

Abstract
Somatic hypermutation (SHM) diversifies the variable region of Ig genes and underlies the process of affinity maturation, in which B lymphocytes producing high affinity antibodies are generated and selected. SHM is triggered in activated B cells by deamination of deoxycytosine residues mediated by activation-induced deaminase (AID). While mistargeting of SHM and AID results in mutations and DNA damage in many non-Ig genes, they act preferentially at Ig loci. The mechanisms responsible for preferential targeting of SHM and AID activity to Ig loci are poorly understood. Using an assay involving a SHM reporter cassette inserted into the Ig light chain locus (IgL) of chicken DT40 B cells, we have identified a 1.9 kb DIVAC (diversificationactivator) element derived from chicken IgL that supports high levels of AID-dependent mutation activity. Systematic deletion analysis reveals that targeting activity is spread throughout much of the sequence and identifies two core regions that are particularly critical for function: a 200 bp region within the IgL enhancer, and a 350 bp 3′ element. Chromatin immunoprecipitation experiments demonstrate that while DIVAC does not alter levels of several epigenetic marks in the mutation cassette, it does increase levels of serine-5 phosphorylated RNA polymerase II in the mutation target region, consistent with an effect on transcriptional elongation/pausing. We propose that multiple, dispersed DNA elements collaborate to recruit and activate the mutational machinery at Ig gene variable regions during SHM.
Introduction
The activation induced deaminase (AID)5, encoded by Aicda, plays a pivotal role in Ig gene diversification in developing and activated B cells. AID is required for somatic hypermutation (SHM) and gene conversion (GCV), which diversify the variable (IgV) region exon of Ig heavy (IgH) and light (IgL) chain genes, and for class switch recombination (CSR), in which recombination between switch regions leads to the replacement of one IgH constant region with another (1–4). AID-mediated IgV region mutation can be used both for creation of the primary Ig repertoire in developing B cells (e.g., GCV in chicken and rabbit) and for Ig gene diversification in activated mature B cells in germinal centers (e.g., SHM in mouse and human), where it can facilitate the generation of B cells expressing high affinity antibodies, a process known as affinity maturation (5).
SHM, GCV, and CSR can be thought of as occurring in two phases. In the first, AID is recruited to IgV or Igswitch regions, where it deaminates cytosine residues in the DNA, converting them to uracil. Deamination requires transcription of the region being acted upon by AID, which is thought to reflect the biochemical specificity of AID for cytosine residues in single stranded DNA (6). Interactions between AID and RNA polymerase II (Pol II) and Pol II-associated factors (7–11) support a model (12) in which AID “piggybacks” with Pol II through the Ig gene and deaminates DNA in a process that is tightly coupled to transcription. In the second phase, the uracils created by AID are processed by mismatch repair and base excision repair enzymes to generate DNA single or double strand break intermediates, which, upon further processing, yield single nucleotide non-templated point mutations in the case of SHM, patches of templated sequence inserted by homologous recombination with pseudo V(ψV) donor sequences in the case of GCV, or DNA deletions in the case of CSR (13, 14). The ability of AID to trigger the formation of DNA mutations, breaks, and deletions in the genome suggests a propensity to contribute to genomic instability (15, 16), the risks of which would be greatly increased if AID acted outside of the Ig loci. Hence, mechanisms to restrict AID action to Ig genes would seem highly desirable.
Results from studies in the last fifteen years indicate that while such mechanisms exist, they are imperfect. AID has been linked to mutations and large scale genome rearrangements (particularly chromosomal translocations) involving both Ig and non-Ig genes in B cell lymphomas (17), and numerous non-Ig genes have been found to be mutated in an AID-dependent manner in normal germinal center B cells (18). A large scale sequencing analysis of germinal center B cells found that approximately one-quarter of the genes analyzed sustained mutations as a result of SHM, and that at least one-half of them were deaminated at detectable levels by AID (19). Ig genes, however, were found to undergo SHM and be deaminated by AID at frequencies ten- to 1000-fold higher than non-Ig genes, consistent with the existence of mechanisms to target AID and/or AID activity preferentially to Ig genes (19). These mechanisms remain poorly defined, leaving a significant gap in our understanding of how the genome is protected from AID-mediated instability.
Numerous studies have pursued the hypothesis that DNA sequences associated with Ig loci function as SHM/AID targeting elements (20–23). Such analyses have ruled out an essential role for the IgV exon itself (24) or the IgV promoter (25), although the IgV promoter can enhance the efficiency of AID-mediated diversification (26). Analyses of the targeting function of Ig gene enhancers have yielded conflicting results, leaving their role as mutation targeting elements uncertain (20–22). The E box sequence CANNTG, as well as the E2a-encoded E box binding proteins E12/E47, have been linked to SHM targeting (19, 27–31), although the magnitude and mechanism of their contribution to AID/SHM targeting remains unknown.
Several groups have taken advantage of the relatively small size of the chicken IgL locus, and the ease of manipulating the genome of the DT40 chicken B cell line (32), to search for IgL-associated DNA elements capable of targeting AID-mediated sequence diversification activity. Collectively, these studies indicate that sequences downstream of the chicken IgL constant region (C) exon function as a diversification activator (DIVAC) element, capable of conferring AID-mediated GCV/SHM on a nearby transcription unit driven by the IgV promoter or a heterologous promoter (33–38). DIVAC is active in multiple locations in the DT40 genome (34, 36) and contains evolutionarily conserved sequence elements (38) and binding sites for a number of transcription factors, some of which have been suggested to be relevant to DIVAC function (35, 37, 38). These studies, however, did not converge on particular sequence motifs or protein factors that account for DIVAC function. Furthermore, the mechanism of action of DIVAC is not understood at even a simplistic level, although it is clear that DIVAC contains functions beyond those of a transcriptional enhancer (38).
In this study, we used an established SHM assay involving a heterologous expression cassette targeted to the IgL locus of DT40 cells (34) to identify DNA sequences downstream of the chicken IgL C exon critical for DIVAC function. This allowed us to define a 1928 bp composite element with strong DIVAC activity, which we refer to as DIVAC 1928. Extensive deletion and mutation analyses revealed that functionally important elements are broadly dispersed throughout DIVAC 1928, and also identified two "core" regions that are particularly important for DIVAC function. Chromatin immunoprecipitation (ChIP) experiments demonstrate that DIVAC increases levels of serine-5 phosphorylated Pol II within the mutating region, but doesnot alter many other parameters of the cassette including levels of activating and repressive epigenetic modifications or the Pol II-associated factor Spt5. The striking dispersion of DIVAC function throughout DIVAC 1928 and the DIVAC core regions strongly suggests that multiple DNA sequences, and possibly multiple protein factors, collaborate to recruit and activate the mutation machinery during SHM/GCV and helps explain why the precise definition of SHM targeting elements has proven so elusive.
Materials and Methods
Targeting Constructs
Targeting constructs were derived by modifying the pIgL(−)GFP2 plasmid (34). Test DNA fragments were PCR amplified using Phusion polymerase (New England Biolabs) and cloned into unique NheI/SpeI sites in pIgL(−)GFP2 (primers and DNA templates, Supplemental Table 1A). Fragment deletions depicted in Figs. 2–7 were assembled by PCR. Primer sets were made to include the sequence 5′ of the deletion (Reaction 1:F1/R1 primer set) and 3′ of the deletion (Reaction 2:F2/R2 primer set) where roughly 15 bp of the R1 and F2 primers overlap (Supplemental Table 1A). PCR was performed using Phusion polymerase and products were separated on a 1% agarose gel. Desired products were excised and extracted using the QIAquick gel extraction kit (Qiagen), and 0.2 µL of the gel extraction products for reaction 1 and reaction 2 were used as template in a third assembly reaction using the F1/R2 primers. Correct orientation and assembly of all test DNA fragments were confirmed by sequencing. 2b-2 fragment multimerization was accomplished by ligating 2b-2 digested with Nhe1/SpeI in the pIgL(−)GFP2 2b-2 vector digested with NheI. The resulting pIgL(−)GFP2 2x 2b-2 vector was then digested with NheI/SpeI to release a 2b-2 dimer, which was then ligated into a pIgL(−)GFP2 2x 2b-2 vector digested with NheI.
Figure 2. Localization of DIVAC activity within the W fragment.
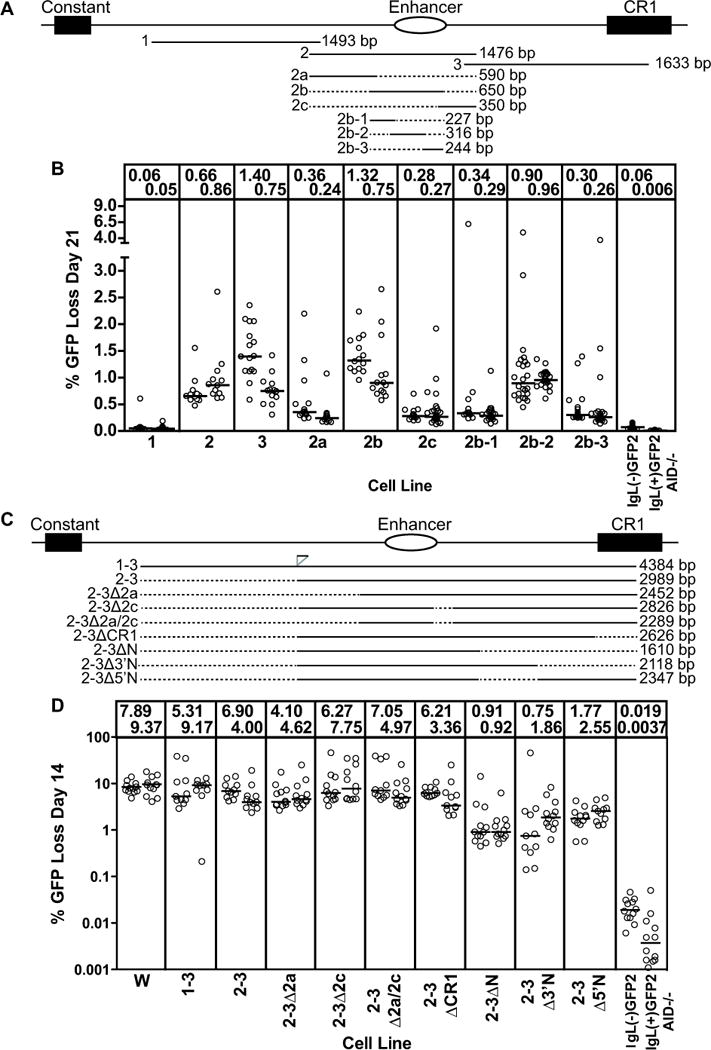
(A,C) Schematic diagram of the region 3′ of the chicken IgL locus. The constant region exon, IgL enhancer, and CR1 element are shown. The position and length of each DNA fragment tested are specified below the locus diagram, with dotted lines indicating missing sequence. Position and size of the subfragments are drawn to scale. Note that fragment 1-3 was discovered to contain a small (approx. 100 bp) duplication of sequences from the 3′ end of fragment 1, inserted at the position indicated in panel (C). (B, D) Fluctuation analysis of GFP loss in subclones. Each circle represents percent GFP loss for one subclone and the median GFP loss value of all subclones from the same primary transfectant is indicated with a horizontal bar and stated numerically at the top of the graph. The name of the test fragment adjacent to the GFP2 cassette, from which each cell line derives its name, is indicated at the bottom of the graphs. Data in (B) and (D) derive from subclones cultured for 21 and 14 days, respectively. Note that the Y axis for (D) and all subsequent plots of % GFP loss are depicted on a log scale. In (D), the end point for the deletion in 2-3ΔN corresponds to the 5′ end of fragment "N" (34), and the deletion removes a region that increased GFP loss activity in that study.
