

Abstract
MicroRNAs (miRNAs) are noncoding RNAs that impact almost every aspect of biology and disease. Until now, the cell-specific effects of miRNAs in cardiovascular system have not been established. In the current study, the cellular functions of miR-221 and miR-222 (miR-221/222) in vascular smooth muscle cells (VSMCs) and vascular endothelial cells (ECs) were compared. In cultured cells, we identified that the effects of miR-221/222 on proliferation, migration, and apoptosis are opposite between VSMCs and ECs. In VSMCs, miR-221/222 had effects of pro-proliferation, pro-migration, and anti-apoptosis. In contrast, miR-221/222 had effects of anti-proliferation, anti-migration, and pro-apoptosis in ECs. The different expression profiles of their target genes, p27(Kip1), p57(kip2), and c-kit between the two cell types might be related to the opposite effects. Finally, the opposite cellular effects of miR-221/222 were verified in vivo in balloon-injured rat carotid artery as demonstrated by different consequences in neointimal growth and re-endothelialization. The results suggest that the biological functions of miR-221/222 in vascular walls are cell-specific. The opposite cellular effects of miR-221/222 on VSMCs and ECs may have important therapeutic applications in many vascular diseases such as atherosclerosis and restenosis after angioplasty.
Keywords: microRNAs, smooth muscle cells, endothelial cells, cell-specific effects, vascular disease
Introduction
MicroRNAs (miRNAs) are a novel class of endogenous, small, noncoding RNAs that regulate gene expression via degradation, translational inhibition, or translational activation of their target mRNAs [1-5]. Functionally, an individual miRNA is important as a transcription factor because it is able to regulate the expression of its multiple target genes. As a group, miRNAs are able to directly regulate at least 30% of genes in a cell. In addition, other genes may also be regulated indirectly by miRNAs. It is therefore not surprising that miRNAs could be the pivotal regulators in normal development, physiology, and pathology [1-5]. Recent studies have identified that miRNAs are highly expressed in cardiovascular system and many of them are dysregulated in diseased vessels and hearts [6]. Thus, identifying the biological functions of miRNAs in cardiovascular system is critical to uncovering miRNAs in the pathogenesis of cardiovascular diseases.
Interestingly, recent studies of cancer cells indicate that the biological functions of an individual miRNA might be cell specific, although the mechanisms are still unknown. For example, mi.R-21 has an antiapoptotic effect on glioblastoma cells, but increases Hela cell apoptosis [7, 8]. However, the cell-specific effects of miRNAs are not established in cardiovascular system.
miR-221 and miR-222 are highly expressed in both vascular smooth muscle cells (VSMCs) and vascular endothelial cells (ECs). In our recent study, we have identified, miR-221/222 have a strong pro-proliferative effect on VSMCs [9]. However, one report revealed that miR-221/222 could inhibit EC migration [10]. Thus, miR-221/222 might have different effects on VSMCs and ECs. In current study, the effects of miR-221/222 on proliferation, migration and apoptosis of VSMCs and ECs, and the potential molecular mechanisms involved were determined.
Materials and Methods
Rat carotid artery balloon injury model
Carotid artery balloon injury was induced in male Sprague-Dawley rats (230 to 300 g) as described in our previous studies [11, 12]. Briefly, rats were anesthetized with ketamine (80 mg/kg, ip)/xylazine(5 mg/kg, ip). The adequacy of anesthesia was tested by tail-clamping with a hemostat, and additional ketamine and xylazine was administered as necessary to prevent response to surgical stimulation. Under a dissecting microscope, the right common carotid artery was exposed through a midline cervical incision. A 2F Fogarty catheter (Baxter Edwards) was introduced via an arteriotomy in the external carotid artery, and then the catheter was advanced to the proximal edge of the omohyoid muscle. To produce carotid artery injury, we inflated the balloon with saline and withdrew it three times from just under the proximal edge of the omohyoid muscle to the carotid bifurcation. After injury, the external carotid artery was permanently ligated with a 6-0 silk suture, and blood flow in the common carotid artery was restored. At the end of the experiments, an overdose injection of pentobarbital (200mg/kg, ip) was used to induce euthanasia. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Medicine and Dentistry of New Jersey and were consistent with the Guide for the Care and Use of Laboratory Animals (NIH publication 85-23, revised 1985).
Local oligo and adenovirus delivery into the injured vascular walls
miR-221 and miR-222 were downregulated in the balloon-injured rat carotid artery via their inhibitor. miR-221 and miR-222 inhibitor (2′OMe-miR-221/222) was synthesized by Integrated DNA Technologies and had the following sequence and structure: 5′-mAmCmCmCmAmGmUmAmGmCmCmAmGmAmUmGmUmAmGmCmU-3′. To deliver 2′OMe-miR-221/222 or control oligonucleotides into the injured vascular tissue and to avoid any potential systemic side effects, we applied an established local oligo delivery model via F-127 pluronic gel as described in our recent reports with little modification [9, 11]. Briefly, immediately after balloon injury of the rat common carotid artery, transfection solutions (60 μl 0.2% transfection reagent in DMEM) were mixed with 2′OMe-miR-221/222 (10 μg), vehicle (DMEM), or control oligonucleotides (10 μg) and infused into the ligated segment of the common carotid artery for 30 min. Then, 90 μg of these oligonucleotides, preloaded into 200 μl 30 % F-127 pluronic gel (Sigma) and 1 % transfection reagent (Qiagen, CA) at 4 °C, was applied locally to the adventitia around injured artery segments.
To upregulate the expression of miR-221 and miR-222 expression in rat carotid arteries, we have generated adenoviruses expressing miR-221 and miR-222 (Ad-miR-221/222) and control adenovirus, Ad-GFP. Ad-miR-221/222 was transfected into the balloon-injured rat carotid artery segments using a method established in our lab as described [12]. Immediately after balloon injury, 50 μl solutions (15 % poloxamer 407 containing 3% sucrose in PBS) of Ad-miR-221/222 (5×109 pfu/ml), Ad-GFP (5×109 pfu/ml), (control adenovirus), or vehicle were infused into the ligated segment of the common carotid artery for 30 min. Then, the external carotid artery was permanently ligated with a 6-0 silk suture and blood flow in the common carotid artery was restored.
