

ABSTRACT
The chronic nature of tuberculosis (TB), its requirement of long duration of treatment, its ability to evade immune intervention, and its propensity to relapse after drug treatment is discontinued are reminiscent of other chronic, biofilm-associated bacterial diseases. Historically, Mycobacterium tuberculosis was grown as a pellicle, a biofilm-like structure, at the liquid-air interface in a variety of synthetic media. Notably, the most widely administered human vaccine, BCG, is grown as a pellicle for vaccine production. However, the molecular requirements for this growth remain ill defined. Here, we demonstrate that keto-mycolic acids (keto-MA) are essential for pellicle growth, and mutants lacking in or depleted of this MA species are unable to form a pellicle. We investigated the role of the pellicle biofilm in the reduction of antibiotic sensitivity known as drug tolerance using the pellicle-defective ΔmmaA4 mutant strain. We discovered that the ΔmmaA4 mutant, which is both pellicle defective and highly sensitive to rifampicin (RIF) under planktonic growth, when incorporated within the wild-type pellicle biofilm, was protected from the bactericidal activity of RIF. The observation that growth within the M. tuberculosis pellicle biofilm can confer drug tolerance to a drug-hypersensitive strain suggests that identifying molecular requirements for pellicle growth could lead to development of novel interventions against mycobacterial infections. Our findings also suggest that a class of drugs that can disrupt M. tuberculosis biofilm formation, when used in conjunction with conventional antibiotics, has the potential to overcome drug tolerance.
IMPORTANCE
Two of the most important questions in tuberculosis (TB) research are (i) how does Mycobacterium tuberculosis persist in the human host for decades in the face of an active immune response and (ii) why does it take six months and four drugs to treat uncomplicated TB. Both these aspects of M. tuberculosis biology are reminiscent of infections caused by organisms capable of forming biofilms. M. tuberculosis is capable of growing as a biofilm-like structure called the pellicle. In this study, we demonstrate that a specific cell wall component, keto-mycolic acid, is essential for pellicle growth. We also demonstrate that a strain of M. tuberculosis that is both drug sensitive and pellicle defective exhibits commensal behavior and becomes drug tolerant by becoming part of a heterogeneous pellicle, a characteristic of multispecies biofilms. These observations could have important implications for identifying novel pathways for M. tuberculosis drug tolerance and the design of new modalities to rapidly treat TB.
Introduction
In the laboratory, bacteria are usually grown and studied as dispersed, planktonic, pure cultures. In nature, however, this is seldom the case. One preferred microbial lifestyle is that of a surface-adherent, multispecies microbial community called biofilm that is physiologically and phenotypically distinct from bacteria growing in a free-swimming planktonic state. The current definition of a bacterial biofilm is that of a structured community of bacterial cells enclosed in a self-produced polymeric matrix and adherent to an inert or living surface (1). Biofilms are strongly implicated in chronicity and transmission of several bacterial infections. Biofilm infections have been shown to occur on abiotic medical implants as well as living tissue, such as in the case of endocarditis (1, 2). The ability of Pseudomonas aeruginosa to grow as a biofilm in patients with cystic fibrosis has been implicated in the chronic nature of cystic fibrosis pneumonia (3, 4). Uropathogenic Escherichia coli, which causes urinary tract infections, has been shown to grow as biofilm-like pods in mice (5). Legionella pneumophila in biofilms was shown to be involved in the transmission of Legionnaires’ disease (6). Vibrio cholerae, streptococci, staphylococci, and several other bacterial pathogens have been shown to form biofilms that have been implicated in their pathogenesis and chronicity (1, 7–10). One of the most important characteristics of bacteria growing within biofilm is their ability to tolerate a wide variety of bactericidal compounds, and this contributes to the refractory nature of biofilm-associated infections to drugs and the chronicity of such infections. Biofilms are also thought to confer protection to bacteria growing within them to immune intervention (5, 11). Genetic experiments have led to the identification of several genes and pathways that are involved in biofilm formation in different bacteria, such as P. aeruginosa, E. coli, Bacillus subtilis, staphylococci, and streptococci (12–15).
Several members of the genus Mycobacterium, such as M. ulcerans, M. avium, M. marinum, and the saprophytic M. smegmatis, have been shown to form biofilms (16–19). The ability of M. smegmatis to form biofilms has been implicated in its resistance to antibiotics (20). Biofilms may also play a role in the transmission and pathogenesis of M. ulcerans, the causative agent of Buruli ulcer, which thrives in aquatic environments, and M. avium, an opportunistic environmental pathogen (17, 21, 22). Some aspects of genetic requirements and molecular events in mycobacterial biofilm formation have been determined. Mutants of M. smegmatis and M. avium with defects in glycopeptidolipid biosynthesis were unable to form biofilms (19, 23). In addition, M. smegmatis mutants altered in both undecaprenyl phosphokinase and mycolic acid (MA) biosynthesis have also shown to be defective in biofilm formation (24, 25). Taken together, mycobacterial pathways involved in cell wall biosynthesis seem to be important for biofilm formation.
It has recently been suggested that M. tuberculosis can form biofilms. These in vitro biofilms were shown to be rich in free mycolic acids that are likely released by the enzymatic hydrolysis of trehalose dimycolate (TDM) and shown to harbor drug-tolerant cells (26, 27).
Several aspects of M. tuberculosis biology and pathogenesis are reminiscent of biofilm behavior. For example, the ability of M. tuberculosis to grow as “cords,” a correlate of virulence and ability to survive within the host, indicates the propensity of this bacterium to be part of a multicellular community (28–30). M. tuberculosis has also been described to be found in a biofilm-like structure in the acellular regions of certain types of human caseating lesions (31). In addition, studies using a guinea pig model of TB revealed that M. tuberculosis strains that survive extended drug treatment were primarily extracellular and confined to an acellular rim around the primary granuloma, which is morphologically similar to what has been described for human lung lesions (32). The chronic nature of TB, its requirement of a long duration of treatment, its ability to evade immune intervention, and its propensity to relapse after drug treatment is discontinued are all also reminiscent of other chronic, biofilm-associated bacterial diseases.
