

Abstract
The catalytic cross-coupling of arylboronic acids with pyridines through single electron oxidation provides efficient access to substituted heterocycles. Although important, there is very little known about the mechanism of the reaction and as a consequence, it is unclear if the full scope of the transformation has been realized. In this communication we present kinetic and spectroscopic evidence showing a high degree of complexity in the reaction system. The mechanism derived from these studies shows the activation of Ag(I) for reduction of persulfate and an off-cycle protodeboronation by pyridine substrate. These results provide key mechanistic insights that enable control of the off-cycle process thus providing higher efficiency and yield.
Catalytic oxidations that proceed through single electron transfer are becoming one of the most important approaches for the formation of C-C bonds in molecules of pharmaceutical and biological importance.1 A great deal of recent effort has focused on two general methods: 1) visible light photoredox catalysis2 and 2) metal-catalyzed oxidations.3 In both approaches, the use of a terminal oxidant is often required. In addition, the use of readily available starting materials employed in these reactions can provide a wide range of related structures important for screening in medicinal chemistry and for the construction of building blocks important in materials chemistry. In this vein, the recent work of Baran on the Ag(I)/persulfate cross-coupling of arylboronic acids with electron deficient pyridines is of fundamental importance.4 The approach developed by Baran overcomes several of the shortcomings of the traditional Minisci reaction,5 most importantly the addition of aryl radicals to an aromatic heterocycle. In addition, the method is procedurally elegant and has broad substrate scope. Although the reaction provides access to a variety of substituted heteroarenes, a second addition of Ag catalyst and stoichiometric oxidant are often required. Given the importance of this bond- forming reaction, it is our supposition that a detailed mechanistic study of the system would facilitate a more efficient approach to reaction design using the Ag(I)/persulfate catalytic system. Herein, we present mechanistic data defining the role of the individual components in the reaction that reveal a great degree of mechanistic complexity, that once understood provides a more effective approach to this catalytic system, thus extending its utility.
To study the system, the reaction of 4-trifluoromethyl pyridine (1, 1 mmol) and p-tolylboronic acid (2, 1.5 mmol) containing 20 mol% AgNO3 (with respect to 1), potassium persulfate (3 mmol) and trifluoroacetic acid (TFA, 1 mmol) in a 1:1 solution of dichloromethane (DCM) and water was examined. Protonation of pyridine by TFA produces a better radicophile and enhances selectivity of the 2-position.6 To maintain controlled reaction conditions, solvents were degassed and the reaction was carried out under an atmosphere of argon as shown in Scheme 1. The exclusion of air from the reaction resulted in a 76% isolated yield, compared to the 81% yield reported by Baran and coworkers, which required a second addition of K2S2O8 and AgNO3.4
Scheme 1.
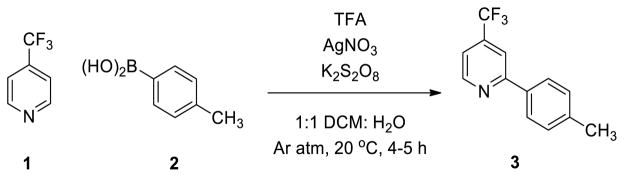
Cross-coupling reaction of electron- deficient pyridine with arylboronic acid
Additionally, toluene was obtained as a side product in roughly 30% yield (with respect to 2). Removal of oxygen and the subsequent increased yield is consistent with the previously proposed free-radical mechanism.4 These higher-yielding conditions were suitable for thorough mechanistic studies.
Kinetic studies were performed in which either the loss of starting material (1) or growth of product was followed over time via gas chromatography (GC). A “same excess” experiment was initiated to determine the stability of catalytic silver nitrate during the course of the reaction7 using the conditions contained in Table 1. If total catalyst concentration remains constant during the course of the reaction, the rates of both reactions would be identical. When rate profiles of Runs 1 and 2 are compared, the decays do not overlay, consistent with a decrease in [AgNO3] during the course of the reaction (Figure 1).
Table 1.
