

Abstract
A challenge in biology is to understand how complex molecular networks in the cell execute sophisticated regulatory functions. Here we explore the idea that there are common and general principles that link network structures to biological functions, principles that constrain the design solutions that evolution can converge upon for accomplishing a given cellular task. We describe approaches for classifying networks based on abstract architectures and functions, rather than on the specific molecular components of the networks. For any common regulatory task, can we define the space of all possible molecular solutions? Such inverse approaches might ultimately allow the assembly of a design table of core molecular algorithms that could serve as a guide for building synthetic networks and modulating disease networks.
In the postgenomic era, we are accumulating a vast amount of data describing the array of molecules in living cells and their web of interactions. Yet even as more and more genomes, proteomes, and network maps appear, one of the remaining great challenges is to make sense of all the data to answer the fundamental question of how complex molecular networks are able to robustly and accurately carry out their physiological functions. Do we need to take into account all of this information to comprehend the mechanism, or are there more salient functional features which we can focus on, and, conversely, other details that we can place less emphasis on? Have such networks evolved as arbitrary and unique solutions, or is there an underlying logic and pattern to how and why networks have the structures that they have? These questions of mapping network structure and function are at the very heart of understanding the mechanistic relationship between genotype and phenotype at the cellular level.
Do Simplifying Design Principles Underlie Complex Biological Networks?
Here we explore the question of whether there might exist simplifying design principles that underlie the structure and evolution of complex cellular regulatory networks. The word design is often considered taboo within the biological community, given its close association with the term intelligent design—the notion that living systems were purposefully constructed by an intelligent force rather than through a random, evolutionary process. We argue that there is an important scientific role for considering design principles and how they influence biological systems. After all, biological systems have evolved under selective pressures to perform certain functions that increase organismal fitness. At the same time, there are physical constraints that limit the ways in which the tool kit of available biomolecular components can be used to solve these functional needs (i.e., limits on diffusion, catalytic or gene expression rates, binding specificity, etc.). Thus it seems critical to ask, for given functions, are there “better” or more accessible designs for how to harness molecular components to perform particular regulatory functions? If so, then such “good” designs would be likely attractors for the search process of evolution—if one could hypothetically replay evolution over repeatedly, one would observe convergence to these same archetypal classes, even if the detailed molecular implementations were very different. Thus when we use the term design principles, we are referring to the underlying landscape within which evolution can explore, and not to the explicit path or process by which a particular complex system came about.
Another way to phrase these questions is to ask whether there are meaningful ways to abstract diverse and complex regulatory networks to understand the common patterns for how they achieve a particular function. As an analogy, take the example of a chair. We can find many examples of chairs throughout cultures and history that differ greatly in their details, but at some abstract level, they all share low-resolution structural commonality that is dictated by how the laws of physics can be used to solve the functional problem of supporting a seated human being (Figure 1A). Similarly, at a molecular scale, in machines such as DNA polymerases, we can recognize common abstract organizational similarities that persist across different examples, in spite of wide sequence variation. These similarities are again linked to the physical constraints on how to perform this particular class of molecular scale mechanical work (Figure 1B). These two analogies are meant to be simply illustrative, and there are many other examples one could use to illustrate the concept of common design principles.
Figure 1. Underlying Design Principles.

(A) Mechanical devices often show core design principles intimately linked to their function. For example, chairs, despite many differences in detail and in origin, usually share common features that are linked to the physical requirements of supporting a seated human being.
(B) Molecular machines also often shown common features, as illustrated by the common organization of diverse DNA polymerases. (Panel is adapted from Steitz, 1999.)
(C) Ways in which complex cellular circuits might be abstracted into simpler core networks. A complex network could potentially be composed of several subnetwork modules, each with a simpler core function.
(D) Three interlinked ways to explore the existence of design rules and constraints: physical/biochemical constraints should prescribe the range of possible network solutions to a functional problem; common functional network solutions are expected to be enriched in natural evolutionary examples; synthetic molecular networks should also obey design rules.
Today, much effort is focused on understanding how regulatory networks allow a cell to process information in complex ways. Thus, a reasonable question is whether we can recognize analogous core organizational rules in cellular networks that are dictated by function. In macroscopic information processing systems, such as electronic circuits or computer programs, there are common architectures and algorithms that are used to solve common problems (Figure 1C). Might this also be the case for cellular information processing systems, and if so, how do we go about recognizing them?
An attractive working concept that a number of researchers have converged upon is the idea of a tool kit of elemental network motifs, each of which can perform common core functions (Buchler et al., 2003; Alon, 2007; Ma et al., 2009; Sneppen et al., 2010; Tyson and Novák, 2010). These motifs could potentially serve as the core framework or elemental building blocks to construct complex cellular functions. This universe of core motifs might be relatively finite, given the physical constraints on the biological molecules used to build them. Although hypothetical, one can easily imagine how such a set of core functional modules could be extraordinarily valuable in deconstructing the logic and mechanism underlying diverse classes of complex biological processes, including cell signaling, development, and metabolism (Hartwell et al., 1999; Tyson et al., 2001; Milo et al., 2002; Wagner, 2005; Ma et al., 2006, 2009; Novák and Tyson, 2008; Sneppen et al., 2010; Peter and Davidson, 2011a; Stephens et al., 2011; Bar-Even et al., 2012; Kholodenko et al., 2012).
There are several ways to explore the validity of this kind of network organizational framework (Figure 1D). If the premise is true that there are a finite number of ways to harness biochemical systems to execute a particular regulatory function, and that these rules govern network structure and evolution, then there are several predictions. First, we would expect that even in highly diverse organisms, there would be evolutionary convergence on particular preferred network motifs that perform common modular functions. Second, we would expect that, if we used molecularly constrained first principles to theoretically scan through the space of all possible networks, we should be able to enumerate and define a finite subset of networks capable of performing certain common, key regulatory functions. Third, we should be able to use these design principles to guide forward engineering in biology—the use of synthetic biology to build cells or organisms with new custom, targeted behaviors—as we would predict that even nonevolved regulatory systems should obey the same design rules.
Evidence of Common Solutions: Enriched Network Motifs
One way to explore whether nature has preferred network designs is to search for network motifs that occur at a higher frequency than expected. One of the most accessible types of networks to examine in this way is transcriptional regulatory networks, which can be mapped using high-throughput methods like chromatin immunoprecipitation studies, which reveal links between specific transcription factors and their downstream targets.