Morphometric analysis for neointimal formation and re-endothelialization
Morphometric analysis via computerized image analysis system (Scion Image CMS-800) was performed on sections stained with Masson's trichrome staining as described [11, 12]. Six sections (5 μm thick) sectioned at equally spaced intervals of injured carotid arteries were used. The medial area was calculated by subtracting the area defined by the internal elastic lamina (IEL) from the area defined by the external elastic lamina (EEL), and the intimal area was determined by subtracting the lumen area from the area defined by the IEL. Finally, the intimal to medial area ratio (I/M) of each section was calculated. The average I/M of the six sections was used as the I/M of this animal.
Re-endothelialization was evaluated by planimetric analysis as described by our previous report [13]. Briefly, the rats were sacrificed at 14d after carotid artery balloon-injury. These rats received an intravenous injection via the femoral vein of 0.5 ml of 0.5% Evans blue dye 30 min before they were sacrificed. The right atrium of rat was dissected quickly, and an 18-gauge catheter connected to the perfusion system was inserted in the left ventricle. All animals were perfused for 5 min with saline at physiological pressure. Planimetric analysis of the photograph of the harvested carotid arteries stained with Evans blue dye to identify the remaining endothelium-denuded site was performed with a computerized sketching program (Scion Image CMS-800) by a technician who was unaware of the treatment regimen. The initially denuded area was defined as the total surface area between the carotid bifurcation and the edge of the omohyoid muscle. The re-endotheliazation area was defined as the area that was not stained with Evans blue.
Cell culture
VSMCs and ECs (Cell Applications, Inc.) were obtained from the aortas of male Sprague-Dawley rats (5 weeks old) by using an enzymatic dissociation method as described [11, 12]. The basic culture medium was DMEM for VSMCs and M199 for ECs. Cells between passage 3 and 6 were used.
Oligo transfection, knockdown of miR-221/222 and c-Kit, and overexpression of miR-221/222 and P27(Kip1) in cultured VSMCs and/or ECs
Oligo transfection was performed according to a pre-established protocol (9). Briefly, cells were transfected using a transfection reagent (Qiagen, Chatsworth, CA) 24 h after seeding into the wells. Transfection complexes were prepared according to the manufacturer's instructions. For miR-221/222 knockdown, 2′OMe-miR-221/222 (Integrated DNA Technologies) was added to the complexes at final oligonucleotide concentration of 100 nmol/L. Vehicle and oligo controls (Ambion, Inc.) were applied. For miR-221/222 or P27(Kip1) overexpression, Ad-221/222 or Ad- P27(Kip1) was added to the culture medium at 30 MOI. Adenovirus expressing GFP (Ad-GFP) was used as an adenovirus control. For c-Kit knockdown, the siRNAs (100 nM, Ambion, Inc.) was used. The transfection medium was replaced 4 h posttransfection by the regular culture medium.
Cell proliferation, migration and apoptosis
VSMC and EC proliferation was determined by cell counting and bromodeoxyuridine (BrdU) incorporation assay [9, 11, 12]. For cell counting, the cells were detached by trypsinization and re-suspended in PBS. The cells were then counted under a microscope. For BrdU incorporation assay, 10 mM BrdU was added to the culture medium for incorporation into the DNA of replicating cells. After 2 h of incubation, cells were fixed, and anti-BrdU antibody (In Situ Cell Proliferation Kit) was added to each well for 45 min. Finally, the proliferative cells were detected under a fluorescence microscope.
Cell migration was determined by a scratch wound assay and a modified Boyden chamber assay [14-16]. For the scratch wound assay, VSMCs and ECs at 100% confluence in six-well plates will be wounded with a sterile pipette tip to generate a cell-free gap of ∼ 1-mm width, and the wound location in the culture dish will be marked as described [14]. Cells will be washed with serum-free DMEM and photographed to record the wound width at 0 h. After that, one group of cells will be cultured in serum-free medium for 24 h as a negative control. Other groups will be treated with 5% FBS. Twenty-four hours later, photographs will be taken again at the marked wound location for migration measurement. For the modified Boyden chamber assay, the upper inserts (8μm pores coated with 0.1% gelatin) containing VSMCs or ECs (1 × 105 cells) were placed in the bottom 24-well chamber containing M131 with stimulants (Invitrogen, USA) for ECs or DMEM with PDGF (10 ng/ml) for VSMCs. After incubation for 6 h at 37°C, the cells passing through the membrane onto the lower side of the chamber were fixed with 4% formaldehyde and stained with 4′, 6-diamidinophenylidole (DAPI). The migrated cells were then counted in nine random fields at × 200 magnifications.
VSMC and EC apoptosis in cultured cells was measured by TUNEL analysis 48 h in serum-free culture as described [11). The VSMCs cultured on coverslips in 24-well plates were fixed in 4% paraformaldehyde. TUNEL staining was done using the In Situ Cell Death Detection Kit (Roche) according to the manufacturer's protocol. The number of TUNEL-positive cells was counted under a fluorescence microscope. Apoptotic cells were quantified by counting the percentage of TUNEL-positive cells against total nucleated cells stained by DAPI.
Luciferase assay
A construct in which a fragment of the 3′-UTR of either p27(Kip1), p57(Kip2), or c-kit mRNA containing the putative miR-221/222 binding sequences was cloned into a firefly luciferase reporter construct and transfected into HEK 293 cells, ECs or VSMCs. For the HEK 293 cells, co-transfection with vehicle (vehicle), an empty plasmid (pDNR-CMV), a plasmid expressing miR-221/222 (pmiR-221/222), or a control plasmid expressing an unrelated miRNA, miR-145 (pmiR-145) was performed. In addition, the constructs with mutated fragment of the 3′-UTR of p27(Kip1) p57(Kip2), c-kit mRNA without the putative miR-221 and miR-222 binding sequences were used as mutated controls. Luciferase assay in VSMCs and ECs was performed as described by Dentelli et al [17]. Ad-miR-221/222 or control virus (Ad-GFP) (30 MOI) was transfected into the VSMCs and ECs. Relative luciferase expression was measured on a scintillation counter by using a dual luciferase reporter system [11, 12).
