

Abstract
Biological processes that drive cell growth are exciting targets for cancer therapy. The fibroblast growth factor (FGF) signaling network plays a ubiquitous role in normal cell growth, survival, differentiation, and angiogenesis, but has also been implicated in tumor development. Elucidation of the roles and relationships within the diverse FGF family and of their links to tumor growth and progression will be critical in designing new drug therapies to target FGF receptor (FGFR) pathways. Recent studies have shown that FGF can act synergistically with vascular endothelial growth factor (VEGF) to amplify tumor angiogenesis, highlighting that targeting of both the FGF and VEGF pathways may be more efficient in suppressing tumor growth and angiogenesis than targeting either factor alone. In addition, through inducing tumor cell survival, FGF has the potential to overcome chemotherapy resistance highlighting that chemotherapy may be more effective when used in combination with FGF inhibitor therapy. Furthermore, FGFRs have variable activity in promoting angiogenesis, with the FGFR-1 subgroup being associated with tumor progression and the FGFR-2 subgroup being associated with either early tumor development or decreased tumor progression. This review highlights the growing knowledge of FGFs in tumor cell growth and survival, including an overview of FGF intracellular signaling pathways, the role of FGFs in angiogenesis, patterns of FGF and FGFR expression in various tumor types, and the role of FGFs in tumor progression.
Keywords: Fibroblast growth factors, gene expression regulation, neoplasms, angiogenesis/neovascularization, molecular targets
INTRODUCTION
Fibroblast growth factors (FGFs) and their receptors form part of a unique and diverse signaling system, which plays a key role in a variety of biological processes. Their diversity is largely evolutional, occurring during a two-phase developmental process [1, 2]. In the first phase, during early metazoan evolution, FGF genes increased from two or three to six genes by gene duplication. In the second phase, during the evolution of early vertebrates, the FGF gene family expanded by two genome duplications. This evolutional process, saw the FGF gene family expand to at least 22 different genes encoding related polypeptides [1, 2]. However, human FGF15 has not been described, and human FGF19 is a possible homolog of the mouse FGF15, raising the possibility that human FGF19 may be identical to mouse Fgf15. The 22 FGFs in humans can be classified into several subgroups, based on their evolutionary relationship (Fig. 1). Several human FGF genes are clustered within the genome, such as FGF-4 and FGF-19 which are located on chromosome 11q13, and FGF6 and FGF23 on chromosome 12p13, which suggests that the FGF gene family was generated by gene and chromosomal duplication, and translocation during the evolutionary process [3].
Fig. 1.
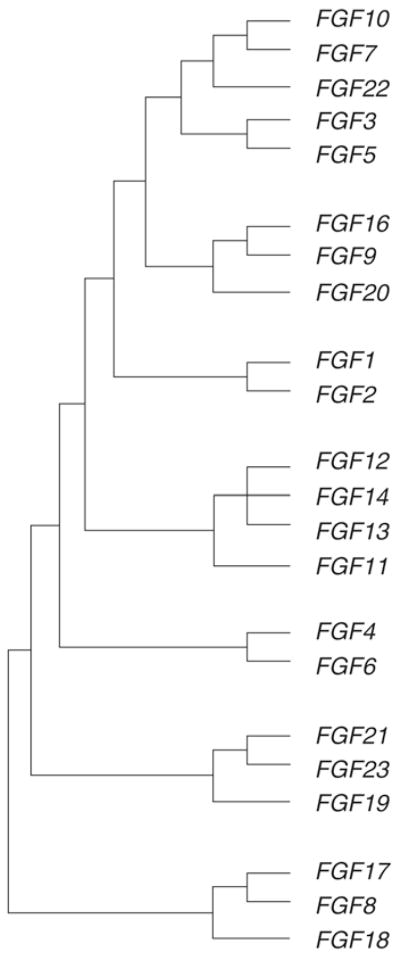
Evolutionary relationships within the human FGF family (reproduced from reference 3).
FGFs interact with a family of distinct, high-affinity tyrosine kinase receptors known as fibroblast growth factors (FGFRs). In contrast to their ligands, only four human FGFRs exist, designated FGFR-1 to −4; however, their specificity is enhanced by a number of processes that give rise to multiple isoforms by alternative initiation, alternative splicing, and C-terminal truncations. Together the FGFs and their receptors form a complex and ubiquitous cellular signaling network that drives many important developmental and physiological processes.
FGFs are widely expressed in both developing and adult tissues and play important roles in a variety of normal and pathological processes, including tissue development, tissue regeneration, angiogenesis, and neoplastic transformation. FGF is considered to be a pleiotropic mitogen, but is also involved in diverse cellular processes including chemotaxis, cell migration, cellular differentiation, and cell survival [2, 4–6]. Although not directly pro-inflammatory, FGFs have been shown to synergistically potentiate inflammatory mediator-induced leukocyte recruitment, at least in part, by enhancing cell adhesion molecule up-regulation [6].
FGF signaling pathways have been implicated in tumor development and progression, and may play a significant role in cancer pathobiology [7]. The effects of increased FGF receptor signaling are wide ranging, involving both tumor cells and the surrounding stroma, including the vasculature, and this dual activity plays an important role in tumorigenesis [8]. The potential of FGFs to promote tumor progression is highly dependent on specific FGFR signaling. FGF-1 (acidic FGF) and FGF-2 (basic FGF), and their receptors promote autocrine and paracrine growth control of malignant tumors [7, 8], while the FGF-2 ligand has been shown to have potent angiogenic activity [9]. FGFs have been shown to increase the motility and invasiveness of a variety of cancers; the FGF signaling network thus makes an attractive anti-neoplastic target across a range of tumor types [8]. FGF-2 is particularly widespread, being expressed in many malignant tumors including melanomas, astrocytomas, and breast, pancreas, non-small lung cell, bladder, head and neck, prostate, and hepatocellular carcinoma (HCC) [9–17]. Prostate cancer is additionally associated with expression of FGF-1, FGF-6, FGF-7, FGF-8, and FGF-9 [8, 18]. In addition, FGF-10 is associated with the development of pulmonary adenomas [19], whereas FGF-3 is overexpressed in non-small cell lung cancer and HCC [20, 21], FGF-4 is overexpressed in Kaposi sarcoma lesions and in testicular cancers[22, 23]. FGF-5 is overexpressed in glioblastoma multiforme and pancreatic cancer [24, 25], FGF-17 is overexpressed in prostate cancer [26, 27], FGF-18 is overexpressed in colorectal cancer [28], and FGF21 is upregulated in HCC [17]. A recent study also suggests that FGF-19 may promote tumor growth in colorectal cancer and hepatocellular carcinoma through its interaction with FGFR4 [29]. In contrast, the expression of FGF-11–16 in malignant tumors has been less extensively investigated.
The FGF System: Ligands, Receptors, and Binding Proteins
FGFRs are cell surface, tyrosine kinase receptors consisting of three extracellular immunoglobulin-like domains—designated IgI, IgII, and IgIII—a single pass transmembrane domain, and an intracellular dimerised tyrosine kinase domain [30]. IgI and IgII are separated by a stretch of acidic residues, sometimes known as the acid box. In contrast to the multiple FGF genes, only four FGFR genes are known to exist; however, receptor diversity is increased because gene expression for FGFR-1 to FGFR-3 can undergo alternative splicing events, thereby generating receptor isoforms with dramatically altered FGF binding specificity [3]. Through the use of splice variants, multiple different receptor types are possible [3], dramatically increasing the selectivity of FGF binding (Fig. 2). Different exon usage produces transcripts that result in the translation of proteins that can be prematurely truncated and lack Ig-like domains or utilize different exons coding regions for the same Ig-like domains. One of the most important mechanisms involves the alternative splicing of the IgIII domain. The IgIII domain of FGFR-1, FGFR-2, and FGFR-3 is comprised of an invariant IgIIIa exon alternatively spliced to either IgIIIb or IgIIIc. The differential expression of IgIIIb and IgIIIc is important in determining FGF signaling specificity. For example, FGF-7 is known to bind to FGFR-2(IIIb), but not to the closely related FGFR-2(IIIc) thus explaining the high level of specificity of this ligand. The ligand specificities of the FGFR family are shown in Table 1.
Fig. 2.
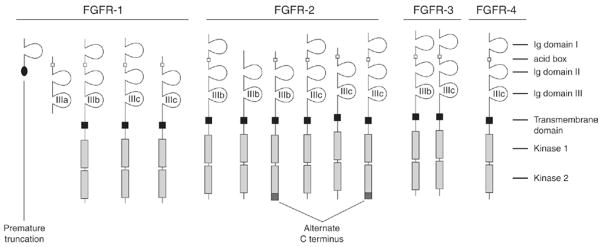
Representative view of FGF transmembrane receptors, showing additional diversity created by use of splice variants (reproduced from reference 30). The alternate C-terminus variants are designated C1 and C3. The open square in the extracellular domain represents the acid box region.
Table 1.
Ligand Specificities of the FGFR Familya
| Gene | Receptor | Specific forb |
|---|---|---|
| FGFR-1 | FGFR-1b | FGF-1, 2, 3, and 10 |
| FGFR-1c | FGF-1, 2, 4, 5, and 6 | |
| FGFR-2 | FGFR-2b | FGF-1, 3, 7, and 10 |
| FGFR-2c | FGF-1, 2, 4, 6, and 9 | |
| FGFR-3 | FGFR-3b | FGF-1 and 9 |
| FGFR-3c | FGF-1, 2, 4, 8, and 9 | |
| FGFR-4 | FGFR-4 | FGF-1, 2, 4, 6, 8, and 9 |
Adapted from reference 1. Alternate splicing has increased the functional diversity of FGFRs. Alternatively spliced forms of immunoglobulin (Ig) domains III (the IIIb and IIIc isoforms of FGFRs are shown
FGF1–10 only shown.
FGFs are small polypeptide molecules that reside mainly in the extracellular matrix where they form complexes with heparin and heparan sulfate proteoglycans. Bound FGFs can act either directly on target cells or be taken up intracellularly upon their mobilization from the matrix by degradative enzymes such as proteases. The discovery of FGF-binding protein, a 17 kDa secreted polypeptide, which can act as a carrier protein for FGFs within the extracellular matrix, points to an additional mechanism for the interaction of these growth factors with their specific cellular receptors. There is controversy regarding the exact manner by which this occurs, although it is thought to involve the induction of receptor dimerization and be heparin-dependent [30]. The release of FGF from the extracellular matrix through the action of FGF-binding protein is one of the critical steps in rendering FGF bioavailable. FGF-binding protein is upregulated in several tumors including squamous cell, pancreatic, colon, and breast, and is associated with early stages of tumor formation, where angiogenesis plays a critical role [31]. Furthermore, the presence of FGF-binding protein can be rate-limiting for tumor growth and serves as an angiogenic switch molecule [32].
