

Abstract
Activated cancer-associated fibroblasts (CAFs) or myofibroblasts not only facilitate tumor growth and spread but also affect tumor response to therapeutic agents. Therefore, it became clear that efficient therapeutic regimens should also take into account the presence of these supportive cells and inhibit their paracrine effects. To this end, we tested the effect of low concentrations of curcumin, a pharmacologically safe natural product, on patient-derived primary breast CAF cells. We have shown that curcumin treatment upregulates p16INK4A and other tumor suppressor proteins while inactivates the JAK2/STAT3 pathway. This reduced the level of alpha-smooth muscle actin (α-SMA) and the migration/invasion abilities of these cells. Furthermore, curcumin suppressed the expression/secretion of stromal cell-derived factor-1 (SDF-1), interleukin-6 (IL-6), matrix metalloproteinase-2 (MMP-2), MMP-9, and transforming growth factor-β, which impeded their paracrine procarcinogenic potential. Intriguingly, these effects were sustained even after curcumin withdrawal and cell splitting. Therefore, using different markers of senescence [senescence-associated β-galactosidase (SA-β-gal) activity, Ki-67 and Lamin B1 levels, and bromodeoxyuridine incorporation], we have shown that curcumin markedly suppresses Lamin B1 and triggers DNA damage-independent senescence in proliferating but not quiescent breast stromal fibroblasts. Importantly, this curcumin-related senescence was p16INK4A-dependent and occurred with no associated inflammatory secretory phenotype. These results indicate the possible inactivation of cancer-associated myofibroblasts and present the first indication that curcumin can trigger DNA damage-independent and safe senescence in stromal fibroblasts.
Introduction
Breast cancer remains the leading cause of morbidity and second leading cause of death in women worldwide [1]. Breast carcinogenesis is a complex process involving molecular and functional alterations in both the epithelial and stromal compartments. Indeed, several lines of evidence indicate that cancer-associated fibroblasts (CAFs), which constitute a major portion of the reactive tumor stroma, actively participate in tumor growth, invasion, and metastasis [2–4]. This involves many chemokines, growth factors, and matrix metalloproteinases (MMPs), which transmit the message in both directions, allowing cooperative crosstalk between cancer cells and their stroma [3,5].
Thereby, it became clear that efficient cancer therapy should take into account the presence of stromal cells, which could be responsible for resistance to treatment and also for tumor recurrence [6]. To this end, nontoxic bioactive dietary components represent a promising strategy for cancer prevention/treatment. Curcumin, the yellow pigment of turmeric, showed diverse biologic and medicinal activities [7]. Epidemiological studies have shown reduced rate of colon carcinogenesis in populations whose diet is rich in curcumin, which suggested anticarcinogenic activities of this molecule [8]. It has been shown in various animal models and human studies that curcumin is extremely safe even at very high doses. Phase I clinical trials showed that curcumin is safe to humans up to 12 g/day when taken orally and caused histologic improvement of precancerous lesions in some patients, suggesting that it is biologically active at these doses [9,10]. Recently, a phase I dose escalation trial of docetaxel plus curcumin was carried out on patients with advanced and metastatic breast cancer and showed safety and tolerability of the combination, with an increase in the intestinal absorption of docetaxel [11].
Currently, most anticancer drugs are designed to kill cancer cells, while inducing other cellular tumor suppressor processes, such as senescence, in tumor cells or their stroma may be more efficient with less side effects [12]. Senescence is a cell fate program characterized by irreversible growth arrest associated with changes in cell morphology, function, and behavior [13–15]. This program is triggered by various stimuli and stresses that prevent aged or abnormal cells from anarchical proliferation. Senescent cells have been shown to accumulate in a variety of aging tissues as well as several premalignant and malignant lesions. Because cellular senescence eliminates the proliferative capacity of precarcinogenic or procarcinogenic cells, it is considered as a potent tumor-suppressing mechanism [16,17]. Senescent human cells exhibit numerous changes in gene expression, many of which relate to the growth arrest [18]. Senescent cells also develop a senescence-associated secretory phenotype (SASP), which is characterized by the secretion of a wide range of growth factors, cytokines, extracellular matrix proteins, and degradative enzymes, most of which can alter the local tissue microenvironment [19–21].
In the present study, we have shown that curcumin inactivates breast CAFs through induction of p16-dependent senescence with no DNA damage.
Materials and Methods
Cells, Cell Culture, and Chemicals
Breast fibroblast cells were obtained and cultured as previously described (Hawsawi et al., 2008). MDA-MB-231 cells were obtained from ATCC (Manassas, VA) and were cultured following the instructions of the company. All supplements were obtained from Sigma (St Louis, MO) except for antibiotics and antimycotic solutions, which were obtained from Gibco (Grand Island, NY).
Cellular Lysate Preparation and Immunoblot Analysis
This has been performed as previously described [22]. Antibodies directed against JAK2 (D2E12), pJAK2 (Tyr1007/1008), STAT3, pSTAT3-Tyr705 (D3A7), P-pRB (Ser780), MMP-2, and MMP-9 were purchased from Cell Signaling Technology (Danvers, MA); SDF1, interleukin-6 (IL-6; 10E5), alpha-smooth muscle actin (α-SMA), transforming growth factor-β (TGF-β; 2AR2), Lamin B1 (119D5-F1), and Lamin A (131C3) from Abcam (Cambridge, MA); p16 from BD Biosciences (San Jose, CA); c-Myc (C-19), survivin (D-8), p21 (F-5), p53 (DO-1), pRB (F-8), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; FL-335) from Santa Cruz Biotechnology (Santa Cruz, CA); and P-pRB (Thr356) from Abnova (Taipei, Taiwan).