Seminal studies by Alon and colleagues revealed that in bacterial transcriptional networks, there are indeed highly enriched motifs within these transcription factor networks (Shen-Orr et al., 2002). Some of the simplest and most prevalent motifs are autoregulatory circuits (Rosenfeld et al., 2002; Isaacs et al., 2003), which can involve direct (e.g., a factor regulating its own expression) or indirect (with intervening links) feedback, and which can be positive or negative (Figure 2A). Examination of example motifs of this type have shown that positive feedback loops are often observed in systems that show switch-like behavior, memory, or bistability (i.e., toggling between fully ON or OFF states) (Alon, 2007). Negative feedback loops are functionally associated with systems that show strong noise resistance to perturbations (Becskei and Serrano, 2000; Alon, 2007; Hsu et al., 2012). Negative feedback loops are also associated with regulatory circuits that show acceleration—a more rapid time constant for reaching a new, input-induced steady state (Rosenfeld et al., 2002). Construction of synthetic positive and negative feedback circuits has validated the ability of these network architectures to robustly achieve these properties (Becskei and Serrano, 2000; Gardner et al., 2000). Molecular nodes, be they promoters or signaling proteins, seem particularly amenable to these kinds of feedback regulation, given the diversity of allosteric or binding mechanisms by which these nodes can be regulated by partners that lie downstream in regulatory pathways.
Figure 2. Enriched Network Motifs.

(A) The most common motifs in the bacterial transcription factor network are positive and negative feedback loops.
(B) Feedforward loops (FFL) are a common three-node motif. The top and bottom nodes (X and Z) are linked by both a direct regulatory path and an indirect one (via node Y). There are eight major subclasses of FFLs, characterized by the signs of their regulatory links. Coherent FFLs have indirect and direct links with the same overall sign. Incoherent FFLs have indirect and direct links with the opposite overall signs.
(C) A type I coherent feedforward loop with an AND gate terminal node can show the behavior of a persistence detector—it will only respond to a longer pulse of input.
(D) Table showing examples of simple functional behaviors and network architectures that are often associated with them (reviewed in Alon, 2007; Sneppen et al., 2010; Tyson and Novák, 2010).
Another, slightly more complex, network architecture that is highly enriched in bacterial transcriptional circuits is feedforward loops (FFLs), in which a single upstream node fans out to regulate two distinct downstream pathway branches of different lengths, but then these branches reconverge on an integrating node further downstream (Alon, 2007) (Figure 2B). Even in the more recently characterized human transcription factor network, there is strong enrichment for FFL motifs among three node networks (Gerstein et al., 2012; Neph et al., 2012). There are two major classes of feedforward loop motifs (each with several subclasses): coherent FFL motifs, in which the long and short branches of the network have the same net sign of action, and incoherent FFL motifs, in which the two branches have different overall signs of action (one branch is positive, one branch is negative) (Figure 2B). These subclasses are associated with distinct functions (Alon, 2007; Goentoro et al., 2009). Examples have been found of coherent feedforward loops that act as persistence detectors—systems that only switch on when the input persists for a minimum stimulation time (Figure 2C). Such motifs have a terminal integrating node that functions as an AND gate (stimulus must come from both branches simultaneously to activate this node). Thus the terminal node will only switch on when the stimulus lasts for as long as the difference in time that it takes for the signal to be transmitted down the two branches of the network. This kind of persistence detecting network module is postulated to filter against induction of a response by spurious, transient stimulation. Other classes of enriched motifs have been well summarized in a number of excellent reviews, and these motifs and their associated functional behaviors are summarized in Figure 2D (Alon, 2007; Sneppen et al., 2010; Tyson and Novák, 2010).
There are several limitations to the approach for searching for enriched motifs. First, in most cases, except for relatively straightforward transcription factor networks, we have relatively little data in which to search for enriched motifs—many proteomic data sets lack information about the directionality or sign of regulatory links. Second, we do not have a good understanding of the degeneracy of regulatory networks, and therefore we can only search for the enrichment of relatively rigidly defined network features. We are learning that in many cases, molecular details of cellular regulatory systems can be remarkably different, even among networks that perform the same overall function and share, at low resolution, the same overall regulatory architecture (Marín et al., 2000; Kitagawa and Hieter, 2001; Dementyeva and Zakian, 2010; Li and Johnson, 2010). Thus our goal of recognizing such potential enriched motifs is made more challenging by the degeneracy caused by evolutionary drift or fine-tuning. In seeking such modules, we must be cognizant that in some cases, similar network designs may be constructed, not only form completely different types of molecules, but also may have extra or equivalent links inserted or deleted. In addition, in some cases, a node that is composed of an individual molecule in one network may correspond to a multimolecular system in a different but functionally similar network. Thus any one function might be performed by a cluster of network architectures, rather than a few specific circuits. Identifying these functionally critical structural patterns and identifying the proper level of granularity with which to view networks will be essential.
Theoretical Exploration of Network Space
The fact that certain simple transcriptional network motifs are observed at higher frequency is consistent with there being an underlying design logic but does not give a complete picture. Particular network motifs might be more prevalent because of historical evolutionary accidents that locked in these types of solutions. A distinct approach for extracting design principles is to try to use molecular first principles combined with computational methods to theoretically explore the full space of possible networks. In this case, one can start, not with a particular network structure, but rather with a target function of interest, and ask the inverse question: given a particular function X or Y, what is the space of possible network motif solutions that can solve these problems (Figure 3A)? In this case, one is not asking what network is observed in any particular organism, but rather, what is the full space of physically plausible ways to achieve a function, given the first-principles biochemical constraints. How big is the space of solutions compatible with this function, and what is the space of networks that are incompatible with function? Is the landscape such that there are only a few clusters of network solutions, or are many solutions scattered about in distinct regions of network space (Figure 3B)?
Figure 3. Theoretically Mapping Network Structure Function.

(A) Ideal goal of exploring how distinct functions (X and Y) map to regions of possible network space. Two functions might correspond to completely distinct or partially overlapping classes of network architectures.
(B) Mapping the complexity of network space for one function. In this case there are two major regions of network space that can show robust performance (fitness) of function X.
(C) Adaptation maps to two general network solutions. Adaptation is observed in many sensory systems, and is defined as when the system output responds transiently to a change in input, but then restores itself at the original steady-state output level in order to allow for response to further changes in input. Searching the space of all three-node enzymatic networks (16,000 possible architectures) for robust solutions for adaptation revealed only two major solution classes: negative feedback loop with buffering node (NFBLB) and incoherent feedforward loop with proportioner node (IFFLP). The architectures require specific parameter ranges for links that control the key regulatory node B (Ma et al., 2009).