“Same excess” experiment reaction conditions
| Run | 1 (M) | 2 (M) | excess (M) | K2S2O8 (M) | excess (M) | AgNO3 (M) |
|---|---|---|---|---|---|---|
| 1–100% | 0.10 | 0.15 | 0.050 | 0.30 | 0.20 | 0.020 |
| 2–50% | 0.050 | 0.10 | 0.050 | 0.25 | 0.20 | 0.020 |
Figure 1.

Rate vs. [1] for same excess experiments.
To determine the rate orders of each of the substrates in the reaction “different excess” experiments were performed. The reaction was approximately first order in 1 and AgNO3. Initial experiments showed that the rate order of K2S2O8 was zero. Given the low solubility of K2S2O8 it is possible that its true rate order could be masked by phase transfer. To examine this physical process, the more soluble Na2S2O8 was employed in the reaction and a rate order of approximately 1 was observed. Surprisingly, an increase in the initial concentration of 2 resulted in an overall decrease in reaction rate (Figure 2). The order of 2 was determined through normalization of −d[1]/dt, defined by eq 1
Figure 2.

Rate vs. [1] for different excess experiments for 2. Run 3 was performed by increasing [2] to 0.30 M and keeping concentrations of all other reactants the same as in Run 2.
| (1) |
where x is the order of 2. Overlay of the two reaction profiles is observed when x = −0.5, indicative of an inverse half order for 2 (Figure 2, inset).
The rate orders for all reaction components are shown in Table 2. The rate orders of persulfate and AgNO3 are consistent with a process where catalytic Ag(I) is oxidized by persulfate. The orders obtained for 1 and 2 are surprising. If the reaction is indeed proceeding through initial oxidation of 2 followed by addition to 1, the first order observed for 1 compared to the inverse half order for 2 suggests a more complicated process.
Table 2.
Observed Rate Orders of Substrates in Cross-Coupling Reaction of 1 and 2
| 1 | 2 | K2S2O8 | AgNO3 |
|---|---|---|---|
| 1 | −0.5 | 0* | 1 |
Due to low solubility of K2S2O8 (4.72 g/100 ml at 20 °C)10; when using Na2S2O8 (54.6 g/100mL), rate order shown to be approximately 1.
The formation of silver-pyridine complexes is well- established in the literature.8 In addition, there is literature precedence describing the activation of Ag(I) by amines toward oxidation by persulfate.9 Bonchev and Aleksiev showed that the addition of suitable nitrogen-containing neutral ligands, such as phenanthroline, ethylenediamine, and pyridine, to Ag(I)/persulfate reactions resulted in the acceleration in the oxidation of Ag(I) to Ag(II).9 This activating effect was attributed to a lowering of the Ag(II)/Ag(I) couple.
To investigate the possible interaction between 1 and AgNO3, 1H NMR studies were carried out in D2O to mimic reaction conditions (Figure 3). Downfield shifts of aromatic protons were observed upon addition of TFA (Figure 3b) or AgNO3 (Figure 3d) to 1 in D2O. Upon addition of TFA and AgNO3 to 1 (Figure 3c), a slight upfield shift of aromatic protons is observed compared to the mixture of 1 and TFA (Figure 3b).
Figure 3.

1H NMR spectra of 1 with additives
The lack of overlay of the spectra is consistent with equilibrium complexation between 1 and Ag(I) under reaction conditions.
