

Background: The role of HDAC6 in mutant SOD1 aggregation and ALS etiology is unclear.
Results: HDAC6 interacted with mutant SOD1 via two SMIR motifs, and HDAC6 knockdown promoted aggregation of mutant SOD1.
Conclusion: Mutant SOD1 can modulate HDAC6 activity and increase tubulin acetylation, which, in turn, facilitates mutant SOD1 aggregation.
Significance: HDAC6 deficiency might be a converging point of various subtypes of ALS.
Keywords: Amyotrophic Lateral Sclerosis (Lou Gehrig's Disease), Histone Deacetylase, Neurodegenerative Diseases, Protein Aggregation, Superoxide Dismutase (SOD), Cu/Zn Superoxide Dismutase (SOD1) (SOD1), Histone Deacetylase 6, p62/Sequestosome 1
Abstract
Histone deacetylase 6 (HDAC6) is a tubulin deacetylase that regulates protein aggregation and turnover. Mutations in Cu/Zn superoxide dismutase (SOD1) linked to familial amyotrophic lateral sclerosis (ALS) make the mutant protein prone to aggregation. However, the role of HDAC6 in mutant SOD1 aggregation and the ALS etiology is unclear. Here we report that HDAC6 knockdown increased mutant SOD1 aggregation in cultured cells. Different from its known role in mediating the degradation of poly-ubiquitinated proteins, HDAC6 selectively interacted with mutant SOD1 via two motifs similar to the SOD1 mutant interaction region (SMIR) that we identified previously in p62/sequestosome 1. Expression of the aggregation-prone mutant SOD1 increased α-tubulin acetylation, and the acetylation-mimicking K40Q α-tubulin mutant promoted mutant SOD1 aggregation. Our results suggest that ALS-linked mutant SOD1 can modulate HDAC6 activity and increase tubulin acetylation, which, in turn, facilitates the microtubule- and retrograde transport-dependent mutant SOD1 aggregation. HDAC6 impairment might be a common feature in various subtypes of ALS.
Introduction
Amyotrophic lateral sclerosis (ALS,2 also known as Lou Gehrig's disease) is a progressive and fatal neurodegenerative disease that primarily affects motor neurons (1). A hallmark of ALS is the appearance of cytoplasmic protein inclusions in affected motor neurons and glial cells (2, 3). To date, well over 100 independent mutations in the copper/zinc superoxide dismutase (SOD1) were reported to cause ALS, accounting for ∼20% of familial ALS (1, 4–6).
In the familial ALS cases caused by SOD1 mutations, the cytoplasmic protein inclusions are positive for SOD1 (3, 7, 8), and most ALS SOD1 mutants are prone to aggregation (9). We have shown that aggregation of mutant SOD1 in cultured cells is facilitated by the dynein-mediated retrograde transport and that aggregation can impair axonal transport (10, 11). We have also shown that p62/sequestosome 1, which plays a critical role in the aggregation and turnover of misfolded proteins (12, 13), selectively interacted with mutant SOD1 (14, 15). The p62-mutant SOD1 interaction is mediated through a motif termed SOD1 mutant interaction region (SMIR) and is critical to recognizing mutant SOD1 and targeting it to autophagosomes for degradation (15).
Histone deacetylase 6 (HDAC6) is critical to the aggregation of poly-ubiquitinated proteins induced by proteasome inhibition (16) as well as the autophagic degradation of such aggregates (17). HDAC6 is a unique member in the HDAC family as it is localized almost exclusively in the cytoplasm instead of in the nucleus (18). Its most abundant known physiological substrate is acetyl-α-tubulin (19–21). The domain structure of HDAC6 is also unusual as it possesses two lysine deacetylase domains, and the second deacetylase domain (DAC2) is responsible for the deacetylation of acetyl-α-tubulin (22–24). HDAC6 was proposed to be an adaptor between poly-ubiquitinated misfolded proteins and dynein motors, facilitating the retrograde transport of such poly-ubiquitinated proteins to form aggresome (16). HDAC6 was shown to promote autophagy by recruiting cortactin to polyubiquitinated protein aggregates, leading to local actin remodeling and stimulation of autophagosome/lysosome fusion (17). In addition, HDAC6 was implicated in the aggregation and degradation of expanded huntingtin (25), α-synuclein oligomers (26), and the tau protein (27–29). However, the role of HDAC6 in mutant SOD1 aggregation and ALS has not yet been examined.
We hypothesized that HDAC6 could play a role in regulating mutant SOD1 protein aggregation and turnover in ALS and set forth to test this hypothesis in a cellular model system. We found that HDAC6 knockdown increased mutant SOD1 aggregation as demonstrated by live cell imaging and cellulose acetate membrane filtration. HDAC6 interacted selectively with mutant SOD1, and the ubiquitin-binding zinc finger of HDAC6 was dispensable for the interaction. Instead, we identified two separate motifs that are highly similar to the SMIR motif of p62/SQSTM1 (15). Our results suggest that HDAC6 and p62/SQSTM1, two proteins involved in protein aggregation and turnover, share a mechanism to recognize aggregation-prone proteins that is independent of their ubiquitin binding domains.
The expression of mutant SOD1 resulted in elevation of the acetylation of α-tubulin, consistent with the inhibition of the tubulin deacetylase activity of HDAC6. HDAC6 was sequestered into mutant SOD1 aggregates, providing a potential mechanism for the inhibition of its deacetylase activity. The coexpression of the acetylation-mimicking K40Q α-tubulin mutant with mutant SOD1 resulted in elevation of the cellulose acetate-filtered amount of SOD1 aggregates. On the basis of the results in this study, we propose that the aggregation of mutant SOD1 is enhanced through the inhibition of the tubulin deacetylase activity of HDAC6, which, in turn, increases tubulin acetylation and enhances retrograde transport of mutant SOD1 to form large inclusions.
MATERIALS AND METHODS
Plasmids
The SOD1 expression constructs have been reported previously (11, 14, 15). The HDAC6 expression constructs used in this study were based on the pcDNA3-FLAG-mouse HDAC6 plasmid (a gift from Dr. Marie Wooten, Auburn University) and were generated using standard cloning techniques. The monomeric and tetrameric DsRed fusions were constructed by cloning HDAC6 fragments into the pDsRed-monomer-C1 and the pDsRed2-C1 vectors (Clontech), respectively. The Myc-tagged WT and K40Q α-tubulin constructs were generated by subcloning the respective tubulin fragments of the GFP-Tubulin WT and K40Q plasmids (Addgene, plasmids 30487 and 32912) (30) into pCMV-Myc (Clontech). The pMAL-C1 plasmid, the mammalian expression construct for maltose binding protein (MBP) fusions was generated by replacing enhanced GFP of pEGFP-C1 (Clontech) with the malE gene from pMAL-c2G (New England Biolabs), preserving the reading frame of pEGFP-C1. All plasmid constructs were verified with sequencing.
Cell Culture and Transfection
The NSC34 and HEK293 cells were cultured in DMEM (Invitrogen, catalog no. 11965) with 10% fetal bovine serum and penicillin-streptomycin at 37 °C in 5% CO2/95% air. The transfections were performed in a 6-well plate format. For the HDAC6 knockdown studies, the cells were transfected using 0.75 μg of plasmid DNA, 20 pmol of HDAC6 siRNA (Dharmacon, siGenome SmartPool M-003499-00-0005) or non-targeting siRNA (Dharmacon, d-001210-02-05) and 5 μl of Lipofectamine 2000 (Invitrogen) per well following the instructions of the manufacturer. For all other studies, HEK293 cells were transfected using polyethylenimine “Max” (Polysciences, Inc.). In the plasmid cotransfection experiments, 0.5μg of both plasmids was transfected unless noted otherwise. The NSC34 cells were transfected using Lipofectamine (Invitrogen).
