

Abstract
Thermodynamic and structural characteristics of the E. coli DnaT protein trimerization reaction have been quantitatively examined using fluorescence anisotropy and analytical ultracentrifugation methods. Binding of magnesium to the DnaT monomers regulates the intrinsic affinity of the DnaT trimerization reaction. Comparison between the DnaT trimer and the isolated N-terminal core domain suggests that magnesium binds to the N-terminal domain but does not associate with the C-terminal region of the protein. The magnesium binding process is complex and involves ~3 Mg2+ cations per protein monomer. The observed effect seems to be specific for Mg2+. In the examined salt concentration range, monovalent cations and anions do not affect the trimer assembly process. However, magnesium affects neither the cooperativity of the trimerization reaction nor the GnHCl-induced trimer dissociation, strongly indicating that Mg2+ indirectly stabilizes the trimer through the induced changes in the monomer structures. Nevertheless, formation of the trimer also involves specific conformational changes of the monomers, which are independent of the presence of magnesium. Binding of Mg2+ cations dramatically changes the thermodynamic functions of the DnaT trimerization, transforming the reaction from the temperature-dependent to the temperature-independent process. Highly cooperative dissociation of the trimer by GnHCl indicates that both interacting sites of the monomer, located on the N-terminal core domain and formed by the small C-terminal region, are intimately integrated with the entire protein structure. In the intact protein, the C-terminal region most probably interacts with the corresponding binding site on the N-terminal domain of the monomer. Functional implications of these findings are discussed.
Keywords: DnaT Protein, Oligomerization, Fluorescence Anisotropy, Fluorescence Titrations, Protein - Protein Interactions, Primosome
The DnaT protein is an essential replication protein in Escherichia coli that plays a fundamental role in the assembly of the primosome, a multiple-protein-nucleic acid complex, which catalyzes priming of the DNA strand during the DNA replication process (1-7). The protein was originally discovered to be a critical factor during synthesis of the complementary DNA strand of phage ϕX174 DNA (1,4). Current data show that the assembly of the primosome is a fundamental step in the restart of the stalled replication fork at the damaged DNA sites (6,7). Subsequent studies indicated that the role of the DnaT protein is to provide a specific recognition marker within the primosome structure for the primary replicative helicase, the DnaB protein, which allows the helicase to enter and become the integral part of the assembly (1,4,5,8,9). The DnaT protein monomer contains 179 amino acids with a molecular weight of ~19,455 (10,11). Although early biochemical data indicated that the native protein forms a homo-trimer, the presence of monomer, dimer, tetramer, and pentamer was also proposed, with the molecular weight of the native protein ranging from ~ ~61,000 to ~110,000, depending on the presence of magnesium (10).
In the accompanying paper, we addressed the fundamental characteristics of the DnaT protein trimerization reaction. In solution, the protein exists as a monomer - trimer equilibrium system, without the presence of any specific oligomeric states larger than trimer. The trimerization reaction is highly cooperative, without the detectable presence of the intervening dimer. Nevertheless, due to the modest affinity, the DnaT protein forms a mixture of the monomer and trimer states with the 3:1 molar ratio, at physiological concentrations in the E. coli cell. The protein monomer is built of a large, N-terminal core domain and a small C-terminal region. It also possesses two structurally different, interacting sites located on the N-terminal core domain and the small C-terminal region, respectively. While the interaction site located on the N-terminal core domain stabilizes the initial dimer, the final attachment of the third monomer in the trimer is achieved through the C-terminal region (accompanying paper). The data indicate that each monomer in the trimer is in contact with the remaining two monomers forming a star-like global structure.
Although the DnaT protein has been recognized as a key factor in the activities of the primosome, little is known about its functional behavior (5,10). Prior to this work, the very functional form of the DnaT in the primosome structure was still under the debate, with both the trimer and the monomer proposed as possible ingredients of the primosome (5). Nevertheless, these early data suggest that oligomerization/disassembly of the protein oligomers may be a specific part of the primosome assembly process. On the other hand, how these processes are controlled by temperature, solution conditions, salt, and magnesium, is unknown. Nothing is known about the role of the protein structure in the stability of the trimer. The discovery of two different interacting sites on the DnaT monomer, which differently stabilize the trimer, adds additional complexity (accompanying paper). Nothing is known about the control exerted by the solution variables on these two very different interactions.
In this communication, we report quantitative analyses of the energetics of the DnaT trimerization process. Specific binding of Mg2+ cations to the DnaT monomers regulates the intrinsic affinity of the DnaT trimerization reaction. Magnesium binds to the N-terminal domain of the protein in the process that involves ~3 Mg2+ cations per DnaT monomer. Mg2+ cations indirectly stabilize the DnaT trimer through the induced changes in the structures of the monomers. Both interacting sites of the DnaT protein are intimately integrated with the entire protein structure. In the intact protein, the C-terminal region constitutively interacts with the binding site on the N-terminal domain.
MATERIALS & METHODS
Reagents and Buffers
All solutions were made with distilled and deionized >18 MΩ (Milli-Q Plus) water. All chemicals were reagent grade. Buffer C is 10 mM sodium cacodylate adjusted to pH 7.0 with HCl, 1 mM DTT, 100 mM NaCl, 5 mM MgCl2, and 25% glycerol (w/v) (12-15). Buffer C, without magnesium, contains 0.1 mM EDTA. The temperature and salt concentrations are indicated in the text.
The DnaT Protein
The E. coli DnaT protein has been isolated as described in the accompanying paper. The protein concentration was spectrophotometrically determined, with the extinction coefficient ε280 = 2.7960 × 104 cm−1M−1 (monomer) obtained using an approach based on Edelhoch’s method (12-15,16,17).
Fluorescence Measurements
All steady-state fluorescence titrations were performed using the ISS PC-1 spectrofluorometer (Urbana, IL) as previously described by us (18-25). The state of the DnaT protein oligomerization has been analyzed, using the steady-state fluorescence anisotropy, r, of the protein tryptophan residues (26-32). The dynamic quenching data were analyzed using the Stern-Volmer equation
| (1) |
where Fo is the fluorescence intensity of the sample prior to the addition of the quencher, F is the intensity in the presence of the quencher, KSV is the Stern-Volmer quenching constant, and [Q] is the molar concentration of the quencher (33-35).
Analytical Ultracentrifugation Measurements
Analytical, equilibrium and velocity ultracentrifugation experiments were performed with an Optima XL-A analytical ultracentrifuge (Beckman Inc., Palo Alto, CA), using double-sector charcoal-filled 12 mm centerpieces, as previously described by us (36-43). The values of sedimentation coefficients, s20,w, were corrected to s°20,w for solvent viscosity and temperature to standard conditions (44).
RESULTS
Magnesium Effect on the DnaT Protein Trimerization
Early studies of the DnaT protein suggested that presence of magnesium in solution might significantly affect the oligomeric state of the protein (see above) (10). Therefore, we address the Mg2+ effect on the protein trimerization reaction. The fluorescence anisotropy dependence upon the total DnaT concentration (monomer) obtained in buffer C (pH 7.0, 20°C), containing 0.1 mM EDTA and no magnesium, is shown in Figure 1a. Similar to the data obtained in the presence of Mg2+ (accompanying paper), the titration curve spans less than ~2 orders of magnitude on the protein total monomer concentration scale between 10% and 90% of the observed signal, indicating that the monomer - trimer reaction is predominantly observed (26). The solid line in Figure 1a is the nonlinear least-squares fit of the experimental curve using the monomer <-> trimer model (accompanying paper, eqs. 3 - 7). The fit provides an excellent description of the experimental curve, indicating that both the exclusive formation of the trimer and the cooperative character of the trimerization process are not affected by the absence of magnesium, and provides, KT = (1.8 ± 0.3) × 1014 M−2, rM = 0.052 ± 0.001, and rT = 0.011 ± 0.001. The values of the fluorescence anisotropies of the monomer and the trimer are also very similar to the same parameters obtained in the presence of Mg2+. Nevertheless, KT has a lower value than in the presence of magnesium (KT = (3.5 ± 0.6) × 1014 M−2, accompanying paper, see below). The dashed line in Figures 1a is the best fit of the experimental titration curve, using the monomer <-> dimer <-> trimer model (accompanying paper, eqs. 8 - 13). Clearly, the thermodynamic model, which assumes the presence of a significant population of the dimer, does not adequately describe the experimental data.
Figure 1.

a. The dependence of the DnaT fluorescence anisotropy upon the total DnaT monomer concentration (λex = 296 nm, λem = 347 nm) in buffer C (pH 7.0, 20°C), containing 0.1 mM EDTA and no magnesium (Materials and Methods). The solid line is the nonlinear least-squares fit of the titration curve, using the trimer model described by eqs. 3 - 7 (accompanying paper), with KT = 1.8 × 1014 M−2, rM = 0.0520, and rT = 0.1133. The dashed line is the best fit of the titration curve using the monomer <-> dimer <-> trimer model described by eqs. 8 - 13 (accompanying paper), with KM = 1.6 × 107 M−1, rM = 0.0520, rD = 0.080, and rT = 0.1133. The symbols  represent the nonlinear least squares fit of the titration curve using the cooperative monomer<-> dimer <-> trimer model (eq. 21a, accompanying paper), with KM = 5 × 105 M−1, σ = 750 ± 100, rM = 0.0520, rD = 0.080, and rT = 0.1133. b. Sedimentation equilibrium concentration profiles of the DnaT protein in the same buffer conditions without magnesium. The concentration of the protein is 2.86 × 10−6 M (monomer). The profiles have been recorded at 280 nm and at 14000 (blue), 16000 (red), and 18000 (green) rpm. The solid lines are nonlinear least-squares fits to a single exponential function (accompanying paper, eq. 2), with a single species having a molecular weight of 62,000 ± 4,000 (Materials and Methods). Lower panel shows the residuals of the fits. c. Sedimentation velocity absorption profiles at 280 nm of the DnaT protein in buffer C (pH 7.0, 25°C), containing 0.1 mM EDTA and no magnesium. The concentration of the DnaT is 4.29 × 10−6 M (monomer); 50000 rpm. d. Apparent sedimentation coefficient distribution, g(s*), as a function of the radial sedimentation coefficient coordinate, s*, obtained from the time derivatives of the DnaT protein sedimentation profiles in panel c. The solid line is the nonlinear least-squares fit of the distribution using the software provided by the manufacture (Materials and Methods).
represent the nonlinear least squares fit of the titration curve using the cooperative monomer<-> dimer <-> trimer model (eq. 21a, accompanying paper), with KM = 5 × 105 M−1, σ = 750 ± 100, rM = 0.0520, rD = 0.080, and rT = 0.1133. b. Sedimentation equilibrium concentration profiles of the DnaT protein in the same buffer conditions without magnesium. The concentration of the protein is 2.86 × 10−6 M (monomer). The profiles have been recorded at 280 nm and at 14000 (blue), 16000 (red), and 18000 (green) rpm. The solid lines are nonlinear least-squares fits to a single exponential function (accompanying paper, eq. 2), with a single species having a molecular weight of 62,000 ± 4,000 (Materials and Methods). Lower panel shows the residuals of the fits. c. Sedimentation velocity absorption profiles at 280 nm of the DnaT protein in buffer C (pH 7.0, 25°C), containing 0.1 mM EDTA and no magnesium. The concentration of the DnaT is 4.29 × 10−6 M (monomer); 50000 rpm. d. Apparent sedimentation coefficient distribution, g(s*), as a function of the radial sedimentation coefficient coordinate, s*, obtained from the time derivatives of the DnaT protein sedimentation profiles in panel c. The solid line is the nonlinear least-squares fit of the distribution using the software provided by the manufacture (Materials and Methods).
