

Abstract
Despite the long held hypothesis that oxidant stress results in accumulated oxidative damage to cellular macromolecules and subsequently to aging and age-related chronic disease, it has been difficult to consistently define and specifically identify markers of oxidant stress that are consistently and directly linked to age and disease status. Inflammation because it is also linked to oxidant stress, aging, and chronic disease also plays an important role in understanding the clinical implications of oxidant stress and relevant markers. Much attention has focused on identifying specific markers of oxidative stress and inflammation that could be measured in easily accessible tissues and fluids (lymphocytes, plasma, serum). The purpose of this review is to discuss markers of oxidant stress used in the field as biomarkers of aging and age-related diseases, highlighting differences observed by race when data is available. We highlight DNA, RNA, protein, and lipid oxidation as measures of oxidative stress, as well as other well-characterized markers of oxidative damage and inflammation and discuss their strengths and limitations. We present the current state of the literature reporting use of these markers in studies of human cohorts in relation to age and age-related disease and also with a special emphasis on differences observed by race when relevant.
Keywords: DNA oxidation, RNA oxidation, Protein oxidation, Single Strand Breaks, CRP
1. Introduction
The specific role of oxidative stress in aging and in the development of age-related disease is an area of active investigation but the exact mechanisms that may define this complex relationship are unclear (Voss and Siems, 2006). Understanding this complex relationship may provide potential biomarkers of oxidative stress, which can be objectively measured as indicators of normal and pathologic processes that result in age-related disease and decrement in cellular function associated with aging.
The Harman Free Radical Theory of Aging states that accumulation of oxidative damage to DNA and other cellular components and tissues over the lifespan leads to aging, disease and death (Harman, 1956). Free radicals can be formed exogenously by environmental sources including ionizing radiation, ultraviolet light, and pollutants (cigarette smoke, emissions from automobiles or factories, asbestos). These sources of oxidative stress, in addition to endogenous enzymatic sources, such as cellular respiration, cell signaling, and inflammation, result in oxidative damage in a biologically relevant manner (Mateos and Bravo, 2007).
1.1. What is Oxidant Stress?
Oxidant stress has been defined as an alteration in the balance between the production of reactive oxygen species (free radicals) and the antioxidant defense system in place to counter them (Halliwell, 1994). Free radicals as well as reactive oxygen species (ROS) are any chemical species with unpaired electrons and are produced through many sources including the environment (from ozone and nitrogen dioxide) and many varied biological and biochemical processes (both deliberately and as by-products). Common examples of free radicals and ROS are the hydroxyl radical (−OH), the superoxide anion (O2−), nitric oxide (NO−), and transition metals.
Free radicals are neutralized by the anti-oxidant system. This system functions at the cellular, membrane, and extracellular levels to protect against free radical attack. Components that comprise this system include members of the catalase, peroxidase, and dismutase families as well as the glutathione system, including superoxide dismutase (SOD) (converts superoxide to hydrogen peroxide), catalase (removes hydrogen peroxide), and glutathione peroxidase (removes hydrogen peroxide generated by SOD).
In addition to the endogenous enzyme antioxidant system, additional antioxidants play a key role in defense against ROS including vitamins A, C, and E. These antioxidants are classified into two groups: those that are hydrophobic (Vitamins A and E) protect membranes from free radical attack and those that are hydrophilic (Vitamin C) interact with free radicals in the blood and cytosol (Sies, 1997) to neutralize the free radicals that are formed.
Free radicals can cause damage in DNA that can potentially lead to mutagenesis and thus cellular transformation and uncontrolled proliferation. In addition, oxidant stress is thought to contribute to the development of human diseases including but not limited to Alzheimer's disease (AD) (Butterfield, 2006; Christen, 2000; Halliwell, 2006; Nunomura et al., 2006; Perry et al., 2002), cardiovascular disease (Aviram, 2000), atherosclerosis (Parthasarathy et al., 2008; Stocker and Keaney, 2004), Parkinsons disease (PD) (Wood-Kaczmar et al., 2006), rheumatoid arthritis (Hitchon and El-Gabalawy, 2004), diabetes (Dav et al., 2005; Giugliano et al., 1996), and motor neuron diseases (Cookson and Shaw, 1999). In this review we address commonly used markers of oxidant stress as they are related to aging and age-related diseases only in the context of human studies. These human cohort studies are summarized in Tables 1 and 2 where we have highlighted the disease state examined, the methodology and the significant findings.
Table 1.
Population Studies using Selected Oxidative and Inflammatory Biomarkers
| Marker | Cohort | Disorder | Methodology | Result | Reference |
|---|---|---|---|---|---|
| 8-oxoGua and 8-oxodG in DNA | n=14 Ages 75–83 |
Alzheimer's Disease | GC/MS of CSF and analysis of intact DNA structures | ↑ 8-oxodG in AD patients DNA (p<0.05) | (Lovell et al., 1999) |
| n=26 Ages 70–78 |
Alzheimer's Disease | HPLC using brain tissues | Correlation of 8-oxodG with age in controls (p<0.0002); ↑ 8-oxodG in mDNA compared with nDNA(p<0.001) | (Mecocci et al., 1994) | |
| n=20 Ages 75–86 |
Alzheimer's Disease | GC/MS using brain tissues | ↑ 8-oxoGua in AD patients (p<0.03) | (Gabbita et al., 1998) | |
| n=76 48–91 |
Alzheimer's Disease | GC/MS using brain tissues | ↑ 8-oxoGua in Occipital lobe of AD patients (p<0.001); ↑ 8-oxoGua levels in Parietal lobe of AD patients (p<0.003) | (Lyras et al., 1997) | |
| n=16 Ages 81–88 |
Alzheimer's Disease | GC/MS-SIM using brain tissues | ↑ 8-oxodG in mDNA and nDNA ofAD patients DNA (p<0.05); ↑ 8-oxodG in mDNA compared with nDNA (p<0.01) | (Wang et al., 2005) | |
| n=20 Ages 71–84 |
Parkinson's Disease | GC/MS using brain tissues | ↑ 8-oxoGua in PD patients (p=0.002) | (Alam et al., 1997) | |
| n=22 Ages 66–83 |
Parkinson's Disease | Immunohistochemistry of brain tissues for 8-oxoG | ↑ 8-oxoGua immunoreactivity in PD patients synaptic neurons (p<0.01) | (Zhang et al., 1999) | |
| n=177 Ages 25–5 |
Healthy | HPLC of using lymphocytes | ↑ 8-oxodG levels in men (p<0.01) and correlation with early CAD (r=0.9) | (Collins et al., 1998) | |
| n=255 Ages ~8–85 |
Healthy | HPLC of using lymphocytes | ↑ in 8-oxodG with age (p<0.01) | (Siomek et al., 2007) | |
| n=21 Ages N.G. |
Patients with carotid stenosis > 70% | Immunohystochemistry of carotid endarterectomy specimens | ↑ 8-oxodG immunoreactivity of affected tissues compared with unaffected tissue (p<0.01) | (Martinet et al., 2002) | |
| n=66 Ages 29–93 |
Healthy | HPLC of DNA isolated from muscle | ↑ levels of 8-oxodG with age (p<0.001) | (Mecocci et al., 1999) | |
| n=117 Ages 30–64 |
Community dwelling healthy females | ELISA using serum | ↑ levels of 8-oxodG with age (p<0.01) | (Noren Hooten et al., 2012) | |
| n=90 Ages 68–86 |
Community-dwelling older adults | ELISA using serum | ↑ levels correlated with ↑ frailty (p=0.03) | (Wu et al., 2009) | |
| DNA SSB | n=55 Ages 35–69 |
Healthy males | ELISA using human lymphocytes | ↑ in SSB in aged population (p=0.039) | (Barnett and King, 1995) |
| n=55 Ages 21–40 and 61–85 |
Healthy | Alkaline COMET assay using lymphocytes | ~2-fold ↑ in endogenous SSB in elderly (p<0.05) | (Mutlu-Turkoglu et al., 2003) | |
| n=80 Ages 21–60 |
Healthy | Alkaline COMET assay using lymphocytes | ↑ in SSB in individuals over 40 years of age*; no correlation to sex or smoking status | (Diem et al., 2002) | |
| n=64 Ages 28–59 |
HBV-related cirrhosis and chronic HCV | Alkaline COMET assay using lymphocytes | ↑ SSB in both HBV (p<0.013) and HCV patients (p<0.016) | (Bolukbas et al., 2006) | |
| n=96 Ages 30–64 |
Community dwelling bi-racial cohort | Alkaline COMET assay using lymphocytes | ↑ SSB for females (p=0.013) | (Trzeciak et al., 2012) | |
| n=97 Ages 20–82 |
Healthy | Alkaline COMET assay using lymphocytes | ↑ SSB and Fpg-sensitive sites in aged (p<0.001) | (Humphreys et al., 2007) | |
| n=147 Ages 20–45 |
Healthy | Alkaline COMET assay using lymphocytes | ↑ Fpg-sensitive sites in African Americans (p<0.01) | (Watters et al., 2008) | |
| n=31 Ages 35–69 and 75–80 |
Healthy | ELISA using lymphocyte samples | Old cohort had similar levels of SSBs as young (p=0.42) | (King et al., 1997) | |
| n=170 Ages 20–64 |
Healthy | Alkaline COMET assay using lymphocytes | No changes in DNA damage with age or sex*; increase in SSB with smoking (p<0.05) | (Kopjar et al., 2006) | |
| n=156 Ages 95, 94, 90, 86 and 40–60 |
Swedish NONA Immune Study | Alkaline COMET assay using lymphocytes | No change in DNA damage with age* | (Hyland et al., 2002) | |
| n=136 Ages 44–70 |
Acute Coronary Syndrome | Alkaline COMET assay using lymphocytes | ↑ DNA damage in ACS patients (p<0.001) | (Demirbag et al., 2005) | |
| n=216 Ages 40–77 |
Twin pairs born 1930–1969 | FADU using lymphocytes | No change in DNA damage with age (p≥0.51) | (Garm et al., 2012) | |
| SSB Repair Capacity | n=96 Ages 30–64 |
Community dwelling bi-racial cohort | Alkaline COMET assay using lymphocytes | Repair is dependent on age (p<0.01 for white females), gender (p<0.01), and race (p<0.002 for females) | (Trzeciak et al., 2008) |
| n=50 Ages N.G.; n=28 Ages 21–55 n=240 Ages 39–70; n=54 Ages 24–75 |
Various cancers; Healthy; NSCLC; Various cancers |
Alkaline COMET assay using lymphocytes | No effect of age, gender, or smoking in repair* | (Marcon et al., 2003; Muller et al., 2002; Muller et al., 2001; Rajaee-Behbahani et al., 2001) | |
| n=96 Ages 30–64 |
Community dwelling bi-racial cohort | Alkaline COMET assay using lymphocytes | Negative correlation between SSB-RC and SSB level (p=0.041) | (Trzeciak et al., 2012) | |
| n=632 Ages 32–86; n=1441 Ages N.G. |
Lung Cancer | Host-cell reactivation assay | ↓ repair capacity in patients (p<0.001), specifically white females (p<0.001)1 | (Spitz et al., 2003; Wei et al., 2000) | |
| n=140 Ages 41–63 | Breast cancer | Alkaline COMET assay using lymphocytes | ↑ DNA damage in cancer patients (p<0.001), but no effect on repair with age* | (Smith et al., 2003) | |
| n=223 Ages 20–60 |
Basal cell carcinoma | Host-cell reactivation assay | ↓ repair across life span (p<0.003); lowest repair in young with BCC (p=0.022) | (Wei et al., 1993) | |
| n=120 Ages N.G. |
Healthy | OGG activity assay from lymphocytes | ↓ OGG1 activity in males age 55 and older (p=0.0064); no effect of smoking status (p=0.84) | (Paz-Elizur et al., 2007) | |
| n=845 Age 85 |
Newcastle 85+ cohort | Automated Fluorimetric Alkaline DNA Unwinding Analysis | No correlation between SSB-RC and frailty (p=0.13) | (Collerton et al., 2012) | |
| n=216 Ages 40–77 |
Twin pairs born 1930–1969 | FADU using lymphocytes | No changes in repair capacity with age (p≥0.7) | (Garm et al., 2012) | |
| DNA DSBs | n=26 Ages 21–71 |
Healthy | Immunocytochemistry of lymphocytes | ↑ number of γ-H2AX foci in older donors compared with younger donors (p<0.01) | (Sedelnikova et al., 2002) |
| n=40 Ages 35–80 |
Healthy | Immunocytochemistry of lymphocytes | ↑ number of γ-H2AX foci with age; ↑ number of γ-H2AX foci in participants over age 57 with hypertension (p=0.037) | (Schurman et al., 2012) | |
| n=66 Ages 20–57 |
Healthy | Neutral COMET assay using sperm | ↑ levels of DSBs in older individuals (p<0.02) | (Singh et al., 2003) | |
| n=80 Ages 22–80 |
Healthy | Neutral COMET assay using sperm | No changes in DSB levels with age (p=0.7) | (Schmid et al., 2007) | |
| Age-related Macular Degeneration (AMD) | Neutral COMET assay using lymphocytes | ↑ levels of DSBs in patients with AMD (p<0.05) | (Szaflik et al., 2009) | ||
| n=216 Ages 40–77 |
Twin pairs born 1930–1969 | Neutral COMET assay using lymphocytes; Immunocytochemistry of lymphocytes | ↓ repair of DBS with age (p<0.01); ↑ number of γ-H2AX foci with age (p<0.01) | (Garm et al., 2012) | |
| 8-OHG in RNA | n=20 Ages 45–70 |
Healthy | HPLC-MS of urine | ↑ levels of 8-OHG compared to DNA oxidation products* | (Weimann et al., 2002) |
| n=33 Ages 53–71 |
Alzheimer's Disease | HPLC-EC of CSF | ↑ 8-OHG in AD patients (p<0.001); no correlation with age | (Abe et al., 2002) | |
| n=47 Ages 57–85 |
Dementia w/Lewy Bodies | Immunohystochemistry of brain tissue | ↑ 8-OHG immunoreactivity in DLB patients (p < 0.01) | (Nunomura et al., 2002) | |
| n=22 Ages 3–93 |
Alzheimer's Disease | Immunohystochemistry of brain tissue | ↑ 8-OHG immunoreactivity in AD patients (p < 0.0001) | (Nunomura et al., 1999b) | |
| n=39 Ages 57–93 |
Alzheimer's Disease | Immunohystochemistry of brain tissue | ↑ 8-OHG immunoreactivity in AD patients (p < 0.0001) | (Nunomura et al., 1999a) | |
| n=23 Ages 57–75 |
Alzheimer's Disease | HPLC-EC of CSF and Serum | ↑ 8-OHG in CSF of AD patients (p>0.001); no correlation with age; no change in 8-OHG in serum of AD patients | (Isobe et al., 2009) | |
| n=16 Ages 65–93 |
Alzheimer's Disease | Immunohystochemistry of brain tissue | Levels of Aβ42 negatively correlated with 8-OHG levels (p<0.02) | (Nunomura et al., 2010) | |
| n=20 Ages 65–86 |
Alzheimer's Disease | Northwestern Blotting | ↑ 8-OHG in AD frontal cortex | (Shan et al., 2003) | |
| n=12 Ages 62–86 |
Alzheimer's Disease | Southern blotting and Semi-quantitative RT-PCR analysis from brain tissues | 50% of mRNAs are oxidized in AD frontal cortices (p<0.001) | (Shan and Lin, 2006) | |
| n=26 Ages 80–93 |
Alzheimer's Disease and Mild Cognitive Impairment | Northern immunoblot of brain tissue | ↑ levels of 8-OHG in the IP of AD and MCI patients (p<0.01); no change in levels from cerebellum | (Ding et al., 2005) | |
| n=70 Ages 49–74 |
Parkinson's Disease | ELISA from serum and CSF | ↑ 8-OHG serum and CFS for PD patients (p<0.001); no correlation with age | (Kikuchi et al., 2002) | |
| n=39 Ages 53–74 |
Parkinson's Disease | HPLC-EC of CSF and serum | ↑ 8-OHG in CSF of PD patients (p>0.001); no change in 8-OHG in serum of PD patients | (Abe et al., 2003) | |
| n=40 Ages N.G. |
Patients with carotid stenosis > 70% | Immunohistochemistry of carotid endarterectomy specimens | ↑ 8-OHG immunoreactivity of affected tissues compared with adjacent unaffected tissue* | (Martinet et al., 2004) | |
| Protein Carbonyls | n=55 Ages 21–40 and 61–85 |
Healthy | Spectrophotometric method for carbonyl assay | ↑ levels in aged (p<0.01) | (Mutlu-Turkoglu et al., 2003) |
| n=29 Ages 15–88 |
Alzheimer's Disease | Spectrophotometric analysis using brain tissues | ↑ levels in aged (p<0.0001) | (Smith et al., 1991) | |
| n=76 48–91 |
Alzheimer's Disease | Spectrophotometric analysis using brain tissues | ↑ levels parietal lobe of AD patients (p<0.01); no correlation with age | (Lyras et al., 1997) | |
| n=76 Ages 32–90 |
Essential arterial hypertension | 2,4-dinitrophenyl hydrazine assay from serum | ↑ in patients with hypertension (p<0.001) | (Kedziora-Kornatowska et al., 2004) | |
| n=194 Ages 18–84 |
Healthy | ELISA of blood plasma | Minor ↑ in levels with age (p=0.34) | (Gil et al., 2006) | |
| n=100 Ages 20–70 |
Healthy | Absorbance measurement from serum | ↑ levels in aged (p<0.05) | (Kasapoglu and Ozben, 2001) | |
| n=84 Ages 23–66 and 92–96 |
Healthy | 2,4-dinitrophenyl hydrazine assay from plasma | ↓ levels in aged (p<0.01) | (Traverso et al., 2003) | |
| GSH | n=194 Ages 18–84 |
Healthy | Colorimetric assay from RBCs | ↓ levels with age (p<0.001) | (Gil et al., 2006) |
| n=119 Ages 19–93 |
Diabetes and Age Related Macular Degeneration | HPLC from plasma | ↓ plasma levels in aged (p<0.01) | (Samiec et al., 1998) | |
| n=76 Ages 32–90 |
Essential arterial hypertension | Colorimetric assay from whole blood | ↓ in patients with hypertension (p<0.01) | (Kedziora-Kornatowska et al., 2004) | |
| n=170 Ages 20–94 |
Healthy | Colorimetric assay from whole blood | ↓ levels in healthy aging adults (p<0.001) | (Lang et al., 1992) | |
| n=53 Over age 65 |
Alzheimer's Disease | HPLC from RBCs, lymphocytes, and plasma | ↓ levels in male AD patients (p<0.05), but no change in female patients* | (Liu et al., 2005) | |
| n=80 Ages 18–85 |
Healthy | Colorimetric assay from RBCs | ↓ in erythrocyte RBC levels with age (p<0.001) | (Rizvi and Maurya, 2007) | |
| n=28 Ages 34–82 |
Healthy | Colorimetric assay from RBCs | No effect on erythrocyte RBC levels with age* | (Kędziora-Kornatowska et al., 2007) | |
| n=100 Ages 20–70 |
Healthy | Measured from RBCs by the method of Fairbanks and Klee | No effect on erythrocyte RBC levels with age* | (Kasapoglu and Ozben, 2001) | |
| Isoprostanes | n=421 Ages 21–89 |
Healthy | GC-MSofCSF | ↑ of F2-isoprostanes over the life span (P,0.001) | (Montine et al., 2011) |
| n=68 Ages 27–71 |