Animals
Transgenic mouse strains overexpressing WT (B6.Cg-Tg(SOD1)2Gur/J) or G93A mutant SOD1 (B6.Cg-Tg(SOD1-G93A)1Gur/J) (31) were bred and maintained as hemizygotes at the University of Kentucky animal facility. Transgenic mice were identified using PCR. The mice were sacrificed at age 90 ± 5 days. Mice were anesthetized with an intraperitoneal injection of 0.1 ml pentobarbital (50 mg/ml, Abbott Laboratories) and perfused transcardially with 0.1 m PBS (pH 7.5) before spinal cords were dissected. All animal procedures were approved by the university IACUC committee.
Fluorescence Microscopy
The live cell imaging of the cells transfected with SOD1-GFP was performed using a Zeiss Axiovert 100 microscope, counting the transfected cells and cells with inclusions in ten random view fields 3 days post-transfection. The confocal microscopy experiments were performed as follows. NSC34 or HEK293 cells were seeded on gelatin-treated glass coverslips and transfected with SOD1-GFP constructs. Two days later, the cells were fixed in −20 °C methanol and then rehydrated in 1× PBS. The cells were permeabilized with 1× PBS supplemented with 0.1% Triton X-100. Spinal cords of 90-day-old transgenic mice were dissected, postfixed in 4% paraformaldehyde in 1× PBS for 3 h, cryopreserved in 30% sucrose overnight, embedded in Tissue-Tek OCT compound (Sakura), and then 12-μm sections were cut. The sections were permeabilized with 1× PBS supplemented with 0.1% Triton X-100. The primary antibodies were mouse anti-acetyl-α-tubulin (clone 6-11B-1, Sigma, catalog no. T6793), mouse anti-cortactin (clone 4F11, Millipore, catalog no. 05-180), sheep anti-human SOD1 (The Binding Site, catalog no. PC077), rabbit anti-human HDAC6 (Santa Cruz Biotechnology, Inc., catalog no. sc-11420), and rabbit anti-mouse HDAC6 (a gift from Dr. Tso-Pang Yao, Duke University). The secondary antibodies were Alexa Fluor 568 donkey anti-mouse (Invitrogen, catalog no. A10037), Alexa Fluor 594 donkey anti-mouse (Invitrogen, catalog no. A21203), Alexa Fluor 568 donkey anti-rabbit (Invitrogen, catalog no. A10042), and Alexa Fluor 488 donkey anti-sheep (Invitrogen, catalog no. A11015). The F-actin staining experiments were performed by fixing the cells in 3.7% paraformaldehyde, permeabilizing with 0.1% Triton X-100, and staining with Texas Red-X phalloidin (Invitrogen, catalog no. T7471). The samples were mounted by applying Vectashield mounting medium (Vector Laboratories) and visualized using a Nikon A1 (acetyl-α-tubulin, HDAC6, and SOD1) or a Leica SP5 (cortactin and F-actin) confocal microscope with a ×60 objective.
Cellulose Acetate Membrane Filtration
The transfected cells were rinsed with 1× PBS, and cell extracts were prepared in 1× radioimmune precipitation assay buffer (Millipore) supplemented with protease inhibitor mixture (Sigma, catalog no. P-8340, 1:500), 1 mm sodium orthovanadate, 1.5 μm trichostatin A, 20 mm nicotinamide, and 20 mm sodium butyrate. The spinal cord extracts were prepared using a dounce homogenizer in the same lysis buffer. The extracts were cleared with two consecutive centrifugations at 800 × g for 10 min at 4 °C. The cellulose acetate membrane (0.2-μm pore size, Whatman, catalog no. 10404131) was prewetted in TBS (100 mm Tris-HCl (pH 7.5), 0.9% NaCl) before use. From each sample, 60 μg of total protein in 150 μl of total volume was filtered through the cellulose acetate membrane using the Bio-Dot apparatus (Bio-Rad) using mild “house” vacuum. The membranes were washed three times with 200 μl of TBS/well and blocked with 5% milk in TBST (TBS with 0.1% Tween 20) before blotting.
Coprecipitation Assays
The immunoprecipitation and pull-down experiments were done 2 days post-transfection. The transfected cell extracts were prepared in 1× radioimmune precipitation assay buffer (Millipore) supplemented with protease inhibitor mixture (Sigma, catalog no. P-8340, 1:500) and 1 mm sodium orthovanadate. The extracts for the HA and FLAG immunoprecipitations were also supplemented with 0.2 mm PMSF and 0.625 mg/ml N-ethylmaleimide. The extracts for the Myc-α-tubulin immunoprecipitations were also supplemented with 1.5 μm trichostatin A, 20 mm nicotinamide, and 20 mm sodium butyrate. The FLAG immunoprecipitations were performed using EZview Red anti-FLAG M2 affinity gel (Sigma, catalog no. F2426). The HA and Myc immunoprecipitations were done using protein G-Sepharose (GE Healthcare, catalog no. 17-0618-01) and mouse anti-HA (Santa Cruz Biotechnology, Inc., catalog no. sc-7392) or mouse anti-Myc (Santa Cruz Biotechnology, Inc., catalog no. sc-40) antibodies, respectively. The MBP pull-downs were performed using amylose resin (New England Biolabs, catalog no. E8021S).
Western Blotting
The nitrocellulose membranes were blocked, and antibodies were applied in 5% milk/TBST with the exception of the DsRed antibody, which was applied in PBST (1× PBS with 0.1% Tween 20). The antibodies used were mouse anti-FLAG M2-HRP (Sigma, catalog no. A8592), rabbit anti-HA (Santa Cruz Biotechnology, Inc., catalog no. sc-805), mouse anti-Myc (clone 9E10, Santa Cruz Biotechnology, Inc., catalog no. sc-40), rabbit anti-DsRed (Clontech, catalog no. 632496), rabbit anti-GFP (Santa Cruz Biotechnology, Inc., catalog no. sc-8334), mouse anti-MBP (New England Biolabs, catalog no. E8032S), mouse anti-α-tubulin (clone DM1A, Santa Cruz Biotechnology, Inc., catalog no. sc-32293), mouse anti-acetyl-α-tubulin (clone 6-11B-1, Sigma, catalog no. T6793), rabbit anti-human HDAC6 (Santa Cruz Biotechnology, Inc., catalog no. sc-11420), and rabbit anti-mouse HDAC6 and goat anti-actin (Santa Cruz Biotechnology, Inc., catalog no. sc-1616).
Native Gel Electrophoresis
The native gel electrophoresis was performed using 4–20% Mini-Protean TGX precast gradient gels (Bio-Rad) under non-denaturing conditions as reported before (15).
Mass Spectrometric Identification of Mutant SOD1 in the MBP-HDAC6 (L816-S859) Pull-down Sample
The MBP pull-down samples were subjected to SDS-PAGE. The gel was stained with Sypro Ruby protein gel stain (Invitrogen), and the respective bands were excised and subjected to in-gel trypsin digestion (32). The tryptic digestion peptides were analyzed using an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific) as described previously (33).
Statistical Analysis
Statistical analyses were performed using Student's t test (two-tailed distribution, two-sample unequal variance). The p values were calculated and are shown where needed.