RNA Purification and Reverse Transcription-Polymerase Chain Reaction
Total RNA was purified using the TRI reagent (Sigma) according to the manufacturer's instructions. The concentration of RNA was determined using NanoDrop ND-1000 Spectrophotometer (Nanodrop Inc, Wilmington, DE). Single-stranded cDNA was obtained from reverse transcription (RT) of 1 µg of RNA using RT-polymerase chain reaction (PCR) Kit (BD Biosciences) and following the manufacturer's protocol. cDNA was then amplified with 1U Taq polymerase, dNTPs (50 mM), and primers (25 pmol each). The mixture was first heated at 94°C for 5 minutes and then 30 cycles at 94°C for 1 minute, 55°C for 1 minute, and 72°C for 1 minute, then 72°C for 10 minutes. PCR products were seen after electrophoresis on ethidium bromide-stained 2% agarose gels. The respective primers were as follows: LMNB1, 5′-AAGCAGCTGGAGTGGTTGTT-3′ (forward) and 5′-TTGGATGCTCTTGGGGTT-3′ (reverse); LMNA, 5′-AGCAAAGTGCGTGAGGAGTT-3′ (forward) and 5′-TCAGGTCACCCTCCTTCTTG-3′ (reverse); GAPDH, 5′-GAGTCCACTGGCGTCTTC-3′ (forward) and 5′-GGGGTGCTAAGCAGTTGGT-3′ (reverse).
Migration and Invasion Assays
Cell migration and invasion were evaluated using 24-well BD BioCoat Matrigel Invasion Chambers as per the manufacturer's guideline (BD Biosciences). In brief, 0.75 ml of migration buffer [serum-free medium (SFM)] or chemoattractants (serum-containing medium) was added to the lower chambers. Cells were washed three times in migration medium and 2 x 105 to 4 x 105 cells were added to the upper wells separated by 8-µm pore size polyethylene terephthalate (PET) membrane with thin layer of matrigel basement membrane matrix (for invasion) or without (for migration) and incubated for 18 hours. The membranes were stained with Diff-Quick stain (Fisher Scientific, Fair Lawn, NJ) after removing the nonmigrated cells from the top of the membrane with Q-tips. After air-drying, the membranes were cut and mounted on slides with oil, and cells that had migrated to the underside of the filter were counted using light microscope (Zeiss Axio Observer) in five randomly selected fields. Each assay was performed in triplicate. The results were expressed as mean ± SD of migrating cells per fields counted.
Transfection
pLKO.1-CDKN2A-shRNA and pIRES-CDKN2A-ORF plasmids were purchased from System Biosciences (Mountain View, CA) and used to prepare the lentiviral supernatant. Medium was removed from the target cells and replaced with lentiviral supernatant and incubated for 24 hours. Transduced cells were selected after 48 hours with puromycin or G418.
Viral Infection
Lentivirus-based vector bearing CDKN2A-ORF (pIRES) as well as its respective control (System Biosciences) was used to prepare the lentiviral supernatant from 293FT cells. Lentiviral supernatants were collected 48 hours post-transfection, filtered, and used for infection. Twenty-four hours later, media were replaced with complete media and cells were grown for 3 days.
Enzyme-Linked Immunosorbent Assay
Conditioned media from 24-hour fibroblast cell cultures either treated or not were harvested, and ELISA was performed according to the manufacturer's instructions (R&D Systems, Minneapolis, MN). The OD was used at 450 nm on a standard ELISA plate reader.
Bromodeoxyuridine and Ki-67 Immunostaining
The proportion of S phase cells was determined by assessing the levels of bromodeoxyuridine (BrdU) and Ki-67 by immunostaining as previously described [23]. Cells were incubated at 37°C with 10 µM BrdU for 18 hours, fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 minutes and then permeabilized with 0.2% Triton X-100 for 10 minutes at room temperature. Subsequently, cells were exposed to the BrdU antibody (Bu20a; Dako, Carpinteria, CA) in 10% fetal calf serum for 2 hours and then washed and incubated with the Alexa Fluor-conjugated antibody (1:500; Molecular Probes, Grand Island, NY).
For Ki-67, cells were fixed in 1:1 acetone/methanol, and a standard indirect immunoperoxidase procedure was applied using Ki-67 antibody (Abcam), followed by peroxidase-conjugated anti-rabbit antibody (Dako). Sites of antibody binding were visualized by the deposition of brown polymer of DAB chromogen (Novocastra Laboratories Ltd, Buffalo Grove, IL).
The percentage of BrdU- or Ki-67-labeled cells (LI) was determined for at least 500 cells per data point and expressed as a mean and standard error of triplicate determinations.
Assessment of SA-β-Galactosidase Activity
Endogenous senescence-associated β-galactosidase (SA-β-gal) activity was assessed histochemically. Cells were fixed in 0.5% glutaraldehyde in PBS for 15 minutes and then permeabilized with 0.02% NP-40 with 0.1% sodium deoxycholate for 15 minutes, followed by incubation (overnight at 37°C) in a 1 mg/ml solution of X-Gal substrate (5-bromo-4-chloro-3-indolyl-bd-galactopyranoside) with 5 mM potassium ferricyanide and 2 mM magnesium chloride together with the above-mentioned detergents, at an acidic pH (6.0). The proportion of β-gal-positive cells was assessed in a total count of 500 cells.