Arthritis | Radioimmunoassay of immunoreactive (RIA) 8-iso-PGF2αin Serum | ↑ levels in arthritic disorders associated with oxidant stress* | (Basu et al., 2001) | |
| n=60 Ages 30–60 |
Diabetes mellitus | GC-MS/NICI from plasma | ↑ levels in individuals with diabetes (p<0.001) | (Gopaul et al., 1995) | |
| n=25 Ages 49–61 |
Renal Failure | GS-MS from urine | ↑ levels in patients in renal failure (p<0.02) | (Holt et al., 1999) | |
| n=25 Ages 49–61 |
Renal Failure | GS-MS from urine | ↑ levels in patients in renal failure (p<0.02) | (Holt et al., 1999) | |
| n=845 Age 85 |
Newcastle 85+ cohort | LC-MS/MS from plasma | No correlation with frailty (p=0.28) | (Collerton et al., 2012) | |
| IL-6 | n=845 Age 85 |
Newcastle 85+ cohort | Electrochemiluminescence | ↑ levels correlated with ↑ frailty (p=0.023) | (Collerton et al., 2012) |
| n=436 Ages 70–79 |
Women's Health and Aging Studies | ELISA from blood | ↑ levels correlated with ↑ frailty (p<0.01) | (Fried et al., 2009) | |
| n=720 Ages 65+ |
InCHIANTI study population | ELISA from serum | ↑ levels correlated with ↓ mobility (p<0.05) | (Bandeen Roche et al., 2009) | |
| n=110 Ages 77–91 |
Healthy and Function impaired | ELISA from plasma | ↑ levels correlated with ↑ frailty (p<0.01) | (Hubbard et al., 2009) | |
| n=30 Ages 74–98 |
Community-dwelling older adults | ELISA from serum | ↑ levels correlated with ↑ frailty (p<0.01) | (Leng et al., 2002) | |
| n=51 Ages 77–98 |
Community-dwelling older adults | ELISA from serum | No association between IL-6 levels and frailty (p=0.26) | (Leng et al., 2004a) | |
| n=22 Ages 77–96 |
Community-dwelling older adults | ELISA from PBMCs | No association between unstimulated IL-6 levels and frailty*; when stimulated ↑ levels correlated with ↑ frailty (p<0.03) | (Leng et al., 2004b) | |
| n=1106 Ages 65–101 |
Women's Health and Aging Studies | ELISA from serum | ↑ levels correlated with ↑ frailty (p<0.001) | (Leng et al., 2007) | |
| n=193 Ages 72–97 |
Community-dwelling older adults | ELISA from serum | ↑ levels correlated with ↑ frailty (p<0.05) | (Leng et al., 2011) | |
| n=724 Ages 70–79 |
Women's Health and Aging Studies | ELISA from plasma | ↑ levels correlated with ↑ frailty (p<0.01) | (Schmaltz et al., 2005) | |
| n=2826 Ages 65–80 |
Cardiovascular Health Study | ELISA from blood | ↑ levels correlated with ↑ frailty (p<0.01) | (Barzilay et al., 2007) | |
| n=1800 Ages 65–79 |
Women's Health Initiative | Plasma biomarker assay | No association between IL-6 levels and frailty (p=0.27) | (Reiner et al., 2009) | |
| CRP | n=845 Age 85 |
Newcastle 85+ cohort | Assayed from blood | ↑ levels correlated with ↑ frailty (p<0.001) | (Collerton et al., 2012) |
| n=90 Ages 68–86 |
Community-dwelling older adults | Roche Tina-quant CRP HS assay | ↑ levels correlated with ↑ frailty (p=0.01) | (Wu et al., 2009) | |
| n=110 Ages 77–91 |
Healthy and Function impaired | Assayed spectrophotometricly from serum | ↑ levels correlated with ↑ frailty (p<0.05) | (Hubbard et al., 2009) | |
| n=1800 Ages 65–79 |
Women's Health Initiative | Plasma biomarker assay | No association between CRP levels and frailty (p=0.95) | (Reiner et al., 2009) | |
| n=2826 Ages 65–80 |
Cardiovascular Health Study | ELISA from blood | ↑ levels correlated with ↑ frailty (p<0.001) | (Barzilay et al., 2007) | |
| n= 4735 Ages 67–79 |
Cardiovascular Health Study | ELISA from blood | ↑ levels correlated with ↑ frailty (p<0.001) | (Walston et al., 2002) | |
| n=720 Ages 65+ |
InCHIANTI study population | ELISA from serum | No significant association between CRP levels and frailty* | (Bandeen Roche et al., 2009) |
No statistical values given
N.G.= Not Given
Same p-values from both studies
Table 2.
Population Studies Correlating Selected Oxidative and Inflammatory Biomarkers
| Marker | Cohort | Disorder | Methodology | Result | Reference |
|---|---|---|---|---|---|
| Fluorescent Heme Degredation Products | n=96 Ages 30–64 |
Community dwelling bi-racial cohort | Alkaline COMET assay using lymphocytes | ↑ SSBs correlates with ↑ heme degradation products in AA males; SSBs correlated with ↑ heme degradation products in AA females; negative correlation between heme degradation products and SSB-RC | (Trzeciak et al., 2012) |
| Protein Carbonyls | n=96 Ages 30–64 |
Community dwelling bi-racial cohort | Alkaline COMET assay using lymphocytes | No relationship between protein carbonyls and SSBs | (Trzeciak et al., 2012) |
| IL-6 | n=730 Ages 22–93 |
Framingham Heart Study | Non-cross-reacting radioimmunoassay from PBMCs | ↑ levels with age and with increasing CRP levels (p<0.001) | (Roubenoff et al., 1998) |
| n=59 Ages 23–80 |
Healthy volunteers | ELISA from plasma | ↑ levels with age, but no correlation to CRP levels* | (Hager et al., 1994) | |
| n=220 Ages 68–75 |
Healthy | ELISA from serum; UPLC MS/MS from urine | No correlation with 8-oxodG levels (p=0.24) or 8-OHG levels (p=0.7) | (Broedbaek et al., 2009) | |
| CRP | n=136 Ages 44–70 |
Acute Coronary Syndrome | Alkaline COMET assay using lymphocytes | Weak correlation with SSB in patients with ACS (r = 0.544, p < 0.001) | (Demirbag et al., 2005) |
| n=130 Ages 46–70 |
Metabolic syndrome | Alkaline COMET assay using lymphocytes | No correlation with SSB levels in healthy and MBS (r=0.098) | (Demirbag et al., 2006) | |
| n=96 Ages 30–64 |
Community dwelling bi-racial cohort | Alkaline COMET assay | Significant interaction between CRP and sex in their effect on residual DNA damage (p=0.002) | (Trzeciak et al., 2012) | |
| n=30 Ages 23–69 |
Renal Disease on dialysis | FISH in flow cytometry | ↑ CRP levels correlate with ↓ telomere length (r=0.74, p=0.007) | (Ramirez et al., 2005) | |
| n=64 Ages 43–55 |
Cardiac X Syndrome | Alkaline COMET assay using lymphocytes | Correlation between CRP levels and lymphocyte DNA damage (p=0.001) | (Gur et al., 2007) | |
| n=32 Ages 36–86 |
End Stage Renal Disease on dialysis | ELISA for 8-oxodG using serum | Positive correlation between CRP and 8-oxodG (ρ=0.4, p<0.02) | (Haghdoost et al., 2006) | |
| n=220 Ages 68–75 |
Healthy | ELISA from serum; UPLC MS/MS from urine | No correlation between levels and 8-oxodG levels (p=0.28) or 8-OHG levels (p=0.11) | (Broedbaek et al., 2009) | |
| n=96 Ages 30–64 |
Community dwelling bi-racial cohort | Alkaline COMET assay using lymphocytes | positive correlation in AA males between SSBs and CRP (p=0.022) | (Trzeciak et al., 2012) | |
| n=117 Ages 30–64 |
Community dwelling healthy females | ELISA from serum | ↑ levels of 8-oxodG with ↑ CRP (p<0.02) | (Noren Hooten et al., 2012) |
2. DNA oxidative lesions as a measure of oxidant stress
DNA is a highly susceptible target of free radicals, resulting in oxidation of DNA bases and the ribose sugar ring leading to sites of base loss and strand breaks. The rate of damage to DNA by free radicals is estimated to be 1,000 to 1,000,000 hits per day in a single cell (Ames et al., 1993). The accumulation of free radical DNA damage can be a lethal event for an organism. An increased baseline level of DNA oxidation (single strand breaks (SSBs) and oxidative base damage) is associated with several age-related diseases including: cardiovascular disease (Collins et al., 1998a), diabetes mellitus (Hannon-Fletcher et al., 2000), cancer (Malins et al., 2001), neurodegenerative disease (Morocz et al., 2002), and end-stage renal disease (Domenici et al., 2005). The level of DNA oxidative lesions depends on a variety of factors, including age (Malins et al., 2001), exposure to environmental hazards and genotoxic factors (Blasiak et al., 2000; Dusinska et al., 2006; Kopjar and Garaj-Vrhovac, 2001; Trzeciak et al., 2000; Wojewodzka et al., 1998), smoking (Piperakis et al., 1998), alcohol intake (Blasiak et al., 2000), and intracellular and extracellular metabolism (Knaapen et al., 2002).
2.1. 8-oxo-7,8-dihydro-guanine (8-oxoGua) and 8-oxo-7,8-dihydro-2'deoxyguanosine (8-oxodG)
Oxidative damage to DNA caused by free radicals can result in a variety of mutagenic lesions including: 2-hydroxy adenine, FapyAdenine, 8-oxoadenine, 5-hydroxycytosine, cytosine glycol, and thymine glycol. The predominant lesion produced is 8-oxo-7,8-dihydro-guanine (8-oxoGua). This lesion is produced by oxidative damage at the C-8 position of guanine, with an estimated number of 100–500 8-oxoGua bases arising daily in the genome (Lindahl, 1993). If left unrepaired this lesion can result in G-to-T transversion events. Oxidation of DNA can also result in 8-oxo-7,8-dihydro-2'deoxyguanosine (8-oxodG) lesions, an alteration to the guanine nucleoside. 8-oxoGua and 8-oxodG are the most mutagenic consequence of oxidant stress, and can be specifically detected from each other. Many assays have been developed to measure quantitatively 8-oxoGua and 8-oxodG bases in human DNA samples, such as HPLC, GC/MS, immunohistochemistry, and ELISA.
The best methodology for assessing 8-oxodG levels has been an area of concern for the oxidant stress field (the results using multiple methodologies are listed in Table 1). In the late 1990's the European Standards Committee on Oxidative DNA Damage (ESCODD) was formed to establish standard protocols for assessing 8-oxodG levels in DNA samples isolated from lymphocytes of healthy volunteers. This group of research laboratories has assessed the various methods available to prepare samples and measure 8-oxodG, as well as define standard units to report damage levels. The ESCODD laboratories have found the background level of oxidative damage to be approximately 0.3–4.2 8-oxodG per 106 Guanines, and are currently working on defining an absolute level of background oxidative damage (Gedik and Collins, 2005).
To date, no perfect method has been identified to determine 8-oxodG levels. Based on trials from member laboratories, it has been concluded that both the CG-MS and HPLC-MS/MS chromatography methods are not reliable for assessing low levels of oxidative damage (Collins et al., 2004). The lowest levels of background damage have been detected through an enzymatic approach utilizing the enzymes Formamidopyrimidine DNA glycosylase (FPG), Endonuclease III (EndoIII), or human 8-hydroxyguanine glycosylase 1 (hOGG1) to convert certain kinds of oxidative damage into AP sites or single stranded DNA breaks (Bjelland and Seeberg, 2003; Boiteux et al., 1992; Smith et al., 2006). The presence of AP sites and strand breaks can be analyzed using the single cell gel electrophoresis (COMET) assay under various conditions to assess the levels of 8-oxodG levels in DNA samples (Collins et al., 1997). Although this approach detects lower levels of damage, it is possible that it is an underestimate, as FPG, Endo III, and hOGG1 may not convert all oxidative lesions to SSBs.
Additional methods are available to measure 8-oxoGua and 8-oxodG levels from human biofluids, such as urine, serum, plasma, and blood. To assess mass spectrometry (MS), electrochemical detection (EC), and ELISA based methodologies for analysis of DNA lesions from urine, the European Standards Committee on Urinary (DNA) Lesion Analysis (ESCULA) was formed. Similar to ESCODD, the member laboratories are working to achieve a consensus between the various methods, as well as determine reference values for the lesions (Cooke et al., 2008). To date, ESCULA has shown that for urine analysis there is strong within technique agreement, as well as strong agreement in the results for both the MS and EC based assays. In contrast, the ELISA based methods had the largest within technique variation (possibly because different urine samples were used in the various studies) and found the highest 8-oxodG levels in urine. The current conclusion of ESCULA is that a greater than expected consensus was achieved for both MS and EC methodologies; however there is concern among the member labs about the use of ELISA methods to assess 8-oxodG levels in urine (Cooke et al., 2009; Evans et al., 2012).
Levels of oxidized bases have been assessed using an LC-MS/MS assay, as a first attempt to define reference ranges in urine. The results of these studies found that females excrete lower levels of 8-oxoGua in urine than males, and that 8-oxoGua levels are affected by age, sex, and hOGG1 polymorphism status (Andreoli et al., 2010; Andreoli et al., 2011; Manini et al., 2009). In addition, it was reported that 8-oxoGua levels are approximately 10 times higher than those of 8-oxodG (Andreoli et al., 2010; Andreoli et al., 2011).
To date a very limited number of studies have attempted to address the issue of 8-oxoGua and 8-oxodG levels in aging. Analysis of 8-oxodG levels from muscle DNA was performed by Mecocci et. al from a cohort of healthy individuals (Mecocci et al., 1999). The HPLC analysis showed an increase in 8-oxodG levels with increasing age (Mecocci et al., 1999). Similarly, analysis of leukocyte DNA from healthy subjects from a different cohort found that with increasing age, the levels of 8-oxodG damage also increased (Siomek et al., 2007). In frail individuals over 65 years of age, increased serum levels of 8-oxodG have been observed (Wu et al., 2009). Recently, we have found that serum levels of 8-oxodG increase with age in a cohort of middle aged women (Noren Hooten et al., 2012). These data support the free radical theory of aging that DNA damage increases with age.
Using many of the above mentioned assays, levels of 8-oxoGua and 8-oxodG have been examined in many disease states (summarized in Tables 1 and 2). Studies in cancer have proposed 8-oxoGua as a potential biomarker, with the supporting evidence that GC to TA transversions (potentially from 8-oxoGua lesions) have been observed within the ras and p53 genes in liver and lung cancers (Cooke et al., 2003; Hussain et al.; Rodin and Rodin, 2005). Although oxidative stress has been implicated in neurological disorders such as AD (Gabbita et al., 1998; Lovell et al., 1999; Lyras et al., 1997; Mecocci et al., 1994; Wang et al., 2005), PD (Alam et al., 1997; Nakabeppu et al., 2007; Zhang et al., 1999), and Huntington's disease (De Luca et al., 2008), it has been difficult to assess the level or determine the direct role of DNA oxidative lesions in these disorders (Alam et al., 2000; Te Koppele et al., 1996). Studies have examined levels of 8-oxoGua relating to cardiovascular disease and found increased levels of 8-oxoGua and 8-oxodG lesions in atherosclerotic plaques (De Flora et al., 1997; Martinet et al., 2002), and one study reported a strong association between increased levels of 8-oxodG and premature coronary artery disease in men (Collins et al., 1998a).
2.2. Single Stranded DNA Breaks
It is well-established that SSBs are a serious threat to genome stability and to cell survival. If SSBs are unrepaired there can be serious consequences such as base substitution mutagenesis, which often results in alteration of multiple bases (Dar and Jorgensen, 1995), as well as giving rise to double strand DNA breaks. Additionally, SSBs have been shown to cause stalling of the cell cycle in G1 (Huang et al., 1996b), G2/M abnormal mitoses (Johnson et al., 2000) and mitotic structures (Ame et al., 2009), and ultimately programmed cell death (Ame et al., 2009; Johnson et al., 2000; Yan et al., 2003).
As previously mentioned, one technique used to measure SSBs is the COMET assay. This assay examines DNA for damage by allowing supercoiled DNA to relax and lengthen to form what looks like tails upon electrophoresis, with the amount of DNA in the tail being indicative of the number of SSBs. The alkaline COMET assay is a sensitive and relatively inexpensive technique used for the detection of DNA damage as well as DNA repair (Singh et al., 1988). The major advantage provided by the alkaline COMET assay is the ability to analyze DNA damage and repair in individual cells. Furthermore, small numbers of cells are required for this assay which is particularly advantageous for analyses performed in samples from human populations. Since this method is very sensitive, even minimal changes in DNA damage levels and DNA repair capacity (DRC) in human populations caused by genetic and demographic variation can be studied.
Although the COMET assay has many advantages, and has been commonly used for measuring DNA damage by some research groups; there are disadvantages as well (Collins, 2002). One main disadvantage of this assay is that it is very labor intensive, often for only a small number of samples. For reproducibility of the assay each experiment must be carefully planned and executed with the proper positive controls to ensure that assays completed at different times can be compared. Also, the calculations made from the primary data must be done properly so that data can be compared between laboratories, as well as with other methods used to measure oxidative damage to DNA. In some instances, the COMET assay may be too sensitive, reaching saturation at low levels of damage and not representing the actual levels of damage. Many reports have used frozen samples for the COMET assay, but they must be preserved properly. Live or frozen cells must be at greater than 90% viability, otherwise erroneously high levels of damage can be detected. Additionally, the presence of alkali labile sites as well as DNA double strand breaks can interfere with the output of the assay.