RESULTS
HDAC6 Regulates the Level of Mutant SOD1 Aggregates
HDAC6 plays a role in both the formation and the degradation of ubiquitin-positive protein aggregates induced by proteasome inhibition (16, 17). To test whether HDAC6 affects the level of mutant SOD1 aggregates, we cotransfected HDAC6 siRNA or control siRNA with plasmids expressing GFP-tagged WT or ALS mutants of SOD1. The percentage of transfected cells that developed visible mutant SOD1 inclusions was assessed using live-cell fluorescence microscopy. The SOD1-GFP levels varied up to 30% with HDAC6 knockdown (Fig. 1D). The percentages of transfected cells with inclusions were normalized with the variation of mutant SOD1 levels and are shown in Fig. 1A. The proportion of cells with GFP-tagged mutant SOD1 inclusions was significantly higher with HDAC6 knockdown in the case of the A4V (p = 0.0076) and G93A (p = 0.0099) mutants but not with the G85R mutant.
FIGURE 1.

The effect of HDAC6 knockdown on the aggregation of mutant SOD1. A, HEK293 cells were cotransfected with plasmids expressing GFP-tagged WT, A4V, G85R, or G93A SOD1 and HDAC6 siRNA or control siRNA. Three days later, the cells with visible SOD1-GFP inclusions were counted in ten random view fields, and the percentage of all transfected cells having visible GFP-positive inclusions was normalized with the expression level variations with or without HDAC6 knockdown (D). Data are mean ± S.E. from all view fields in three independent experiments. B, cellulose acetate membrane filtration assay of SOD1-GFP aggregates from cells treated as in A. The membrane was blotted with GFP antibody. C, densitometric quantification of B. ImageJ was used to analyze the densitometric intensities of the dots that were normalized to the A4V/control siRNA dot and normalized with the expression level variations. Data are mean ± S.E. from three independent experiments. D, protein extracts from the above samples. IB, immunoblot.
The amount of aggregated SOD1 with or without HDAC6 knockdown was also examined using the cellulose acetate membrane filtration method. The cellulose acetate membrane filtration data (Fig. 1B) were also normalized with the variation of SOD1-GFP levels and are shown in Fig. 1C. The amount of aggregated mutant SOD1 captured by the membrane was significantly higher when the plasmids were cotransfected with HDAC6 siRNA in the case of all three tested mutants (A4V, p = 0.041; G85R, p = 0.0076; and G93A, p = 0.034).
The Western blotting of total protein extracts showed successful HDAC6 knockdown and corresponding accumulation of a major HDAC6 substrate acetyl-α-tubulin (Fig. 1D). The α-tubulin and actin protein levels were comparable in all cells with or without HDAC6 siRNA (Fig. 1D). The above results suggest that HDAC6 can modulate the level of mutant SOD1 aggregates.
HDAC6 Selectively Interacts with Mutant SOD1
We next tested whether HDAC6 was capable of discriminating ALS mutants of SOD1 from the WT protein. HA-tagged WT or mutant SOD1 was cotransfected with FLAG-tagged HDAC6, and FLAG immunoprecipitations were performed. HDAC6 selectively coprecipitated with SOD1 ALS mutants but not with the WT protein from the motor neuron-like NSC34 cells (Fig. 2A) and HEK293 cells (supplemental Fig. S1A). It is noted that the SOD1 mutants coprecipitated with HDAC6 were at the expected monomeric size in denaturing gel electrophoresis and not as polyubiquitinated proteins (Fig. 2A and supplemental Fig. S1A). This is remarkably similar to our previous observations about the mutant SOD1-p62 interaction (14, 15). We also performed the reverse immunoprecipitation experiment and demonstrated the coprecipitation of endogenous HDAC6 with FLAG-tagged mutant SOD1 (supplemental Fig. S1B).
FIGURE 2.

Mutant SOD1 interacts with HDAC6 through a SMIR motif between the DAC2 and SE14 domains. A, HDAC6 interacts with the A4V SOD1 mutant but not WT SOD1. FLAG-HDAC6 and the respective SOD1–3xHA were cotransfected into NSC34 cells. FLAG-HDAC6 immunoprecipitations (IP) were performed followed by Western blot analysis (IB) using the indicated antibodies. B–E, mapping the SOD1 mutant interaction region within HDAC6. Either WT or A4V mutant SOD1 and various HDAC6 constructs were cotransfected into HEK293 cells. SOD1 immunoprecipitations were performed, followed by Western blot analysis using the indicated antibodies. Protein levels in cell extracts were also examined as controls. Ext, extract; Vec, vector control. B, the SE14 domain is required for the efficient binding of mutant SOD1 to HDAC6 because deleting SE14 domain weakened the interaction. FL, full-length. C, identification of the SMIR motif located C-terminally to DAC2 of HDAC6. The isolated SE14 domain did not interact with mutant SOD1. The Leu-816 to Leu-835 segment was the minimal sequence that was sufficient to interact with mutant SOD1. The asterisks indicate nonspecific bands or potential proteolytic fragments. D, alignments of the Leu-816 to Leu-835 motif of HDAC6 and p62 SMIR. +, positively charged; Ω, aromatic, Φ, hydrophobic amino acid residues. E, mutational analysis of the C-SMIR motif showed that the conserved residues were critical for the HDAC6-mutant SOD1 interaction.
To understand the nature of the mutant SOD1-HDAC6 interaction, we generated a series of HDAC6 mutants and tested them in coimmunoprecipitation studies (see the HDAC6 domain structure in Fig. 2B). We generated an enzymatically inactive HDAC6 mutant, H215A/H610A (22), and found that the H215A/H610A mutant retained its ability to bind to mutant SOD1 (Fig. 2B, lane 5). Another mutant lacking the ubiquitin-binding zinc finger domain of HDAC6 (M1-Q1,035) also bound mutant SOD1 (Fig. 2B, lane 6). Thus, the enzymatic activity and the ubiquitin-binding zinc finger of HDAC6 are dispensable for its interaction with mutant SOD1.
Truncation of the SE14 domain (Ala-883 to Pro-962) of HDAC6 led to significantly impaired binding of mutant SOD1 (Fig. 2B, lane 8). Likewise, the in-frame deletion of the SE14 domain in the context of full-length HDAC6 also weakened the coprecipitation with mutant SOD1 (Fig. 2B, lane 9). However, an N-terminally monomeric DsRed (DsRedM)-tagged SE14 domain (Ala-883 to Pro-962) failed to interact with mutant SOD1 (Fig. 2C, lane 3). Thus, the SE14 domain is essential but not sufficient for the HDAC6-mutant SOD1 interaction.
HDAC6 Interacts with Mutant SOD1 through a SMIR Motif
We hypothesized that sequence element(s) between the second deacetylase domain (DAC2) and the SE14 domain of HDAC6 might also play a role in the binding of mutant SOD1. Indeed, the DsRedM-HDAC6 (Leu-816 to Pro-962) fusion construct interacted with mutant SOD1 (Fig. 2C, lane 9). We next generated a series of deletion constructs from both the N-terminal and C-terminal directions to map the determinant(s) of the HDAC6 (Leu-816 to Pro-962)-mutant SOD1 interaction. The constructs containing the short Leu-816 to Leu-835 segment coprecipitated with mutant SOD1 (Fig. 2C, lanes 6–9), whereas the constructs lacking Leu-816 to Leu-835 did not coprecipitate with mutant SOD1 (Fig. 2C, lanes 3–5). Thus, the Leu-816 to Leu-835 segment is essential and sufficient for the HDAC6-mutant SOD1 interaction.