The air-liquid interface is an advantageous niche for aerobes due to increased accessibility to oxygen. Many aerobic bacteria exhibit an aerotactic response over a range of preferred oxygen tension, resulting in migration to the air-liquid interface and formation of a biofilm-like structure composed of cells and an extracellular matrix (ECM), known as a pellicle (33, 34). Historically, M. tuberculosis was predominantly grown as a pellicle at the liquid-air interphase in a variety of synthetic media, resulting in luxuriant, hypha-like growth at the surface (35). The name “mycobacteria,” which means “fungus-like bacteria,” in fact, reflects this observation (36). The hydrophobic nature of the mycobacterial cell wall, the preference for an aerobic lifestyle, and the presence of zinc ions (Zn2+) have all been shown to contribute to this phenotype (37). However, the molecular basis of this mode of growth remains ill defined. Although a role for biofilms in M. tuberculosis pathogenesis remains unclear, the recent finding that M. tuberculosis pellicles harbor drug-tolerant cells, a trait commonly associated with biofilms (27), suggests that understanding the molecular events involved in the transition from planktonic to pellicle growth might provide useful insights into mechanisms by which M. tuberculosis acquires drug tolerance.
We hypothesized that M. tuberculosis pellicles may represent an adaptive diversification of the organism in response to certain stimuli, into a structured environment. Pellicles may hence be regarded as analogs of biofilms. In this study, we demonstrate that the ability to synthesize keto-mycolic acids (keto-MAs) is essential for bacterial organization into this ordered structure. We also demonstrate that growth within the pellicle confers drug tolerance to otherwise drug-susceptible bacilli.
RESULTS
M. tuberculosis pellicle is an organized structure distinct from cording.
In the laboratory, several model systems have been used to study biofilms (38). Submerged biofilms such as those seen in aquatic environments are modeled using flow cells (39), biofilms that form on surfaces are assayed based on their ability to adhere to abiotic polyvinyl chloride or polystyrene surfaces (12, 40) or simply studied as bacterial colonies on agar plates (41), and biofilms that form at the air-liquid interface are studied as pellicles (42, 43).
Growth as pellicles requires a high degree of self-organization owing to the lack of a solid surface on which growth can be initiated. Scanning electron microscopy (SEM) of M. tuberculosis pellicles following processing with osmium tetroxide vapor, which allows preservation of the ultrastructure, showed that the bacteria in the pellicle are encased in an extracellular matrix (Fig. 1A). A more conventional processing of the pellicle using potassium ferrocyanide resulted in the stripping of the matrix and revealed a structured organization of the bacterial cells within this matrix (Fig. 1B). These observations are reminiscent of other bacterial biofilms, which have been shown to confer drug tolerance and contribute to persistence (1, 44). M. tuberculosis is known to display another form of organized growth referred to as cording. The ability to cord has been associated with the virulence of M. tuberculosis, and TDM has been identified to be the cord factor (29, 45). Modification of the mycolic acid constituents of TDM has been found to be required for cording, specifically cyclopropanation catalyzed by PcaA (30). However, the M. tuberculosis ΔpcaA mutant (Table 1) is fully capable of forming a pellicle (Fig. 1C). This suggests that the pellicle phenotype is distinct from cording. This was further confirmed by the observation that an unrelated cording mutant of M. tuberculosis, H37RvΔkasB (46), is also competent for pellicle growth (Fig. 1C).
FIG 1 .

M. tuberculosis pellicle is an organized structure. SEM of M. tuberculosis pellicle using osmium tetroxide vapor (A) or potassium ferrocyanide (B) processing shows an organized bacterial community. Scale bars represent 10 µm in ×1,000 and 5 µm in ×3,000 magnification. (C) Pellicle phenotype is distinct from cording. ΔpcaA and ΔkasB mutants (both defective in cording) were inoculated into Sauton’s medium without Tween 80 and incubated for 3 weeks under pellicle-promoting conditions. Both ΔpcaA and ΔkasB mutants are capable of growing as a pellicle, indicating that the ability to cord is not a prerequisite for pellicle growth.
TABLE 1 .
Plasmids and bacterial strains used in this study
| Plasmid, strain, or phage | Description | Reference or source |
|---|---|---|
| pMV361-mmaA3 | Hygr, single-copy integrating vector, overexpressing M. tuberculosis MmaA3 | (75) |
| Bacterial strains | ||
| mc2 2 | Wild-type H37Rv | Jacobs Lab, obtained from Trudeau Institute |
| mc2 7051 | H37RvΔmmaA4 | (60) |
| mc2 7052 | H37RvΔmmaA3 | (60) |
| mc2 7053 | H37Rv::pMV361::mmaA3 | This study |
| mc2 5859 | H37RvΔkasB | Jacobs Lab, Catherine Vilchèze |
| mc2 7054 | H37RvΔpcaA | Gift from M. Glickman |
| mc2 7055 | H37RvΔmmaA4::mmaA4 | (60) |
It has been recently demonstrated that the ability of M. tuberculosis to form a mycolate-rich extracellular matrix is central to the ability to form pellicles, of which a major constituent is free methoxy-MA (27). However, M. bovis BCG Pasteur, which is grown as a pellicle for vaccine production, is inherently defective in production of methoxy-MA due to a point mutation in the methoxy-MA synthase mmaA3 (47). This led us to hypothesize that other MA species may play pivotal roles in pellicle growth.
Construction of M. tuberculosis strains defective in specific mycolic acids.
The defining feature of the mycobacterial cell envelope is the mycolic acids. They are among the most abundant components of the mycobacterial cell wall, comprising around 57% of the cell wall dry weight, and contribute to the pathogenicity of M. tuberculosis in a variety of ways (48, 49). Three major structural classes of MAs are found in M. tuberculosis—alpha (α), methoxy, and keto, characterized by their functional groups. Mycolic acid methyltransferases are involved in the addition of functional groups that distinguish the three species of MAs (48), such that strains lacking these genes would be predicted to be lacking in specific MA species (Table 2).
TABLE 2 .
Effect of genotype on mycolic acid production
| Genotype | Production of:a |
||
|---|---|---|---|
| α-Mycolates | Keto-mycolates | Methoxy-mycolates | |
| Wild type | + | + | + |
| ΔkasB | + | + | + |
| ΔpcaA | − | + | + |
| ΔmmaA3 | + | + | − |
| ΔmmaA4 | + | − | − |
| pMV361::mmaA3 | + | − | + |
| ΔmmaA4::mmaA4 | + | + | + |
+, produced; −, not produced.