This approach requires a search through the large space of possible networks. In principle, one could generate all possible molecular networks, and then test them for their ability to perform the target function. In practice, however, this is not possible, given both the huge size of possible networks (dependent on the number of nodes, types of nodes, and parameters necessary to describe each node) and the computational cost of functionally evaluating each network. Thus, realistically, such an approach inherently requires some form of coarse-grained approximation to be computationally feasible. All approaches also require mathematically defining a relatively simple fitness function that can be used as a metric for how well a network performs the target function. There are then several distinct strategies, each with distinct advantages and compromises, that have been used to attack the challenging problem of searching network space to find the highest-performing functions.
Evolutionary Search Algorithms
One search strategy is based on in silico evolution—a starting set of random networks is permuted and tracked using genetic algorithms (François and Hakim, 2004; François and Siggia, 2008, 2010; Warmflash et al., 2012). At each round of evolution, these networks are tested for the target function, and a fraction of the best-performing networks are selected, then used as the pool that is subjected to further mutation (addition/deletion of nodes, addition/deletion of links, change in parameters). After multiple rounds of mutation and selection (often hundreds of cycles), convergence on particular network structures can often be observed. This type of simulation can be run many independent times. This strategy, because it follows individual network evolution trajectories, has the advantage of highlighting networks that are “evolvable”—ones for which there is a theoretically assessable path (i.e., a path of monotonically increasing fitness). At the same time, such a strategy is more likely to get trapped in local fitness maxima than to give a more unbiased view of the global landscape.
Network Enumeration Approaches
A distinct but complementary search strategy is based on enumerating all possible architectures within a complete network space and evaluating their ability to perform the function (without any rounds of simulated evolution) (Schuster et al., 1994; Li et al., 1996; Wagner, 2005; Ma et al., 2006, 2009; Lau et al., 2007; Hornung and Barkai, 2008). This strategy should in principle give a more unbiased (path-independent) picture of the network space, including plausible solutions that might be more difficult to reach through an evolutionary process. However, in this case, one must compromise by clearly delimiting the space to be searched by coarse-graining the features of the network. By fixing features such as the number of nodes and the types of nodes (e.g., enzymatic versus transcriptional), one can define a finite space of networks that is computationally feasible to analyze. At this point it is feasible to search a space of around three nodes, which corresponds to 104 possible network architectures. Each of these nodes, however, has multiple parameters associated with it, and it is impossible to perform a full scan through parameter space for all networks. Thus a common strategy is to analyze each network architecture with a large sample of parameters (e.g., 104). Parameters include features such as node concentrations, kinetic parameters like kcat and KM for enzymes, hill coefficients for ultrasensitive nodes, and link strengths (how much activity of upstream node alters activity of downstream node).
Although the ability of a network architecture to perform a function (its absolute fitness) may vary greatly dependent on the exact parameters associated with that network, one can evaluate each network architecture by its robustness, defined as the fraction of the sampled parameters for which it can perform the target function above some threshold score. The advantage of analyzing the network architectures by robustness is that it gives a picture of the fitness landscape for that architecture—what is the probability that this solution could be found in an evolutionary search, and what is the probability that the solution would be evolutionarily stable, i.e., able to perform the function in the face of random evolutionary drift of parameters?
Searching for Solutions to Common Functional Problems
These two computational search strategies have been used to explore the solution space for a number of common biological regulatory problems. These include dynamic behaviors observed in biological regulation such as the following: bistability, the ability of a system to switch in an all-or-none fashion between two distinct states, often with memory, as is observed in cell fate switches (Shah and Sarkar, 2011); adaptation, the ability of a system to transiently respond after input stimulus, but then to reset itself back to its original steady-state output level in order to allow for detection of further stimuli, a linear control system behavior that is observed in many sensory systems or homeostatic systems (François and Siggia, 2008; Ma et al., 2009); and oscillation, the ability of a system to stably fluctuate between distinct states, as is the case in the circadian clock or in wave-like signaling systems (Wagner, 2005; Markevich et al., 2006; Tsai et al., 2008; Muñoz-García and Kholodenko, 2010). These network search approaches have also been used to search for solutions to spatial regulation problems, such as the following: developmental patterning, networks that can form polar boundaries (Ma et al., 2006) and networks that can interpret a transient gradient to give an array of cells that form repeated stripes or a distinct series of segments, as observed in development (François and Siggia, 2010); and cell polarization, networks that can drive self-organized symmetry breaking to yield cells with molecularly distinct poles (Chau et al., 2012).
These types of target problems or behaviors have been chosen because they represent examples of what we currently consider as primitive regulatory functions that are prevalent throughout biology. Although simple, we can also see how these core functions could serve as building blocks to assemble higher-order function. We also know of and understand at least some natural systems that perform these functions (i.e., we have positive controls).
These computational search strategies have been extremely enlightening. First, they almost always yield networks that are capable of performing the target function, and identify known solutions that have been observed in real biology. Second, the fact that in silico evolutionary searches have been able to find networks that can perform these complex functions is fundamentally important evidence supporting the plausibility of evolution of complex behaviors (François and Siggia, 2008). It is remarkable that a random evolutionary search process, using only a set of simple regulatory modules (like promoter/transcription factors with varying affinities and hill coefficients) can lead to complex “biological” functions. Third, in nearly all cases, such searches have found a relatively finite cluster of solutions for each functional task, indicating that there may be some truth to the notion that there are a small number of “good” network solutions for a given function.
An example of a specific biological function that has been analyzed by multiple theoretical approaches is that of adaptation. As described earlier, adaptation is a dynamic behavior observed in many sensory systems, ranging from vision to bacterial chemotaxis, in which the system responds transiently to a change in input but then resets itself back to its original steady state. Notably, both evolutionary algorithms (François and Siggia, 2008) and circuit enumeration (Ma et al., 2009) approaches show convergence on a very small set of basic circuit families, including the architecture that has been well characterized for adaptation in bacterial chemotaxis.
In the enumeration analysis, all possible three-node network architectures (limited to Michaelis-Menten enzyme nodes that regulate one another) were tested for adaptation behavior, using 10,000 parameter sets for each (Figure 3C) (Ma et al., 2009). Out of the 16,000 possible networks, ~400 were found to show robust adaptation (i.e., performed behavior under a reasonable number of parameter sets). Analysis of these networks revealed that all 400 robust networks mapped to only two fundamental classes of network architectures. The first architecture is a negative feedback architecture in which the feedback node buffers against change in output—analogous to the methylation-based feedback node observed in bacterial chemotaxis that allows it to achieve adaptation to chemorepellent or attractant input. The second architecture is an incoherent feedforward architecture in which the slower feedforward node responds in a proportional way to compensate for input and resets the output node to the original steady state. It can be analytically shown, in this case, that these are the only two general solutions to yield a system that will return to the same output steady-state value in the face of input perturbations.