Figure 7. Analysis of F2 Core and F3 Core DIVAC elements.
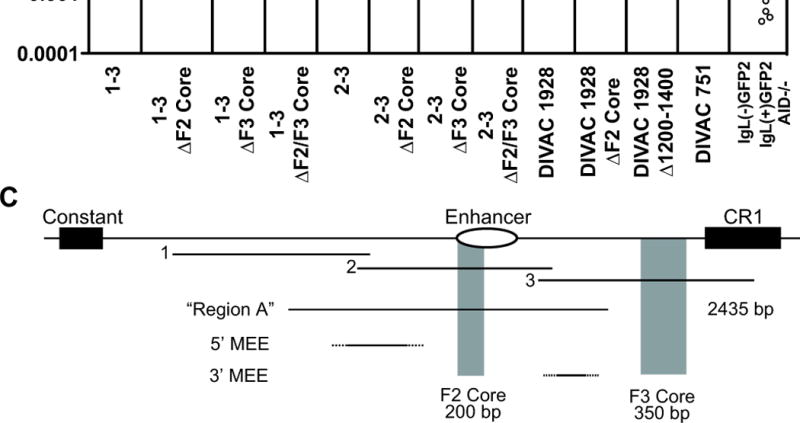
(A) Schematic diagram of the position of the F2 core and F3 core regions in the region 3′ of the IgLlocus (drawn to scale). The core elements were either singly (ΔF2 Core or ΔF3 Core) or doubly (ΔF2/F3 Core) deleted from DIVAC 1-3 or DIVAC 2-3, and individually deleted from DIVAC 1928 (the equivalent of the DIVAC 1703 Δ1201-1400 deletion was performed for DIVAC 1928 in place of the ΔF3 Core deletion). Deleted DIVAC core regions are depicted with gray shading. The Δ1201-1400 equivalent deletion is shown below DIVAC 1928. The end points of DIVAC 751, which is made up primarily of the two core elements, is also shown. (B) Fluctuation analysis of GFP loss in subclones with data depicted as in Fig. 2D. The DIVAC 1-3, 2-3 and 1928 datasets are the same as those in Figure 2D, and 6B, respectively, and are shown for ease of comparison. (C) Schematic diagram of the 3′ portion of the IgL locus, drawn approximately to scale, indicating the location of fragments 1, 2, and 3, the F2 and F3 cores, region A from (35, 37) and the 5′ mutational enhancer element (5′ MEE) and 3′ MEE from (38). The 222 bp element identified as functionally important in (38) lies within the 3′MEE. Dashed lines are used to indicate that the boundaries of the MEEs have not been defined.
Cell Culture
Cells were cultured in RPMI-1640 with 10% fetal bovine serum, 1% chicken serum, 2 mM L-glutamine, 0.1 mM β-mercaptoethanol and penicillin/streptomycin at 41°C with 5% CO2. Transfections were performed by electroporating 107 cells with 40 µg linearized plasmid DNA at 25 μF and 700 V (Bio-Rad Gene Pulser). Stable transfectants were selected with 10-15 μg/ml blasticidin (Invitrogen) for 6–8 days and subsequently screened by duplicate plating in 1 μg/ml puromycin (Sigma) to confirm deletion of the puromycin resistance gene within the rearranged IgL locus. Genotypes of clones with targeted integration were confirmed by PCR of the targeting construct and VJ intervening sequence of the unrearranged locus as previously described (34). The IgL(−)GFP2 (ψV−IgL−,GFP2), IgL(−)GFP2 W (ψV−IgLW,GFP2), and IgL(+)GFP2 AID−/− (ψV−IgLGFP2), and ψV−IgL−puroR DT40 cells have been described previously (34).
Flow Cytometry
GFP levels for each targeting construct were assessed by FACS (FACS Calibur, BD Bioscience) for at least two independent targeted transfectants. These primary targeted transfectants were subcloned by limiting dilution and at least 12 subclones for each primary transfectant were analyzed on day 14 post-subcloning (with the exception of subclones in Figs. 2b, and 4, which were analyzed on day 21 post-subcloning). At least 100,000 events were collected, and the gate for GFP low/negative cells was drawn one log below the mean fluorescence intensity for the main population of green fluorescent cells (FlowJo software). Subclones where more than 50% of live cell events fell into the GFP low or negative gate were excluded from analysis due to the possibility of expansion of a precursor cell expressing mutated GFP at the time of subcloning (34).
Figure 4. Deletion analysis of fragment 3.
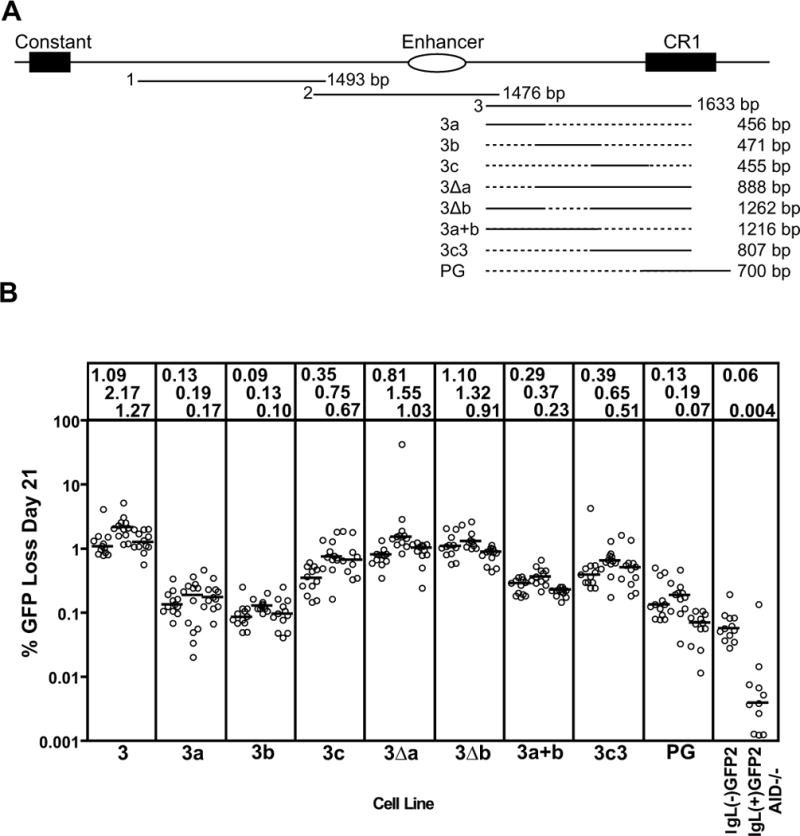
(A) Schematic diagram of the location of fragments 1, 2, and 3 in the region 3′ of the chicken IgL locus. The structures and lengths of the various fragment 3 deletion mutants analyzed are indicated, with dotted lines indicating missing sequence. (B) Fluctuation analysis of GFP loss in subclones, with data depicted as in Fig. 2D.
Chromatin Immunoprecipitation
Antibodies used for ChIP analysis were: H3K4me3 (04-745, Millipore), H4ac (06-866, Millipore), H3K9ac (06-942, Millipore), H3K9me3 (ab8898, Abcam), Ub-H2B (05-1312, Millipore), H3K79me (ab3594, Abcam), Pol II (sc-899x, Santa Cruz Biotechnology), RNA Pol II CTD PhosphoS2 (ab5095, Abcam), RNA Pol II CTD PhosphoS5 (ab5131, Abcam), Cdk9 (sc-8338, Santa Cruz Biotechnology), and normal rabbit IgG (12-370; Millipore). A polyclonal rabbit anti-Spt5 antibody (39) was a generous gift from Dr. Yuki Yamaguchi, Tokyo Institute of Technology, Japan.
ChIP was performed as described previously (40), with modifications. Briefly, cells were cross linked with 1% formaldehyde, quenched with 0.125 M glycine, then washed and resuspended in RIPA buffer (10 mM Tris [pH 7.4], 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, and 0.1% SDS) containing 0.5 M NaCl. Sonication was performed using a water bath sonicator (Diagenode), precleared with Protein G Dynabeads (Invitrogen), and chromatin from roughly 15 × 106 cells was incubated with specific antibody or normal rabbit IgG overnight at 4°C (an aliquot of 10% of the chromatin volume of an IP was set aside as the input sample). Immune complexes were isolated after a 3 hour incubation at 4°C with Protein G Dynabeads and extensive washing [twice for 10 minutes with each of the following buffers: RIPA, 0.125 M NaCl RIPA, 0.5 M NaCl RIPA, LiCl buffer (0.25M LiCl, 0.5% NP-40, 0.5% sodium deoxycholate), and TE (10 mM Tris-Cl [pH 7.5], 1 mM EDTA)]. After crosslink reversal and DNA purification, duplicate Taqman qPCR reactions were performed with PerfeCTa qPCR supermix with Ung (Quanta Biosciences) using company-specified cycling parameters with a 3000 XP thermocycler (Stratagene) (primer pairs and probes, Supplemental Table 1A). IP/Inputcorr was calculated as [(IPsp − IPrIg)/Input] × 1000, where IPsp and IPrIg are the amount of DNA recovered in IPs with specific antibody and rabbit IgG, respectively. Normalized signal was calculated by dividing the IP/Inputcorr for a test PCR region by the IP/Inputcorr calculated for γ-Actin with the same antibody. Two-tailed unpaired t tests were used in Fig. 8 to compare data for IgL(−)GFP2 versus IgL(−)GFP2 W and IgL(+)GFP2 AID−/− at PCR regions a–f and to compare data for region e versus region f using the combined data for IgL(−)GFP2 W and IgL(+)GFP2 AID−/−.