Signal Transduction
FGF receptors, like other receptor tyrosine kinases, transmit extracellular signals to various cytoplasmic signal transduction pathways through the process of tyrosine phosphorylation (Fig. 3) [33]. Activation of FGFRs by tyrosine phosphorylation leads to signal transduction through multiple pathways, including phospholipase Cγ (PLCγ) [34], phosphatidylinositol 3-kinase (PI3K) [34], mitogen-activated protein kinases (MAPK) [35], protein kinase C (PKC), and signal transducers and activators of transcription (STATs) [36]. The biological outcomes of FGF/FGFR activation are dependent on a complex network of signaling and transcriptional events regulated by multiple factors. The specific biological response modulated, be it proliferative, apoptotic, or migratory, depends entirely on the interplay between these factors. These include the presence or abundance of a specific signal transduction molecule, the existence of specific feedback loops, crosstalk with other signaling networks, and the availability of target genes to be activated or repressed. Thus the MAPK signaling cascade is implicated in cell growth and differentiation, the PI3K/Akt signaling cascade is involved in cell survival and cell fate determination, while the PI3K and PKC signaling cascades have a function in the control of cell polarity.
Fig. 3.
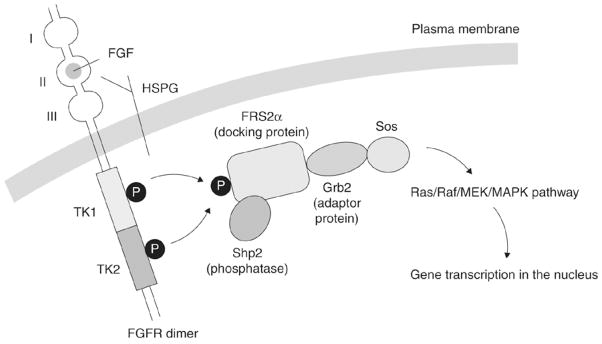
Intracellular signaling pathways for the FGF/FGFR complex (adapted from reference 33).
Several feedback inhibitors of FGF signaling have now been identified and include members of the Sprouty (Spry) family and similar expression to FGF (Sef) [37, 38]. Spry inhibits FGF signaling by interfering with extracellular signal-regulated kinases (ERK) activation either by binding to and sequestering growth factor receptor-bound protein 2 (Grb2) or by binding to Raf1 and preventing Raf1 activation [39]. Spry1 and Spry2 are downregulated in several tumor types including breast [40], prostate [41–43], liver [44], and lung [45]. This downregulation seems to correlate with an aggressive tumor phenotype. The mechanisms for Spry regulation appear to be tumor-type specific and include both epigenetic and non-epigenetic mechanisms. Sef is a transmembrane protein with homology to the interleukin (IL)-17 receptor family. Sef binds to FGFR-1 and FGFR-2 and prevents activation of the receptor tyrosine kinase [46]. Sef is also downregulated in several tumor types of epithelial origin including breast, thyroid, prostate, and ovarian cancers [47–49]. The downregulation of feedback inhibitors of FGF signaling in tumor cells, coupled with an increased expression of FGF ligands, will lead to enhanced tumor growth, progression, and metastasis.
Roles of FGFs indicated by knockout mice
Several FGF knockout mice have been generated to investigate the function of FGF genes (reviewed in reference [50]). Phenotypes range from early embryonic lethality to subtle phenotypic alterations in adult mice. FGF knockout mice, such as FGF 4, 8, 9, 10, 15, 16, and 17, that die in the embryonic or early postnatal stages of development indicate crucial roles for these genes in various development processes. By contrast, knockout mice that survive with subtle phenotypic alterations, such as FGF 2, 3, 5, 6, 7, 12, 16, 17, 21, may have functions that are redundant in mice. Moreover, FGFR-1 knockout mice die prior to gastrulation [51], whereas chimeric mice for FGFR-1 deficiency that bypass the gastrulation block exhibit multiple limb and neuronal malformations [51]. Similarly, FGFR-2 knockout mice die in utero due to an inability to form the chorioallantoic fusion and/or portions of the placenta [52]. By contrast, FGFR-3 knockout mice exhibit increased growth of their long bones in association with kyphosis and pulmonary dysfunction [53], FGFR4 knockout mice are generally normal and exhibit slightly attenuated growth [54], whereas double mutants that are devoid of FGFR-3 and FGFR-4 exhibit marked defects in their pulmonary alveoli [54].
FGF and Mechanisms of Tumorigenesis
The FGF receptor system is associated with multiple biological activities including cellular proliferation, differentiation, invasiveness, and motility that demonstrate the potential to initiate and promote tumorigenesis. Recent research into the oncogenic nature of this signaling network is increasing our understanding of the specific cellular processes at work and implicates an important role for aberrant FGF signaling in the natural history of some common human cancers [55].
The FGFR-1-mediated MAPK kinase pathway is an important inducer of cell differentiation and growth and has been implicated in tumor cell proliferation [55]. Activated FGFR-1 can couple to Ras through two adaptor proteins, fibroblast growth factor receptor substrate 2 (FRS2), which acts as a docking protein, and src-homology-2-containing protein (Shc). However, the involvement of these pathways varies, with both adaptors being activated in the presence of proliferating cells, but only Shc activation occurring when cells are differentiating [56]. Indeed, it has been demonstrated that FGF-stimulated murine endothelial cells are capable of sustaining elevated MAPK levels for considerable periods of time in both proliferating and differentiating cells [56]. However, their differing response to treatment with a mitogen-activated protein kinase or extracellular signal-regulated kinase (MEK) inhibitor suggests that only for proliferating cells is sustained MAPK elevation a strict requirement. In contrast, Src kinase activity is a prerequisite for cellular differentiation. These data indicate that the Ras pathway is routed according to the cellular function required, with blockage of the Shc route attenuating differentiation.
Tumor Suppression
Evidence for a tumor suppressive role of FGFR was demonstrated in mice lacking epithelial FGFR-2b. These mice exhibited heightened sensitivity to chemical carcinogenic insult, displayed several oncogenic Ha-Ras mutations, and developed papillomas and squamous cell carcinomas [57]. These changes occurred in conjunction with an epidermal pro-inflammatory profile and decreased expression of Serpin a3b, a potential tumor suppressor. In a study of the effects of FGFR-1-IIIb expression in human pancreatic cancer mice models, FGFR-1-IIIb expression was shown to inhibit single-cell movement and in vitro invasion of pancreatic tissue as well as in vivo tumor formation and growth in nude mice [58]. Immunohistochemical analysis of the xenograft tumors revealed reduced Ki-67 labeling and a lower amount of tumor necrosis in FGFR-1-IIIb-expressing tumors. In contrast to FGFR-1-IIIc, FGFR-1-IIIb inhibited cell proliferation in this study, thus providing the first reported finding of a naturally occurring FGFR-1 variant that inhibits the growth of epithelial cell types. There is also some suggestion that a switch from FGFR-1-IIIc to -IIIb expression acts as progression from non-malignancy to invasive tumor, however, this is yet to be proven.
Cell Survival
Tumor cell survival pathways are an adaptive mechanism by which the tumor escapes the body’s natural defenses, and are responsible, in part, for the development of drug resistance. In small cell lung cancer (SCLC) FGF-2 increases the expression of antiapoptotic proteins, XIAP and Bcl-X(L), and triggers chemoresistance. These effects are mediated through the formation of a specific multiprotein complex comprising B-Raf, PKCγ and S6K2, S6K1, Raf-1, and other PKC isoforms [59]. There is also some evidence to suggest that FGF-2 is a potent stimulator of breast cancer cell survival as it counteracts the apoptotic activity of the C2 ceramide analog and various chemotherapeutic agents, such as 5-fluorouracil, camptothecin, and etoposide, and has been found to be a predictor of poor prognosis for node-negative patients in a study of 111 patients with primary breast tumors [60, 61]. The antiapoptotic effect of FGF-2 is thought to be mediated through nuclear factor-κB activation induced via interaction between Akt and IκB kinase-beta in breast cancer cells [60]. However, paradoxically, FGF-2 has also been found to inhibit proliferation and promote programmed cell death/apoptosis in a number of human breast cancer cell lines, including MCF-7 and MB-134 [62–65]. Overexpression of FGF-2 in breast cancer cells has been linked to good prognostic indicators and better patient outcomes in a large study of 1307 patients with primary breast cancers [66], while other research in breast cancer cell lines has linked the development of a less malignant FGF-2-associated breast cancer phenotype to the effects of FGF-2 in reducing cell motility and invasion and in inducing a more differentiated phenotype [67, 68]. This process has been attributed to the downregulation of Bcl-2 and to the loss of laminin 5 expression, but no complete explanation of the uncharacteristic and conflicting activities of FGF-2 in breast cancer has yet been found [64, 69]. It may well be that differential expression of FGFR isoforms within breast cancer cells could result in either growth promoting or growth inhibitory effects depending on the individual isoforms expressed, which could explain this paradoxical effect. In experimental models, FGF-2, and to a lesser extent, FGF-1, have been shown to be involved in chemoresistance to drugs with diverse structures and mechanisms of action, including paclitaxel, doxorubicin, and 5-fluorouracil. This effect is not associated with reduced levels of drug in the cell or altered cell proliferation [70]. Elevated FGF-2 expression has also been shown to be a strong indicator of paclitaxel resistance in tumors from patients with bladder, breast, head and neck, ovarian, and prostate cancers [71].
FGF Signaling and Tumor Development
Overexpression of FGFs leads to progression, invasion, and metastasis in a variety of human tumors. However, in general, while FGF expression is associated with tumor progression (although uncharacteristically not always in breast cancer as previously discussed), FGFR expression is more selective. FGFR-1 expression is associated with tumorigenesis while FGFR-2 expression is associated with decreased tumor progression in some tumors, such as astrocytomas, bladder cancer, prostate cancer, and thyroid carcinomas [72–75]. In other tumor types, such as skin carcinoma and oral carcinomas, FGFR-2 expression has been found to be associated with early tumor development [57, 76]. However, in one study of human mammary epithelial cells, aberrant expression of alternatively spliced FGFR-2 was related to tumor progression [77]. FGFR-3 can also be expressed as two different splice isoforms—FGFR-3-IIIb and –IIIc. However, there is no apparent link of either of these isoforms with cellular responses, which appear to be associated with the cell type or its differentiation status [78]. In addition, somatic mutations in FGFR-3 have also been linked to bladder cancer, cervical cancer, and multiple myeloma [78].
FGFR expression in bladder cancers is particularly well characterized and some intriguing aberrations in FGF signaling pathways have been observed, indicating that FGFRs play a significant role in bladder cancer development [79]. FGFR-2 has been demonstrated to have tumor suppressor properties and loss of FGFR-2-IIIb expression is associated with decreased tumor formation in vivo, and with poor prognosis in bladder carcinoma [80–82]. A high incidence of FGFR-3 activating point mutations in exons 7, 10, and 15 of the FGFR-3 gene has also been observed in bladder carcinomas (approximately 40%), particularly those of the epithelium, with a number of different missense mutations identified [83]. The most common mutations found in bladder cancers, S249C and Y375C, represent highly activated forms of the receptor, generating an additional cysteine residue which results in ligand-independent dimerization of the receptor. Antibodies targeted against FGFR-3-IIIc have demonstrated anti-mitogenic effects [84]. FGFR-3 has also been found to be overexpressed in some invasive bladder carcinomas [85].
