

SUMMARY
Targeted gene regulation on a genome-wide scale is a powerful strategy for interrogating, perturbing, and engineering cellular systems. Here, we develop a method for controlling gene expression based on Cas9, an RNA-guided DNA endonuclease from a type II CRISPR system. We show that a catalytically dead Cas9 lacking endonuclease activity, when coexpressed with a guide RNA, generates a DNA recognition complex that can specifically interfere with transcriptional elongation, RNA polymerase binding, or transcription factor binding. This system, which we call CRISPR interference (CRISPRi), can efficiently repress expression of targeted genes in Escherichia coli, with no detectable off-target effects. CRISPRi can be used to repress multiple target genes simultaneously, and its effects are reversible. We also show evidence that the system can be adapted for gene repression in mammalian cells. This RNA-guided DNA recognition platform provides a simple approach for selectively perturbing gene expression on a genome-wide scale.
INTRODUCTION
The systematic interrogation of genomes and the genetic reprogramming of cells require methods to precisely and predictably target sets of genes for expression or repression. RNA interference (RNAi) and engineered DNA-binding proteins such as zinc finger or transcription-activator-like effector (TALE) proteins, have emerged as powerful technologies for targeted gene regulation (Hannon, 2002; Beerli and Barbas, 2002; Zhang et al., 2011). RNAi can be used in a relatively straightforward manner to knock down expression of targeted genes. RNAi, however, is limited to particular organisms that have the proper host machinery and can sometimes exhibit significant off-target effects and toxicity. In addition, custom DNA-binding proteins, such as zinc finger or TALE proteins, remain somewhat difficult and expensive to design, develop, and empirically test in the cellular context (Klug, 2010). As a consequence, it remains challenging to use DNA-binding proteins for simultaneous modulation of multiple genes and implementation of large-scale genetic programs.
The CRISPR (clustered regularly interspaced short palindromic repeats) system provides a potential platform for targeted gene regulation (Barrangou et al., 2007). About 40% of bacteria and 90% of archaea possess CRISPR/CRISPR-associated (Cas) systems to confer resistance to foreign DNA elements (Makarova et al., 2011). CRISPR systems use small base-pairing RNAs to target and cleave foreign DNA elements in a sequence-specific manner (Wiedenheft et al., 2012). There are diverse CRISPR systems in different organisms, and one of the simplest is the type II CRISPR system from Streptococcus pyogenes: only a single gene encoding the Cas9 protein and two RNAs, a mature CRISPR RNA (crRNA) and a partially complementary trans-acting RNA (tracrRNA), are necessary and sufficient for RNA-guided silencing of foreign DNAs (Figure S1 available online) (Jinek et al., 2012; Gasiunas et al., 2012). Maturation of crRNA requires tracrRNA and RNase III (Deltcheva et al., 2011). However, this requirement can be bypassed by using an engineered small guide RNA (sgRNA) containing a designed hairpin that mimics the tracrRNA-crRNA complex (Jinek et al., 2012). Base pairing between the sgRNA and target DNA causes double-strand breaks (DSBs) due to the endonuclease activity of Cas9. Binding specificity is determined by both sgRNA-DNA base pairing and a short DNA motif (protospacer adjacent motif [PAM] sequence: NGG) juxtaposed to the DNA complementary region (Marraffini and Sontheimer, 2010). Thus, the CRISPR system only requires a minimal set of two molecules—the Cas9 protein and the sgRNA—and therefore holds the potential to be used as a host-independent gene-targeting platform. Very recently, it has been demonstrated that the Cas9/CRISPR can be harnessed for site-selective RNA-guided genome editing (Figure 1A) (Mali et al., 2013; Cong et al., 2013; Jinek et al., 2013; Jiang et al., 2013; Hwang et al., 2013; Cho et al., 2013).
Figure 1. Design of the CRISPR Interference System.

(A) The minimal interference system consists of a single protein and a designed sgRNA chimera. The sgRNA chimera consists of three domains (boxed region): a 20 nt complementary region for specific DNA binding, a 42 nt hairpin for Cas9 binding (Cas9 handle), and a 40 nt transcription terminator derived from S. pyogenes. The wild-type Cas9 protein contains the nuclease activity. The dCas9 protein is defective in nuclease activity.
(B) The wild-type Cas9 protein binds to the sgRNA and forms a protein-RNA complex. The complex binds to specific DNA targets by Watson-Crick base pairing between the sgRNA and the DNA target. In the case of wild-type Cas9, the DNA will be cleaved due to the nuclease activity of the Cas9 protein. We hypothesize that the dCas9 is still able to form a complex with the sgRNA and bind to specific DNA target. When the targeting occurs on the protein-coding region, it could block RNA polymerase and transcript elongation.
See also Figure S1.
To repurpose the Cas9/CRISPR for genome regulation instead of genome editing, here we demonstrate that a catalytically inactive version of Cas9 can be repurposed as a platform for RNA-guided transcription regulation. The transcription of arbitrary genes can be modified by the mutant Cas9 without genetically altering the target sequence (Figure 1B). To implement the system, we transferred the minimal Cas9/CRISPR system from S. pyogenes to Escherichia coli (E. coli). Using a modified Cas9 protein lacking endonucleolytic activity, we generated an RNA-guided DNA recognition platform. Coexpression of the mutant Cas9 with an sgRNA designed with a 20 base pair (bp) complementary region yields specific silencing of a gene of interest without off-target effects. We call this modified system CRISPR interference (CRISPRi). We show that CRISPRi silencing occurs by blocking transcription and is highly efficient with up to 1,000-fold repression. We characterize determinants of the regulatory efficiency, including target loci, length, and mismatches within the sgRNA base-pairing region. We also show that multiple sgRNAs can be used simultaneously to regulate multiple genes. Furthermore, we demonstrate that the CRISPRi system can be used to knock down endogenous genes and to profile cis regulatory elements for transcription factor binding in the lactose regulatory network. Finally, we show that the CRISPRi system can also be used to knock down gene expression in mammalian cells. The CRISPRi sequence-specific targeting platform thus holds promise as a general approach for modulating gene expression in a broad range of host cells.