Figure 8. ChIP analysis of histone modifications and the transcription apparatus associated with GFP2.
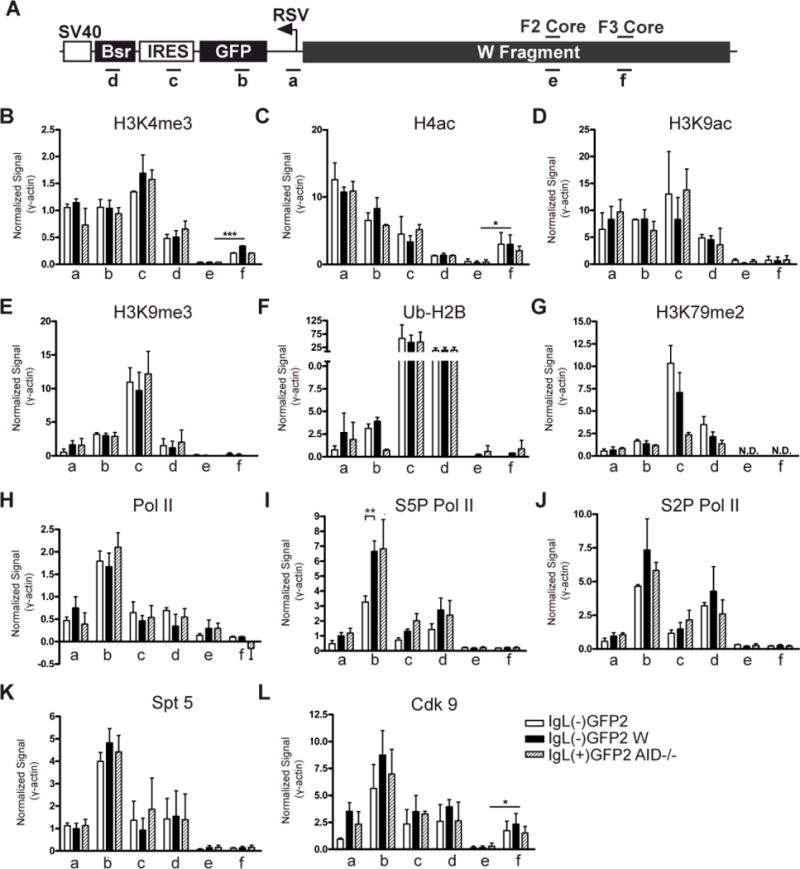
(A) Schematic diagram of an allele containing the GFP2 expression cassette flanked by the W fragment. Regions analyzed by PCR are indicated below and the DIVAC core elements are noted above. (B-L) Levels of each modified histone or transcriptional component (denoted at the top of each graph) were assessed by ChIP in the IgL(−)GFP2, IgL(−)GFP2 W and IgL(+)GFP2 AID−/− cell lines at the regions specified. IP/Inputcorr values have been corrected for background and normalized to the input signal as described in Materials and Methods, and then were divided by the IP/Inputcorr value obtained at γ-actin in the same ChIP. Each bar indicates the mean value with SEM shown. Two-tailed unpaired t tests were used to compare the data for the IgL(−)GFP2 cell line to that from the IgL(−)GFP2 W and IgL(+)GFP2 AID−/− cell lines for each PCR region, and to compare signals for PCR regions e and f. For this latter analysis, data from the cell lines containing the W region on the rearranged IgL allele (IgL(−)GFP2 W and IgL(+)GFP2 AID−/−) were pooled for each region and then compared. Data with statistically significant differences are noted (P<0.001 (***), P<0.01 (**), P<0.05 (*)). (B) H3K4me3 (n=5); (C) H4ac (n=2); (D) H3K9ac (n=2); (E) H3K9me3 (n=2); (F) Ub-H2B (n=2); (G) H3K79me2 (n=2); (H) Pol II (n=3); (I) S5P Pol II (n=4); (J) S2P Pol II (n=3); (K) Spt 5 (n=5); (L) Cdk9 (n=3).
RT-PCR Analysis
Total RNA was isolated from 5 × 106 DT40 cells using the RNeasy Mini Kit (Qiagen). RNA concentration was measured using a NanoDrop spectrophotometer (Thermo Scientific) and 1 μg RNA was treated with RNase-free DNase I (Invitrogen) and reverse transcribed using Superscript II (Invitrogen) primed with random hexamers. Taqman qPCR reactions were performed in duplicate using PerfeCTa qPCR supermix with Ung (Quanta Biosciences) and company-specified cycling parameters with 50 ng of first-strand product (primers and probes, Supplemental Table 1B). The starting quantity of the initial cDNA sample was calculated from primer-specific standard curves with the 3000 XP thermocycler (Stratagene) data-analysis software. cDNA values were normalized by dividing the calculated amount of input cDNA for GFP by the calculated input cDNA of 18S rRNA in each sample.
Results
DIVAC Assay
To assess DNA sequences for DIVAC function, we took advantage of a previously developed assay in which a heterologous expression cassette, flanked by the DNA sequence of interest, is inserted into the IgL locus of DT40 cells (34). The expression cassette, termed GFP2, consists of the strong, enhancer-independent Rous Sarcoma Virus promoter driving expression of GFP, followed by an internal ribosome entry site, the blasticidin resistance gene and an SV40 virus polyadenylation signal (Fig. 1A). To perform the assay, a targeting vector containing GFP2, the test fragment, and appropriate homology arms, is transfected into the ψV−IgL−puror DT40 cell line, in which the entirety of the chicken IgL locus has been deleted and replaced by a puromycin resistance gene (Fig. 1A), and which constitutively expresses AID (34). Homologous recombinants are identified as blasticidin-resistant, puromycin-sensitive clones, with proper insertion into the recombined IgL allele confirmed by PCR. At least two independent primary transfectants for each targeting vector were subcloned, and GFP expression assessed by flow cytometry in multiple subclones 14–21 days later. The frequency of cells with decreased GFP fluorescence (hereafter, “GFP loss”) provides a sensitive measure of the mutation frequency of GFP (34).
Figure 1. The DT40 DIVAC experimental system.
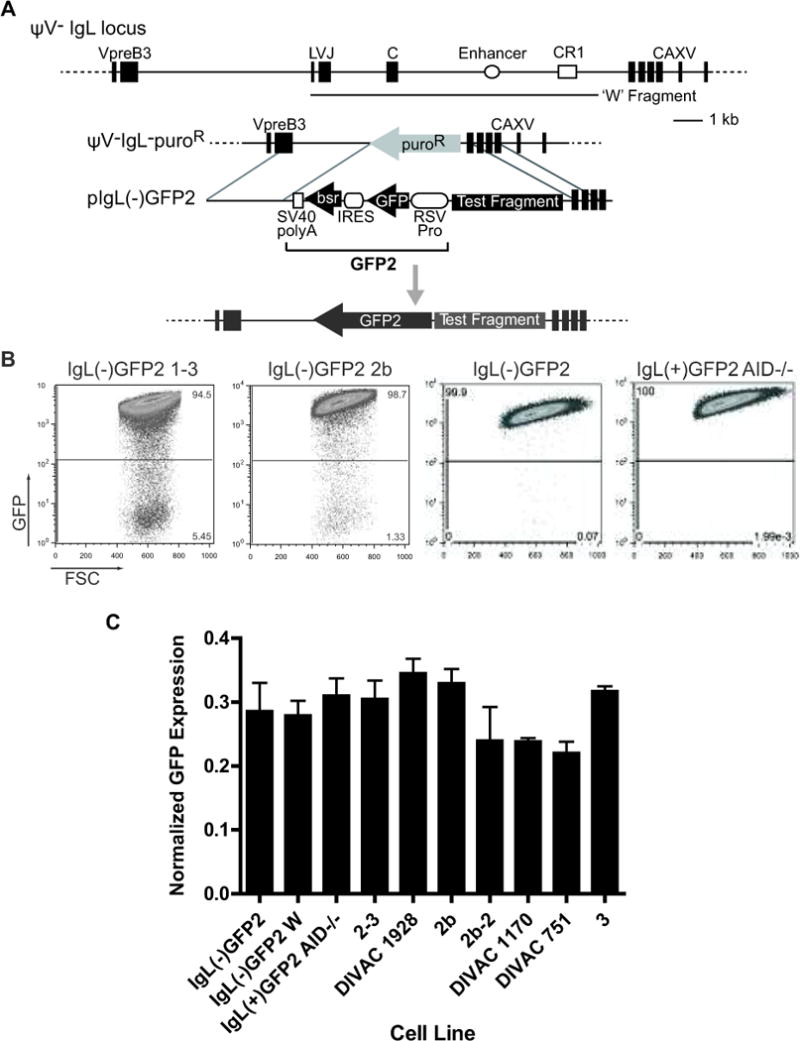
(A) Upper part: Schematic diagram of the ψV−IgL locus. Exons for IgL (leader (L), VJ, and constant (C)) and the surrounding genes VpreB and CAXV (carbonic anhydrase XV; predicted) are depicted as black boxes. The IgL enhancer and chicken repeat region 1(CR1) are shown as an open circle and open square, respectively. The location of the W Fragment (34) (GenBank accession no. FJ482234) is depicted with a line below the main diagram. The position and size of each of these elements are drawn to scale. Lower part: Map of the ‘empty’ IgL locus in the ψV−Ig−puroR cell line, targeting construct including the GFP2 reporter, and IgL locus after targeted insertion of the GFP2 reporter are shown. (B) FACS analysis of representative primary transfectants (21 days of culture) for the indicated targeting constructs. (C) Transcript levels of the GFP coding region assayed by Taqman quantitative RT-PCR. Bars show average signals for GFP transcripts from two independently derived RNA samples from each cell line, with standard error of the mean (SEM) indicated. Transcript levels were normalized to the 18S rRNA signal for each respective sample.