As we discuss in accompanying paper, a more complex model, includes two binding parameters, the intrinsic binding constant, KM, and cooperativity parameter, σ (accompanying paper, eq. 21a). However, the high cooperativity of the system precludes any unique and independent determination of KM and σ, due to their highly correlated values and the analysis, using the model, renders only the maximum possible value of the intrinsic association constant, KM, and the minimum value of σ. The nonlinear least-squares fit of the titration curve using the model defined by eq. 21a (accompanying paper) is included in Figure 1a. The plot has been obtained by setting a given value of KM and fitting σ and rD. The fit provides the maximum possible value of KM = (5 ± 1) × 105 M−1 and the minimum value of σ = 750 ± 100.
The sedimentation equilibrium profiles of the DnaT protein in buffer C (pH 7.0, 20°C), containing 0.1 mM EDTA and no magnesium, recorded at the protein absorption band (280 nm), at different rotational speeds, are shown in Figure 1b (36-43). The selected protein concentrations (2.86 × 10−6 M (monomer)) correspond to the final plateaus observed in the fluorescence anisotropy titrations (Figure 1a). The smooth solid lines in Figure 1b are the nonlinear least-squares fits, using the single exponential function (accompanying paper, eq. 2). The fits indicate the presence of a single molecular species with a molecular weight of 62,000 ± 4000. Adding additional exponents does not improve the statistics of the fits (data not shown). With the molecular weight of the DnaT monomer of ~19,455, these results establish that, at high protein concentration and in the absence of magnesium, the DnaT protein exists in solution as a stable trimer, which is in full agreement with the fluorescence anisotropy data (Figure 1a).
Global Conformation of the DnaT Protein Trimer in the Absence of Magnesium
The sedimentation velocity profiles (monitored at 280 nm) of the DnaT protein in buffer C (pH 7.0, 25°C), containing 0.1 mM EDTA and no magnesium, are shown in Figure 1c. The concentration of the protein is 4.29 × 10−6 M (monomer). The profiles clearly show that there is a single moving boundary, indicating the presence of single molecular entity (41-44). The sedimentation coefficient of the protein, s20,w, has been obtained using the time-derivative approach as shown in Figure 1b (45,46, accompanying paper). Similarly to the buffer conditions containing magnesium, the value of s20,w shows little, if any, dependence upon the protein concentration, in the protein concentrations range where only the stable trimer is present (data not shown). The obtained value of the sedimentation coefficient of the DnaT protein in the absence of magnesium is: s°20,w = 3.5 ± 0.1 S, i.e., within experimental accuracy, the same as obtained in the presence of magnesium (accompanying paper). In other words, Mg2+ cations have no effect on the global conformation of the DnaT trimer (accompanying paper) (see Discussion).
Dynamic Fluorescence Quenching of the DnaT Monomer and Trimer in the Absence and Presence of Magnesium
Both acrylamide and iodide anions, I−, are efficient dynamic quenchers of the protein tryptophan fluorescence (33-35). While neutral acrylamide provides information about the steric access of the tryptophan residues in the protein, the negatively charged iodide accesses tryptophans on the protein surface and senses the local environment of the fluorophore (33-35). The quenching studies have been performed with the DnaT solutions of 3 × 10−8 M (monomer), where the protein is predominantly in the monomer state, and with the solution of 3.25 × 10−6 M (monomer), where the DnaT forms a stable trimer (Figures 1a and 1b).
Stern-Volmer plots of the DnaT protein monomer and the trimer for the acrylamide as the dynamic quencher, in the absence and presence of Mg2+, are shown in Figure 2a and 2b. For comparison, the Stern-Volmer plots for NATA, in corresponding solution conditions, are also included in the figures. Within experimental accuracy, the plots are linear. The solid lines in Figures 2a and 2b are the linear least-squares fits to eq. 1, which provide the Stern-Volmer quenching constants, KSV = 12.3 ± 1 M−1 for the DnaT monomer and KSV = 4.5 ± 0.5 M−1 for the trimer, respectively, independently of the presence or absence of magnesium. Thus, magnesium does not change the acrylamide accessibility of the tryptophan residues in the monomer and trimer (see Discussion). Nevertheless, the data show that the tryptophan residues are significantly more accessible to acrylamide in the monomer than in the trimer. In fact, the accessibility of the tryptophans in the DnaT monomer is practically the same as the accessibility of the free NATA in solution (KSV = 12.3 ± 1 M−1) (Figure 2a and 2b).
Figure 2.

a. Stern-Volmer plots of the DnaT fluorescence quenching in buffer C (pH 7.0, 20°C), containing 0.1 mM EDTA and no magnesium, with acrylamide as the dynamic fluorescence quencher. The protein concentrations are:  3.0 × 10−8 M (monomer),
3.0 × 10−8 M (monomer),  3.18 × 10−6 M (monomer). b. Stern-Volmer plots (Materials and Methods) of the DnaT fluorescence quenching in buffer C (pH 7.0, 20°C) with acrylamide as the dynamic fluorescence quencher (λex = 296 nm, λem = 347 nm). The protein concentrations are:
3.18 × 10−6 M (monomer). b. Stern-Volmer plots (Materials and Methods) of the DnaT fluorescence quenching in buffer C (pH 7.0, 20°C) with acrylamide as the dynamic fluorescence quencher (λex = 296 nm, λem = 347 nm). The protein concentrations are:  3.0 × 10−8 M (monomer),
3.0 × 10−8 M (monomer),  3.25 × 10−6 M (monomer). In both panels, Stern-Volmer plots of NATA (3.18 × 10−6 M) fluorescence quenching in the corresponding buffer conditions,
3.25 × 10−6 M (monomer). In both panels, Stern-Volmer plots of NATA (3.18 × 10−6 M) fluorescence quenching in the corresponding buffer conditions,  . c. Stern-Volmer plots of the DnaT fluorescence quenching in buffer C (pH 7.0, 20°C), containing 0.1 mM EDTA and no magnesium, with I− as the dynamic fluorescence quencher. The protein concentrations are:
. c. Stern-Volmer plots of the DnaT fluorescence quenching in buffer C (pH 7.0, 20°C), containing 0.1 mM EDTA and no magnesium, with I− as the dynamic fluorescence quencher. The protein concentrations are:  3.0 × 10−8 M (monomer),
3.0 × 10−8 M (monomer),  3.18 × 10−6 M (monomer). d. Stern-Volmer plots of the DnaT fluorescence quenching in buffer C (pH 7.0, 20°C) with I− as the dynamic fluorescence quencher. The protein concentrations are:
3.18 × 10−6 M (monomer). d. Stern-Volmer plots of the DnaT fluorescence quenching in buffer C (pH 7.0, 20°C) with I− as the dynamic fluorescence quencher. The protein concentrations are:  3.0 × 10−8 M (monomer),
3.0 × 10−8 M (monomer),  3.25 × 10−6 M (monomer). In both panels, Stern-Volmer plots of NATA (3.18 × 10−6 M) fluorescence quenching in the corresponding buffer conditions,
3.25 × 10−6 M (monomer). In both panels, Stern-Volmer plots of NATA (3.18 × 10−6 M) fluorescence quenching in the corresponding buffer conditions,  .
.
The situation is different in the case of the quenching by iodide anions, with corresponding Stern-Volmer plots shown in Figures 2c and 2d. In the absence of Mg2+, the Stern-Volmer quenching constants are, KSV = 14.1 ± 1.2 M−1 for the monomer and KSV = 7.2 ± 0.7 M−1 for the trimer, respectively. In the presence of Mg2+, the corresponding Stern-Volmer constants are KSV = 10.2 ± 1.1 M−1 and KSV = 7.8 ± 0.7 M−1. First, the tryptophan residues are still considerably more accessible to I− in the monomer than in the trimer. Second, magnesium significantly changes the iodide accessibility of the tryptophan residues of the monomer, while the accessibility of the residues in the trimer remains, within experimental accuracy, unaffected by Mg2+. The fluorescence quenching data provide the first indication that magnesium cations bind to the DnaT monomers prior to their entry into the trimer structure (see below). Interestingly, the quenching of the tryptophan residues in the monomer by I−, in the absence of Mg2+, is even more efficient than that observed for free NATA (KSV = 9.1 ± 0.9 M−1), in the same buffer conditions (Figure 2c). The presence of magnesium (Figure 2d) reduces the quenching of the tryptophan residues in the monomer to that of NATA (KSV = 9.8 ± 0.9 M−1) (see Discussion).
Magnesium Effect on the Free Energy of the DnaT Trimerization
Analysis of the temperature effect on the DnaT trimerization reaction showed that the presence of Mg2+ cations mostly affects the free energy of trimerization at the highest examined temperature (see below). Therefore, to increase the resolution, we address the effect of magnesium on the trimerization constant, KT, at 35°C. Figure 3 shows the dependence of KT upon the logarithm of the magnesium concentration. The plot clearly indicates a complex behavior of the examined system. The value of KT remains unchanged from 0 M to ~3 × 10−5 M MgCl2. However, above [MgCl2] ~1 × 10−4 M, KT steeply increases and reaches the plateau at ~5 × 10−4 M MgCl2, indicating that the binding process saturates at higher [MgCl2]. Because NaCl does not affect the DnaT trimerization in the same or higher salt concentration range (data not shown), neither monovalent cations nor anions participate in the reaction and the observed effect must be due to the specific binding of magnesium cations.
Figure 3.