FGF expression may also have a prognostic value. Detection of high levels of secreted FGF-2 at diagnosis has been linked to poor outcome in a study of patients with SCLC and bladder carcinomas; however, the levels of FGF-2 were not correlated with age, sex, performance status, or disease stage. High levels of FGF-2 were possibly a reflection of active angiogenesis and rapid tumor growth; patients with extensive disease and high FGF-2 were found to have a particularly poor prognosis [79, 86]. FGFR-1 expression and immunoreactivity are elevated in a number of human brain tumors including gliomas, meningiomas, and astrocytomas, possibly reflecting the prominent neovascularization observed in cerebral tumor growth [87–89]. However, in tissue samples from patients with glioblastoma multiforme, FGFR-1 expression was found to be confined to tumor cells, suggesting a direct effect of FGF on tumor growth [87]. The relative lack of FGFR-1 expression in endothelial cells from these patients suggests that angiogenesis occurs in a FGF-independent manner in this tumor type. The FGF-2/FGFR-1 axis in autonomous glioma was also found to promote cell growth and malignant progression [88, 89].
FGF signaling is involved in a diverse range of biological functions in the liver, including homeostasis, and tissue regeneration, as well as tumor development [90]. The mRNAs of all four FGFRs have been reported in adult liver [91, 92]. Expression of FGF-1, FGF-2, and FGFR-1 mRNA is elevated in the livers of patients with HCC as are their activators FGF-1 and FGF-2. These results suggest that FGF may play an important role in the development and progression of HCC via an autocrine mechanism [93]. In cooperation with an initiator, sustained FGFR-1 activity in hepatocytes is a strong promoter of HCC by initially driving cell proliferation and subsequently enhancing neoangiogenesis at late stages of tumor progression [94].
Multiple FGFs are expressed either as autocrine or paracrine growth factors in prostate cancer tissue. High levels of expression of FGF-2 are present in metastatic prostate cancer cells, and patients with prostate cancer have elevated levels of serum FGF-2 [95]. Furthermore, FGF-2 concentrations are almost 2.5-fold higher in clinically localized cancer tissue compared with normal prostate tissue [96]. Increased expression of FGF-2 is associated with transformation and progressive loss of differentiation [97]. More than 80% of prostate cancers express FGF-1 in the cancer cells and that strong expression is correlated with increased Gleason score [97]. FGFR-1 is expressed in approximately 20% of moderately differentiated cancers and 40% of poorly differentiated clinically localized cancers based on immunohistochemistry and Western blotting of prostate cancer extracts, but is not detected in well-differentiated cancers [98]. In oral squamous cell carcinomas, evidence suggests the expression of FGF-2 and FGFR-1 in fibroblasts at the invasive front indicating a potential for interaction of the cancer cells with the host stroma. FGF-2 has been shown to enhance the ability of host fibroblasts to produce a variety of growth factors including hepatocyte growth factor, transforming growth factor-beta, and matrix metalloproteinase, thus providing a possible explanation of their increased invasiveness [99].
At least several FGFs (FGF-1, FGF-2, FGF-5, keratinocyte growth factor [KGF]) are overexpressed in pancreatic ductal adenocarcinoma (PDAC), a cancer that has a very poor prognosis [13, 25, 100, 101]. In addition, the 2-Ig form of FGFR-1, isoforms FGFR-1-IIIb and -IIIc, KGFR and FGFR-2 are overexpressed in this cancer [101–102]. The important role in PDAC of the aberrant FGF-FGFR autocrine and paracrine pathways that are activated by these overexpressed ligands and receptors is evidenced by the observation that blocking FGFR signaling by dominant negative strategies markedly attenuates in vitro and in vivo growth of pancreatic cancer cells [103, 104], and that overexpression of these FGFRs in pancreatic cells leads to enhanced activation of mitogenic signaling pathways and promotion of in vivo tumorigenicity [101, 102]. In addition, correlative studies have shown that increased FGF-2 expression is associated with decreased patient survival in PDAC [13], and that enhanced expression of KGF and KGFR (FGFR2IIIb) is associated with venous invasion by the tumor, enhanced VEGF-A expression, and poor prognosis [105].
Angiogenesis is also increasingly seen as relevant to the study of hematological malignancies. In multiple myeloma, the progression from undifferentiated gammopathy to active disease is marked by an increase in both bone marrow microvascular density and plasma levels of FGF-2, while increased levels of angiogenesis are also seen in acute lymphocytic leukemia, chronic lymphocytic leukemia, acute myelogenous leukemia chronic myelogenous leukemia, myelodysplastic syndrome, and non-Hodgkin’s lymphoma [106].
Deregulation of the FGF/FGFR system appears to be part of the development of hematopoietic malignancies, and current understanding of this area is growing. FGF signaling has been shown to be antimitogenic and apoptotic in multiple myeloma [107]. Evidence exists of a paracrine interaction between myeloma cells and the bone marrow through mutual stimulation of FGF-2 and IL-6, while IL-6 has been shown to interact with the FGFR-3 signaling pathway [108, 109]. FGFR-3 is known to be overexpressed in t(4;14) multiple myeloma and to be oncogenic when targeted by FGF ligands in the bone marrow microenvironment. Recent research has also shown that FGFR-3 may play a Ras-like role in tumorigenesis, perhaps through the down-stream activation of macrophage inflammatory protein (MIP)-1alpha [110, 111]. The t(4;14) translocation also involves the multiple myeloma SET domain (MMSET), which is universally overexpressed in t(4;14) multiple myeloma, whereas FGFR-3 expression is lost in one third of cases. Thus, the relative impact of each gene set is unclear [112]. The lack of correlation between plasma FGF-2 levels and bone marrow vascularity indicates the presence of additional growth factors too.
Role of FGF and FGFR in Tumor-Stroma Interactions
Normal homeostasis of many organs is dependent upon reciprocal communication between the epithelial and stromal compartments. Signals from the stroma to the epithelium contribute to this homeostasis. FGF7 and FGF10 are stromal-derived FGFs in the prostate that signal to FGFR2IIIb in the epithelium. FGF9 produced by the epithelium signals to the some stromal cell types but not others [113]. These results suggest that epithelial control of the stroma may be one determinant as to whether prostate tumors progress to malignancy or remain dormant. FGF9, as well other FGFs (FGF2 and FGF17), have been reported to be expressed in the stroma. Thus bidirectional signaling is mediated by multiple FGFs and in the case of FGF9, the response is likely dictated by the expression of FGFR3 which is expressed exclusively in the stromal compartment [8]. Studies in a melanoma model of progression and metastasis indicate that FGF-2 induces VEGF-A-dependent angiogenesis in the stroma surrounding nascent tumors which supports increased tumor growth and metastasis [114]. Additional studies indicate that TGFβ may also play a role in tumor stroma interactions through TβRII/Smad3-dependent upregulation of FGF2 resulting in increased tumor mass and vascularity [115]. Therefore, multiple FGFs play roles in tumor-stroma interactions which determine the rate of tumor growth and extent of metastasis.
FGF Signaling in Epithelial-Mesenchymal Transition
Epithelial-Mesenchymal Transition (EMT), a process where epithelial cells acquire a motile, invasive mesenchymal-like phenotype, plays a critical role in cancer progression and metastasis. Epithelial cell characteristics are altered during EMT, including changes in the expression of extracellular matrix proteins, proteins involved in cell adhesion and cell-cell interactions and transcription factors [116]. Key among these changes is the upregulation of the transcriptional repressor Slug and its target E-cadherin. FGF1 induces Slug expression in an in vitro model of EMT using the NBT-II bladder carcinoma cell line, in which desmosomes dissociate, and cells begin to spread and migrate [117]. Furthermore, a down stream effector of the FGFR and PI3-kinase pathway, P70 S6 kinase mediates increased expression of Slug and decreased expression of E-cadherin [118]. During EMT when E-cadherin expression is decreased, there is a concomitant increase in N-cadherin expression [118, 119]. Interestingly, N-cadherin and FGF2 act synergistically in breast cancer cells to increase cell invasion and migration, and to enhance the expression of matrix metalloproteinase 9. N-cadherin prevents ligand mediated down regulation of FGFR1, leading to increased receptor signaling, sustained ERK activation and suggests a mechanism whereby the FGF signaling system participates in EMT and establishment of the metastatic phenotype.
Role of FGF Signaling in Tumor Angiogenesis
Angiogenesis, or neovascularization, plays a key role in the growth and metastasis of tumors. The local, uncontrolled release of angiogenic growth factors is responsible for the uncontrolled endothelial cell proliferation that takes place during tumor neovascularization. FGFs are among the earliest described angiogenic factors [120, 121] and are known to be involved in stimulating the proliferation, migration, and differentiation of vascular cells. Experimental evidence suggests that FGFs and their receptors can induce a “pro-angiogenic” phenotype to create a favorable environment for vasculature growth. This is achieved through modulation of endothelial cell proliferation and migration, the production of proteases, promotion of integrin and cadherin receptor expression, and communication across intercellular gap junctions [122].
FGF has a direct effect on tumor angiogenesis by promoting cell proliferation in FGFR-expressing endothelial cells. FGFR-1 is the main FGFR to be expressed on endothelial cells, although FGFR-2 is also present in small amounts [122–124]. There has been some suggestion that the role of FGFR-2 on endothelial cells is limited to cell motility, however, this has not been proven [122]. The expression of FGFR-3 or FGFR-4 has not been reported in endothelium. There is a suggestion that FGF-binding protein can serve as an angiogenic switch for different tumor cells [32]. In mouse models of squamous cell and colon carcinomas, the growth and angiogenesis of human xenograft tumors was decreased linearly with the reduction of FGF-binding protein.
FGFs and other proangiogenic factors have also been implicated in the emerging phenomenon of resistance to vascular endothelial growth factor receptor-2 (VEGFR-2) inhibition. Phenotypic resistance to VEGFR-2 blockade in late-stage tumors has been demonstrated in vivo in mouse models of pancreatic islet cell carcinoma, as tumors regrew following an initial period of growth suppression. This loss of activity has been linked to reactivation of tumor angiogenesis. FGF inhibition by adenoviral mediated expression of soluble FGFR-2 in this model was found to re-establish tumor inhibition, producing a significant reduction in tumor burden compared with the anti-VEGFR-2 treatment alone [125].
Cross Talk
Individual tumors produce a variety of angiogenic factors, whose relative production can change over time. Evidence suggests that expression of pro- and anti-angiogenic factors changes with tumor progression, and that tumor cells express distinct levels of angiogenic factors at different stages of tumor progression [126]. VEGF is a prominent regulator of angiogenesis. An intimate cross talk is thought to exist among FGF-2 and the different members of the VEGF family during angiogenesis. It is thought that FGF-2 and VEGF synergistically stimulate vascularization, but with distinct effects on vessel functionality and tumor survival. VEGF is thought to appear earlier in the initiation of angiogenesis than FGF, although FGF shows both early and late phase angiogenic stimulation (Fig. 4) [127]. It is known that simultaneous expression of FGF-2 and VEGF results in fast-growing tumor xenografts in nude mice with high vessel density, patency, and permeability, but that inhibition of FGF-2 production decreases vessel density [128].
Fig. 4.
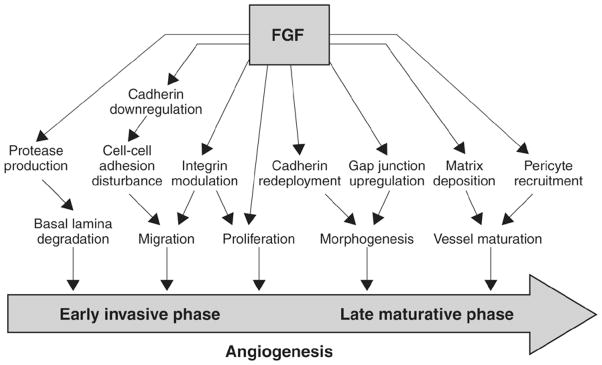
FGFR signal transduction contributes to both early and late phase tumor angiogenesis (adapted from reference 127).