We confirmed that cells containingGFP2 without any flanking IgL sequences (cell line IgL(−)GFP2) exhibit very little GFP loss, with median frequencies <0.1% 21 days after subcloning (Figs. 1B and 2B) and <0.06% 14 days after subcloning (Fig. 2D). The data presented here were derived from cells cultured for 14 days after subcloning except that of Figs. 1B, 2B, and 4B, which derive from 21 day cultured cells. We observed a reproducible approx. two-fold increase in median GFP loss frequencies between days 14 and 21 of culture (data for 2b-2 in Figs. 2B and 3B, and data not shown). If GFP2 is flanked by the 9.8 kb “W” fragment (spanning IgL sequences downstream of the VL promoter; Fig. 1A), or by a subfragment of W with strong DIVAC function (e.g., the first panel of Fig. 1B), median GFP loss is increased at least 100 fold (Fig. 2D). In the absence of AID, GFP loss is reduced to extremely low levels (<0.005% at 14 days of culture) even when GFP2 is flanked by the entirety of the W fragment (IgL(+)GFP2 AID−/− in Fig. 1B and Fig. 2B and D). Our data, combined with that reported previously (34), demonstrate that in this assay system, mutation of GFP and GFP loss are strongly dependent on DIVAC sequences and AID.
Figure 3. Deletion and mutation analysis of fragment 2b-2.
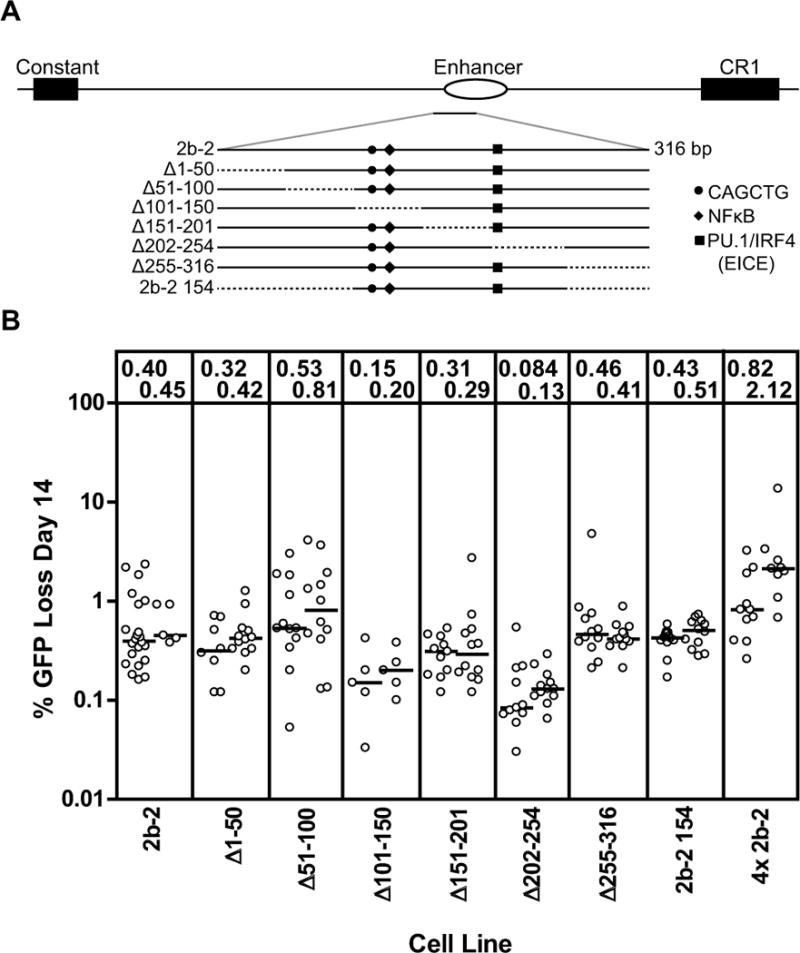
(A) Schematic diagram of the location of fragment 2b-2 in the region 3′ of the chicken IgL locus. The 2b-2 fragment is enlarged below and the structures of the various deletion mutants analyzed are indicated, with dotted lines indicating missing sequence. The positions of DNA motifs of interest are indicated with symbols, as indicated in the legend to the right. (B) Fluctuation analysis of GFP loss in subclones with data depicted as in Fig. 2D.
Importantly, the effects of DIVAC and AID on GFP loss and GFP mutation are not due to changes in levels of GFP transcription. GFP fluorescence levels of the main GFP+ population do not vary substantially depending on the presence or absence of a test fragment flanking GFP2, the length or nature of the test fragment, or the presence or absence of AID ((34), Fig. 1B, and data not shown). Furthermore, direct measurement of GFP transcript levels showed no differences between IgL(−)GFP2 cells (no test fragment), IgL(−)GFP2 W cells (W as a test fragment), and IgL(+)GFP2 AID−/− cells (W sequences flanking GFP2 but no AID) (Fig. 1C, first three bars). GFP transcript levels showed at most small variations in the other transfectants examined (Fig. 1C), and these variations did not correlate with DIVAC function or with the particular test fragment they contained. Finally, GFP transcript levels were previously shown to be equivalent between sorted GFP-high and GFP-low populations, demonstrating that GFP loss is not due to down regulation of GFP transcription (34).
Strong DIVAC function arises from synergy between discrete DNA elements
We focused our analysis on a 4.4 kb region starting downstream of the IgL C exon and terminating within a CR1 retrotransposon element (Fig. 2A) based on a previous low resolution deletion analysis of the W fragment (34). The 4.4 kb region was divided into three 1.5–1.6 kb fragments with 100 bp overlaps at their ends (fragments 1, 2, and 3; Fig. 2A), which were individually tested for DIVAC function. Fragment 1 was devoid of activity, while fragments 2 and 3 were active (Fig. 2B) but at levels well below that seen for W (Fig. 2D). Strikingly, either the entire 4.4 kb region (DIVAC 1–3) or a combination of fragments 2 and 3 (DIVAC 2–3), yielded levels of GFP loss that were nearly equivalent to that supported by the entire W fragment (Fig. 2D) and were substantially higher than the sum of the activities of the individual fragments (note that the results for the individual fragments in Fig. 2B are from day 21 while those for the combined fragments in Fig. 2D are from day 14). These results indicate that sequences in fragments 2 and 3 synergize with one another to yield strong DIVAC function.
To confirm that the high levels of GFP loss driven by DIVAC 2–3 were accompanied by frequent point mutations in GFP, we cultured IgL(−)GFP2 2–3 and IgL(−)GFP2 cell lines for 21 days and sequenced a 1217 bp region spanning GFP. As expected, IgL(−)GFP2 cells (which contain no DIVAC element) showed little evidence for mutation (5.7 × 10−5 mutations/bp), while IgL(−)GFP2 2–3 cells exhibited a mutation frequency of 1.28 × 10−3 mut/bp, similar to the frequency of 1.24 × 10−3 mut/bp reported previously for IgL(+)GFP2 cells (which contain all W sequences) cultured for six weeks (34) (Fig. S1). The mutation spectrum in IgL(−)GFP2 2–3 cells was similar to that of IgL(+)GFP2 cells, with almost all mutations occurring at G and C residues and C mutations being biased toward C→G transversions. DIVAC2-3 supported an increased frequency of G→A mutations compared to W (Fig. S1), which might be due to the smaller sample size in our analysis and differences in the size of the region sequenced. We conclude that DIVAC 2–3 has activity roughly comparable to that of W as assessed by GFP loss and point mutation of GFP.
Deletion analysis to identify minimal DIVAC sequences
We then performed deletion experiments to localize DIVAC function in fragments 2 and 3. Fragment 2 was divided into three overlapping fragments (2a, 2b, and 2c), and essentially all GFP loss activity was found associated with 2b (Fig. 2B). This suggests that fragments 2a and 2c lack functionally important elements, a conclusion further supported by the finding that deletion 2a, 2c, or 2a and 2c had no effect on the activity of DIVAC 2–3 (samples 2–3Δ2a, 2–3Δ2c, and 2–3Δ2a/2c, Fig. 2D). Subdivision of 2b into fragments 2b-1, 2b-2, and 2b-3 revealed that most activity resided in 2b-2, which, like fragment 2b, was as active as the entirety of fragment 2 (Fig. 2B). Fragment 2b-2 is therefore a small (316 bp) region with detectable DIVAC function (> 10x above background), although it contains only about 10% as much activity as DIVAC 2–3.
When a series of six approx. 50 bp deletions were made through the entirety of the 2b-2 element (Fig. 3A), GFP loss activity was found to be most reduced in the Δ101–150 and Δ202–254 deletions (Fig. 3B). A 154 bp fragment spanning these two deletion intervals and the intervening DNA (2b-2 154) was as active as 2b-2 (Fig. 3B), and therefore contains elements with DIVAC function. This 154 bp sequence contains an E-box motif (CAGCTG), an NF-κB binding site, and a binding site for the transcription factors PU.1 and IRF4 termed the Ets-IRF composite element (EICE) (Fig. 3A). This NF-κB site (35) and EICE element (37) have previously been suggested to contribute to DIVAC function, and considerable data link E boxes to SHM (see Introduction). Further analysis of the role of these individual motifs was difficult because of the low level of GFP loss associated with the 2b-2 fragment. We are currently developing a more sensitive DIVAC assay with which to identify functionally important motifs in DNA fragments with low activity. Interestingly, a 4x multimer of the 2b-2 fragment exhibited an average of approx. 1.5% GFP loss, more than three-fold higher than the 0.4–0.45% GFP loss seen with a single copy of 2b-2 (Fig. 3B). This suggests that DIVAC function can be modulated in an additive fashion, with the number of active DNA motifs, not just the presence of the motifs, being important.
Since much of 2b-2 lies within the chicken IgL enhancer, we tested mouse Igκ locus enhancers (iEκ, 3′Eκ)and mouse Igλ locus enhancers (Eλ2-4, Eλ3-1) for DIVAC function. These enhancers supported low levels of GFP loss, ranging from slightly above background to similar to that of 2b-2 in the case of 3′Eκ (Fig. S2). This argues that the murine enhancers are not strong DIVAC elements in DT40 cells by themselves, but leaves open the possibility that they can collaborate with other sequences to target SHM to Ig light chain genes.