The dependence of the DnaT trimerization constant, KT, upon the logarithm of MgCl2 concentration in solution, in buffer C (pH 7.0, 35°C). The solid red line is the nonlinear least-squares fit of the plot to eq. 6 with n = 3 and Kn = 2.3 × 1012 M−3. For comparison, the best fits using n = 2 (turquoise) and n = 4 (green) are also included.
Notice, magnesium does not affect the global structure of the DnaT trimer (Figures 1c and 1d). Also, the fluorescence quenching data indicate that the presence of Mg2+ cations only affect the monomer structure prior to trimerization reaction (Figure 2). Therefore, the simplest thermodynamic model, which describes the magnesium effect on KT, includes the binding of Mg2+ cations to the monomer, which indirectly affects the monomer-monomer interactions in the trimer (see below) (47-52). The system is described by the following equilibria:
| (2) |
| (3) |
| (4) |
The trimerization constant, KT, in the absence of Mg2+ is described by eq. 4 in the accompanying paper. The magnesium binding constant to the monomer, Kn, and the trimerization constant, KTn, in the presence of Mg2+, are defined as
| (5a) |
and
| (5b) |
The observed, overall trimerization constant, KTobs, at any [MgCl2] is then (47-52)
| (6) |
At a high magnesium concentration ([MgCl2] -> ∞), KTobs = KTn = 3.5 × 1014 M−2 (accompanying paper). The value of KT in the absence of Mg2+ at 35°C is also known (KT = 6 × 1013 M−2) (Figure 3). Therefore, there are only two independent parameters, Kn and n, in eq. 6, which have to be determined. The solid red line in Figure 3 is the nonlinear least-squares fit of eq. 6 to the observed dependence of the trimerization constant upon [MgCl2], which provides, n = 3 ± 1 and Kn = (2.3 ± 0.7) × 1012 M−3. The apparent, intrinsic binding constant of Mg2+, Knint = (Kn)1/3 = (1.3 ± 0.3) × 104 M−1. For comparison, the best fits using n = 2 (turquoise) and n = 4 (green) are also shown in Figure 3 (see Discussion).
Temperature Effect on the DnaT Protein Trimerization in the Presence and Absence of Magnesium
We address the nature of the DnaT trimerization process, by examining the temperature effect on the reaction. The dependence of the DnaT protein fluorescence anisotropy upon the total monomer concentration, in buffer C (pH 7.0), obtained at different temperatures, is shown in Figure 4a. The steady-state anisotropy of the monomer becomes lower at higher temperatures, while the steady-state anisotropy of the trimer, as well as the mid point of the titration curves, are only slightly affected by the temperature. The solid lines in Figure 4a are the nonlinear least-squares fits of the experimental titration curves, using the monomer <-> trimer model (accompanying paper, eqs. 3 - 7). Analogous anisotropy titrations, obtained in the absence of magnesium, are shown in Figure 4b. Similarly, the increase of the temperature predominantly affects the steady-state anisotropy of the monomer. Although the changing plateaus of the plots obscure the visual evaluation of the behavior of the system, the midpoints of the titration curves are more pronouncedly shifted toward higher protein concentrations than in the presence of Mg2+, indicating a lower affinity at the higher temperature.
Figure 4.

a. The dependence of the DnaT fluorescence anisotropy upon the total DnaT monomer concentration (λex = 296 nm, λem = 347 nm) in buffer C (pH 7.0), recorded at different temperature:  20°C,
20°C,  25°C,
25°C,  30°C, and
30°C, and  35°C. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). b. The dependence of the DnaT fluorescence anisotropy upon the total DnaT monomer concentration (λex = 296 nm, λem = 347 nm) in buffer C (pH 7.0), containing 0.1 mM EDTA and no magnesium, recorded at different temperature:
35°C. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). b. The dependence of the DnaT fluorescence anisotropy upon the total DnaT monomer concentration (λex = 296 nm, λem = 347 nm) in buffer C (pH 7.0), containing 0.1 mM EDTA and no magnesium, recorded at different temperature:  20°C,
20°C,  25°C,
25°C,  30°C, and
30°C, and  35°C. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). c. The dependences of the trimerization constant, KT, upon the reciprocal of temperature (Kelvin) (van’t Hoff plot), in the presence
35°C. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). c. The dependences of the trimerization constant, KT, upon the reciprocal of temperature (Kelvin) (van’t Hoff plot), in the presence  and absence
and absence  of magnesium. The solid lines are linear least-squares fits to the van’t Hoff equation (details in text).
of magnesium. The solid lines are linear least-squares fits to the van’t Hoff equation (details in text).
The dependences of the trimerization constant, KT, upon the reciprocal of the temperature (Kelvin) (van’t Hoff plot), in the presence and absence of magnesium, are shown in Figure 4c (19,20,53). The plots are linear in the examined temperature range. The solid lines are linear least-squares fits to the van’t Hoff equation, ∂lnKT/d(1/T) = −ΔH/R. Thus, in the presence of magnesium, the trimer formation is characterized by the apparent enthalpy change ΔH° = 0 ± 2 kcal/mol. In other words, the interactions are completely driven by the apparent entropy change, ΔS°. The situation is very different in the absence of magnesium, where the trimerization process is characterized by the negative apparent enthalpy change, ΔH° = −15 ± 2 kcal/mol, indicating that the trimerization reaction is driven by the apparent enthalpy. Using the standard thermodynamic formulas, ΔG° = −RTlnKT, at 20°C and ΔS° = (−ΔG° + ΔH°)/T, one obtains the apparent entropy changes, characterizing the trimerization process in the presence and absence of magnesium, as ΔS° ≈ 67 cal/mol deg and ΔS° ≈ −116 cal/mol deg, respectively (see Discussion).
Solvent Effect on the DnaT Protein Trimerization in the Presence and Absence of Magnesium
To obtain further insight into the nature of the DnaT oligomerization reaction, we examined the solvent effect on the protein trimer formation. The fluorescence anisotropy dependence upon the total DnaT concentration (monomer) obtained in buffer C (pH 7.0, 25°C), containing different concentrations of the neutral solute, glycerol (w/v %), is shown in Figure 5a. Corresponding fluorescence anisotropy titrations obtained in the absence of Mg2+ are shown in Figure 5b. In the presence of magnesium, increasing the glycerol concentration, from 10% to 25% (w/v), has only a slight effect on the mid-point of the titration curves, indicating similar affinities in the examined solute concentration range. In the absence of Mg2+, the titration curves shift toward higher total protein concentrations at higher [glycerol], indicating a significant decrease of the monomer-monomer affinity. Analogous titrations have been performed in the presence of another neutral solute, sucrose, which showed very similar effects on the fluorescence anisotropy titration curves, in the presence and absence of Mg2+, respectively (data not shown). The solid lines in Figures 5a and 5b are nonlinear least-squares fits of the experimental titration curves to the monomer <-> trimer model (accompanying paper, eqs. 3 - 7), with the trimerization binding constant, KT as a fitting parameter.
Figure 5.

a. The dependence of the DnaT fluorescence anisotropy upon the total DnaT monomer concentration (λex = 296 nm, λem = 347 nm) in buffer C (pH 7.0, 25°C) containing different concentrations of glycerol (w/v):  10 %,
10 %,  15 %,
15 %,  20 %, and
20 %, and  25 %. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). b. The dependence of the DnaT fluorescence anisotropy upon the total DnaT monomer concentration (λex = 296 nm, λem = 347 nm) in buffer C (pH 7.0, 25 °C) containing 0.1 mM EDTA and no magnesium, and different concentrations of glycerol (w/v):
25 %. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). b. The dependence of the DnaT fluorescence anisotropy upon the total DnaT monomer concentration (λex = 296 nm, λem = 347 nm) in buffer C (pH 7.0, 25 °C) containing 0.1 mM EDTA and no magnesium, and different concentrations of glycerol (w/v):  10 %,
10 %,  15 %,
15 %,  20 %, and
20 %, and  25 %. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). c. The dependences of the trimerization constant, KT, upon the water concentration in the sample in the presence
25 %. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). c. The dependences of the trimerization constant, KT, upon the water concentration in the sample in the presence  and absence
and absence  of magnesium, respectively. The solid lines are linear least-squares fits with the slopes: ∂logKT/∂log[H2O] = 0 in the presence of Mg2+ and ∂logKT/∂log[H2O] = 11.1 in the absence of Mg2+, respectively (details in text).
of magnesium, respectively. The solid lines are linear least-squares fits with the slopes: ∂logKT/∂log[H2O] = 0 in the presence of Mg2+ and ∂logKT/∂log[H2O] = 11.1 in the absence of Mg2+, respectively (details in text).
The effect of the “neutral” solute on the macromolecular interactions can be complicated by the presence of preferential interactions (54,55). However, the very similar effect of two different solutes on the same interactions strongly suggests that the selected solutes do not affect the DnaT oligomerization through preferential interactions with the protein (see below) (54,55). The simplest thermodynamic model of the observed solute effect is that the trimerization is affected by the changes of the water concentration in the sample and the reaction can be described by a general linkage equation, 3M <-> T ± n H2O (47-52). It should be stressed that, even at the maximum concentration of the applied solutes (25% (w/v) glycerol or sucrose), the water activity coefficient is practically ~1 and the water concentration, instead of its activity, can be used in the analysis (56,57). Moreover, very small effects of the selected solutes on the water activity are completely absorbed by the error in the determination of the trimerization constant, which is 10-15% and is included in errors of the determined slopes of the log-log plots (see below) (47,50,51).
The dependences of the logarithm of the trimerization constant, KT upon the logarithm of [H2O] (log-log plots), determined in the presence and absence of magnesium and with glycerol as the neutral solute, are shown in Figure 5c (58,59). The plots are linear in examined solute concentration ranges. The slopes of the plots are ∂logKT/∂log[H2O] = 0 ± 1.6 in the presence of Mg2+ and ∂logKT/∂log[H2O] = 11.1 ± 3.5, in the absence of Mg2+, respectively. The values of the analogous slopes of the log-log plots, obtained in the presence of sucrose, are ∂logKT/∂log[H2O] = 1 ± 1.6 and ∂logKT/∂log[H2O] = 10.3 ± 2.8 (data not shown). The very similar values of the slopes of the log-log plots, obtained for different solutes, reinforce the conclusion that the water concentration is the dominant factor in the observed solute effects (54,55). The results indicate that the DnaT trimerization, in the presence of magnesium, is not accompanied by any significant, apparent water uptake/release, while in the absence of magnesium, the formation of the trimer is accompanied by the apparent, net uptake of ~11 water molecules (58,59) (see Discussion).