γ-H2AX Immunofluorescence Microscopy
Cells were fixed with 4% paraformaldehyde in PBS for 10 minutes and permeabilized with 0.2% Triton X-100 for 10 minutes at room temperature. Cells were blocked in 10% fetal calf serum (in PBS) and incubated with γ-H2AX (Ser-139) antibody (Upstate Biotechnology, New York, NY). Cells were then washed and incubated with Alexa Fluor-conjugated antibody (1:500; Molecular Probes). Nuclear foci were detected using Optika microscope.
Conditioned Media
Cells were cultured in medium ± serum for 24 hours, and then the medium was collected and centrifuged. The resulting supernatant was either used immediately or frozen at -80°C until needed.
Quantification of Protein and RNA Expression Levels
The expression levels of the immunoblotted proteins were measured using the densitometer (BIO-RAD GS-800 Calibrated Densitometer) as previously described [22].
Statistical Analysis
Statistical analysis was performed by Student's t test and P values of .05 and less were considered as statistically significant.
Results
Curcumin Upregulates p16, p21, and p53 and Inhibits STAT3-Related Secretory Phenotype in CAFs
We have previously shown that p16, p21, and p53 proteins are downregulated in breast CAFs compared to their tumor counterpart fibroblasts (TCFs), and these tumor suppressor proteins negatively regulate the expression of several procarcinogenic cytokines, which are highly expressed in these cells [2,24,25]. Therefore, we sought to investigate the possible up-regulation of these three tumor suppressor proteins and the inhibition of the procarcinogenic paracrine effects of these cells using curcumin. To this end, CAF-87 cells were challenged with curcumin (10 µM) for different periods of time (0, 4, 8, 18, and 24 hours), and protein extracts were prepared and used for immunoblot analysis using specific antibodies. Figure 1A shows that the basal levels of p16, p21, and p53 were indeed very low at time 0 and then increased several folds upon curcumin treatment. This increase was sustained during the whole treatment period (Figure 1A). Interestingly, the accumulation of these tumor suppressor proteins was accompanied by a sharp decrease in the level of α-SMA, a major marker of active stromal fibroblasts, which became undetectable after 8 hours of treatment (Figure 1B). Likewise, the levels of stromal cell-derived factor-1 (SDF-1) and TGF-β, which are also highly expressed in active fibroblasts, decreased following curcumin treatment (Figure 1B). Furthermore, curcumin strongly reduced the expression of IL-6, MMP-2, and MMP-9, reaching undetectable levels after only 8 hours of treatment (Figure 1C). To further show this, we assessed the level of secreted cytokines in the media upon curcumin treatment (10 µM for 24 hours) using ELISA. Figure 1D shows that curcumin significantly reduced the secreted levels of MMP-2, MMP-9, IL-6, SDF-1, and TGF-β. Similar results were obtained for other CAF cells (data not shown). These results indicate that curcumin can normalize active breast CAFs.
Figure 1.

Curcumin inactivates CAFs. (A–C and E) CAF-87 cells were challenged with curcumin (10 µM) for the indicated periods of time. Whole-cell lysates were prepared and 50 µg of proteins were used for immunoblot analysis using the indicated antibodies. The numbers below the bands represent fold of change compared to the level at time 0, upon normalization against GAPDH, used as internal control. (D) CAF-87 cells were either DMSO-treated (-) or challenged with curcumin (10 µM) for 24 hours (+). Secreted levels of proteins were determined by ELISA and shown in the histograms. Error bars represent means ± SD. *P value < .05.
In an effort to elucidate the molecular mechanism underlying curcumin-dependent repression of these genes, we investigated the possible role of curcumin in inhibiting the important transcription factor STAT3 that regulates the expression of all these genes in addition to p21 and p53 [26,27]. Figure 1E shows that curcumin markedly reduced the active form of JAK2 and its downstream effector STAT3, with no effect on their inactive forms. This indicates that curcumin suppresses the phosphorylation of these two proteins in breast stromal fibroblasts. To confirm this, we investigated the effect of curcumin on some STAT3 targets. Figure 1E shows that c-Myc and survivin were downregulated following curcumin treatment. Therefore, curcumin represses cancer-prone cytokines through inhibiting the STAT3 pathway.
Curcumin Inhibits the Migration/Invasion Abilities of Breast Stromal Fibroblasts
To further show the ability of curcumin in inactivating CAFs, we investigated the migration/invasion abilities of curcumin-treated cells. To this end, CAF-87 cells were cultured in SFM and were either sham-treated (DMSO) or challenged with different doses of curcumin (1, 5, and 10 µM) for 24 hours. Media were removed and curcumin-CAF-87 as well as DMSO-CAF-87 cells were collected and seeded with SFM into BioCoat Matrigel-coated invasion chambers and uncoated chambers for migration. Complete medium (CpM) was added to the lower chambers of the inserts as chemoattractant. After 18 hours of incubation, cells were stained with Diff-Quick stain and then counted. Figure 2A (upper panel) shows the invading and migrating breast stromal fibroblasts, with higher abilities for those treated with DMSO compared to those treated with curcumin (10 µM). Indeed, the invasiveness and the migratory abilities of curcumin-treated cells were reduced in a dose-dependent manner reaching levels 5-fold and 25-fold lower compared to their respective control cells (Figure 2A, lower panel). This shows that curcumin represses the migration/invasion abilities of active breast stromal fibroblasts.