FGF-2 may also induce neovascularization indirectly by activation of the VEGF/VEGFR pathway. Surprisingly, when VEGFR-2 antagonists were administered to VEGF and FGF-2 mouse models of angiogenesis, both VEGF- and FGF-2-induced angiogenesis were inhibited in vitro and in vivo [129]. Expression of dominant-negative FGFR-1 or FGFR-2 in glioma cells also results in a decrease in tumor vascularization paralleled by VEGF downregulation [130]. In mouse models, both endogenous and exogenous FGF-2 modulated VEGF expression in endothelial cells, and in the mouse cornea, systemic administration of anti-VEGF-A neutralizing antibodies dramatically reduced FGF-2-induced vascularization [131]. VEGFR-1-blocking antibodies or the expression of a dominant-negative VEGFR-1 have also been found to result in a significant reduction of FGF-2-induced cell extensions and capillary morphogenesis [132], while FGF-2 has been shown to upregulate the expression of both FGFRs and VEGFRs in endothelial cells [133].
Further evidence of cross talk is provided through studies of the indirect effects of FGF-2 on vascular smooth muscle cell (VSMC) migration, which also induces the VEGF pathway. Neuropilin-1 (NRP-1) is a co-receptor for VEGF, enhancing VEGF interactions with VEGFRs. FGF-2 increases NRP-1 expression in human VSMCs that in turn enhance the effect of VEGF on cell migration and promote neovascularization [134]. In T47D breast cancer cells, FGF-2 has demonstrated interactions with VEGF pathways by augmenting hypoxia inducible factor (HIF)-1-alpha expression and subsequent VEGF release [135]. FGF-2 also promotes lymphangiogenesis by activating the Akt/mammalian target of rapamycin (mTOR)/p70S6 kinase pathway in lymphatic endothelial cells [136]. In addition to its VEGF modulatory role, FGF-2 acts as a sensitizer for endothelial cells, enhancing their ability to respond to platelet-derived growth factor (PDGF)-BB, which feeds back to VSMCs to enhance their responses to FGF-2 stimulation [137]. The underlying mechanisms of this reciprocal interaction involve upregulation of PDGFR-α and -β expression at the transcriptional level in endothelial cells by FGF-2 and upregulation of FGFR-1 expression in VSMCs by PDGF-BB [137]. The biological consequence of such a reciprocal interaction in tumors is manifested by hyper-neovascularization and a disorganized, primitive tumor vasculature, which is poorly coated with pericytes and VSMCs. These alterations of tumor blood vessels lead to an accelerated tumor growth rate and metastasis [137].
FGF Signaling and genetic diseases
Disrupted FGF signaling plays a role in several human inherited diseases. For example, germline activating mutations of FGFRs 1–3 are associated with a range of skeletal disorders, including the common forms of dwarfism [138, 139]. Mutations in FGF-1 and -8 are associated with hypogonadism [140, 141]. In addition, overexpression, or mutations in FGF-23 are associated with hypophosphatemic diseases such as hypophosphatemic rickets, tumor-induced osteomalacia and McCune Albright syndrome [142]. Understanding the underlying role of FGFs/FGFRs in these diseases will ultimately lead to better treatment.
CONCLUSION
The FGF signaling network has an important role in tumor development and progression through its effects on cancer cell proliferation, migration, and survival and its pro-angiogenic activity, and therefore presents a significant therapeutic opportunity. FGF can act synergistically with VEGF to amplify tumor angiogenesis by direct regulation of VEGF expression, via induction of VEGFR, or indirectly by promoting HIF-1. Thus, targeting both FGF and VEGF pathways may be more efficient in suppressing tumor growth and angiogenesis than targeting either factor alone. Furthermore, FGF has the potential to overcome chemotherapy resistance by inducing tumor cell survival pathways. This may have important clinical applications in that chemotherapy may be more effective when combined with FGF inhibitor therapy. In addition, FGFRs demonstrate variable activity in promoting tumorigenesis. FGFR-1 is associated with tumor progression, whereas FGFR-2 is associated with either early tumor development or decreased tumor progression; however, mutations in FGFR-2 have the ability to drive tumor progression in some tumor types. These activities may have important implications for targeting specific tumor types that overexpress FGFRs.
FUTURE DIRECTIONS
Certain cancers appear to have usurped, through a variety of mechanisms, various components of the FGF receptor signaling network to promote tumor growth and metastasis. In such cancers, it is likely that inhibiting relevant FGF receptors and their downstream signaling pathways will allow for targeting these cancers in a highly specific manner. Such an approach may include sequestering FGFs with soluble receptors or binding proteins, targeting the cell-surface receptors with high specific monoclonal antibodies and/or small molecule inhibitors, abrogating the function of co-receptors that facilitate ligand-receptor interactions, blocking signaling pathways that are downstream of FGF receptors, and activating mechanisms that inhibit FGF receptor signaling. In conjunction with proteomic and genomic approaches to delineate tumor markers related to FGF receptors and ligands, such targeted therapies will allow for the design of individualized protocols that will be more effective than current therapeutic regimens, while potentially minimizing any associated side effects. Targeted therapies could also be combined with conventional chemotherapy and or radiotherapy, immune therapy, or additional anti-angiogenic therapy, in order to maximally prolong patient survival, eliminate residual cancer cells, prevent cancer cell metastasis, or induce a state of tumor dormancy.
As we improve our understanding of the pathobiology of human cancers and of the connection between microRNAs and other non-coding RNAs and their relationships to classical FGF receptor signaling cascades, it is possible to imagine that targeting oncogenic microRNAs in conjunction with targeting FGFs and their receptors could lead to novel therapeutic strategies. Moreover, it is conceivable that advances in nanotechnology could lead to improved drug delivery systems that will eliminate systemic side effects. Further studies are necessary to explore these exciting possibilities.
Acknowledgments
This study was supported in part by the US Public Health Service grants CA-750509 and CA-101306 to M. Korc, and DK-73871 and RR-15555 to Robert Friesel. Editorial support was provided by M. English, PhD, of PAREXEL, and was funded by Bristol-Myers Squibb.
ABBREVIATIONS
- ERK
Extracellular signal-regulated kinases
- FGF
Fibroblast growth factor
- FGFR
Fibroblast growth factor receptor
- FRS2
Fibroblast growth factor receptor substrate 2
- Grb2
Growth factor receptor-bound protein 2
- HCC
Hepatocellular carcinoma
- HIF
Hypoxia inducible factor
- HSPG
Heparin sulfate proteoglycan
- IL
Interleukin
- KGF
Keratinocyte growth factor
- MAPK
Mitogen-activated protein kinase
- MEK
Mitogen-activated protein kinase or extracellular signal-regulated kinase
- MIP
Macrophage inflammatory protein
- MMSET
Multiple myeloma SET domain
- mTOR
Mammalian tartget of rapamycin
- PDAC
Pancreatic ductal adenocarcinoma
- PDGF
Platelet-derived growth factor
- PI3K
Phosphatidylinositol 3-kinase
- PLCγ
Phospholipase Cγ
- SCLC
Small cell lung cancer
- Sef
Similar expression to FGF
- Shc
Src-homology-2-containing protein
- Spry
Sprouty
- STATs
Signal transducers and activators of transcription
- TK
Tyrosine kinase
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor
- VSMC
Vascular smooth muscle cell
References
- 1.Itoh N, Ornitz DM. Evolution of the FGF and FGFR Gene Families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Katoh M, Katoh M. FGF Signaling Network in the Gastrointestinal Tract (Review) Int J Oncol. 2006;29:163–168. [PubMed] [Google Scholar]
- 3.Ornitz DM, Itoh N. Fibroblast Growth Factors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. Reviews 3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolitho C, Xu W, Zoellner H. Negative Feedback for Endothelial Apoptosis: A Potential Physiological Role for Fibroblast Growth Factor. J Vasc Res. 2008;45:193–204. doi: 10.1159/000111072. [DOI] [PubMed] [Google Scholar]
- 5.Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous Expression of N-Cadherin in Breast Cancer Cells Induces Cell Migration, Invasion, and Metastasis. J Cell Biol. 2000;148:779–790. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zittermann SI, Issekutz AC. Basic Fibroblast Growth Factor (BFGF, FGF-2) Potentiates Leukocyte Recruitment to Inflammation by Enhancing Endothelial Adhesion Molecule Expression. Am J Pathol. 2006;168:835–846. doi: 10.2353/ajpath.2006.050479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi JA, Igarashi K, Oda K, Kikuchi H, Hatanaka M. Correlation of Basic Fibroblast Growth Factor Expression Levels With the Degree of Malignancy and Vascularity in Human Gliomas. J Neurosurg. 1992;76:792–798. doi: 10.3171/jns.1992.76.5.0792. [DOI] [PubMed] [Google Scholar]
- 8.Kwabi-Addo B, Ozen M, Ittmann M. The Role of Fibroblast Growth Factors and Their Receptors in Prostate Cancer. Endocr Relat Cancer. 2004;11:709–724. doi: 10.1677/erc.1.00535. [DOI] [PubMed] [Google Scholar]