Importantly, architecture alone is not sufficient to specify this function. First, each solution class really represents a cluster of network architectures that embody a range of different precise architectures that mathematically perform the same operation (i.e., negative feedback loop via node B can involve different linkages A→B–|A, C→B–|A, or C→B–|C). Second, while most parameters are relatively unconstrained in both of these general solution classes, each has a handful of absolutely critical parameter values for key nodes. In these cases, the critical regulatory nodes must have KM values that cause the enzyme to function in either the linear or the saturated regimes. Thus, in reality, it is perhaps best to consider these two general network solutions as being a cluster of networks encompassed by a set of connectivity and parameter constraints.
It is quite remarkable that all of the functional networks for adaptation, as well as other functional examples, cluster into so few major classes, showing that there are a finite number of ways to solve this functional problem with basic molecular nodes. Thus, such studies suggest that it may be possible to determine a set of basic architectures that are preferred biochemical solutions for particular tasks.
Experimentally Exploring Network Space with Synthetic Biology
A parallel, and empirical, way to explore network space is using the emerging approach of synthetic biology. Although synthetic biology is often associated primarily with specific applications, such as the design of novel biomanufacturing pathways, the large-scale rewiring of biological regulatory networks actually offers a remarkably powerful way to explore basic science questions about the design logic of regulatory networks (Marshall, 2008; Rafelski and Marshall, 2008; Mukherji and van Oudenaarden, 2009; Weber and Fussenegger, 2009; Bashor et al., 2010; Elowitz and Lim, 2010; Liu et al., 2011; Nandagopal and Elowitz, 2011; Randall et al., 2011; Miller et al., 2012; Slusarczyk and Weiss, 2012; Slusarczyk et al., 2012). An exciting approach is to empirically explore network space by building new or altered synthetic circuits. If there are indeed a finite number of possible core networks that can perform a key function, then the same design rules should govern the construction of networks composed of nonnative components and generated through a nonevolutionary process. In fact, rebuilding a minimal network that can perform a function of interest using completely nonnative components can be viewed as one of the strongest proofs of particular design rules (much like how the synthesis of organic molecules was viewed as the ultimate proof of their molecular structure). Moreover, synthetic networks, because of their minimal and streamlined designs, are often experimentally easier to tune and scan parameter space for. Thus they may allow more systematic probing of the boundaries of parameter space that are required for performing the target function. In this sense, a synthetic biology approach is in many ways a philosophical extension of the much older biochemical reconstitution approach—the goal is to minimize and simplify the system to systematically explore the key requirements for function.
There are several distinct approaches to using synthetic biology to explore design principles. Some researchers have used completely nonnative molecular platforms to build networks from scratch. One powerful system of this type is networks of specially designed interacting nucleic acid molecules, whereby the presence of a single-strand molecule input can catalyze stand displacement reactions that can, if properly designed, propagate as a cascade through an in vitro molecular network. The cascades in these strand displacement systems are catalytically controlled (i.e., activated strands act as enzymes) but are energetically powered by the presence of “fuel” molecules that base pair with leftover strands, acting as a LeChatlier’s sink to push the reactions forward (Seelig et al., 2006). As far as we know, these types of reactions are not used in evolved living systems, but they represent an orthogonal molecular communication system that is analogous to the distributed molecular communication systems of cells. Such nucleic acid strand displacement reactions have been used to construct systems that show network behaviors, including logic operations, cascades, amplification, and feedback (Kim et al., 2006; Zhang et al., 2007; Qian and Winfree, 2011; Qian et al., 2011). Theoretical analysis indicates that arbitrary chemical reaction networks can be encoded using this type of nucleic acid component framework (Soloveichik et al., 2010). Thus a goal in this field is to ask whether such nucleic acid strand displacement circuits can be used as a platform to explore fundamental molecular rules about what distributed molecular systems can or cannot compute.
Other synthetic biology approaches aim to use natural biomolecular components, but in novel arrangements to construct circuits capable of target functions. One of the classic examples of a synthetic network is the synthetic oscillator (Elowitz and Leibler, 2000), built from a simple ring of three interlinked transcriptional repressors (Figure 4A). This original synthetic repressor, though functional, showed relatively poor performance, displaying inconsistent amplitudes and periods. Since then, many researchers have used synthetic oscillator as a model for iterative network improvement (Figure 4B). These synthetic biology efforts, combined with complementary computational analysis, have resulted in dramatic improvements in performance by incorporating additional network elements such as strong positive feedback on key nodes. Minimal designs have been identified that yield robust oscillations with either tunable amplitude or frequency, and these match architectures observed in natural oscillator systems. Thus iterative synthetic cycles have been useful in defining the space of oscillatory networks, and in distinguishing bare bones oscillator designs from slightly more complex designs that show far more robust behaviors or more specialized classes of behaviors (Atkinson et al., 2003; Fung et al., 2005; Stricker et al., 2008; Tsai et al., 2008; Tigges et al., 2009, 2010; Aubel and Fussenegger, 2010).
Figure 4. Using Synthetic Biology to Empirically Map Network Space.

(A) Improvements in transcriptional oscillator design. The original repressilator design, a minimal three-ring negative feedback loop made of three repressors, functioned but did not show consistent periods or amplitudes (Elowitz and Leibler, 2000).
(B) Many iterative improvements have been made in oscillator design, as exemplified by a two-repressor negative feedback loop coupled with positive and negative self-feedback loops, which yields a robust, tunable oscillator (Stricker et al., 2008).
(C and D) Exploration of multicellular patterning using synthetic circuits. Cell-cell communication circuits in bacteria have been used to generate fields of many cells that show developmental-like, static as well as expanding patterns such as ring/stripe formation (Basu et al., 2005; Liu et al., 2011).
(E) Probing circuits that can yield intracellular spatial self-organization. Parallel computational and synthetic studies were performed to construct circuits that can robustly generate self-organized cell polarization. The most robust network involves a combination of less functional minimal motifs, and was used to build a circuit that generated artificial phosphoinositide (3,4,5) tris-phosphate (PIP3) poles in yeast (Chau et al., 2012).