Figure 2.

Curcumin inhibits the migration/invasion of CAF cells and their procarcinogenic effects. (A) CAF-87 cells were first treated with DMSO or different doses of curcumin for 24 hours and then were cultured on the upper compartments of BioCoat Matrigel chambers in the presence of SFM. After 18 hours of incubation, cells were stained with Diff-Quick stain and then counted. (Upper panel) Representative photographs showing invasive and migrated cells. Scale bars represent 50 µm. Histograms are depicting average numbers of invasive and migrated cells. (B) SFCM were collected from CAF-87 cells either sham-treated or challenged with curcumin (10 µM) for 24 hours; SFM and CpM were also used as control. These media were added separately into the lower compartments of the 24-well BD BioCoat plates. MDA-MB-231 cells (1 x 104) were seeded onto the upper compartment of the migration (noncoated chambers) and invasion (Matrigel Invasion Chamber) plates and incubated for 18 hours. Average numbers of migrated/invaded cells are depicted in the histograms. Error bars represent means ± SD.
Curcumin Suppresses the Paracrine Proinvasive/Migratory Effects of CAFs on Breast Cancer Cells
We have next studied the effect of curcumin on the ability of stromal fibroblasts in enhancing the migration and invasion of human breast cancer cells. Therefore, CAF-87 cells were either sham-treated (DMSO) or challenged with curcumin (10 µM) for 24 hours; thereafter, the culture medium was replaced by SFM and cells were reincubated for another 24 hours. Subsequently, serum-free conditioned medium (SFCM) was collected and used to assess the effect on breast cancer cells. We used BioCoat Matrigel-coated invasion chambers and uncoated chambers for migration. CpM, SFM, and SFCM were added to the upper wells with MDA-MB-231 cells, and SFM was also added separately to the lower compartment of each chamber. After 18 hours of incubation, cells were stained with Diff-Quick stain and then counted. Figure 2B shows that the invasion ability of breast cancer cells was six-fold lower in the presence of SFCM from curcumin-treated cells (curcumin-SFCM) than in the presence of SFM conditioned with DMSO-treated CAF-87 cells (DMSO-SFCM). Interestingly, the invasion of MDA-MB-231 cells in the presence of curcumin-SFCM was similar to that obtained in the presence of SFM (Figure 2B). This indicates that curcumin completely suppressed the proinvasive secretions from CAF-87 cells. Similar effects were obtained for the migration of MDA-MB-231 cells under the same conditions (Figure 2B). These results show that curcumin suppresses breast stromal fibroblast-dependent induction of the migratory and invasiveness abilities of breast cancer cells.
Curcumin Effects on Breast Stromal Fibroblasts Are Persistent
Next, we sought to test whether the effect of curcumin on breast stromal fibroblasts is transient or persistent. To this end, CAF-87 cells were either sham-treated (DMSO) or challenged with curcumin (10 µM) for 24 hours, and then cells were either immediately harvested (24) or were reincubated in fresh medium for 48 hours and then were either harvested (48) or split and reincubated for another 48 hours (split). Subsequently, cell lysates were prepared from all these cells and the levels of various proteins were assessed by immunoblot analysis. Figure 3A shows that the levels of p16, p21, and p53 increased in response to curcumin and remained at high levels or slightly increased, despite the removal of curcumin from the media (48) and even after splitting cells. Interestingly, this effect was accompanied by a persistent sharp decrease in the level of SDF-1 and IL-6 (Figure 3A). This clearly shows that the effect of curcumin on breast stromal fibroblasts is persistent.
Figure 3.

Curcumin triggers senescence in CAF cells. CAF-87 cells were either sham-treated (DMSO) or challenged with curcumin (10 µM) for 24 hours (A) cells were either immediately harvested (24) or were reincubated in fresh media for 48 hours and then were either harvested (48) or split and reincubated for another 48 hours (split). Subsequently, cell lysates were prepared from all these cells and the levels of various proteins were assessed by immunoblot analysis. (B) Cells were analyzed by phase contrast microscopy and Ki-67 immunostaining. Scale bars represent 50 µm. (C) Labeling index for Ki-67 and BrdU staining was determined for at least 500 cells per data point and expressed as mean ± SD of triplicate determinations. (D) Immunofluorescence staining using BrdU antibody. Scale bars represent 50 µm. (E) Curcumin-treated cells were split and reincubated for 48 hours in curcumin-free medium, and then SA-β-gal activity was analyzed on cells before and after split. Scale bars represent 50 µm. (F) Total RNA was prepared and used for RT-PCR using primers for the indicated genes.