Guanidine Hydrochloride Effect on the DnaT Trimer Stability in the Presence and Absence of Magnesium
The analysis has so far focused on the reaction of the DnaT trimer formation. In the next set of experiments, we address the role of the DnaT structure in the trimer stability, by examining the dissociation reaction of the trimer in the presence and absence of magnesium. The DnaT trimer dissociation has been induced using guanidine hydrochloride (GnHCl), which is a potent denaturant of the protein secondary and tertiary structures (60-62). The dependence of the fluorescence anisotropy of the DnaT protein (2.91 × 10−6 M (monomer)) upon the concentration of GnHCl in buffer C (pH 7.0, 25°C) is shown in Figure 6a. At this protein concentration, in the absence of GnHCl, the DnaT protein primarily exists as a trimer (accompanying paper). The value of the fluorescence anisotropy, corresponding to the trimer (r ~0.111), remains unchanged up to [GnHCl] ~4 M. As [GnHCl] increases, the anisotropy steeply decreases reaching the plateau (r ~0.088) at [GnHCl] ≈ 4.8 M, indicating a highly cooperative transition (47-52, 60-62). The corresponding plot, obtained in the absence of Mg2+, is shown in Figure 6b. The dependence of the DnaT protein anisotropy upon [GnHCl] is similar to that obtained in the presence of Mg2+. However, the observed transition is much less cooperative, as compared to the transition observed in the buffer containing Mg2+ (see below) (47-52,60-62).
Figure 6.

a. The dependence of the fluorescence anisotropy of the DnaT protein (2.91 × 10−6 M (monomer) upon the concentration of GnHCl in buffer C (pH 7.0, 25°C). The solid line is the nonlinear least-squares fit of the experimental titration curve to eqs. 7 - 14, with, p = 18 and KG = 4.0 × 10−11 M−18. b. The dependence of the fluorescence anisotropy of the DnaT protein (2.91 × 10−6 M (monomer) upon the concentration of GnHCl in buffer C (pH 7.0, 25°C), containing 0.1 mM EDTA and no magnesium. The solid line is the nonlinear least-squares fit of the experimental titration curve to eqs. 7 - 14, with p = 10 and KG = 3.4 × 10−6 M−10 (details in text). c. Sedimentation equilibrium concentration profiles of the DnaT protein in buffer C (pH 7.0, 25°C). The concentration of the protein is 3.25 × 10−6 M (monomer). The profile has been recorded at 280 nm and at 27000 rpm. The smooth solid line is the nonlinear least-squares fit to a single exponential function (accompanying paper, eq. 2), with a single species having a molecular weight of 18,950 (Materials and Methods). Lower panel shows the residuals of the fit. d. Sedimentation equilibrium concentration profiles of the DnaT protein in buffer C (pH 7.0, 25°C). The concentration of the protein is: 3.25 × 10−6 M (monomer). The profile has been recorded at 280 nm and at 27000 rpm. The smooth solid line is the nonlinear least-squares fit to a single exponential function (accompanying paper, eq. 2), with a single species having a molecular weight of 19500 (Materials and Methods). Lower panel shows the residuals of the fit.
It should be noted that the anisotropies of the system at plateaus, obtained at the high [GnHCl], are significantly higher than the anisotropies (~0.05) of the native DnaT monomer (Figures 1a, 4a, and 4b). This is true both in the presence and absence of magnesium (Figures 6a and 6b). Therefore, the oligomeric state of the protein at high [GnHCl] has been addressed using the sedimentation equilibrium method (accompanying paper). Figure 6c shows the sedimentation equilibrium profile of the DnaT protein in buffer C (pH 7.0, 25°C), containing 5.4 M GnHCl recorded at the protein absorption band (280 nm). An analogous profile recorded in the absence of magnesium is shown in Figure 6d. The protein concentrations are 3.25 × 10−6 M (monomer)). The smooth solid lines in Figure 6a and 6b are nonlinear least-squares fits, using the single exponential function (accompanying paper, eqs. 2). The fits provide excellent descriptions of the experimental sedimentation equilibrium profiles, indicating the presence of a single species with the molecular weight of 18,950 ± 2000 and 19,500 ± 2000, in the presence and absence of Mg2+, respectively. Thus, it is clear that the molecular entity observed at high [GnHCl] (Figures 6c and 6d) is the denatured DnaT monomer, characterized by the higher fluorescence anisotropy than the native monomer (see Discussion).
There are several aspects of the observed transitions, which are peculiar to the examined system. The locations of the mid points of the titration curves at high [GnHCl] are where the protein secondary structure is usually denatured (Figures 6a and 6b) (60-62). High cooperativities of the observed GnHCl-induced transitions are striking and indicate the lack of any intermediate states of the trimer. The cooperativity of the transition is particularly pronounced in the presence of magnesium cations, which are bound to the monomers, i.e., they do not directly stabilize the trimer (Figure 2) (see Discussion). Finally, the protein is exclusively in the denatured monomer state, at the saturating [GnHCl]. All these features strongly suggest that the dissociation occurs practically as a single step, without any trimer or dimer interventions, and results from the denaturation of the native (secondary and tertiary) structures of the DnaT monomers. Thus, the simplest thermodynamic model of the observed GnHCl effect on the DnaT trimer dissociation includes the following equilibria (47-52):
| (7a) |
and
| (7b) |
The trimer dissociation constant in the absence of GnHCl is 1/KT and the binding constant of GnHCl to the monomer, KG, is defined as
| (8) |
The total DnaT monomer concentration in the sample is defined by the mass conservation expression, as
| (9a) |
and
| (9b) |
However, the analysis is complicated by the fact that there are two independent variables, [M]F and [GnHCl], two simultaneous equilibria, and the analytical expression for the concentration of the free DnaT monomer cannot be obtained. Below, we present an algorithm, which allows the analysis of these simultaneous equilibria. Moreover, the presented algorithm can be applied to more complex situations.
The concentration of the monomer state at any [GnHCl] is defined as a function of the sum of concentrations, [M]F + [M]G. The sum, [M]F + [M]G, is then treated as the independent variable. Because the concentration of the free trimer, [T]F = {[M]T − ([M]F + [M]G)}/3, the observed, overall trimer dissociation constant, KTD, in the presence of [GnHCl] is
| (10) |
The same overall trimer dissociation constant, KTD, can exclusively be expressed using [GnHCl], as
| (11) |
From eq. 11, one obtains [GnHCl] as a function of KTD as
| (12) |
The anisotropy of the sample is defined as
| (13) |
where rMF rMG, and rT are fluorescence anisotropies of the free native monomer, denatured monomer associated with GnHCl, and the trimer. The quantities, fMF, fMG, and fT are the corresponding fractional contributions of the three species to the total emission, FT, of the sample defined as
| (14a) |
Then,
| (14b) |
| (14c) |
and
| (14d) |
where FMF, FMG, and FTr are molar fluorescence intensities of the monomer states and the trimer.
The values of rMF rMG, rT, FMF, FMG, FTr, KT are known. Also, [M]T is known. Thus, there are only two unknown parameters in eqs. 8 - 14, KG and p. The algorithm for the simulation and fitting is the following: 1. KTD is calculated from eq. 10, using the sum, [M]F + [M]G, as the independent variable. 2. [GnHCl] is calculated, using the obtained KTD, from eq. 12. 3. [T]F is calculated as [T]F = {[M]T − ([M]F + [M]G)}/3. 4. [M]F is calculated using the obtained [T]F and the known trimerization constant, KT (accompanying paper). 5. [M]G is calculated from eq. 9a. 6. FT, fMF, fMG, and fT are calculated from eqs. 14a - 14d. 7. Finally, the fluorescence anisotropy, r, is calculated from eq. 13.
The solid lines in Figures 6a and 6b are nonlinear least-squares fits of the experimental DnaT dissociation curves to eqs. 7 - 14, which provide, p = 18 ± 2 and KG = (4.0 ± 1.5) × 10−11 M−18, in the presence of Mg2+, and p = 10 ± 2, and KG = (3.4 ± 1.1) × 10−6 M−10, in the absence of magnesium, respectively. Thus, the apparent intrinsic affinities of GnHCl for the DnaT monomer is (4 × 10−11 M−18)1/18 ≈ 0.26 M−1 and (3.4 × 10−6 M−10)1/10 = 0.28 M−1 are virtually the same in the presence and absence of Mg2+, respectively. Nevertheless, in the presence of Mg2+, the system is significantly more cooperative than in the absence of magnesium, as reflected by the considerably larger value of p (see Discussion).
Temperature Effect on the DnaT N-Terminal Core Domain Dimerization in the Presence and Absence of Magnesium
The dependence of the fluorescence anisotropy of the DnaT N-terminal core domain upon the total, domain monomer concentration in buffer C (pH 7.0), obtained at different temperatures, is shown in Figure 7a. Similar to the behavior of the intact DnaT trimer, the steady-state anisotropy of the domain monomer becomes lower at higher temperatures, while the steady-state anisotropy of the domain dimer is only slightly affected by the temperature. Analogous anisotropy titrations, obtained in the absence of magnesium, are shown in Figure 7b. Correspondingly, the increase of the temperature predominantly affects the steady-state anisotropy of the domain monomer, although to lesser extent than in the presence of magnesium. As a result of changing plateaus, visual inspection of the titration curves does not provide an easy visual evaluation of the behavior of the system (see above). The solid lines in Figure 7a are the nonlinear least-squares fits of the experimental titration curves, using the monomer <-> dimer model, with the dimerization constant, KD, as the fitting parameter (accompanying paper, eqs. 16 - 20).
Figure 7.

a. The dependence of the fluorescence anisotropy of the DnaT N-terminal core domain upon the total domain monomer concentration (λex = 296 nm, λem = 347 nm) in buffer C (pH 7.0), recorded at different temperature:  20°C,
20°C,  25°C,
25°C,  30°C, and
30°C, and  35°C. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). b. The dependence of the DnaT fluorescence anisotropy upon the total DnaT monomer concentration (λex = 295 nm, λem = 347 nm) in buffer C (pH 7.0), containing 0.1 mM EDTA and no magnesium, recorded at different temperature:
35°C. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model described by eqs. 3 - 7 (accompanying paper). b. The dependence of the DnaT fluorescence anisotropy upon the total DnaT monomer concentration (λex = 295 nm, λem = 347 nm) in buffer C (pH 7.0), containing 0.1 mM EDTA and no magnesium, recorded at different temperature:  20°C,
20°C,  25°C,
25°C, 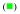 30°C, and
30°C, and 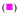 35°C. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model (accompanying paper, eqs. 3 - 7). c. The dependences of the trimerization constant, KT, upon the reciprocal of the temperature (Kelvin) (van’t Hoff plot), in the presence
35°C. The solid lines are the nonlinear least-squares fits of the titration curves, using the trimer model (accompanying paper, eqs. 3 - 7). c. The dependences of the trimerization constant, KT, upon the reciprocal of the temperature (Kelvin) (van’t Hoff plot), in the presence  and absence
and absence  of magnesium, respectively. The solid lines are linear least-squares fits to the van’t Hoff equation (details in text).
of magnesium, respectively. The solid lines are linear least-squares fits to the van’t Hoff equation (details in text).