- 9.Lindner V, Majack RA, Reidy MA. Basic Fibroblast Growth Factor Stimulates Endothelial Regrowth and Proliferation in Denuded Arteries. J Clin Invest. 1990;85:2004–2008. doi: 10.1172/JCI114665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halaban R. Growth Factors and Melanomas. Semin Oncol. 1996;23:673–681. [PubMed] [Google Scholar]
- 11.Bian XW, Du LL, Shi JQ, Cheng YS, Liu FX. Correlation of BFGF, FGFR-1 and VEGF Expression With Vascularity and Malignancy of Human Astrocytomas. Anal Quant Cytol Histol. 2000;22:267–274. [PubMed] [Google Scholar]
- 12.Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, Moghaddam A, Whitehouse R, Bicknell R, Harris AL. Expression of the Angiogenic Factors Vascular Endothelial Cell Growth Factor, Acidic and Basic Fibroblast Growth Factor, Tumor Growth Factor Beta-1, Platelet-Derived Endothelial Cell Growth Factor, Placenta Growth Factor, and Pleiotrophin in Human Primary Breast Cancer and its Relation to Angiogenesis. Cancer Res. 1997;57:963–969. [PubMed] [Google Scholar]
- 13.Yamanaka Y, Friess H, Buchler M, Beger HG, Uchida E, Onda M, Kobrin MS, Korc M. Overexpression of Acidic and Basic Fibroblast Growth Factors in Human Pancreatic Cancer Correlates With Advanced Tumor Stage. Cancer Res. 1993;53:5289–5296. [PubMed] [Google Scholar]
- 14.Berger W, Setinek U, Mohr T, Kindas-Mugge I, Vetterlein M, Dekan G, Eckersberger F, Caldas C, Micksche M. Evidence for a Role of FGF-2 and FGF Receptors in the Proliferation of Non-Small Cell Lung Cancer Cells. Int J Cancer. 1999;83:415–423. doi: 10.1002/(sici)1097-0215(19991029)83:3<415::aid-ijc19>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 15.Gazzaniga P, Gandini O, Gradilone A, Silvestri I, Giuliani L, Magnanti M, Gallucci M, Saccani G, Frati L, Agliano AM. Detection of Basic Fibroblast Growth Factor mRNA in Urinary Bladder Cancer: Correlation With Local Relapses. Int J Oncol. 1999;14:1123–1127. doi: 10.3892/ijo.14.6.1123. [DOI] [PubMed] [Google Scholar]
- 16.Dellacono FR, Spiro J, Eisma R, Kreutzer D. Expression of Basic Fibroblast Growth Factor and its Receptors by Head and Neck Squamous Carcinoma Tumor and Vascular Endothelial Cells. Am J Surg. 1997;174:540–544. doi: 10.1016/s0002-9610(97)00169-4. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, Yu C, Jin C, Yang C, Xie R, Cao D, Wang F, McKeehan WL. Forced Expression of Hepatocyte-Specific Fibroblast Growth Factor 21 Delays Initiation of Chemically Induced Hepatocarcinogenesis. Mol Carcinog. 2006;45:934–942. doi: 10.1002/mc.20241. [DOI] [PubMed] [Google Scholar]
- 18.Jin C, Wang F, Wu X, Yu C, Luo Y, McKeehan WL. Directionally Specific Paracrine Communication Mediated by Epithelial FGF9 to Stromal FGFR3 in Two-Compartment Premalignant Prostate Tumors. Cancer Res. 2004;64:4555–4562. doi: 10.1158/0008-5472.CAN-03-3752. [DOI] [PubMed] [Google Scholar]
- 19.Clark JC, Tichelaar JW, Wert SE, Itoh N, Perl AK, Stahlman MT, Whitsett JA. FGF-10 Disrupts Lung Morphogenesis and Causes Pulmonary Adenomas in Vivo. Am J Physiol Lung Cell Mol Physiol. 2001;280:L705–L715. doi: 10.1152/ajplung.2001.280.4.L705. [DOI] [PubMed] [Google Scholar]
- 20.Tai AL, Sham JS, Xie D, Fang Y, Wu YL, Hu L, Deng W, Tsao GS, Qiao GB, Cheung AL, Guan XY. Co-Overexpression of Fibroblast Growth Factor 3 and Epidermal Growth Factor Receptor Is Correlated With the Development of Nonsmall Cell Lung Carcinoma. Cancer. 2006;106:146–155. doi: 10.1002/cncr.21581. [DOI] [PubMed] [Google Scholar]
- 21.Hu L, Sham JS, Xie D, Wen JM, Wang WS, Wang Y, Guan XY. Up-Regulation of Fibroblast Growth Factor 3 Is Associated With Tumor Metastasis and Recurrence in Human Hepatocellular Carcinoma. Cancer Lett. 2007;252:36–42. doi: 10.1016/j.canlet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Kiuru-Kuhlefelt S, Sarlomo-Rikala M, Larramendy ML, Soderlund M, Hedman K, Miettinen M, Knuutila S. FGF4 and INT2 Oncogenes Are Amplified and Expressed in Kaposi’s Sarcoma. Mod Pathol. 2000;13:433–437. doi: 10.1038/modpathol.3880074. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki K, Tokue A, Kamiakito T, Kuriki K, Saito K, Tanaka A. Predominant Expression of Fibroblast Growth Factor (FGF) 8, FGF4, and FGF Receptor 1 in Nonseminomatous and Highly Proliferative Components of Testicular Germ Cell Tumors. Virchows Arch. 2001;439:616–621. doi: 10.1007/s004280100437. [DOI] [PubMed] [Google Scholar]
- 24.Allerstorfer S, Sonvilla G, Fischer H, Spiegl-Kreinecker S, Gauglhofer C, Setinek U, Czech T, Marosi C, Buchroithner J, Pichler J, Silye R, Mohr T, Holzmann K, Grasl-Kraupp B, Marian B, Grusch M, Fischer J, Micksche M, Berger W. FGF5 as an Oncogenic Factor in Human Glioblastoma Multiforme: Autocrine and Paracrine Activities. Oncogene. 2008;27:4180–4190. doi: 10.1038/onc.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kornmann M, Ishiwata T, Beger HG, Korc M. Fibroblast Growth Factor-5 Stimulates Mitogenic Signaling and Is Overexpressed in Human Pancreatic Cancer: Evidence for Autocrine and Paracrine Actions. Oncogene. 1997;15:1417–1424. doi: 10.1038/sj.onc.1201307. [DOI] [PubMed] [Google Scholar]
- 26.Heer R, Douglas D, Mathers ME, Robson CN, Leung HY. Fibroblast Growth Factor 17 Is Over-Expressed in Human Prostate Cancer. J Pathol. 2004;204:578–586. doi: 10.1002/path.1668. [DOI] [PubMed] [Google Scholar]
- 27.Polnaszek N, Kwabi-Addo B, Wang J, Ittmann M. FGF17 Is an Autocrine Prostatic Epithelial Growth Factor and Is Upregulated in Benign Prostatic Hyperplasia. Prostate. 2004;60:18–24. doi: 10.1002/pros.20026. [DOI] [PubMed] [Google Scholar]
- 28.Sonvilla G, Allerstorfer S, Stattner S, Karner J, Klimpfinger M, Fischer H, Grasl-Kraupp B, Holzmann K, Berger W, Wrba F, Marian B, Grusch M. FGF18 in Colorectal Tumour Cells: Autocrine and Paracrine Effects. Carcinogenesis. 2008;29:15–24. doi: 10.1093/carcin/bgm202. [DOI] [PubMed] [Google Scholar]
- 29.Desnoyers LR, Pai R, Ferrando RE, Hotzel K, Le T, Ross J, Carano R, D’Souza A, Qing J, Mohtashemi I, Ashkenazi A, French DM. Targeting FGF19 Inhibits Tumor Growth in Colon Cancer Xenograft and FGF19 Transgenic Hepatocellular Carcinoma Models. Oncogene. 2008;27:85–97. doi: 10.1038/sj.onc.1210623. [DOI] [PubMed] [Google Scholar]
- 30.Powers CJ, McLeskey SW, Wellstein A. Fibroblast Growth Factors, Their Receptors and Signaling. Endocr Relat Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 31.Tassi E, Al Attar A, Aigner A, Swift MR, McDonnell K, Karavanov A, Wellstein A. Enhancement of Fibroblast Growth Factor (FGF) Activity by an FGF-Binding Protein. J Biol Chem. 2001;276:40247–40253. doi: 10.1074/jbc.M104933200. [DOI] [PubMed] [Google Scholar]
- 32.Abuharbeid S, Czubayko F, Aigner A. The Fibroblast Growth Factor-Binding Protein FGF-BP. Int J Biochem Cell Biol. 2006;38:1463–1468. doi: 10.1016/j.biocel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms Underlying Differential Responses to FGF Signaling. Cytokine Growth Factor Rev. 2005;16:233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Hart KC, Robertson SC, Donoghue DJ. Identification of Tyrosine Residues in Constitutively Activated Fibroblast Growth Factor Receptor 3 Involved in Mitogenesis, Stat Activation, and Phosphatidylinositol 3-Kinase Activation. Mol Biol Cell. 2001;12:931–942. doi: 10.1091/mbc.12.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadari YR, Gotoh N, Kouhara H, Lax I, Schlessinger J. Critical Role for the Docking-Protein FRS2 Alpha in FGF Receptor-Mediated Signal Transduction Pathways. Proc Natl Acad Sci U S A. 2001;98:8578–8583. doi: 10.1073/pnas.161259898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Udayakumar TS, Stratton MS, Nagle RB, Bowden GT. Fibroblast Growth Factor-1 Induced Promatrilysin Expression Through the Activation of Extracellular-Regulated Kinases and STAT3. Neoplasia. 2002;4:60–67. doi: 10.1038/sj.neo.7900207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsang M, Dawid IB. Promotion and Attenuation of FGF Signaling Through the Ras-MAPK Pathway. Sci STKE. 2004;2004:e17. doi: 10.1126/stke.2282004pe17. [DOI] [PubMed] [Google Scholar]
- 38.Cabrita MA, Christofori G. Sprouty Proteins, Masterminds of Receptor Tyrosine Kinase Signaling. Angiogenesis. 2008;11:53–62. doi: 10.1007/s10456-008-9089-1. [DOI] [PubMed] [Google Scholar]
- 39.Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty Proteins: Multifaceted Negative-Feedback Regulators of Receptor Tyrosine Kinase Signaling. Trends Cell Biol. 2006;16:45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Lo TL, Yusoff P, Fong CW, Guo K, McCaw BJ, Phillips WA, Yang H, Wong ES, Leong HF, Zeng Q, Putti TC, Guy GR. The Ras/Mitogen-Activated Protein Kinase Pathway Inhibitor and Likely Tumor Suppressor Proteins, Sprouty 1 and Sprouty 2 Are Deregulated in Breast Cancer. Cancer Res. 2004;64:6127–6136. doi: 10.1158/0008-5472.CAN-04-1207. [DOI] [PubMed] [Google Scholar]
- 41.Kwabi-Addo B, Wang J, Erdem H, Vaid A, Castro P, Ayala G, Ittmann M. The Expression of Sprouty1, an Inhibitor of Fibroblast Growth Factor Signal Transduction, Is Decreased in Human Prostate Cancer. Cancer Res. 2004;64:4728–4735. doi: 10.1158/0008-5472.CAN-03-3759. [DOI] [PubMed] [Google Scholar]
- 42.Fritzsche S, Kenzelmann M, Hoffmann MJ, Muller M, Engers R, Grone HJ, Schulz WA. Concomitant Down-Regulation of SPRY1 and SPRY2 in Prostate Carcinoma. Endocr Relat Cancer. 2006;13:839–849. doi: 10.1677/erc.1.01190. [DOI] [PubMed] [Google Scholar]