Curcumin Triggers Senescence in Breast Stromal Fibroblasts
Because the persistent effect included great increase in three important senescence-related genes (p16, p21, and p53), we sought to investigate the possible curcumin-dependent induction of senescence in these stromal fibroblasts. Therefore, we first investigated the effect of curcumin on the master regulator of cell proliferation and the pRB protein. Figure 3A shows that pRB and its phosphorylated form at serine-780 and serine-356 were persistently downregulated upon treatment with curcumin, which suggests strong inhibition of cell proliferation. Next, cells were either sham-treated (DMSO) or challenged with curcumin (10 µM) for 24 hours, and then different senescence features were investigated. Interestingly, curcumin treatment changed themorphology of cells, which became much larger and flat (Figure 3B). Subsequently, we assessed cell proliferation by measuring the level of Ki-67 and the incorporation of BrdU (two important markers of cell proliferation) in control as well as curcumin-treated cells. Figure 3B shows that the expression of Ki-67 is much lower in curcumin-treated cells than in their corresponding control cells. Indeed, the labeling index of Ki-67 was more than five-fold lower in curcumin-treated cells (Figure 3C). Similarly, curcumin treatment strongly reduced (10-fold) the incorporation of BrdU (Figure 3, C and D). This clearly shows curcumin-dependent inhibition of breast stromal fibroblast proliferation. To confirm curcumin-related induction of senescence, we measured the SA-β-gal activity in the same cells. Figure 3E shows that curcumin-treated giant cells stained positive for SA-β-gal before and after splitting cells. Similar results were obtained on other breast stromal fibroblasts (Figure W1). To further show curcumin-related induction of senescence, we made use of the recently discovered senescence marker, Lamin B1 [28]. To this end, total RNA was purified from control as well as curcumin-treated cells and was used for assessing the level of Lamin B1 and Lamin A by RT-PCR. Figure 3F shows that curcumin significantly reduced the level of Lamin B1 compared to control cells, while the level of Lamin A remained constant. Together, these results show curcumin-dependent induction of senescence in breast stromal fibroblasts.
Dose- and Time-Dependent Induction of p16 and Senescence in Response to Curcumin
Next, we investigated the effect of low curcumin doses on p16 up-regulation and the induction of senescence. Therefore, cells were challenged with increasing doses of curcumin (0, 1, 5, and 10 µM) for 24 hours and then cell extracts were prepared and used for immunoblot analysis. Figure 4A shows that curcumin upregulated p16 in a dose-dependent manner, and this increase started with a curcumin dose as low as 1 µM. Concomitantly, a dose-dependent decrease in the level of Lamin B1 but not Lamin A was observed (Figure 4A). This suggested the induction of senescence by low doses of curcumin. Indeed, 1 µM curcumin strongly reduced the level of Lamin B1 (Figure 4A). To confirm the induction of senescence at low curcumin doses, we measured the SA-β-gal activity in the same cells in response to the same doses. Figure 4B shows dose-dependent increase in the SA-β-gal activity, which was also observed at 1 µM curcumin. This confirms the induction of senescence at low curcumin doses and shows clear link between curcumin doses, p16 and Lamin B1 expression levels, and SA-β-gal activity.
Figure 4.
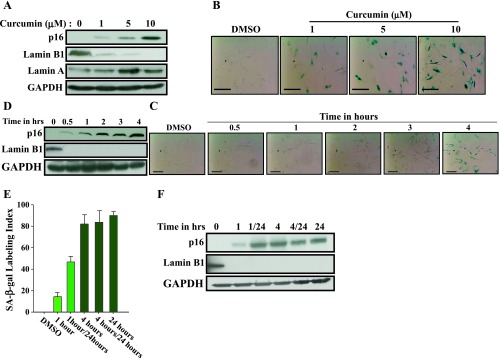
Dose- and time-dependent induction of senescence in response to curcumin. CAF-87 cells were treated with curcumin (10 µM) for 24 hours or as indicated and then were harvested. (A, D, F) Cell lysates were prepared for immunoblot analysis using the indicated antibodies. (B, C, E) Cells were fixed and the SA-β-gal activity was assessed. Scale bars represent 50 µm.
We have also investigated time-dependent effect of curcumin (10 µM) on p16 expression and senescence by treating cells for different periods of time (0–4 hours). Figure 4C shows clear increase in the SA-β-gal activity only after 4 hours of treatment. This senescence was accompanied by marked increase in the expression of p16 and strong decrease in the senescence marker Lamin B1, which became undetectable (Figure 4D). It is noteworthy that Lamin B1 disappeared during the first 30 minutes of curcumin treatment, before the strong upregulation of p16 (Figure 4D). This indicates that 4 hours of curcumin treatment is sufficient to trigger senescence in these cells and that Lamin B1 down-regulation precedes, and may induce, curcumin-related senescence.
To further elucidate the kinetics of curcumin-dependent induction of senescence in breast stromal fibroblasts, we treated CAF-87 cells with curcumin (10 µM) for 1 or 4 hours, and then cells were split into two subpopulations, one was immediately withdrawn for analysis, while the other one was reincubated in curcumin-free media for another 23 or 20 hours, respectively. As control, cells were either treated with DMSO or with curcumin (10 µM) for 24 hours. Senescence was evaluated by measuring the SA-β-gal activity. Figure 4E shows that while 1 hour of treatment did not affect the SA-β-gal activity, this activity was markedly increased and cells exhibited senescent features after 4 hours of treatment. The reincubation of cells in curcumin-free media increased slightly the SA-β-gal activity in the 1 hour-treated cells but did not affect this activity in the 4 hour-treated cells, which showed an activity similar to those treated for 24 hours (Figure 4E). To further elucidate this, we investigated the effect on the expression of the tumor suppressor and senescence-related genes p16 and Lamin B1. Figure 4F shows clear increase in p16 level in cells treated for only 1 hour. However, the increase in the p16 level was much stronger in cells treated for 4 or 24 hours and also those who were reincubated up to 24 hours in curcumin-free media (Figure 4F). This indicates that curcumin treatment for only 1 hour is enough to initiate the senescence process, which will take more time to be active, even in the absence of curcumin.