To determine the role of specific MA species in pellicle growth, strains of M. tuberculosis H37Rv individually lacking specific methyl transferases were tested (Table 1). In addition, a strain defective in pcaA, which codes for a cyclopropane synthase required for α-MA synthesis, was also examined (30). Strains lacking the MA methyl transferases MmaA3 or MmaA4 had defects in MA synthesis (Fig. 2). The H37RvΔmmaA3 mutant did not produce methoxy-MA, whereas the H37RvΔmmaA4 mutant was unable to synthesize methoxy- and keto-MA. It has previously been shown that the overproduction of MmaA3 in a wild-type genetic background results in the loss of keto-MA without accumulation of the aberrant α-MA (50). We constructed a strain of H37Rv overexpressing MmaA3, and this strain was indeed deficient in keto-MA (Fig. 2). The availability of strains lacking individual MA species now allowed us to determine the role of individual MA in pellicle growth.
FIG 2 .

Mutants defective in specific MA species. Two-dimensional argentation TLC of mycolic acids from H37Rv, ΔmmaA4, ΔmmaA3, ΔmmaA4::Comp, and H37Rv::mmaA3 strains grown planktonically in Middlebrook 7H9 medium. Samples were run using hexane-ethyl acetate (95:5 [vol/vol]) in the first dimension (1) and petroleum ether-diethyl ether (85:15 [vol/vol]) in the second (2). O, origin; α1–4, α-MA; K, keto-MA; M, methoxy-MA; H, hydroxy MA.
Keto-MA is essential for pellicle growth.
H37RvΔmmaA3 (Fig. 3) and H37RvΔpcaA (Fig. 1C) mutants were both capable of forming pellicles that were indistinguishable from those of the parental strain, suggesting that neither the loss of methoxy-MA, the major lipid constituent of the mycobacterial pellicle matrix (27), nor α-MA (30), the predominant MA species in M. tuberculosis under planktonic growth, interferes with pellicle formation. In sharp contrast, the H37RvΔmmaA4 mutant was defective in pellicle formation, and this defect was rescued upon genetic complementation of the deleted locus (Fig. 3). Taken together, these data suggest either both species of oxygenated MA or keto-MA alone is required for pellicle formation. Alternatively, the accumulation of aberrant α-MA (51) in the ΔmmaA4 mutant (Fig. 2) could be inhibitory. To distinguish these possibilities, the strain of H37Rv that overexpresses MmaA3, characterized by the production of α-MA and methoxy-MA only (Fig. 2), was examined for its ability to grow as a pellicle. This strain was indeed defective for pellicle formation, indicating that the lack of keto-MA, but not production of aberrant α-MA, is responsible for the inability to form pellicles (Fig. 3).
FIG 3 .

Keto-mycolates are essential for M. tuberculosis pellicle growth. H37Rv, ΔmmaA4, ΔmmaA4, ΔmmaA4::Comp, and H37Rv::mmaA3 strains were inoculated into Sauton’s medium without Tween 80 to assess pellicle formation. Pellicle formation was recorded at 4 weeks postinoculation.
Furthermore, coculturing the two keto-MA-deficient strains (the ΔmmaA4 and H37RV::mmaA3 strains) did not restore pellicle formation, whereas coculturing a keto-MA-deficient strain with a keto-MA-producing strain (ΔmmaA4 and ΔmmaA3 strains; ΔmmaA4 and H37Rv strains) does restore pellicle formation, demonstrating the absolute requirement of keto-MA for pellicle formation and suggesting that the arrest in pellicle formation was not due to accumulation of repressive products in the mutants (Fig. 4).
FIG 4 .

Coculturing keto-MA-deficient strains does not restore pellicle growth. H37Rv, ΔmmaA4, and H37Rv::mmaA3 strains, individually and in combinations—ΔmmaA4 and H37RV coculture, H37RV::mmaA3 and H37Rv coculture, H37RV::mmaA3 and ΔmmaA4 coculture—were inoculated into Sauton’s medium without Tween 80 and incubated for 3 weeks under pellicle-promoting conditions.
Mycolic acid fluctuations under pellicle growth conditions.
It has previously been demonstrated that growth under pellicle-promoting conditions results in formation of an extracellular matrix enriched in free methoxy-MA (27). Following the method described by Ojha et al. (27), apolar detergent-extractable lipids from wild-type and pellicle-defective strains, grown under pellicle-promoting conditions, were isolated, purified, and analyzed by chromatography. Indeed, wild-type M. tuberculosis accumulated methoxy-MA. The pellicle-defective mmaA4 mutant, on the other hand, did not accumulate methoxy-MA, instead accumulating α-MA in the detergent-extractable fraction, and this defect was reversed in the complemented strain (Fig. 5A). However, the pellicle-defective H37Rv::mmaA3 extract contained methoxy-MA at levels comparable to that of the wild type even though it does not form a pellicle (Fig. 5B). These results suggest that methoxy-MA accumulation is probably a function of the fluctuating levels of MA in response to a pellicle growth condition, and its accumulation in the wild-type pellicle is secondary to initiation of pellicle growth. Indeed, MAs are known to undergo fluctuations in relative abundance in response to changing environmental conditions, such as in vivo growth and growth under hypoxic conditions (50, 52). The common denominator in both the pellicle-defective strains was the lack of keto-MA, suggesting that keto-MA is a molecular determinant of M. tuberculosis pellicle growth.
FIG 5 .
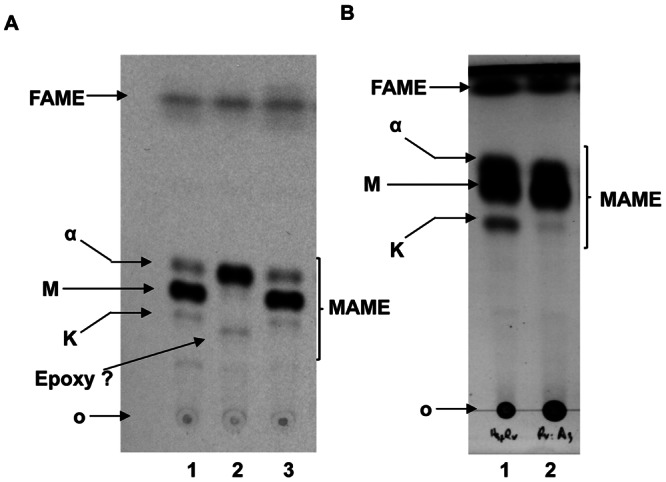
Analysis of mycolic acids under pellicle-promoting conditions. Major apolar lipids from cells grown as pellicles were derivatized as methyl esters and resolved on TLC using petroleum ether-acetone (95:5 [vol/vol]). (A) Methoxy-MA (M) is enriched in the wild-type pellicle (lane 1). The mmaA4 extract lacks both keto-MA (K) and methoxy-MA (lane 2). This defect is relieved in the complemented strain (lane 3). (B) The H37Rv::mmaA3 extract (lane 2) is deficient in keto-mycolates relative to the wild-type H37Rv (lane 1) but retains wild-type levels of methoxy-MA. O, origin; FAME, fatty acid methyl esters; MAME, mycolic acid methyl esters. This shows that methoxy mycolate, despite being a major constituent of mycobacterial pellicle, is dispensable for growth as a pellicle, and keto-mycolate, despite being a minor constituent, is essential.