Fragment 3 was also subjected to deletion analysis to identify regions important for DIVAC function. Analysis of three overlapping fragments spanning all of fragment 3 except CR1 sequences (3a, 3b, and 3c; Fig. 4A) revealed that only fragment 3c exhibited substantial activity, but at levels well below the entirety of fragment 3 (Fig. 4B). Deletion of 3a (3Δa) or 3b (3Δb) from fragment 3yielded regions with slightly more activity than 3c, while deletion of 3c and CR1 sequences (3a+b) resulted in loss of most activity. (Fig. 4B). Adding CR1 sequences to 3c (3c3) did not enhance activity, and a fragment spanning all of CR1 (PG) did not have substantial activity (Fig. 4B). Together, these results indicate that functionally relevant DNA sequences are dispersed in fragment 3, with fragment 3c containing particularly important DNA elements that require other sequences, likely within both fragments 3a and 3b, to attain the full DIVAC function found in fragment 3. The idea that functionally important elements are dispersed in fragment 3 is supported by deletion analyses carried out in the context of DIVAC 2–3, where deletion of most of fragment 3 (2–3ΔN), or the 3′ portion of fragment 3 (2–3Δ3′N), or 5′ sequences of fragment 3 (2–3Δ5′N), resulted in a substantial drop in GFP loss, while deletion of only the CR1 sequences (2–3ΔCR1) did not (Fig. 2D).
Broad dispersion of the elements contributing to DIVAC function
The data described above indicate that DIVAC 2–3 requires sequences in both fragments 2 and 3 for strong activity, that the activity of fragment 2 resides within the central part of fragment 2b, and that sequences from regions 3a, 3b, and 3c are important for the DIVAC function of fragment 3. In an attempt to create a smaller fragment that retained strong DIVAC function, the unique sequences of fragment 2b (those not contained in the overlap with 2a or 2c) were combined with most of fragment 3 (lacking the overlap with fragment 2 as well as CR1 sequences) to create DIVAC 1703 (Fig. 5A and Fig. S3A). In Fig. 5A and Fig. 6A, certain transcription factor recognition motifs are depicted to serve as landmarks. This includes E boxes, which are broken down into those conforming to the motif CASSTG (S=G or C), which has been demonstrated to be the preferential binding site for E2a-encoded proteins (41), and those that do not conform to this motif.
Figure 5. Deletion analysisof DIVAC 1703.
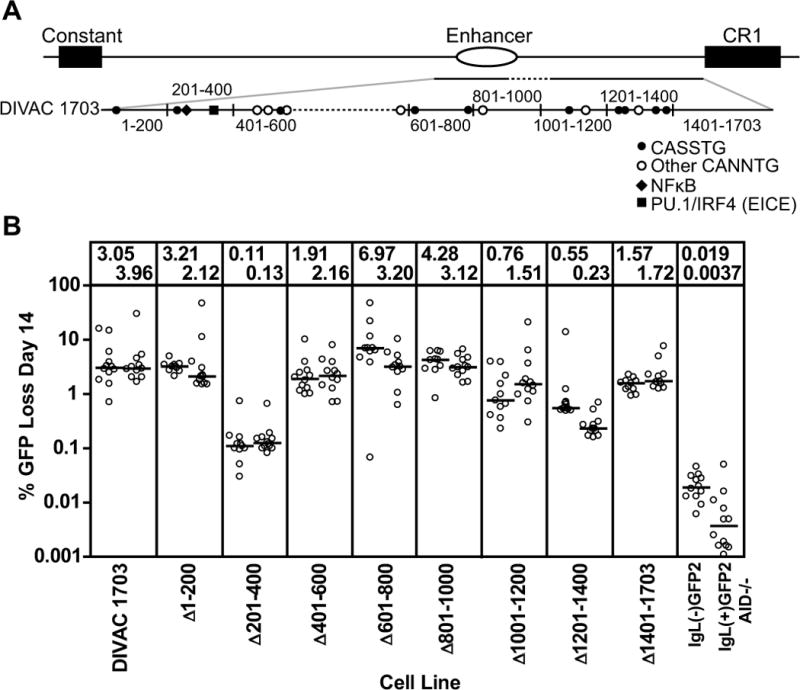
(A) Schematic diagram of the location of the DIVAC 1703 fragment in the region 3′ of the IgL locus. The DIVAC 1703 fragment is enlarged below to illustrate the end points of deletions (black vertical lines) and relevant DNA motifs (see key; S=G or C). Dotted lines indicate missing sequence. (B) Fluctuation analysis of GFP loss in subclones with data depicted as in Fig. 2D. The cell lines and test fragments are named for the roughly 200 bp segment that was deleted from DIVAC 1703.
Figure 6. Deletion analysisof DIVAC 1928.
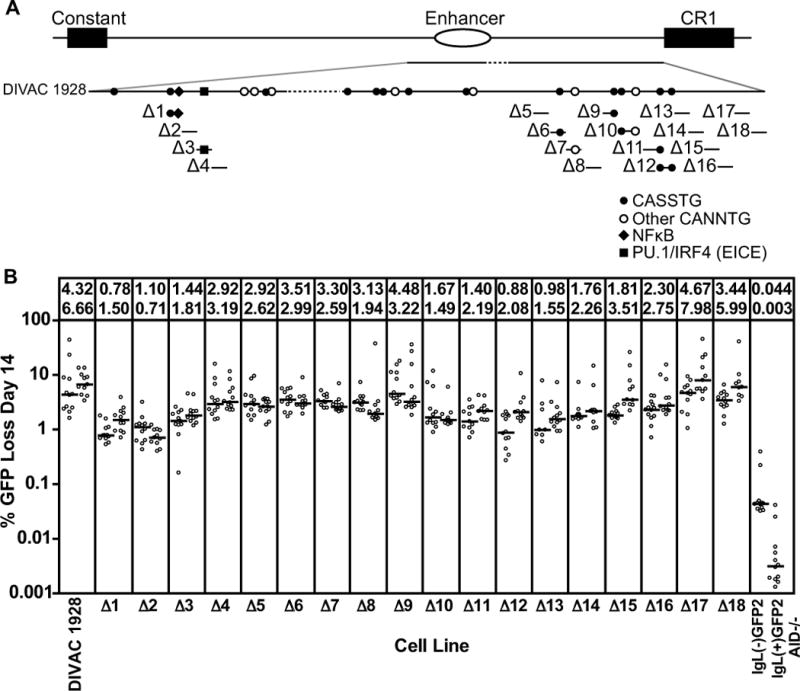
(A) Schematic diagram of the DIVAC 1928 element with positions of DNA motifs indicated. Each bar below the DIVAC 1928 line (Δ1-Δ18) shows a 50 bp DNA fragment and associated DNA motifs deleted from the DIVAC 1928 element. (B) Fluctuation analysis of GFP loss in subclones with data depicted as in Fig. 2D. The cell lines and test fragments are named for the 50 bp segment that was deleted from DIVAC 1928.
DIVAC 1703 was found to confer 3–4% GFP loss in our assay (Fig. 5B), less than the 4–7% observed with DIVAC 2–3. A series of approx. 200 bp deletions was created across the entirety of the 1703 bp fragment to localize important DNA motifs. Two deletions strongly affected GFP loss: Δ201–400, with a 25–30 fold reduction in activity, and Δ1201–1400, with an approx.8-fold reduction in activity (Fig. 5B). Intriguingly, theΔ201–400 deletion spans the 2b-2 154 fragment, illustrating that this small region not only has activity on its own, but is also critical for the function of a larger, more active element. The relatively strong effect of the Δ1201–1400 deletion, which includes the 5′ 150 bp of fragment 3c, suggests that this portion of 3c contains important DNA sequences. However, decreased activity was also seen with Δ1001–1200 (removing part of 3b) and Δ1401–1703 (removing the 3′ part of 3c), again supporting the idea that multiple elements in fragment 3 contribute to DIVAC function. The Δ401–600 deletion also reduced activity somewhat. The strong loss of activity seen with Δ201–400 and Δ1201–1400 further supports the idea that sequences from fragments 2 and 3 work together synergistically to constitute DIVAC function. However, the data argue that DNA motifs outside of Δ201–400 and Δ1201–1400 also contribute to DIVAC function.
In an attempt to create a fragment smaller than DIVAC 2–3 but that retained its full activity, we joined all of fragment 2b (including the overlap with 2a and 2c) to fragment 3 (lacking CR1 sequences), to create DIVAC 1928 (Fig. 6A and Fig. S3B). DIVAC 1928 (4.3–6.7% GFP loss; Fig. 6B) was found to be as active DIVAC 2–3 (4.0–6.9% GFP loss; Fig. 2D). In an attempt to identify with greater resolution the DNA motifs responsible for DIVAC function, a series of eighteen 50 bp deletions were made across DIVAC 1928 (Fig. 6A), focusing on the regions suggested by the data of Fig. 5B to be most important for function. Remarkably, almost every 50 bp deletion caused at least a moderate drop in the frequency of GFP loss (Fig. 6B), suggesting that functionally relevant sequences are distributed broadly in DIVAC 1928. Two regions were identified in which deletions caused the greatest loss of DIVAC function: Δ1–Δ3 and Δ10–Δ16. Δ1–Δ3 corresponds closely to2b-2 154 (Fig. 3A) and lies within the Δ201–400 interval (Fig. 5A), whileΔ10–Δ16 lies within fragment 3c (Fig. 4A) and the Δ1201–1400 and Δ1401–1703 intervals (Fig. 5A). Based on these data, we defined functionally important “core” regions of fragments 2 and 3 as the F2 core (Δ201–400) and the F3 core (350 bp spanned by Δ10–Δ16).
The analysis of DIVAC 1703 indicated that sequences from 1–200 and 601–1000 contributed little to DIVAC function in this context (Fig. 5B), while the analysis of DIVAC 1928 indicated that Δ17 and Δ18 did not reduce activity substantially (Fig. 6B). However, deletion of all of these apparently unimportant sequences from DIVAC 1928 reduced activity substantially, to approx. 2.1–2.7% GFP loss (data not shown). We have thus far not been able to identify a DNA fragment substantially smaller than DIVAC 1928 that retains high levels of activity.