RESULTS
A Minimal CRISPRi System Consists of a Single Protein and RNA and Can Effectively Silence Transcription Initiation and Elongation
To implement such a CRISPRi platform in E. coli, we first expressed the wild-type S. pyogenes cas9 gene and an sgRNA from bacterial vectors to determine whether it could perturb gene expression at a targeted locus (Figure 2A). The S. pyogenes CRISPR system is orthogonal to the native E. coli system (Jinek et al., 2012). The Cas9 protein is expressed from an anhydrotetracycline (aTc)-inducible promoter on a plasmid containing a p15A replication origin, and the sgRNA is expressed from a minimal constitutive promoter on a plasmid containing a ColE1 replication origin. Cas9 has been shown to have strong nuclease activity. Thus, we expected that cotransformation of wild-type Cas9 and an sgRNA that targets the E. coli genome would direct double-stranded breaks at the target site (Mali et al., 2013; Jiang et al., 2013). As an alternative strategy, we used a catalytically dead Cas9 mutant (dCas9), which is defective in DNA cleavage, hypothesizing that this form of Cas9 might still act as a simple RNA-guided DNA-binding complex.
Figure 2. CRISPRi Effectively Silences Transcription Elongation and Initiation.

(A) The CRISPRi system consists of an inducible Cas9 protein and a designed sgRNA chimera. The dCas9 contains mutations of the RuvC1 and HNH nuclease domains. The sgRNA chimera contains three functional domains, as described in Figure 1.
(B) Sequence of designed sgRNA (NT1) and the DNA target. NT1 targets the nontemplate DNA strand of the mRFP-coding region. Only the region surrounding the base-pairing motif (20 nt) is shown. Base-pairing nucleotides are shown in orange, and the dCas9-binding hairpin is in blue. The PAM sequence is shown in red.
(C) CRISPRi blocks transcription elongation in a strand-specific manner. A synthetic fluorescence-based reporter system containing an mRFP-coding gene is inserted into the E. coli MG1655 genome (the nsfA locus). Six sgRNAs that bind to either the template DNA strand or the nontemplate DNA strand are coexpressed with the dCas9 protein, with their effects on the target mRFP measured by in vivo fluorescence assay. Only sgRNAs that bind to the nontemplate DNA strand showed silencing (10- to 300-fold). The control shows fluorescence of the cells with dCas9 protein but without the sgRNA.
(D) CRISPRi blocks transcription initiation. Five sgRNAs are designed to bind to different regions around an E. coli promoter (J23119). The transcription start site is labeled as +1. The dotted oval shows the initial RNAP complex that covers a 75 bp region from −55 to +20. Only sgRNAs targeting regions inside of the initial RNAP complex show repression (P1–P4). Unlike transcription elongation block, silencing is independent of the targeted DNA strand.
(E) CRISPRi regulation is reversible. Both dCas9 and sgRNA (NT1) are under the control of an aTc-inducible promoter. Cell culture was maintained during exponential phase. At time T = 0, 1 μM of aTc was supplemented to cells with OD = 0.001. Repression of target mRFP starts within 10 min. The fluorescence signal decays in a way that is consistent with cell growth, suggesting that the decay is due to cell division. In 240 min, the fluorescence reaches the fully repressed level. At T = 370 min, aTc is washed away from the growth media, and cells are diluted back to OD = 0.001. Fluorescence starts to increase after 50 min and takes about 300 min to rise to the same level as the positive control. Positive control: always without the inducer; negative control: always with 1 μM aTc inducer.
Fluorescence results in (C)–(E) represent average and SEM of at least three biological replicates. See also Figures S2 and S3.
The sgRNA molecules coexpressed with Cas9 each consist of three segments: a 20 nucleotide (nt) target-specific complementary region, a 42 nt Cas9-binding hairpin (Cas9 handle), and a 40 nt transcription terminator derived from S. pyogenes (Figure 2B). We constructed a red fluorescent protein (mRFP)-based reporter system (Campbell et al., 2002) and inserted it into the E. coli MG1655 genome.
Coexpression of the wild-type Cas9 protein and an sgRNA (NT1) targeted to the mRFP coding sequence dramatically decreased transformation efficiency (see Data S1 for sgRNA sequences), likely due to Cas9-induced double-stranded breaks on the genome (Figure S2A). Sequencing of a few survivor colonies showed that they all had sequence rearrangements around the target mRFP site on the genome, suggesting that there was strong selection against expression of wild-type Cas9 and an sgRNA targeted to a host sequence. The dCas9 mutant gene (noncleaving), which contained two silencing mutations of the RuvC1 and HNH nuclease domains (D10A and H841A) (Jinek et al., 2012), alleviated this lethality, as verified by transformation efficiency and E. coli growth rates (Figures S2A and S2B).
To test whether the dCas9:sgRNA complex could yield highly efficient repression of gene expression, we designed sgRNAs complementary to different regions of the mRFP coding sequence, either binding to the template DNA strand or to the nontemplate DNA strand. Our results indicated that sgRNAs targeting the nontemplate DNA strand demonstrated effective gene silencing (10- to 300-fold of repression), whereas those targeting the template strand showed little effect (Figure 2C). The system exhibited similar repression effects for genes that were within the E. coli genome or on a high-copy plasmid (Figure S3). Furthermore, targeting to the promoter region also led to effective gene silencing (Figure 2D). Targeting of the sgRNA to the −35 box significantly knocked down gene expression (P1, ~100-fold of repression), whereas targeting to other adjacent regions showed a dampened effect (P2–P4). Targeting sequences about 100 bp upstream of the promoter showed no effects (P5). Unlike targeting the coding sequence, when targeting the promoter, the efficiency of silencing is independent of the DNA strand; targeting of template or nontemplate strands is equally effective (P2 and P3).