With some insight into the possible role of 1 we next focused on the mechanistic role of 2. An inverse rate order is typically indicative of two possible scenarios: 1) The presence of the reaction component shifts an equilibrium, decreasing the concentration of the intermediate prior to a rate determining step, or 2) the component behaves as an antagonist acting outside the desired pathway that leads to product. Given the long history of the Ag(I)/persulfate oxidation system, it is instructive to examine the literature carefully for possible precedent to explain the unusual observations obtained from the kinetic studies. Studies on the interaction between AgNO3 and boronic acids in ammoniacal solution dates back to the 1880s.11 These early investigations showed that while alkyl boronic acids reduce Ag(I) through an intermediate Ag- alkyl, aromatic boronic acids form an insoluble salt. Heating of this salt led to hydrolytic cleavage producing an arene, boric acid and Ag2O.11
To examine the Ag(I)-initiated hydrolysis under reaction conditions, stoichiometric quantities of 2 and AgNO3 were stirred in 1:1 DCM:H2O solution. After approximately five minutes, the formation of a gray/silver colored precipitate was observed consistent with silver oxide described in previous studies. The formation of toluene as a by-product of this reaction was confirmed by gas chromatography-mass spectrometry (GC-MS). To further probe the system, two reactions were carried out. The first involved reacting 1.5 mmol of 2 with 1.0 mmol AgNO3 in 1:1 DCM:H2O solution. A second reaction was run under the same conditions with 3.0 mmol of 1. Approximately 0.84 mmol of toluene formed when 1 was included in the reaction mixture, compared to 0.23 mmol of toluene formed when 1 was excluded, a finding consistent with inhibition of AgNO3 and consumption of 2 outside of the desired reaction pathway. The increase in toluene formation in the presence of 1 is also consistent with interaction between 1 and AgNO3 and the classic studies on the reaction of AgNO3 and boronic acids in ammoniacal solutions.8,11
Based on the observed kinetic and spectroscopic data, the proposed reaction mechanism involves: i) a pre-equilibrium step in which 1 and Ag(I) form a complex, ii) the reduction of S2O82− by the Ag(I)-1 complex, which is the rate-determining step, and iii) an off-cycle step involving protodeboronation of 2 accelerated by the Ag(I)-1 complex (Scheme 2).
Scheme 2.
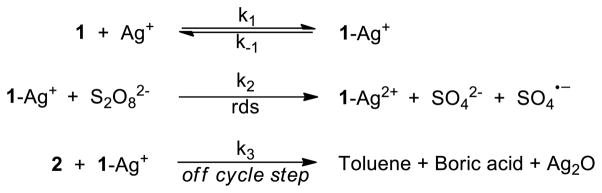
Mechanism of Ag(I)/Persulfate-Catalysis in Coupling of Arylboronic acid and Electron-deficient Pyridine
Assuming steady state approximation for the Ag(I)-1 complex, and accounting for all the states of Ag(I), the rate of reaction can be expressed as eq 2 (for full derivation refer to Supporting Information).
| (2) |
The derived rate law can be compared to the empirical rate law (eq 3), in which 1, S2O82−, and Ag(I) are first order and 2 is inverse half order. The reaction orders in the empirical rate law can be equated to the first order of 1 and Ag(I), positive additive order of S2O82−, and overall inverse fractional order of 2, as shown in the derived rate law.
| (3) |
Aside from providing insight into the reaction mechanism, these results provide a means to optimize reaction conditions and increase the yield of the reaction. The inverse half order of 2 indicates that increasing its concentration is deleterious to reaction progress. Furthermore, addition of a reagent capable of preventing formation of catalytically inactive Ag(I) to its active form should be beneficial to reaction progress (See Supporting Information). To test this supposition, a reaction was initiated using 0.5 M HNO3 to prevent the formation of catalytically inactive Ag(I).11c Employing these modified conditions enabled the reduction of catalyst and oxidant loading to 10 mol% AgNO3 and 2 equiv of persulfate, respectively in a 1:1 solution of DCM and water under Ar atmosphere overnight, leading to an isolated yield of 90%. Additionally, toluene side-product formation was reduced to 9% (with respect to 2).