- 43.McKie AB, Douglas DA, Olijslagers S, Graham J, Omar MM, Heer R, Gnanapragasam VJ, Robson CN, Leung HY. Epigenetic Inactivation of the Human Sprouty2 (HSPRY2) Homologue in Prostate Cancer. Oncogene. 2005;24:2166–2174. doi: 10.1038/sj.onc.1208371. [DOI] [PubMed] [Google Scholar]
- 44.Lee SA, Ho C, Roy R, Kosinski C, Patil MA, Tward AD, Fridlyand J, Chen X. Integration of Genomic Analysis and in Vivo Transfection to Identify Sprouty 2 as a Candidate Tumor Suppressor in Liver Cancer. Hepatology. 2008;47:1200–1210. doi: 10.1002/hep.22169. [DOI] [PubMed] [Google Scholar]
- 45.Sutterluty H, Mayer CE, Setinek U, Attems J, Ovtcharov S, Mikula M, Mikulits W, Micksche M, Berger W. Down-Regulation of Sprouty2 in Non-Small Cell Lung Cancer Contributes to Tumor Malignancy Via Extracellular Signal-Regulated Kinase Pathway-Dependent and -Independent Mechanisms. Mol Cancer Res. 2007;5:509–520. doi: 10.1158/1541-7786.MCR-06-0273. [DOI] [PubMed] [Google Scholar]
- 46.Kovalenko D, Yang X, Nadeau RJ, Harkins LK, Friesel R. Sef Inhibits Fibroblast Growth Factor Signaling by Inhibiting FGFR1 Tyrosine Phosphorylation and Subsequent ERK Activation. J Biol Chem. 2003;278:14087–14091. doi: 10.1074/jbc.C200606200. [DOI] [PubMed] [Google Scholar]
- 47.Zisman-Rozen S, Fink D, Ben Izhak O, Fuchs Y, Brodski A, Kraus MH, Bejar J, Ron D. Downregulation of Sef, an Inhibitor of Receptor Tyrosine Kinase Signaling, Is Common to a Variety of Human Carcinomas. Oncogene. 2007;26:6093–6098. doi: 10.1038/sj.onc.1210424. [DOI] [PubMed] [Google Scholar]
- 48.Yang RB, Ng CK, Wasserman SM, Komuves LG, Gerritsen ME, Topper JN. A Novel Interleukin-17 Receptor-Like Protein Identified in Human Umbilical Vein Endothelial Cells Antagonizes Basic Fibroblast Growth Factor-Induced Signaling. J Biol Chem. 2003;278:33232–33238. doi: 10.1074/jbc.M305022200. [DOI] [PubMed] [Google Scholar]
- 49.Darby S, Sahadevan K, Khan MM, Robson CN, Leung HY, Gnanapragasam VJ. Loss of Sef (Similar Expression to FGF) Expression Is Associated With High Grade and Metastatic Prostate Cancer. Oncogene. 2006;25:4122–4127. doi: 10.1038/sj.onc.1209428. [DOI] [PubMed] [Google Scholar]
- 50.Funakami Y, Hata T, Itoh E, Itano S. Effects of Some Beta-Adrenoceptor Antagonists on Orthostatic Hypotension in Repeatedly Cold- (SART-) Stressed Rats. Biol Pharm Bull. 2007;30:303–308. doi: 10.1248/bpb.30.303. [DOI] [PubMed] [Google Scholar]
- 51.Deng C, Bedford M, Li C, Xu X, Yang X, Dunmore J, Leder P. Fibroblast Growth Factor Receptor-1 (FGFR-1) Is Essential for Normal Neural Tube and Limb Development. Dev Biol. 1997;185:42–54. doi: 10.1006/dbio.1997.8553. [DOI] [PubMed] [Google Scholar]
- 52.Xu X, Weinstein M, Li C, Naski M, Cohen RI, Ornitz DM, Leder P, Deng C. Fibroblast Growth Factor Receptor 2 (FGFR2)-Mediated Reciprocal Regulation Loop Between FGF8 and FGF10 Is Essential for Limb Induction. Development. 1998;125:753–765. doi: 10.1242/dev.125.4.753. [DOI] [PubMed] [Google Scholar]
- 53.Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast Growth Factor Receptor 3 Is a Negative Regulator of Bone Growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 54.Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 Function Cooperatively to Direct Alveogenesis in the Murine Lung. Development. 1998;125:3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- 55.Grose R, Dickson C. Fibroblast Growth Factor Signaling in Tumorigenesis. Cytokine Growth Factor Rev. 2005;16:179–186. doi: 10.1016/j.cytogfr.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 56.Klint P, Kanda S, Kloog Y, Claesson-Welsh L. Contribution of Src and Ras Pathways in FGF-2 Induced Endothelial Cell Differentiation. Oncogene. 1999;18:3354–3364. doi: 10.1038/sj.onc.1202680. [DOI] [PubMed] [Google Scholar]
- 57.Grose R, Fantl V, Werner S, Chioni AM, Jarosz M, Rudling R, Cross B, Hart IR, Dickson C. The Role of Fibroblast Growth Factor Receptor 2b in Skin Homeostasis and Cancer Development. EMBO J. 2007;26:1268–1278. doi: 10.1038/sj.emboj.7601583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Z, Neiss N, Zhou S, Henne-Bruns D, Korc M, Bachem M, Kornmann M. Identification of a Fibroblast Growth Factor Receptor 1 Splice Variant That Inhibits Pancreatic Cancer Cell Growth. Cancer Res. 2007;67:2712–2719. doi: 10.1158/0008-5472.CAN-06-3843. [DOI] [PubMed] [Google Scholar]
- 59.Pardo OE, Wellbrock C, Khanzada UK, Aubert M, Arozarena I, Davidson S, Bowen F, Parker PJ, Filonenko VV, Gout IT, Sebire N, Marais R, Downward J, Seckl MJ. FGF-2 Protects Small Cell Lung Cancer Cells From Apoptosis Through a Complex Involving PKCepsilon, B-Raf and S6K2. EMBO J. 2006;25:3078–3088. doi: 10.1038/sj.emboj.7601198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vandermoere F, Yazidi-Belkoura I, Adriaenssens E, Lemoine J, Hondermarck H. The Antiapoptotic Effect of Fibroblast Growth Factor-2 Is Mediated Through Nuclear Factor-KappaB Activation Induced Via Interaction Between Akt and IkappaB Kinase-Beta in Breast Cancer Cells. Oncogene. 2005;24:5482–5491. doi: 10.1038/sj.onc.1208713. [DOI] [PubMed] [Google Scholar]
- 61.Faridi A, Rudlowski C, Biesterfeld S, Schuh S, Rath W, Schroder W. Long-Term Follow-Up and Prognostic Significance of Angiogenic Basic Fibroblast Growth Factor (BFGF) Expression in Patients With Breast Cancer. Pathol Res Pract. 2002;198:1–5. doi: 10.1078/0344-0338-00176. [DOI] [PubMed] [Google Scholar]
- 62.McLeskey SW, Ding IY, Lippman ME, Kern FG. MDA-MB-134 Breast Carcinoma Cells Overexpress Fibroblast Growth Factor (FGF) Receptors and Are Growth-Inhibited by FGF Ligands. Cancer Res. 1994;54:523–530. [PubMed] [Google Scholar]
- 63.Fenig E, Wieder R, Paglin S, Wang H, Persaud R, Haimovitz-Friedman A, Fuks Z, Yahalom J. Basic Fibroblast Growth Factor Confers Growth Inhibition and Mitogen-Activated Protein Kinase Activation in Human Breast Cancer Cells. Clin Cancer Res. 1997;3:135–142. [PubMed] [Google Scholar]
- 64.Maloof P, Wang Q, Wang H, Stein D, Denny TN, Yahalom J, Fenig E, Wieder R. Overexpression of Basic Fibroblast Growth Factor (FGF-2) Downregulates Bcl-2 and Promotes Apoptosis in MCF-7 Human Breast Cancer Cells. Breast Cancer Res Treat. 1999;56:153–167. doi: 10.1023/a:1006258510381. [DOI] [PubMed] [Google Scholar]
- 65.Wang H, Rubin M, Fenig E, DeBlasio A, Mendelsohn J, Yahalom J, Wieder R. Basic Fibroblast Growth Factor Causes Growth Arrest in MCF-7 Human Breast Cancer Cells While Inducing Both Mitogenic and Inhibitory G1 Events. Cancer Res. 1997;57:1750–1757. [PubMed] [Google Scholar]
- 66.Linderholm BK, Lindh B, Beckman L, Erlanson M, Edin K, Travelin B, Bergh J, Grankvist K, Henriksson R. Prognostic Correlation of Basic Fibroblast Growth Factor and Vascular Endothelial Growth Factor in 1307 Primary Breast Cancers. Clin Breast Cancer. 2003;4:340–347. doi: 10.3816/cbc.2003.n.039. [DOI] [PubMed] [Google Scholar]
- 67.Korah RM, Sysounthone V, Golowa Y, Wieder R. Basic Fibroblast Growth Factor Confers a Less Malignant Phenotype in MDA-MB-231 Human Breast Cancer Cells. Cancer Res. 2000;60:733–740. [PubMed] [Google Scholar]
- 68.Korah R, Choi L, Barrios J, Wieder R. Expression of FGF-2 Alters Focal Adhesion Dynamics in Migration-Restricted MDA-MB-231 Breast Cancer Cells. Breast Cancer Res Treat. 2004;88:17–28. doi: 10.1007/s10459-004-6006-2. [DOI] [PubMed] [Google Scholar]
- 69.Korah R, Das K, Lindy ME, Hameed M, Wieder R. Coordinate Loss of Fibroblast Growth Factor 2 and Laminin 5 Expression During Neoplastic Progression of Mammary Duct Epithelium. Hum Pathol. 2007;38:154–160. doi: 10.1016/j.humpath.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 70.Song S, Wientjes MG, Gan Y, Au JL. Fibroblast Growth Factors: An Epigenetic Mechanism of Broad Spectrum Resistance to Anticancer Drugs. Proc Natl Acad Sci U S A. 2000;97:8658–8663. doi: 10.1073/pnas.140210697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gan Y, Wientjes MG, Au JL. Expression of Basic Fibroblast Growth Factor Correlates With Resistance to Paclitaxel in Human Patient Tumors. Pharm Res. 2006;23:1324–1331. doi: 10.1007/s11095-006-0136-6. [DOI] [PubMed] [Google Scholar]
- 72.Fujisawa H, Kurrer M, Reis RM, Yonekawa Y, Kleihues P, Ohgaki H. Acquisition of the Glioblastoma Phenotype During Astrocytoma Progression Is Associated With Loss of Heterozygosity on 10q25-Qter. Am J Pathol. 1999;155:387–394. doi: 10.1016/S0002-9440(10)65135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bernard-Pierrot I, Ricol D, Cassidy A, Graham A, Elvin P, Caillault A, Lair S, Broet P, Thiery JP, Radvanyi F. Inhibition of Human Bladder Tumour Cell Growth by Fibroblast Growth Factor Receptor 2b Is Independent of its Kinase Activity. Involvement of the Carboxy-Terminal Region of the Receptor. Oncogene. 2004;23:9201–9211. doi: 10.1038/sj.onc.1208150. [DOI] [PubMed] [Google Scholar]
- 74.Naimi B, Latil A, Fournier G, Mangin P, Cussenot O, Berthon P. Down-Regulation of (IIIb) and (IIIc) Isoforms of Fibroblast Growth Factor Receptor 2 (FGFR2) Is Associated With Malignant Progression in Human Prostate. Prostate. 2002;52:245–252. doi: 10.1002/pros.10104. [DOI] [PubMed] [Google Scholar]
- 75.Kondo T, Zheng L, Liu W, Kurebayashi J, Asa SL, Ezzat S. Epigenetically Controlled Fibroblast Growth Factor Receptor 2 Signaling Imposes on the RAS/BRAF/Mitogen-Activated Protein Kinase Pathway to Modulate Thyroid Cancer Progression. Cancer Res. 2007;67:5461–5470. doi: 10.1158/0008-5472.CAN-06-4477. [DOI] [PubMed] [Google Scholar]