Interestingly, the Leu-816 to Leu-835 segment of HDAC6 revealed a motif highly reminiscent of the SMIR in p62 (15). As shown in Fig. 2D, the similarities included a conserved histidine and a leucine residue, four positively charged amino acid positions, and two adjacent hydrophobic residues, one of which is a tryptophan. To assess the significance of the conserved SMIR residues in the HDAC6 SMIR-mutant SOD1 interaction, we generated a set of mutants in the DsRedM -HDAC6 (Leu-816 to Ser-859) construct and tested their ability to coprecipitate with mutant SOD1 (Fig. 2E). The Y829A/W830A double mutation that removes two adjacent aromatic side chains and the R827A/K828A/R831A/R834A quadruple mutation (“AAAA”) that removes the positively charged side chains both severely impaired the binding of mutant SOD1. The L833A mutation suppressed the binding as well. The H826A mutation appeared to abolish the interaction, but the protein expression of this mutant was significantly lower. Therefore, the interpretation of the effect of the H826A mutation should be performed with caution. Thus, the conserved aromatic and positively charged residues are critical to the interaction.
In addition, we performed reverse coprecipitations using mammalian expression constructs for MBP fused to HDAC6. When the MBP-HDAC6 (Leu-816 to Ser-859) pull-down samples were analyzed by SDS-PAGE and Sypro Ruby protein stain, the only apparent difference between the WT and A4V SOD1 lanes was the presence of a band at the expected size of FLAG-tagged A4V SOD1 in the A4V SOD1 lane (supplemental Fig. S2). Proteomic analysis of the band verified it as SOD1. The corresponding gel slice in the WT SOD1 lane was also analyzed, but no evidence of any SOD1 fragment was found. The above results demonstrate that HDAC6 interacts with mutant SOD1 through a SMIR motif located immediately C-terminally to DAC2.
Identification of a Second SMIR Motif in HDAC6
Further sequence analysis identified a second possible SMIR motif (His-275 to Gly-301) in HDAC6, which is in the middle of the first deacetylase (DAC1) domain (Fig. 3A). The three-dimensional structure of HDAC6 has not yet been reported. To assess the surface accessibility of the putative SMIR motif in DAC1, we generated a homology model using the SWISS-MODEL server (34) on the basis of 36% sequence identity with the catalytic domain of human HDAC7 (PDB code 3c10, Ref. 35). According to the homology model, the putative SMIR (His-275 to Gly-301) is part of a surface-exposed loop (supplemental Fig. S3). Intriguingly, the putative SMIR is located in close proximity to the active center of the deacetylase domain. In addition, the corresponding region in DAC2 (His-671 to Gly-697) does not show SMIR-like sequence features (Fig. 3A).
FIGURE 3.

Identification of a second SMIR motif in DAC1 of HDAC6. A, sequence alignment of the p62 SMIR and the two SMIR motifs in HDAC6. The His-275 to Gly-301 segment harbors a SMIR-like motif that represents a surface-exposed loop in DAC1 on the basis of homology modeling. The corresponding loop, His-671 to Gly-697, in DAC2 does not have the SMIR features. +, positively charged; Ω, aromatic, Φ, hydrophobic amino acid residues. B, the tetrameric (Tetra) DsRed-tagged His-275 to Gly-301 loop from DAC-1 interacted with A4V mutant SOD1, but the His-671 to Gly-697 loop in DAC-2 did not. FLAG-SOD1 immunoprecipitations were performed, and the immunoprecipitation (IP) products and the total cell extracts (Ext) were blotted (IB) with the indicated antibodies. Mono, monomeric DsRed-tagged HDAC6 fragments. C, the Phe-282/Trp-283 positions of the N-SMIR are critical for the binding of mutant SOD1 as the F282A/W283A double mutant abolished the interaction.
To test whether the putative second SMIR motif in DAC1 can interact with mutant SOD1, we generated monomeric and tetrameric DsRed-tagged HDAC6 H275-G301. The corresponding loop in DAC2 (His-671 to Gly-697) was also tagged with DsRed as a negative control. As shown in Fig. 3B, the tetrameric DsRed-HDAC6 (His-275 to Gly-301) coprecipitated with mutant SOD1, but its monomeric version did not. Neither tetrameric nor monomeric DsRed-tagged HDAC6 (His-671 to Gly-697) interacted with mutant SOD1. We generated the F282A/W283A double mutation into DsRedT-HDAC6 (His-275 to Gly-301) and found that the mutation severely impacted the interaction with mutant SOD1 (Fig. 3C).
We conclude that the second putative SMIR motif identified in DAC1 is truly functional (Fig. 3), and thus termed it “N-SMIR.” The first SMIR that is immediately C-terminal to DAC2 (Fig. 2) is termed as “C-SMIR.”
The C-SMIR of HDAC6 Is Capable of Oligomerization
There is an intriguing difference between the HDAC6 C-SMIR and the HDAC6 N-SMIR or the p62 SMIR. The monomeric DsRed-tagged HDAC6 C-SMIR interacted with mutant SOD1 (Fig. 2C), whereas only the tetrameric DsRed-tagged HDAC6 N-SMIR interacted with mutant SOD1 (Fig. 3B). On the basis of our understanding of the p62 SMIR, that it requires oligomeric status to interact with mutant SOD1 (15), we hypothesized that the isolated HDAC6 C-SMIR might be able to oligomerize itself, explaining why it did not require an oligomeric fusion partner to interact with mutant SOD1. To test this hypothesis, we first examined whether the isolated C-SMIR could interact with full-length HDAC6. Indeed, the isolated C-SMIR (Leu-816 to Leu-835) was the minimal segment capable of interacting with the full-length HDAC6 (Fig. 4A, lane 6). Three additional constructs containing the C-SMIR also interacted with the full-length HDAC6 (Fig. 4A, lanes 3–5). Two constructs lacking the C-SMIR did not interact with HDAC6 (Fig. 4A, lanes 1–2). We next examined whether the C-SMIR could interact with itself using the DsRedM- and MBP-tagged C-SMIR. As shown in Fig. 4B, the MBP-tagged C-SMIR (Leu-816 to Leu-835) efficiently coprecipitated the DsRedM-tagged C-SMIR. We lastly examined the oligomeric status of the DsRedM-tagged C-SMIR using native gel electrophoresis. As shown in Fig. 4C, the DsRedM-tagged C-SMIR (Leu-816 to Leu-835) showed very slow migration in native gel (lane 3), likely corresponding to oligomers. Native gel electrophoresis of the DsRedM- and DsRedT-tagged N-SMIR was performed as a control and showed a significant shift between the migration of the DsRedM- and DsRedT-tagged versions (Fig. 4C, lanes 1 and 2). This shift is similar to the shift we observed between the DsRedM- and DsRedT-tagged p62 SMIR (15). These results support that the C-SMIR of HDAC6 can self-oligomerize and the N-SMIR cannot.
FIGURE 4.