Equipped with a rate expression for the system, the question that still remains unanswered is: which species is oxidizing the boronic acid, the Ag(II) or SO4−? The classic work of Kochi and others has shown that metastable Ag(II) is responsible for decarboxylation of carboxylic acids to produce radicals by Ag(I)/persulfate.12 This work was applied by Minisci and coworkers, in which the alkyl radical, generated from decarboxylation of an acid adds to a heteroarene to form substituted heterocycles.5 In the present reaction, Baran proposed that persulfate radical anion addition to the aryl boronic acid was responsible for intermediate aryl radical formation.4
To test the question posed above, we first examined the use of potassium 4-methylphenyl trifluoroborate (4) in place of 2. The reaction was performed under the unoptimized conditions shown in Scheme 1 and provided a 60% isolated yield of 3. Aryl trifluoroborates are known to hydrolyze under basic conditions.13 The stability of 4 under reaction conditions was examined in aqueous acidic media by monitoring the 11B NMR spectrum as described by Lloyd-Jones.13 No hydrolysis of 4 to 2 was observed. Next, the reaction with 2 was carried out in the presence of allyl acetate, a well-known radical trap for SO4−.14 If SO4− is acting as the oxidizing agent in the reaction, the addition of allyl acetate would be deleterious to reaction progress. Interestingly, addition of 6 equiv of allyl acetate had no impact on yield and the rate of reaction increased slightly. This observation suggests that by decreasing the concentration of SO4− through capture by allyl acetate, the reaction is being driven forward toward the formation of product, presumably through the reduction of S2O82− by Ag(I) producing Ag(II). These additional experiments show that 4 is not hydrolyzed (i.e. remains quaternized) under reaction conditions, and reactions with 2 proceed even when SO4− is sequestered by allyl acetate. As a consequence, these experiments are consistent with a process where the oxidation proceeds through a Ag(II)-mediated process. Since quaternized boron is more susceptible to single electron oxidation,15 we propose that water (solvent) or pyridine interacts with the aryl boronic acid to facilitate oxidation by Ag(II). A proposed mechanism for the reaction is shown in Scheme 3.
Scheme 3.
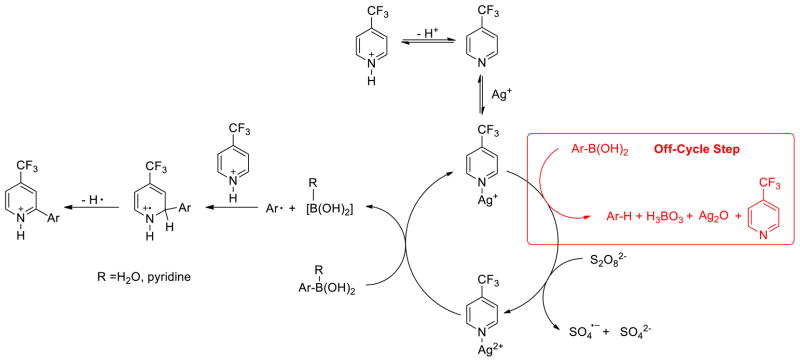
Proposed Mechanism of the Ag(I)/Persulfate-Catalyzed Arylation of Electron-deficient Pyridine
Under the conditions of the reaction, pyridine coordinates to Ag(I), followed by persulfate oxidation in the rate-limiting step. The resulting Ag(II)-pyridine complex oxidizes the arylboronic acid, producing an aryl radical, which can then add to the pyridinium ion leading to product. An off-cycle step is also involved outside of the desired pathway, in which the arylboronic acid is protodeboronated, leading to unwanted side products.
The mechanistic experiments described herein show an un-expected degree of complexity in the Ag(I)/persulfate-catalyzed cross-coupling of arylboronic acids and pyridines, information that could not otherwise be extracted from the use of simple empirical models or studies based on product distributions. The mechanism derived from spectroscopic and kinetic studies shows that Ag(I) is activated for reduction of persulfate and an off-cycle protodeboronation by pyridine. These results provide the key mechanistic insight that enables control of the off-cycle process thus providing higher efficiency and yield. In addition, literature precedent and evidence provided herein demonstrate that Ag(II) is the likely oxidant responsible for formation of aryl radicals from aryl boronic acids. While the studies presented clarify several mechanistic details of the Ag(I)/persulfate catalyzed cross-coupling of arylboronic acids and pyridines, the results may have impact in the design and refinement of other radical-based additions proceeding through catalytic oxidations mediated by Ag(I)/persulfate.