Modifications to the original assay have been made that allow for the detection of oxidized bases (Collins et al., 1993). In addition human samples may be studied in retrospective longitudinal studies using a modification that permits accurate and reproducible analysis of cryopreserved peripheral blood mononuclear cells (PBMCs) (Trzeciak et al., 2009). Applications of the alkaline COMET assay in biomonitoring include analysis of nutrient and micronutrient effects on the level of DNA damage (reviewed in (Hoelzl et al.)), examination of DNA damage levels associated with exposure to genotoxic factors (Blasiak et al., 2000; Dusinska et al., 2006; Kopjar and Garaj-Vrhovac, 2001), and oxidative stress connected with human pathology (such as infectious diseases, diabetes mellitus, cardiovascular disease, and hemodialysis in renal failure patients) (Collins et al., 1998a; Collins et al., 1998b; Domenici et al., 2005; Hannon-Fletcher et al., 2000). Additionally, COMET has been extensively used to assess background levels of DNA damage in human populations (Kopjar et al., 2006; Smith et al., 2003; Stoyanova et al.). Recently, we have shown that sex and race affect the background level of SSBs in PBMCs from both whites and African-Americans (Trzeciak et al., 2012). We observed a significant increase in SSB levels in females in the overall cohort; however when stratified by race, the increase was specifically among African-American females. No effect was observed for the background levels of SSBs in relation to age in this cohort, which may be due to the fact that the age range of this cohort is younger (30–64 yrs) compared to other studies examining similar parameters.
Additional studies have been performed that examined the presence of DNA damage with increasing age and have yielded varying results. Using the COMET assay, increased DNA damage and FPG-sensitive sites were observed in older cohorts as compared to younger ones (Barnett and King, 1995; Dusinska et al., 2006; Humphreys et al., 2007; Kruszewski et al., 1998; Moller, 2006; Mutlu-Turkoglu et al., 2003), as well as in African Americans compared to whites (Watters et al., 2008). In contrast, additional studies have found that there is no correlation between increased age and frequency of DNA damage (Dusinska et al., 2006; Garm et al., 2012; Hyland et al., 2002; King et al., 1997; Wojewodzka et al., 1998). These different findings may have been influenced by the small cohort size or by the age distribution of the cohort studied. In Table 1, we have included information about cohort size and age range to further compare these different studies and their outcomes.
Another epidemiologic application of the assay is in determination of inter-individual variation in DRC (Popanda et al., 2003; Rajeswari et al., 2000; Smith et al., 2003). Analysis of DRC in human populations is an attractive biomarker to pursue for clinical investigators because alterations in several DNA repair pathways are linked with both heritable and sporadically occurring age associated diseases. The capacity of cells to repair DNA damage is an important factor, which affects the level of DNA damage present in cells. SSB repair can be expressed as the logarithm of the initial rate of DNA repair, the logarithm of the half-time of DNA repair, or the residual DNA damage after 30 and 60 min (Trzeciak et al., 2008). The logarithm of the initial rate of DNA repair is directly proportional to DRC, whereas other DNA parameters are inversely proportional to DRC. Studies have been completed on healthy individuals and cancer patients and found that there is decreased SSB repair capacity (SSB-RC) with increasing age (Spitz et al., 2003; Wei et al., 2000; Wei et al., 1993). However, other studies have reported no effect on SSB-RC by age, gender, smoking status, or frailty (Collerton et al., 2012; Garm et al., 2012; Marcon et al., 2003; Muller et al., 2002; Muller et al., 2001; Paz-Elizur et al., 2007; Rajaee-Behbahani et al., 2001; Smith et al., 2003).
We have found that SSB-RC is dependent on age, sex and race in cohort studies (Trzeciak et al., 2008; Trzeciak et al., 2012) using a modified COMET assay that evaluated the SSB-RC in PBMCs (Trzeciak et al., 2009). The results of these studies found a significant positive correlation in the entire cohort between the logarithm of the half-time of DNA repair and SSBs, as well as between the residual DNA damage after 30 min and SSBs. Additionally, we found a negative correlation between SSB-RC and the level of SSBs in the overall cohort. When stratified by sex, we found a significant increase in the fast SSB-RC in white females with age, while a decrease was observed in African-American females. Given that sex and race had significant changes in the various parameters is suggestive that SSB-RC has the potential to become a clinical biomarker. These results are the first steps to help us understand the relationship between DRC and oxidative damage to DNA and the role they play in human health and disease.
2.3. Double Stranded DNA Breaks
One of the most toxic lesions to DNA is the double-strand break (DSB). DSBs can be induced by ionizing radiation, stalled replication forks, meiotic recombination, V(D)J recombination, class switch recombination, and can be generated as a consequence of normal cellular processes like oxidative respiration that generates ROS. DSBs are formed when both DNA strands encounter DNA damage, within 10–20 base pairs of each other, resulting in a break of the phosphodiester bond. The presence of only a few DSBs (1–10 breaks) can induce p53-dependent G1 arrest and cell death (Huang et al., 1996a), explaining the toxicity of these lesions when unrepaired. DSBs cannot be repaired by the numerous template-directed repair systems, as both DNA strands are broken. DSBs can be repaired in two ways: by direct rejoining of the DNA ends, a process called non-homologous end joining or by use of the undamaged sister chromosome as the template for repair, a process called homologous recombination.
One of the first cellular responses to DSBs is phosphorylation of histone H2AX (Rogakou et al., 1998). Thousands of H2AX molecules adjacent to the break site become phosphorylated within minutes of the generation of a DSB, resulting in the formation of gamma-H2AX (γ-H2AX) foci (Rogakou et al., 1999) that most likely represent single DSBs (Pilch et al., 2003; Sedelnikova et al., 2002). By use of antibodies available against γ-H2AX, staining of γ-H2AX foci has become a valuable tool for the detection and evaluation of single DSBs in nuclei. Detection of γ-H2AX foci has been used as a biomarker for various cancers and cancer progression (reviewed in (Ivashkevich et al.)), and detection of foci at eroded telomere ends is used as a marker of aging (Nakamura et al., 2008; Takai et al., 2003).
Detection of γ-H2AX foci can be done in a number of ways, including staining and quantifying γ-H2AX foci by immunofluorescence microscopy, fluoresence-activated cell sorting (FACS) or immunoblotting to detect whole cell levels of γ-H2AX. Using lymphocytes isolated from human blood samples, Sedelnikova et al. found that levels of endogenous γ-H2AX foci increase with age (Sedelnikova et al., 2002). Similarly, using lymphocytes from individuals in the Baltimore Longitudinal Study on Aging, it was found that γ-H2AX foci increase with age, and increased γ-H2AX foci were also present in individuals with hypertension (Schurman et al., 2012). Additionally, it has been shown that the γ-H2AX response decreases with age in a non-significant manner (Garm et al., 2012).
An additional methodology to assess the level of DNA DSBs is by the neutral COMET assay. This assay is performed in a similar manner as the alkaline COMET assay (previously described) under neutral pH conditions, and the DNA in the tail formed during electrophoresis represents the presence of DNA DSBs. There are a very limited number of studies that have used this technique to examine DSBs in aging and age-related diseases. However, the neutral COMET assay has been used to detect DSBs in male sperm. The results have been contradictory finding no change in DSB levels (Schmid et al., 2007) as well as increases in DSB level with age (Singh et al., 2003). In addition, analysis of DSBs in age-related macular degeneration found no increase in DNA DSBs with age (Szaflik et al., 2009), while a recent study by Garm et. al found that DSB-RC decreases with age (Garm et al., 2012). The results of these studies, coupled with the fact that neutral conditions for the COMET assay may not just measure DSBs (Collins et al., 2008), contribute to the thought that further improvement of this technique is needed to accurately measure DSBs.
3. RNA oxidative lesions as a measure of oxidant stress
The human genome consists of more than 21,000 genes, encompassing only 1.5% of the total bases of the genome, yet approximately 80% of the DNA is transcribed into RNA (Birney et al., 2007). Recent data indicates that many of the non-gene encoding but transcribed RNAs have functional roles; however, the focus on nucleic acid oxidation research has been centered on DNA. The vast number and types of RNAs in the body provide a sizable target for ROS. There are many reasons that RNA is more prone to persistant oxidative damage than DNA including: 1) RNA is mainly single stranded, leaving it easily accessible to ROS 2) there is no identified active repair mechanism for oxidized RNA 3) RNA is less protected by proteins compared to DNA and 4) cytoplasmic RNAs are near the mitochondria where many ROS are produced. Indeed, it has been shown that oxidative damage to RNA is more prevalent than oxidative damage to DNA in humans (Henriksen et al., 2009; Hofer et al., 2005; Shen et al., 2000; Weimann et al., 2002), and that RNA oxidation is influenced by environment not by genetics as shown through twin studies (Broedbaek et al., 2011a). Multiple studies have shown that increased RNA oxidization products are present in diseases related to aging including: dementia with Lewy bodies (DLB) (Nunomura et al., 2002), AD (Abe et al., 2002; Isobe et al., 2009; Nunomura et al., 1999a; Nunomura et al., 1999b), PD (Kikuchi et al., 2002; Zhang et al., 1999), atherosclerosis (Martinet et al., 2004), and myopathies (Tateyama et al., 2003). In addition, increased RNA oxidation has been observed in hemochromastosis (Broedbaek et al., 2009), Down syndrome (Nunomura et al., 1999a), prion disease (Guentchev et al., 2002; Petersen et al., 2005), and xeroderma pigmentosum (Hayashi et al., 2005).
3.1. Types of Oxidized RNAs
ROS oxidation of RNA subtypes has not been thoroughly studied. Not surprisingly, it has been shown that both ribosomal (rRNA) and messenger (mRNA) RNAs can be oxidized. Studies of both mild cognitive impairment (MCI) and in AD patients have found increased rRNA oxidation in the inferior parietal lobe (Ding et al., 2005) and as a consequence of RNA-bound iron oxidized by the Fenton reaction (Honda et al., 2005). It has been found that in AD patients oxidation occurs in up to 50% of mRNA of the frontal cortices (Shan and Lin, 2006), and that some mRNA species are more susceptible to oxidation than others (Nunomura et al., 2006; Shan et al., 2003). Oxidation of other non-coding RNAs, including microRNAs, transfer RNAs, small nuclear RNAs, long non-coding RNAs, small nucleolar RNAs has not been investigated. Given the important roles of these different RNAs in gene function and regulation, it will be important in the future to investigate whether these different RNAs are susceptible to oxidative damage and whether this changes the role of these RNAs in aging and/or disease. Although we have just begun to investigate the outcomes of RNA oxidation, currently known consequences of RNA oxidation include premature termination of reverse transcription (Rhee et al., 1995), ribosomal dysfunction resulting in decreased protein synthesis (Ding et al., 2005), translation of nonfunctional or truncated proteins (Tanaka et al., 2007), and proteins containing mutations that results in misfolding or protein aggregation (Lee et al., 2006; Nunomura et al., 2010; van Leeuwen et al., 1998).
3.2. 7,8-dihydro-8-oxo-guanosine (8-OHG)
The similarity between human DNA and RNA molecules would lead to the idea that the oxidative lesions observed in DNA could also be observed on the corresponding bases in RNA (Barciszewski et al., 1999; Bellacosa and Moss, 2003). However, at present only the oxidized product of guanosine, 7,8-dihydro-8-oxo-guanosine (8-OHG), has been actively investigated (current studies summarized in Table 1). 8-OHG is the most examined of the RNA oxidation products due to its similarity to the 8-oxoG lesion in DNA, and the ability to use many of the same methodologies verified on DNA substrates for the analysis of RNA oxidation products. These methodologies include: chromatography and immunological (antibody based) detection (the two most often used methods), primer extension and reverse transcription (Rhee et al., 1995), aldehyde reactive probes (Cooke, 2009; Tanaka et al.), and Southern blotting (first creating cDNA followed by DIG-labeling of sUTPs) (Shan and Lin, 2006).
The various chromatography methods utilized to detect oxidized RNA products are highly specific and include HPLC methods coupled with UV (Park et al., 1992), electrochemistry (EC) (Hofer et al., 2006), or mass spectrometry (MS) (Andreoli et al., 2010; Andreoli et al., 2011; Henriksen et al., 2009; Weimann et al., 2002), as well as UPLC separation and detection by tandem mass spectrometry (UPLC-MS/MS) (Henriksen et al., 2009). Of these, the HPLC-EC method is able to detect 8-OHG bases at 20 fmol and the HPLC-MS procedure can detect 8-OHG bases at 12.5 fmol (Hofer et al., 2006; Weimann et al., 2002). As previously discussed, the verification efforts made by the ESCODD and ESCULA groups for the 8-oxoG and 8-oxodG DNA oxidation products has facilitated and promoted the use of the same chromatography methods for RNA oxidation products, although the studies performed by both groups have not yet looked specifically at 8-OHG. Currently, these procedures have been used for human urine and cerebral spinal fluid (CSF) samples, but for organ or site specific disease analyses tissue samples would be preferred. However, at this time large tissue samples (50–100 mg) are required for analysis making these studies difficult or impractical to perform.
Employing the chromatography protocols, studies on healthy cohorts have found that there are higher levels of 8-OHG oxidation in RNA than 8-oxodG oxidation in DNA (Henriksen et al., 2009; Park et al., 1992; Weimann et al., 2002), that men secrete higher levels of 8-OHG than females, and that levels of the 8-OHG oxidation product increase with age (Andreoli et al., 2010). Abe et al found that 8-OHG levels were increased in the CSF of both AD and PD patients, but that the increased levels in AD patients were not recapitulated in measurements from the serum, indicating the oxidation levels in CSF may more accurately represent oxidation observed in brain tissues (Abe et al., 2003; Abe et al., 2002; Isobe et al., 2009). Similarly, increased levels of 8-OHG were identified in urine samples from individuals with Lewy Body dementia with (Nunomura et al., 2002).
The immunological methodologies used to identify RNA oxidation products employ the use of two monoclonal antibodies, 15A3 and 1F7, which detect 8-OHG lesions (Hofer et al., 2006; Yin et al., 1995). Several groups have conducted studies examining post-mortem brain tissues from individuals with various neurological disorders through antibody based assays. These studies have found increased levels of 8-OHG lesions to be associated with AD (Ding et al., 2005; Honda et al., 2005; Nunomura et al., 1999a; Nunomura et al., 1999b; Nunomura et al.; Shan and Lin, 2006), PD (Kikuchi et al., 2002; Zhang et al., 1999), Down Syndrome (Nunomura et al., 1999a; Nunomura et al., 2000), and dementia with Lewy Bodies (Nunomura et al., 2002). Similarly, using both immunohistochemistry and ELISA assays it has been shown that increased 8-OHG levels are present in individuals with atherosclerosis (Martinet et al., 2004; Martinet et al., 2002). Although experimentation using antibodies to detect 8-OHG has shown increased levels of RNA oxidation products, the antibody specificity is often still questioned. Many experiments using the immunological methods require validation by other more specific methods.
4. Protein oxidation as a measure of oxidant stress
Another target of free radical attack is proteins, which can be both oxidized and cross-linked. These alterations to proteins can result in inhibition of function of various cellular proteins, in some cases permanently. It is estimated that 30–50% of cellular proteins are altered or dysfunctional in cells of older animals due to free radicals (Levine and Stadtman, 2001; Stadtman, 1995). Furthermore, oxidation of proteins has been demonstrated to affect the enzymatic activity of certain proteins in other species.
4.1. Protein Carbonyls
Oxidation of protein can lead to protein carbonyls (Garrison et al., 1962), which are most often formed on the amino acids lysine, arginine, proline, and threonine and by the fragmentation products of peptide bond cleavage reactions. Protein carbonyls are produced by the addition of carbonyl groups (such as aldehydes and ketones) as side chains on these amino acids. The assay detecting protein carbonyls is one of the most frequently used methodologies to examine levels of protein oxidation caused by oxidative stress (Beal, 2002; Chevion et al., 2000; Shacter, 2000; Stadtman and Berlett, 1997). One key feature for the use of protein carbonyls to assess oxidative damage is the fact that they are chemically stable and can be stored at −80°C for 3 months without changes in detectability (Griffiths, 2000).
Several studies have shown age-related increases in protein carbonyl levels for healthy human subjects (all reviewed in (Voss and Siems, 2006)), particularly in heart, muscle, brain, and plasma (Floyd and Hensley, 2002; Gianni et al., 2004; Gil et al., 2006; Grune et al., 2001; Stadtman, 2001). Elevated levels of protein carbonyls in serum, plasma, and tissues are observed in diabetes (Dominguez et al., 1998; Telci et al., 2000), inflammatory bowel disease (Lih-Brody et al., 1996), AD (Conrad et al., 2000; Korolainen et al., 2002; Smith et al., 1991), Werner Syndrome (Oliver et al., 1987), PD (Floor and Wetzel, 1998), Cystic Fibrosis (Range et al., 1999), essential arterial hypertension (Kedziora-Kornatowska et al., 2004), and rheumatoid arthritis (Mantle et al., 1999)(for review see (Dalle-Donne et al., 2006; Stadtman, 2001)). Most studies have agreed that increased levels of protein carbonyls correlate with age. Various methods (Spectrophotometric, ELISA based, and protein oxidation assays) using plasma, serum, and brain tissues have found this positive correlation of protein carbonyl levels and age (Gil et al., 2006; Kasapoglu and Ozben, 2001; Mutlu-Turkoglu et al., 2003; Smith et al., 1991; Traverso et al., 2003) (summarized in Table 1). An additional study by Howard et al found that protein carbonyl levels correlated with poor grip strength in women in the Women's Health and Aging Study (WHAS), implying that oxidative stress may contribute to reduced grip strength and concordant loss of muscle strength in aging (Howard et al., 2007). In a bi-racial cohort, we observed no significant relationship between the levels of protein carbonyls in plasma and oxidative DNA lesions (Trzeciak et al., 2012).
5. Oxidant Stress of Red Blood Cells (RBCs)
Red blood cells are the most abundant cells in the human body and primarily act to transport O2 and CO2 between the lungs and tissues of the body. RBCs pass through the lungs approximately once per minute, where they are exposed to conditions of oxidative stress. A unique feature of RBCs is an effective antioxidant system that protects these cells as well as other organs and tissues from free radical attack. However, despite the presence of an antioxidant system, RBCs are highly susceptible to oxidative damage.