Curcumin-related Induction of Senescence Is p16-Dependent and DNA Damage-Independent
Because p16 up-regulation was sustained upon treatment with curcumin, which also induced senescence, we sought to investigate the possible implication of p16 in curcumin-induced senescence in active breast stromal fibroblasts. To this end, we ectopically expressed p16 in CAF-87 cells (Figure 5A), and then we assessed SA-β-gal activation in the absence of curcumin treatment. Interestingly, p16 expression changed the cellular morphology and increased SA-β-gal activity compared to control cells (Figure 5B), suggesting that p16 up-regulation can indeed trigger senescence in these cells. To confirm this, we have shown that while the Lamin A level was only slightly affected, the Lamin B1 level decreased sharply in cells expressing CDKN2A-ORF compared to those expressing control plasmid (Figure 5A). However, p16 down-regulation using specific shRNA (Figure 5A) upregulated Lamin B1 and suppressed curcumin-dependent induction of senescence, while senescence was observed in curcumin treated control cells wherein Lamin B1 was downregulated (Figure 5, A and C). This indicates that curcumin-related induction of senescence in breast stromal fibroblasts is p16-dependent.
Figure 5.

Curcumin-dependent induction of senescence is p16-related. (A) CAF-87 cells were transfected with the indicated constructions and then were treated or not with curcumin (10 µM for 24 hours) as indicated. Subsequently, cell lysates were prepared and were used for immunoblot analysis. (B) Senescence was analyzed by measuring SA-β-gal activity on the indicated cells. (C) CAF-87 cells expressing either control or CDKN2A-shRNA were either sham-treated (DMSO) or challenged with curcumin (10 µM) for 24 hours, and then the SA-β-gal activity was assessed. Scale bars represent 50 µm. (D) Cells were incubated at 3% oxygen, and then SA-β-gal activity was analyzed. Scale bars represent 50 µm. (E) Cells were either sham-treated (DMSO) or challenged with curcumin (10 µM) for 24 hours or with γ-rays (30 Gy). Thereafter, the expression of γ-H2AX expression was investigated by immunofluorescence. Scale bars represent 25 µm.
Because senescence can also result from oxidative DNA damage, curcumin-treated cells (10 µM) were reincubated for 24 hours at 3% oxygen, and then we assessed the SA-β-gal activity. Figure 5D shows clear change in the cellular morphology with senescent phenotype and also increase in the SA-β-gal activity in curcumin-treated cells, showing that curcumin-dependent induction of senescence is not due to high oxygen-related oxidative damage. To show lack of DNA damage in curcumin-treated cells, cells were either sham-treated (DMSO) or challenged with curcumin (10 µM) for 24 hours or with γ-rays (30 Gy) used as positive control. Thereafter, the expression of γ-H2AX (a marker of induced DNA double-strand breaks [29]) was investigated by immunofluorescence. Figure 5E shows that while γ-rays induced γ-H2AX expression (immunostaining), curcumin and DMSO did not. This indicates that while γ-rays induced DNA double-strand breaks, curcumin did not, suggesting that curcumin triggers senescence in a DNA damage-independent manner.
Curcumin Does Not Trigger Senescence in Quiescent Stromal Fibroblasts
In adult animals, most organs are composed of nonreplicative quiescent cells, which is not the case for tumor-composing cells. Thereby, we sought to investigate the effect of curcumin on nonreplicative quiescent breast fibroblasts. To this end, 100% confluent cells were incubated in SFM for 24 hours, and then they were either sham-treated (DMSO) or challenged with curcumin (10 µM) for another 24 hours in CpM. Figure 6A shows very low and similar activity of the SA-β-gal in both DMSO- and curcumin-treated cells, reflecting the quiescence state of these cells [30–32]. This suggested that curcumin did not trigger senescence in these cells. To confirm this assumption, we assessed the level of the senescence marker Lamin B1 and have shown that the level of this protein, like Lamin A, did not decrease upon curcumin treatment (Figure 6B). Furthermore, the levels of p16 and p53 were very low to undetectable and did not increase following curcumin treatment (Figure 6B). In the contrary, p53 level decreased upon curcumin treatment (Figure 6B). This confirms that curcumin did not induce senescence in quiescent breast stromal fibroblasts.
Figure 6.

Curcumin does not trigger senescence in quiescent cells. Confluent CAF-87 cells were incubated in SFM for 24 hours, and then they were either sham-treated (DMSO) or challenged with curcumin (10 µM) for another 24 hours in CpM. (A) Cells were fixed and the SA-β-gal activity was assessed. Scale bars represent 50 µm. (B) Immunoblot analysis using the indicated antibodies.