RNA levels were determined by qRT-PCR
Briefly, RNAs from cells and rat carotid arteries were isolated with a RNA Isolation Kit (Ambion, Inc.). qRT-PCR for miR-221and miR-222 was performed on cDNA generated from 50 ng of total RNA using the protocol of the mirVana qRT-PCR miRNA Detection Kit (Ambion, Inc). qRT-PCR for p27(Kip1), p57(Kip1), and c-Kit was performed on cDNA generated from 200 ng of total RNA using the protocol of a qRT-PCR mRNA Detection Kit (Roche). Amplification and detection of specific products were performed with a Roche Lightcycler 480 Detection System. As an internal control, U6 was used for miRNA template normalization and GADPH was used for other template normalizations. Fluorescent signals were normalized to an internal reference, and the threshold cycle (Ct) was set within the exponential phase of the PCR. The relative gene expression was calculated by comparing cycle times for each target PCR. The target PCR Ct values were normalized by subtracting the U6 or GADPH Ct value, which provided the ΔCt value. The relative expression level between treatments was then calculated using the following equation: relative gene expression = 2−(ΔCtsample−ΔCtcontrol) [11, 12].
Western blot analysis
Proteins were isolated from cultured cells and carotid arteries and protein levels were determined by western blot analysis. Briefly, equal amounts of protein were subjected to SDS-PAGE. Standard western blot analysis was conducted using p27 (Kip1), p57 (Kip2) and c-Kit antibodies (Santa Cruz Biotechnology). GADPH antibody (1:5000 dilution; Cell Signaling) was used as a loading control.
Construction of the adenoviruses
The adenoviruses expressing miR-221 and miR-222 (Ad-miR-221/222), p27 (kip1) (Ad-p27 (kip1)) and control viruses expressing GFP (Ad-GFP) were generated using the Adeno-X™ Expression Systems 2 kit (Clontech, CA) according to the manufacturer's protocols as described previously [12]. The resulting adenoviruses were further amplified by infection of HEK293A cells and purified by cesium chloride gradient ultracentrifugation. The Ad-miR-221/222, Ad-27(kip1) and Ad-GFP were titrated using a standard plaque assay.
In situ hybridization
In situ hybridization of miR-222 was performed in 10-μm vessel sections as described [9, 12]. Tissue sections from balloon-injured or uninjured rat carotid arteries were cut using a cryostat and transferred to SuperFrost/plus slides (Fisher). Vessel sections were fixed in 4% paraformaldehyde and acetylated in acetic anhydride/triethanolamine, followed by washing in PBS. Sections were then pre-hybridized in hybridization solution (50% formamide, 5× SSC, 0.5 mg/mL yeast tRNA, 1× Denhardt's solution) at 25°C below the predicted T m value of the LNA probe for 30 min. Probes of miR-222 (3 pmol) (LNA miRCURY probe; Exiqon) were DIG-labeled (DIG Oligonucleotide 3′ Tailing Kit; Roche Applied Sciences) and hybridized to the sections for 1 h at the same temperature as pre-hybridization. After washing in 0.1× SSC at 55°C, the ISH signals were detected using the tyramide signal amplification system (PerkinElmer) according to the manufacturer's instructions. Slides were mounted in Prolong Gold containing DAPI (Invitrogen). Sections without miR-222 probes were used as the negative controls. All the fluorescence images were analyzed with a Nikon microscope equipped with a CCD camera and image software.
Immunofluorescence
ECs in vessel sections were identified by using the immunofluorescence of CD31. In addition, VSMCs in vessels were detected by immunofluorescence of smooth muscle (SM) α-actin (SM α-actin).The frozen sections of vessels were incubated with anti-CD31 antibodies (1:200 dilution, Abcam) followed by fluorescein conjugated secondary antibodies (1:200 dilution, Vector Laboratories). For SM α-actin staining, the sections were incubated with anti-SM α-actin (1:200 dilution, Abcam) followed by Texas red conjugated secondary antibodies (1:200 dilution, Vector Laboratories). Cell nuclear was stained by DAPI (blue color). The fluorescence was observed via a Nikon Eclipse 80i immunofluorscence microscope.
Statistics
All data are presented as mean ± standard error. For relative gene expression, the mean value of vehicle control group is defined as 100% or 1. Two-tailed unpaired Student's t tests and ANOVA were used for statistical evaluation of the data. Sigma stat statistical analysis program was used for data analysis. A p value < 0.05 was considered significant.
Results
Opposite effect of miR-221/222 on VSMC and EC proliferation
To determine the potential cellular effects of miR-221/222 on VSMCs and ECs, gain-of-function and loss-of-function approaches were applied. The expression of miR-221/222 was downregulated by their inhibitor, 2′OMe-miR-221/222, and was upregulated by Ad-miR-221/222. Cell proliferation was determined using cell counting and BrdU incorporation assay. As shown in Fig. 1A-E, VSMC proliferation was decreased by 2′OMe-miR-221/222, but increased by Ad-miR-221/222. The pro-proliferative effect of miR-221/22 was consistent with our recent report [9]. In contrast, EC proliferation was increased by 2′OMe-miR-221/222, but decreased by Ad-miR-221/222 (Fig. 1F-J). Clearly, miR-221/222 are anti-proliferative genes in ECs.
Fig. 1. Opposite effect of miR-221/222 on VSMC and EC proliferation.

(A-E) The effect of miR-221/222 on VSMC proliferation. Knockdown of miR-221/222 via 2′OMe-miR-221/222 decreased cell numbers (A) and BrdU incorporation (C) at 48 h after culture with DMEM containing 10% FBS. Overexpression of miR-221/22 via Ad-miR-221/222 increased cell numbers (B) and BrdU incorporation (D). Representative BrdU-stained VSMC photomicrographs from different groups (E). (F-J) The effect of miR-221/222 on EC proliferation. 2′OMe-miR-221/222 increased cell numbers (F) and BrdU incorporation (H) at 48 h after culture with DMEM containing 10% FBS. Overexpression of miR-221/222 via Ad-miR-221/222 decreased cell numbers (G) and BrdU incorporation (I). Representative BrdU-stained EC photomicrographs from different groups (J). Note: green color is BrdU positive cell; Blue color is nucleus stained by DAPI; n=6; *P<0.05 compared with control.
Opposite effect of miR-221/222 on VSMC and EC migration
VSMC and EC migration was measured by the scratch wound assay as described in our previous studies [14]. As shown in Fig. 2A&B, VSMC migration was decreased by 2′OMe-miR-221/222, but increased by Ad-miR-221/222. In contrast, EC migration was increased by 2′OMe-miR-221/222, but decreased by Ad-miR-221/222 (Fig. 2C&D). Clearly, miR-221/222 are pro-migratory genes in VSMCs, but anti-migratory genes in ECs. To further confirm the different biological effects of miR-221/222 on EC and VSMC migration, the modified Boyden chamber assay was performed in cells pretreated with vehicle, control oligos (100 n M), or 2′OMe-miR-221/222 (100 nM). As shown in Fig. 2E&F, inhibition of miR-221/222 decreased the number of migrated VSMCs, but increased the number of migrated ECs.