The major protein in RBCs is hemoglobin, which constitutes nearly 90% of the dry weight of the cell. The abundance of hemoglobin and the constant exposure to oxygen in RBCs create the ideal conditions for oxidation of hemoglobin. In the body, free iron is readily oxidized to Fe (III), which cannot bind oxygen; however, in hemoglobin Fe (II) is much more stable. Nevertheless, 3% of the hemoglobin is oxidized in a 24-hr period via autoxidation, producing Fe(III)hemoglobin (metHb) and a superoxide anion radical (Rifkind et al., 2004). Although there are enzymatic systems in place to reduce the Fe (III) back to the functional Fe (II) form, approximately 1% of the Fe (III) form is present in the blood at steady state. The production of superoxide anions oxidation of hemoglobin is thought to be the major source of oxidative stress in red blood cells, and contribute to additional damage in other tissues (Rifkind et al., 1993).
5.1. Fluorescent Heme Degradation Products
A novel marker of protein oxidation, fluorescent heme degradation products (hemoglobin band 1), are end products formed as a result of ROS attack of hemoglobin and hemoglobin autoxidation (Nagababu and Rifkind, 1998). When autoxidation of hemoglobin or ROS attack on RBCs occurs, hemoglobin is broken down and heme degradation products are produced. These products can be released into the circulation and the presence of heme degradation products in fresh blood samples reflect the pool of non-neutralized ROS generated that have escaped from the RBC antioxidant system. Since red cells have no enzymatic system in place to remove heme degradation products, it is believed that they may be a relevant biomarker of oxidative stress originating from the red cell (Nagababu and Rifkind, 2004). It has been shown that the fluorescent heme band is formed by the reaction of membrane bound hemoglobin with H2O2 or hydroperoxide which is not eliminated by the cytoplasmic antioxidant systems (Nagababu et al., 2010).
Levels of red blood cell oxidative stress may be influenced by many factors including race. African Americans had higher RBC oxidative stress as measured by fluorescent heme degradation products than Whites (Szanton et al., 2011). Though this is the first time that RBC oxidative stress has been studied in an African American cohort, this is not the first time that measures of oxidative stress have been found to be elevated in African Americans. Levels of plasma carbonyls, nitric oxide, and oxidized glutathione were found to be higher in African Americans in comparison to whites by in vivo assays and in some cases in in vitro measurements as well (Feairheller et al.; Morris et al., 2012). In addition, work underway has shown that heme degradation products may be related to race. (Evans MK et al, manuscript in preparation).
There is some evidence in the literature that psychological stress may also play a role in generating oxidative stress. Szanton and colleagues found that perceived racial discrimination reported by both African Americans and whites correlated with higher levels of RBC oxidative stress as measured by fluorescent heme degradation products (Szanton et al., 2011). Notably, when this cohort was stratified by race, the association was only significant for African Americans. These early findings may suggest a link between oxidative stress and psychological stress which might play a role in the disproportionate burden of age-associated diseases like cardiovascular disease, cancer and diabetes mellitus among minority populations.
In a bi-racial cohort we have investigated the relationship between heme degredation products, SSB levels and SSB-RC (Trzeciak et al., 2012). We observed increased SSB levels with increasing heme degradation product levels in African-American males. In contrast, African-American females showed a decrease in SSBs with levels of heme degradation products. We also observed a negative correlation between the level of heme degradation products and SSB-RC as measured by the residual DNA damage after 60 min, as measured by the comet assay, in the entire cohort. These results provide what we believe is the first evidence of a relationship between DNA damage in PBMCs and heme degradation products, and may in the future result in a clinically useful marker for oxidative damage.
6. Other Markers of Oxidant Stress
6.1. Glutathione (GSH)
The glutathione system is pivotal and thought to be a critical safeguard in the cellular defense against oxidative stress. GSH plays a central role as a non-enzymatic antioxidant and functions to eliminate peroxides and maintain the thiol/disulfide redox state of proteins that are critical for proper biological function. GSH also maintains the redox state of ascorbate enhancing its function as a non-enzymatic antioxidant (Jones, 2006).
Several laboratories have reported that reduced glutathione levels with age are associated with a decrease in antioxidant capacity (Erden-İnal et al., 2002; Gil et al., 2006; Matsubara and Machado, 1991). It has been established that a decrease of whole blood, plasma, and lymphoblast GSH concentration may be associated with aging (Lang et al., 1992; Samiec et al., 1998), as well as rheumatoid arthritis (Gambhir et al., 1997), AIDS (Pirmohamed et al., 1996), AD (Cecchi et al., 1999), respiratory distress syndrome (Rahman and MacNee, 2000), Werner syndrome (Pagano et al., 2005), ALS (Bonnefont-Rousselot et al., 2000), alcoholic liver disease (Hadi Yasa et al., 1999), diabetes (Samiec et al., 1998), essential arterial hypertension (Kedziora-Kornatowska et al., 2004), and cataract genesis (Lou and Dickerson, 1992).
RBCs require glutathione to maintain the native structure of hemoglobin and of enzymes and membrane proteins (Kosower and Kosower, 1978), as well as protecting the cells from endogenous and exogenous toxins (Meister and Anderson, 1983). In addition, the level of GSH in RBCs is approximately 1000 times greater than the levels found in plasma (Beutler and Gelbart, 1985), making it easier to assess smaller changes in levels. Several studies have analyzed GSH levels specifically from RBCs, and have found varying results summarized in Table 1. Two studies performed on groups of either healthy men or healthy women have found that there is no change in RBC GSH with age (Kasapoglu and Ozben, 2001; Kędziora-Kornatowska et al., 2007). Contradictory to this, studies of both healthy individuals and AD patients have found reduced GSH levels in RBCs with age (Liu et al., 2005; Rizvi and Maurya, 2007). The limited number of studies examining RBC GSH levels indicate that further investigation is needed to determine whether RBC GSH can be used as a valid marker of oxidative stress with age and in age-related diseases.
We have assessed the relationship between RBC GSH levels, SSB levels, SSB-RC, race, and sex in participants from the HANDLS study (Trzeciak et al., 2012). We found that the RBC GSH concentration is positively correlated with the level of SSB in white females, but observed no effect in while males or African-Americans of either sex. In white females, we also found that the RBC GSH concentration is negatively correlated with SSB repair capacity as measured by both the logarithm of the initial rate of repair and the logarithm of the half-time of repair. A very significant correlation between RBC GSH concentration and the level of Endo III-labile sites was observed in white males and females, but not in African Americans. In addition, the entire cohort showed a positive correlation between the levels of RBC GSH and heme degradation products. Given the range of study findings, plasma, serum and/or RBC GSH levels when used alone may not be reliable or consistently correlative markers of oxidant stress and its association with aging.
6.2. Lipid peroxidation
Peroxidation of lipids is one of the most extensively studied free radical induced reactions in the body. ROS induced lipid peroxidation occurs when a reactive hydrogen atom is extracted from the methylene group of an unsaturated fatty acid. Once this process has begun, lipid peroxidation spreads as a ROS-induced chain reaction until the levels of peroxidation are sufficiently high to result in the production of a non-radical molecule. The peroxidation of lipids can result in damage to the cell membrane due to the high concentration of lipids present. In addition, the end products of lipid peroxidation can be both mutagenic and carcinogenic, and play a role in aging and disease progression.
Here we discuss F2 isoprostanes as a marker of lipid peroxidation. In recent years, F2 isoprostanes have proven to be the among the most sensitive and reliable biomarkers for the investigation of lipid peroxidation (Kadiiska et al., 2005; Morrow et al., 1990). They have been measured as parts of clinical trials and observation studies to determine the role of lipid peroxidation in aging and disease.
6.2.1. Isoprostanes
Isoprostanes are prostaglandin-like substances produced in vivo by esterification of arachidonic acid (Morrow et al., 1990; Pryor et al., 1976). There are many cellular functions of prostaglandins including: activation of the inflammatory response, where they are produced by white blood cells; regulating blood pressure; increasing blood flow in the kidneys; and promoting the bronchial constriction associated with asthma (Miller, 2006). It has also been shown that oxidative stress induces the production of prostaglandins in human cells (Malek et al., 2001) and that some prostaglandins can produce intracellular stress (Kondo et al., 2001).
Levels of isoprostanes change very little on a day to day basis in both healthy and disease states (Bachi et al., 1996; Reilly et al., 1996) and are most commonly measured in urine, as isoprostanes are chemically stable in this biofluid and artificial auto-oxidation does not occur in urine to produce false positives (Cracowski et al., 2002). Products of the isoprostane pathway have biological actions and may play a pivotal role in human disease progression (Morrow and Roberts, 1997). It has been shown that isoprostanes can act as a vasoconstrictor in tissues including the gastrointestinal tract, kidney, blood vessels, lymphatic vessels (Oguogho et al., 1998; Sinzinger et al., 1997), the bronchi (Janssen et al., 2000), and the uterus (Crankshaw, 1995). Isoprostanes are believed to be involved in various disease states of the lung (Janssen, 2008) and have been shown to prevent aggregation of platelets (Cracowski and Durand, 2006).
There are various forms of isoprostanes, which can be formed from the same initial substrate in vivo, the D2, E2, F2, and H2 families each containing compounds of various lengths and conformations (Morrow and Roberts, 1997). The letter corresponding to the various isoprostane compounds (D, E, F, G, and H) indicate the type of cyclopentane ring that comprises the molecule (Rokach et al., 1997). The first of these discovered, the F2-isoprostanes, are considered the best available biomarkers of lipid oxidation and oxidative stress in vivo (Roberts and Morrow, 2000). F2-isoprostanes are detectable in liquid form in all bodily fluids and in their esterified form in biological tissues, indicating physiological levels of oxidative stress (Morrow and Roberts, 1997; Pratico et al., 1998a; Roberts and Morrow, 2000). Measurement of F2-isoprostanes has many advantages over other potential biomarkers of oxidative stress including: they are chemically stable; a specific product of oxidation; formed in vivo; increased levels are observed with oxidant injury; a baseline level is definable; and levels are unaffected by diet (Gopaul et al., 2000; Montuschi et al., 2007; Roberts and Morrow, 2000). One of the pitfalls of using F2-isoprostanes as a marker is the fact that once released into the circulation they are rapidly metabolized and eliminated, thus plasma levels may vary significantly and not be entirely reflective of actual levels.
An increase in F2-isoprostane levels is an early event in asthma (Dworski et al., 1999; Montuschi et al., 1999), hepatic cirrhosis (Pratico et al., 1998b), scleroderma (Cracowski et al., 2001), and AD (Montine et al., 2002; Pratico et al., 2002) implying a role for oxidative stress in these diseases. Additionally, elevated levels of F2-isoprostanes are detected in atherosclerosis (reviewed in (Davies and Roberts Ii, 2011)), arthritic diseases (Basu et al., 2001), diabetes (Gopaul et al., 1995), Huntington's disease (Montine et al., 1999), smoking (Morrow et al., 1995; Reilly et al., 1996), and renal failure (Holt et al., 1999; Moore et al., 1998). An investigation of cerebral spinal fluid from healthy volunteers found that there was a positive correlation between increased F2-isoprostanes and aging (Montine et al., 2011). However, it has recently been shown that there is no correlation between levels of isoprostanes and frailty, as a measure of aging (Collerton et al., 2012).
7. Inflammation as a form of oxidant stress
In the format of this review, we are unable to present a complete review of inflammation. However, we feel that it is important to mention inflammation because of its association with oxidative stress and with aging and age-related diseases.
Inflammation is the body's reaction to both endogenous and exogenous harmful stimuli and the initiation of the healing process, which can be classified into two types, acute and chronic. Chronic inflammation has cellular side effects, including production of free radicals that can result in oxidative damage and depletion of antioxidants (Hold and El-Omar, 2008). Macrophages, one of the main components of the inflammatory response, produce ROS in the forms of superoxide, hydrogen peroxide, hydroxyl radical, nitric oxide, hydrochlorous acid, and peroxynitrite (Fialkow et al., 2007; Maeda and Akaike, 1998). Production of ROS by the immune system results in production of pro-inflammatory cytokines and chemokines (Costa et al., 2008; Ryan et al., 2004). There are numerous cytokines activated by inflammation these include IL-1, IL-6, TNF-α, and IFN-γ which have been shown to generate ROS (Chapple, 1997).
Several studies hypothesize that IL-6 is a central mediator of the inflammatory response (Fasshauer and Paschke, 2003; McCarty, 1999). Many studies suggest that IL-6 levels are also an indicator of increased frailty (Bandeen Roche et al., 2009; Barzilay et al., 2007; Collerton et al., 2012; Fried et al., 2009; Hubbard et al., 2009; Leng et al., 2002; Leng et al., 2011; Leng et al., 2007; Schmaltz et al., 2005); however there are a few reports showing no association (Leng et al., 2004a; Leng et al., 2004b; Reiner et al., 2009). A positive correlation was found between SSBs and IL-6 levels under extreme exercise conditions (Mastaloudis et al., 2004a; Mastaloudis et al., 2004b), as well as in a healthy aged population (Broedbaek et al., 2011b). No correlation was observed between the RNA oxidation product 8-OHG and IL-6 in elderly patients with low-grade inflammation (Broedbaek et al., 2009). Conflicting reports of IL-6 levels in AD have been reported as both unchanged (März et al., 1997) or decreased (Hampel et al., 1998).
High-sensitivity C-reactive protein (CRP) is an acute phase inflammatory protein, belonging to the pentraxin family, which consists of five non-covalently bound subunits arranged in a disk (Thompson et al., 1999). Production of CRP is stimulated by cytokines in response to infection or inflammation in blood vessels or tissues (Clyne and Olshaker, 1999; Libby and Ridker, 2004). In relation to aging, few groups have examined the relationship between CRP levels and frailty. These studies have found both a positive correlation (Barzilay et al., 2007; Collerton et al., 2012; Hubbard et al., 2009; Walston et al., 2002; Wu et al., 2009) and no correlation (Reiner et al., 2009). Most studies examining the presence of DNA damage in relation to CRP levels have found a positive correlation. A weak, but positive correlation was observed between increased SSBs and increased CRP levels in patients with Acute Coronary Syndrome (Demirbag et al., 2005), while similar correlations were observed in Cardiac X Syndrome and End Stage Renal Disease for lymphocyte damage and serum levels of 8-oxodG lesions respectively (Gur et al., 2007; Haghdoost et al., 2006). Analysis of the urine from elderly patients with low grade inflammation found no correlation between CRP levels and the levels of the RNA oxidation product 8-OHG excreted in urine.
In a bi-racial cohort we examined the relationship between CRP and the levels of oxidative DNA lesions and SSB-RC (Trzeciak et al., 2012). We found a statistically significant increase in SSB level with increasing CRP concentration in African-American males. In the overall cohort, a significant positive correlation between sex, the logarithm of CRP concentration, and SSB-RC as measured by the residual DNA damage after 30 min and 60 min was observed; however in females this significant correlation was negative. These same correlations were true when the cohort was stratified by race. We observed a positive correlation between the CRP concentration in serum and RBC GSH concentration, but found no significant relationship between the levels of CRP and heme degradation products. Interestingly, we recently completed a study examining 8-oxodG levels in women with low (<3 mg/L) CRP, mid (>3–20 mg/L), and high (>20 mg/L) CRP. Increasing levels of 8-oxodG were observed in the mid and high CRP groups compared to the low CRP group, indicating a significant relationship between an inflammatory and oxidative stress marker in women at risk for cardiovascular disease (Noren Hooten et al., 2012). As several studies have also associated CRP with 8-oxodG levels in various disease states (Bolukbas et al., 2006; Demirbag et al., 2005; Haghdoost et al., 2006), it will be important in the future to further investigate the promising relationship between this inflammatory marker and 8-oxodG, as well as other oxidative stress markers.
8. Closing Remarks and Perspectives
In the more than 50 years since the introduction of the Harman Free Radical Theory on Aging, great strides have been made to understand the role of free radicals in human health and disease. Our schematic model (Figure 1) highlights pathology, age, race and genotype (non-modifiable risk factors) as well as modifiable risk factors (diet, socioeconomic status, environment and behavior) may interact with oxidant stress and inflammatory processes to contribute to aging and age related disease. These interactions result in oxidative modifications of cellular macromolecules DNA, RNA, lipid and proteins. These modifications are associated with a number of bi-products or end-products that are linked to the oxidatively modified macromolecules (DNA adducts, strand-breaks, heme degradation, protein carbonyls and isoprostanes). These modifications may alone or in association with other biologic factors lead to epigenetic changes, changes in gene expression and/or DNA repair capacity or mitochondrial and membrane dysfunction. Ultimately, these factors over time and in association with a lifetime of exposures result in aging and age related disease and the pathologies that are present. We would like to point out that we have not addressed the role of mitochondria in oxidant stress or mitochondrial DNA damage in this review. It is known that mitochondria contribute to oxidant stress to DNA, accumulate with age, and are thought to contribute to the aging process. Therefore, it is possible that mitochondria could be potential markers of oxidant stress related to aging and age-related diseases. The studies to support this possible role should be the focus of a future literature review.
Figure 1. The role of oxidant stress in aging and age-related disease.
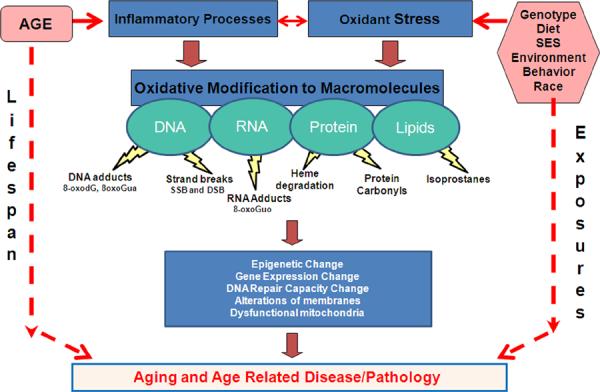
Both non-modifiable risk factors (age, race, genotype) and modifiable risk factors (diet, socioeconomic status, environment) have the propensity to interact with and affect the inflammatory processes and affect and be affected by oxidant stress. The interaction of all of these parameters can result in oxidative modifications of cellular macromolecules DNA, RNA, lipid and proteins. These oxidative modifications alone or in association with other biologic factors can lead to epigenetic changes, changes in both gene expression and DNA repair capacity, or mitochondrial and membrane dysfunction.