In addition to oscillators, synthetic biology approaches have been used to explore the construction of systems performing a range of other functional behaviors, including bistable memory switches, logic gate operations, population control, multicellular patterning, multicellular boundary formation, and cell polarization (Figures 4D and 4E). This impressive array of efforts has shown that it is possible to build minimal systems that recapitulate complex dynamic and spatial biological behaviors. One of the clearest and most important points to emerge is that it is often possible to use very different types of components—be they transcriptional, signaling, metabolic, or RNAi—to achieve the same class of behavior. In addition, these synthetic circuits have usually been constructed in a manner that allows some aspect of combinatorial parameter tuning, and thus have provided a way to empirically explore parameter space to define the boundaries that constrain function, as well as bifurcations (boundaries in parameter space in which function qualitatively changes). In the future, synthetic network combinatorial libraries combined with sophisticated functional screens are likely to provide a powerful empirical way not just to create a network with a particular target function, but to more fully define the architectural and parameter constraints for that function.
Looking Forward
We have explored the concept that complex molecular networks can be deconstructed into simpler network motifs that underlie function. Several lines of evidence support this conceptual simplification of biological regulatory networks. First, within transcriptional networks that have been explored in a high-throughput manner, there are clearly certain network motifs that are enriched, consistent with convergent evolution. These motifs are also associated with classes of regulatory functions. Second, a number of theoretical searches of network space suggest that for given regulatory functions such as adaptation or oscillation there are a finite number of core network solutions. Such studies are supported by synthetic biology reconstruction experiments in which different solutions are built and the key parameters and links tested. These studies suggest that there may be utility in trying to catalog the key subroutines that are necessary for life.
A Design Table of Modular Network Functions
These findings support the intriguing notion of organizing all plausible network architectures into a “biological design table.” One way to view this framework is through an imperfect but instructive analogy to the periodic table of elements (Figure 5). Prior to the periodic table, many analytical measurements of elements had been collected, but there was no sensible way to understand the properties and reactivity of each element. The era of Mendeleev was one in which researchers took this large amount of confusing data and tried to organize it in different ways in attempts to recognize meaningful patterns. This is similar to the stage that we are at in understanding the mechanisms of biological regulatory networks (Figure 5). One of the great insights provided by the periodic table was to classify the physical properties of elements in a way that accurately predicted their chemical reactivity and behavior. Instead of focusing on all properties describing each element, the periodic table, in the end, sorted them based on atomic number and valence—the key abstract properties that determined their ability to bond with other atoms. The organization of the periodic table is really a reflection of the physics of electron orbitals (how they are filled and how they determine bonding), but the abstract classification of atoms as simple models with constrained valency and bonding properties essentially allows one to skip over the detailed physics to understand, at a very practical level, the universe of possible higher-order chemical structures that this atom can participate in.
Figure 5. Concept of a Design Table for Biological Regulatory Networks.
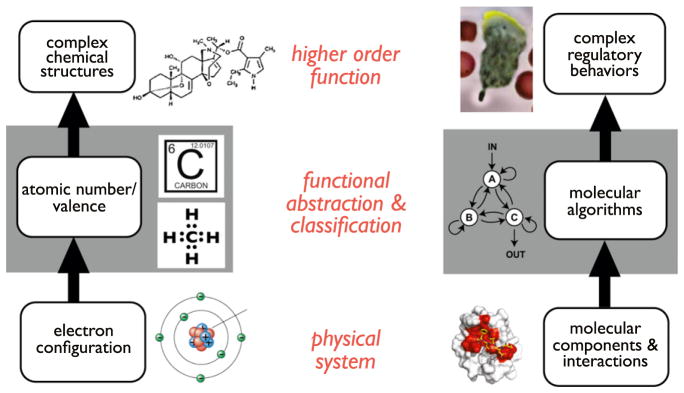
The periodic table abstracts the complex electronic structure of a particular atom and organizes it according to atomic number and valence. These features are in turn the functionally most important features relevant to understanding how the atom forms bonds to generate higher-order chemical structures. Analogously, a hypothetical design table of core regulatory network motifs might encompass and abstract the general/common solutions to core biological functions (which result from the constraints on molecular systems). This organization and classification might prove useful in understanding how evolution or engineering can build higher-order networks that show particular complex behaviors.
In an analogous way, there may be ways to abstract and classify particular cellular network architectures that focus on the properties that are most salient for their function, and the way in which they might be used as modules in evolution to build higher-order networks. If we did have such a design table for biological regulatory networks, it would be a powerful way to deconstruct complex biological or disease networks and to understand how they function. Might the observation of particular patterns of network architectures and parameters suggest functional hypotheses, much the way that observation of sequence homology does today? Such a design table would also be extremely useful to guide the design of novel synthetic circuits for many applications.
But, even if we assume that this model of classifying network motifs is correct, there remain many more questions than answers. Many of the most important and broad sets of questions concern how to search for and map core network modules for this hypothetical design table, discussed below.
Classification: What Is the Set of Primitive Molecular Subroutines that Are Most Critical for Cellular Systems?
Most theoretical studies searching for network space have been limited to a relatively small set of behaviors (bistability, oscillation, adaptation, etc.), and it is both challenging and fascinating to think more fundamentally about what are the core set of subroutines that are necessary to build and keep an organism alive. Most work has focused on simple information-processing functions, but such approaches could clearly be important in understanding more complex spatial organization, such as development (Davies, 2008; Peter and Davidson, 2011b, 2011a; Peter et al., 2012). Similar concepts may also operate in the design and evolution of complex metabolic pathways, as there may be analogous rules governing which chemicals are chosen as precursors, how they are linked by chemical paths and branchpoints, and how the enzymes are regulated as a network (Bar-Even et al., 2012).
Metrics: What Are the Best Ways to Search for and Evaluate Networks?
There remain different strategies for searching network space—which makes the most sense within the context of biology—searching through an evolutionary process (genetic algorithms) or through full enumeration of a coarse-grained space? Moreover, what is the best way to constrain and coarse-grain network space in such studies? How many nodes should be allowed, and what node types should be used, given the many different possible forms of regulatory molecules (e.g., enzymatic nodes, transcriptional nodes, miRNA nodes, etc.). Are these mechanistic differences (which require different mathematical models for node behavior) fine details, or do they significantly constrain solution space in different ways? Finally, in evaluating the function of networks in these search problems, most researchers have focused primarily on identifying robust networks (solutions that work in a larger range of parameter space), as opposed to the ones with the highest performance. This strategy is focused more on what is an evolutionary plausible solution—what is a solution that could potentially be found through an evolutionary process, and one that would be stable to drift in exact parameters. But is this assumption correct, or is it possible that nature may have found higher-performance solutions that are distinct from the robust solutions?
From Abstract to Concrete: How Do We Identify Functional Networks from Experimental Proteomic Data?