Discussion
Traditionally, cancer treatments targeted principally tumor cells. However, studies over the past few decades have demonstrated that cancers do not only contain tumor cells but are very complex entities with multiple components involved in tumor growth, invasion, and metastasis. Thereby, an efficient treatment should take into account these different compounds and also make the milieu unfavorable for tumor growth and spread. Natural and nontoxic agents that can inhibit cancer-stroma crosstalk by normalizing the tumor microenvironment may boost the traditional tumor cell-directed therapy. Indeed, it has been shown that targeting CAFs improves cancer chemotherapy by increasing intratumoral drug uptake [33]. Furthermore, using a mouse model of cervical cancer, it has been shown that blockade of platelet-derived growth factor (PDGF) receptor signaling in CAFs inhibited progression of premalignant cervical lesions and the growth of invasive carcinomas [1]. In this line of research, and on the basis of the fundamental role of p16, p21, and p53 in the behavior of stromal fibroblasts, we have shown that low concentrations of curcumin increase the level of these three tumor suppressor proteins and consequently inhibits the procarcinogenic effects of stromal fibroblasts by reducing the secretion of IL-6, MMP-2, MMP-9, TGF-β, and SDF-1 and suppressing the invasion/migration of breast cancer cells. This indicates that curcumin could be used to normalize the expression of important tumor suppressor genes and consequently inactivates breast CAFs. Indeed, curcumin-treated active breast fibroblasts expressed less α-SMA, SDF1, and TGF-β (three major markers of active fibroblasts) and reduced their invasive and migratory abilities. Together, these results demonstrate that active stromal fibroblasts can be inactivated (normalized) and provide proof of principle that the application of natural products such as curcumin might be used to normalize breast cancer-related stromal fibroblasts and suppress their procarcinogenic effects. Therefore, curcumin could constitute an efficient fibroblast-directed therapeutic approach.
Intriguingly, curcumin effect on breast stromal fibroblasts persisted after the removal of the drug and even after splitting of cells. The change in the cellular morphology and the clear decrease in proliferation rate as well as the up-regulation of three major senescence-related proteins, p16, p21, and p53, prompted us to investigate the possible induction of senescence in these primary noncancerous cells. Using different markers of senescence (SA-β-gal, Ki-67, BrdU, and Lamin B1), we have demonstrated curcumin-dependent induction of senescence in CAF cells. This effect was dose-dependent starting from 1 µM for 24 hours. However, using 10 µM curcumin only, 4 hours of treatment were sufficient to induce senescence and strong p16 up-regulation. Interestingly, 30 minutes of treatment increased p16 level and wiped up Lamin B1, in the absence of other senescence features (cellular shape and increase in the SA-β-gal activity), showing that curcumin modulates p16 and Lamin B1 expression in the absence of senescence. It is noteworthy that the time-dependent increase in p16 level upon curcumin treatment correlated very well with the increase in the SA-β-gal activity, showing the importance of p16 accumulation for senescence induction. However, Lamin B1 disappeared during the first 30 minutes of treatment, suggesting an important role of this protein in the initiation of the senescence process, which requires further time to be active. Indeed, while 1 hour of treatment had only slight effect on the SA-β-gal activity, exposing cells for 1 hour followed by 23 hours in curcumin-free medium increased three-fold the activity of SA-β-gal and also the expression of p16. However, this effect was not as strong as curcumin treatment for 4 or 24 hours (Figure 4, E and F).
It has been previously shown that breast stromal fibroblast cells enter senescence in response to X-rays (10 Gy) and reincubation for 7 days [34]. Several other genotoxic anticancer agents were also used to trigger senescence but mainly in cancer cells. However, the doses and the treatment conditions varied between cell lines and the agents used to induce senescence [35,36].
Importantly, curcumin-related senescence was p16-dependent, but DNA damage-independent. This parallels the reduced secretory phenotype observed upon curcumin treatment shown in Figure 1D. In fact, it has been recently shown the possible p16-dependent induction of senescence without the associated inflammatory secretory phenotype [37]. This indicated that SASP is associated with the accumulation of DNA damage, which is not the case upon curcumin treatment.
Therefore, the present results further confirm the possible induction of senescence without SASP, which has generally procarcinogenic effects [34,38]. In the contrary, curcumin reduced the secretion of several procarcinogenic cytokines such as IL-6, SDF-1, MMP-2, MMP-9, and TGF-β through inhibiting their major transactivator STAT3. Indeed, curcumin markedly inactivated JAK2 and STAT3, which led to down-regulation in the level of their downstream targets c-Myc and survivin (Figure 1E). The curcumin-dependent inhibition of the JAK2/STAT3 pathway may also explain the increase in the expression of the tumor suppressor proteins p21 and p53, known to be under negative control of STAT3 [26,27]. Interestingly, we have also shown that curcumin does not induce senescence in quiescent cells. This may explain lack of this effect in adult animals and human beings, because most cells in adult organisms are resting and therefore curcumin cannot cause senescence. Likewise, it has been recently shown that DNA-damaging agents (doxorubicin and etoposide) and also p53 induction do not cause senescence in quiescent cells [39]. This indicates that quiescent cells are refractory to both DNA damage-dependent and DNA damage-independent senescence. This could be due to the fact that curcumin-dependent changes in senescence-related genes do not take place, or are only minimal, in quiescent cells. Indeed, unlike in proliferating cells, p21 and p53 were not upregulated in curcumin-treated arrested cells (Figure 6B). This could be related to chromatin structure and effects on gene expression.
In summary, the present results demonstrate curcumin-dependent induction of senescence in breast CAFs, which inactivates these cells and reduce their procarcinogenic potential. This constitutes an important step toward stromal fibroblast-directed therapy, which may improve the outcome of the classic therapeutic regimens directed principally toward tumor cells.
Supplementary Material
Acknowledgments
We are grateful to the Research Center administration for their continuous help.
