samples showed marked overexpression of IGF2 mainly those histologically-advanced tumors, and this event was significantly associated with IGF-1R activation. We found that IGF2 overexpression involved a strong reactivation of fetal promoters, which are usually silenced during adulthood(7;8). In addition, we observed relevant losses of IGF2R in 25% of cases, a tumor suppressor gene that removes IGF-2 proteins from the extracellular matrix, which correlated with a decrease in IGF2R expression. This molecular abnormality was identified by others as being more prevalent(9;10). This could be explained by the cell admixture of neoplastic cells with endothelial and non-parenchymal cells. Finally, we also observed a third factor that could be involved in IGF-1R activation such as downregulation of IGFBP3. It is of note that all these aberrations were described in the so-called clinically early HCC (most cases are BCLC class 0 or A). It could be expected that in a more clinically advanced context, increased cross-talks among pathways and chromosomal instability would contribute to additional IGF signaling deregulation.
Regarding IGF-1, molecular epidemiological studies associate increased IGF-1 circulating levels with higher risk of development of several neoplasms. Nonetheless, IGF1 expression is significantly decreased in our tumor samples, which is consistent with previous studies in HCC(27). These data suggest that the mitogenic activity of IGF-1 does not have a determinant role in the progression of HCC.
Our group recently reported a novel molecular classification of HCC in 5 different subclasses: Wnt-CTNNB1 Proliferation, IFN-related, polysomy of chromosome 7 and unannotated class(16). Interestingly, most of the samples with aberrant activation of IGF-1R and increased levels of IGF2 mRNA were significantly enriched in the Proliferation subclass (p<0.001). This suggests an important role of IGF signaling in these molecularly distinct tumors, also characterized by increased levels of α- fetoprotein and activation of downstream signaling molecules such as Akt and RPS6. Consistently, association between IGF-1R activation and mTOR (RPS6 phosphorylation) was also observed.
Emerging data regarding aberrant expression of miRNA in HCC suggests a role for this novel class of gene regulators in this neoplasia. miR-100 has been found to be deregulated in other malignancies(28; 29). The noticeable decreased expression of miR-100 observed in HCC samples exhibiting activation of IGF-1R suggests that this miRNA could play a role in IGF-1R activation. Supporting this hypothesis, in vitro inhibition of miR-100 expression enhanced IGF-1R phosphorylation, however the mechanism that would explain this association needs further investigation.
Sorafenib expands survival in patients with advanced HCC(2) representing the first effective systemic therapy and a first step in the combination of molecular therapies leading to cancer curation(30). Based on the data reported herein, targeted disruption of IGF-1R activation appears as an attractive therapeutic strategy for the treatment of HCC patients. However, manipulation of IGF-1R signaling in HCC animal models has been scarcely tested(26. Our results showed that the treatment with the monoclonal antibody A12 decreased cell viability and proliferation and blocked ligand-induced receptor activation. However, as reported in other cancer types, A12 in vitro did not induce significant apoptosis or cell cycle arrest(31). Whether A12 induces antibody-dependent cellular cytotoxicity remains elusive, but antitumoral effects observed in vitro with antibodies against IGF-1R, such as 19D12(32;33), have been attributed to antibody-dependent cell cytotoxicity.
The in vivo study showed effective antitumoral activity of A12 without evidence of toxicity. Immunohistochemical analysis revealed that A12 induced a slight increase of apoptosis and necrosis, decreased cell proliferation and effectively blocked signaling initiated through IGF-1R. IGF signaling is involved in VEGF production, and therapies blocking IGF-1R decreased VEGF levels and angiogenesis in some experimental models(34). Thus, decreased blood supply resulting from VEGF abrogation could be responsible for extended necrotic areas observed after A12 therapy.
Considering all these pre-clinical and translational data, we conclude that there is rationale for testing monoclonal antibodies against IGF-1R in early phases of clinical trials in HCC. These therapies are more likely to be used in combination with the standard of care, sorafenib, or with other molecular therapies. For instance, since IGF- 1R activation has been involved in acquired resistance to EGFR inhibition in several cancer types(6), there is rationale for co-targeting IGF-1R to restore sensitivity to anti- EGFR therapies(35). Finally, based on our results, IGF-2-mediated activation of IGF- 1R acts as a key driver in the Proliferation subclass of HCC patients. The identification of this oncogenic loop in patients can be ultimately critical for the achievement of relevant anti-tumoral outcomes using a more personalized approach with IGF-1R inhibitors.
Supplementary Material
Acknowledgments
We thank Dr. Rubini for kindly providing the antibody anti-IGF-1R pY1316. ImClone Systems Incorporated supplied the monoclonal antibody A12 used in this study.