Fig. 2. Opposite effect of miR-221/222 on VSMC and EC migration.
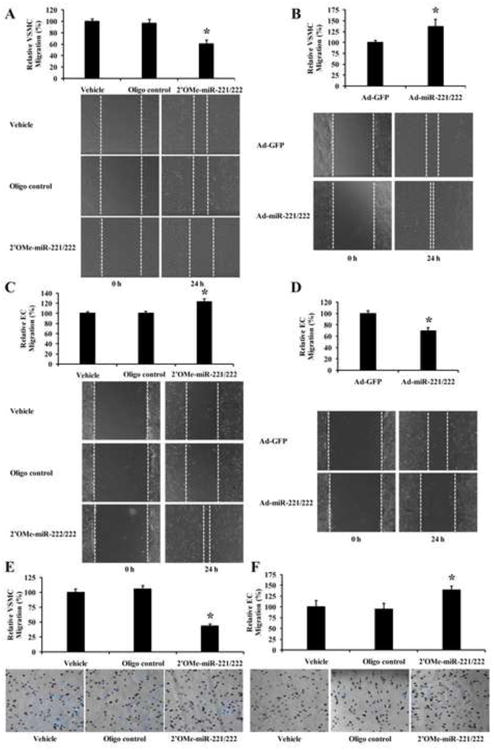
(A) Knockdown of miR-221/222 via 2′OMe-miR-221/222 decreased VSMC migration determined by the scratch wound assay. Bottom panel: Representative photomicrographs of VSMC migration. (B) Overexpression of miR-221/222 via Ad-miR-221/222 increased VSMC migration determined by the scratch wound assay. Bottom panel: Representative photomicrographs of VSMC migration. (C) Knockdown of miR-221/222 increased EC migration determined by the scratch wound assay. Bottom panel: Representative photomicrographs of EC migration. (D) Overexpression of miR-221/222 decreased EC migration determined by the scratch wound assay. Bottom panel: Representative photomicrographs of EC migration. (E) Knockdown of miR-221/222 decreased VSMC migration determined by the modified Boyden chamber assay. Bottom panel: Representative photomicrographs of VSMC migration. (F) Knockdown of miR-221/222 increased EC migration determined by the modified Boyden chamber assay. Bottom panel: Representative photomicrographs of EC migration. Note: n=6; *P<0.05 compared with control.
Opposite effect of miR-221/222 on VSMC and EC apoptosis
In this experiment, VSMCs or ECs were treated with vehicle, control oligos (100 n M), or 2′OMe-miR-221/222 (100 n M), Ad-GFP (30 MOI), or Ad-miR-221/222 (30 MOI) for 4h followed by 48 h of serum deprivation as described in our recent study [10]. Cell apoptosis was determined by TUNEL assay [18, 19]. As shown in Fig. 3A-3C, serum-deprivation-mediated VSMC apoptosis was increased by 2′OMe-miR-221/222, but decreased by Ad-miR-221/222. In contrast, EC apoptosis was decreased by 2′OMe-miR-221/222, but increased by Ad-miR-221/222 (Fig. 3D-3F).
Fig. 3. Opposite effect of miR-221/222 on VSMC and EC apoptosis.
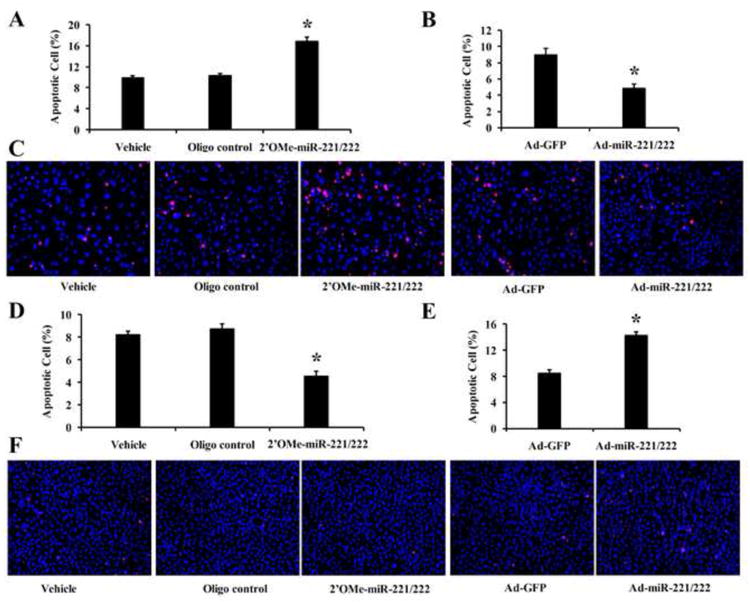
(A) Knockdown of miR-221/222 via 2′OMe-miR-221/222 increased VSMC apoptosis. (B) Overexpression of miR-221/222 via Ad-miR-221/222 decreased VSMC apoptosis. (C) Representative photomicrographs of VSMC apoptosis. (D) Knockdown of miR-221/222 decreased EC apoptosis. (E) Overexpression of miR-221/222 increased EC apoptosis. (F) Representative photomicrographs of EC apoptosis Note: n=6; *P<0.05 compared with control.