The van’t Hoff plots of the dimerization constant, KD, as a function of the reciprocal temperature (Kelvin) in the presence and absence of magnesium, are shown in Figure 7c. The solid lines are linear least-squares fits to the van’t Hoff equation, ∂lnKT/d(1/T) = −ΔH/R. In the presence of magnesium, the formation of the domain dimer is characterized by the small, positive apparent enthalpy change ΔH° = 2.2 ± 2 kcal/mol. Thus, the interactions are driven by the apparent entropy change, ΔS°, as observed for the trimer formation (Figure 4c). In the absence of magnesium, the N-terminal core domain dimerization is characterized by the negative apparent enthalpy change, ΔH° = −4.6 ± 2 kcal/mol. This value is significantly lower than ΔH° = −15 ± 2 kcal/mol, obtained for the trimerization reaction (Figure 4c). The corresponding apparent entropy changes, characterizing the dimerization process of the isolated, N-terminal core domain in the presence and absence of magnesium, are: ΔS° ≈ 26 cal/mol deg and ΔS° ≈ 17 cal/mol deg, respectively (see Discussion).
Guanidine Hydrochloride Effect on the Stability of the DnaT N-Terminal Core Domain Dimer
The dependence of the fluorescence anisotropy of the DnaT N-terminal core domain (1.18 × 10−6 M (monomer)) upon the concentration of GnHCl in buffer C (pH 7.0, 25°C) is shown in Figure 8a. In the presence of magnesium, the observed effect of GnHCl on the fluorescence anisotropy of the isolated domain dimer is very similar to the effect observed in the case of the protein trimer (Figure 6a). The value of the anisotropy, corresponding to the dimer (r ~0.105), remains unchanged up to [GnHCl] ~4 M. Further, an increase of [GnHCl] causes the anisotropy to steeply decrease and reach the plateau (r ~0.085) at [GnHCl] ≈ 5 M. Sedimentation equilibrium data recorded at 5.4 M GnHCl indicate the presence of the a single species with the molecular weight of 18,950 ± 2000 (data not shown). Thus, the molecular entity observed at high [GnHCl] is the monomer of the denatured, N-terminal core domain (see Discussion).
Figure 8.

a. The dependence of the fluorescence anisotropy of the DnaT N-terminal core domain (1.18 × 10−6 M (monomer)) upon the concentration of GnHCl in buffer C (pH 7.0, 25°C). The solid line is the nonlinear least-squares fit of the experimental titration curve to eqs. 7 - 14, with, p = 16 and KG = 1.7 × 10−10 M−16 rMF = 0.055, rMG = 0.085, and rD = 0.113. b. The dependence of the fluorescence anisotropy of the DnaT protein (1.18 × 10−6 M (monomer)) upon the concentration of GnHCl in buffer C (pH 7.0, 25°C), containing 0.1 mM EDTA and no magnesium. The smooth solid line is the nonlinear least-squares fit of the experimental titration curve to eqs. 21 - 25, with m = 5, q = 12, with KM = 1.1 × 10−1 M−1, KQ = 2.1 × 10−8 M−12, rMF = 0.054, rMM = 0.0925, rMQ = 0.070, and rD = 0.1133 (details in text). c. Sedimentation equilibrium concentration profiles of the DnaT N-terminal core domain in buffer C (pH 7.0, 25°C), containing 0.1 mM EDTA and no magnesium, and 3.36 M GnHCl. The concentration of the protein is 3.25 × 10−6 M (monomer). The profile has been recorded at 280 nm and at 27000 rpm. The smooth solid line is the nonlinear least-squares fit to a single exponential function (accompanying paper, eq. 2), with a single species having a molecular weight of 16,467 (Materials and Methods). Lower panel shows the residuals of the fit.
In the presence of magnesium, analogously to the effect of GnHCl on the trimerization reaction (see above), the simplest thermodynamic model of the observed GnHCl effect on the dimer dissociation of the N-terminal core domain includes the following equilibria (47-52):
| (15) |
and
| (16) |
The dimer dissociation constant in the absence of GnHCl is 1/KD (accompanying paper, eq. 17) and the binding constant of GnHCl to the domain monomer, KG, is defined by eq. 8. The total, domain monomer concentration in the sample is then described by the mass conservation expression, as
| (17a) |
and
| (17b) |
Unlike in the case of the trimer, the above expression can be analytically solved, providing the concentration of the free domain monomer as a function of [GnHCl], i.e.,
| (18) |
The anisotropy of the sample is then defined as
| (19) |
where rMF, rMG, and rD are fluorescence anisotropies of the free native monomer, denatured monomer associated with GnHCl, and the dimer. The quantities, fMF, fMG, and fD are the corresponding fractional contributions of corresponding species to the total emission, FT, of the sample defined as
| (20a) |
with,
| (20b) |
| (20c) |
and
| (20d) |
where FMF, FMG, and FD are molar fluorescence intensities of the two monomer states and the dimer. In the above expressions, rMF rMG, rD, FMF, FMG, FD, KD, and [M]T are known thevalue of (KG)1/p can be estimated from the midpoint of the titration curve, leaving only one unknown parameter, p. Nevertheless, we treated KG also as a fitting parameter. The solid line in Figure 8a is the nonlinear-least squares fit of the experimental dissociation curve of the N-terminal domain dimer by GnHCl in the presence of magnesium to eqs. 15 - 20, which provides, p = 16 ± 2 and KG = (1.7 ± 0.5) × 10−10 M−16. Thus, the value of p and the apparent intrinsic affinity of GnHCl for the N-terminal domain monomer, (1.7 × 10−10)1/16 ≈ 0.25 M−1, are very similar the corresponding parameters obtained in the case of the intact DnaT protein trimer (Figure 6a) (see Discussion).
Very different behavior is observed in the absence of magnesium. The dependence of the fluorescence anisotropy of the DnaT N-terminal core domain upon the concentration of GnHCl in the absence of magnesium is shown in Figure 8b. There are clearly two transitions induced by GnHCl with the midpoints at ~1.9 M and ~4.5 M GnHCl, respectively. Figure 8c shows the sedimentation equilibrium profile of the DnaT N-terminal domain in the presence of 3.36 M GnHCl, recorded at the protein absorption band (280 nm). The selected concentration of GnHCl corresponds to the first plateau separating the two transitions in Figure 8b. The smooth solid line in Figure 8c is the nonlinear least-squares fit, using the single exponential function (accompanying paper, eq. 2). The fit provides an excellent description of the experimental sedimentation profile, indicating the presence of a single species with the molecular weight of 16,500 ± 1,500. Thus, in spite of the high fluorescence anisotropy, the molecular entity, induced in the first transition at low [GnHCl] in absence of Mg2+, is the monomer of the N-terminal domain (Figure 8c) (see Discussion). Analogous sedimentation equilibrium experiments in the presence of 5.5 M GnHCl and in the absence of magnesium showed that the plateau at high [GnHCl] also corresponds to the isolated, N-terminal domain monomer (data not shown).
In the absence of magnesium, we must consider two transitions both leading to the dissociation of the domain dimer. The simplest thermodynamic model of the observed GnHCl effect, which can account for the observed behavior, includes the following equilibria (47-52):
| (21a) |
| (21b) |
and
| (21c) |
The GnHCl binding constants to MF and MM, KM and KQ, respectively, are defined by expressions analogous to eq. 8. The total concentration of the domain monomer in the sample is defined by the mass conservation expression, as
| (22a) |
and
| (22b) |
The analytical solution for the concentration of the free domain monomer as a function of [GnHCl] is
| (23a) |
where
| (23b) |
The anisotropy of the sample is defined as
| (24) |
where rMF, rMM, rMQ, and rD are fluorescence anisotropies of the corresponding monomer states and the dimer. The quantities, fMF, fMM, fMQ, and fD are the corresponding fractional contributions of all species to the total emission, FT, i.e.,
| (25a) |
with
| (25b) |
| (25c) |
| (25d) |
and
| (25e) |
where FMF, FMM, FMQ, and FD are molar fluorescence intensities of the monomer states and the dimer. In the above expressions, rMF, rMM, rMQ, rD, KD, and [M]T are known. FMF, FMM, FMQ, and FD are also known from fluorescence spectra of the system at corresponding [GnHCl] (see above). The values of (KM)1/m and (KQ)1/q can be estimated from the midpoints of the both transitions, leaving m and q as independent, unknown parameters. The solid line in Figure 8b is the nonlinear-least squares fit of the experimental dissociation curve of the N-terminal domain dimer in the absence of magnesium with m and q as fitting parameters to eqs. 21 - 25. The fit provides a remarkably good description of the experimental titration curve with m = 5 ± 1, q = 12 ± 2, with KM = (1.1 ± 0.3) × 10−1 M−1, and KQ = (2.1 ± 0.6) × 10−8 M−12, respectively. The apparent intrinsic affinity of GnHCl for the N-terminal domain monomer in the first transition is (KM)1/m ≈ 0.64 M−1, while in the second transition (KQ)1/q ≈ 0.23 M−1 (see Discussion). Thus, although the apparent affinity of the GnHCl is higher for the first transition, the analogous quantity for the second transition is very similar to the value obtained for the intact protein trimerization in the absence of magnesium (see Discussion).
DISCUSSION
Binding of Magnesium Cations to the DnaT Monomers Regulates the Intrinsic Affinity of the DnaT Trimerization Reaction Without Affecting the Cooperativity of the Assembly Reaction
A characteristic aspect of the magnesium effect on the DnaT trimerization reaction is its specificity. NaCl does not affect the trimerization to any extent, in the same salt concentration range (data not shown), strongly suggesting that monovalent cations and anions do not participate in the trimer assembly process. Moreover, the specificity is also reflected in the high, apparent intrinsic affinity, which is of the order of ~1.3 × 104 M−1, indicating that the DnaT monomer possesses specific Mg2+ cation-binding sites (Figure 3). A peculiar feature of the observed effect is that the presence of magnesium does not affect the cooperativity of the trimerization reaction. Consequently, the dimer population is undetectable, both in the absence and presence of Mg2+ (Figure 1a, accompanying paper). The independence of the cooperativity of the trimerization reaction upon magnesium cations provides the first indication that Mg2+ cations do not directly stabilize the DnaT trimer. Otherwise, the cooperativity of the trimerization would be significantly diminished in the absence of magnesium, which is not experimentally observed (Figure 1a) (see below).