Footnotes
This work was performed under RAC Proposal No. 2080009. The authors declare no conflict of interest.
This article refers to supplementary material, which is designated by Figure W1 and is available online at www.neoplasia.com.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Aboussekhra A. Role of cancer-associated fibroblasts in breast cancer development and prognosis. Int J Dev Biol. 2011;55:841–849. doi: 10.1387/ijdb.113362aa. [DOI] [PubMed] [Google Scholar]
- 3.Franco OE, Shaw AK, Strand DW, Hayward SW. Cancer associated fibroblasts in cancer pathogenesis. Semin Cell Dev Biol. 2009;21:33–39. doi: 10.1016/j.semcdb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimoda M, Mellody KT, Orimo A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin Cell Dev Biol. 2009;21:19–25. doi: 10.1016/j.semcdb.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 6.Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 7.Anand P, Sundaram C, Jhurani S, Kunnumakkara AB, Aggarwal BB. Curcumin and cancer: an “old-age” disease with an “age-old” solution. Cancer Lett. 2008;267:133–164. doi: 10.1016/j.canlet.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Gescher AJ, Sharma RA, Steward WP. Cancer chemoprevention by dietary constituents: a tale of failure and promise. Lancet Oncol. 2001;2:371–379. doi: 10.1016/S1470-2045(00)00392-2. [DOI] [PubMed] [Google Scholar]
- 9.Strimpakos AS, Sharma RA. Curcumin: preventive and therapeutic properties in laboratory studies and clinical trials. Antioxid Redox Signal. 2008;10:511–545. doi: 10.1089/ars.2007.1769. [DOI] [PubMed] [Google Scholar]
- 10.Goel A, Kunnumakkara AB, Aggarwal BB. Curcumin as “curecumin”: from kitchen to clinic. Biochem Pharmacol. 2007;19:19. doi: 10.1016/j.bcp.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Bayet-Robert M, Kwiatkowski F, Leheurteur M, Gachon F, Planchat E, Abrial C, Mouret-Reynier MA, Durando X, Barthomeuf C, Chollet P. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol Ther. 2010);9:8–14. doi: 10.4161/cbt.9.1.10392. [DOI] [PubMed] [Google Scholar]
- 12.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 14.Campisi J. Replicative senescence: an old lives' tale? Cell. 1996;84:497–500. doi: 10.1016/s0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- 15.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 16.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2012;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Mohanna MA, Al-Khalaf HH, Al-Yousef N, Aboussekhra A. The p16INK4a tumor suppressor controls p21WAF1 induction in response to ultraviolet light. Nucleic Acids Res. 2007;35:223–233. doi: 10.1093/nar/gkl1075. Epub 2006 Dec 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bond JA, Haughton MF, Rowson JM, Smith PJ, Gire V, Wynford-Thomas D, Wyllie FS. Control of replicative life span in human cells: barriers to clonal expansion intermediate between M1 senescence and M2 crisis. Mol Cell Biol. 1999;19:3103–3114. doi: 10.1128/mcb.19.4.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Ansari MM, Hendrayani SF, Shehata AI, Aboussekhra A. p16INK4A represses the paracrine tumor-promoting effects of breast stromal fibroblasts. Oncogene. 2012 doi: 10.1038/onc.2012.270. E-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hawsawi NM, Ghebeh H, Hendrayani SF, Tulbah A, Al-Eid M, Al-Tweigeri T, Ajarim D, Alaiya A, Dermime S, Aboussekhra A. Breast carcinoma-associated fibroblasts and their counterparts display neoplastic-specific changes. Cancer Res. 2008;68:2717–2725. doi: 10.1158/0008-5472.CAN-08-0192. [DOI] [PubMed] [Google Scholar]
- 26.Niu G, Wright KL, Ma Y, Wright GM, Huang M, Irby R, Briggs J, Karras J, Cress WD, Pardoll D, et al. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol. 2005;25:7432–7440. doi: 10.1128/MCB.25.17.7432-7440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alvarez JV, Frank DA. Genome-wide analysis of STAT target genes: elucidating the mechanism of STAT-mediated oncogenesis. Cancer Biol Ther. 2004;3:1045–1050. doi: 10.4161/cbt.3.11.1172. [DOI] [PubMed] [Google Scholar]
- 28.Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066–2075. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plesca D, Mazumder S, Almasan A. DNA damage response and apoptosis. Methods Enzymol. 2008;446:107–122. doi: 10.1016/S0076-6879(08)01606-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yegorov YE, Chernov DN, Akimov SS, Akhmalisheva AK, Smirnova YB, Shinkarev DB, Semenova IV, Yegorova IN, Zelenin AV. Blockade of telomerase function by nucleoside analogs. Biochemistry (Mosc) 1997;62:1296–1305. [PubMed] [Google Scholar]
- 31.Severino J, Allen RG, Balin S, Balin A, Cristofalo VJ. Is β-galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res. 2000;257:162–171. doi: 10.1006/excr.2000.4875. [DOI] [PubMed] [Google Scholar]
- 32.Cho S, Hwang ES. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol Cells. 2012;33:597–604. doi: 10.1007/s10059-012-0042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116:1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–496. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K, Roninson IB. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59:3761–3767. [PubMed] [Google Scholar]
- 36.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–2715. [PubMed] [Google Scholar]
- 37.Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16INK4a induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–36403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16:238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging. 2010;2:924–935. doi: 10.18632/aging.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.