CRISPRi Gene Knockdown Is Inducible and Reversible
Unlike gene knockout methods, one advantage of using CRISPRi-based knockdown of gene expression is the fact that this perturbation should be reversible. To test whether CRISPRi regulation could be induced and subsequently reversed, we encoded both dCas9 and mRFP-specific sgRNA (NT1) under the control of the aTc-inducible promoter and performed time course measurement of CRISPRi-mediated regulation of mRFP in response to inducers (Figure 2E). At time zero, cell culture that grew to the early exponential phase without inducers was supplemented with 1 μM of aTc. Our data indicated that the system could quickly respond to the presence of inducers—the fluorescent reporter protein signal started to decrease within 10 min of the addition of the inducer molecule. Because the mRFP protein is stable (Campbell et al., 2002), the rate of fluorescence signal decrease is limited by protein dilution due to cell growth, as seen by a similar cell doubling time and loss of fluorescence half-time (both ~36 min). At 240 min, all cells were uniformly repressed to the same level as the negative control. At 370 min, the inducer was washed away from the growth media, and cells were diluted back to a lower OD. After a delay of 50 min, mRFP fluorescence started to increase. It took a total 300 min for single-cell fluorescence to increase to the same level as the positive control. The 50 min delay is most likely determined by the dCas9/sgRNA turnover rate offset by dilution by cell growth and division. In summary, these results demonstrate that the silencing effects of dCas9-sgRNA can be induced and reversed.
Native Elongating Transcript Sequencing Confirms that CRISPRi Functions by Blocking Transcription
We hypothesized that dCas9 was functioning as an RNA-guided DNA-binding complex that could block RNA polymerase (RNAP) binding during transcription elongation. Because the non-template DNA strand shares the same sequence identity as the transcribed mRNA and only sgRNAs that bind to the non-template DNA strand exhibited silencing, it remains a possibility that the dCas9:sgRNA complex interacts with mRNA and alters its translation or stability. To distinguish these possibilities, we applied a recently described native elongating transcript sequencing (NET-seq) approach to E. coli, which could be used to globally profile the positions of elongating RNA polymerases and monitor the effect of the dCas9:sgRNA complex on transcription (Churchman and Weissman, 2011). In this NET-seq method, we transformed the CRISPRi system into an E. coli MG1655-derived strain that contained a FLAG-tagged RNAP. The CRISPRi contained an sgRNA (NT1) that binds to the mRFP-coding region. In vitro immunopurification of the tagged RNAP followed by sequencing of the nascent transcripts associated with elongating RNAPs allowed us to distinguish the pause sites of the RNAP.
Our experiment demonstrated that the sgRNA induced strong transcriptional pausing upstream of the sgRNA target locus (Figure 3A). The distance between the pause site and the target site is 19 bp, which is in perfect accordance with the previously reported ~18 bp distance between the nucleotide incorporation of RNAP and its front edge (Nudler et al., 1994). This finding is consistent with a mechanism of CRISPRi in which the transcription block is due to physical collision between the elongating RNAP and the dCas9:sgRNA complex (Figure 3B). Binding of the dCas9:sgRNA complex to the template strand had little repressive effect, suggesting that RNAP was able to read through the complex in this particular orientation. In this case, the sgRNA faces the RNAP, which might be unzipped by the helicase activity of RNAP. Though more structural studies are required to provide a comprehensive understanding of the system in vivo, we have demonstrated that CRISPRi utilizes RNAs to directly block transcription. This mechanism is distinct from that of RNAi, for which knockdown of gene expression requires the destruction of already transcribed messenger RNAs prior to their translation (Zamore et al., 2000).
Figure 3. CRISPRi Functions by Blocking Transcription Elongation.
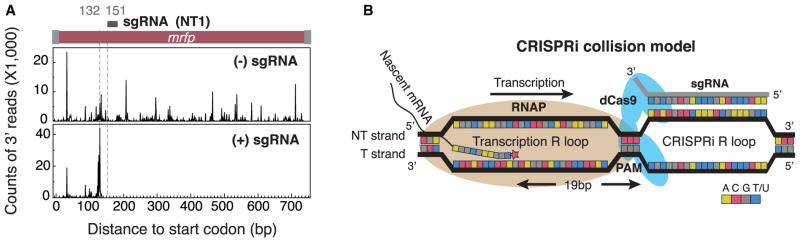
(A) FLAG-tagged RNAP molecules were immunoprecipitated, and the associated nascent mRNA transcripts were sequenced. (Top) Sequencing results of the nascent mRFP transcript in cells without sgRNA. (Bottom) Results in cells with sgRNA. In the presence of sgRNA, a strong transcriptional pause is observed 19 bp upstream of the target site, after which the number of sequencing reads drops precipitously.
(B) A proposed CRISPRi mechanism based on physical collision between RNAP and dCas9-sgRNA. The distance from the center of RNAP to its front edge is ~19 bp, which matches well with our measured distance between the transcription pause site and 3′ of sgRNA base-pairing region. The paused RNAP aborts transcription elongation upon encountering the dCas9-sgRNA roadblock.
CRISPRi sgRNA-Guided Gene Silencing Is Highly Specific
To evaluate the specificity of CRISPRi on a genome-wide scale, we performed whole-transcriptome shotgun sequencing (RNA-seq) of dCas9-transformed cells with and without sgRNA coexpression (Figure 4A) (Mortazavi et al., 2008). In the presence of the sgRNA targeted to mRFP (NT1), the mRFP transcript was the sole gene exhibiting a decrease in abundance. No other genes showed significant change in expression upon addition of the sgRNA, within sequencing errors. We also performed RNA-seq on cells with different sgRNAs that target different genes. None of these experiments showed significant changes of genes besides the targeted gene (Figure S4). Thus, sgRNA-guided gene targeting and regulation are highly specific and do not have significant off-target effects.
Figure 4. Targeting Specificity of the CRISPRi System.