A critical role for the F2 and F3 core regions
To determine the extent to which the F2 and F3 core regions contribute to the activity of large, highly active DIVAC elements, they were deleted either individually or in combination from DIVAC 1–3 and DIVAC 2–3 (Fig. 7A). In both contexts, deletion of either core alone reduced activity, and deletion of both cores resulted in a nearly complete loss of activity (Fig. 7B). Similarly, deletion of the F2 core or a critical portion of the F3 core (the 1201–1400 region) from DIVAC 1928 reduced the frequency of GFP loss (the double core deletion was not tested in this context) (Fig. 7A, B). In all three contexts, deletion of the F3 core caused a greater loss of activity than deletion of the F2 core. These results demonstrate that when the core regions are removed, the remaining sequences (nearly 3.9 kb in the case of DIVAC 1–3) have no ability to support AID-mediated sequence diversification, and hence that the core regions play essential roles in DIVAC function. To determine whether the core regions are sufficient for strong DIVAC function in a smaller fragment, they were combined together with some flanking DNA sequences to yield DIVAC 751 (Fig. 7A). DIVAC 751 (2.6–3.0% GFP loss) was approximately halfas active as DIVAC 2–3 (Fig. 7B). We conclude that sequences outside of the core regions contribute to DIVAC function.
DIVAC and epigenetic modifications
To begin to explore the mechanism by which DIVAC functions, we tested the hypothesis that DIVAC alters levels of epigenetic modifications within the mutation cassette, taking advantage of the availability of cell lines harboring identical GFP2 transcription units either flanked or not flanked by W sequences. ChIP experiments were performed using three cell lines: IgL(−)GFP2 (no DIVAC sequences), IgL(−)GFP2 W (W sequences flanking GFP2), and IgL(+)GFP2 AID−/− (W sequences flanking GFP2, lacking AID). Six different regions of the locus were analyzed (Fig. 8A), four in the GFP2 cassette and two within the F2 or F3 cores. (Note that in IgL(−)GFP2 cells, W sequences are assessed only on the unrearranged IgL allele, while in IgL(−)GFP2 W and IgL(+)GFP2 AID−/− cells, W sequences are present on both the GFP2 knockin allele and the non-rearranged IgL allele.) We analyzed six histone modifications—H3K4me3, acetylated H4 (H4ac), acetylated H3 lysine 9 (H3K9ac), H3 lysine 9 trimethylation (H3K9me3), mono-ubiquitinated H2B (Ub-H2B), and H3 lysine 79 dimethylation (H3K79me2)—based on previous data linking them to AID-mediated diversification processes and transcription (29, 42–49). Values were normalized to the signal obtained from the γ-actin gene, a well expressed gene whose modifications should be similar in the three cell lines. Statistical analyses (see Materials and Methods) were used to compare signals from IgL(−)GFP2 cells to both the IgL(−)GFP2 W and IgL(+)GFP2 AID−/− cells, and to compare the signals obtained at the two core regions for cell lines containing W sequences adjacent to GFP2.
H3K4me3, H4ac, and H3K9ac are marks of open, transcriptionally active chromatin that have been suggested to enhance AID-mediated diversification processes (29, 42–45). We did not observe significant differences in the levels of these marks at any region of GFP2 between IgL(−)GFP2 cells (white bars) and IgL(−)GFP2 W (black bars) or IgL(+)GFP2 AID−/− cells (hatched bars) (Fig. 8B, C, D). Levels of H3K4me3 were, however, higher at the F3 core than the F2 core (Fig. 8B), as were levels of H4ac (Fig. 8C). H3K9me3, a repressive mark generally found in heterochromatin, is found in S regions undergoing CSR (46) where it is thought to facilitate the recruitment of AID (47). No DIVAC-dependent differences in levels of H3K9me3 were detected in GFP2, and this mark was virtually undetectable at the F2 and F3 core regions (Fig. 8E). Ub-H2B is a consequence of transcriptional activation (50) and affects the movement of Pol II through a coding region by directing di- and tri-methylation of H3K4 and H3K79 (51). Ub-H2B and Ub-H2A were previously found associated with AID-targeted loci in a human cell line and with S regions in stimulated primary mouse B cells (48). Low levels of Ub-H2B were found at the promoter and GFP coding region while higher levels were seen at the IRES and Bsr segments of GFP2; however, none of the signals exhibited a dependence on the presence of DIVAC (Fig. 8F). H3K79me2 marks intragenic regions of actively transcribed genes and is associated with transcription elongation (49). Relatively low and comparable levels of H3K79me2 were seen at the promoter and GFPcoding region in the three cell lines, with higher levels detected at the downstream elements of the GFP2 cassette, particularly IRES (Fig. 8G). In addition, IRES displayed significantly higher levels of H3K79me2 in the absence of W as compared to when W was present (Fig. 9G), raising the possibility that transcription elongation within IRES is altered by W sequences. It is unclear what feature of the IRES element dictates its particularly high levels of Ub-H2B and H3K79me2.
In summary, the analysis of six different histone marks failed to reveal any DIVAC-dependent differences at the promoter or at the GFP gene, where DIVAC-dependent mutagenesis occurs.
DIVAC and the transcription apparatus
Because of the tight link between transcription and the action of AID, we used ChIP to investigate the hypothesis that DIVAC mediates changes in Pol II recruitment, promoter clearance, or transcriptional elongation. The C-terminal domain (CTD) of Pol II is hypophosphorylated upon initial recruitment to a promoter and undergoes phosphorylation at serine 5 (S5P) during promoter clearance by general transcription factors (52). S5P Pol II complexes accumulate approx. 40 nt downstream of the transcription start site, partially due to association with the negative-acting elongation factor complex (NELF) and the DRB-sensitivity inducing complex (DSIF; composed of Spt4 and Spt5) (53). The release of Pol II pausing occurs following a second phosphorylation event on serine 2 (S2P) of the CTD that is mediated by P-TEFb, a complex containing the kinase Cdk9 (54). P-TEFb also phosphorylates DSIF, and the combination of S2P Pol II and phosphorylated DSIF causes the dissociation of NELF and activation of elongation.
We first used an antibody to the N-terminal portion of Pol II to measure total Pol II levels at the GFP2 cassette and the F2 and F3 core regions. Total Pol II levels were highest in GFP and did not vary significantly between the cell lines containing (black or shaded bars) or lacking DIVAC (white bars) at any of the regions tested (Fig. 8H), consistent with the similar levels of GFP transcripts detected in these lines (Fig. 1C). We also assessed levels of S5P and S2P Pol II and found that the ChIP signals for these two modifications differed significantly between the cell lines only within the GFP coding region, where S5P Pol II was elevated about two-fold in the presence of DIVAC as compared to its absence, and S2P Pol II was also increased in a DIVAC-dependent manner, although only reaching statistical significance in one of the two DIVAC-containing cell lines (Fig. 8I, J). The DSIF component Spt5 was recently shown to facilitate interactions between AID and Pol II at sites of Pol II stalling (8), making it of particular interest in relation to the mechanism by which DIVAC recruits SHM. There were, however, no significant DIVAC-dependent differences in Spt5 levels in any of the regions analyzed (Fig. 8K). The P-TEFb component Cdk9 was also analyzed, revealing a consistent trend of higher levels of Cdk9 in the presence of DIVAC as compared to its absence, although none of the individual differences were statistically significant (Fig. 8L).
Overall, these experiments indicate that, in the presence of DIVAC, the GFP mutation target region displays increased levels of the stalled (S5P) form of Pol II but not of the Pol II-associated factor Spt5. They also raise the possibility that the presence of DIVAC is associated with an increase in the pause release kinase Cdk9 and its product S2P Pol II.
Discussion
Properties of DIVAC
We have performed an extensive deletion analysis of DIVAC, a region of the chicken IgL locus previously implicated in supporting efficient AID-mediated mutation of a flanking GFP transcription unit (34). The findings reported here reveal a number of novel features of DIVAC. First, the nearly 10 kb W fragment can be reduced in size to less than 2 kb (DIVAC 1928) with only modest loss of activity. Second, sequence elements that contribute to DIVAC function are not found in one small region but instead are widely dispersed. This is most clearly illustrated by our observation that many different 50 bp deletions in DIVAC 1928 cause a measurable drop in activity (Fig. 6), but is also apparent from deletion analyses in the context of relatively small fragments (Figs. 2, 3, and 4) and another large fragment (Fig. 5). Third, despite the dispersal of functional elements, two relatively small regions of particular functional significance exist: the 200 bp F2 core spanning the 5′ half of the canonical IgL enhancer, and the 350 bp F3 core that lies downstream of the enhancer, about 1.2 kb away. In several different contexts, deletions affecting the cores are more detrimental to activity than comparably sized deletions in other regions. Strikingly, while removal of one of the two cores usually has only a modest effect on activity, deletion of both cores, even in the context of quite large fragments, virtually eliminates GFP loss activity. These observations indicate that while other sequences in the fragments analyzed here can partially compensate for loss of one core, they cannot compensate for the loss of both. We cannot rule out the possibility that sequences outside of the 4.4 kb DIVAC 1–3 region could compensate for deletion of both cores. It is also apparent that the consequences of deletion of the F2 core are context dependent, causing a relatively mild (2–3 fold) drop in activity in the context of DIVAC 1–3, DIVAC 2–3, and DIVAC 1928 (Fig. 7B), but a severe (30-fold) decrease in the context of DIVAC 1703 (Fig. 5B). This indicates that the 225 bp present in DIVAC 1928 but missing from DIVAC 1703 play an important role in compensating for the loss of the F2 core. The existence of sequences with redundant function with the F2 core is further highlighted by the finding that the IgL enhancer (which spans the entirety of the F2 core) was dispensable for GCV/SHM in the context of the entire IgL locus (26).
Fourth, small fragments spanning the individual cores (e.g., 2b-2 and 3c) have quite weak DIVAC function (about 10% that DIVAC 2–3), but considerably more when juxtaposed in DIVAC 751 (50% of DIVAC 2–3). This suggests synergy between the F2 and F3 cores, a notion consistent with our finding that DIVAC 2–3 has much more activity than the sum of the activities of fragments 2 and 3 (Fig. 2). Such synergy stands in contrast to the roughly additive increase in DIVAC function seen with a tetramer of fragment 2b-2 (Fig. 3B). Together, our findings suggest that full DIVAC function is achieved through the additive action of multiple DNA elements with similar function combined with the synergistic action of elements with distinct, complementary functions.