p27(Kip1), p57(Kip2), and c-kit are target genes of miR-221/222 in both VSMCs and ECs
Our computational and bioinformatical analysis indicates that p27(Kip1), p57(Kip2) and c-kit are three potential target genes of miR-221/222 that are related to cell differentiation, proliferation, migration and apoptosis. To verify that p27(Kip1), p57(Kip2) and c-kit are the target genes of miR-221/222 in VSMCs and ECs, both gain-of-function and loss-of-function approaches were applied. We found that p27(Kip1), p57(Kip2), and c-kit were upregulated by 2′OMe-miR-221/222 and were downregulated by Ad-miR-221/222 both in VSMCs (Fig. 4A) and in ECs (Fig. 4B) as determined by western blot analysis. To determine whether miR-221/222 are able to directly bind to p27(Kip1), p57(Kip2), and c-kit and inhibit their expression, a construct in which a fragment of the 3′-UTR of either p27(Kip1), p57(Kip2), or c-kit mRNA containing the putative miR-221/222 binding sequences was cloned into a firefly luciferase reporter construct and transfected into HEK 293 cells together with either vehicle (vehicle), an empty plasmid (pDNR-CMV), a plasmid expressing miR-221/222 (pmiR-221/222), or a control plasmid expressing an unrelated miRNA, miR-145 (pmiR-145), following the transfection procedures. In addition, the constructs with mutated fragment of the 3′-UTR of p27(Kip1) p57(Kip2), c-kit mRNA without the putative miR-221 and miR-222 binding sequences were used as mutated controls. As expected, pmiR-221/222, but not pDNR-CMV or pmiR-145, inhibited luciferase activity (Fig. 4C). In the mutated control groups, the inhibitory effect of pmiR-221/222 disappeared. To further verify that miR-221/222 are able to directly bind to these genes and inhibit their expression in ECs and VSMCs, we transfected these fragments of 3′-UTR into ECs and VSMCs pretreated with Ad-miR-221/222 (30 MOI) for 24h. As shown in Fig. 4D&E, the luciferase activity was significantly inhibited by Ad-miR-221/222 both in ECs and VSMCs. The results suggested that miR-221/222 are able to bind to p27(Kip1), p57(Kip2) and c-Kit directly and inhibit their expression.
Fig. 4. p27(Kip1), p57(Kip2), and c-Kit are target genes of miR-221/222.
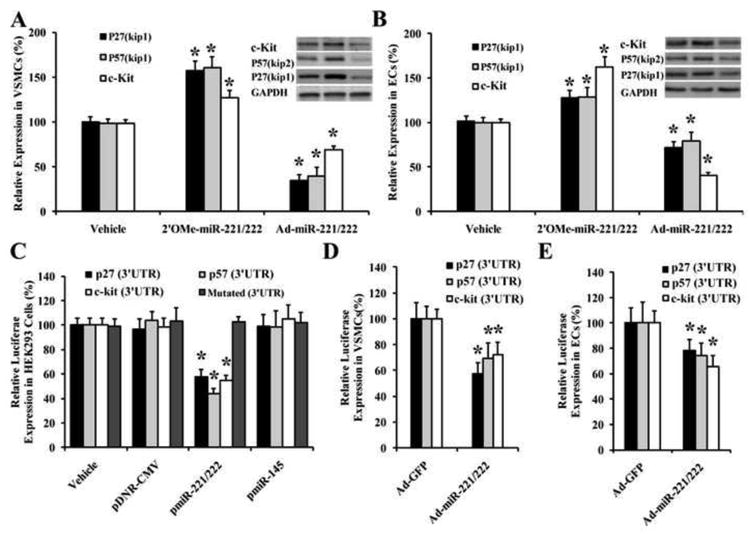
(A) p27(Kip1), p57(Kip2), and c-Kit were upregulated by 2′OMe-miR-221/222 and were downregulated by Ad-miR-221/222 in VSMCs as determined by western blot analysis. (B) p27(Kip1), p57(Kip2), and c-Kit were upregulated by 2′OMe-miR-221/222 and were downregulated by Ad-miR-221/222 in ECs as determined by western blot analysis. (C) pmiR-221/222, but not the control pDNR-CMV or pmiR-145, could bind to and inhibit the expression of p27(Kip1), p57(Kip2), and c-Kit determined by luciferase assay in HEK 293. In the mutated control groups, the inhibitory effect of pmiR-221/miR-222 disappeared. (D) Ad-miR-221/222 decreased the luciferase activity of p27(Kip1), p57(Kip2), and c-Kit transfected into the VSMCs. (E) Ad-miR-221/222 decreased the luciferase activity of p27(Kip1), p57(Kip2), and c-Kit transfected into the ECs. Note: n=6; *P<0.05 compared with controls.
The expression profiles of the target genes, p27(Kip1), p57(Kip2), and c-kit are different between VSMCs and ECs. Moreover, opposite cellular effects between c-kit and other two target genes are identified
The relative mRNA and protein expression of these target genes in VSMCs and ECs was determined. GAPDH was used as the control. Interestingly, as shown in Fig. 5A & 5B, we found that p27(Kip1) and p57(Kip2) are highly expressed in VSMCs, but not in ECs at both mRNA and protein levels. In contrast, c-kit was highly expressed in ECs, but not in VSMCs (Fig. 5A & 5B). To modify the profile of target gene expression in VSMCs, c-kit was knocked down by its siRNAs (Fig. 5C & 5D). As expected, we found that VSMC proliferation was attenuated via knockdown of c-kit. (Fig. 5E). Similarly, the EC proliferation was also decreased after knockdown of c-kit (Fig. 5F-5H), although the inhibitory effect was much stronger compared with that in VSMCs (Fig. 5E & 5H).
Fig. 5. The expression profiles of the target genes of miR-221/222 and functional analysis.
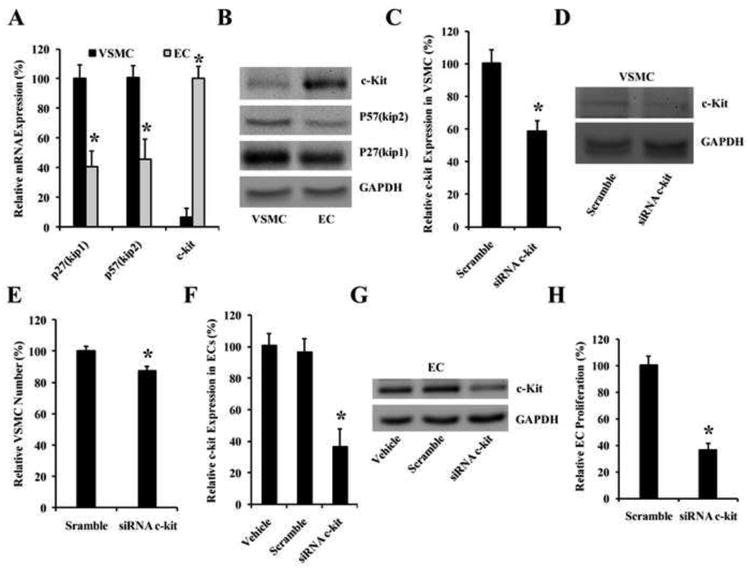
(A) The relative expression of these target genes in VSMCs and ECs at mRNA level. Note: n=5; *P<0.05 compared with that in VSMCs. (B) The relative expression of these target genes in VSMCs and ECs at protein level. (B-D) The effect of c-kit on VSMC proliferation. (B) c-kit was knocked down by its siRNAs (siRNA c-Kit) (100 nM) at mRNA level as determined by qRT-PCR. (C) c-kit was knocked down by its siRNAs (siRNA c-Kit) (100 nM) at protein level as determined by western blot. (D) VSMC proliferation was attenuated by knockdown of c-Kit expression. Note: n=6; *P<0.05 compared with control. (F-H) The effect of c-kit on EC proliferation. (F) c-kit was knocked down by its siRNAs (siRNA c-Kit) (100 nM) at mRNA level as determined by qRT-PCR. (G) c-kit was knocked down by its siRNAs (siRNA c-Kit) (100 nM) at protein level as determined by western blot. (H) EC proliferation was attenuated by knockdown of c-Kit expression. Note: n=6; *P<0.05 compared with control.