To effectively study the various stages of this molecular progression, methodologies must be developed to assess the types and levels of damage to cellular components, and the results need to be verified by studies in different laboratories as has been done by ESCOD and ESCULA. This type of collaborative scientific work pushes the field forward. The studies we have discussed employ the most commonly used methodologies to measure damage to DNA, RNA, protein, and lipids that is a result of oxidative stress and various inflammatory processes. After a thorough review of the literature, we feel that some of the markers of oxidative stress we discussed have inconsistent results with respect to aging and age-related disease (Tables 1 and 2). It appears that further investigation is needed before the field can come to a consensus as to the best and most consistent measure of DNA oxidation, especially in relation to the use of these measures of oxidative damage as a clinically relevant marker for aging or age-related disease. That being said, studies investigating measures of RNA oxidation, protein oxidation, and lipid oxidation show somewhat more consistency in regard to age and age-related disease. To insure the best results all studies must increase cohort size to improve the statistical power of future analyses. Further studies and validation of all markers of oxidative damage would greatly enhance our understanding of the role oxidative stress plays in aging and disease.
Highlights
A review of the current status of oxidative stress markers in aging and age-related diseases
This review discusses the use of different methodologies to assess oxidative stress levels
Particular focus on the oxidation of DNA, RNA and protein in human cohort studies with respect to aging
Acknowledgements
We thank Dr. David Wilson III and Dr. Joseph Rifkind for critical reading of the manuscript. Research in this laboratory is supported by the Intramural Research Program of the NIH, National Institute on Aging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe T, Isobe C, Murata T, Sato C, Tohgi H. Alteration of 8-hydroxyguanosine concentrations in the cerebrospinal fluid and serum from patients with Parkinson's disease. Neurosci Lett. 2003;336:105–108. doi: 10.1016/s0304-3940(02)01259-4. [DOI] [PubMed] [Google Scholar]
- Abe T, Tohgi H, Isobe C, Murata T, Sato C. Remarkable increase in the concentration of 8-hydroxyguanosine in cerebrospinal fluid from patients with Alzheimer's disease. J Neurosci Res. 2002;70:447–450. doi: 10.1002/jnr.10349. [DOI] [PubMed] [Google Scholar]
- Alam ZI, Halliwell B, Jenner P. No evidence for increased oxidative damage to lipids, proteins, or DNA in Huntington's disease. J Neurochem. 2000;75:840–846. doi: 10.1046/j.1471-4159.2000.0750840.x. [DOI] [PubMed] [Google Scholar]
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- Ame J-C, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, Schreiber V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J Cell Sci. 2009;122:1990–2002. doi: 10.1242/jcs.039115. [DOI] [PubMed] [Google Scholar]
- Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreoli R, Manini P, Palma GD, Alinovi R, Goldoni M, Niessen WMA, Mutti A. Quantitative determination of urinary 8-oxo-7,8-dihydro-2'-deoxyguanosine, 8-oxo-7,8-dihydroguanine, 8-oxo-7,8-dihydroguanosine, and their non-oxidized forms: daily concentration profile in healthy volunteers. Biomarkers. 2010;15:221–231. doi: 10.3109/13547500903434501. [DOI] [PubMed] [Google Scholar]
- Andreoli R, Mutti A, Goldoni M, Manini P, Apostoli P, De Palma G. Reference ranges of urinary biomarkers of oxidized guanine in (2'-deoxy)ribonucleotides and nucleic acids. Free Radical Biology and Medicine. 2011;50:254–261. doi: 10.1016/j.freeradbiomed.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Aviram M. Review of human studies on oxidative damage and antioxidant protection related to cardiovascular diseases. Free Radic Res. 2000;33(Suppl):S85–97. [PubMed] [Google Scholar]
- Bachi A, Zuccato E, Baraldi M, Fanelli R, Chiabrando C. Measurement of urinary 8-Epi-prostaglandin F2alpha, a novel index of lipid peroxidation in vivo, by immunoaffinity extraction/gas chromatography-mass spectrometry. Basal levels in smokers and nonsmokers. Free Radic Biol Med. 1996;20:619–624. doi: 10.1016/0891-5849(95)02087-x. [DOI] [PubMed] [Google Scholar]
- Bandeen Roche K, Walston J, Huang Y, Semba R, Ferrucci L. Measuring systemic inflammatory regulation in older adults: evidence and utility. Rejuvenation research. 2009;12:403–410. doi: 10.1089/rej.2009.0883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barciszewski J, Barciszewska MZ, Siboska G, Rattan SI, Clark BF. Some unusual nucleic acid bases are products of hydroxyl radical oxidation of DNA and RNA. Mol Biol Rep. 1999;26:231–238. doi: 10.1023/a:1007058602594. [DOI] [PubMed] [Google Scholar]
- Barnett YA, King CM. An investigation of antioxidant status, DNA repair capacity and mutation as a function of age in humans. Mutat Res. 1995;338:115–128. doi: 10.1016/0921-8734(95)00017-z. [DOI] [PubMed] [Google Scholar]
- Barzilay JI, Blaum C, Moore T, Xue QL, Hirsch CH, Walson J, Fried LP. Insulin resistance and inflammation as precursors of frailty: The cardiovascular health study. Archives of Internal Medicine. 2007;167:635–641. doi: 10.1001/archinte.167.7.635. [DOI] [PubMed] [Google Scholar]
- Basu S, Whiteman M, Mattey DL, Halliwell B. Raised levels of F2-isoprostanes and prostaglandin F2α in different rheumatic diseases. Annals of the Rheumatic Diseases. 2001;60:627–631. doi: 10.1136/ard.60.6.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Oxidatively modified proteins in aging and disease. Free Radic Biol Med. 2002;32:797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Moss EG. RNA repair: damage control. Curr Biol. 2003;13:R482–484. doi: 10.1016/s0960-9822(03)00408-1. [DOI] [PubMed] [Google Scholar]
- Beutler E, Gelbart T. Plasma glutathione in health and in patients with malignant disease. Journal of Laboratory and Clinical Medicine. 1985;105:581–584. [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermuller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, Gilbert J, Drenkow J, Bell I, Zhao X, Srinivasan KG, Sung WK, Ooi HS, Chiu KP, Foissac S, Alioto T, Brent M, Pachter L, Tress ML, Valencia A, Choo SW, Choo CY, Ucla C, Manzano C, Wyss C, Cheung E, Clark TG, Brown JB, Ganesh M, Patel S, Tammana H, Chrast J, Henrichsen CN, Kai C, Kawai J, Nagalakshmi U, Wu J, Lian Z, Lian J, Newburger P, Zhang X, Bickel P, Mattick JS, Carninci P, Hayashizaki Y, Weissman S, Hubbard T, Myers RM, Rogers J, Stadler PF, Lowe TM, Wei CL, Ruan Y, Struhl K, Gerstein M, Antonarakis SE, Fu Y, Green ED, Karaoz U, Siepel A, Taylor J, Liefer LA, Wetterstrand KA, Good PJ, Feingold EA, Guyer MS, Cooper GM, Asimenos G, Dewey CN, Hou M, Nikolaev S, Montoya-Burgos JI, Loytynoja A, Whelan S, Pardi F, Massingham T, Huang H, Zhang NR, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Seringhaus M, Church D, Rosenbloom K, Kent WJ, Stone EA, Batzoglou S, Goldman N, Hardison RC, Haussler D, Miller W, Sidow A, Trinklein ND, Zhang ZD, Barrera L, Stuart R, King DC, Ameur A, Enroth S, Bieda MC, Kim J, Bhinge AA, Jiang N, Liu J, Yao F, Vega VB, Lee CW, Ng P, Yang A, Moqtaderi Z, Zhu Z, Xu X, Squazzo S, Oberley MJ, Inman D, Singer MA, Richmond TA, Munn KJ, Rada-Iglesias A, Wallerman O, Komorowski J, Fowler JC, Couttet P, Bruce AW, Dovey OM, Ellis PD, Langford CF, Nix DA, Euskirchen G, Hartman S, Urban AE, Kraus P, Van Calcar S, Heintzman N, Kim TH, Wang K, Qu C, Hon G, Luna R, Glass CK, Rosenfeld MG, Aldred SF, Cooper SJ, Halees A, Lin JM, Shulha HP, Xu M, Haidar JN, Yu Y, Iyer VR, Green RD, Wadelius C, Farnham PJ, Ren B, Harte RA, Hinrichs AS, Trumbower H, Clawson H, Hillman-Jackson J, Zweig AS, Smith K, Thakkapallayil A, Barber G, Kuhn RM, Karolchik D, Armengol L, Bird CP, de Bakker PI, Kern AD, Lopez-Bigas N, Martin JD, Stranger BE, Woodroffe A, Davydov E, Dimas A, Eyras E, Hallgrimsdottir IB, Huppert J, Zody MC, Abecasis GR, Estivill X, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VV, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffe DB, Chang JL, Lindblad-Toh K, Lander ES, Koriabine M, Nefedov M, Osoegawa K, Yoshinaga Y, Zhu B, de Jong PJ. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res. 2003;531:37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Blasiak J, Trzeciak A, Malecka-Panas E, Drzewoski J, Wojewodzka M. In vitro genotoxicity of ethanol and acetaldehyde in human lymphocytes and the gastrointestinal tract mucosa cells. Toxicol In Vitro. 2000;14:287–295. doi: 10.1016/s0887-2333(00)00022-9. [DOI] [PubMed] [Google Scholar]
- Boiteux S, Gajewski E, Laval J, Dizdaroglu M. Substrate specificity of the Escherichia coli Fpg protein (formamidopyrimidine-DNA glycosylase): excision of purine lesions in DNA produced by ionizing radiation or photosensitization. Biochemistry. 1992;31:106–110. doi: 10.1021/bi00116a016. [DOI] [PubMed] [Google Scholar]
- Bolukbas C, Bolukbas FF, Kocyigit A, Aslan M, Selek S, Bitiren M, Ulukanligil M. Relationship between levels of DNA damage in lymphocytes and histopathological severity of chronic hepatitis C and various clinical forms of hepatitis B. Journal of Gastroenterology and Hepatology. 2006;21:610–616. doi: 10.1111/j.1440-1746.2005.04069.x. [DOI] [PubMed] [Google Scholar]
- Bonnefont-Rousselot D, Lacomblez L, Jaudon M, Lepage S, Salachas F, Bensimon G, Bizard C, Doppler V, Delattre J, Meininger V. Blood oxidative stress in amyotrophic lateral sclerosis. J Neurol Sci. 2000;178:57–62. doi: 10.1016/s0022-510x(00)00365-8. [DOI] [PubMed] [Google Scholar]
- Broedbaek K, Poulsen HE, Weimann A, Kom GD, Schwedhelm E, Nielsen P, Boger RH. Urinary excretion of biomarkers of oxidatively damaged DNA and RNA in hereditary hemochromatosis. Free Radic Biol Med. 2009;47:1230–1233. doi: 10.1016/j.freeradbiomed.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Broedbaek K, Ribel-Madsen R, Henriksen T, Weimann A, Petersen M, Andersen JT, Afzal S, Hjelvang B, Roberts LJ, 2nd, Vaag A, Poulsen P, Poulsen HE. Genetic and environmental influences on oxidative damage assessed in elderly Danish twins. Free Radic Biol Med. 2011a;50:1488–1491. doi: 10.1016/j.freeradbiomed.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broedbaek K, Siersma V, Andersen JT, Petersen M, Afzal S, Hjelvang B, Weimann A, Semba RD, Ferrucci L, Poulsen HE. The association between low-grade inflammation, iron status and nucleic acid oxidation in the elderly. Free Radic Res. 2011b;45:409–416. doi: 10.3109/10715762.2010.538391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA. Oxidative stress in neurodegenerative disorders. Antioxid Redox Signal. 2006;8:1971–1973. doi: 10.1089/ars.2006.8.1971. [DOI] [PubMed] [Google Scholar]
- Cecchi C, Latorraca S, Sorbi S, Iantomasi T, Favilli F, Vincenzini MT, Liguri G. Gluthatione level is altered in lymphoblasts from patients with familial Alzheimer's disease. Neurosci Lett. 1999;275:152–154. doi: 10.1016/s0304-3940(99)00751-x. [DOI] [PubMed] [Google Scholar]
- Chapple IL. Reactive oxygen species and antioxidants in inflammatory diseases. J Clin Periodontol. 1997;24:287–296. doi: 10.1111/j.1600-051x.1997.tb00760.x. [DOI] [PubMed] [Google Scholar]
- Chevion M, Berenshtein E, Stadtman ER. Human studies related to protein oxidation: protein carbonyl content as a marker of damage. Free Radic Res. 2000;33(Suppl):S99–108. [PubMed] [Google Scholar]
- Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr. 2000;71:621s–629. doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- Clyne B, Olshaker JS. The C-reactive protein. J Emerg Med. 1999;17:1019–1025. doi: 10.1016/s0736-4679(99)00135-3. [DOI] [PubMed] [Google Scholar]
- Collerton J, Martin-Ruiz C, Davies K, Hilkens CM, Isaacs J, Kolenda C, Parker C, Dunn M, Catt M, Jagger C, von Zglinicki T, Kirkwood TBL. Frailty and the role of inflammation, immunosenescence and cellular ageing in the very old: Cross-sectional findings from the Newcastle 85+ Study. Mechanisms of Ageing and Development. 2012;133:456–466. doi: 10.1016/j.mad.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Collins AR. The Comet Assay. 2002:163–177. doi: 10.1385/1-59259-179-5:163. [DOI] [PubMed] [Google Scholar]
- Collins AR, Cadet J, Moller L, Poulsen HE, Vina J. Are we sure we know how to measure 8-oxo-7,8-dihydroguanine in DNA from human cells? Arch Biochem Biophys. 2004;423:57–65. doi: 10.1016/j.abb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Collins AR, Dobson VL, Dusinska M, Kennedy G, Stetina R. The comet assay: what can it really tell us? Mutat Res. 1997;375:183–193. doi: 10.1016/s0027-5107(97)00013-4. [DOI] [PubMed] [Google Scholar]
- Collins AR, Duthie SJ, Dobson VL. Direct enzymic detection of endogenous oxidative base damage in human lymphocyte DNA. Carcinogenesis. 1993;14:1733–1735. doi: 10.1093/carcin/14.9.1733. [DOI] [PubMed] [Google Scholar]
- Collins AR, Gedik CM, Olmedilla B, Southon S, Bellizzi M. Oxidative DNA damage measured in human lymphocytes: large differences between sexes and between countries, and correlations with heart disease mortality rates. FASEB J. 1998a;12:1397–1400. [PubMed] [Google Scholar]
- Collins AR, Oscoz AA, Brunborg G, Gaivão I, Giovannelli L, Kruszewski M, Smith CC, Å tÄ❯tina R. The comet assay: topical issues. Mutagenesis. 2008;23:143–151. doi: 10.1093/mutage/gem051. [DOI] [PubMed] [Google Scholar]
- Collins AR, Raslova K, Somorovska M, Petrovska H, Ondrusova A, Vohnout B, Fabry R, Dusinska M. DNA damage in diabetes: correlation with a clinical marker. Free Radic Biol Med. 1998b;25:373–377. doi: 10.1016/s0891-5849(98)00053-7. [DOI] [PubMed] [Google Scholar]
- Conrad CC, Marshall PL, Talent JM, Malakowsky CA, Choi J, Gracy RW. Oxidized proteins in Alzheimer's plasma. Biochem Biophys Res Commun. 2000;275:678–681. doi: 10.1006/bbrc.2000.3356. [DOI] [PubMed] [Google Scholar]
- Cooke MS. A commentary on “Urea, the most abundant component in urine, cross-reacts with a commercial 8-OH-dG ELISA kit and contributes to overestimation of urinary 8-OH-dG”. What is ELISA detecting? Free Radic Biol Med. 2009;47:30–31. doi: 10.1016/j.freeradbiomed.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Barregard L, Mistry V, Potdar N, Rozalski R, Gackowski D, Siomek A, Foksinski M, Svoboda P, Kasai H, Konje JC, Sallsten G, Evans MD, Olinski R. Interlaboratory comparison of methodologies for the measurement of urinary 8-oxo-7,8-dihydro-2'-deoxyguanosine. Biomarkers. 2009;14:103–110. doi: 10.1080/13547500802706012. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Olinski R, Loft S. Measurement and Meaning of Oxidatively Modified DNA Lesions in Urine. Cancer Epidemiology Biomarkers & Prevention. 2008;17:3–14. doi: 10.1158/1055-9965.EPI-07-0751. [DOI] [PubMed] [Google Scholar]
- Cookson MR, Shaw PJ. Oxidative stress and motor neurone disease. Brain pathology. 1999;9:165–186. doi: 10.1111/j.1750-3639.1999.tb00217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AD, Pierre SV, Cohen MV, Downey JM, Garlid KD. cGMP signalling in pre- and post-conditioning: the role of mitochondria. Cardiovasc Res. 2008;77:344–352. doi: 10.1093/cvr/cvm050. [DOI] [PubMed] [Google Scholar]
- Cracowski J-L, Durand T. Cardiovascular pharmacology and physiology of the isoprostanes. Fundamental & clinical pharmacology. 2006;20:417–427. doi: 10.1111/j.1472-8206.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- Cracowski JL, Durand T, Bessard G. Isoprostanes as a biomarker of lipid peroxidation in humans: physiology, pharmacology and clinical implications. Trends Pharmacol Sci. 2002;23:360–366. doi: 10.1016/s0165-6147(02)02053-9. [DOI] [PubMed] [Google Scholar]
- Cracowski JL, Marpeau C, Carpentier PH, Imbert B, Hunt M, Stanke-Labesque F, Bessard G. Enhanced in vivo lipid peroxidation in scleroderma spectrum disorders. Arthritis Rheum. 2001;44:1143–1148. doi: 10.1002/1529-0131(200105)44:5<1143::AID-ANR196>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Crankshaw D. Effects of the isoprostane, 8-epi-prostaglandin F2 alpha, on the contractility of the human myometrium in vitro. European Journal of Pharmacology. 1995;285:151–158. doi: 10.1016/0014-2999(95)00398-5. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of Oxidative Damage in Human Disease. Clin Chem. 2006;52:601–623. doi: 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- Dar ME, Jorgensen TJ. Deletions at short direct repeats and base substitutions are characteristic mutations for bleomycin-induced double- and single-strand breaks, respectively, in a human shuttle vector system. Nucl. Acids Res. 1995;23:3224–3230. doi: 10.1093/nar/23.16.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dav G, Falco A, Patrono C. Lipid peroxidation in diabetes mellitus. Antioxidants & redox signalling. 2005;7:256–268. doi: 10.1089/ars.2005.7.256. [DOI] [PubMed] [Google Scholar]