Beyond simple transcriptional networks, there are very few ways to take current proteomic data and translate it into a functional and directional network map. Thus we lack sufficient tractable experimental data. How can we address this gap and build a bridge that can link theoretical network analysis to experimental high-throughput systems biology data? There is currently a flood of functional genomic data that can identify sets of genes that are functionally linked, and yield some information about whether they are linked in series or in parallel processes via differential epistatic relationships (Ryan et al., 2012) (also see review by Fraser et al., 2013, in this issue of Molecular Cell). How do we take these functional genomic maps, as well as protein-protein interaction maps, and turn them into functional network maps, given the ambiguity of linkage, directionality, and sign of regulation between nodes?
Can Biological Regulation Be Accurately Represented by Simpler Networks?
Perhaps the most fundamental question concerning the logic of biological networks is whether this type of abstract and hierarchical model of complex cellular networks is an accurate approximation. Can we actually divide circuits into clearly distinct functional classes? To some degree, we already know that the assumption that network architecture alone is sufficient to determine function is wrong—the same network topology can qualitatively change function in different regions of parameter space. But are these parameter “phase diagrams” for a particular network architecture relatively simple so that we can define clear network/parameter regions that correspond to distinct functions? Or alternatively, are the functional classes of networks so degenerate and overlapping such that there are not distinct definable boundaries between circuits of different functional classes? It is possible that there is a class of networks architectures that are monofunctional (locked into one particular type of function, despite parameter variation), whereas other classes are polyfunctional and can move from one functional regime to another based on subtle parameter changes or changes of a few links.
Another fundamental question is whether network motifs really can be thought of as modules that can be used to build more complex function (Hartwell et al., 1999). Even if an isolated network can perform a function, does it still behave the same when it is linked to other upstream and downstream modules? Or does the network behavior change when you place this kind of functional load on it (i.e., can downstream effectors compete with feedback or feedforward interactions with an output node?) (Jiang et al., 2011)?
A Question of Utility
Ultimately, like other abstract theoretical constructs such as the periodic table and valency in chemistry, the bottom-line question concerns the utility of this framework. As the statistician George Box wrote, “All models are wrong, but some are useful” (Box, 1976). At some resolution, the abstract classification of networks is incorrect—each regulatory system will have somewhat different links, composition, and parameters, and these will alter function of the system in some way. But how good an approximation is this?
As discussed earlier, we believe that this conceptual framework of a design table of network motifs will be useful for forward engineering. Some molecular networks will be easier to recognize, build, and work with in a reliable and predictable way, and thus will have immediate and exciting utility in the construction and engineering of biological systems. We may be able to design optimized modules that can repeatedly and reliably execute desired subfunctions, and include features like tunability and insulation. Thus this design framework is likely to be of use in synthetic biology, whether it be in the design of organisms, optimized production of fuels, nutrients, or chemicals, or in the design of smart cell-based therapies that use designed signaling networks to make complex therapeutic decisions. One of the more interesting questions will be whether the synthetic solutions we converge on for particular functions are the same as those that evolution has settled on. It is possible that evolutionary solutions may be more limited because of stochastic constraints on exploring the solution space that are not limiting for first principles-based design.
Another emerging area in which a systematic understanding of network design principles may be of great utility is systems pharmacology. Systems pharmacology can be thought of as an example of network engineering in that one wants to take a disease network and strategically figure out how to tune or modulate the network so that it restores function or stability. One of the intriguing possibilities of network-based medicine is the idea that some strategies might not focus on simply blocking one malfunctioning protein with a drug, but that instead one might modulate different nodes in a network to redirect the network toward a robust and stable region of function space (Yang et al., 2008). Importantly, it is possible that such stable regions of function space that we want to drive disease networks toward may be “synthetic” in the sense that they are different from the wild-type (nondisease) network structure.
As we move forward with the maturation of systems and synthetic biology, we shall see if this kind of conceptual framework of idealized network motifs is useful in mechanistically deconstructing what are, for now, often impenetrably complex molecular networks. The biggest payoff would indeed be achieving a more intimate and fundamental understanding of the mysteries of how living systems harness ensembles of genetically encoded molecules to execute complex phenotypic functions, and the landscape of physically plausible network structure/function relationships that evolution operates within. Then we could look forward to a day when we do not view the complexity of biological networks as a source of confusion and mystery, but instead as a system that we have logical command of, and which we can tune and harness in treating disease and solving other biotechnological challenges.
Acknowledgments
We thank Matthew Thomson, Hana El-Samad, and colleagues in the UCSF Center for Systems and Synthetic Biology for valuable discussions and feedback on this review. This work was supported by the NIGMS P50 Center grant GM081879.
References
- Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- Atkinson MR, Savageau MA, Myers JT, Ninfa AJ. Development of genetic circuitry exhibiting toggle switch or oscillatory behavior in Escherichia coli. Cell. 2003;113:597–607. doi: 10.1016/s0092-8674(03)00346-5. [DOI] [PubMed] [Google Scholar]
- Aubel D, Fussenegger M. Watch the clock-engineering biological systems to be on time. Curr Opin Genet Dev. 2010;20:634–643. doi: 10.1016/j.gde.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Bar-Even A, Flamholz A, Noor E, Milo R. Rethinking glycolysis: on the biochemical logic of metabolic pathways. Nat Chem Biol. 2012;8:509–517. doi: 10.1038/nchembio.971. [DOI] [PubMed] [Google Scholar]