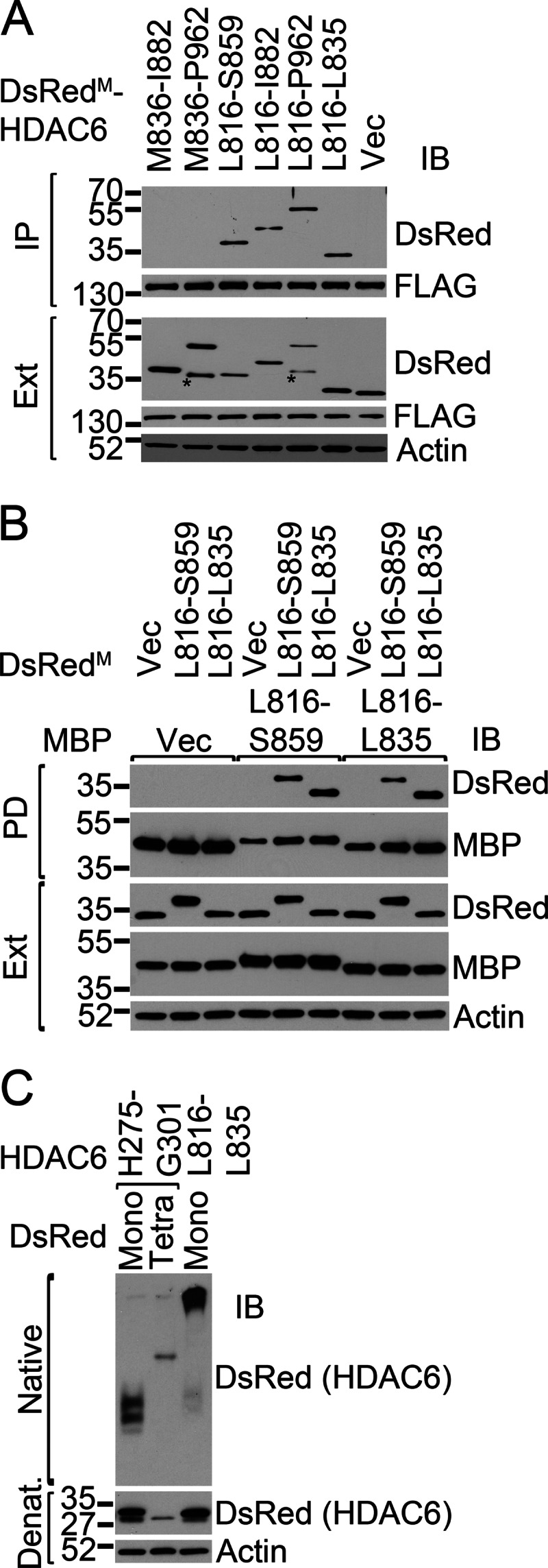
The HDAC6 C-SMIR is capable of oligomerization. A, the C-SMIR interacts with full-length HDAC6. Immunoprecipitations (IP) of FLAG-tagged full-length HDAC6 were followed by Western blot analysis (IB) with the indicated antibodies. The asterisks indicate potential proteolytic fragments. Ext, extracts. B, the MBP- and DsRed-tagged C-SMIR can interact with each other. MBP-tagged or DsRedM-tagged Leu-816 to Ser-859 or Leu-816 to Leu-835 HDAC6 segments were coexpressed in HEK293 cells. MBP pull-downs (PD) were performed followed by Western blot analysis with the indicated antibodies. Vec, vector control. C, oligomeric status of monomeric (Mono) or tetrameric (Tetra) DsRed-tagged HDAC6 SMIRs were examined by native and denaturing (Denat) gel electrophoresis.
Mutant SOD1 Causes an Elevated Level of α-Tubulin Acetylation
The N-SMIR motif is located in close proximity to the active site of DAC1 (His-215) in the homology model of HDAC6 (supplemental Fig. S3). The C-SMIR is also immediately C-terminal to DAC2 (Fig. 2). We hypothesized that the binding of mutant SOD1 to HDAC6 through the two SMIR motifs could modulate the enzymatic activity of HDAC6. Because HDAC6 is the dominant α-tubulin deacetylase in mammalian cells (19, 20, 36), we next examined whether expression of mutant SOD1 would change the acetylation of α-tubulin. Myc-tagged α-tubulin was cotransfected with either WT or A4V mutant SOD1, and the acetylation status of Myc-α-tubulin was assessed by Myc immunoprecipitation followed by acetyl-tubulin Western blotting. As shown in Fig. 5, A and B, the acetylation level of Myc-α-tubulin was ∼3-fold higher in the presence of A4V SOD1 than with WT SOD1 expression (p = 0.013). Coexpression of WT SOD1 did not significantly change tubulin acetylation as compared with the vector control.
FIGURE 5.

Acetylated α-tubulin increases in the presence of mutant SOD1. A, Myc-tagged α-tubulin (0.1 μg of plasmid) and WT or A4V SOD1–3xFLAG or FLAG vector control (0.9 μg of plasmid) (Vec) were coexpressed in HEK293 cells. Myc (α-tubulin) immunoprecipitations (IP) were performed, followed by Western blot analysis (IB) with acetyl-tubulin and other antibodies as indicated. The α-tubulin immunoblot analysis of the cell lysate is shown with a shorter and a longer exposure because the transfected Myc-α-tubulin was far less abundant than the endogenous α-tubulin. The asterisks indicate the immunoprecipitation antibody. Ext, extracts. B, densitometric quantification of the acetylated and total tubulin bands in the immunoprecipitation samples as shown in A. The acetyl-α-tubulin bands were normalized with total (Myc) tubulin bands, and the results are shown as mean ± S.E. of three independent experiments. C, confocal analysis of GFP-tagged WT or A4V mutant SOD1 and acetylated α-tubulin (red) in NSC34 cells. Cells with diffused A4V SOD1 showed significantly higher acetyl-α-tubulin signals (solid arrows), whereas cells with large (center panels) or small (bottom panels) A4V SOD1 inclusions in the same view field showed significantly lower acetyl-α-tubulin signals. Scale bars = 25 μm.
Confocal microscopy showed significantly higher acetyl-α-tubulin immunostaining signals in NSC34 cells transfected with A4V mutant SOD1 as compared with cells transfected with WT SOD1 and the untransfected cells (Fig. 5C). Interestingly, the elevated acetyl-α-tubulin signal was primarily seen in cells with a diffused SOD1-GFP pattern (Fig. 5C, solid arrows) and not in the cells with large mutant SOD1 inclusions (open arrows). Our results suggest that mutant SOD1 can suppress the tubulin deacetylase activity of HDAC6 and result in an elevated level of tubulin acetylation.
HDAC6 Is Sequestrated into Mutant SOD1 Aggregates and Inclusions
We hypothesized that mutant SOD1 might cause hyperacetylation of α-tubulin by sequestrating HDAC6 into mutant SOD1 aggregates. To test this hypothesis, we cotransfected FLAG-tagged HDAC6 and EGFP-tagged WT or mutant forms of SOD1 and performed cellulose acetate filtration. We detected a significantly higher amount of aggregated HDAC6 with all tested mutants than with WT SOD1 (Fig. 6, A and B; A4V, p = 0.00033; G85R, p = 0.00011; G93A, p = 0.0042). We also observed a significantly higher amount of cellulose acetate-filtered HDAC6 aggregates in the spinal cord of 90-day-old G93A SOD1 transgenic mice than in the age-matched WT SOD1 transgenic mice (Fig. 6, C and D, p = 0.0013). Moreover, immunofluorescence microscopy also showed the presence of HDAC6 in mutant SOD1 inclusions in cell culture models as well as in spinal cord motor neurons of G93A SOD1 transgenic mice (Fig. 6E). Thus, we conclude that the likely mechanism of HDAC6 inhibition by mutant SOD1 is sequestration of HDAC6 into inclusions.
FIGURE 6.

Sequestration of HDAC6 in mutant SOD1 inclusions. A, HEK293 cells were cotransfected with plasmids expressing GFP-tagged WT, A4V, G85R, or G93A SOD1 and FLAG-tagged HDAC6. Cellular extracts (Ext) were prepared and subjected to a cellulose acetate (CA) membrane filtration assay. The cellulose acetate membrane was blotted (IB) with FLAG antibody. Protein levels in cell extracts were also examined as controls. B, densitometric quantification of the cellulose acetate membrane blots in A. C, spinal cord extracts were prepared from 90-day-old WT and G93A SOD1 transgenic mice and subjected to a cellulose acetate membrane filtration assay. The cellulose acetate membrane was blotted with HDAC6 antibody. Protein levels in the extracts were also examined as controls. D, densitometric quantification of the cellulose acetate membrane blots in C. ImageJ was used in B and D to analyze the densitometric intensities of the dots that were normalized to the WT SOD1 dot. The mean ± S.E. of three experiments is shown. E, confocal analysis of HDAC6 immunofluorescence in HEK293 (top panels) and NSC34 (center panels) cells transfected with GFP-tagged A4V SOD1. The bottom panel shows HDAC6 and SOD1 immunofluorescence in spinal cord motor neurons of 90-day-old G93A SOD1 transgenic mice. The sequestration of HDAC6 in mutant SOD1 inclusions are illustrated by arrows. The inset in the lower left of the merged image shows the magnified inclusions. Scale bars = 10 μm.