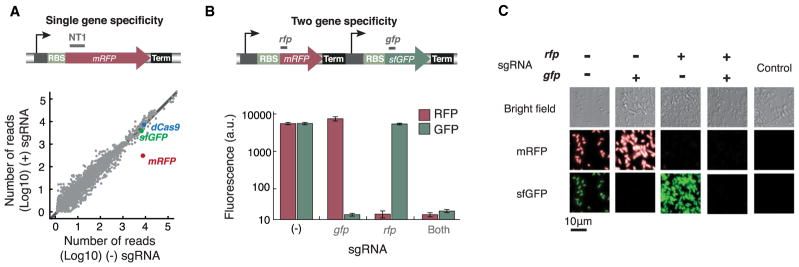
(A) Genome-scale mRNA sequencing (RNA-seq) confirms that CRISPRi targeting has no off-target effects. The sgRNA NT1 that binds to the mRFP coding region is used. The dCas9, mRFP, and sfGFP genes are highlighted.
(B) Multiple sgRNAs can independently silence two fluorescent protein reporters in the same cell. Each sgRNA specifically represses its cognate gene, but not the other gene. When both sgRNAs are present, both genes are silenced. Error bars represent SEM from at least three biological replicates.
(C) Microscopic images for using two sgRNAs to control two fluorescent proteins. (Top) Bright-field images of the E. coli cells; (middle) RFP channel; (bottom) GFP channel. Coexpression of one sgRNA and dCas9 only silences the cognate fluorescent protein, but not the other. The knockdown effect is strong, as almost no fluorescence is observed from cells with certain fluorescent protein silenced. Scale bar, 10 μm. Control shows cells without any fluorescent protein reporters.
Fluorescence results represent average and SEM of at least three biological replicates. See also Figure S4.
CRISPRi Can Be Used to Simultaneously Regulate Multiple Genes
We next asked whether the CRISPRi system could allow control of multiple genes independently without crosstalk. We devised a dual-color fluorescence reporter system based on mRFP and sfGFP (Pédelacq et al., 2006). Two sgRNAs with distinct complementary regions to each gene were designed. Expression of each sgRNA only silenced the cognate gene and had no effect on the other. Coexpression of two sgRNAs knocked down both genes (Figures 4B and 4C). These results suggest that the sgRNA-guided targeting is specific, with the specificity dictated by its sequence identity, and is not impacted by the presence of other sgRNAs. This behavior should enable multiplex control of multiple genes simultaneously by CRISPRi.
Factors that Determine CRISPRi Silencing Efficiency
To find determinants of CRISPRi targeting efficiency, we investigated the roles of length, sequence complementarity, and position on silencing efficiency (Figure 5A). As suggested in Figure 2C, the location of the sgRNA target sequence along the gene was important for efficiency. We further designed sgRNAs to cover the full length of the coding regions for both mRFP and sfGFP (see Data S1 for sgRNA sequences). In all cases, repression was inversely correlated with the target distance from the transcription start site (Figure 5B). A strong linear correlation was observed for mRFP. A similar but slightly weaker correlation was observed when sfGFP was used as the target, perhaps indicating varying kinetics of the RNA polymerase during different points in elongation of this gene.
Figure 5. Characterization of Factors that Affect Silencing Efficiency.

(A) We measured the silencing effects of sgRNAs with different targeting loci on the same gene (distance from the translation start codon) and sgRNAs with different lengths of the base-pairing region to the same target locus (based on NT1).
(B) The silencing efficiency is inversely correlated with the target distance from the translation start codon (orange, mRFP; green, sfGFP). The relative repression activity is calculated by normalizing repression of each sgRNA to that of the sgRNA with the highest repression fold change. Error bars represent SEM from three biological replicates.
(C) The length of the Watson-Crick base-pairing region between the sgRNA and the target DNA affects repression efficiency. Extensions of the base-pairing region all exhibit strong silencing effect, and truncations dramatically decrease repression. The minimal length of the base-pairing region for detectable repression is 12 bp. Error bars represent SEM from three biological replicates.
(D) We introduced single mismatches into every nucleotide on sgRNA (NT1; Figure 2B) and measured how single mismatches affect repression efficiency. Three subregions with distinct importance to the overall silencing can be discerned. They show a step function. The first 7 nt region is critical for silencing and likely constitutes a “seed” region for probing sgRNAs binding to the DNA target. The PAM sequence (NGG) is indispensible for silencing. Error bars represent SEM from three biological replicates.
(E) Silencing effects of sgRNAs with adjacent double mismatches. The relative repression activity of single-mismatched sgRNAs is shown in gray, with the mismatch position labeled on the bottom. Experimentally measured activity of double-mismatched sgRNAs is shown in blue. Calculated activity by multiplying the effects of two single-mismatched sgRNAs is shown in white and labeled with “Com.” In most cases, the silencing activity of a double-mismatched sgRNA is simply a multiplication of the activities of single-mismatched sgRNAs (except Figure S5B), suggesting an independent relationship between single mismatches. Error bars represent SEM from three biological replicates.
(F) Combinatorial silencing effects of using double sgRNAs to target a single mRFP gene. Using two sgRNAs that target the same gene, the overall knockdown effect can be improved to almost 1,000-fold. When two sgRNAs bind to nonoverlapping sequences of the same gene, repression is augmented. When two sgRNAs target overlapping regions, repression is suppressed. Error bars represent SEM from three biological replicates.
Fluorescence results represent average and SEM of at least three biological replicates. See also Figures S5 and S6.
The sgRNA contains a 20 bp region that is complementary to the target. To identify the importance of this base-pairing region, we altered the length of sgRNA NT1 (Figure 5C). Whereas extension of the region from the 5′ end did not affect silencing, truncation of the region severely decreased repression. The minimal length of the base-pairing region needed for gene silencing was 12 bp, with further truncation leading to complete loss of function. We also introduced single mutations into the base-pairing region of sgRNA NT1 and tested how mismatches affected overall silencing. From our results, three subregions could be discerned, each with a distinct contribution to the overall binding and silencing (Figure 5D). Importantly, any single mutation of the first 7 nt dramatically decreased repression, suggesting that this sequence constitutes a “seed region” for binding, as noted previously for both the type I and type II CRISPR systems (Wiedenheft et al., 2011; Jinek et al., 2012; Gasiunas et al., 2012). We further mutated adjacent nucleotides in pairs (Figures 5E and S5). In most cases, the relative repression activity due to a double mutation was multiplicative relative to the effects of the single mutants, suggesting an independent relationship between the mismatches. Furthermore, in agreement with previous results on the importance of the PAM sequence, an incorrect PAM totally abolished silencing even with a 20 bp perfect binding region (Figure 5E). Thus, we conclude that the specificity of the CRISPRi system is determined jointly by the PAM (2 bp) and at least a 12 bp sgRNA-DNA stretch, the space of which is large enough to cover most bacterial genomes for unique target sites.