Taken together, these genetic and biochemical studies demonstrate an essential role for keto-MA in M. tuberculosis pellicle growth. The fact that keto-MA, a minor MA component in both the planktonic and pellicle growth conditions assayed, dictates the ability to grow, as a pellicle confirms the vital role of this MA in organizing the mycobacterial outer membrane. Studies of the conformation adopted by the MAs have revealed keto-MA to be an extraordinarily stable W-shaped molecule in which the meromycolate chain is folded back on itself (53, 54). The stability of this conformation, which is not matched by the methoxy- and α-MA, may underpin the unique requirement for keto-MA for pellicle formation. Additionally, the presence of the nonbonded electron pair on the oxygen moiety could act as an anchoring point for hydrogen bonding of loosely associated cell wall components (55).
Growth within pellicle restores drug tolerance.
Interestingly, the H37RvΔmmaA4 mutant exhibited a loss of tolerance to rifampicin (RIF) under planktonic growth conditions (Fig. 6A), and this decreased tolerance to a lipophilic drug (RIF) is likely not due to increased permeability (56). The availability of a mutant (the ΔmmaA4 strain) that is not only defective in pellicle formation but also inherently hypersensitive to a drug (RIF) provides us with a tool to address the question of pellicle-associated drug tolerance. First, wild-type H37Rv and the ΔmmaA4 mutant were grown under pellicle-promoting conditions for three weeks, at which point the wild type forms a mature pellicle, and then the cultures were treated with RIF for seven days. As previously described by Ojha et al. (27), RIF was ineffective against wild-type H37Rv in the pellicle, whereas the bactericidal activity of the drug against the mutant was retained (Fig. 6B). This suggests that it is the growth within the pellicle and not merely the conditions of growth that results in tolerance to the drug. Second, we cocultured the wild-type and mutant strains under pellicle-promoting conditions. This resulted in the incorporation of the mutant into the wild-type pellicle, as observed by confocal microscopy (see Fig. S1 in the supplemental material). We then treated the mixed pellicle composed of H37Rv and the ΔmmaA4 mutant with RIF for seven days, after which the pellicle was harvested and plated on solid medium containing hygromycin (ΔmmaA4 selectable marker) or with no drug added. The ΔmmaA4 mutant could be recovered from the treated mixed pellicle at proportions similar to those from the untreated control and at levels similar to those of the wild type from the treated mixed pellicles (Fig. 6C). These results imply that growth within the pellicle environment confers drug tolerance, and identifying pathways involved in this adaptation could result in the discovery of mechanisms contributing to tolerance and survival under stress.
FIG 6 .

Growth within the pellicle confers protection to drug-intolerant M. tuberculosis strains. (A) H37Rv (upside down triangle), H37RvΔmmaA4 (diamond), and the complemented strain (star), grown planktonically in Middlebrook 7H9 medium containing RIF (2 µg/ml). Untreated controls of H37Rv (circle), H37RvΔmmaA4 (square), and the complemented strain (triangle) were included. CFUs were enumerated at the indicated time points by plating aliquots onto Middlebrook 7H10 agar plates. Results are representative of at least two independent experiments. (B) H37Rv (black bar) and H37RvΔmmaA4 (gray bar) strains were grown in Sauton’s medium under pellicle-promoting conditions for 3 weeks, followed by RIF (5 µg/ml) treatment for 1 week. CFU were determined by plating aliquots onto Middlebrook 7H10 agar plates. (C) H37Rv (black bar) and H37RvΔmmaA4 (Gray bar) strains were grown in coculture in Sauton’s medium under pellicle-promoting conditions for 3 weeks, followed by treatment with RIF (5 µg/ml) for 1 week. CFU proportions of H37Rv and H37RvΔmmaA4 strains in the mixed pellicle were determined by plating aliquots onto Middlebrook 7H10 agar plates with or without hygromycin (50 µg/ml), H37RvΔmmaA4 selectable marker. Experiments were performed in triplicate.
DISCUSSION
Mycobacterial persistence—against drug and immune intervention—is the most significant challenge to the control of TB. Persistence is a complex phenotype that allows the cells to enter into a physiological state that can resist killing induced by drugs (57) or a bactericidal immune response (58). While persistence in vitro may be induced by a heterogeneous set of cues, such as hypoxia or starvation, this work has focused on the ability of M. tuberculosis to grow as a pellicle, a form of biofilm.
Our studies on in vitro growth of M. tuberculosis as a pellicle have demonstrated that keto-MA is required for M. tuberculosis to adapt to pellicle growth. An essential role for keto-MA in growth within the natural host cells has been described (50). Indeed, there is no known naturally occurring, slow-growing mycobacterial strain that is defective exclusively in keto-MA, suggesting an important role for this MA species in mycobacterial pathogenesis. This view is supported by the fact that mmaA4-knockout strains of M. tuberculosis are highly attenuated in vivo (56). Interestingly, previous studies on transcriptional profiling of M. tuberculosis in the lungs of human TB patients have shown that the mmaA4 gene is upregulated in vivo (59). A recent study demonstrated that mycolic acid modification by MmaA4 renders M. tuberculosis capable of repressing interleukin 12 (IL-12) production, thereby assisting the bacterium in establishing chronic infection, since the attenuation depends on IL-12p40-mediated immunity (60). Taken together, these studies strongly suggest that identifying molecules targeting the keto-MA biosynthetic pathway may result in the development of new therapeutics against M. tuberculosis.