In our circulation research article, we have identified that p27(Kip1) is a major target gene that responsible for miR-221/222-mediated pro-proliferative effect on VSMCs [9]. To determine the potential role of p27(Kip1) in ECs, the cultured ECs were treated with Ad-p27(Kip1) (30 MOI) or Ad-GFP (30 MOI). As shown in Fig. 6A, the expression of p27(Kip1) was successfully increased by Ad-p27(Kip1). The proliferation of ECs was inhibited by overexpression of p27(Kip1) (Fig. 6B). Thus, no difference was found in p27(Kip1)-mediated effect on cell proliferation in VSMCs and ECs. In addition, the migration of ECs was inhibited by overexpression of p27(Kip1) (Fig. 6C-6D). In contrast, the apoptosis of ECs was significantly increased by p27(Kip1) overexpression (Fig. 6E).
Fig. 6. The cellular functions of P27(Kip1) in cultured ECs.
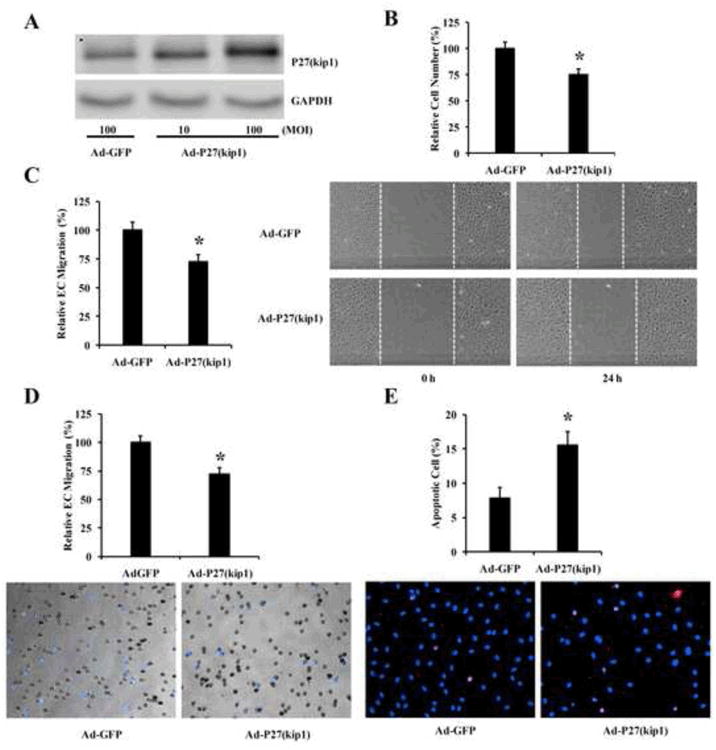
(A) Upregulation of P27(Kip1) by Ad-P27(Kip1) (30 MOI). (B) The effect of P27(Kip1) overexpression on EC proliferation. (C) The effect of P27(Kip1) overexpression on EC migration determined by the scratch wound assay. Left panel: Representative photomicrographs of EC migration. (D) The effect of P27(Kip1) overexpression on EC migration determined by the modified Boyden chamber assay. Bottom panel: Representative photomicrographs of EC migration. (E) The effect of P27(Kip1) overexpression on EC apoptosis. Bottom panel: Representative photomicrographs of EC apoptosis. Note: n=6; *P<0.05 compared with control.
Downregulation of miR-221/222 decreases neointimal formation, but increases re-endothelialization in rat carotid artery after angioplasty. In contrast, upregulation of miR-221/222 increases neointimal formation, but decreases re-endothelialization
In our recent study, we have identified that miR-2221/222 were upregulated in rat carotid arteries after balloon-injury [9]. To further determine the distribution of miR-221/22 in vascular walls, In situ hybridization of miR-222 was performed in normal uninjured and balloon-injured rat carotid arteries. As shown in Fig.7A, miR-222 (green color) was expressed in endothelium and media of the normal vessels and its expression was increased in balloon-injured vessels both in neointima and in media of the vessels. No obvious signal was identified in vessels without miR-222 probe (negative control w/o probe). To further provide cellular distribution of miR-222 in these vessels, double immunostaining of EC marker, CD31 (green color) and VSMC marker, SM α-action (red color) was performed (Fig. 7A).
Fig. 7. The effects of miR-221/222 on neointimal formation and re-endothelialization in rat carotid arteries after angioplasty.