Acetylation-mimicking α-Tubulin Enhances Mutant SOD1 Aggregation
We next tested whether tubulin acetylation could affect mutant SOD1 aggregation using the acetylation-mimicking K40Q mutation of tubulin (30). Myc-tagged WT or K40Q α-tubulin was cotransfected with FLAG-tagged WT or A4V mutant SOD1. Cellulose acetate membrane filtration was performed to assess the aggregation of mutant SOD1. As shown in Fig. 7, A and B, significant accumulation of aggregated A4V SOD1 was observed in the presence of K40Q α-tubulin as compared with WT tubulin (p = 0.0072). The expression levels of SOD1 were apparently unchanged with coexpression of either WT or K40Q α-tubulin (Fig. 7C). No aggregation of WT SOD1 was observed with coexpression of either WT or K40Q α-tubulin (Fig. 7A). We conclude that elevated levels of tubulin acetylation, as mimicked by the expression of the K40Q mutant α-tubulin, enhanced mutant SOD1 aggregation.
FIGURE 7.

The effect of tubulin acetylation on mutant SOD1 aggregation. The amount of A4V mutant SOD1 (0.95 μg of plasmid) aggregates in the presence of WT or the acetylation-mimicking K40Q mutant α-tubulin (0.05 μg of plasmid) was assessed by cellulose acetate membrane filtration assay. The experiment was performed in triplicate, and a representative image is shown. B, densitometric quantification of mutant SOD1 aggregates shown in A. The intensities of the mutant SOD1 aggregate dots in the presence of K40Q tubulin were compared with those with WT tubulin. The mean ± S.E. of three experiments is shown. C, Western blot analyses (IB) of the respective cellular extracts showing that the expression levels of A4V SOD1 cotransfected with WT or K40Q Myc-α-tubulin were comparable.
DISCUSSION
HDAC6 Regulates the Aggregation of Mutant SOD1 by a Mechanism Different from Modulating the Aggregation of MG132-induced Poly-ubiquitinated Proteins
The familial ALS mutants of SOD1 are prone to aggregation, and the cytoplasmic inclusions are characteristic in relevant human patients as well as disease model systems (3, 4, 7–10). HDAC6 has been shown to regulate the formation of poly-ubiquitin-positive inclusions (16), but its role in mutant SOD1 aggregation has not yet been reported yet. In this work, we establish that HDAC6 plays an important role in the regulation of mutant SOD1 aggregation, and we explore the underlying mechanism.
The knockdown of HDAC6 significantly enhanced the formation of large aggresome-like inclusions of mutant SOD1 (Fig. 1A, A4V and G93A). The results were confirmed using an independent cellulose acetate membrane filtration assay for all three tested mutants: A4V, G85R, and G93A (Fig. 1, B and C). Similar observations were reported for the effect of HDAC6 knockdown on the aggregation of expanded huntingtin (25). However, this observation is apparently different from the report that the aggresomes of the MG132-induced poly-ubiquitinated proteins were dependent on HDAC6 (16). We propose that HDAC6 regulates mutant SOD1 aggregation differently from the case of MG132-induced poly-ubiquitinated proteins. Firstly, the polyubiquitin content is different. The mutant SOD1 aggregates were barely poly-ubiquitinated as compared with other aggresome-forming proteins (37). In contrast, the MG132-induced protein aggregates are heavily poly-ubiquitinated (16). Secondly, the C-terminal ubiquitin-binding domain of HDAC6 was dispensable for the binding of mutant SOD1 (Fig. 2B) but essential for the binding of poly-ubiquitinated CFTR-ΔF508 (16). This is remarkably similar to the p62-mutant SOD1 interaction that was also independent of the ubiquitin binding domain of p62 (15). Moreover, the ubiquitin-binding domain of HDAC6 was similarly dispensable in the HDAC6-tau and HDAC6-MyD88 interactions (27, 38).
Another important difference is that the MG132-induced protein aggregates recruited cortactin in an HDAC6-dependent manner, resulting in F-actin remodeling around the aggregates (17). In contrast, no accumulation of cortactin or F-actin was observed in the mutant SOD1 inclusions (supplemental Fig. S4). These results consistently suggest that although HDAC6 plays an important role in regulating mutant SOD1 aggregation, the underlying mechanism is different from that seen in the case of heavily poly-ubiquitinated protein aggregates.
HDAC6 Recognizes Mutant SOD1 through p62-like SMIR Motifs
We found that HDAC6 selectively interacted with ALS mutants of SOD1 but not with the WT SOD1 (Fig. 2A and supplemental Fig. S1). We found that neither the enzymatic activity nor the C-terminal ubiquitin-binding domain of HDAC6 was required for the interaction (Fig. 2B). HDAC6 was reported to interact with tau and MyD88 and to regulate their aggregation. The HDAC6-tau and HDAC6-MyD88 interactions were also independent of the enzymatic activity and the ubiquitin-binding domain of HDAC6 (27, 38). We also examined the SE14 domain of HDAC6, which was reported to be required for the HDAC6-tau interaction (27). Our results showed that the SE14 domain was essential (Fig. 2B) but not sufficient (Fig. 2C) for the HDAC6-mutant SOD1 interaction. The study on tau did not examine whether the SE14 domain alone was sufficient for the HDAC6-tau interaction (27).
Our study later determined two regions that are essential and sufficient for the HDAC6-mutant SOD1 interaction. Both regions resemble the SMIR in p62 we described previously (15). One is within the DAC1 (termed N-SMIR, Fig. 3 and supplemental Fig. S3), and the other one is immediately C-terminal to DAC2 (termed C-SMIR, Fig. 2 and supplemental Fig. S2). Mutational analysis of the conserved residues within both N- and C-SMIR motifs showed that the aromatic residues (Phe-282/Trp-283 in the N-SMIR and Tyr-829/Trp-830 in the C-SMIR) and the positively charged residues (Arg-827/Lys-828/Arg-831/Lys-834 in the C-SMIR) were critical for the binding of mutant SOD1 (Figs. 2E and 3C).
Our results also demonstrate that the SMIR motif needs to be in oligomeric state to interact with mutant SOD1. Similar to the p62 SMIR, the HDAC6 N-SMIR needed to be in tetrameric DsRed fusion to interact with mutant SOD1 (Fig. 3B). However, the HDAC6 C-SMIR interacted with mutant SOD1 even in monomeric DsRed fusion (Fig. 2C). We next showed that the isolated C-SMIR segment indeed interacted with itself and the full-length HDAC6 (Fig. 4, A and B). The native gel electrophoresis also suggested that the C-SMIR was likely capable of self-oligomerization, whereas the N-SMIR required tetrameric DsRed to form oligomers (Fig. 4C).
All results taken together, we found two SMIR motifs in HDAC6 that share sequence similarity with the p62 SMIR. Each of the SMIR motifs, as defined by the name, is essential and sufficient for interacting with mutant SOD1. The isolated SMIR motif also requires an oligomeric status to interact with mutant SOD1, which is also consistent with the characteristics of the first SMIR we reported previously in p62 (15).