Financial support: Augusto Villanueva is the recipient of a Sheila Sherlock fellowship. Beatriz Minguez is supported by a grant from Instituto de Salud Carlos III (FISCM04/ 00044). Vincenzo Mazzaferro and Sara Toffanin are supported by the Italian Association for Cancer Research and the Italian National Ministry of Health. Jordi Bruix is supported by a grant from Instituto Carlos III (ISCIII/FIS PI 05-0150). Josep M. Llovet is supported by grants from the U.S. National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK076986-01), the Samuel Waxman Cancer Research Foundation and the Spanish National Health Institute (SAF-2007-61898).
Financial disclosure: There are no financial disclosures relevant to this manuscript.
Abbreviations
- HCC
hepatocellular carcinoma
- IGFBP-3
IGF binding protein 3
- HCV
hepatitis C virus
- RPS6
ribosomal protein S6
- mTOR
mechanistic target of rapamycin
- GSEA
gene set enrichment analysis
- miR-
microRNA
- VEGF
vascular endothelial growth factor.
Footnotes
This study was presented in the Plenary Session I in the 43rd EASL Annual Conference (Milan 2008).
REFERENCES
- 1.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 3.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:76–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 4.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 5.Peruzzi F, Prisco M, Dews M, Salomoni P, Grassilli E, Romano G, et al. Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol Cell Biol. 1999;19:7203–7215. doi: 10.1128/mcb.19.10.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu Y, Zi X, Pollak M. Molecular mechanisms underlying IGF-I-induced attenuation of the growth-inhibitory activity of trastuzumab (Herceptin) on SKBR3 breast cancer cells. Int J Cancer. 2004;108:334–341. doi: 10.1002/ijc.11445. [DOI] [PubMed] [Google Scholar]
- 7.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 8.Tang SH, Yang DH, Huang W, Zhou HK, Lu XH, Ye G. Hypomethylated P4 promoter induces expression of the insulin-like growth factor-II gene in hepatocellular carcinoma in a Chinese population. Clin Cancer Res. 2006;12:4171–4177. doi: 10.1158/1078-0432.CCR-05-2261. [DOI] [PubMed] [Google Scholar]
- 9.De Souza AT, Hankins GR, Washington MK, Orton TC, Jirtle RL. M6P/IGF2R gene is mutated in human hepatocellular carcinomas with loss of heterozygosity. Nat Genet. 1995;11:447–449. doi: 10.1038/ng1295-447. [DOI] [PubMed] [Google Scholar]
- 10.Oka Y, Waterland RA, Killian JK, Nolan CM, Jang HS, Tohara K, et al. M6P/IGF2R tumor suppressor gene mutated in hepatocellular carcinomas in Japan. Hepatology. 2002;35:1153–1163. doi: 10.1053/jhep.2002.32669. [DOI] [PubMed] [Google Scholar]
- 11.Hanafusa T, Yumoto Y, Nouso K, Nakatsukasa H, Onishi T, Fujikawa T, et al. Reduced expression of insulin-like growth factor binding protein-3 and its promoter hypermethylation in human hepatocellular carcinoma. Cancer Lett. 2002;176:149–158. doi: 10.1016/s0304-3835(01)00736-4. [DOI] [PubMed] [Google Scholar]
- 12.Kim SO, Park JG, Lee YI. Increased expression of the insulin-like growth factor I (IGF-I) receptor gene in hepatocellular carcinoma cell lines: implications of IGF-I receptor gene activation by hepatitis B virus X gene product. Cancer Res. 1996;56:3831–3836. [PubMed] [Google Scholar]
- 13.Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res. 2003;63:8912–8921. [PubMed] [Google Scholar]
- 14.Rowinsky EK, Youssoufian H, Tonra JR, Solomon P, Burtrum D, Ludwig DL. IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin Cancer Res. 2007;13:5549s–5555s. doi: 10.1158/1078-0432.CCR-07-1109. [DOI] [PubMed] [Google Scholar]
- 15.Higano CS, Yu EY, Whiting SH, Gordon MS, LoRusso P, Fox F, et al. A phase-I, first in man study of weekly IMC-A12, a fully human insulin like growth factor-I receptor IgG1 monoclonal antibody, in patients with advanced solid tumors. [Abstract] Journal of Clinical Oncology. 2007;25(Suppl):3505. [Google Scholar]
- 16.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–6788. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armengol C, Tarafa G, Boix L, Sole M, Queralt R, Costa D, et al. Orthotopic implantation of human hepatocellular carcinoma in mice: analysis of tumor progression and establishment of the BCLC-9 cell line. Clin Cancer Res. 2004;10:2150–2157. doi: 10.1158/1078-0432.ccr-03-1028. [DOI] [PubMed] [Google Scholar]