We also demonstrate that growth within the pellicle biofilm confers drug tolerance. The modality of this tolerance is still unclear. One possible mechanism for biofilm-mediated drug tolerance is the inability of antibiotics/drugs to penetrate the biofilm, which in itself is a formidable physical barrier (61). This has been shown to be true in some cases, such as the penetration of piperacillin into P. aeruginosa biofilm (62). However, in other cases, such as in S. epidermidis and E. coli biofilms, penetration of RIF, vancomycin, and tetracycline did not seem to be affected (63, 64). The ability of the biofilm environment to inactivate a drug at a rate that is faster than the diffusion rate of the drugs is another physical way by which the biofilm matrix can act as a barrier. Examples include the enhanced levels of beta-lactamases in Klebsiella pneumoniae biofilms, which results in the deactivation of beta-lactam antibiotics, such as ampicillin (65), and the inability of oxygen radicals to effectively penetrate the biofilm barrier (66, 67). Another model for biofilm drug tolerance proposes that bacteria within biofilms are in a nutrient-starved environment, causing the bacteria to slow down replication and enter a dormant state. As most antibacterial compounds are dependent on active replication and metabolism for their action, cells in a nongrowing state would therefore be refractory to such drugs (68). Indeed, it has recently been shown that active starvation response rather than passive growth arrest mediates antibiotic tolerance in P. aeruginosa biofilms (69). A third model for biofilm drug tolerance posits that the chemical and physiological heterogeneity within biofilms results in distinct bacterial populations with various levels of growth and metabolism (70). This would suggest that even though some of the bacteria within biofilms are in a state conducive to drug action and thus would be killed, some bacteria that exist in a state nonconducive to drug action would survive. Finally, it is thought that biofilms harbor specialized persisters, cells that are phenotypically drug resistant, and that the biofilm environment may also enrich the formation of persister cells (71, 72). Recently, it has been shown that persisters are enriched in P. aeruginosa biofilms in patients with cystic fibrosis, strongly suggesting that the recalcitrance to drug and immune elimination of P. aeruginosa infections in these patients could be due to these persisters (73). It is also possible that phenotypic resistance to antibiotics exhibited by bacteria within biofilms is multifactorial and is due to more than one of the modalities discussed above.
Nevertheless, our finding that growth within the pellicle contributes to drug tolerance suggests that the M. tuberculosis pellicle biofilm phenotype can be a convenient surrogate to identify novel in vivo-active TB drugs that can kill drug-tolerant bacteria and may help identify targets and pathways relevant to drug tolerance and survival under stress.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Planktonic cultures of mycobacteria were grown at 37°C in Middlebrook 7H9 supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC; Difco), 0.5% glycerol, and 0.05% Tween 80 or in Sauton’s liquid medium supplemented with 3.5 µM ZnSO4. Escherichia coli strains were grown at 37°C in Luria-Bertani broth (Difco). Selection of gene deletion mutants was carried out in the presence of 50 mg/liter hygromycin. Wherever necessary, mycobacteria were grown on solid Middlebrook 7H10 medium containing 10% OADC, 0.5% glycerol, and appropriate antibiotics and supplements. E. coli on solid medium was grown using Luria-Bertani agar (Difco). Mycobacterial pellicles were grown in polystyrene or polyvinyl chloride multiwell cell culture plates (Corning) by inoculating Sauton’s medium (without Tween 80) with the appropriate mycobacterial strain (with supplements where necessary) and incubating without shaking at 37°C under 5% CO2 for 3 to 5 weeks (27).
Planktonic drug-killing assay.
Exponentially growing cultures of mycobacteria were diluted in fresh medium to an optical density at 600 nm (OD600) of 0.1 to 0.2. RIF (Sigma) was added at a final concentration of 2 µg/ml. The number of CFU at the start of the experiment was estimated by plating the appropriate dilution of the culture onto 7H10 agar plates. The effect of the drug treatment on viability was monitored by plating for CFU at the various indicated time points. All experiments were carried out in triplicates.
Pellicle biofilm drug-killing assay.
Exponentially growing mycobacteria were used to inoculate biofilm medium (1:100). Pellicle biofilm was allowed to establish for three weeks, at which time RIF (5 µg/ml) was injected into the biofilm using a tuberculin syringe. CFU in pellicle biofilms were assessed by harvesting the pellicle, resuspending in 1 ml of phosphate-buffered saline (PBS) containing 0.5% (vol/vol) Tween 80, sonication, and plating appropriate dilutions on 7H10 agar plates after 7 days of treatment. All experiments were carried out in triplicate.
Mycolic acids analysis.
Cultures of M. tuberculosis in exponential growth phase (OD600 of ~0.6) were labeled with [1-14C]acetate (10 µCi; Moravek Biochemicals) for 24 h. Cells were centrifuged, washed twice with water, resuspended in 1 ml of water, and added to 1 ml of 40% of tetrabutylammonium hydroxide. The suspension was heated at 100°C for 20 h. After the suspension cooled, 100 µl of methyl iodide in 2 ml of methylene chloride was added. The mixture was stirred for 1 h at room temperature, and the organic phase was washed first with a 3 N aqueous HCl solution followed by water. The organic phase was dried. Along with the mycolic acids, the shorter-chain fatty acids are also extracted and methylated during this process (fatty acid methyl esters [FAMEs]). Samples were then resuspended in 200 µl methylene chloride (CH2Cl2), and equal cpm amounts of different samples were loaded onto a silica gel 60 F254 plate. The samples were run with hexane-ethyl acetate (95:5 [vol/vol]; 3 elutions). For two-dimensional separation of mycolic acids, TLC plates were impregnated with 10% silver nitrate solution and activated. Samples were spotted onto the activated plate and run in the first dimension using hexane-ethyl acetate (95:5 [vol/vol]) and in the second dimension using petroleum ether-diethyl ether (85:15 [vol/vol]), and the mycolic acids were visualized using autoradiography (74).
Apolar lipid analysis.
Mycobacteria grown planktonically to an OD600 of 0.5 to 0.6 or as pellicle for 3 weeks were labeled with [1-14C]acetate (Moravek Biochemicals) for 24 h (final concentration of 10 µCi), harvested by centrifugation, and washed in sterile water. Cells were resuspended in 2 ml of methanol-0.3% NaCl (10:1) and mixed with 1 ml of petroleum ether (PET) for 15 min at room temperature. The upper PET phase containing apolar lipids was collected in a glass vial. The residual solution was once again extracted of remaining apolar lipids by being mixed with PET for 15 min. The PET phase was pooled with the extract from the earlier step. The extract was dried under nitrogen at 50°C.
Apolar lipids from planktonic and pellicle cells were dissolved in 0.5 ml of dichloromethane and spotted onto a silica gel 60 F254 plate using a capillary tube. The major apolar lipids in biofilm cells which were further methylated to form methyl esters were spotted onto the TLC plates and resolved using PET-acetone (95:5 [vol/vol]).
Scanning electron microscopy.
Pellicles were fixed for 24 h with 5% glutaraldehyde, 0.2 M sodium cacodylate, 0.4 M sucrose, and 10 mM MgCl2 (pH 7.4) (mixed 1:1 with growth medium).
Osmium tetroxide vapor.