- Davies SS, Roberts Ii LJ. F2-isoprostanes as an indicator and risk factor for coronary heart disease. Free Radical Biology and Medicine. 2011;50:559–566. doi: 10.1016/j.freeradbiomed.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Flora S, Izzotti A, Walsh D, Degan P, Petrilli GL, Lewtas J. Molecular epidemiology of atherosclerosis. The FASEB Journal. 1997;11:1021–1031. [PubMed] [Google Scholar]
- De Luca G, Russo MT, Degan P, Tiveron C, Zijno A, Meccia E, Ventura I, Mattei E, Nakabeppu Y, Crescenzi M, Pepponi R, Pèzzola A, Popoli P, Bignami M. A Role for Oxidized DNA Precursors in Huntington's Disease‚ÄìLike Striatal Neurodegeneration. PLoS Genet. 2008;4:e1000266. doi: 10.1371/journal.pgen.1000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirbag R, Yilmaz R, Gur M, Kocyigit A, Celik H, Guzel S, Selek S. Lymphocyte DNA damage in patients with acute coronary syndrome and its relationship with severity of acute coronary syndrome. Mutat Res. 2005;578:298–307. doi: 10.1016/j.mrfmmm.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer's disease. J Neurosci. 2005;25:9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenici FA, Vannucchi MT, Jordao AA, Jr., Meirelles MS, Vannucchi H. DNA oxidative damage in patients with dialysis treatment. Ren Fail. 2005;27:689–694. doi: 10.1080/08860220500242678. [DOI] [PubMed] [Google Scholar]
- Dominguez C, Ruiz E, Gussinye M, Carrascosa A. Oxidative stress at onset and in early stages of type 1 diabetes in children and adolescents. Diabetes Care. 1998;21:1736–1742. doi: 10.2337/diacare.21.10.1736. [DOI] [PubMed] [Google Scholar]
- Dusinska M, Dzupinkova Z, Wsolova L, Harrington V, Collins AR. Possible involvement of XPA in repair of oxidative DNA damage deduced from analysis of damage, repair and genotype in a human population study. Mutagenesis. 2006;21:205–211. doi: 10.1093/mutage/gel016. [DOI] [PubMed] [Google Scholar]
- Dworski R, Murray JJ, Roberts LJ, 2nd, Oates JA, Morrow JD, Fisher L, Sheller JR. Allergen-induced synthesis of F(2)-isoprostanes in atopic asthmatics. Evidence for oxidant stress. Am J Respir Crit Care Med. 1999;160:1947–1951. doi: 10.1164/ajrccm.160.6.9903064. [DOI] [PubMed] [Google Scholar]
- Erden-İnal M, Sunal E, Kanbak G. Age-related changes in the glutathione redox system. Cell Biochemistry and Function. 2002;20:61–66. doi: 10.1002/cbf.937. [DOI] [PubMed] [Google Scholar]
- Evans MD, Olinski R, Loft S, Cooke MS. Toward consensus in the analysis of urinary 8-oxo-7,8-dihydro-2'-deoxyguanosine as a noninvasive biomarker of oxidative stress. The FASEB Journal. 2010;24:1249–1260. doi: 10.1096/fj.09-147124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer M, Paschke R. Regulation of adipocytokines and insulin resistance. Diabetologia. 2003;46:1594–1603. doi: 10.1007/s00125-003-1228-z. [DOI] [PubMed] [Google Scholar]
- Feairheller DL, Park JY, Sturgeon KM, Williamson ST, Diaz KM, Veerabhadrappa P, Brown MD. Racial differences in oxidative stress and inflammation: in vitro and in vivo. Clin Transl Sci. 4:32–37. doi: 10.1111/j.1752-8062.2011.00264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fialkow L, Wang Y, Downey GP. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic Biol Med. 2007;42:153–164. doi: 10.1016/j.freeradbiomed.2006.09.030. [DOI] [PubMed] [Google Scholar]
- Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem. 1998;70:268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- Floyd RA, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23:795–807. doi: 10.1016/s0197-4580(02)00019-2. [DOI] [PubMed] [Google Scholar]
- Fried LP, Xue Q-L, Cappola AR, Ferrucci L, Chaves P, Varadhan R, Guralnik JM, Leng SX, Semba RD, Walston JD, Blaum CS, Bandeen-Roche K. Nonlinear Multisystem Physiological Dysregulation Associated With Frailty in Older Women: Implications for Etiology and Treatment. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2009;64A:1049–1057. doi: 10.1093/gerona/glp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbita SP, Lovell MA, Markesbery WR. Increased Nuclear DNA Oxidation in the Brain in Alzheimer's Disease. Journal of Neurochemistry. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- Gambhir JK, Lali P, Jain AK. Correlation between blood antioxidant levels and lipid peroxidation in rheumatoid arthritis. Clin Biochem. 1997;30:351–355. doi: 10.1016/s0009-9120(96)00007-0. [DOI] [PubMed] [Google Scholar]
- Garm C, Moreno-Villanueva M, Bürkle A, Petersen I, Bohr VA, Christensen K, Stevnsner T. Age and gender effects on DNA strand break repair in peripheral blood mononuclear cells. Aging Cell. 2012:n/a–n/a. doi: 10.1111/acel.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison WM, Jayko ME, Bennett W. Radiation-induced oxidation of protein in aqueous solution. Radiat Res. 1962;16:483–502. [PubMed] [Google Scholar]
- Gedik CM, Collins A. Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J. 2005;19:82–84. doi: 10.1096/fj.04-1767fje. [DOI] [PubMed] [Google Scholar]
- Gianni P, Jan KJ, Douglas MJ, Stuart PM, Tarnopolsky MA. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Exp Gerontol. 2004;39:1391–1400. doi: 10.1016/j.exger.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Gil L, Siems W, Mazurek B, Gross J, Schroeder P, Voss P, Grune T. Age-associated analysis of oxidative stress parameters in human plasma and erythrocytes. Free Radical Research. 2006;40:495–505. doi: 10.1080/10715760600592962. [DOI] [PubMed] [Google Scholar]
- Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- Gopaul NK, Anggard EE, Mallet AI, Betteridge DJ, Wolff SP, Nourooz-Zadeh J. Plasma 8-epi-PGF2 alpha levels are elevated in individuals with non-insulin dependent diabetes mellitus. FEBS Lett. 1995;368:225–229. doi: 10.1016/0014-5793(95)00649-t. [DOI] [PubMed] [Google Scholar]
- Gopaul NK, Halliwell B, Anggrd EE. Measurement of plasma F2-isoprostanes as an index of lipid peroxidation does not appear to be confounded by diet. Free Radical Research. 2000;33:115–127. doi: 10.1080/10715760000300671. [DOI] [PubMed] [Google Scholar]
- Griffiths HR. Antioxidants and protein oxidation. Free Radic Res. 2000;33(Suppl):S47–58. [PubMed] [Google Scholar]
- Grune T, Shringarpure R, Sitte N, Davies K. Age-related changes in protein oxidation and proteolysis in mammalian cells. The Journals of Gerontology; Series A; Biological sciences and medical sciences. 2001;56:B459–B467. doi: 10.1093/gerona/56.11.b459. [DOI] [PubMed] [Google Scholar]
- Guentchev M, Siedlak SL, Jarius C, Tagliavini F, Castellani RJ, Perry G, Smith MA, Budka H. Oxidative damage to nucleic acids in human prion disease. Neurobiol Dis. 2002;9:275–281. doi: 10.1006/nbdi.2002.0477. [DOI] [PubMed] [Google Scholar]
- Gur M, Yildiz A, Demirbag R, Yilmaz R, Kocyigit A, Celik H, Aksoy N. Increased lymphocyte deoxyribonucleic acid damage in patients with cardiac syndrome X. Mutat Res. 2007;617:8–15. doi: 10.1016/j.mrfmmm.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Hadi Yasa M, Kacmaz M, Serda Ozturk H, Durak I. Antioxidant status of erythrocytes from patients with cirrhosis. Hepatogastroenterology. 1999;46:2460–2463. [PubMed] [Google Scholar]
- Haghdoost S, Maruyama Y, Pecoits-Filho R, Heimburger O, Seeberger A, Anderstam B, Suliman ME, Czene S, Lindholm B, Stenvinkel P, Harms-Ringdahl M. Elevated serum 8-oxo-dG in hemodialysis patients: a marker of systemic inflammation? Antioxid Redox Signal. 2006;8:2169–2173. doi: 10.1089/ars.2006.8.2169. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause, or consequence? Lancet. 1994;344:721–724. doi: 10.1016/s0140-6736(94)92211-x. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Hampel H, Sunderland T, Kötter HU, Schneider C, Teipel SJ, Padberg F, Dukoff R, Levy J, Möller H-J. Decreased soluble interleukin-6 receptor in cerebrospinal fluid of patients with Alzheimer's disease. Brain Research. 1998;780:356–359. doi: 10.1016/s0006-8993(97)01355-3. [DOI] [PubMed] [Google Scholar]
- Hannon-Fletcher MP, O'Kane MJ, Moles KW, Weatherup C, Barnett CR, Barnett YA. Levels of peripheral blood cell DNA damage in insulin dependent diabetes mellitus human subjects. Mutat Res. 2000;460:53–60. doi: 10.1016/s0921-8777(00)00013-6. [DOI] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Araki S, Kohyama J, Shioda K, Fukatsu R. Oxidative nucleotide damage and superoxide dismutase expression in the brains of xeroderma pigmentosum group A and Cockayne syndrome. Brain Dev. 2005;27:34–38. doi: 10.1016/j.braindev.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Henriksen T, Hillestrom PR, Poulsen HE, Weimann A. Automated method for the direct analysis of 8-oxo-guanosine and 8-oxo-2'-deoxyguanosine in human urine using ultraperformance liquid chromatography and tandem mass spectrometry. Free Radic Biol Med. 2009;47:629–635. doi: 10.1016/j.freeradbiomed.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Hitchon C, El-Gabalawy H. Oxidation in rheumatoid arthritis. Arthritis Res Ther. 2004;6:265–278. doi: 10.1186/ar1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelzl C, Knasmüller S, Misík M, Collins A, Dusinská M, Nersesyan A. Use of single cell gel electrophoresis assays for the detection of DNA-protective effects of dietary factors in humans: Recent results and trends. Mutation Research/Reviews in Mutation Research. 2009;681:68–79. doi: 10.1016/j.mrrev.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Hofer T, Badouard C, Bajak E, Ravanat JL, Mattsson A, Cotgreave IA. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol Chem. 2005;386:333–337. doi: 10.1515/BC.2005.040. [DOI] [PubMed] [Google Scholar]
- Hofer T, Seo AY, Prudencio M, Leeuwenburgh C. A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: greater RNA than DNA oxidation in rat liver after doxorubicin administration. Biol Chem. 2006;387:103–111. doi: 10.1515/BC.2006.014. [DOI] [PubMed] [Google Scholar]
- Hold GL, El-Omar EM. Genetic aspects of inflammation and cancer. Biochem J. 2008;410:225–235. doi: 10.1042/BJ20071341. [DOI] [PubMed] [Google Scholar]
- Holt S, Reeder B, Wilson M, Harvey S, Morrow JD, Roberts LJ, 2nd, Moore K. Increased lipid peroxidation in patients with rhabdomyolysis. Lancet. 1999;353:1241. doi: 10.1016/S0140-6736(98)05768-7. [DOI] [PubMed] [Google Scholar]
- Honda K, Smith MA, Zhu X, Baus D, Merrick WC, Tartakoff AM, Hattier T, Harris PL, Siedlak SL, Fujioka H, Liu Q, Moreira PI, Miller FP, Nunomura A, Shimohama S, Perry G. Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol Chem. 2005;280:20978–20986. doi: 10.1074/jbc.M500526200. [DOI] [PubMed] [Google Scholar]
- Howard C, Ferrucci L, Sun K, Fried LP, Walston J, Varadhan R, Guralnik JM, Semba RD. Oxidative protein damage is associated with poor grip strength among older women living in the community. J Appl Physiol. 2007;103:17–20. doi: 10.1152/japplphysiol.00133.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LC, Clarkin KC, Wahl GM. Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc Natl Acad Sci U S A. 1996a;93:4827–4832. doi: 10.1073/pnas.93.10.4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LC, Clarkin KC, Wahl GM. Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proceedings of the National Academy of Sciences of the United States of America. 1996b;93:4827–4832. doi: 10.1073/pnas.93.10.4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard RE, O'Mahony MS, Savva GM, Calver BL, Woodhouse KW. Inflammation and frailty measures in older people. Journal of Cellular and Molecular Medicine. 2009;13:3103–3109. doi: 10.1111/j.1582-4934.2009.00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys V, Martin RM, Ratcliffe B, Duthie S, Wood S, Gunnell D, Collins AR. Age-related increases in DNA repair and antioxidant protection: a comparison of the Boyd Orr Cohort of elderly subjects with a younger population sample. Age and Ageing. 2007;36:521–526. doi: 10.1093/ageing/afm107. [DOI] [PubMed] [Google Scholar]
- Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26:2166–2176. doi: 10.1038/sj.onc.1210279. [DOI] [PubMed] [Google Scholar]
- Hyland P, Duggan O, Turbitt J, Coulter J, Wikby A, Johansson B, Tompa A, Barnett C, Barnett Y. Nonagenarians from the Swedish NONA Immune Study have increased plasma antioxidant capacity and similar levels of DNA damage in peripheral blood mononuclear cells compared to younger control subjects. Experimental Gerontology. 2002;37:465–473. doi: 10.1016/s0531-5565(01)00216-9. [DOI] [PubMed] [Google Scholar]
- Isobe C, Abe T, Terayama Y. Homocysteine may contribute to pathogenesis of RNA damage in brains with Alzheimer's disease. Neurodegener Dis. 2009;6:252–257. doi: 10.1159/000262443. [DOI] [PubMed] [Google Scholar]
- Ivashkevich A, Redon CE, Nakamura AJ, Martin RF, Martin OA. Use of the Î-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Letters. 327:123–133. doi: 10.1016/j.canlet.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen LJ. Isoprostanes and Lung Vascular Pathology. Am. J. Respir. Cell Mol. Biol. 2008;39:383–389. doi: 10.1165/rcmb.2008-0109TR. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Premji M, Netherton S, Catalli A, Cox G, Keshavjee S, Crankshaw DJ. Excitatory and Inhibitory Actions of Isoprostanes in Human and Canine Airway Smooth Muscle. Journal of Pharmacology and Experimental Therapeutics. 2000;295:506–511. [PubMed] [Google Scholar]
- Johnson PA, Clements P, Hudson K, Caldecott KW. The mitotic spindle and DNA damage-induced apoptosis. Toxicology Letters. 2000;112–113:59–67. doi: 10.1016/s0378-4274(99)00245-3. [DOI] [PubMed] [Google Scholar]
- Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- Kadiiska MB, Gladen BC, Baird DD, Germolec D, Graham LB, Parker CE, Nyska A, Wachsman JT, Ames BN, Basu S, Brot N, Fitzgerald GA, Floyd RA, George M, Heinecke JW, Hatch GE, Hensley K, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, Rokach J, Shigenaga MK, Sohal RS, Sun J, Tice RR, Van Thiel DH, Wellner D, Walter PB, Tomer KB, Mason RP, Barrett JC. Biomarkers of oxidative stress study II: are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free radical biology & medicine. 2005;38:698–710. doi: 10.1016/j.freeradbiomed.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Kasapoglu M, Ozben T. Alterations of antioxidant enzymes and oxidative stress markers in aging. Exp Gerontol. 2001;36:209–220. doi: 10.1016/s0531-5565(00)00198-4. [DOI] [PubMed] [Google Scholar]
- Kedziora-Kornatowska K, Czuczejko J, Pawluk H, Kornatowski T, Motyl J, Szadujkis-Szadurski L, Szewczyk-Golec K, Kedziora J. The markers of oxidative stress and activity of the antioxidant system in the blood of elderly patients with essential arterial hypertension. Cellular & Molecular Biology Letters. 2004;9:635–641. [PubMed] [Google Scholar]
- Kędziora-Kornatowska K, Szewczyk-Golec K, Czuczejko J, Lumen K.v.M.d., Pawluk H, Motyl J, Karasek M, Kędziora J. Effect of melatonin on the oxidative stress in erythrocytes of healthy young and elderly subjects. Journal of Pineal Research. 2007;42:153–158. doi: 10.1111/j.1600-079X.2006.00394.x. [DOI] [PubMed] [Google Scholar]
- Kikuchi A, Takeda A, Onodera H, Kimpara T, Hisanaga K, Sato N, Nunomura A, Castellani RJ, Perry G, Smith MA, Itoyama Y. Systemic increase of oxidative nucleic acid damage in Parkinson's disease and multiple system atrophy. Neurobiol Dis. 2002;9:244–248. doi: 10.1006/nbdi.2002.0466. [DOI] [PubMed] [Google Scholar]
- King CM, Bristow-Craig HE, Gillespie ES, Barnett YA. In vivo antioxidant status, DNA damage, mutation and DNA repair capacity in cultured lymphocytes from healthy 75- to 80-year-old humans. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 1997;377:137–147. doi: 10.1016/s0027-5107(97)00072-9. [DOI] [PubMed] [Google Scholar]
- Knaapen AM, Schins RP, Polat D, Becker A, Borm PJ. Mechanisms of neutrophil-induced DNA damage in respiratory tract epithelial cells. Mol Cell Biochem. 2002;234–235:143–151. [PubMed] [Google Scholar]
- Kondo M, Oya-Ito T, Kumagai T, Osawa T, Uchida K. Cyclopentenone Prostaglandins as Potential Inducers of Intracellular Oxidative Stress. The Journal of Biological Chemistry. 2001;276:12076–12083. doi: 10.1074/jbc.M009630200. [DOI] [PubMed] [Google Scholar]
- Kopjar N, Garaj-Vrhovac V. Application of the alkaline comet assay in human biomonitoring for genotoxicity: a study on Croatian medical personnel handling antineoplastic drugs. Mutagenesis. 2001;16:71–78. doi: 10.1093/mutage/16.1.71. [DOI] [PubMed] [Google Scholar]
- Kopjar N, Zeljezic D, Garaj-Vrhovac V. Evaluation of DNA damage in white blood cells of healthy human volunteers using the alkaline comet assay and the chromosome aberration test. Acta Biochim Pol. 2006;53:321–336. [PubMed] [Google Scholar]