We tested whether the efficiency of CRISPRi could be enhanced by using two sgRNAs both targeted against the same gene (Figures 5F and S6). Depending on the relative positioning of multiple sgRNAs, we observed distinct combinatorial effects. Combining two sgRNAs—each with about 300-fold repression—allowed us to increase the overall silencing to up to 1,000-fold. Combining two weaker sgRNAs (~5-fold) showed multiplicative effects when used together. We also observed suppressive combinatorial effects using two sgRNAs whose targets overlapped. This was probably due to competition of both sgRNAs for binding to the same region.
Interrogating an Endogenous Regulatory Network Using CRISPRi Gene Knockdown
We next tested whether the CRISPRi system could be used as a gene knockdown platform to interrogate endogenous gene networks. Previous methods to interrogate microbial gene networks have mostly relied on laborious and costly genomic engineering and knockout procedures. By contrast, gene knockdown with CRISPRi requires only the design and synthesis of a small sgRNA bearing a 20 bp complementary region to the desired genes. To demonstrate this, we applied CRISPRi to create E. coli knockdown strains by designing sgRNAs (see Data S1 for sgRNA sequences) to systematically perturb genes that were part of the well-characterized E. coli lactose regulatory pathway (Figure 6A). We performed β-galactosidase assays to measure LacZ expression from the knockdown strains, with and without isopropyl β-D-1-thiogalactopyranoside (IPTG), a chemical that inhibits the lac repressor (LacI) (Lewis, 2005). In wild-type cells, addition of IPTG induced LacZ expression. Our results showed that a lacZ-specific sgRNA could strongly repress LacZ expression (Figure 6B). Conversely, an sgRNA targeting the lacI gene led to activation of LacZ expression even in the absence of IPTG, as would be expected for silencing a direct repressor of LacZ expression.
Figure 6. Functional Profiling of a Complex Regulatory Network Using CRISPRi Gene Knockdown.

(A) sgRNAs are designed and used to knock down genes (cya, crp, lacI, lacZ, lacY, and lacA) in the lac regulatory pathway or block transcriptional operator sites (A/P/O). LacI is a repressor of the lacZYA operon by binding to a transcription operator site (O site). The lacZ gene encodes an enzyme that catalyzes lactose into glucose. A few trans-acting host genes such as cya and crp are involved in the activation of the lacZYA system. The cAMP-CRP complex binds to a transcription operator site (A site) and recruits RNA polymerase binding to the P site, which initiates transcription of lacZYA. IPTG, a chemical that inhibits LacI function, induces LacZ expression.
(B) β-galactosidase assay of the knockdown strains without (white) and with (gray) IPTG. Control shows that the wild-type cells without CRISPRi perturbation can be induced by addition of IPTG. The sgRNA that targets LacZ strongly represses LacZ expression even in the presence of IPTG. When LacI is targeted, LacZ expression is high even without IPTG. Targeting cya and crp genes leads to decreased LacZ expression level in the presence of IPTG. Presence of 1 mM cAMP rescues cya knockdown, but not crp knockdown. Blocking the transcription operator sites results in LacZ repression, suggesting that these are important cis-acting regulatory sites for LacZ. Upon perturbation, decreased (red arrows) and increased (green arrows) expression of LacZ are indicated. Error bars represent SEM from three biological replicates.
(C) The knockdown experiments allow us to profile the roles of regulators in the lac regulatory circuit. The data is shown on a two-dimensional (2D) graph, with x axis showing LacZ activity without IPTG and y axis showing its activity with IPTG. The spreading of ovals along each axis shows the SEM of three biological replicates.
The β-galactosidase assay results represent average and SEM of three biological replicates. For RNA-seq data on LacI and LacZ targeting, see also Figure S4.
It is known that cAMP-CRP is an essential activator of LacZ expression by binding to a cis regulatory site upstream of the promoter (A site). Consistently, the sgRNA that was targeted to the crp gene or to the A site in the LacZ promoter led to repression, demonstrating a means to link a regulator to its cis-regulatory sequence using CRISPRi experiments. Targeting the adenylate cylase gene (cya), which is necessary to produce the cAMP that makes CRP more effective at the LacZ promoter, only led to partial repression. Addition of 1 mM cAMP to the growth media complemented the effects for cya knockdown, but not for crp knockdown, suggesting that cya is an indirect regulator of LacZ. Furthermore, targeting the LacI cis-regulatory site (O site) with an sgRNA led to inhibition, presumably because Cas9 complex binding at this site sterically blocks RNA polymerase, mimicking the behavior of the LacI transcription repressor. Targeting the known RNAP-binding site (P site) also blocked expression. In summary, these studies demonstrate that the CRISPRi-based gene knockdown method provides a rapid and effective approach for interrogating the regulatory functions (activating or repressing) of genes and cis elements in a complex regulatory network (Figure 6C).