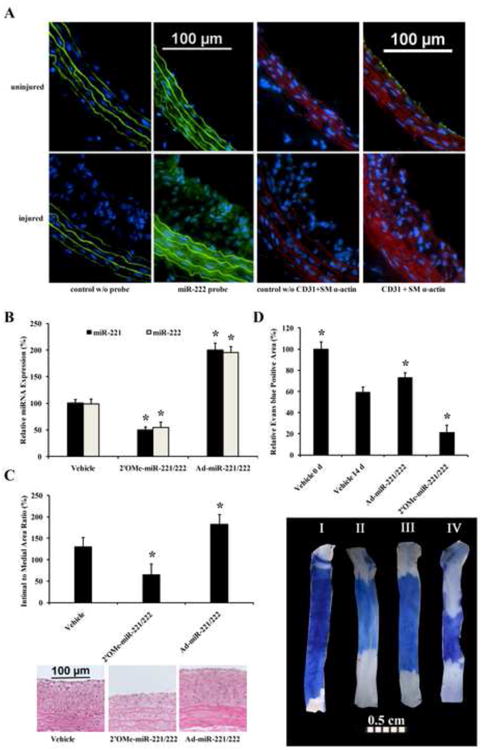
(A) In situ hybridization of miR-222 (green color) and immunofluorescence of CD31 (green color) and SM α-actin (red color) in normal uninjured and balloon-injured rat carotid arteries. Note: control w/o probe, negative control without miR-222 probe; control w/o CD31+ SM α-actin; negative control without CD31and SM α-actin antibodies; Blue color is nucleus stained by DAPI. (B) The expression of miR-221/222 was downregulated by 2′OMe-miR-221/222, and upregulated by Ad-miR-221/222 in balloon-injured rat carotid arteries. (C) Neointimal formation was decreased by knockdown of miR-221/222. In contrast, the neointimal formation was increased by overexpression of miR-221/222. Representative H-E stained photomicrographs of rat carotid arteries from different groups were shown in the bottom panel of C. (D) Re-endothelialization was increased by knockdown of miR-221/222, but was decreased by overexpression of miR-221/222. Representative Evans blue stained photomicrographs of rat carotid arteries immediately after balloon injury and at 14 days after injury from different groups were shown in the bottom panel of D. Note: n=8; *P<0.05 compared with control.
It is well established that the increased VSMC proliferation, migration, survival, and the decreased EC proliferation, migration, and survival are responsible for the increased restenosis after angioplasty and stent implantation. Thus, inhibition of miR-221/222 should result in the decreased neointimal lesion formation, but increased re-endothelialization after angioplasty. To test the hypothesis, the expression of miR-221/222 in balloon-injured rat carotid arteries was knocked down by their inhibitor, 2′OMe-miR-221/222. The inhibitor and control oligonucleotides were applied into the vascular walls using local oligo delivery model via F-127 pluronic gel as described in our recent reports [9, 11]. As shown in Fig. 7B, miR-221and miR-222 were successfully knocked down in balloon-injured arteries. Compared with vehicle-treated vessels, downregulation of miR-221/222 resulted in nearly 40% decrease in neointimal formation at 14 days after angioplasty (Fig. 7C). More excitingly, re-endothelialization was significantly increased by miR-221/222 inhibition (Fig. 7D)
To further verify the opposite effect of miR-221/222 on VSMCs and ECs in the vascular walls, gain-of-function approaches were applied, in which the expression of miR-221/222 in balloon-injured rat carotid arteries was upregulated by local delivery of Ad-miR-221/222 as described in our previous study (9) (Fig. 7B). We found, compared with control adenovirus (Ad-GFP)-treated vessels, upregulation of miR-221/222 resulted in the increased neointimal formation at 14 days after angioplasty (Fig. 7C). However, re-endothelialization was significantly decreased by Ad-miR-221/222 (Fig. 7D)
Discussion
The recent discovery of miRNAs is an important breakthrough in molecular biology. Indeed, miRNAs are involved in almost every aspect of biology and disease [1-6]. Identifying biological functions of an individual miRNA is therefore an important research area in biomedical field. Recent research advances in cancer cells indicate that the biological functions of miRNAs might be cell-specific [7, 8]. If the cell-specific effect of miRNAs also occurs in cardiovascular system, it may have big impact on miRNA studies and on their therapeutic applications in cardiovascular diseases. For example, it is well known that the impaired or delayed growth of ECs, and enhanced growth of VSMCs cause restenosis after angioplasty and stent implantation. In addition, the impaired EC repair and enhanced VSMC growth are also critical cellular events in the pathogenesis of atherosclerosis. Unfortunately, all the current used anti-restenosis and anti-atherosclerosis drugs are nonselective. In addition to inhibiting VSMC proliferation, migration and survival, which is their therapeutic effect, they also suppress EC growth, mobility and survival that results in side effects such as the delayed or impaired re-endothelialization, late thrombosis and restenosis [20-22]. The undesired side effect is obviously the key challenge to both clinical cardiologist and basic vascular biologist, and a critical barrier in interventional cardiology [20-25].
miR-221/222 are highly expressed in both VSMCs and ECs [9, 10]. In VSMCs, miR-221/222 were found to have a pro-proliferative effect and a pro-migratory effect by targeting p27(Kip1) p57(Kip2) [9, 26]. In ECs, an anti-migratory effect was reported by targeting c-kit, transcription 5A, endothelial nitric oxide synthase, and Ets-1 [10, 16, 17, 27, 28]. In the current study, we compared the cellular functions of miR-221/222 in VSMCs with these in ECs. We are excited to find that their biological functions are opposite between VSMCs and ECs as demonstrated in cell proliferation, migration, and apoptosis.
The molecular mechanisms responsible for miR-221/222-mediated opposite cellular effects are currently unclear. In the current study, we identified that p27(Kip1), p57(Kip2), and c-Kit are target genes of miR-221/222 in both VSMCs and ECs as demonstrated by luciferase assay in HEK 293 cells, ECs and VSMCs and the expression assay of these target genes in VSMCs and ECs via gain-of-function and loss-of-function of miR-221/222. However, the expression profiles of these target genes are different between VSMCs and ECs. p27(Kip1), p57(Kip2) are highly expressed in VSMCs, but not in ECs. In contrast, c-Kit was highly expressed in ECs, but not in VSMCs. Moreover, the cellular effects of these target genes might be opposite. For example, p27(Kip1) and p57(Kip2) have an anti-proliferative effect on VSMCs and ECs. In contrast, c-Kit is a pro-proliferative gene in these vascular cells. Thus, different availability of these target gene mRNAs in VSMCs and ECs might be related to, at least in part, the different cellular effects of miR-221/222 in these two cell types. It is well established that the gene expression profiles are different among the different cells. The cell-specific effects of miRNAs might be a common feature of miRNA-mediated biological functions due to the different availability of their target genes. If it were true, the new finding from the current study might have big impact on the whole miRNA studies. The cell specific effects of other miRNAs in cardiovascular system are still an unexplored research area. Our unpublished study on miR-145 has suggested that miR-145 is a VSMC specific miRNA. miR-145 has a strong anti-proliferative effect on VSMCs, but not on ECs. It should be noted that, not only the biological functions of miR-221/222, but also the regulatory mechanisms of their expression are different between VSMCs and ECs. For example, serum and growth factors could increase the expression of miR-221/222 in VSMCs, but decrease their expression in ECs [9, 17].