Magnesium Cations Affect the Structure of the DnaT Monomers Prior to the Trimer Assembly
Neither global structure of the DnaT trimer (Figures 1c and 1d) nor the local accessibility of protein tryptophan residues in the trimer are affected by the presence of magnesium, as tested using two different dynamic quenchers (Figure 2). On the other hand, Mg2+ cations induce significant changes in the monomer conformation. Assessment of the acrylamide and I− quenching data of the monomer indicates that Mg2+ binding does not change the solvent accessibility of the tryptophan residues in the monomer, reflected in the unchanged acrylamide accessibility, but changes the structure of the protein matrix surrounding them (Figure 2). In the absence of magnesium, the tryptophan residues in the monomer are characterized by the iodide quenching constant even higher than that of the free NATA, indicating the presence of an excess of positive charges around the tryptophan residues, which attract negatively charged iodide anions (33-35).
Binding of Mg2+ reduces the quenching of the monomer tryptophan fluorescence by I− to that of NATA. The effect is not due to the simple presence of bound Mg2+ cations, which would increase the positive charge and lead to the amplification, not to the reduction of the iodide quenching efficiency. Thus, magnesium binding to the monomer induces changes in the structure of the protein matrix, which, in turn, favor the formation of the trimer. These data together with the fact that cooperativity of the trimerization reaction is not affected by magnesium, i.e., Mg2+ cations do not directly stabilize the trimer (see above), and the lack of any Mg2+ effect on the trimer global structure indicate that Mg2+ cations bind to the DnaT monomers prior to the assembly of the trimer.
Formation of the DnaT Trimer Involves Specific Conformational Changes of the Monomers Which Are Independent of the Presence of Magnesium
While acrylamide accessibility of the tryptophans in the DnaT monomer are as high as that of the free NATA in solution, the accessibility of the same residues in the trimer is strongly diminished. Similar behavior is observed for the fluorescence quenching in the presence of iodide anions. Thus, the fluorescence quenching results show very different structures of the protein matrix surrounding the tryptophan residues in the monomer, as compared to the trimer (Figure 2). This is true both in the presence and absence of magnesium, indicating that the decreased accessibility of the tryptophan residues in the trimer is induced by the trimer formation, not by magnesium binding to the monomers. This conclusion is also strongly supported by the results on the N-terminal core domain dimerization. First, analogous fluorescence quenching data of the truncated domain monomer and the dimer are very similar to that obtained for the intact monomer and the trimer (data not shown). Second, conformation changes leading to the trimer formation include the C-terminal region of the protein, which does not bind magnesium cations (accompanying paper, see below).
Magnesium Dramatically Changes the Thermodynamic Functions of the DnaT Trimerization
A striking result is the magnesium effect on the thermodynamic functions characterizing the trimerization reaction (Figure 4c). In the presence Mg2+, the trimerization reaction is driven by the large, apparent entropy change, while in the absence of magnesium the same reaction is driven by the large, apparent enthalpy change. The situation is even more complicated by the fact that in the presence of magnesium, there is no detectable participation of the water molecules in the trimerization process (Figures 5a and 5c). A large net release of water molecules would rather be expected, which would contribute to the observed, large and positive change of entropy (58,59). For instance, in the absence of Mg2+, the trimerization reaction is accompanied by an uptake of ~11 water molecules, which accounts for a part of the observed, large, and negative entropy change (Figures 5b and 5c).
The observed behavior is very complex and must include compensatory effects between protein binding processes at two different interacting sites, magnesium binding to monomers, and conformational changes of the monomers accompanying the trimer formation (see above). A plausible explanation of this peculiar behavior is based on the fact that binding of magnesium is characterized by a high affinity, the trimerization process is characterized by a negative enthalpy in the absence of Mg2+, and there is a net uptake of water molecules. Because magnesium cations are not present, the negative enthalpy change includes intrinsic protein association and magnesium-independent, conformational changes of the monomers accompanying the trimerization (Figure 4c). The intrinsic protein - protein associations would be also characterized by a positive change of entropy, masked by a large net uptake of water molecules (Figure 5c),
On the other hand, high intrinsic affinity of magnesium cations indicates that some cations can form inner-sphere complexes, i.e., they release their hydration layers, a process characterized by a large positive enthalpy change (63). Therefore, in the presence of magnesium, conformational changes of the monomers induced by the trimer formation may also induce an inner-sphere complex formation by some of the bound magnesium cations. The positive enthalpy of the cation dehydration would compensate for the intrinsic negative enthalpy of the protein - protein interactions in the trimerization reaction, observed in the absence of magnesium. In such a case, the water molecules released from the dehydrated cations would compensate the intrinsic water uptake accompanying the intrinsic association process. As a result, in the presence of magnesium, the apparent ΔH° ≈ 0, ΔS° is positive, and dLogKT/dLog[H2O] ≈ 0, as experimentally observed (Figures 4c and 5c).
Highly Cooperative Dissociation of the DnaT Trimer by GnHCl Indicates a Significant Integration of the DnaT Monomer Interacting Sites with the Entire Structure of the Protein Stabilized by Magnesium
Analogously to the high cooperativity of the trimerization process, the dissociation of the DnaT trimer by the protein denaturant, GnHCl, is also characterized by a very high cooperativity (Figures 6a and 6b). Moreover, the dissociation process of the DnaT trimer, as well as the N-terminal core domain dimer occurs in the [GnHCl] range where the secondary structure of the protein is destabilized (60-62). It should be stressed that although we use binding models to describe the effect of GnHCl on the dissociation of the DnaT trimer and the N-terminal domain dimer, the values of the obtained exponents are measures of the structural cooperativity of the protein, analogously to the well-known Hill coefficient in the binding studies and not necessarily mirror the average numbers of the bound GnHCl molecules (52).
First, very high cooperativity of the dissociation process indicates that a large cooperative unit of the protein structure, i.e., a large structural part of the protein responding to GnHCl, is subjected to destabilization by GnHCl, without any detectable intervening intermediates (52,60-62). The only and final product is the denatured monomer, characterized by the higher fluorescence anisotropy than the fluorescence anisotropy of the native monomer. These data indicate that the DnaT dissociation by GnHCl occurs as a result of denaturation of most of the secondary structures of the monomers constituting the trimer. Second, both interacting sites of the DnaT monomer, located on the N-terminal core domain and formed by the small C-terminal region, must be intimately integrated with the entire protein structure. If the sites were to any extent structurally autonomous, then a trimer intermediate with partially denatured monomers would be a part of the transition process, which is not experimentally observed.
Third, the apparent affinity of GnHCl for the protein is not affected by the presence of magnesium, although the cooperativity of the trimer dissociation is (Figures 6a and 6b). Notice, the midpoints of the titrations curves in Figures 6a and 6b are, within experimental accuracy, at the same locations on the [GnHCl] scale. These results provide an additional strong support for the conclusion that Mg2+ cations do not directly stabilize the trimer, i.e., they do not bind at the monomer - monomer interfaces in the trimer, otherwise the titration curve would be shifted toward a high [GnHCl] in the presence of magnesium, which is not observed (Figures 6a and 6b). Rather, binding of magnesium results in an increase of the size of the protein cooperative unit within each monomer subjected to denaturation. Such a process can occur without affecting the apparent GnHCl affinity.
The results on the effect of GnHCl on the N-terminal core domain dimer provide further strong support for the conclusion that the binding site on the N-terminal domain is an integral part of the protein structure without any significant structural autonomy from the protein. Thus, in the presence of magnesium, the dimer disintegrates directly to denatured monomers, without the appearance of any detectable dimer intermediate(s). The dissociation occurs in the same concentration of GnHCl as the trimer dissociation, where the secondary structure of the protein is destabilized by the denaturant (60-62). The monomer cooperative unit is characterized by p ≈ 16, i.e., very similar to the value of p ≈ 18, obtained for the monomers in the trimer. A collateral result of the very similar behavior of the monomers in the trimer and the isolated domain dimer in the presence of magnesium is that Mg2+ cations do not seem to bind to the C-terminal region of the protein but only to the large N-terminal core domain.
In the absence of magnesium, the lack of the C-terminal region leads to the disintegration of the domain dimer into monomers already at a significantly lower [GnHCl] (Figure 8b). Thus, even limited changes in the protein structure, induced at the lower [GnHCl], affect the interacting site of the N-terminal domain. The first transition is much less cooperative (m ≈ 5) and the monomers possess a higher fluorescence anisotropy (r ≈ 0.09) than that of the native monomers (r ≈ 0.05) (Figure 1a), most probably preserving their secondary structure. As the [GnHCl] increases, the secondary structure of the monomers is further destabilized, reaching the fully denatured state at high [GnHCl] (Figure 8b). The cooperative unit of the second transition is characterized by q ≈ 12, i.e., very similar to p ≈ 10 obtained for the analogous transition of the intact monomer in the trimer (Figures 8b and 6b).
The Small C-Terminal Region of the DnaT Protein May Engage in Interactions the N-Terminal Domain Prior to the Formation of the Trimer
Formation of the isolated, N-terminal core domain dimer is characterized by the apparent enthalpy change ΔH° ≈ 2.2 kcal/mol and ΔH° ≈ −4.6 kcal/mol, in the presence and absence of magnesium, respectively (Figure 7c). Thus, in both solution conditions, the values of enthalpy changes are more positive than those observed in the case of the trimerization reaction of the intact protein, although the difference is more pronounced in the absence of magnesium (Figure 4c). Nevertheless, these data strongly suggest that the lack of the C-terminal region does not affect the major characteristics of the monomer - monomer interactions in the domain dimer, as compared to the trimer, i.e., the enthalpy change in the presence of magnesium is much more positive than in the absence of Mg2+ cations.
Recall, the affinity of the monomers in the domain dimer is significantly higher than the affinity among the monomers in the trimer, i.e., the flexible, small C-terminal region must hinder interactions between monomers through the sites located on the corresponding N-terminal domains, prior to the trimer formation (accompanying paper). The data suggest the simplest explanation of the same direction in the observed enthalpy changes in the presence and absence of magnesium, in the domain dimer formation. In the intact protein, the C-terminal region interacts with the corresponding binding site on the N-terminal domain of the monomer. A simple steric hindrance would not contribute to the enthalpy change. Separation of the C-terminal regions from the N-terminal domain, formation of the additional interacting sites by the regions, and engagement of the sites in interactions with the third monomer in the trimer contribute an unfavorable enthalpy change to the overall enthalpy of the trimer formation and to the accompanying entropy changes.