CRISPRi Can Knock Down Targeted Gene Expression in Human Cells
To test the generality of the CRISPRi approach for using the dCas9-sgRNA complex to repress transcription, we tested the system in the HEK293 mammalian cells. The dCas9 protein was codon optimized, fused to three copies of a nuclear localization sequence (NLS), and expressed from a murine stem cell virus (MSCV) retroviral vector. The same sgRNA design shown in Figure 2B was used to express sgRNAs from the RNA polymerase III U6 promoter. A reporter HEK293 cell line expressing EGFP under the SV40 promoter was created by viral infection. Using an sgRNA (eNT2) that targeted the nontemplate DNA strand of the EGFP-coding region (see Data S1 for sgRNA sequences), we observed moderate but reproducible knockdown of gene expression (46% repression; Figure 7A). The repression is dependent on both the dCas9 protein and sgRNA, implying that repression is due to the dCas9-sgRNA complex and RNA-guided targeting. The same sgRNA exhibited better repression on the same gene when transiently expressed from a plasmid (63% repression; Figure S7). Consistent with the bacterial system, only sgRNAs targeted to the nontemplate strand exhibited repression. The regulatory effects in mammalian cells, however, appear to be more dependent on the targeting locus, as only two out of seven tested sgRNAs showed moderate repression (Figure 7B). Factors such as the distance from the transcription start and the local chromatin state may be critical parameters in determining repression efficiency (Figure S7). Optimization of dCas9 and sgRNA expression, stability, nuclear localization, and interaction will likely allow for further improvement of CRISPRi efficiency in mammalian cells.
Figure 7. CRISPRi Can Repress Gene Expression in Human Cells.

(A) A CRISPRi system in HEK293 cells. The SV40-EGFP expression cassette is inserted into the genome via retroviral infection. The dCas9 protein is codon-optimized and fused with three copies of NLS sequence. The sgRNA is expressed from an RNA polymerase III U6 vector. Cotransfection of dCas9 and an sgRNA (eNT2) that targets the nontemplate DNA strand of EGFP decreases fluorescence (~46%), whereas the expression of either dCas9 or sgRNA alone shows no effect.
(B) The dCas9:sgRNA-mediated repression is dependent on the target loci. Seven sgRNAs are designed to target different regions of the EGFP-coding sequence on the template or nontemplate strand. Only eNT2 and eNT5 show moderate repression. EGFP fluorescence (targeted reporter) is shown in green, whereas mCherry fluorescence (control reporter) is shown in pink.
Fluorescence results from (A) and (B) represent average and error of two biological replicates. See also Figure S7.
DISCUSSION
CRISPRi Efficiently and Selectively Represses Transcription of Target Genes
The CRISPRi system that we report here is a relatively simple platform for targeted gene regulation. CRISPRi does not rely on the presence of complex host factors but instead only requires the dCas9 protein and guide RNAs and thus is flexible and highly designable. Our results have demonstrated that the system can efficiently silence genes in bacteria. The silencing is very specific; we observe no detectable off-target effects. Furthermore, the efficiency of the knockdown can be tuned by changing the target loci and the degree of base pairing between the sgRNA and the target gene. This will make it possible to create allelic series of hypomorphs, a feature that will be especially useful for the study of essential genes. The system functions by directly blocking transcription in a manner that can be easily programmed by designing sgRNAs. To our knowledge, this is one of the first examples of utilizing a targeted protein-RNA complex that directly blocks transcription elongation within protein-coding regions. Mechanistically, this is distinct from RNAi-based silencing, which requires the destruction of already transcribed mRNAs.
In addition, these dCas9:sgRNA complexes can also modulate transcription by targeting key cis-acting motifs within any promoter, sterically blocking the association of their cognate trans-acting transcription factors. Thus, in addition to its use as a gene knockdown tool, CRISPRi could be used for functional mapping of promoters and other genomic regulatory modules.
CRISPRi Is Amenable to Genome-Scale Analysis and Regulation
The CRISPRi method is based on the use of sgRNAs, and only the 20 bp matching region needs to be designed for specific gene targets. With the advances of large-scale DNA oligonucleotide synthesis technology, generating large sets of oligonucleotides that contain unique 20 bp regions for genome targeting is fast and inexpensive. These oligonucleotide libraries could potentially allow us to target large numbers of individual genes to infer gene function or to target gene pairs to map genetic interactions. Furthermore, CRISPRi could be used to simultaneously modulate the expression of large sets of genes, as the small size of sgRNAs allows one to concatenate multiple elements into the same expression vector (Qi et al., 2012).
CRISPRi Provides Tools for Manipulating Microbial Genomes
Because the CRISPRi platform is compact and self-contained, it has the potential to be adapted for different organisms. CRISPRi could thus be a powerful tool for studying nonmodel organisms for which genetic engineering methods are not well developed, including pathogens or industrially useful organisms. Unlike most eukaryotes, most bacteria lack the RNAi machinery. As a consequence, regulation of endogenous genes using designed synthetic RNAs is currently limited. CRISPRi could provide an RNAi-like method for gene perturbation in microbes.
CRISPRi as a Platform for Engineering Transcriptional Regulatory Networks
The CRISPRi has the potential to be utilized as a flexible framework for engineering transcriptional regulatory networks. Because it is essentially an RNA-guided DNA-binding complex, the CRISPRi platform also provides a potentially flexible scaffold for directing diverse regulatory machinery to specific sites in the genome. Beyond simply blocking transcription of target genes, it may be possible to couple the dCas9 protein with numerous regulatory domains to modulate different biological processes and to generate different functional outcomes (e.g., transcriptional activation and chromatin modifications).
While our paper was under review, several very recent studies reported the use of wild-type Cas9/CRISPR for targeted genome editing in diverse organisms (Mali et al., 2013; Cong et al., 2013; Jinek et al., 2013; Jiang et al., 2013; Hwang et al., 2013; Cho et al., 2013). Different engineered forms of the CRISPR platforms can address complementary applications. The wild-type CRISPR system with its endogenous nuclease activity is ideal for targeted modification of genome sequences such as gene mutagenesis, deletion, and insertion. In contrast, the CRISPRi system reported here, based on the catalytically dead Cas9, is ideal for targeted gene regulation such as transcription repression and activation without altering the target sequence.
In the CRISPRi system, it may also be possible to link multiple sgRNAs into transcriptional circuits in which one upstream sgRNA controls the expression of a different downstream sgRNA (Lucks et al., 2011). As RNA molecules in microorganisms tend to be short-lived, we suspect that the genetic programs regulated by sgRNAs might show rapid kinetics distinct from circuits that involve slow processes such as protein expression and degradation. Furthermore, the sgRNAs can also act as inputs to control arbitrary genes in the genome, thus providing a way to directly interface with the host genome without altering the target sequence.