One limitation of our study is the difficulty in distinguishing between effects of a deletion caused by altering the spacing of flanking elements versus effects due to loss of the deleted sequences themselves. Given the numerous different contexts in which deletion of some or all of the F2 core or the F3 core causes a substantial drop in activity, it is unlikely that the cores function primarily as spacers; rather, it is likely that the core sequences are themselves important for DIVAC function. This is supported by our finding that DIVAC 751, which contains little besides the cores, has substantial activity. This result with DIVAC 751 also suggests that the natural spacing between the two cores is not essential for their ability to cooperate to some extent with one another. Other data, however, are consistent with the notion that a minimal inter-core spacing is important for maximal activity. Almost any manipulation that reduces the intercore distance below that seen in DIVAC 1928 results in a drop in activity, from approx. 5% GFP loss in DIVAC 1928 to 2.5–3.5% GFP loss in Δ5–Δ9 (Fig. 6B), DIVAC 1703 (Fig. 5B), and DIVAC 751 (Fig. 7B). This raises the possibility that DIVAC 1928 contains a favorable, minimal spacing between the core regions that is disrupted by deletions between them.
Comparisons with other studies of the DIVAC region
Several previous studies have searched the chicken IgL locus for sequences capable of directing AID-mediated sequence diversification (26, 33–38). The results of these previous studies are in general agreement with those reported here in localizing DIVAC function to the region 3′ of the IgL C region. In one set of studies, the most important region was found to span the enhancer (“Region A” in (35, 37)). Another study identified an important 3′ regulatory region (3′RR) consisting of the 4 kb 3′ of the enhancer (33), while a follow up analysis identified mutation enhancer elements (MEEs) both 5′ and 3′ of the enhancer(5′MEE and 3′MEE) (38). The relationship between these regions and the sequences identified as important in our experiments is summarized in Fig. 7C. Our finding that DIVAC is dispersed over many sequences helps explain the failure of previous studies to converge on a single functional element.
Previous studies implicated NF-κB subunits, IRF4, and the NF-κB and EICE binding sites located in the IgL enhancer as important for GCV in DT40 cells (35, 37). The results of our deletion analysis of 2b-2 (Fig. 3) and the location of the NF-κB and EICE sites within the F2 core, are consistent with the idea that these binding sites are relevant to DIVAC function. Numerous studies have implicated the E box sequence CANNTG and the E box binding proteins encoded by the E2a locus in SHM/GCV, both in mouse germinal center B cells and chicken DT40 cells (19, 27–31). Our data are consistent with this, but also suggest that the relationship between E boxes and DIVAC function is not a simple one. The Δ401–600 and Δ1201–1400 deletions each remove five E boxes from DIVAC 1703, but the latter reduces GFP loss activity almost ten fold while the former has less than a two fold effect (Fig. 5); and in the context of DIVAC 1928, 50 bp deletions that remove one or more E boxes typically reduce activity, but two of the deletions with the strongest deleterious effects (Δ2 and Δ13) do not contain E boxes (Fig. 6).
A recent study by Kothapalli et al. identified a 222 bp region downstream of the IgL enhancer that substantially increased the frequency of GCV in a stop codon reversion assay (although it had only a marginal effect on mutation frequency as assessed by DNA sequencing) (38). This 222 bp region, located within the 3′MEE (38) (Fig. 7C), lies between the F2 and F3 core elements and spans the junction between the Δ601–800 and Δ801–1000 deletion intervals, neither of which had a substantial effect on DIVAC function (Fig. 5B). Hence we do not detect a major, nonredundant role for the 222 bp region in our assay. Nor does our data lead us to assign substantial DIVAC function to DNA sequences in the area designated as the 5′MEE (38) (Fig. 7C). While the MEEs do not correspond to either the F2 core or the F3 core, the precise degree of overlap remains to be determined since the boundaries of the MEEs have not been mapped.
Methodological differences might help explain the apparent discrepancy between the sequences that we assign as the DIVAC cores and those assigned as the MEEs. One important difference is that Kothapalli et al. (38), as well as several other relevant studies in DT40 cells (35–37), utilized a transcription unit driven by the IgL promoter. This promoter is strongly dependent on distal sequences for its transcriptional activity (33), while the GFP2 cassette used here and previously (34) does not require other sequences to drive high level transcription. In some experiments utilizing the IgL promoter, mutations in putative GCV/SHM regulatory elements altered transcription levels, making effects on GCV/SHM more complicated to interpret (35, 37, 38). We did not observe substantial differences in transcription or expression of GFP in the different constructs analyzed, allowing us to determine the DIVAC function of a particular DNA sequence directly from the GFP loss activity it supported. Our approach, however, has the disadvantage that it would not detect potential IgL promoter-specific contributions to DIVAC function. Such promoter-specific effects might exist given the finding that the IgL promoter supports higher levels of IgL V region diversification than two heterologous promoters when normalized for transcription levels (26). One possibility is that the MEEs defined by Kothapalli et al. (38) have an important function in targeting SHM/GCV specifically in the context of the IgL promoter. We note that test fragments in our assay are located upstream of the GFP2 promoter, while they lie downstream of the transcription unit in IgL. However, this seems unlikely to be a relevant parameter given that GFP loss frequencies were similar when GFP2 was inserted 15 kb upstream or 26 kb downstream of the chicken IgL locus (34).
We observed low levels of DIVAC function associated with mouse Ig light chain enhancers (Fig. S2). Interestingly, the mouse 3′Eκ enhancer showed activity comparable to that of fragment 2 and small fragments derived from fragment 2 (e.g., 2b-2 and 2b-2 154), which span or represent a portion of the chickenIgL enhancer (Figs. 2B and 3). Given that fragment 2, or portions of it, are able to synergize with other sequences to create a highly active DIVAC element, it is possible that mouse Ig light chain enhancers also play a significant role, in concert with other sequences, in directing SHM to mouse Ig V regions. A recent study found that the mouse iEκ and 3′Eκ enhancers did not have substantial GCV/SHM targeting activity when assessed in conjunction with the IgL V promoter in DT40 cells (55). Our ability to detect DIVAC activity associated with theses sequences might be due to the greater sensitivity of our assay. We have not been able to identify regions of extended sequence similarity between chicken DIVAC sequences and mouse or human Ig loci. The possibility that mouse light chain gene enhancers contribute to DIVAC function warrants further investigation.
While our analysis represents by far the most extensive deletion and mutation analysis of DIVAC sequences to date, it does not exclude the possibility that sequences outside of DIVAC 1-3 also have DIVAC (mutation enhancer) activity. Indeed, our finding that sequence elements that contribute to DIVAC function are broadly dispersed renders this possibility likely.
Mechanism of action of DIVAC
DIVAC might act to increase AID-mediated diversification of a nearby transcription unit by multiple different mechanisms, including AID recruitment, AID activation, rendering the transcribed DNA a better substrate for AID, and/ortilting the balance of deoxyuracil repair in favor of pathways that have a mutagenic outcome over those that result in high-fidelity repair. Despite extensive efforts, we have thus far been unable to detect AID binding reproducibly within the GFP2 cassette by ChIP (in the same cell lines used for the ChIP analyses of Fig. 8), and hence have not been able to evaluate the hypothesis that DIVAC acts to recruit AID. A previous study detected a small (approx. 2 fold) increase in AID recruitment to the chicken IgL locus attributable to Fragment A (Fig. 7C) using an assay involving an AID-DAM methylase fusion protein (36). We note that IgL(−)GFP2 cells (which express AID) exhibit GFP loss frequencies that, while very low, are consistently an order of magnitude higher than in IgL(+)GFP2 AID−/− cells (Figs. 2, 4, 5, 6, 7). This suggests that AID is recruited to GFP2, at least at low levels, in the absence of DIVAC, and is consistent with the finding that AID associates at low levels with thousands of active promoters in ex vivo activated splenic B cells (11).
How does DIVAC facilitate mutation of a nearby transcription unit? Our data indicate that in our system it does not function by increasing transcription, as measured by steady state RNA levels, GFP fluorescence levels, or total Pol II association. Nor does it modulate the levels of six different covalent histone modifications within GFP that have previously been linked to SHM/CSR. We also do not see DIVAC-dependent differences in the levels of Spt5 associated with GFP2, which indicates that DIVAC is unlikely to act by increasing Spt5 recruitment. While epigenetic marks and Spt5 are likely to be important for SHM/GCV, our data indicate that an active transcription unit associated with Spt5, H3K4me3, and H3 and H4 acetylation is not sufficient for efficient SHM. We do observe that levels of S5P Pol II, the form associated with transcriptional pausing, increase significantly in GFP in a DIVAC-dependent manner. The DIVAC-mediated increase in S5P Pol II does not appear to be a consequence of the action of AID since it is observed equally in AID-sufficient and AID-deficient cells (Fig. 8I). Given the growing link between the action of AID, paused or stalled RNA Pol II, and single stranded regions of substrate DNA (7, 8, 11, 42, 56–59), it is conceivable that the increase in S5P Pol II reflects DIVAC-dependent perturbations of transcriptional elongation that assist in the generation of a suitable substrate for AID.
One appealing model is that DIVAC acts through the recruitment of trans-acting factors. If this is the case, then our data suggest that there are likely multiple relevant binding sites dispersed over the DIVAC region. There are 20 E boxes in DIVAC 1928, and as noted above, some deletions that reduce activity contain E boxes and others do not (Fig. 6). An important challenge for the future will be to determine to what extent E boxes, and non-E box sequences, contribute to DIVAC function. While our data are broadly consistent with an additive contribution of multiple DNA motifs to DIVAC function (e.g., the increase in activity seen upon multimerization of 2b-2; Fig. 3B), they are also suggestive of regions with distinct and synergistic activities (e.g., fragments 2 and 3; Fig. 2). In this regard, it is noteworthy that the F2 and F3 cores were found to immunoprecipitate with distinct factors and epigenetic marks—the F2 core with RNA Pol II, and the F3 core with Cdk9, acetylated H4, H3K4me3—perhaps reflecting distinct contributions to DIVAC function. An interesting possibility raised by our findings is that DIVAC is constructed from well known transcription factor binding sites, with Ig locus specificity achieved through the arrangement, number, and density of these binding motifs. A corollary of this is that the preferential mistargeting of SHM to particular non-Ig loci (19) might be the result of the presence of "degenerate" DIVAC elements in such loci.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Y. Yamaguchi for providing anti-Spt5 antibodies, Dr. M. Scharff and Dr. F. L. Kuang for reagents and advice, and members of the Schatz laboratory for advice and assistance during the course of this study.
Footnotes
This work was supported by the Howard Hughes Medical Institute. KMK and JJM were supported in part by Training Grant T32AI07019 from the National Institutes of Health. JLD was supported in part by the PhRMA Foundation and NIH Grant T15 LM07056 from the National Library of Medicine.