Functional Implications
In spite of the key physiological role of the DnaT protein in the primosome assembly and the primosome activities, the mechanistic details of exactly how the protein fulfills its role are unknown (3,5,7,10). As we mentioned before, both the monomer and the trimer have been suggested as the oligomeric state of the DnaT protein in the functional primosome (5). Results, described in this, and the accompanying paper, show that the protein exists as 3:1 molar mixture of the monomer and trimer forms in the cell at all physiological temperatures. Thus, both oligomeric states are physiological, states of the DnaT protein.
Our current thinking, based on the obtained results, is that these two physiological oligomeric states have different and specific functions at different stages of the primosome activities. The discovered highly cooperative character of the DnaT trimerization reaction strongly suggests that in the primosome assembly processes, the trimer is the major state in which the DnaT protein enters the assembling process of the primosome scaffold. Moreover, the trimer would also provide the protein with an increased number of interacting sites necessary to efficiently recognize the formed PriA - PriB - DNA complex (12). In this context, the engagement of both interacting sites, one located on the N-terminal core domain and another one on the C-terminal region, in the stabilization of the trimer structure, indicates that the DnaT molecule possesses another binding site(s) that serves to recognize the ingredients of the primosome scaffold.
However, once in the primosome, the strong dependence of the DnaT trimer integrity on the conformational states of the constituting monomers controlled by magnesium binding, suggests that strong interactions with the primosome ingredients affect the oligomeric state of the bound protein. High cooperativity of the trimer dissociation and the rather modest intrinsic affinity among the monomers in the trimer assure an efficient transformation into the monomer state modulated by magnesium binding. Thus, the trimer would be a transient form of the DnaT protein, in terms of the primosome functions, which would be operational in the recognition stages of the primosome assembly reaction. The remaining monomeric protein within the primosome scaffold now has two interacting areas, on the N-terminal core domain and the C-terminal region, free to engage the DnaB - DnaC complex. Consequently, the monomer form would be the DnaT state in the post-assembly activities of the primosome (5). Our laboratory is currently examining these possibilities.
ACKNOWLEDGMENT
We wish to thank Mrs. Gloria Drennan Bellard for reading the manuscript.
Abbreviations
- NATA
N-acethyl-L-tryprophanamide
- EDTA
Ethylendiaminetetraacetic acid
- GnHCl
Guanidine hydrochloride
Footnotes
This work was supported by NIH Grant GM46679 (to W. B.). M. R. S. was partially supported by J. B. Kempner postdoctoral fellowship.
REFERENCES
- 1.Kornberg A, Baker TA. DNA Replication. Freeman; San Francisco: 1992. pp. 275–293. [Google Scholar]
- 2.Sangler SJ. Requirements for Replication Restart Proteins During Constitutive Stable DNA Replication in Escherichia coli K-12. Genetics. 2005;169:1799–1806. doi: 10.1534/genetics.104.036962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen GC, Kornberg A. Assembly of the Primosome of DNA Replication in Escherichia coli. J. Biol. Chem. 1993;268:19204–19209. [PubMed] [Google Scholar]
- 4.Arai K, Arai N, Shlomai J, Kornberg A. Replication of the Duplex DNA of Phage PhiX174 Reconstituted with Purified Enzymes. Proc. Acad. Sci. USA. 1981;77:3322–3326. doi: 10.1073/pnas.77.6.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng JY, Marians KJ. The Ordered Assembly of the fX174 Primosome. II. Preservation Primosome Composition From Assembly Through Replication. J. Biol. Chem. 1996;271:15649–15655. doi: 10.1074/jbc.271.26.15649. [DOI] [PubMed] [Google Scholar]
- 6.Heller RC, Marians KJ. Replication Fork Reactivation Downstream of the Blocked Nascent Leading Strand. Nature. 2006;439:557–562. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 7.Heller RC, Marians KJ. Non-Replicative Helicases at the Replication Fork. DNA Repair. 2007;6:945–952. doi: 10.1016/j.dnarep.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Marians KJ. PriA: at the Crossroads of DNA Replication and Recombination. Prog. Nucleic Acid Res., Mol. Biol. 1999;63:39–67. doi: 10.1016/s0079-6603(08)60719-9. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Nurse P, Marians K. The Ordered Assembly of the ϕX174 Primosome. III. PriB Facilitates Complex Formation Between PriA and DnaT. J. Biol. Chem. 1996;271:15656–15661. doi: 10.1074/jbc.271.26.15656. [DOI] [PubMed] [Google Scholar]
- 10.Arai K, McMacken R, Yasuda S, Kornberg A. Purification and Properties of Escherichia coli Protein i, a Prepriming Protein in ϕX174 DNA Replication. J. Biol. Chem. 1981;256:5281–5286. [PubMed] [Google Scholar]
- 11.Masai H, Bond M, Arai K. Cloning of the Escherichia coli gene for Primosomal Protein i: The Relationship to DnaT, Essential for Chromosomal DNA Replication. Proc. Natl. Acad. Sci. USA. 1986;83:1256–1260. doi: 10.1073/pnas.83.5.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szymanski MR, Jezewska MJ, Bujalowski W. Binding of Two PriA - PriB Complexes to the Primosome Assembly Site Initiates the Primosome Formation. J. Mol. Biol. 2011;411:123–142. doi: 10.1016/j.jmb.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 13.Szymanski MR, Jezewska MJ, Bujalowski W. The E. coli PriA Helicase Specifically Recognizes Gapped DNA Substrates. Effect of the Two Nucleotide-Binding Sites of the Enzyme on the Recognition Process. J. Biol. Chem. 2010;285:9683–9696. doi: 10.1074/jbc.M109.094789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szymanski MR, Jezewska MJ, Bujalowski W. The Escherichia coli PriA Helicase - Double-Stranded DNA Complex. Location of the Strong DNA-Binding Subsite on the Helicase Domain of the Protein and the Affinity Control By the Two Nucleotide-Binding Sites of the Enzyme. J. Mol. Biol. 2010;402:344–362. doi: 10.1016/j.jmb.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szymanski MR, Jezewska MJ, Bujalowski W. Interactions of the Escherichia coli Primosomal PriB Protein with the Single-Stranded DNA. Stoichiometries, Intrinsic Affinities, Cooperativities, and Base Specificities. J. Mol. Biol. 2010;398:8–25. doi: 10.1016/j.jmb.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edeldoch H. Spectroscopic Determination of Tryptophan and Tyrosine in Proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 17.Gill SC, von Hippel PH. Calculation of Protein Extinction Coefficients From Amino Acid Sequence Data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 18.Jezewska MJ, Rajendran S, Bujalowski W. Functional and Structural Heterogeneity of the DNA Binding Site of the Escherichia coli Primary Replicative Helicase DnaB Protein. J. Biol. Chem. 1998;273:9058–9069. doi: 10.1074/jbc.273.15.9058. [DOI] [PubMed] [Google Scholar]
- 19.Jezewska MJ, Szymanski MR, Bujalowski W. The Primary DNA-Binding Subsite of the Rat Pol β. Energetics of Interactions of the 8-kDa Domain of the Enzyme With the ssDNA. Biophys. Chem. 2011;156:115–127. doi: 10.1016/j.bpc.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jezewska MJ, Bujalowski PJ, Bujalowski W. Interactions of the DNA Polymerase X of African Swine Fever Virus with Double-Stranded DNA. Functional Structure of the Complex. J. Mol. Biol. 2007;373:75–95. doi: 10.1016/j.jmb.2007.06.054. [DOI] [PubMed] [Google Scholar]
- 21.Marcinowicz A, Jezewska MJ, Bujalowski PJ, Bujalowski W. The Structure of the Tertiary Complex of the RepA Hexameric Helicase of Plasmid RSF1010 with the ssDNA and Nucleotide Cofactors in Solution. Biochemistry. 2007;46:13279–13296. doi: 10.1021/bi700729k. [DOI] [PubMed] [Google Scholar]
- 22.Jezewska MJ, Bujalowski PJ, Bujalowski W. Interactions of the DNA Polymerase X From African Swine Fever Virus with Gapped DNA Substrates. Quantitative Analysis of Functional Structures of the Formed Complexes. Biochemistry. 2007;46:12909–12924. doi: 10.1021/bi700677j. [DOI] [PubMed] [Google Scholar]
- 23.Jezewska MJ, Rajendran S, Bujalowska D, Bujalowski W. Does ssDNA Pass Through the Inner Channel of the Protein Hexamer in the Complex with the E. coli DnaB Helicase? Fluorescence Energy Transfer Studies. J. Biol. Chem. 1998;273:10515–10529. doi: 10.1074/jbc.273.17.10515. [DOI] [PubMed] [Google Scholar]
- 24.Jezewska MJ, Galletto R, Bujalowski W. Rat Polymerase β Gapped DNA Interactions: Antagonistic Effects of the 5′ Terminal PO4− Group and Magnesium on the Enzyme Binding to the Gapped DNAs with Different ssDNA Gaps. Cell Biochem. and Biophys. 2003;38:125–160. doi: 10.1385/CBB:38:2:125. [DOI] [PubMed] [Google Scholar]
- 25.Andreeva IE, Roychowdhury A, Szymanski MR, Jezewska MJ, Bujalowski W. Mechanisms of Nucleotide Cofactor Interactions with the RepA Protein of Plasmid RSF1010. Binding Dynamics Studied Using Fluorescence Stopped-Flow Method. Biochemistry. 2009;48:10620–10636. doi: 10.1021/bi900940q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bujalowski W, Lohman TM. Monomer-Tetramer Equilibrium of the E. coli SSB-1 Mutant Single Strand Binding Protein. J. Biol.Chem. 1991;266:1616–1626. [PubMed] [Google Scholar]