Pieces of the fixed pellicles were lifted from the fixative and placed on carbon adhesive tabs (Electron Microscopy Sciences). The tabs were placed above 0.22-µm-pore-size filter paper and were allowed to air dry (~6 h). Samples were then placed in a closed chamber with 0.25 g osmium tetroxide for 16 h.
Potassium ferrocyanide.
Pieces of the fixed pellicles were processed in suspension on a rotator. The pellicles were postfixed with 1% osmium tetroxide, 0.7% potassium ferrocyanide, 0.1 M sodium cacodylate, 0.2 M sucrose, and 5 mM MgCl2 (pH 7.4) for 30 min. Samples were buffer rinsed and dehydrated through a graded series of ethanol. They were critical point dried with hexamethyldisilazane (HMDS) (Polysciences) (4 washes: 3 times 5 minutes and the 4th overnight under a hood) until all HMDS was evaporated.
The samples processed using the above-described methods were then placed on aluminum SEM stubs, sputter coated with gold-palladium in a Denton Vacuum desk-2 sputter coater (Cherry Hill, NJ), and examined under a JEOL JSM6400 scanning electron microscope (Peabody, MA), using an accelerating voltage of 10 kV.
SUPPLEMENTAL MATERIAL
Incorporation of the ΔmmaA4 mutant within the wild-type pellicle. H37Rv::egfp (green fluorescence) and H37RVΔmmaA4::mcherry (red fluorescence) were grown individually or in coculture in Sauton’s medium under pellicle-promoting conditions. Single-strain pellicles were imaged at 3 weeks, and mixed-strain pellicles were imaged at 3 weeks and 5 weeks using confocal fluorescent microscope (Zeiss). Download
ACKNOWLEDGMENTS
We thank the personnel at the Analytical Imaging Facility (AIF) at the Albert Einstein College of Medicine for their help with the confocal microscopy and SEM and Michael Glickman for the pcaA mutant.
This work was partially supported by National Institutes of Health award R37AI026170-23 and Center for AIDS Research award P30 A151519 (W.R.J.) and 5R01AI064494-05 (University of Pittsburgh).
Footnotes
Citation Sambandan D, Dao DN, Weinrick BC, Vilchèze C, Gurcha SS, Ojha A, Kremer L, Besra GS, Hatfull GF, Jacobs WR. 2013. Keto-mycolic acid-dependent pellicle formation confers tolerance to drug-sensitive Mycobacterium tuberculosis. mBio 4(3):e00222-13. doi:10.1128/mBio.00222-13.
REFERENCES
- 1. Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322 [DOI] [PubMed] [Google Scholar]
- 2. Hall-Stoodley L, Stoodley P. 2005. Biofilm formation and dispersal and the transmission of human pathogens. Trends Microbiol. 13:7–10 [DOI] [PubMed] [Google Scholar]
- 3. Lam J, Chan R, Lam K, Costerton JW. 1980. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect. Immun. 28:546–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kulczycki LL, Murphy TM, Bellanti JA. 1978. Pseudomonas colonization in cystic fibrosis. A study of 160 patients. JAMA 240:30–34 [PubMed] [Google Scholar]
- 5. Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. 2003. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 301:105–107 [DOI] [PubMed] [Google Scholar]
- 6. Fraser DW, Tsai TR, Orenstein W, Parkin WE, Beecham HJ, Sharrar RG, Harris J, Mallison GF, Martin SM, McDade JE, Shepard CC, Brachman PS. 1977. Legionnaires’ disease: description of an epidemic of pneumonia. N. Engl. J. Med. 297:1189–1197 [DOI] [PubMed] [Google Scholar]
- 7. Higgins DA, Pomianek ME, Kraml CM, Taylor RK, Semmelhack MF, Bassler BL. 2007. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature 450:883–886 [DOI] [PubMed] [Google Scholar]
- 8. Purdy AE, Watnick PI. 2011. Spatially selective colonization of the arthropod intestine through activation of Vibrio cholerae biofilm formation. Proc. Natl. Acad. Sci. U. S. A. 108:19737–19742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oggioni MR, Trappetti C, Kadioglu A, Cassone M, Iannelli F, Ricci S, Andrew PW, Pozzi G. 2006. Switch from planktonic to sessile life: a major event in pneumococcal pathogenesis. Mol. Microbiol. 61:1196–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Archer NK, Mazaitis MJ, Costerton JW, Leid JG, Powers ME, Shirtliff ME. 2011. Staphylococcus aureus biofilms: properties, regulation, and roles in human disease. Virulence 2:445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Begun J, Gaiani JM, Rohde H, Mack D, Calderwood SB, Ausubel FM, Sifri CD. 2007. Staphylococcal biofilm exopolysaccharide protects against Caenorhabditis elegans immune defenses. PLoS Pathog. 3:e57 http://dx.doi.org/10.1371/journal.ppat.0030057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O’Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449–461 [DOI] [PubMed] [Google Scholar]
- 13. Kearns DB, Chu F, Branda SS, Kolter R, Losick R. 2005. A master regulator for biofilm formation by Bacillus subtilis. Mol. Microbiol. 55:739–749 [DOI] [PubMed] [Google Scholar]
- 14. Tu Quoc PH, Genevaux P, Pajunen M, Savilahti H, Georgopoulos C, Schrenzel J, Kelley WL. 2007. Isolation and characterization of biofilm formation-defective mutants of Staphylococcus aureus. Infect. Immun. 75:1079–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loo CY, Corliss DA, Ganeshkumar N. 2000. Streptococcus gordonii biofilm formation: identification of genes that code for biofilm phenotypes. J. Bacteriol. 182:1374–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marsollier L, Stinear T, Aubry J, Saint André JP, Robert R, Legras P, Manceau AL, Audrain C, Bourdon S, Kouakou H, Carbonnelle B. 2004. Aquatic plants stimulate the growth of and biofilm formation by Mycobacterium ulcerans in axenic culture and harbor these bacteria in the environment. Appl. Environ. Microbiol. 70:1097–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamazaki Y, Danelishvili L, Wu M, Hidaka E, Katsuyama T, Stang B, Petrofsky M, Bildfell R, Bermudez LE. 2006. The ability to form biofilm influences Mycobacterium avium invasion and translocation of bronchial epithelial cells. Cell. Microbiol. 8:806–814 [DOI] [PubMed] [Google Scholar]
- 18. Hall-Stoodley L, Brun OS, Polshyna G, Barker LP. 2006. Mycobacterium marinum biofilm formation reveals cording morphology. FEMS Microbiol. Lett. 257:43–49 [DOI] [PubMed] [Google Scholar]