- 18.Wurmbach E, Chen YB, Khitrov G, Zhang W, Roayaie S, Schwartz M, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;45:938–947. doi: 10.1002/hep.21622. [DOI] [PubMed] [Google Scholar]
- 19.Nardone G, Romano M, Calabro A, Pedone PV, de SI, Persico M, et al. Activation of fetal promoters of insulinlike growth factors II gene in hepatitis C virus-related chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Hepatology. 1996;23:1304–1312. doi: 10.1002/hep.510230602. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly MB, et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104:19971–19976. doi: 10.1073/pnas.0709313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HC, Tian B, Sedivy JM, Wands JR, Kim M. Loss of Raf kinase inhibitor protein promotes cell proliferation and migration of human hepatoma cells. Gastroenterology. 2006;131:1208–1217. doi: 10.1053/j.gastro.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubini M, D'Ambrosio C, Carturan S, Yumet G, Catalano E, Shan S, et al. Characterization of an antibody that can detect an activated IGF-I receptor in human cancers. Exp Cell Res. 1999;251:22–32. doi: 10.1006/excr.1999.4562. [DOI] [PubMed] [Google Scholar]
- 24.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972–1983. doi: 10.1053/j.gastro.2008.08.008. 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nussbaum T, Samarin J, Ehemann V, Bissinger M, Ryschich E, Khamidjanov A, et al. Autocrine insulin-like growth factor-II stimulation of tumor cell migration is a progression step in human hepatocarcinogenesis. Hepatology. 2008;48:146–156. doi: 10.1002/hep.22297. [DOI] [PubMed] [Google Scholar]
- 27.Mazziotti G, Sorvillo F, Morisco F, Carbone A, Rotondi M, Stornaiuolo G, et al. Serum insulin-like growth factor I evaluation as a useful tool for predicting the risk of developing hepatocellular carcinoma in patients with hepatitis C virus-related cirrhosis: a prospective study. Cancer. 2002;95:2539–2545. doi: 10.1002/cncr.11002. [DOI] [PubMed] [Google Scholar]
- 28.Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim JH, et al. MicroRNA expression profiles in serous ovarian carcinoma. Clin Cancer Res. 2008;14:2690–2695. doi: 10.1158/1078-0432.CCR-07-1731. [DOI] [PubMed] [Google Scholar]
- 29.Wong TS, Liu XB, Wong BY, Ng RW, Yuen AP, Wei WI. Mature miR-184 as Potential Oncogenic microRNA of Squamous Cell Carcinoma of Tongue. Clin Cancer Res. 2008;14:2588–2592. doi: 10.1158/1078-0432.CCR-07-0666. [DOI] [PubMed] [Google Scholar]
- 30.Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, Zhu AX, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:698–711. doi: 10.1093/jnci/djn134. [DOI] [PubMed] [Google Scholar]
- 31.Yeh J, Litz J, Hauck P, Ludwig DL, Krystal GW. Selective inhibition of SCLC growth by the A12 anti-IGF-1R monoclonal antibody correlates with inhibition of Akt. Lung Cancer. 2008;60:166–174. doi: 10.1016/j.lungcan.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Goetsch L, Gonzalez A, Leger O, Beck A, Pauwels PJ, Haeuw JF, et al. A recombinant humanized anti-insulin-like growth factor receptor type I antibody (h7C10) enhances the antitumor activity of vinorelbine and anti-epidermal growth factor receptor therapy against human cancer xenografts. Int J Cancer. 2005;113:316–328. doi: 10.1002/ijc.20543. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Hailey J, Williams D, Wang Y, Lipari P, Malkowski M, et al. Inhibition of insulin-like growth factor-I receptor (IGF-IR) signaling and tumor cell growth by a fully human neutralizing anti-IGF-IR antibody. Mol Cancer Ther. 2005;4:1214–1221. doi: 10.1158/1535-7163.MCT-05-0048. [DOI] [PubMed] [Google Scholar]
- 34.Wu KD, Zhou L, Burtrum D, Ludwig DL, Moore MA. Antibody targeting of the insulin-like growth factor I receptor enhances the anti-tumor response of multiple myeloma to chemotherapy through inhibition of tumor proliferation and angiogenesis. Cancer Immunol Immunother. 2007;56:343–357. doi: 10.1007/s00262-006-0196-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desbois-Mouthon C, Cacheux W, Blivet-Van Eggelpoel MJ, Barbu V, Fartoux L, Poupon R, et al. Impact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinib. Int J Cancer. 2006;119:2557–2566. doi: 10.1002/ijc.22221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.