- Korolainen MA, Goldsteins G, Alafuzoff I, Koistinaho J, Pirttila T. Proteomic analysis of protein oxidation in Alzheimer's disease brain. Electrophoresis. 2002;23:3428–3433. doi: 10.1002/1522-2683(200210)23:19<3428::AID-ELPS3428>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Kosower NS, Kosower EM. The glutathione status of cells. International Review of Cytology. 1978;54:109–160. doi: 10.1016/s0074-7696(08)60166-7. [DOI] [PubMed] [Google Scholar]
- Kruszewski M, Wojewodzka M, Iwanenko T, Collins AR, Szumiel I. Application of the comet assay for monitoring DNA damage in workers exposed to chronic low-dose irradiation. II. Base damage. Mutat Res. 1998;416:37–57. doi: 10.1016/s1383-5718(98)00074-6. [DOI] [PubMed] [Google Scholar]
- Lang CA, Naryshkin S, Schneider DL, Mills BJ, Lindeman RD. Low blood glutathione levels in healthy aging adults. J Lab Clin Med. 1992;120:720–725. [PubMed] [Google Scholar]
- Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P, Ackerman SL. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- Leng S, Cappola A, Andersen R, Blackman M, Koenig K, Blair M, Walston J. Serum levels of insulin-like growth factor-I (IGF-I) and dehydroepiandrosterone sulfate (DHEAS), and their relationships with serum interleukin-6, in the geriatric syndrome of frailty. Aging Clinical and Experimental Research. 2004a;16:153–157. doi: 10.1007/BF03324545. [DOI] [PubMed] [Google Scholar]
- Leng S, Chaves P, Koenig K, Walston J. Serum Interleukin-6 and Hemoglobin as Physiological Correlates in the Geriatric Syndrome of Frailty: A Pilot Study. Journal of the American Geriatrics Society. 2002;50:1268–1271. doi: 10.1046/j.1532-5415.2002.50315.x. [DOI] [PubMed] [Google Scholar]
- Leng S, Yang H, Walston J. Decreased cell proliferation and altered cytokine production in frail older adults. Aging Clinical and Experimental Research. 2004b;16:249–252. doi: 10.1007/BF03327392. [DOI] [PubMed] [Google Scholar]
- Leng SX, Tian X, Matteini A, Li H, Hughes J, Jain A, Walston JD, Fedarko NS. IL-6-independent association of elevated serum neopterin levels with prevalent frailty in community-dwelling older adults. Age and Ageing. 2011;40:475–481. doi: 10.1093/ageing/afr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng SX, Xue Q-L, Tian J, Walston JD, Fried LP. Inflammation and Frailty in Older Women. Journal of the American Geriatrics Society. 2007;55:864–871. doi: 10.1111/j.1532-5415.2007.01186.x. [DOI] [PubMed] [Google Scholar]
- Levine RL, Stadtman ER. Oxidative modification of proteins during aging. Exp Gerontol. 2001;36:1495–1502. doi: 10.1016/s0531-5565(01)00135-8. [DOI] [PubMed] [Google Scholar]
- Libby P, Ridker PM. Inflammation and atherosclerosis: role of C-reactive protein in risk assessment. Am J Med. 2004;116(Suppl 6A):9S–16S. doi: 10.1016/j.amjmed.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Lih-Brody L, Powell SR, Collier KP, Reddy GM, Cerchia R, Kahn E, Weissman GS, Katz S, Floyd RA, McKinley MJ, Fisher SE, Mullin GE. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci. 1996;41:2078–2086. doi: 10.1007/BF02093613. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Liu H, Harrell LE, Shenvi S, Hagen T, Liu RM. Gender differences in glutathione metabolism in Alzheimer's disease. J Neurosci Res. 2005;79:861–867. doi: 10.1002/jnr.20424. [DOI] [PubMed] [Google Scholar]
- Lou MF, Dickerson JE., Jr. Protein-thiol mixed disulfides in human lens. Exp Eye Res. 1992;55:889–896. doi: 10.1016/0014-4835(92)90015-k. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Gabbita SP, Markesbery WR. Increased DNA oxidation and decreased levels of repair products in Alzheimer's disease ventricular CSF. J Neurochem. 1999;72:771–776. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. An Assessment of Oxidative Damage to Proteins, Lipids, and DNA in Brain from Patients with Alzheimer's Disease. Journal of Neurochemistry. 1997;68:2061–2069. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc) 1998;63:854–865. [PubMed] [Google Scholar]
- Malek A, Sager R, Schneider H. Effect of Hypoxia, Oxidative Stress and Lipopolysaccharides on the Release of Prostaglandins and Cytokines from Human Term Placental Explants. Placenta. 2001;22:S45–S50. doi: 10.1053/plac.2001.0635. [DOI] [PubMed] [Google Scholar]
- Malins DC, Johnson PM, Wheeler TM, Barker EA, Polissar NL, Vinson MA. Age-related radical-induced DNA damage is linked to prostate cancer. Cancer Res. 2001;61:6025–6028. [PubMed] [Google Scholar]
- Manini P, De Palma G, Andreoli R, Marczynski B, Hanova M, Mozzoni P, Naccarati A, Vodickova L, Hlavac P, Mutti A, Vodicka P. Biomarkers of nucleic acid oxidation, polymorphism in, and expression of, hOGG1 gene in styrene-exposed workers. Toxicology Letters. 2009;190:41–47. doi: 10.1016/j.toxlet.2009.06.862. [DOI] [PubMed] [Google Scholar]
- Mantle D, Falkous G, Walker D. Quantification of protease activities in synovial fluid from rheumatoid and osteoarthritis cases: comparison with antioxidant and free radical damage markers. Clin Chim Acta. 1999;284:45–58. doi: 10.1016/s0009-8981(99)00055-8. [DOI] [PubMed] [Google Scholar]
- Marcon F, Andreoli C, Rossi S, Verdina A, Galati R, Crebelli R. Assessment of individual sensitivity to ionizing radiation and DNA repair efficiency in a healthy population. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 2003;541:1–8. doi: 10.1016/s1383-5718(03)00171-2. [DOI] [PubMed] [Google Scholar]
- Martinet W, de Meyer GR, Herman AG, Kockx MM. Reactive oxygen species induce RNA damage in human atherosclerosis. Eur J Clin Invest. 2004;34:323–327. doi: 10.1111/j.1365-2362.2004.01343.x. [DOI] [PubMed] [Google Scholar]
- Martinet W, Knaapen MWM, De Meyer GRY, Herman AG, Kockx MM. Elevated Levels of Oxidative DNA Damage and DNA Repair Enzymes in Human Atherosclerotic Plaques. Circulation. 2002;106:927–932. doi: 10.1161/01.cir.0000026393.47805.21. [DOI] [PubMed] [Google Scholar]
- März P, Heese K, Hock C, Golombowski S, Müller-Spahn F, Rose-John S, Otten U. Interleukin-6 (IL-6) and soluble forms of IL-6 receptors are not altered in cerebrospinal fluid of Alzheimer's disease patients. Neuroscience Letters. 1997;239:29–32. doi: 10.1016/s0304-3940(97)00886-0. [DOI] [PubMed] [Google Scholar]
- Mastaloudis A, Morrow JD, Hopkins DW, Devaraj S, Traber MG. Antioxidant supplementation prevents exercise-induced lipid peroxidation, but not inflammation, in ultramarathon runners. Free Radical Biology and Medicine. 2004a;36:1329–1341. doi: 10.1016/j.freeradbiomed.2004.02.069. [DOI] [PubMed] [Google Scholar]
- Mastaloudis A, Yu T-W, O'Donnell RP, Frei B, Dashwood RH, Traber MG. Endurance exercise results in DNA damage as detected by the comet assay. Free Radical Biology and Medicine. 2004b;36:966–975. doi: 10.1016/j.freeradbiomed.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Mateos R, Bravo L. Chromatographic and electrophoretic methods for the analysis of biomarkers of oxidative damage to macromolecules (DNA, lipids, and proteins) J Sep Sci. 2007;30:175–191. doi: 10.1002/jssc.200600314. [DOI] [PubMed] [Google Scholar]
- Matsubara LS, Machado PE. Age-related changes of glutathione content, glutathione reductase and glutathione peroxidase activity of human erythrocytes. Brazilian journal of medical and biological research. 1991;24:449–454. [PubMed] [Google Scholar]
- McCarty MF. Interleukin-6 as a central mediator of cardiovascular risk associated with chronic inflammation, smoking, diabetes, and visceral obesity: down-regulation with essential fatty acids, ethanol and pentoxifylline. Med Hypotheses. 1999;52:465–477. doi: 10.1054/mehy.1997.0684. [DOI] [PubMed] [Google Scholar]
- Mecocci P, Fanó G, Fulle S, MacGarvey U, Shinobu L, Polidori MC, Cherubini A, Vecchiet J, Senin U, Beal MF. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radical Biology and Medicine. 1999;26:303–308. doi: 10.1016/s0891-5849(98)00208-1. [DOI] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Annals of Neurology. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annual Review of Biochemistry. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Miller SB. Prostaglandins in health and disease: an overview. Semin Arthritis Rheum. 2006;36:37–49. doi: 10.1016/j.semarthrit.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Moller P. Assessment of reference values for DNA damage detected by the comet assay in human blood cell DNA. Mutat Res. 2006;612:84–104. doi: 10.1016/j.mrrev.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Montine T, Peskind E, Quinn J, Wilson A, Montine K, Galasko D. Increased Cerebrospinal Fluid F2-Isoprostanes are Associated with Aging and Latent Alzheimer's Disease as Identified by Biomarkers. NeuroMolecular Medicine. 2011;13:37–43. doi: 10.1007/s12017-010-8126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Beal MF, Robertson D, Cudkowicz ME, Biaggioni I, O'Donnell H, Zackert WE, Roberts LJ, Morrow JD. Cerebrospinal fluid F2-isoprostanes are elevated in Huntington's disease. Neurology. 1999;52:1104–1105. doi: 10.1212/wnl.52.5.1104. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD. Lipid peroxidation in aging brain and Alzheimer's disease. Free Radic Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- Montuschi P, Barnes P, Roberts LJ., 2nd Insights into oxidative stress: the isoprostanes. Curr Med Chem. 2007;14:703–717. doi: 10.2174/092986707780059607. [DOI] [PubMed] [Google Scholar]
- Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov SA, Barnes PJ. Increased 8-isoprostane, a marker of oxidative stress, in exhaled condensate of asthma patients. Am J Respir Crit Care Med. 1999;160:216–220. doi: 10.1164/ajrccm.160.1.9809140. [DOI] [PubMed] [Google Scholar]
- Moore KP, Holt SG, Patel RP, Svistunenko DA, Zackert W, Goodier D, Reeder BJ, Clozel M, Anand R, Cooper CE, Morrow JD, Wilson MT, Darley-Usmar V, Roberts LJ., 2nd A causative role for redox cycling of myoglobin and its inhibition by alkalinization in the pathogenesis and treatment of rhabdomyolysis-induced renal failure. J Biol Chem. 1998;273:31731–31737. doi: 10.1074/jbc.273.48.31731. [DOI] [PubMed] [Google Scholar]
- Morocz M, Kalman J, Juhasz A, Sinko I, McGlynn AP, Downes CS, Janka Z, Rasko I. Elevated levels of oxidative DNA damage in lymphocytes from patients with Alzheimer's disease. Neurobiol Aging. 2002;23:47–53. doi: 10.1016/s0197-4580(01)00257-3. [DOI] [PubMed] [Google Scholar]
- Morris AA, Zhao L, Patel RS, Jones DP, Ahmed Y, Stoyanova N, Gibbons GH, Vaccarino V, Din-Dzietham R, Quyyumi AA. Differences in systemic oxidative stress based on race and the metabolic syndrome: the Morehouse and Emory Team up to Eliminate Health Disparities (META-Health) study. Metab Syndr Relat Disord. 2012;10:252–259. doi: 10.1089/met.2011.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JD, Frei B, Longmire AW, Gaziano JM, Lynch SM, Shyr Y, Strauss WE, Oates JA, Roberts LJ., 2nd Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med. 1995;332:1198–1203. doi: 10.1056/NEJM199505043321804. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., 2nd A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ. The isoprostanes: unique bioactive products of lipid peroxidation. Prog Lipid Res. 1997;36:1–21. doi: 10.1016/s0163-7827(97)00001-5. [DOI] [PubMed] [Google Scholar]
- Muller WU, Bauch T, Streffer C, Von Mallek D. Does radiotherapy affect the outcome of the comet assay? British Journal of Radiology. 2002;75:608–614. doi: 10.1259/bjr.75.895.750608. [DOI] [PubMed] [Google Scholar]
- Muller WU, Bauch T, Stüben G, Sack H, Streffer C. Radiation sensitivity of lymphocytes from healthy individuals and cancer patients as measured by the comet assay. Radiation and Environmental Biophysics. 2001;40:83–89. doi: 10.1007/s004110000087. [DOI] [PubMed] [Google Scholar]
- Mutlu-Turkoglu U, Ilhan E, Oztezcan S, Kuru A, Aykac-Toker G, Uysal M. Age-related increases in plasma malondialdehyde and protein carbonyl levels and lymphocyte DNA damage in elderly subjects. Clin Biochem. 2003;36:397–400. doi: 10.1016/s0009-9120(03)00035-3. [DOI] [PubMed] [Google Scholar]
- Nagababu E, Mohanty JG, Bhamidipaty S, Ostera GR, Rifkind JM. Role of the membrane in the formation of heme degradation products in red blood cells. Life Sciences. 2010;86:133–138. doi: 10.1016/j.lfs.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagababu E, Rifkind JM. Formation of fluorescent heme degradation products during the oxidation of hemoglobin by hydrogen peroxide. Biochem Biophys Res Commun. 1998;247:592–596. doi: 10.1006/bbrc.1998.8846. [DOI] [PubMed] [Google Scholar]
- Nagababu E, Rifkind JM. Heme degradation by reactive oxygen species. Antioxid Redox Signal. 2004;6:967–978. doi: 10.1089/ars.2004.6.967. [DOI] [PubMed] [Google Scholar]
- Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K. Oxidative damage in nucleic acids and Parkinson's disease. Journal of Neuroscience Research. 2007;85:919–934. doi: 10.1002/jnr.21191. [DOI] [PubMed] [Google Scholar]
- Nakamura AJ, Chiang YJ, Hathcock KS, Horikawa I, Sedelnikova OA, Hodes RJ, Bonner WM. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin. 2008;1:6. doi: 10.1186/1756-8935-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren Hooten N, Ejiogu N, Zonderman AB, Evans MK. Association of Oxidative DNA Damage and C-Reactive Protein in Women at Risk for Cardiovascular Disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:2776–2784. doi: 10.1161/ATVBAHA.112.300276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Castellani R, Zhu X, Moreira P, Perry G, Smith M. Involvement of oxidative stress in Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2006;65:631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Chiba S, Kosaka K, Takeda A, Castellani RJ, Smith MA, Perry G. Neuronal RNA oxidation is a prominent feature of dementia with Lewy bodies. Neuroreport. 2002;13:2035–2039. doi: 10.1097/00001756-200211150-00009. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Hirai K, Aliev G, Takeda A, Chiba S, Smith MA. Neuronal RNA oxidation in Alzheimer's disease and Down's syndrome. Ann N Y Acad Sci. 1999a;893:362–364. doi: 10.1111/j.1749-6632.1999.tb07855.x. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S, Smith MA. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol. 2000;59:1011–1017. doi: 10.1093/jnen/59.11.1011. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999b;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Tamaoki T, Tanaka K, Motohashi N, Nakamura M, Hayashi T, Yamaguchi H, Shimohama S, Lee HG, Zhu X, Smith MA, Perry G. Intraneuronal amyloid beta accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol Dis. 2010;37:731–737. doi: 10.1016/j.nbd.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguogho A, Kaliman J, Sinzinger H. Levels of eicosanoids (6-oxo-PGF1 alpha and 8-epi-PGF2 alpha) in human and porcine lymphatics and lymph. Lymphology. 1998;31:186–189. [PubMed] [Google Scholar]
- Oliver CN, Ahn BW, Moerman EJ, Goldstein S, Stadtman ER. Age-related changes in oxidized proteins. Journal of Biological Chemistry. 1987;262:5488–5491. [PubMed] [Google Scholar]
- Pagano G, Zatterale A, Degan P, d'Ischia M, Kelly F, Pallardó F, Kodama S. Multiple Involvement of Oxidative Stress in Werner Syndrome Phenotype. Biogerontology. 2005;6:233–243. doi: 10.1007/s10522-005-2624-1. [DOI] [PubMed] [Google Scholar]
- Park EM, Shigenaga MK, Degan P, Korn TS, Kitzler JW, Wehr CM, Kolachana P, Ames BN. Assay of excised oxidative DNA lesions: isolation of 8-oxoguanine and its nucleoside derivatives from biological fluids with a monoclonal antibody column. Proc Natl Acad Sci U S A. 1992;89:3375–3379. doi: 10.1073/pnas.89.8.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy S, Litvinov D, Selvarajan K, Garelnabi M. Lipid peroxidation and decomposition--conflicting roles in plaque vulnerability and stability. Biochim Biophys Acta. 2008;1781:221–231. doi: 10.1016/j.bbalip.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Elizur T, Elinger D, Leitner-Dagan Y, Blumenstein S, Krupsky M, Berrebi A, Schechtman E, Livneh Z. Development of an enzymatic DNA repair assay for molecular epidemiology studies: Distribution of OGG activity in healthy individuals. DNA Repair. 2007;6:45–60. doi: 10.1016/j.dnarep.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Perry G, Nunomura A, Hirai K, Zhu X, Perez M, Avila J, Castellani RJ, Atwood CS, Aliev G, Sayre LM, Takeda A, Smith MA. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer's and other neurodegenerative diseases? Free Radic Biol Med. 2002;33:1475–1479. doi: 10.1016/s0891-5849(02)01113-9. [DOI] [PubMed] [Google Scholar]