Relationship of HDAC6 Activity, Tubulin Acetylation, and Mutant SOD1 Aggregation
The N-SMIR of HDAC6 is in close proximity to the active-site His-215 residue in DAC1 (supplemental Fig. S3), and the C-SMIR is located immediately C-terminal to DAC2 (Fig. 2, C and D). Accordingly, we hypothesized that mutant SOD1 might modulate the enzymatic activity of HDAC6. We found that the expression of mutant SOD1 indeed caused a significant elevation in the level of α-tubulin acetylation (Fig. 5), indicating that HDAC6 activity was suppressed in the presence of mutant SOD1. In addition, we found that HDAC6 was sequestrated into mutant SOD1 inclusions in cellular and mouse models (Fig. 6), likely depleting the active cytosolic HDAC6 pool.
Hyperacetylation of tubulin, as mimicked by the expression of the K40Q mutant of α-tubulin, caused a significant elevation of aggregated mutant SOD1 (Fig. 7, A and B). We and others (10, 11, 37) reported previously that the sequestration of mutant SOD1 into cytoplasmic aggresomes was dependent on dynein/dynactin-mediated retrograde transport along microtubules. The acetylation of α-tubulin is known to promote the association of microtubules with dynein and kinesin motors, resulting in increased motor processivity and transport flux in neurons (39–41). Conversely, decreased acetylation of α-tubulin caused severe axonal transport deficits, and inhibition of HDAC6 could correct such deficiency (42). On the basis of the findings in this study and the above reports, we propose a model for the role of HDAC6 in the regulation of mutant SOD1 aggregation (Fig. 8). In the model, HDAC6 is sequestrated by mutant SOD1 oligomers and small aggregates in the cytoplasm. Depletion of the active HDAC6 causes decreased tubulin deacetylase activity, increased tubulin acetylation, and enhanced retrograde transport of the mutant SOD1 oligomers and smaller aggregates to large aggregates and inclusions. Our results suggest that the normal tubulin acetylation level can be restored upon the clearance of smaller aggregates and the formation of larger inclusions (Fig. 5C), which is illustrated at the left of Fig. 8. However, the sustained generation of aggregated proteins might lead to chronic HDAC6 inhibition, which is a conceivable scenario, as shown at the right of Fig. 8. This model can also explain why the knockdown of HDAC6 resulted in enhanced mutant SOD1 aggregation (Fig. 1).
FIGURE 8.

Proposed model of the role of HDAC6 in the aggregation of mutant SOD1.
Potential Role of HDAC6 and Tubulin Hyperacetylation in ALS and Related Diseases
Frontotemporal lobar degeneration and ALS represent a clinicopathological spectrum of neurodegenerative diseases (43). It was reported that the frontotemporal lobar degeneration protein tau bound to HDAC6 (27, 29) and inhibited its function, increasing tubulin acetylation (29). The ALS proteins TDP-43 and Fused in Sarcoma (FUS) were reported to bind the HDAC6 mRNA, and their knockdown resulted in decreased levels of HDAC6 and the accumulation of acetyl-α-tubulin (44, 45). Taken together, the alteration of HDAC6 by the culprit proteins may represent a common molecular trait in the SOD1, TDP-43, FUS, and tau pathologies.
During the revision of this manuscript, a new study was published, reporting that deletion of the HDAC6 gene delayed disease progression in the G93A SOD1 mouse model of ALS (46). This new study provides direct evidence that HDAC6 has functional relevance in ALS. However, HDAC6 can be involved in multiple cellular processes, and other studies found that HDAC6 inhibition can have detrimental effects. For instance, HDAC6 inhibition could slow down axonal growth in cultured hippocampal neurons (47) and reduced neurite outgrowth in SH-SY5Y cells (48). Taes et al. suggested that the inhibition of the tubulin deacetylase activity of HDAC6 produced a beneficial outcome in G93A mice through enhancing axonal transport via hyperacetylation of tubulin (46). In other words, the up-regulation of axonal transport was more important than other potential detrimental effects of HDAC6 inhibition in the G93A mice in their study. Additional studies are needed to further test this hypothesis and to exploit the potential benefits of HDAC6 inhibition in other ALS animal models. The translational value of HDAC6 in ALS therapy is also yet to be further evaluated.
Supplementary Material
Acknowledgments
We thank Dr. Tso-Pang Yao for the anti-mouse HDAC6 antibody and Dr. Marie Wooten for the pcDNA3-FLAG-mouse HDAC6 plasmid. We also thank Dr. Ping Shi, Wei-si Fu, and Tyler Frailie (supported by the National Science Foundation Research Experiences for Undergraduates program) for technical assistance and Drs. Edward Kasarskis, Daniel Noonan, and Qingjun Wang for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01NS049126, R01NS077284, and R21AG032567 (to H. Z.); NCRR Grant P20RR020171-09; and NIGMS Grant P20GM103486-09. This work was also supported by High-End Instrumentation Grant S10RR029127 (to H. Z.) and by the federally funded work-study (summer research) program in the College of Medicine (to E. B.).

This article contains supplemental Figs. S1–S4.
- ALS
- amyotrophic lateral sclerosis
- SOD1
- Cu/Zn superoxide dismutase 1
- SMIR
- Cu/Zn superoxide dismutase 1 mutant interaction region
- HDAC
- histone deacetylase
- MBP
- maltose binding protein.
REFERENCES
- 1. Wijesekera L. C., Leigh P. N. (2009) Amyotrophic lateral sclerosis. Orphanet. J. Rare Dis. 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piao Y. S., Wakabayashi K., Kakita A., Yamada M., Hayashi S., Morita T., Ikuta F., Oyanagi K., Takahashi H. (2003) Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis. 102 autopsy cases examined between 1962 and 2000. Brain Pathol. 13, 10–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Watanabe M., Dykes-Hoberg M., Culotta V. C., Price D. L., Wong P. C., Rothstein J. D. (2001) Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol. Dis. 8, 933–941 [DOI] [PubMed] [Google Scholar]
- 4. Valentine J. S., Doucette P. A., Zittin Potter S. (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593 [DOI] [PubMed] [Google Scholar]
- 5. Chattopadhyay M., Valentine J. S. (2009) Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid. Redox. Signal 11, 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X., Rahmani Z., Krizus A., McKenna-Yasek D., Cayabyab A., Gaston S. M., Berger R., Tanzi R. E., Halperin J. J., Herzfeldt B., Vandenbergh R., Hung W.-Y., Bird T., Deng G., Mulder D. W., Smyth C., Laing N. G., Soriano E., Pericak-Vance M. A., Haines J., Rouleau G. A., Guesella J. S., Horvitz H. R., Brown R. H. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 [DOI] [PubMed] [Google Scholar]
- 7. Shibata N., Asayama K., Hirano A., Kobayashi M. (1996) Immunohistochemical study on superoxide dismutases in spinal cords from autopsied patients with amyotrophic lateral sclerosis. Dev. Neurosci. 18, 492–498 [DOI] [PubMed] [Google Scholar]
- 8. Shibata N., Hirano A., Kobayashi M., Siddique T., Deng H. X., Hung W. Y., Kato T., Asayama K. (1996) Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J. Neuropathol. Exp. Neurol. 55, 481–490 [DOI] [PubMed] [Google Scholar]
- 9. Hart P. J. (2006) Pathogenic superoxide dismutase structure, folding, aggregation and turnover. Curr. Opin. Chem. Biol. 10, 131–138 [DOI] [PubMed] [Google Scholar]
- 10. Ström A. L., Shi P., Zhang F., Gal J., Kilty R., Hayward L. J., Zhu H. (2008) Interaction of amyotrophic lateral sclerosis (ALS)-related mutant copper-zinc superoxide dismutase with the dynein-dynactin complex contributes to inclusion formation. J. Biol. Chem. 283, 22795–22805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang F., Ström A. L., Fukada K., Lee S., Hayward L. J., Zhu H. (2007) Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J. Biol. Chem. 282, 16691–16699 [DOI] [PubMed] [Google Scholar]