The different expressions of the target genes in VSMCs and ECs despite the high expression of miR-221/22 in both cells suggests that the basal levels of the target genes such as p27(kip1), p57(kip2) and c-kit might be determined majorly by factors at the transcription level. However, miR-221/222 may play a regulatory role in the expression of these target genes at the post transcription level as demonstrated by miR-221/222 loss-of-function and gain-of-function experiments.
The opposite biological functions of miR-221/222 in VSMCs and ECs may have therapeutic applications in vascular diseases such as restenosis after angioplasty. Indeed, in the current study, we have demonstrated that inhibition of miR-221/222 is able to enhance re-endothelialization. In contrast, the neointimal formation was significantly reduced by inhibition of miR-221/222. Thus, miR-221/222 could be ideal therapeutic targets in patients with atherosclerosis, angioplasty, stent implantation, or artery bypass. On the other hand, the cell specific effect of miR-221/222 also suggests that miR-221/222-based therapy may have unexpected side-effects. For example, anti-221/222 therapy has an inhibitory effect on cancer cell growth. However, its pro-proliferative effect on ECs and angiogenesis may limit its therapeutic effect on cancer.
Research Highlights.
The cellular effects of miR-221/222 are opposite between VSMCs and ECs.
miR-221/222 increases neointimal growth, but decreases re-endothelialization.
The cell-specific effects are related to expression profiles of their target genes.
Acknowledgments
The author's research was supported by National Institutes of Health Grant HL080133, HL095707, and a grant from American Heart Association 09GRNT2250567.
Footnotes
Conflicts of interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113:673–76. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 2.Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, et al. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–21. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 3.Pasquinelli AE, Hunter S, Bracht J. MicroRNAs: a developing story. Curr Opin Genet Dev. 2005;15:200–05. doi: 10.1016/j.gde.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Ambros V. The functions of animal microRNAs. Nature. 2004;431 doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C. Novel functions for small RNA molecules. Curr Opin Mol Ther. 2009;11:541–51. [PMC free article] [PubMed] [Google Scholar]
- 7.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–33. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 8.Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33:1290–97. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-222 and miR-221 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476–87. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poliseno L, Tuccoli A, Mariani L, Evangelista M, Citti L, Woods K, et al. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108:3068–71. doi: 10.1182/blood-2006-01-012369. [DOI] [PubMed] [Google Scholar]
- 11.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, et al. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of microRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–88. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, et al. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105:158–66. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C, Yang J, Jennings LK. Attenuation of neointima formation through the inhibition of DNA repair enzyme PARP-1 in balloon-injured rat carotid artery. Am J Physiol Heart Circ Physiol. 2004;287:H659–66. doi: 10.1152/ajpheart.00162.2004. [DOI] [PubMed] [Google Scholar]
- 14.Kotha J, Zhang C, Longhurst CM, Lu Y, Jacobs J, Cheng Y, et al. Functional relevance of tetraspanin CD9 in vascular smooth muscle cell injury phenotypes: A novel target for the prevention of neointimal hyperplasia. Atherosclerosis. 2009;203:377–86. doi: 10.1016/j.atherosclerosis.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 15.Ashino T, Sudhahar V, Urao N, Oshikawa J, Chen GF, Wang H, et al. Unexpected role of the copper transporter ATP7A in PDGF-induced vascular smooth muscle cell migration. Circ Res. 2010;107:787–799. doi: 10.1161/CIRCRESAHA.110.225334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res. 2007;101:59–68. doi: 10.1161/CIRCRESAHA.107.153916. [DOI] [PubMed] [Google Scholar]
- 17.Dentelli P, Rosso A, Orso F, Olgasi C, Taverna D, Brizzi MF. microRNA-222 controls neovascularization by regulating signal transducer and activator of transcription 5A expression. Arterioscler Thromb Vasc Biol. 2010;30:1562–68. doi: 10.1161/ATVBAHA.110.206201. [DOI] [PubMed] [Google Scholar]
- 18.Lin Y, Liu X, Cheng Y, Yang J, Huo Y, Zhang C. Involvement of microRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. J Biol Chem. 2009;284:7903–13. doi: 10.1074/jbc.M806920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, et al. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. 2009;284:29514–25. doi: 10.1074/jbc.M109.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen JP. Safety and efficacy of the drug-eluting stent: a double-edged sword? South Med J. 2008;101:123–124. doi: 10.1097/01.smj.0000286752.67205.e0. [DOI] [PubMed] [Google Scholar]
- 21.Austin D, Pell JP, Oldroyd KG. Drug-eluting stents: a review of current evidence on clinical effectiveness and late complications. Scott Med J. 2008;53:16–24. doi: 10.1258/RSMSMJ.53.1.16. [DOI] [PubMed] [Google Scholar]
- 22.Flores-Ríos X, Marzoa-Rivas R, Abugattás-de Torres JP, Piñón-Esteban P, Aldama-López G, Salgado-Fernández J, et al. Late thrombosis of paclitaxel-eluting stents: long-term incidence, clinical consequences, and risk factors in a cohort of 604 patients. Am Heart J. 2008;155:648–53. doi: 10.1016/j.ahj.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 23.Agostoni P, Vermeersch P. Bare-metal versus drug-eluting coronary stents. N Engl J Med. 2008;358:2516–17. [PubMed] [Google Scholar]
- 24.Pendyala L, Jabara R, Shinke T, Chronos N, Robinson K, Li J, et al. Pendyala LDrug-eluting stents: present and future. Cardiovasc Hematol Agents Med Chem. 2008;6:105–15. doi: 10.2174/187152508783955051. [DOI] [PubMed] [Google Scholar]
- 25.Saia F, Marzocchi A, Serruys PW. Drug-eluting stents. The third revolution in percutaneous coronary intervention. Ital Heart J. 2005;6:289–03. [PubMed] [Google Scholar]
- 26.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284:3728–38. doi: 10.1074/jbc.M808788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suárez Y, Fernández-Hernando C, Yu J, Gerber SA, Harrison KD, Pober JS, et al. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100:1164–73. doi: 10.1161/01.RES.0000265065.26744.17. [DOI] [PubMed] [Google Scholar]
- 28.Zhu N, Zhang D, Chen S, Liu X, Lin L, Huang X, et al. Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis. 2011;215:286–93. doi: 10.1016/j.atherosclerosis.2010.12.024. [DOI] [PubMed] [Google Scholar]