- Bashor CJ, Horwitz AA, Peisajovich SG, Lim WA. Rewiring cells: synthetic biology as a tool to interrogate the organizational principles of living systems. Annu Rev Biophys. 2010;39:515–537. doi: 10.1146/annurev.biophys.050708.133652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Gerchman Y, Collins CH, Arnold FH, Weiss R. A synthetic multicellular system for programmed pattern formation. Nature. 2005;434:1130–1134. doi: 10.1038/nature03461. [DOI] [PubMed] [Google Scholar]
- Becskei A, Serrano L. Engineering stability in gene networks by autoregulation. Nature. 2000;405:590–593. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- Box GEP. Science and statistics. J Am Stat Assoc. 1976;71:791–799. [Google Scholar]
- Buchler NE, Gerland U, Hwa T. On schemes of combinatorial transcription logic. Proc Natl Acad Sci USA. 2003;100:5136–5141. doi: 10.1073/pnas.0930314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau AH, Walter JM, Gerardin J, Tang C, Lim WA. Designing synthetic regulatory networks capable of self-organizing cell polarization. Cell. 2012;151:320–332. doi: 10.1016/j.cell.2012.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JA. Synthetic morphology: prospects for engineered, self-constructing anatomies. J Anat. 2008;212:707–719. doi: 10.1111/j.1469-7580.2008.00896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dementyeva EV, Zakian SM. Dosage compensation of sex chromosome genes in eukaryotes. Acta Naturae. 2010;2:36–43. [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Leibler S. A synthetic oscillatory network of transcriptional regulators. Nature. 2000;403:335–338. doi: 10.1038/35002125. [DOI] [PubMed] [Google Scholar]
- Elowitz M, Lim WA. Build life to understand it. Nature. 2010;468:889–890. doi: 10.1038/468889a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François P, Hakim V. Design of genetic networks with specified functions by evolution in silico. Proc Natl Acad Sci USA. 2004;101:580–585. doi: 10.1073/pnas.0304532101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François P, Siggia ED. A case study of evolutionary computation of biochemical adaptation. Phys Biol. 2008;5:026009. doi: 10.1088/1478-3975/5/2/026009. http://dx.doi.org/10.1088/1478-3975/5/2/026009. [DOI] [PubMed] [Google Scholar]
- François P, Siggia ED. Predicting embryonic patterning using mutual entropy fitness and in silico evolution. Development. 2010;137:2385–2395. doi: 10.1242/dev.048033. [DOI] [PubMed] [Google Scholar]
- Fraser JS, Gross JD, Krogan NJ. From systems to structure: bridging networks and mechanism. Mol Cell. 2013;49:222–231. doi: 10.1016/j.molcel.2013.01.003. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung E, Wong WW, Suen JK, Bulter T, Lee SG, Liao JC. A synthetic gene-metabolic oscillator. Nature. 2005;435:118–122. doi: 10.1038/nature03508. [DOI] [PubMed] [Google Scholar]
- Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;488:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goentoro L, Shoval O, Kirschner MW, Alon U. The incoherent feedforward loop can provide fold-change detection in gene regulation. Mol Cell. 2009;36:894–899. doi: 10.1016/j.molcel.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402(6761, Suppl):C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- Hornung G, Barkai N. Noise propagation and signaling sensitivity in biological networks: a role for positive feedback. PLoS Comput Biol. 2008;4:e8. doi: 10.1371/journal.pcbi.0040008. http://dx.doi.org/10.1371/journal.pcbi.0040008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C, Scherrer S, Buetti-Dinh A, Ratna P, Pizzolato J, Jaquet V, Becskei A. Stochastic signalling rewires the interaction map of a multiple feedback network during yeast evolution. Nat Commun. 2012;3:682. doi: 10.1038/ncomms1687. http://dx.doi.org/10.1038/ncomms1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs FJ, Hasty J, Cantor CR, Collins JJ. Prediction and measurement of an autoregulatory genetic module. Proc Natl Acad Sci USA. 2003;100:7714–7719. doi: 10.1073/pnas.1332628100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Ventura AC, Sontag ED, Merajver SD, Ninfa AJ, Del Vecchio D. Load-induced modulation of signal transduction networks. Sci Signal. 2011;4:ra67. doi: 10.1126/scisignal.2002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodenko B, Yaffe MB, Kolch W. Computational approaches for analyzing information flow in biological networks. Sci Signal. 2012;5:re1. doi: 10.1126/scisignal.2002961. http://dx.doi.org/10.1126/scisignal.2002961. [DOI] [PubMed] [Google Scholar]
- Kim J, White KS, Winfree E. Construction of an in vitro bistable circuit from synthetic transcriptional switches. Mol Syst Biol. 2006;2:68. doi: 10.1038/msb4100099. http://dx/10.1038/msb4100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Hieter P. Evolutionary conservation between budding yeast and human kinetochores. Nat Rev Mol Cell Biol. 2001;2:678–687. doi: 10.1038/35089568. [DOI] [PubMed] [Google Scholar]
- Lau K-Y, Ganguli S, Tang C. Function constrains network architecture and dynamics: a case study on the yeast cell cycle Boolean network. Phys Rev E Stat Nonlin Soft Matter Phys. 2007;75:051907. doi: 10.1103/PhysRevE.75.051907. http://dx.doi.org/10.1103/PhysRevE.75.051907. [DOI] [PubMed] [Google Scholar]
- Li H, Johnson AD. Evolution of transcription networks—lessons from yeasts. Curr Biol. 2010;20:R746–R753. doi: 10.1016/j.cub.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Helling R, Tang C, Wingreen N. Emergence of preferred structures in a simple model of protein folding. Science. 1996;273:666–669. doi: 10.1126/science.273.5275.666. [DOI] [PubMed] [Google Scholar]
- Liu C, Fu X, Liu L, Ren X, Chau CKL, Li S, Xiang L, Zeng H, Chen G, Tang LH, et al. Sequential establishment of stripe patterns in an expanding cell population. Science. 2011;334:238–241. doi: 10.1126/science.1209042. [DOI] [PubMed] [Google Scholar]
- Ma W, Lai L, Ouyang Q, Tang C. Robustness and modular design of the Drosophila segment polarity network. Mol Syst Biol. 2006;2:70. doi: 10.1038/msb4100111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Trusina A, El-Samad H, Lim WA, Tang C. Defining network topologies that can achieve biochemical adaptation. Cell. 2009;138:760–773. doi: 10.1016/j.cell.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín I, Siegal ML, Baker BS. The evolution of dosage-compensation mechanisms. Bioessays. 2000;22:1106–1114. doi: 10.1002/1521-1878(200012)22:12<1106::AID-BIES8>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Markevich NI, Tsyganov MA, Hoek JB, Kholodenko BN. Long-range signaling by phosphoprotein waves arising from bistability in protein kinase cascades. Mol Syst Biol. 2006;2:61. doi: 10.1038/msb4100108. http://dx.doi.org/10.1038/msb4100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WF. Engineering design principles for organelle size control systems. Semin Cell Dev Biol. 2008;19:520–524. doi: 10.1016/j.semcdb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Miller M, Hafner M, Sontag E, Davidsohn N, Subramanian S, Purnick PEM, Lauffenburger D, Weiss R. Modular design of artificial tissue homeostasis: robust control through synthetic cellular heterogeneity. PLoS Comput Biol. 2012;8:e1002579. doi: 10.1371/journal.pcbi.1002579. http://dx.doi.org/10.1371/journal.pcbi.1002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R, Shen-Orr SS, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- Mukherji S, van Oudenaarden A. Synthetic biology: understanding biological design from synthetic circuits. Nat Rev Genet. 2009;10:859–871. doi: 10.1038/nrg2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-García J, Kholodenko BN. Signalling over a distance: gradient patterns and phosphorylation waves within single cells. Biochem Soc Trans. 2010;38:1235–1241. doi: 10.1042/BST0381235. [DOI] [PubMed] [Google Scholar]