- 76.Vairaktaris E, Ragos V, Yapijakis C, Derka S, Vassiliou S, Nkenke E, Yannopoulos A, Spyridonidou S, Vylliotis A, Papakosta V, Loukeri S, Lazaris A, Tesseromatis C, Tsigris C, Patsouris E. FGFR-2 and -3 Play an Important Role in Initial Stages of Oral Oncogenesis. Anticancer Res. 2006;26:4217–4221. [PubMed] [Google Scholar]
- 77.Moffa AB, Ethier SP. Differential Signal Transduction of Alternatively Spliced FGFR2 Variants Expressed in Human Mammary Epithelial Cells. J Cell Physiol. 2007;210:720–731. doi: 10.1002/jcp.20880. [DOI] [PubMed] [Google Scholar]
- 78.L’Hote CG, Knowles MA. Cell Responses to FGFR3 Signalling: Growth, Differentiation and Apoptosis. Exp Cell Res. 2005;304:417–431. doi: 10.1016/j.yexcr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 79.Chaffer CL, Dopheide B, Savagner P, Thompson EW, Williams ED. Aberrant Fibroblast Growth Factor Receptor Signaling in Bladder and Other Cancers. Differentiation. 2007;75:831–842. doi: 10.1111/j.1432-0436.2007.00210.x. [DOI] [PubMed] [Google Scholar]
- 80.Ricol D, Cappellen D, El Marjou A, Gil-Diez-de-Medina S, Girault JM, Yoshida T, Ferry G, Tucker G, Poupon MF, Chopin D, Thiery JP, Radvanyi F. Tumour Suppressive Properties of Fibroblast Growth Factor Receptor 2-IIIb in Human Bladder Cancer. Oncogene. 1999;18:7234–7243. doi: 10.1038/sj.onc.1203186. [DOI] [PubMed] [Google Scholar]
- 81.Finch PW, Rubin JS. Keratinocyte Growth Factor Expression and Activity in Cancer: Implications for Use in Patients With Solid Tumors. J Natl Cancer Inst. 2006;98:812–824. doi: 10.1093/jnci/djj228. [DOI] [PubMed] [Google Scholar]
- 82.Diez de Medina SG, Chopin D, El Marjou A, Delouvee A, LaRochelle WJ, Hoznek A, Abbou C, Aaronson SA, Thiery JP, Radvanyi F. Decreased Expression of Keratinocyte Growth Factor Receptor in a Subset of Human Transitional Cell Bladder Carcinomas. Oncogene. 1997;14:323–330. doi: 10.1038/sj.onc.1200830. [DOI] [PubMed] [Google Scholar]
- 83.Knowles MA. Novel Therapeutic Targets in Bladder Cancer: Mutation and Expression of FGF Receptors. Future Oncol. 2008;4:71–83. doi: 10.2217/14796694.4.1.71. [DOI] [PubMed] [Google Scholar]
- 84.Martinez-Torrecuadrada J, Cifuentes G, Lopez-Serra P, Saenz P, Martinez A, Casal JI. Targeting the Extracellular Domain of Fibroblast Growth Factor Receptor 3 With Human Single-Chain Fv Antibodies Inhibits Bladder Carcinoma Cell Line Proliferation. Clin Cancer Res. 2005;11:6280–6290. doi: 10.1158/1078-0432.CCR-05-0282. [DOI] [PubMed] [Google Scholar]
- 85.Matsumoto M, Ohtsuki Y, Ochii K, Seike Y, Iseda N, Sasaki T, Okada Y, Kurabayashi A, Furihata M. Fibroblast Growth Factor Receptor 3 Protein Expression in Urothelial Carcinoma of the Urinary Bladder, Exhibiting No Association With Low-Grade and/or Non-Invasive Lesions. Oncol Rep. 2004;12:967–971. [PubMed] [Google Scholar]
- 86.Ruotsalainen T, Joensuu H, Mattson K, Salven P. High Pretreatment Serum Concentration of Basic Fibroblast Growth Factor Is a Predictor of Poor Prognosis in Small Cell Lung Cancer. Cancer Epidemiol Biomarkers Prev. 2002;11:1492–1495. [PubMed] [Google Scholar]
- 87.Morrison RS, Yamaguchi F, Bruner JM, Tang M, McKeehan W, Berger MS. Fibroblast Growth Factor Receptor Gene Expression and Immunoreactivity Are Elevated in Human Glioblastoma Multiforme. Cancer Res. 1994;54:2794–2799. [PubMed] [Google Scholar]
- 88.Takahashi JA, Mori H, Fukumoto M, Igarashi K, Jaye M, Oda Y, Kikuchi H, Hatanaka M. Gene Expression of Fibroblast Growth Factors in Human Gliomas and Meningiomas: Demonstration of Cellular Source of Basic Fibroblast Growth Factor mRNA and Peptide in Tumor Tissues. Proc Natl Acad Sci US A. 1990;87:5710–5714. doi: 10.1073/pnas.87.15.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zagzag D, Miller DC, Sato Y, Rifkin DB, Burstein DE. Immunohistochemical Localization of Basic Fibroblast Growth Factor in Astrocytomas. Cancer Res. 1990;50:7393–7398. [PubMed] [Google Scholar]
- 90.McKeehan WL, Wang F, Kan M. The Heparan Sulfate-Fibroblast Growth Factor Family: Diversity of Structure and Function. Prog Nucleic Acid Res Mol Biol. 1998;59:135–176. doi: 10.1016/s0079-6603(08)61031-4. [DOI] [PubMed] [Google Scholar]
- 91.Hu Z, Evarts RP, Fujio K, Marsden ER, Thorgeirsson SS. Expression of Fibroblast Growth Factor Receptors flg and bek During Hepatic Ontogenesis and Regeneration in the Rat. Cell Growth Differ. 1995;6:1019–1025. [PubMed] [Google Scholar]
- 92.Steiling H, Wustefeld T, Bugnon P, Brauchle M, Fassler R, Teupser D, Thiery J, Gordon JI, Trautwein C, Werner S. Fibroblast Growth Factor Receptor Signalling Is Crucial for Liver Homeostasis and Regeneration. Oncogene. 2003;22:4380–4388. doi: 10.1038/sj.onc.1206499. [DOI] [PubMed] [Google Scholar]
- 93.Kin M, Sata M, Ueno T, Torimura T, Inuzuka S, Tsuji R, Sujaku K, Sakamoto M, Sugawara H, Tamaki S, Tanikawa K. Basic Fibroblast Growth Factor Regulates Proliferation and Motility of Human Hepatoma Cells by an Autocrine Mechanism. J Hepatol. 1997;27:677–687. doi: 10.1016/s0168-8278(97)80085-2. [DOI] [PubMed] [Google Scholar]
- 94.Huang X, Yu C, Jin C, Kobayashi M, Bowles CA, Wang F, McKeehan WL. Ectopic activity of fibroblast growth factor receptor 1 in hepatocytes accelerates hepatocarcinogenesis by driving proliferation and vascular endothelial growth factor-induced angiogenesis. Cancer Res. 2006;66:1481–1490. doi: 10.1158/0008-5472.CAN-05-2412. [DOI] [PubMed] [Google Scholar]
- 95.Dorkin TJ, Robinson MC, Marsh C, Neal DE, Leung HY. AFGF Immunoreactivity in Prostate Cancer and its Co-Localization With BFGF and FGF8. J Pathol. 1999;189:564–569. doi: 10.1002/(SICI)1096-9896(199912)189:4<564::AID-PATH480>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 96.Cronauer MV, Hittmair A, Eder IE, Hobisch A, Culig Z, Ramoner R, Zhang J, Bartsch G, Reissigl A, Radmayr C, Thurnher M, Klocker H. Basic Fibroblast Growth Factor Levels in Cancer Cells and in Sera of Patients Suffering From Proliferative Disorders of the Prostate. Prostate. 1997;31:223–233. doi: 10.1002/(sici)1097-0045(19970601)31:4<223::aid-pros3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 97.Giri D, Ropiquet F, Ittmann M. Alterations in Expression of Basic Fibroblast Growth Factor (FGF) 2 and its Receptor FGFR-1 in Human Prostate Cancer. Clin Cancer Res. 1999;5:1063–1071. [PubMed] [Google Scholar]
- 98.Takahashi H. Studies on the Expression of Fibroblast Growth Factors and Fibroblast Growth Factor Receptors in Human Prostate Cancer. Nippon Hinyokika Gakkai Zasshi. 1998;89:836–845. doi: 10.5980/jpnjurol1989.89.836. [DOI] [PubMed] [Google Scholar]
- 99.Hase T, Kawashiri S, Tanaka A, Nozaki S, Noguchi N, Kato K, Nakaya H, Nakagawa K. Correlation of Basic Fibroblast Growth Factor Expression With the Invasion and the Prognosis of Oral Squamous Cell Carcinoma. J Oral Pathol Med. 2006;35:136–139. doi: 10.1111/j.1600-0714.2006.00397.x. [DOI] [PubMed] [Google Scholar]
- 100.Siddiqi I, Funatomi H, Kobrin MS, Friess H, Büchler MW, Korc M. Increased expression of keratinocyte growth factor in human pancreatic cancer. Biochem Biophys Res Commun. 1995;215:309–315. doi: 10.1006/bbrc.1995.2467. [DOI] [PubMed] [Google Scholar]
- 101.Ishiwata T, Friess H, Buchler MW, Lopez ME, Korc M. Characterization of Keratinocyte Growth Factor and Receptor Expression in Human Pancreatic Cancer. Am J Pathol. 1998;153:213–222. doi: 10.1016/S0002-9440(10)65562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu Z, Ishiwata T, Zhou S, Maier S, Henne-Bruns D, Korc M, Bachem M, Kornmann M. Human Fibroblast Growth Factor Receptor 1-IIIb Is a Functional Fibroblast Growth Factor Receptor Expressed in the Pancreas and Involved in Proliferation and Movement of Pancreatic Ductal Cells. Pancreas. 2007;35:147–157. doi: 10.1097/mpa.0b013e318053e7e3. [DOI] [PubMed] [Google Scholar]
- 103.Wagner M, Lopez ME, Cahn M, Korc M. Suppression of Fibroblast Growth Factor Receptor Signaling Inhibits Pancreatic Cancer Growth in Vitro and in Vivo. Gastroenterology. 1998;114:798–807. doi: 10.1016/s0016-5085(98)70594-3. [DOI] [PubMed] [Google Scholar]
- 104.Kleeff J, Kothari NH, Friess H, Fan H, Korc M. Adenovirus-Mediated Transfer of a Truncated Fibroblast Growth Factor (FGF) Type I Receptor Blocks FGF-2 Signaling in Multiple Pancreatic Cancer Cell Lines. Pancreas. 2004;28:25–30. doi: 10.1097/00006676-200401000-00004. [DOI] [PubMed] [Google Scholar]
- 105.Cho K, Ishiwata T, Uchida E, Nakazawa N, Korc M, Naito Z, Tajiri T. Enhanced Expression of Keratinocyte Growth Factor and its Receptor Correlates With Venous Invasion in Pancreatic Cancer. Am J Pathol. 2007;170:1964–1974. doi: 10.2353/ajpath.2007.060935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ribatti D, Vacca A, Rusnati M, Presta M. The Discovery of Basic Fibroblast Growth Factor/Fibroblast Growth Factor-2 and its Role in Haematological Malignancies. Cytokine Growth Factor Rev. 2007;18:327–334. doi: 10.1016/j.cytogfr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 107.Firme L, Bush AB. FGF Signaling Inhibits the Proliferation of Human Myeloma Cells and Reduces c-myc Expression. BMC Cell Biol. 2003;4:17. doi: 10.1186/1471-2121-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ishikawa H, Tsuyama N, Liu S, Abroun S, Li FJ, Otsuyama K, Zheng X, Ma Z, Maki Y, Iqbal MS, Obata M, Kawano MM. Accelerated Proliferation of Myeloma Cells by Interleukin-6 Cooperating With Fibroblast Growth Factor Receptor 3-Mediated Signals. Oncogene. 2005;24:6328–6332. doi: 10.1038/sj.onc.1208782. [DOI] [PubMed] [Google Scholar]