- 12. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Komatsu M., Waguri S., Koike M., Sou Y. S., Ueno T., Hara T., Mizushima N., Iwata J., Ezaki J., Murata S., Hamazaki J., Nishito Y., Iemura S., Natsume T., Yanagawa T., Uwayama J., Warabi E., Yoshida H., Ishii T., Kobayashi A., Yamamoto M., Yue Z., Uchiyama Y., Kominami E., Tanaka K. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 [DOI] [PubMed] [Google Scholar]
- 14. Gal J., Ström A. L., Kilty R., Zhang F., Zhu H. (2007) p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J. Biol. Chem. 282, 11068–11077 [DOI] [PubMed] [Google Scholar]
- 15. Gal J., Ström A. L., Kwinter D. M., Kilty R., Zhang J., Shi P., Fu W., Wooten M. W., Zhu H. (2009) Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J. Neurochem. 111, 1062–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawaguchi Y., Kovacs J. J., McLaurin A., Vance J. M., Ito A., Yao T. P. (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 [DOI] [PubMed] [Google Scholar]
- 17. Lee J. Y., Koga H., Kawaguchi Y., Tang W., Wong E., Gao Y. S., Pandey U. B., Kaushik S., Tresse E., Lu J., Taylor J. P., Cuervo A. M., Yao T. P. (2010) HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verdel A., Curtet S., Brocard M. P., Rousseaux S., Lemercier C., Yoshida M., Khochbin S. (2000) Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr. Biol. 10, 747–749 [DOI] [PubMed] [Google Scholar]
- 19. Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X. F., Yao T. P. (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 [DOI] [PubMed] [Google Scholar]
- 20. Matsuyama A., Shimazu T., Sumida Y., Saito A., Yoshimatsu Y., Seigneurin-Berny D., Osada H., Komatsu Y., Nishino N., Khochbin S., Horinouchi S., Yoshida M. (2002) In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 21, 6820–6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y., Li N., Caron C., Matthias G., Hess D., Khochbin S., Matthias P. (2003) HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 22, 1168–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haggarty S. J., Koeller K. M., Wong J. C., Grozinger C. M., Schreiber S. L. (2003) Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U.S.A. 100, 4389–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zou H., Wu Y., Navre M., Sang B. C. (2006) Characterization of the two catalytic domains in histone deacetylase 6. Biochem. Biophys. Res. Commun. 341, 45–50 [DOI] [PubMed] [Google Scholar]
- 24. Zhang Y., Gilquin B., Khochbin S., Matthias P. (2006) Two catalytic domains are required for protein deacetylation. J. Biol. Chem. 281, 2401–2404 [DOI] [PubMed] [Google Scholar]
- 25. Iwata A., Riley B. E., Johnston J. A., Kopito R. R. (2005) HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 280, 40282–40292 [DOI] [PubMed] [Google Scholar]
- 26. Du G., Liu X., Chen X., Song M., Yan Y., Jiao R., Wang C. C. (2010) Drosophila histone deacetylase 6 protects dopaminergic neurons against α-synuclein toxicity by promoting inclusion formation. Mol. Biol. Cell 21, 2128–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ding H., Dolan P. J., Johnson G. V. (2008) Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 106, 2119–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guthrie C. R., Kraemer B. C. (2011) Proteasome inhibition drives HDAC6-dependent recruitment of tau to aggresomes. J. Mol. Neurosci. 45, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perez M., Santa-Maria I., Gomez de Barreda E., Zhu X., Cuadros R., Cabrero J. R., Sanchez-Madrid F., Dawson H. N., Vitek M. P., Perry G., Smith M. A., Avila J. (2009) Tau. An inhibitor of deacetylase HDAC6 function. J. Neurochem. 109, 1756–1766 [DOI] [PubMed] [Google Scholar]
- 30. Gao Y. S., Hubbert C. C., Yao T. P. (2010) The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J. Biol. Chem. 285, 11219–11226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., Deng H. X., Chen W. J., Zhai P., Sufit R. L., Siddique T. (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 [DOI] [PubMed] [Google Scholar]
- 32. Zhai J., Ström A. L., Kilty R., Venkatakrishnan P., White J., Everson W. V., Smart E. J., Zhu H. (2009) Proteomic characterization of lipid raft proteins in amyotrophic lateral sclerosis mouse spinal cord. FEBS J. 276, 3308–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuo K. L., Zhu H., McNamara P. J., Leggas M. (2012) Localization and functional characterization of the rat Oatp4c1 transporter in an in vitro cell system and rat tissues. PloS ONE 7, e39641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 37, D387–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schuetz A., Min J., Allali-Hassani A., Schapira M., Shuen M., Loppnau P., Mazitschek R., Kwiatkowski N. P., Lewis T. A., Maglathin R. L., McLean T. H., Bochkarev A., Plotnikov A. N., Vedadi M., Arrowsmith C. H. (2008) Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 283, 11355–11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao Y. S., Hubbert C. C., Lu J., Lee Y. S., Lee J. Y., Yao T. P. (2007) Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol. Cell Biol. 27, 8637–8647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnston J. A., Dalton M. J., Gurney M. E., Kopito R. R. (2000) Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 97, 12571–12576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Into T., Inomata M., Niida S., Murakami Y., Shibata K. (2010) Regulation of MyD88 aggregation and the MyD88-dependent signaling pathway by sequestosome 1 and histone deacetylase 6. J. Biol. Chem. 285, 35759–35769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dompierre J. P., Godin J. D., Charrin B. C., Cordelières F. P., King S. J., Humbert S., Saudou F. (2007) Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J. Neurosci. 27, 3571–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reed N. A., Cai D., Blasius T. L., Jih G. T., Meyhofer E., Gaertig J., Verhey K. J. (2006) Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 16, 2166–2172 [DOI] [PubMed] [Google Scholar]
- 41. Chen S., Owens G. C., Makarenkova H., Edelman D. B. (2010) HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS ONE 5, e10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. d'Ydewalle C., Krishnan J., Chiheb D. M., Van Damme P., Irobi J., Kozikowski A. P., Vanden Berghe P., Timmerman V., Robberecht W., Van Den Bosch L. (2011) HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 17, 968–974 [DOI] [PubMed] [Google Scholar]
- 43. van Langenhove T., van der Zee J., van Broeckhoven C. (2012) The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann. Med. 44, 817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim S. H., Shanware N. P., Bowler M. J., Tibbetts R. S. (2010) Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 285, 34097–34105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fiesel F. C., Voigt A., Weber S. S., Van den Haute C., Waldenmaier A., Görner K., Walter M., Anderson M. L., Kern J. V., Rasse T. M., Schmidt T., Springer W., Kirchner R., Bonin M., Neumann M., Baekelandt V., Alunni-Fabbroni M., Schulz J. B., Kahle P. J. (2010) Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 29, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Taes I., Timmers M., Hersmus N., Bento-Abreu A., Van Den Bosch L., Van Damme P., Auwerx J., Robberecht W. (2013) Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum. Mol. Genet. 22, 1783–1790 [DOI] [PubMed] [Google Scholar]
- 47. Tapia M., Wandosell F., Garrido J. J. (2010) Impaired function of HDAC6 slows down axonal growth and interferes with axon initial segment development. PLoS ONE 5, e12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fiesel F. C., Schurr C., Weber S. S., Kahle P. J. (2011) TDP-43 knockdown impairs neurite outgrowth dependent on its target histone deacetylase 6. Mol. Neurodegener. 6, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.