In summary, the CRISPRi system holds great promise as a general genetic programming platform that is suitable for a variety of biomedical research and clinical applications, including genome-scale functional profiling, microbial metabolic engineering, and cell reprogramming.
EXPERIMENTAL PROCEDURES
Strains and Media
The Escherichia coli (E. coli) K-12 strain MG1655 was used as the host strain for the in vivo fluorescence measurements. An E. coli MG1655-derived strain that endogenously expresses a variant of RNAP with a 3 ×FLAG epitope tag attached to the C-terminal end of the RpoC subunit was used for all NET-sequencing experiments. EZ-rich defined media (EZ-RDM, Teknoka) was used as the growth media for in vivo fluorescence assays. Genetic transformation and verification of transformation were done as previously described (Qi et al., 2012), using AmpR, CmR, or KanR genes as selectable markers.
Plasmid Construction and E. coli Genome Cloning
The Cas9 and dCas9 genes were cloned from the previously described vectors pMJ806 and pMJ841, respectively (Jinek et al., 2012). The genes were PCR amplified and inserted into a vector containing an aTc-inducible promoter PLtetO-1 (Lutz and Bujard, 1997), a chloramphenicol-selectable marker, and a p15A replication origin. The sgRNA template was cloned into a vector containing a minimal synthetic promoter (J23119) with an annotated transcription start site, an ampicillin-selectable marker, and a ColE1 replication origin (see Data S1 for sgRNA sequences). Inverse PCR was used to generate sgRNA cassettes with new 20 bp complementary regions. To insert fluorescent reporter genes into E. coli genomes, the fluorescent gene was first cloned onto an entry vector, which was then PCR amplified to generate linearized DNA fragments that contained nsfA 5′/3′ UTR sequences, the fluorescent gene, and a KanR-selectable marker. The E. coli MG1655 strain was transformed with a temperature-sensitive plasmid pKD46 that contained λ-Red recombination proteins (Exo, Beta, and Gam) (Wang et al., 2009). Cell cultures were grown at 30°C to an OD (600 nm) of ~0.5, and 0.2% arabinose was added to induce expression of the λ-Red recombination proteins for 1 hr. The cells were harvested at 4°C and were used for transformation of the linearized DNA fragments by electroporation. Cells that contain correct genome insertions were selected by using 50 μg/ml kanamycin.
Flow Cytometry and Analysis
Strains were cultivated in EZ-RDM containing 100 μg/ml carbenicillin and 34 μg/ml chloramphenicol in 2 ml 96-well deep well plates (Costar 3960) overnight at 37°C and 1200 rpm. 1 μL of this overnight culture was then added to 249 μl of fresh EZ-RDM with the same antibiotic concentrations, with 2 μM aTc supplemented to induce production of the dCas9 protein. When cells were grown to midlog phase (~4 hr), the levels of fluorescence protein were determined using the LSRII flow cytometer (BD Biosciences) equipped with a high-throughput sampler. Cells were sampled with a low flow rate until at least 20,000 cells had been collected. Data were analyzed using FCS Express (De Novo Software) by gating on a polygonal region containing 60% cell population in the forward scatter-side scatter plot. For each experiment, triplicate cultures were measured, and their standard deviation was indicated as the error bar.
β-Galactosidase Assay
To perform β-galactosidase assay, we added 1 μl of overnight culture prepared as above to 249 μl of fresh EZ-RDM with the same antibiotic concentrations with 2 μM aTc, with or without 1 mM IPTG. Cells were grown to midlog phase. The LacZ activity of 100 ul of this culture was measured using the yeast β-galactosidase assay kit (Pierce) following the instructions.
Extraction and Purification of Total RNA
For each sample, a monoclonal culture of E. coli was grown at 37°C from an OD (600 nm) 0.1 in 500 ml of EZ-RDM to early logphase (OD 0.45 ± 0.05), at which point the cells were harvested by filtration over 0.22 μm nitrocellulose filters (GE) and frozen in liquid nitrogen to simultaneously halt all transcriptional progress. Frozen cells (100 μg) were pulverized on a QIAGEN TissueLyser II mixer mill six times at 15 Hz for 3 min in the presence of 500 μl frozen lysis buffer (20 mM Tris [pH 8], 0.4% Triton X-100, 0.1% NP-40, 100 mM NH4Cl, 50 U/ml SUPERase·In (Ambion), and 1×protease inhibitor cocktail (Complete, EDTA-free, Roche), supplemented with 10 mM MnCl2 and 15 μM Tagetin transcriptional inhibitor (Epicenter).
The lysate was resuspended on ice by pipetting. RQ1 DNase I (110 U total, Promega) was added and incubated for 20 min on ice. The reaction was quenched with EDTA (25 mM final) and the lysate clarified at 4°C by centrifugation at 20,000 × g for 10 min. The lysate was loaded onto a PD MiniTrap G-25 column (GE Healthcare) and eluted with lysis buffer supplemented with 1 mM EDTA.
Total mRNA Purification
Total RNA was purified from the clarified lysate using the miRNeasy kit (QIAGEN). 1 μg of RNA in 20 μl of 10 mM Tris (pH 7) was mixed with an equal volume of 2× alkaline fragmentation solution (2 mM EDTA, 10 mM Na2CO3, and 90 mM NaHCO3 [pH 9.3]) and incubated for ~25 min at 95°C to generate fragments ranging from 30 to 100 nt. The fragmentation reaction was stopped by adding 0.56 ml of ice-cold precipitation solution (300 mM NaOAc [pH 5.5] plus GlycoBlue [Ambion]), and the RNA was purified by a standard isopropanol precipitation. The fragmented mRNA was then dephosphorylated in a 50 μl reaction with 25 U T4 PNK (NEB) in 1× PNK buffer (without ATP) plus 0.5 U SUPERase·In, and precipitated with GlycoBlue via standard isopropanol precipitation methods.