- Nandagopal N, Elowitz MB. Synthetic biology: integrated gene circuits. Science. 2011;333:1244–1248. doi: 10.1126/science.1207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012;150:1274–1286. doi: 10.1016/j.cell.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák B, Tyson JJ. Design principles of biochemical oscillators. Nat Rev Mol Cell Biol. 2008;9:981–991. doi: 10.1038/nrm2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter IS, Davidson EH. A gene regulatory network controlling the embryonic specification of endoderm. Nature. 2011a;474:635–639. doi: 10.1038/nature10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter IS, Davidson EH. Evolution of gene regulatory networks controlling body plan development. Cell. 2011b;144:970–985. doi: 10.1016/j.cell.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter IS, Faure E, Davidson EH. Predictive computation of genomic logic processing functions in embryonic development. Proc Natl Acad Sci USA. 2012;109:16434–16442. doi: 10.1073/pnas.1207852109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Winfree E. Scaling up digital circuit computation with DNA strand displacement cascades. Science. 2011;332:1196–1201. doi: 10.1126/science.1200520. [DOI] [PubMed] [Google Scholar]
- Qian L, Winfree E, Bruck J. Neural network computation with DNA strand displacement cascades. Nature. 2011;475:368–372. doi: 10.1038/nature10262. [DOI] [PubMed] [Google Scholar]
- Rafelski SM, Marshall WF. Building the cell: design principles of cellular architecture. Nat Rev Mol Cell Biol. 2008;9:593–602. doi: 10.1038/nrm2460. [DOI] [PubMed] [Google Scholar]
- Randall A, Guye P, Gupta S, Duportet X, Weiss R. Design and connection of robust genetic circuits. Methods Enzymol. 2011;497:159–186. doi: 10.1016/B978-0-12-385075-1.00007-X. [DOI] [PubMed] [Google Scholar]
- Rosenfeld N, Elowitz MB, Alon U. Negative autoregulation speeds the response times of transcription networks. J Mol Biol. 2002;323:785–793. doi: 10.1016/s0022-2836(02)00994-4. [DOI] [PubMed] [Google Scholar]
- Ryan CJ, Roguev A, Patrick K, Xu J, Jahari H, Tong Z, Beltrao P, Shales M, Qu H, Collins SR, et al. Hierarchical modularity and the evolution of genetic interactomes across species. Mol Cell. 2012;46:691–704. doi: 10.1016/j.molcel.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster P, Fontana W, Stadler PF, Hofacker IL. From sequences to shapes and back: a case study in RNA secondary structures. Proc Biol Sci. 1994;255:279–284. doi: 10.1098/rspb.1994.0040. [DOI] [PubMed] [Google Scholar]
- Seelig G, Soloveichik D, Zhang DY, Winfree E. Enzyme-free nucleic acid logic circuits. Science. 2006;314:1585–1588. doi: 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- Shah NA, Sarkar CA. Robust network topologies for generating switch-like cellular responses. PLoS Comput Biol. 2011;7:e1002085. doi: 10.1371/journal.pcbi.1002085. http://dx.doi.org/10.1371/journal.pcbi.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr SS, Milo R, Mangan S, Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- Slusarczyk AL, Weiss R. Understanding signaling dynamics through synthesis. Sci Signal. 2012;5:pe16. doi: 10.1126/scisignal.2003092. [DOI] [PubMed] [Google Scholar]
- Slusarczyk AL, Lin A, Weiss R. Foundations for the design and implementation of synthetic genetic circuits. Nat Rev Genet. 2012;13:406–420. doi: 10.1038/nrg3227. [DOI] [PubMed] [Google Scholar]
- Sneppen K, Krishna S, Semsey S. Simplified models of biological networks. Annu Rev Biophys. 2010;39:43–59. doi: 10.1146/annurev.biophys.093008.131241. [DOI] [PubMed] [Google Scholar]
- Soloveichik D, Seelig G, Winfree E. DNA as a universal substrate for chemical kinetics. Proc Natl Acad Sci USA. 2010;107:5393–5398. doi: 10.1073/pnas.0909380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- Stephens GJ, Osborne LC, Bialek W. Searching for simplicity in the analysis of neurons and behavior. Proc Natl Acad Sci USA. 2011;108(Suppl 3):15565–15571. doi: 10.1073/pnas.1010868108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker J, Cookson S, Bennett MR, Mather WH, Tsimring LS, Hasty J. A fast, robust and tunable synthetic gene oscillator. Nature. 2008;456:516–519. doi: 10.1038/nature07389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigges M, Marquez-Lago TT, Stelling J, Fussenegger M. A tunable synthetic mammalian oscillator. Nature. 2009;457:309–312. doi: 10.1038/nature07616. [DOI] [PubMed] [Google Scholar]
- Tigges M, Dénervaud N, Greber D, Stelling J, Fussenegger M. A synthetic low-frequency mammalian oscillator. Nucleic Acids Res. 2010;38:2702–2711. doi: 10.1093/nar/gkq121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai TYC, Choi YS, Ma W, Pomerening JR, Tang C, Ferrell JE., Jr Robust, tunable biological oscillations from interlinked positive and negative feedback loops. Science. 2008;321:126–129. doi: 10.1126/science.1156951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Novák B. Functional motifs in biochemical reaction networks. Annu Rev Phys Chem. 2010;61:219–240. doi: 10.1146/annurev.physchem.012809.103457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Chen K, Novak B. Network dynamics and cell physiology. Nat Rev Mol Cell Biol. 2001;2:908–916. doi: 10.1038/35103078. [DOI] [PubMed] [Google Scholar]
- Wagner A. Circuit topology and the evolution of robustness in two-gene circadian oscillators. Proc Natl Acad Sci USA. 2005;102:11775–11780. doi: 10.1073/pnas.0501094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmflash A, François P, Siggia ED. Pareto evolution of gene networks: an algorithm to optimize multiple fitness objectives. Phys Biol. 2012;9:056001. doi: 10.1088/1478-3975/9/5/056001. http://dx.doi.org/10.1088/1478-3975/9/5/056001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber W, Fussenegger M. Engineering of synthetic mammalian gene networks. Chem Biol. 2009;16:287–297. doi: 10.1016/j.chembiol.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Yang K, Bai H, Ouyang Q, Lai L, Tang C. Finding multiple target optimal intervention in disease-related molecular network. Mol Syst Biol. 2008;4:228. doi: 10.1038/msb.2008.60. http://dx.doi.org/10.1038/msb.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DY, Turberfield AJ, Yurke B, Winfree E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 2007;318:1121–1125. doi: 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]