- 109.Bisping G, Leo R, Wenning D, Dankbar B, Padro T, Kropff M, Scheffold C, Kroger M, Mesters RM, Berdel WE, Kienast J. Paracrine Interactions of Basic Fibroblast Growth Factor and Interleukin-6 in Multiple Myeloma. Blood. 2003;101:2775–2783. doi: 10.1182/blood-2002-09-2907. [DOI] [PubMed] [Google Scholar]
- 110.Masih-Khan E, Trudel S, Heise C, Li Z, Paterson J, Nadeem V, Wei E, Roodman D, Claudio JO, Bergsagel PL, Stewart AK. MIP-1alpha (CCL3) Is a Downstream Target of FGFR3 and RAS-MAPK Signaling in Multiple Myeloma. Blood. 2006;108:3465–3471. doi: 10.1182/blood-2006-04-017087. [DOI] [PubMed] [Google Scholar]
- 111.Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, Kuehl WM, Bergsagel PL. Activated Fibroblast Growth Factor Receptor 3 Is an Oncogene That Contributes to Tumor Progression in Multiple Myeloma. Blood. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- 112.Lauring J, Abukhdeir AM, Konishi H, Garay JP, Gustin JP, Wang Q, Arceci RJ, Matsui W, Park BH. The Multiple Myeloma Associated MMSET Gene Contributes to Cellular Adhesion, Clonogenic Growth, and Tumorigenicity. Blood. 2008;111:856–864. doi: 10.1182/blood-2007-05-088674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wu X, Jin C, Wang F, Yu C, McKeehan WL. Stromal Cell Heterogeneity in Fibroblast Growth Factor-Mediated Stromal-Epithelial Cell Cross-Talk in Premalignant Prostate Tumors. Cancer Res. 2003;63:4936–4944. [PubMed] [Google Scholar]
- 114.Tsunoda S, Nakamura T, Sakurai H, Saiki I. Fibroblast Growth Factor-2-Induced Host Stroma Reaction During Initial Tumor Growth Promotes Progression of Mouse Melanoma Via Vascular Endothelial Growth Factor A-Dependent Neovascularization. Cancer Sci. 2007;98:541–548. doi: 10.1111/j.1349-7006.2007.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang F, Strand DW, Rowley DR. Fibroblast Growth Factor-2 Mediates Transforming Growth Factor-Beta Action in Prostate Cancer Reactive Stroma. Oncogene. 2008;27:450–459. doi: 10.1038/sj.onc.1210663. [DOI] [PubMed] [Google Scholar]
- 116.Alves CC, Carneiro F, Hoefler H, Becker KF. Role of the Epithelial-Mesenchymal Transition Regulator Slug in Primary Human Cancers. Front Biosci. 2009;14:3035–3050. doi: 10.2741/3433. [DOI] [PubMed] [Google Scholar]
- 117.Savagner P, Yamada KM, Thiery JP. The Zinc-Finger Protein Slug Causes Desmosome Dissociation, an Initial and Necessary Step for Growth Factor-Induced Epithelial-Mesenchymal Transition. J Cell Biol. 1997;137:1403–1419. doi: 10.1083/jcb.137.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pon YL, Zhou HY, Cheung AN, Ngan HY, Wong AS. p70 S6 Kinase Promotes Epithelial to Mesenchymal Transition Through Snail Induction in Ovarian Cancer Cells. Cancer Res. 2008;68:6524–6532. doi: 10.1158/0008-5472.CAN-07-6302. [DOI] [PubMed] [Google Scholar]
- 119.Suyama K, Shapiro I, Guttman M, Hazan RB. A Signaling Pathway Leading to Metastasis Is Controlled by N-Cadherin and the FGF Receptor. Cancer Cell. 2002;2:301–314. doi: 10.1016/s1535-6108(02)00150-2. [DOI] [PubMed] [Google Scholar]
- 120.Abraham JA, Mergia A, Whang JL, Tumolo A, Friedman J, Hjerrild KA, Gospodarowicz D, Fiddes JC. Nucleotide Sequence of a Bovine Clone Encoding the Angiogenic Protein, Basic Fibroblast Growth Factor. Science. 1986;233:545–548. doi: 10.1126/science.2425435. [DOI] [PubMed] [Google Scholar]
- 121.Maciag T, Mehlman T, Friesel R, Schreiber AB. Heparin Binds Endothelial Cell Growth Factor, the Principal Endothelial Cell Mitogen in Bovine Brain. Science. 1984;225:932–935. doi: 10.1126/science.6382607. [DOI] [PubMed] [Google Scholar]
- 122.Javerzat S, Auguste P, Bikfalvi A. The Role of Fibroblast Growth Factors in Vascular Development. Trends Mol Med. 2002;8:483–489. doi: 10.1016/s1471-4914(02)02394-8. [DOI] [PubMed] [Google Scholar]
- 123.Bastaki M, Nelli EE, Dell’Era P, Rusnati M, Molinari-Tosatti MP, Parolini S, Auerbach R, Ruco LP, Possati L, Presta M. Basic Fibroblast Growth Factor-Induced Angiogenic Phenotype in Mouse Endothelium: A Study of Aortic and Microvascular Endothelial Cell Lines. Arterioscler Thromb Vasc Biol. 1997;17:454–464. doi: 10.1161/01.atv.17.3.454. [DOI] [PubMed] [Google Scholar]
- 124.Dell’Era P, Belleri M, Stabile H, Massrdi ML, Ribatti D, Presta M. Paracrine and Autocrine Effects of Fibroblast Growth Factor-4 in Endothelial Cells. Oncogene. 2001;20:2655–2663. doi: 10.1038/sj.onc.1204368. [DOI] [PubMed] [Google Scholar]
- 125.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug Resistance by Evasion of Antiangiogenic Targeting of VEGF Signaling in Late-Stage Pancreatic Islet Tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 126.Jubb AM, Oates AJ, Holden S, Koeppen H. Predicting Benefit From Anti- Angiogenic Agents in Malignancy. Nat Rev Cancer. 2006;6:626–635. doi: 10.1038/nrc1946. [DOI] [PubMed] [Google Scholar]
- 127.Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast Growth Factor/Fibroblast Growth Factor Receptor System in Angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 128.Giavazzi R, Sennino B, Coltrini D, Garofalo A, Dossi R, Ronca R, Tosatti MPM, Presta M. Distinct Role of Fibroblast Growth Factor-2 and Vascular Endothelial Growth Factor on Tumor Growth and Angiogenesis. Am J Pathol. 2003;162:1913–1926. doi: 10.1016/S0002-9440(10)64325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tille JC, Wood J, Mandriota SJ, Schnell C, Ferrari S, Mestan J, Zhu Z, Witte L, Pepper MS. Vascular Endothelial Growth Factor (VEGF) Receptor-2 Antagonists Inhibit VEGF- and Basic Fibroblast Growth Factor-Induced Angiogenesis in Vivo and in Vitro. J Pharmacol Exp Ther. 2001;299:1073–1085. [PubMed] [Google Scholar]
- 130.Auguste P, Gursel DB, Lemiere S, Reimers D, Cuevas P, Carceller F, Di Santo JP, Bikfalvi A. Inhibition of Fibroblast Growth Factor/Fibroblast Growth Factor Receptor Activity in Glioma Cells Impedes Tumor Growth by Both Angiogenesis-Dependent and -Independent Mechanisms. Cancer Res. 2001;61:1717–1726. [PubMed] [Google Scholar]
- 131.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P. Fibroblast Growth Factor-2 (FGF-2) Induces Vascular Endothelial Growth Factor (VEGF) Expression in the Endothelial Cells of Forming Capillaries: An Autocrine Mechanism Contributing to Angiogenesis. J Cell Biol. 1998;141:1659–1673. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kanda S, Miyata Y, Kanetake H. Fibroblast Growth Factor-2-Mediated Capillary Morphogenesis of Endothelial Cells Requires Signals Via Flt-1/Vascular Endothelial Growth Factor Receptor-1: Possible Involvement of c-Akt. J Biol Chem. 2004;279:4007–4016. doi: 10.1074/jbc.M307569200. [DOI] [PubMed] [Google Scholar]
- 133.Gabler C, Plath-Gabler A, Killian GJ, Berisha B, Schams D. Expression Pattern of Fibroblast Growth Factor (FGF) and Vascular Endothelial Growth Factor (VEGF) System Members in Bovine Corpus Luteum Endothelial Cells During Treatment With FGF-2: VEGF or Oestradiol. Reprod Domest Anim. 2004;39:321–327. doi: 10.1111/j.1439-0531.2004.00517.x. [DOI] [PubMed] [Google Scholar]
- 134.Liu W, Parikh AA, Stoeltzing O, Fan F, McCarty MF, Wey J, Hicklin DJ, Ellis LM. Upregulation of Neuropilin-1 by Basic Fibroblast Growth Factor Enhances Vascular Smooth Muscle Cell Migration in Response to VEGF. Cytokine. 2005;32:206–212. doi: 10.1016/j.cyto.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 135.Shi YH, Bingle L, Gong LH, Wang YX, Corke KP, Fang WG. Basic FGF Augments Hypoxia Induced HIF-1-Alpha Expression and VEGF Release in T47D Breast Cancer Cells. Pathology. 2007;39:396–400. doi: 10.1080/00313020701444549. [DOI] [PubMed] [Google Scholar]
- 136.Matsuo M, Yamada S, Koizumi K, Sakurai H, Saiki I. Tumour-Derived Fibroblast Growth Factor-2 Exerts Lymphangiogenic Effects Through Akt/mTOR/p70S6kinase Pathway in Rat Lymphatic Endothelial Cells. Eur J Cancer. 2007;43:1748–1754. doi: 10.1016/j.ejca.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 137.Nissen LJ, Cao R, Hedlund EM, Wang Z, Zhao X, Wetterskog D, Funa K, Brakenhielm E, Cao Y. Angiogenic Factors FGF2 and PDGF-BB Synergistically Promote Murine Tumor Neovascularization and Metastasis. J Clin Invest. 2007;117:2766–2777. doi: 10.1172/JCI32479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Webster MK, Donoghue DJ. FGFR Activation in Skeletal Disorders: Too Much of a Good Thing. Trends Genet. 1997;13:178–182. doi: 10.1016/s0168-9525(97)01131-1. [DOI] [PubMed] [Google Scholar]
- 139.Passos-Bueno MR, Wilcox WR, Jabs EW, Sertie AL, Alonso LG, Kitoh H. Clinical Spectrum of Fibroblast Growth Factor Receptor Mutations. Hum Mutat. 1999;14:115–125. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 140.Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of- Function Mutations in FGFR1 Cause Autosomal Dominant Kallmann Syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 141.Pitteloud N, Acierno JS, Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, Yialamas M, Hall JE, Grant E, Mohammadi M, Crowley WF., Jr Mutations in Fibroblast Growth Factor Receptor 1 Cause Both Kallmann Syndrome and Normosmic Idiopathic Hypogonadotropic Hypogonadism. Proc Natl Acad Sci U S A. 2006;103:6281–6286. doi: 10.1073/pnas.0600962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. Decreased FGF8 Signaling Causes Deficiency of Gonadotropin-Releasing Hormone in Humans and Mice. J Clin Invest. 2008;118:2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]