- 27.Lakowicz JR. Principle of Fluorescence Spectroscopy. Kluwer Academc/Plenum Publishers; New York: 1999. pp. 291–319. [Google Scholar]
- 28.Bailey MF, Van der Schans EJ, Millar DP. Dimerization of the Klenow Fragment of Escherichia coli DNA Polymerase I Is Linked to Its Mode of DNA Binding. Biochemistry. 2007;46:885–899. doi: 10.1021/bi6024148. [DOI] [PubMed] [Google Scholar]
- 29.LiCata VJ, Wowor AJ. Applications of Fluorescence Anisotropy to the Studies of Protein-DNA Interactions. Methods Cell. Biol. 2008;84:243–262. doi: 10.1016/S0091-679X(07)84009-X. [DOI] [PubMed] [Google Scholar]
- 30.Royer CA, Scarlata SF. Fluorescence Approaches to Quantifying Biological Interactions. Meth. Enzym. 2008;450:79–106. doi: 10.1016/S0076-6879(08)03405-8. [DOI] [PubMed] [Google Scholar]
- 31.Bujalowski W, Klonowska MM. Structural Characteristics of the Nucleotide Binding Site of E. Coli Primary Replicative Helicase DnaB Protein. Studies with Ribose and Base-Modified Fluorescent Nucleotide Analogs. Biochemistry. 1994;33:4682–4694. doi: 10.1021/bi00181a028. [DOI] [PubMed] [Google Scholar]
- 32.Galletto R, Maillard R, Jezewska MJ, Bujalowski W. Global Conformation of the Escherichia coli Replication Factor DnaC Protein in Absence and Presence of Nucleotide Cofactors. Biochemistry. 2004;43:10988–11001. doi: 10.1021/bi049377y. [DOI] [PubMed] [Google Scholar]
- 33.De Kroon AIPM, Soekarjo MW, De Gier J, Kruijff B. The Role of Charge and Hydrophobicity in Peptide-Lipid Interaction: A Comparative Study Based on Tryptophan, Fluorescence Measurements Combined with the Use of Aqueous and Hydrophobic Quenchers. Biochemistry. 1990;29:8229–8240. doi: 10.1021/bi00488a006. [DOI] [PubMed] [Google Scholar]
- 34.Alston RW, Lasagna M, Grimsley GR, Scholtz JM, Reinhart GD, Pace CN. Tryptophan Fluorescence Reveals the Presence of Long-Range Interactions in the Denatured State of Ribonuclease Sa. Biophys. J. 2008;94:2288–2296. doi: 10.1529/biophysj.107.116954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bujalowski W, Klonowska MM. Close Proximity of Tryptophan Residues and ATP Binding Site in Escherichia coli Primary Replicative Helicase DnaB Protein. Molecular Topography of the Enzyme. J. Biol. Chem. 1994;269:31359–31371. [PubMed] [Google Scholar]
- 36.Galletto R, Jezewska MJ, Bujalowski W. Interactions of the E. coli DnaB Helicase Hexamer with the Replication Factor the DnaC Protein. Effect of Nucleotide Cofactors and the ssDNA on Protein-Protein Interactions and the Topology of the Complex. J. Mol. Biol. 2003;329:441–465. doi: 10.1016/s0022-2836(03)00435-2. [DOI] [PubMed] [Google Scholar]
- 37.Marcinowicz A, Jezewska MJ, Bujalowski W. Multiple Global Conformational States of the Hexameric RepA Helicase of Plasmid RSF1010 with Different ssDNA-Binding Capabilities Are Induced By Different Numbers of Bound Nucleotides. Analytical Ultracentrifugation and Dynamic Light Scattering Studies. J. Mol. Biol. 2008;375:386–408. doi: 10.1016/j.jmb.2007.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roychowdhury A, Szymanski MR, Jezewska MJ, Bujalowski W. Interactions of the E. coli DnaB - DnaC Protein Complex with Nucleotide Cofactors. 1. Allosteric Conformational Transitions of the Complex. Biochemistry. 2009;48:6712–6729. doi: 10.1021/bi900050x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roychowdhury A, Szymanski MR, Jezewska MJ, Bujalowski W. The Escherichia coli DnaB Helicase - DnaC Protein Complex: Allosteric Effects of the Nucleotides on the Nucleic Acid Binding and the Kinetic Mechanism of NTP Hydrolysis. 3. Biochemistry. 2009;48:6747–6763. doi: 10.1021/bi9000535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jezewska MJ, Rajendran S, Bujalowski W. Escherichia coli Replicative Helicase PriA Protein - Single-Stranded DNA Complex. Stoichiometries, Free Energy of Binding, and Cooperativities. J. Biol. Chem. 2000;275:27865–27873. doi: 10.1074/jbc.M004104200. [DOI] [PubMed] [Google Scholar]
- 41.Lucius AL, Jezewska MJ, Bujalowski W. The Escherichia coli PriA Helicase Has Two Nucleotide-Binding Sites Differing in Their Affinities for Nucleotide Cofactors. 1. Intrinsic Affinities, Cooperativities, and Base Specificity of Nucleotide Cofactor Binding. Biochemistry. 2006;45:7202–7216. doi: 10.1021/bi051826m. [DOI] [PubMed] [Google Scholar]
- 42.Bujalowski W, Klonowska MM, Jezewska MJ. Oligomeric Structure of Escherichia coli Primary Replicative Helicase DnaB Protein. J. Biol. Chem. 1994;269:31350–31358. [PubMed] [Google Scholar]
- 43.Jezewska MJ, Bujalowski W. Global Conformational Transitions in E. coli Primary Replicative DnaB Protein Induced by ATP, ADP and Single-Stranded DNA Binding. J. Biol. Chem. 1996;271:4261–4265. doi: 10.1074/jbc.271.8.4261. [DOI] [PubMed] [Google Scholar]
- 44.Cantor RC, Schimmel PR. Biophysical Chemistry. Vol. II. W. H. Freeman; New York: 1980. pp. 591–641. [Google Scholar]
- 45.Stafford W., III Boundary Analysis in Sedimentation Transport Experiments: A Procedure for Obtaining Sedimentation Coefficient Distributions Using the Time Derivative of the Concentration Profile. Anal. Biochem. 1992;203:295–301. doi: 10.1016/0003-2697(92)90316-y. [DOI] [PubMed] [Google Scholar]
- 46.Correia JJ, Chacko BM, Lam SS, Lin K. Sedimentation Studies Reveal a Direct role of Phosphorylation in Smad3:Smad4 Homo- and Hetero-Dimerization. Biochemistry. 2001;40:1473–1482. doi: 10.1021/bi0019343. [DOI] [PubMed] [Google Scholar]
- 47.Bujalowski W. Thermodynamic and Kinetic Methods of Analyses of Protein – Nucleic Acid Interactions. From Simpler to More Complex Systems. Chem. Rev. 2006;106:556–606. doi: 10.1021/cr040462l. [DOI] [PubMed] [Google Scholar]
- 48.Lohman TM, Bujalowski W. Thermodynamic Methods for Model-Independent Determination of Equilibrium Binding Isotherms for Protein-DNA Interactions: Spectroscopic Approaches to Monitor Binding. Meth. Enzym. 1991;208:258–290. doi: 10.1016/0076-6879(91)08017-c. [DOI] [PubMed] [Google Scholar]
- 49.Jezewska MJ, Bujalowski W. A General Method of Analysis of Ligand Binding to Competing Macromolecules Using the Spectroscopic Signal Originating from a Reference Macromolecule. Application to Escherichia coli Replicative Helicase DnaB Protein-Nucleic Acid Interactions. Biochemistry. 1998;35:2117–2128. doi: 10.1021/bi952344l. [DOI] [PubMed] [Google Scholar]
- 50.Bujalowski W, Jezewska MJ. Thermodynamic Analysis of the Structure-Function Relationship in the Total DNA-Binding Site of Enzyme - DNA Complexes. Meth. Enzym. 2009;466:294–324. doi: 10.1016/S0076-6879(09)66013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bujalowski W, Jezewska MJ. Macromolecular Competition Titration Method: Accessing Thermodynamics of the Unmodified Macromolecule-Ligand Interactions Through Spectroscopic Titrations of Fluorescent Analogs. Meth. Enzym. 2011;488:17–57. doi: 10.1016/B978-0-12-381268-1.00002-1. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hill TL. Cooperativity Theory in Biochemistry. Steady State and Equilibrium Systems. Springer; Verlag, New York: 1985. pp. 167–234. [Google Scholar]
- 53.Connors KA. Chemical Kinetics. The Study of Reaction Rates in Solution. VCH Publishers; New York: 1990. pp. 187–200. [Google Scholar]
- 54.Timasheff SN. Protein - Solvent Interactions, Protein Hydration, and Modulation of Biochemical Reactions by Solvent Components. Proc. Natl. Acad. Sci. USA. 2002;99:9721–9726. doi: 10.1073/pnas.122225399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parsegian VA, Rand RP, Rau DC. Osmotic Stress, Crowding, Preferential Hydration, and Binding: A Comparison of Perspectives. Proc. Natl. Acad. Sci. USA. 2000;97:3987–3992. doi: 10.1073/pnas.97.8.3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marcolli C, Peter T. Water Activity in Polyol/Water Systems: New UNIFAC Parameterization. Atmos. Chem. Phys. Discuss. 2005;5:1501–1527. [Google Scholar]
- 57.Sereno AM, Hubinger MD, Comesana JF, Correa A. Prediction of Water Activity of Osmotic Solutions. J. Food Eng. 2001;49:103–1014. [Google Scholar]
- 58.Record MT, Jr., Anderson CF, Lohman TM. Thermodynamic Analysis of Ion Effects on the Binding and Conformational Equilibria of Proteins and Nucleic Acids: the Roles of Ion Association or Release, Screening, and Ion Effects on Water Activity. Q. Rev. Biophys. 1978;11:103–178. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- 59.Record MT, Jr., Lohman TM, deHaseth PL. Ion Effects on Ligand - Nucleic Acid Interactions. J. Mol. Biol. 1976;107:145–158. doi: 10.1016/s0022-2836(76)80023-x. [DOI] [PubMed] [Google Scholar]
- 60.Lim WK, Rosgen J, Englander SW. Urea But Not Guanidinium, Destabilizes Proteins by Forming Hydrogen Bonds to the Peptide Group. Proc. Natl. Acad. Sci. USA. 2008;106:2595–2600. doi: 10.1073/pnas.0812588106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greene RF, Pace CN. Urea and Guanidine hydrochloride Denaturation of Ribonuclease, Lysozyme, Alpha-chymotrypsin, and Beta-Lactoglobulin. J. Biol. Chem. 1974;249:5388–5393. [PubMed] [Google Scholar]
- 62.Tanford C. Protein Denaturation. Adv. Protein Chem. 1968;23:121–282. doi: 10.1016/s0065-3233(08)60401-5. [DOI] [PubMed] [Google Scholar]
- 63.Coffy G, Olofsson G. The Standard Enthalpy of Formation of Aqueous Magnesium Ion at 298.15 K. J. Chem. Therm. 1979;11:141–144. [Google Scholar]