Nascent RNA Purification
For nascent RNA purification, we added the clarified lysate to 0.5 ml anti-FLAG M2 affinity gel (Sigma Aldrich) as described previously (Churchman and Weissman, 2011). The affinity gel was washed twice with lysis buffer supplemented with 1 mM EDTA before incubation with the clarified lysate at 4°C for 2.5 hr with nutation. The immunoprecipitation was washed 4 × 10 ml with lysis buffer supplemented with 300 mM KCl, and bound RNAP was eluted twice with lysis buffer supplemented with 1 mM EDTA and 2 mg/ml 3× FLAG peptide (Sigma Aldrich). Nascent RNA was purified from the eluate using the miRNeasy kit (QIAGEN) and was converted to DNA using a previously established library generation protocol (Churchman and Weissman, 2011).
DNA Library Preparation and DNA Sequencing
The DNA library was sequenced on an Illumina HiSeq 2000. Reads were processed using the HTSeq Python package, and other custom software written in Python. The 3′ end of the sequenced transcript was aligned to the reference genome using Bowtie (http://bowtie-bio.sourceforge.net/) and the RNAP profiles were generated in MochiView (http://johnsonlab.ucsf.edu/mochi.html).
Plasmid Design and Construction for CRISPRi in Human Cells
The sequence encoding mammalian-codon-optimized Streptococcus pyogenes Cas9 (DNA 2.0) was fused with three C-terminal SV40 nuclear localization sequences (NLS) or to tagBFP flanked by two NLS. Using standard ligation-independent cloning, we cloned these two fusion proteins into MSCV-Puro (Clontech). Guide sgRNAs were expressed using a lentiviral U6-based expression vector derived from pSico, which coexpresses mCherry from a CMV promoter. The sgRNA expression plasmids were cloned by inserting annealed primers into the lentiviral U6-based expression vector that was digested by BstXI and XhoI.
Cell Culture, DNA Transfections, and Fluorescence Measurements for CRISPRi in Human Cells
HEK293 cells were maintained in Dulbecco’s modified Eagle medium (DMEM) in 10% FBS, 2 mM glutamine, 100 units/ml streptomycin, and 100 μg/ml penicillin. HEK293 were infected with a GFP-expressing MSCV retrovirus using standard protocols and were sorted by flow cytometry using a BD FACS Aria2 for stable GFP expression. GFP-expressing HEK293 cells were transiently transfected using TransIT-LT1 transfection reagent (Mirus) with the manufacturers recommended protocol in 24-well plates using 0.5 μg of the dCas9 expression plasmid and 0.5 μg of the RNA expression plasmid (with 0.25 μg of GFP reporter plasmid for Figure S7). At 72 hr following transfection, cells were trypsinized to a single cell suspension. The U6 vector contains a constitutive CMV promoter driving an mCherry gene. GFP expression was analyzed using a BD LSRII FACS machine by gating on the mCherry-positive populations (>10-fold brighter mCherry over the negative control cells).
Supplementary Material
Acknowledgments
The authors thank Martin Jinek for discussion and distribution of Cas9 and dCas9 genes and Connie Lee for discussion and critical reading of the manuscript. The authors also thank Leonardo Morsut and Esteban Toro for technical advice and help. L.S.Q. acknowledges support from the UCSF Center for Systems and Synthetic Biology. This work was supported by NIH P50 GM081879 (L.S.Q. and W.A.L.), Howard Hughes Medical Institute (L.A.G., J.A.D., J.S.W., and W.A.L.), NSF SynBERC EEC-0540879 (A.P.A. and W.A.L.), a Howard Hughes Collaborative Initiative Award (J.S.W.), and a Ruth L. Kirschstein National Research Service Award (M.H.L.). J.A.D. is a founder of Caribou Biosciences and a member of its scientific advisory board. None of the other authors have a financial interest related to this work. The authors have filed a patent related to this work.
Footnotes
Supplemental Information includes seven figures and one data file and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2013.02.022.
References
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Beerli RR, Barbas CF., 3rd Engineering polydactyl zinc-finger transcription factors. Nat Biotechnol. 2002;20:135–141. doi: 10.1038/nbt0202-135. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim J-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013 doi: 10.1038/nbt.2507. Published online January 29, 2013. http://dx.doi.org/10.1038/nbt.2507. [DOI] [PubMed]
- Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh J-RJ, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013 doi: 10.1038/nbt.2501. Published online January 29, 2013. http://dx.doi.org/10.1038/nbt.2501. [DOI] [PMC free article] [PubMed]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013 doi: 10.1038/nbt.2508. Published online January 29, 2013. http://dx.doi.org/10.1038/nbt.2508. [DOI] [PMC free article] [PubMed]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem. 2010;79:213–231. doi: 10.1146/annurev-biochem-010909-095056. [DOI] [PubMed] [Google Scholar]
- Lewis M. The lac repressor. C R Biol. 2005;328:521–548. doi: 10.1016/j.crvi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Lucks JB, Mortimer SA, Trapnell C, Luo S, Aviran S, Schroth GP, Pachter L, Doudna JA, Arkin AP. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq) Proc Natl Acad Sci USA. 2011;108:11063–11068. doi: 10.1073/pnas.1106501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1–I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica FJM, Wolf YI, Yakunin AF, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, Dicarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. 2010;11:181–190. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nudler E, Goldfarb A, Kashlev M. Discontinuous mechanism of transcription elongation. Science. 1994;265:793–796. doi: 10.1126/science.8047884. [DOI] [PubMed] [Google Scholar]
- Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- Qi L, Haurwitz RE, Shao W, Doudna JA, Arkin AP. RNA processing enables predictable programming of gene expression. Nat Biotechnol. 2012;30:1002–1006. doi: 10.1038/nbt.2355. [DOI] [PubMed] [Google Scholar]
- Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJJ, van der Oost J, Doudna JA, Nogales E. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011;477:486–489. doi: 10.1038/nature10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol. 2011;29:149–153. doi: 10.1038/nbt.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.