- 19. Recht J, Kolter R. 2001. Glycopeptidolipid acetylation affects sliding motility and biofilm formation in Mycobacterium smegmatis. J. Bacteriol. 183:5718–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teng R, Dick T. 2003. Isoniazid resistance of exponentially growing Mycobacterium smegmatis biofilm culture. FEMS Microbiol. Lett. 227:171–174 [DOI] [PubMed] [Google Scholar]
- 21. Marsollier L, Brodin P, Jackson M, Korduláková J, Tafelmeyer P, Carbonnelle E, Aubry J, Milon G, Legras P, André JP, Leroy C, Cottin J, Guillou ML, Reysset G, Cole ST. 2007. Impact of Mycobacterium ulcerans biofilm on transmissibility to ecological niches and Buruli ulcer pathogenesis. PLoS Pathog. 3:e62 http://dx.doi.org/10.1371/journal.ppat.0030062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feazel LM, Baumgartner LK, Peterson KL, Frank DN, Harris JK, Pace NR. 2009. Opportunistic pathogens enriched in showerhead biofilms. Proc. Natl. Acad. Sci. U. S. A. 106:16393–16399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamazaki Y, Danelishvili L, Wu M, Macnab M, Bermudez LE. 2006. Mycobacterium avium genes associated with the ability to form a biofilm. Appl. Environ. Microbiol. 72:819–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Röse L, Kaufmann SH, Daugelat S. 2004. Involvement of Mycobacterium smegmatis undecaprenyl phosphokinase in biofilm and smegma formation. Microbes Infect. 6:965–971 [DOI] [PubMed] [Google Scholar]
- 25. Ojha A, Anand M, Bhatt A, Kremer L, Jacobs WR, Hatfull GF. 2005. GroEL1: a dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell 123:861–873 [DOI] [PubMed] [Google Scholar]
- 26. Ojha AK, Trivelli X, Guerardel Y, Kremer L, Hatfull GF. 2010. Enzymatic hydrolysis of trehalose dimycolate releases free mycolic acids during mycobacterial growth in biofilms. J. Biol. Chem. 285:17380–17389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, Guerardel Y, Alahari A, Kremer L, Jacobs WR, Hatfull GF. 2008. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol. Microbiol. 69:164–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Darzins E, Fahr G. 1956. Cord-forming property, lethality and pathogenicity of Mycobacteria. Dis. Chest 30:642–648 [DOI] [PubMed] [Google Scholar]
- 29. Middlebrook G, Dubos RJ, Pierce C. 1947. Virulence and morphological characteristics of mammalian tubercle bacilli. J. Exp. Med. 86:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glickman MS, Cox JS, Jacobs WR., Jr. 2000. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol. Cell 5:717–727 [DOI] [PubMed] [Google Scholar]
- 31. Canetti G. 1954. The tubercle bacillus in the pulmonary lesion of man. Springer Publishing Company, New York, NY. [Google Scholar]
- 32. Lenaerts AJ, Hoff D, Aly S, Ehlers S, Andries K, Cantarero L, Orme IM, Basaraba RJ. 2007. Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob. Agents Chemother. 51:3338–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamamoto K, Arai H, Ishii M, Igarashi Y. 2011. Trade-off between oxygen and iron acquisition in bacterial cells at the air-liquid interface. FEMS Microbiol. Ecol. 77:83–94 [DOI] [PubMed] [Google Scholar]
- 34. Fenchel T. 2002. Microbial behavior in a heterogeneous world. Science 296:1068–1071 [DOI] [PubMed] [Google Scholar]
- 35. Wayne LG. 1994. Cultivation of Mycobacterium tuberculosis for research purposes, p 73–83 In Bloom BR, Tuberculosis: pathogenesis, protection, and control. ASM Press, Washington, DC [Google Scholar]
- 36. Lehmann KB, Neumann R. 1896. Atlas und grundriss der bacteriologie und lehrbuch der speziellen bacteriologischen diagnostik, 1st ed. J. F. Lehmann, München, Germany [Google Scholar]
- 37. Darzins E. 1958. The bacteriology of tuberculosis. University of Minnesota Press, Minneapolis, MN. [Google Scholar]
- 38. Branda SS, Vik S, Friedman L, Kolter R. 2005. Biofilms: the matrix revisited. Trends Microbiol. 13:20–26 [DOI] [PubMed] [Google Scholar]
- 39. Christensen BB, Sternberg C, Andersen JB, Palmer RJ, Nielsen AT, Givskov M, Molin S. 1999. Molecular tools for study of biofilm physiology. Methods Enzymol. 310:20–42 [DOI] [PubMed] [Google Scholar]
- 40. O’Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30:295–304 [DOI] [PubMed] [Google Scholar]
- 41. Friedman L, Kolter R. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 51:675–690 [DOI] [PubMed] [Google Scholar]
- 42. Kobayashi K. 2007. Bacillus subtilis pellicle formation proceeds through genetically defined morphological changes. J. Bacteriol. 189:4920–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Spiers AJ, Bohannon J, Gehrig SM, Rainey PB. 2003. Biofilm formation at the air-liquid interface by the Pseudomonas fluorescens SBW25 wrinkly spreader requires an acetylated form of cellulose. Mol. Microbiol. 50:15–27 [DOI] [PubMed] [Google Scholar]
- 44. Mah TF, O’Toole GA. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 9:34–39 [DOI] [PubMed] [Google Scholar]
- 45. Noll H, Bloch H, Asselineau J, Lederer E. 1956. The chemical structure of the cord factor of Mycobacterium tuberculosis. Biochim. Biophys. Acta 20:299–309 [DOI] [PubMed] [Google Scholar]
- 46. Bhatt A, Fujiwara N, Bhatt K, Gurcha SS, Kremer L, Chen B, Chan J, Porcelli SA, Kobayashi K, Besra GS, Jacobs WR. 2007. Deletion of kasB in mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc. Natl. Acad. Sci. U. S. A. 104:5157–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Behr MA, Schroeder BG, Brinkman JN, Slayden RA, Barry CE. 2000. A point mutation in the mma3 gene is responsible for impaired methoxymycolic acid production in Mycobacterium bovis BCG strains obtained after 1927. J. Bacteriol. 182:3394–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takayama K, Wang C, Besra GS. 2005. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 18:81–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Verschoor JA, Baird MS, Grooten J. 2012. Towards understanding the functional diversity of cell wall mycolic acids of Mycobacterium tuberculosis. Prog. Lipid Res. 51:325–339 [DOI] [PubMed] [Google Scholar]