Abbreviations used in this paper: AID, activation induced deaminase; DIVAC, diversification activator; SHM, somatic hypermutation; GCV, Ig gene conversion; IgV, Ig variable region; IgL, Ig light chain; CSR, class switch recombination; Pol II, RNA polymerase II; ChIP, chromatin immunoprecipitation; ΨV, pseudo-variable gene segment.
References
- 1.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 2.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 3.Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295:1301–1306. doi: 10.1126/science.1067308. [DOI] [PubMed] [Google Scholar]
- 4.Harris RS, Sale JE, Petersen-Mahrt SK, Neuberger MS. AID is essential for immunoglobulin V gene conversion in a cultured B cell line. Curr Biol. 2002;12:435–438. doi: 10.1016/s0960-9822(02)00717-0. [DOI] [PubMed] [Google Scholar]
- 5.Flajnik MF. Comparative analyses of immunoglobulin genes: surprises and portents. Nat Rev Immunol. 2002;2:688–698. doi: 10.1038/nri889. [DOI] [PubMed] [Google Scholar]
- 6.Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, Scharff MD. The biochemistry of somatic hypermutation. Annu Rev Immunol. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- 7.Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, Januszyk K, Gregory RI, Deng H, Lima CD, Alt FW. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, Nieland TJ, Root DE, Casellas R, Nussenzweig MC. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Besmer E, Market E, Papavasiliou FN. Transcription elongation complex directs activation-induced cytidine deaminase-mediated DNA deamination. Mol Cell Biol. 2006;26:4378–4385. doi: 10.1128/MCB.02375-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 11.Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2010;12:62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 13.Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 14.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 15.Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, Neuberg D, Monti S, Giallourakis CC, Gostissa M, Alt FW. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, Casellas R, Nussenzweig MC. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141:27–38. doi: 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu M, Schatz DG. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. 2009;30:173–181. doi: 10.1016/j.it.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 20.Kothapalli NR, Fugmann SD. Targeting of AID-mediated sequence diversification to immunoglobulin genes. Curr Opin Immunol. 2011;23:184–189. doi: 10.1016/j.coi.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang SY, Schatz DG. Targeting of AID-Mediated Sequence Diversification by cis-Acting Determinants. Adv Immunol. 2007;94:109–125. doi: 10.1016/S0065-2776(06)94004-8. [DOI] [PubMed] [Google Scholar]
- 22.Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol. 2006;6:573–583. doi: 10.1038/nri1896. [DOI] [PubMed] [Google Scholar]
- 23.Longerich S, Basu U, Alt F, Storb U. AID in somatic hypermutation and class switch recombination. Curr Opin Immunol. 2006;18:164–174. doi: 10.1016/j.coi.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 24.Yelamos J, Klix N, Goyenechea B, Lozano F, Chui YL, Gonzalez Fernandez A, Pannell R, Neuberger MS, Milstein C. Targeting of non-Ig sequences in place of the V segment by somatic hypermutation. Nature. 1995;376:225–229. doi: 10.1038/376225a0. [DOI] [PubMed] [Google Scholar]
- 25.Betz AG, Milstein C, Gonzalez-Fernandez A, Pannell R, Larson T, Neuberger MS. Elements regulating somatic hypermutation of an immunoglobulin kappa gene: critical role for the intron enhancer/matrix attachment region. Cell. 1994;77:239–248. doi: 10.1016/0092-8674(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 26.Yang SY, Fugmann SD, Schatz DG. Control of gene conversion and somatic hypermutation by immunoglobulin promoter and enhancer sequences. J Exp Med. 2006;203:2919–2928. doi: 10.1084/jem.20061835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michael N, Shen HM, Longerich S, Kim N, Longacre A, Storb U. The E box motif CAGGTG enhances somatic hypermutation without enhancing transcription. Immunity. 2003;19:235–242. doi: 10.1016/s1074-7613(03)00204-8. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka A, Shen HM, Ratnam S, Kodgire P, Storb U. Attracting AID to targets of somatic hypermutation. J Exp Med. 2010;207:405–415. doi: 10.1084/jem.20090821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitao H, Kimura M, Yamamoto K, Seo H, Namikoshi K, Agata Y, Ohta K, Takata M. Regulation of histone H4 acetylation by transcription factor E2A in Ig gene conversion. Int Immunol. 2008;20:277–284. doi: 10.1093/intimm/dxm140. [DOI] [PubMed] [Google Scholar]
- 30.Schoetz U, Cervelli M, Wang YD, Fiedler P, Buerstedde JM. E2A Expression Stimulates Ig Hypermutation. J Immunol. 2006;177:395–400. doi: 10.4049/jimmunol.177.1.395. [DOI] [PubMed] [Google Scholar]
- 31.Yabuki M, Ordinario EC, Cummings WJ, Fujii MM, Maizels N. E2A Acts in cis in G(1) Phase of Cell Cycle to Promote Ig Gene Diversification. J Immunol. 2009;182:408–415. doi: 10.4049/jimmunol.182.1.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arakawa H, Buerstedde JM. Activation-induced cytidine deaminase-mediated hypermutation in the DT40 cell line. Philos Trans R Soc Lond. 2009;364:639–644. doi: 10.1098/rstb.2008.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kothapalli N, Norton DD, Fugmann SD. Cutting edge: a cis-acting DNA element targets AID-mediated sequence diversification to the chicken Ig light chain gene locus. J Immunol. 2008;180:2019–2023. doi: 10.4049/jimmunol.180.4.2019. [DOI] [PubMed] [Google Scholar]
- 34.Blagodatski A, Batrak V, Schmidl S, Schoetz U, Caldwell RB, Arakawan H, Buerstedde JM. A cis-acting diversification activator both necessary and sufficient for AID-mediated hypermutation. PLoS Genet. 2009;5:e1000332. doi: 10.1371/journal.pgen.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim Y, Tian M. NF-kappaB family of transcription factor facilitates gene conversion in chicken B cells. Mol Immunol. 2009;46:3283–3291. doi: 10.1016/j.molimm.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 36.Kim Y, Tian M. The recruitment of activation induced cytidine deaminase to the immunoglobulin locus by a regulatory element. Mol Immunol. 2010;47:1860–1865. doi: 10.1016/j.molimm.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 37.Luo H, Tian M. Transcription factors PU.1 and IRF4 regulate activation induced cytidine deaminase in chicken B cells. Mol Immunol. 2010;47:1383–1395. doi: 10.1016/j.molimm.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 38.Kothapalli NR, Collura KM, Norton DD, Fugmann SD. Separation of mutational and transcriptional enhancers in Ig genes. J Immunol. 2011;187:3247–3255. doi: 10.4049/jimmunol.1101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y, Yamaguchi Y, Tsugeno Y, Yamamoto J, Yamada T, Nakamura M, Hisatake K, Handa H. DSIF, the Paf1 complex, and Tat-SF1 have nonredundant, cooperative roles in RNA polymerase II elongation. Genes Dev. 2009;23:2765–2777. doi: 10.1101/gad.1834709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell. 2010;141:419–431. doi: 10.1016/j.cell.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, Ideker T, Glass CK, Murre C. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med. 2009;206:1817–1830. doi: 10.1084/jem.20081678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang LL, Whang N, Wuerffel R, Kenter AL. AID-dependent histone acetylation is detected in immunoglobulin S regions. J Exp Med. 2006;203:215–226. doi: 10.1084/jem.20051774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woo CJ, Martin A, Scharff MD. Induction of somatic hypermutation is associated with modifications in immunoglobulin variable region chromatin. Immunity. 2003;19:479–489. doi: 10.1016/s1074-7613(03)00261-9. [DOI] [PubMed] [Google Scholar]
- 45.Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc Natl Acad Sci USA. 2010;107:22190–22195. doi: 10.1073/pnas.1016923108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuang FL, Luo ZH, Scharff MD. H3 trimethyl K9 and H3 acetyl K9 chromatin modifications are associated with class switch recombination. Proc Natl Acad Sci USA. 2009;106:5288–5293. doi: 10.1073/pnas.0901368106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, Alt FW, Losson R, Reina-San-Martin B. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med. 2011;208:1649–1660. doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borchert GM, Holton NW, Edwards KA, Vogel LA, Larson ED. Histone H2A and H2B are monoubiquitinated at AID-targeted loci. PLoS ONE. 2010;5:e11641. doi: 10.1371/journal.pone.0011641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 50.Wyrick JJ, Parra MA. The role of histone H2A and H2B post-translational modifications in transcription: a genomic perspective. Biochim Biophys Acta. 2009;1789:37–44. doi: 10.1016/j.bbagrm.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Chandrasekharan MB, Huang F, Sun ZW. Ubiquitination of histone H2B regulates chromatin dynamics by enhancing nucleosome stability. Proc Natl Acad Sci USA. 2009;106:16686–16691. doi: 10.1073/pnas.0907862106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Selth LA, Sigurdsson S, Svejstrup JQ. Transcript elongation by RNA Polymerase II. Annu Rev Biochem. 2010;79:271–293. doi: 10.1146/annurev.biochem.78.062807.091425. [DOI] [PubMed] [Google Scholar]
- 53.Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008;319:1791–1792. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bres V, Yoh SM, Jones KA. The multi-tasking P-TEFb complex. Curr Opin Cell Biol. 2008;20:334–340. doi: 10.1016/j.ceb.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kothapalli NR, Norton DD, Fugmann SD. Classical Mus musculus Igkappa Enhancers Support Transcription but not High Level Somatic Hypermutation from a V-Lambda Promoter in Chicken DT40 Cells. PLoS ONE. 2011;6:e18955. doi: 10.1371/journal.pone.0018955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R, Gearhart PJ. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med. 2009;206:1237–1244. doi: 10.1084/jem.20082514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ronai D, Iglesias-Ussel MD, Fan MX, Li ZQ, Martin A, Scharff MD. Detection of chromatin-associated single-stranded DNA in regions targeted for somatic hypermutation. J Exp Med. 2007;204:181–190. doi: 10.1084/jem.20062032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parsa JY, Ramachandran S, Zaheen A, Nepal RM, Kapelnikov A, Belcheva A, Berru M, Ronai D, Martin A. Negative Supercoiling Creates Single-Stranded Patches of DNA That Are Substrates for AID-Mediated Mutagenesis. PLoS Genet. 2012;8:e1002518. doi: 10.1371/journal.pgen.1002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen HM, Storb U. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc Natl Acad Sci USA. 2004;101:12997–13002. doi: 10.1073/pnas.0404974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.