Supplementary Material
Acknowledgments
R.A.F. is grateful to the National Institutes of Health (GM075960-03) for support of this work. We thank Dr. James Devery and Dr. Lawrence Courtney for useful discussion and aid in the elucidation of the reaction mechanism, and Dr. Brian Casey for initial work on this project
Footnotes
Experimental procedures, kinetic data, and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) Jahn U. Top Curr Chem. 2012;320:121–452. doi: 10.1007/128_2011_261. [DOI] [PubMed] [Google Scholar]; (b) Kozlowski MC, Morgan BJ, Linton EC. Chem Soc Rev. 2009;38:3193–3207. doi: 10.1039/b821092f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; (b) Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- 3.(a) John A, Nicholas KM. J Org Chem. 2011;76:4158–4162. doi: 10.1021/jo200409h. [DOI] [PubMed] [Google Scholar]; (b) Chen X, Hao X, Goodhue CE, Yu J. J Am Chem Soc. 2006:6790–6791. doi: 10.1021/ja061715q. [DOI] [PubMed] [Google Scholar]; (c) Zhang C, Jiao N. J Am Chem Soc. 2010;132:28–29. doi: 10.1021/ja908911n. [DOI] [PubMed] [Google Scholar]; (d) Fujiwara Y, Domingo V, Seiple IB, Gianatassio R, Del Bel M. J Am Chem Soc. 2011;133:3292–3295. doi: 10.1021/ja111152z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seiple IB, Su S, Rodriguez RA, Gianatassio R, Fujiwara Y, Sobel AL, Baran PS. J Am Chem Soc. 2010;132:13194–13196. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minisci F, Bernardi R, Galli R, Perchinummo M. Tetrahedron. 1971;27:3575–3579. [Google Scholar]
- 6.Minisci F, Vismara E, Fontana F, Morini G, Serravalle M. J Org Chem. 1986;51:4411–4416. [Google Scholar]
- 7.(a) Mathew JS, Klussman M, Iwamura H, Valera F, Futran A, Emanuelsson EAC, Blackmond DG. J Org Chem. 2006;71:4711–4722. doi: 10.1021/jo052409i. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG. Angew Chem Int Ed. 2005;44:4302–4320. doi: 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]; (c) Devery JJ, III, Conrad JC, MacMillan DWC, Flowers RA., II Angew Chem Int Ed. 2010;49:6106–6110. doi: 10.1002/anie.201001673. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Choquette KA, Sadasivam DV, Flowers RA., II J Am Chem Soc. 2011;133:10655–10661. doi: 10.1021/ja204287n. [DOI] [PubMed] [Google Scholar]
- 8.(a) Bruehlman RJ, Verhoek FH. J Am Chem Soc. 1948;70:1401–1404. [Google Scholar]; (b) Bjerrum J. Acta Chem Scand. 1972;26:2734–2742. [Google Scholar]; (c) Vosburgh WC, Cogswell SA. J Am Chem Soc. 1943;65:2412–2413. [Google Scholar]
- 9.(a) Alexiev A, Bontchev PR. Mikrochim Acta. 1970:13–19. [Google Scholar]; (b) Bonchev PR, Aleksiev AA. Theor Exp Chem. 1975;9:144–147. [Google Scholar]
- 10.Apelblat A, Korin E, Manzurola E. J Chem Thermodynamics. 2001;33:61–69. [Google Scholar]
- 11.(a) Michaelis, Becker Chem Ber. 1882;15:181. [Google Scholar]; (b) Johnson JR, Van Campen MG, Grummitt O. J Am Chem Soc. 1938;60:111–115. [Google Scholar]; (c) Seaman W, Johnson JR. J Am Chem Soc. 1931;53:711–723. [Google Scholar]
- 12.(a) Anderson JM, Kochi JK. J Am Chem Soc. 1970;92:1651–1659. [Google Scholar]; (b) Anderson JM, Kochi JK. J Org Chem. 1970;35:986–989. [Google Scholar]
- 13.Butters M, Harvey JN, Jover J, Lennox AJJ, Lloyd-Jones GC, Murray PM. Angew Chem Int Ed. 2010;49:5156–5160. doi: 10.1002/anie.201001522. [DOI] [PubMed] [Google Scholar]
- 14.Kalb AJ, Allen TL. J Am Chem Soc. 1964;86:5107–5112. [Google Scholar]
- 15.Shundrin LA, Bardin VV, Frohn HJ. Z Anorg Allg Chem. 2004;630:1253–1257. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.