- Petersen RB, Siedlak SL, Lee HG, Kim YS, Nunomura A, Tagliavini F, Ghetti B, Cras P, Moreira PI, Castellani RJ, Guentchev M, Budka H, Ironside JW, Gambetti P, Smith MA, Perry G. Redox metals and oxidative abnormalities in human prion diseases. Acta Neuropathol. 2005;110:232–238. doi: 10.1007/s00401-005-1034-4. [DOI] [PubMed] [Google Scholar]
- Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol. 2003;81:123–129. doi: 10.1139/o03-042. [DOI] [PubMed] [Google Scholar]
- Piperakis SM, Visvardis EE, Sagnou M, Tassiou AM. Effects of smoking and aging on oxidative DNA damage of human lymphocytes. Carcinogenesis. 1998;19:695–698. doi: 10.1093/carcin/19.4.695. [DOI] [PubMed] [Google Scholar]
- Pirmohamed M, Williams D, Tingle MD, Barry M, Khoo SH, O'Mahony C, Wilkins EG, Breckenridge AM, Park BK. Intracellular glutathione in the peripheral blood cells of HIV-infected patients: failure to show a deficiency. AIDS. 1996;10:501–507. doi: 10.1097/00002030-199605000-00008. [DOI] [PubMed] [Google Scholar]
- Popanda O, Ebbeler R, Twardella D, Helmbold I, Gotzes F, Schmezer P, Thielmann HW, von Fournier D, Haase W, Sautter-Bihl ML, Wenz F, Bartsch H, Chang-Claude J. Radiation-induced DNA damage and repair in lymphocytes from breast cancer patients and their correlation with acute skin reactions to radiotherapy. Int J Radiat Oncol Biol Phys. 2003;55:1216–1225. doi: 10.1016/s0360-3016(02)04415-2. [DOI] [PubMed] [Google Scholar]
- Pratico D, Barry OP, Lawson JA, Adiyaman M, Hwang SW, Khanapure SP, Iuliano L, Rokach J, FitzGerald GA. IPF2alpha-I: an index of lipid peroxidation in humans. Proc Natl Acad Sci U S A. 1998a;95:3449–3454. doi: 10.1073/pnas.95.7.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratico D, Clark CM, Liun F, Rokach J, Lee VY, Trojanowski JQ. Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of Alzheimer disease. Arch Neurol. 2002;59:972–976. doi: 10.1001/archneur.59.6.972. [DOI] [PubMed] [Google Scholar]
- Pratico D, Iuliano L, Basili S, Ferro D, Camastra C, Cordova C, FitzGerald GA, Violi F. Enhanced lipid peroxidation in hepatic cirrhosis. J Investig Med. 1998b;46:51–57. [PubMed] [Google Scholar]
- Pryor WA, Stanley JP, Blair E. Autoxidation of polyunsaturated fatty acids: II. A suggested mechanism for the formation of TBA-reactive materials from prostaglandin-like endoperoxides. Lipids. 1976;11:370–379. doi: 10.1007/BF02532843. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. 2000;16:534–554. doi: 10.1034/j.1399-3003.2000.016003534.x. [DOI] [PubMed] [Google Scholar]
- Rajaee-Behbahani N, Schmezer P, Risch A, Rittgen W, Kayser KW, Dienemann H, Schulz V, Drings P, Thiel S, Bartsch H. Altered DNA repair capacity and bleomycin sensitivity as risk markers for non-small cell lung cancer. International Journal of Cancer. 2001;95:86–91. doi: 10.1002/1097-0215(20010320)95:2<86::aid-ijc1015>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Rajeswari N, Ahuja YR, Malini U, Chandrashekar S, Balakrishna N, Rao KV, Khar A. Risk assessment in first degree female relatives of breast cancer patients using the alkaline Comet assay. Carcinogenesis. 2000;21:557–561. doi: 10.1093/carcin/21.4.557. [DOI] [PubMed] [Google Scholar]
- Range SP, Dunster C, Knox AJ, Kelly FJ. Treatment of pulmonary exacerbations of cystic fibrosis leads to improved antioxidant status. Eur Respir J. 1999;13:560–564. doi: 10.1183/09031936.99.13356099. [DOI] [PubMed] [Google Scholar]
- Reilly M, Delanty N, Lawson JA, FitzGerald GA. Modulation of oxidant stress in vivo in chronic cigarette smokers. Circulation. 1996;94:19–25. doi: 10.1161/01.cir.94.1.19. [DOI] [PubMed] [Google Scholar]
- Reiner AP, Aragaki AK, Gray SL, Wactawski-Wende J, Cauley JA, Cochrane BB, Kooperberg CL, Woods NF, LaCroix AZ. Inflammation and Thrombosis Biomarkers and Incident Frailty in Postmenopausal Women. The American Journal of Medicine. 2009;122:947–954. doi: 10.1016/j.amjmed.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee Y, Valentine MR, Termini J. Oxidative base damage in RNA detected by reverse transcriptase. Nucleic Acids Res. 1995;23:3275–3282. doi: 10.1093/nar/23.16.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkind J, Abugo O, Levy A, Monticone R. Formation of free radicals under hypoxia. In: Hochachka PW, Lutz PL, Sick T, Rosenthal M, van den Thillart G, editors. Surviving Hypoxia: Mechanisms of Control and Adaptation. CRC Press; Boca Raton, FL: 1993. [Google Scholar]
- Rifkind JM, Ramasamy S, Manoharan PT, Nagababu E, Mohanty JG. Redox reactions of hemoglobin. Antioxid Redox Signal. 2004;6:657–666. doi: 10.1089/152308604773934422. [DOI] [PubMed] [Google Scholar]
- Rizvi SI, Maurya PK. Markers of Oxidative Stress in Erythrocytes during Aging in Humans. Annals of the New York Academy of Sciences. 2007;1100:373–382. doi: 10.1196/annals.1395.041. [DOI] [PubMed] [Google Scholar]
- Roberts LJ, Morrow JD. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free Radic Biol Med. 2000;28:505–513. doi: 10.1016/s0891-5849(99)00264-6. [DOI] [PubMed] [Google Scholar]
- Rodin SN, Rodin AS. Origins and selection of p53 mutations in lung carcinogenesis. Seminars in Cancer Biology. 2005;15:103–112. doi: 10.1016/j.semcancer.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rokach J, Khanapure SP, Hwang SW, Adiyaman M, Lawson JA, FitzGerald GA. Nomenclature of isoprostanes: a proposal. Prostaglandins. 1997;54:853–873. doi: 10.1016/s0090-6980(97)00184-6. [DOI] [PubMed] [Google Scholar]
- Ryan KA, Smith MF, Jr., Sanders MK, Ernst PB. Reactive oxygen and nitrogen species differentially regulate Toll-like receptor 4-mediated activation of NF-kappa B and interleukin-8 expression. Infect Immun. 2004;72:2123–2130. doi: 10.1128/IAI.72.4.2123-2130.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samiec PS, Drews-Botsch C, Flagg EW, Kurtz JC, Sternberg P, Jr., Reed RL, Jones DP. Glutathione in human plasma: decline in association with aging, age-related macular degeneration, and diabetes. Free Radic Biol Med. 1998;24:699–704. doi: 10.1016/s0891-5849(97)00286-4. [DOI] [PubMed] [Google Scholar]
- Schmaltz HN, Fried LP, Xue Q-L, Walston J, Leng SX, Semba RD. Chronic Cytomegalovirus Infection and Inflammation Are Associated with Prevalent Frailty in Community-Dwelling Older Women. Journal of the American Geriatrics Society. 2005;53:747–754. doi: 10.1111/j.1532-5415.2005.53250.x. [DOI] [PubMed] [Google Scholar]
- Schmid TE, Eskenazi B, Baumgartner A, Marchetti F, Young S, Weldon R, Anderson D, Wyrobek AJ. The effects of male age on sperm DNA damage in healthy non-smokers. Human Reproduction. 2007;22:180–187. doi: 10.1093/humrep/del338. [DOI] [PubMed] [Google Scholar]
- Schurman SH, Dunn CA, Greaves R, Yu B, Ferrucci L, Croteau DL, Seidman MM, Bohr VA. Age-Related Disease Association of Endogenous gamma-H2AX Foci in Mononuclear Cells Derived from Leukapheresis. PLoS One. 2012;7:e45728. doi: 10.1371/journal.pone.0045728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat Res. 2002;158:486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Shacter E. Quantification and significance of protein oxidation in biological samples. Drug Metab Rev. 2000;32:307–326. doi: 10.1081/dmr-100102336. [DOI] [PubMed] [Google Scholar]
- Shan X, Lin CL. Quantification of oxidized RNAs in Alzheimer's disease. Neurobiol Aging. 2006;27:657–662. doi: 10.1016/j.neurobiolaging.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Shan X, Tashiro H, Lin CL. The identification and characterization of oxidized RNAs in Alzheimer's disease. J Neurosci. 2003;23:4913–4921. doi: 10.1523/JNEUROSCI.23-12-04913.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Wu W, Hazen SL. Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical. Biochemistry. 2000;39:5474–5482. doi: 10.1021/bi992809y. [DOI] [PubMed] [Google Scholar]
- Sies H. Oxidative stress: oxidants and antioxidants. Experimental Physiology. 1997;82:291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- Singh NP, Muller CH, Berger RE. Effects of age on DNA double-strand breaks and apoptosis in human sperm. Fertility and Sterility. 2003;80:1420–1430. doi: 10.1016/j.fertnstert.2003.04.002. [DOI] [PubMed] [Google Scholar]
- Sinzinger H, Oguogho A, Kaliman J. Isoprostane 8-epi-prostaglandin F2 alpha is a potent contractor of human peripheral lymphatics. Lymphology. 1997;30:155–159. [PubMed] [Google Scholar]
- Siomek A, Gackowski D, Rozalski R, Dziaman T, Szpila A, Guz J, Olinski R. Higher leukocyte 8-oxo-7,8-dihydro-2'-deoxyguanosine and lower plasma ascorbate in aging humans? Antioxidants & redox signaling. 2007;9:143–150. doi: 10.1089/ars.2007.9.143. [DOI] [PubMed] [Google Scholar]
- Smith CC, O'Donovan MR, Martin EA. hOGG1 recognizes oxidative damage using the comet assay with greater specificity than FPG or ENDOIII. Mutagenesis. 2006;21:185–190. doi: 10.1093/mutage/gel019. [DOI] [PubMed] [Google Scholar]
- Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TR, Miller MS, Lohman KK, Case LD, Hu JJ. DNA damage and breast cancer risk. Carcinogenesis. 2003;24:883–889. doi: 10.1093/carcin/bgg037. [DOI] [PubMed] [Google Scholar]
- Spitz MR, Wei Q, Dong Q, Amos CI, Wu X. Genetic susceptibility to lung cancer: The role of DNA damage and repair. Cancer Epidemiology Biomarkers and Prevention. 2003;12:689–698. [PubMed] [Google Scholar]
- Stadtman ER. Role of oxidized amino acids in protein breakdown and stability. Methods Enzymol. 1995;258:379–393. doi: 10.1016/0076-6879(95)58057-3. [DOI] [PubMed] [Google Scholar]
- Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x. [DOI] [PubMed] [Google Scholar]
- Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Chem Res Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- Stocker R, Keaney JF., Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- Stoyanova E, Sandoval SB, Zúñiga LA, El-Yamani N, Coll E, Pastor S, Reyes J, Andrés E, Ballarin J, Xamena N, Marcos R. Oxidative DNA damage in chronic renal failure patients. Nephrology Dialysis Transplantation. 2010;25:879–885. doi: 10.1093/ndt/gfp575. [DOI] [PubMed] [Google Scholar]
- Szaflik JP, Janik-Papis K, Synowiec E, Ksiazek D, Zaras M, Wozniak K, Szaflik J, Blasiak J. DNA damage and repair in age-related macular degeneration. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2009;669:169–176. doi: 10.1016/j.mrfmmm.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Szanton SL, Rifkind JM, Mohanty JG, Miller ER, 3rd, Thorpe RJ, Nagababu E, Epel ES, Zonderman AB, Evans MK. Racial Discrimination Is Associated with a Measure of Red Blood Cell Oxidative Stress: A Potential Pathway for Racial Health Disparities. Int J Behav Med. 2011 doi: 10.1007/s12529-011-9188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Chock PB, Stadtman ER. Oxidized messenger RNA induces translation errors. Proc Natl Acad Sci U S A. 2007;104:66–71. doi: 10.1073/pnas.0609737104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Han S, Kupfer PA, Leumann CJ, Sonntag WE. An assay for RNA oxidation induced abasic sites using the Aldehyde Reactive Probe. Free Radic Res. 2011;45:237–247. doi: 10.3109/10715762.2010.535529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateyama M, Takeda A, Onodera Y, Matsuzaki M, Hasegawa T, Nunomura A, Hirai K, Perry G, Smith MA, Itoyama Y. Oxidative stress and predominant Abeta42(43) deposition in myopathies with rimmed vacuoles. Acta Neuropathol. 2003;105:581–585. doi: 10.1007/s00401-003-0685-2. [DOI] [PubMed] [Google Scholar]
- Te Koppele JM, Lucassen PJ, Sakkee AN, Van Asten JG, Ravid R, Swaab DF, Van Bezooijen CF. 8OHdG levels in brain do not indicate oxidative DNA damage in Alzheimer's disease. Neurobiol Aging. 1996;17:819–826. doi: 10.1016/s0197-4580(96)00165-0. [DOI] [PubMed] [Google Scholar]
- Telci A, Cakatay U, Salman S, Satman I, Sivas A. Oxidative protein damage in early stage Type 1 diabetic patients. Diabetes Res Clin Pract. 2000;50:213–223. doi: 10.1016/s0168-8227(00)00197-2. [DOI] [PubMed] [Google Scholar]
- Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999;7:169–177. doi: 10.1016/S0969-2126(99)80023-9. [DOI] [PubMed] [Google Scholar]
- Traverso N, Patriarca S, Balbis E, Furfaro AL, Cottalasso D, Pronzato MA, Carlier P, Botta F, Marinari UM, Fontana L. Anti malondialdehyde-adduct immunological response as a possible marker of successful aging. Experimental Gerontology. 2003;38:1129–1135. doi: 10.1016/s0531-5565(03)00188-8. [DOI] [PubMed] [Google Scholar]
- Trzeciak A, Kowalik J, Malecka-Panas E, Drzewoski J, Wojewodzka M, Iwanenko T, Blasiak J. Genotoxicity of chromium in human gastric mucosa cells and peripheral blood lymphocytes evaluated by the single cell gel electrophoresis (comet assay) Med Sci Monit. 2000;6:24–29. [PubMed] [Google Scholar]
- Trzeciak AR, Barnes J, Ejiogu N, Foster K, Brant LJ, Zonderman AB, Evans MK. Age, sex, and race influence single-strand break repair capacity in a human population. Free Radical Biology and Medicine. 2008;45:1631–1641. doi: 10.1016/j.freeradbiomed.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzeciak AR, Barnes J, Evans MK. A Modified Alkaline Comet Assay for Measuring DNA Repair Capacity in Human Populations. Radiation Research. 2009;169:110–121. doi: 10.1667/RR1101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzeciak AR, Mohanty JG, Jacob KD, Barnes J, Ejiogu N, Lohani A, Zonderman AB, Rifkind JM, Evans MK. Oxidative damage to DNA and single strand break repair capacity: Relationship to other measures of oxidative stress in a population cohort. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2012;736:93–103. doi: 10.1016/j.mrfmmm.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Koycu S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hol EM. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science. 1998;279:242–247. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- Voss P, Siems W. Clinical oxidation parameters of aging. Free Radic Res. 2006;40:1339–1349. doi: 10.1080/10715760600953859. [DOI] [PubMed] [Google Scholar]
- Walston J, McBurnie M, Newman A, Tracy RP, Kop WJ, Hirsch CH, Gottdiener JS, Fried LP. Frailty and activation of the inflammation and coagulation systems with and without clinical comorbidities: Results from the cardiovascular health study. Archives of Internal Medicine. 2002;162:2333–2341. doi: 10.1001/archinte.162.20.2333. [DOI] [PubMed] [Google Scholar]
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. Journal of Neurochemistry. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- Watters JL, Satia JA, Kupper LL. Correlates of antioxidant nutrients and oxidative DNA damage differ by race in a cross-sectional study of healthy African American and white adults. Nutr Res. 2008;28:565–576. doi: 10.1016/j.nutres.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Cheng L, Amos CI, Wang LE, Guo Z, Hong WK, Spitz MR. Repair of tobacco carcinogen-induced DNA adducts and lung cancer risk: A molecular epidemiologic study. Journal of the National Cancer Institute. 2000;92:1764–1772. doi: 10.1093/jnci/92.21.1764. [DOI] [PubMed] [Google Scholar]
- Wei Q, Matanoski GM, Farmer ER, Hedayati MA, Grossman L. DNA repair and aging in basal cell carcinoma: A molecular epidemiology study. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:1614–1618. doi: 10.1073/pnas.90.4.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimann A, Belling D, Poulsen HE. Quantification of 8-oxo-guanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high-performance liquid chromatography-electrospray tandem mass spectrometry. Nucleic Acids Research. 2002;30:e7. doi: 10.1093/nar/30.2.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojewodzka M, Kruszewski M, Iwanenko T, Collins AR, Szumiel I. Application of the comet assay for monitoring DNA damage in workers exposed to chronic low-dose irradiation. I. Strand breakage. Mutat Res. 1998;416:21–35. doi: 10.1016/s1383-5718(98)00073-4. [DOI] [PubMed] [Google Scholar]
- Wood-Kaczmar A, Gandhi S, Wood NW. Understanding the molecular causes of Parkinson's disease. Trends in molecular medicine. 2006;12:521–528. doi: 10.1016/j.molmed.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Wu IC, Shiesh S-C, Kuo P-H, Lin X-Z. High Oxidative Stress Is Correlated with Frailty in Elderly Chinese. Journal of the American Geriatrics Society. 2009;57:1666–1671. doi: 10.1111/j.1532-5415.2009.02392.x. [DOI] [PubMed] [Google Scholar]
- Yan T, Berry SE, Desai AB, Kinsella TJ. DNA Mismatch Repair (MMR) Mediates 6-Thioguanine Genotoxicity by Introducing Single-strand Breaks to Signal a G2-M Arrest in MMR-proficient RKO Cells. Clinical Cancer Research. 2003;9:2327–2334. [PubMed] [Google Scholar]
- Yin B, Whyatt RM, Perera FP, Randall MC, Cooper TB, Santella RM. Determination of 8-hydroxydeoxyguanosine by an immunoaffinity chromatography-monoclonal antibody-based ELISA. Free Radic Biol Med. 1995;18:1023–1032. doi: 10.1016/0891-5849(95)00003-g. [DOI] [PubMed] [Google Scholar]
- Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]