- 50. Yuan Y, Zhu Y, Crane DD, Barry CE. 1998. The effect of oxygenated mycolic acid composition on cell wall function and macrophage growth in Mycobacterium tuberculosis. Mol. Microbiol. 29:1449–1458 [DOI] [PubMed] [Google Scholar]
- 51. Dinadayala P, Laval F, Raynaud C, Lemassu A, Laneelle MA, Laneelle G, Daffe M. 2003. Tracking the putative biosynthetic precursors of oxygenated mycolates of Mycobacterium tuberculosis. Structural analysis of fatty acids of a mutant strain deviod of methoxy- and ketomycolates. J. Biol. Chem. 278:7310–7319 [DOI] [PubMed] [Google Scholar]
- 52. Davidson LA, Draper P, Minnikin DE. 1982. Studies on the mycolic acids from the walls of Mycobacterium microti. J. Gen. Microbiol. 128:823–828 [DOI] [PubMed] [Google Scholar]
- 53. Villeneuve M, Kawai M, Kanashima H, Watanabe M, Minnikin DE, Nakahara H. 2005. Temperature dependence of the Langmuir monolayer packing of mycolic acids from Mycobacterium tuberculosis. Biochim. Biophys. Acta 1715:71–80 [DOI] [PubMed] [Google Scholar]
- 54. Villeneuve M, Kawai M, Watanabe M, Aoyagi Y, Hitotsuyanagi Y, Takeya K, Gouda H, Hirono S, Minnikin DE, Nakahara H. 2007. Conformational behavior of oxygenated mycobacterial mycolic acids from Mycobacterium bovis BCG. Biochim. Biophys. Acta 1768:1717–1726 [DOI] [PubMed] [Google Scholar]
- 55. Minnikin DE. 1982. Lipids: complex lipids, their chemistry, biosynthesis and roles, p 95–184 In Ratledge C, Standford J, The biology of the mycobacteria. Academic Press, London, United Kingdom [Google Scholar]
- 56. Dubnau E, Chan J, Raynaud C, Mohan VP, Lanéelle MA, Yu K, Quémard A, Smith I, Daffé M. 2000. Oxygenated mycolic acids are necessary for virulence of Mycobacterium tuberculosis in mice. Mol. Microbiol. 36:630–637 [DOI] [PubMed] [Google Scholar]
- 57. McCune RM, Jr, Tompsett R. 1956. Fate of mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. I. The persistence of drug-susceptible tubercle bacilli in the tissues despite prolonged antimicrobial therapy. J. Exp. Med. 104:737–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Flynn JL, Chan J. 2001. Tuberculosis: latency and reactivation. Infect. Immun. 69:4195–4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rachman H, Strong M, Ulrichs T, Grode L, Schuchhardt J, Mollenkopf H, Kosmiadi GA, Eisenberg D, Kaufmann SH. 2006. Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect. Immun. 74:1233–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dao DN, Sweeney K, Hsu T, Gurcha SS, Nascimento IP, Roshevsky D, Besra GS, Chan J, Porcelli SA, Jacobs WR. 2008. Mycolic acid modification by the mmaA4 gene of M. tuberculosis modulates IL-12 production. PLoS Pathog. 4:e1000081 http://dx.doi.org/10.1371/journal.ppat.1000081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stewart PS. 2003. Diffusion in biofilms. J. Bacteriol. 185:1485–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hoyle BD, Alcantara J, Costerton JW. 1992. Pseudomonas aeruginosa biofilm as a diffusion barrier to piperacillin. Antimicrob. Agents Chemother. 36:2054–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dunne WM, Jr, Mason EO, Jr, Kaplan SL. 1993. Diffusion of rifampin and vancomycin through a Staphylococcus epidermidis biofilm. Antimicrob. Agents Chemother. 37:2522–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stone G, Wood P, Dixon L, Keyhan M, Matin A. 2002. Tetracycline rapidly reaches all the constituent cells of uropathogenic Escherichia coli biofilms. Antimicrob. Agents Chemother. 46:2458–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Anderl JN, Franklin MJ, Stewart PS. 2000. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 44:1818–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. De Beer D, Srinivasan R, Stewart PS. 1994. Direct measurement of chlorine penetration into biofilms during disinfection. Appl. Environ. Microbiol. 60:4339–4344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu X, Stewart PS, Chen X. 1996. Transport limitation of chlorine disinfection of Pseudomonas aeruginosa entrapped in alginate beads. Biotechnol. Bioeng. 49:93–100 [DOI] [PubMed] [Google Scholar]
- 68. Brown MR, Allison DG, Gilbert P. 1988. Resistance of bacterial biofilms to antibiotics: a growth-rate related effect? J. Antimicrob. Chemother. 22:777–780 [DOI] [PubMed] [Google Scholar]
- 69. Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, Britigan BE, Singh PK. 2011. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334:982–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Boles BR, Thoendel M, Singh PK. 2004. Self-generated diversity produces “insurance effects” in biofilm communities. Proc. Natl. Acad. Sci. U. S. A. 101:16630–16635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lewis K. 2005. Persister cells and the riddle of biofilm survival. Biochemistry 70:267–274 [DOI] [PubMed] [Google Scholar]
- 72. Roberts ME, Stewart PS. 2005. Modelling protection from antimicrobial agents in biofilms through the formation of persister cells. Microbiology 151:75–80 [DOI] [PubMed] [Google Scholar]
- 73. Mulcahy LR, Burns JL, Lory S, Lewis K. 2010. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 192:6191–6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vilchèze C, Jacobs WR. 2007. Isolation and analysis of Mycobacterium tuberculosis mycolic acids. Curr. Protoc. Microbiol. Chapter 10:Unit 10A.3 [DOI] [PubMed] [Google Scholar]
- 75. Alahari A, Alibaud L, Trivelli X, Gupta R, Lamichhane G, Reynolds RC, Bishai WR, Guerardel Y, Kremer L. 2009. Mycolic acid methyltransferase, MmaA4, is necessary for thiacetazone susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 71:1263–1277 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Incorporation of the ΔmmaA4 mutant within the wild-type pellicle. H37Rv::egfp (green fluorescence) and H37RVΔmmaA4::mcherry (red fluorescence) were grown individually or in coculture in Sauton’s medium under pellicle-promoting conditions. Single-strain pellicles were imaged at 3 weeks, and mixed-strain pellicles were imaged at 3 weeks and 5 weeks using confocal fluorescent microscope (Zeiss). Download