

Summary
Chemotaxis, the environment-specific swimming behavior of a bacterial cell is controlled by flagellar rotation. The steady-state level of the phosphorylated or activated form of the response regulator CheY dictates the direction of flagellar rotation. CheY phosphorylation is regulated by a fine equilibrium of three phosphotransfer activities: phosphorylation by the kinase CheA, its auto-dephosphorylation and dephosphorylation by its phosphatase CheZ. Efficient dephosphorylation of CheY by CheZ requires two spatially distinct protein-protein contacts: tethering of the two proteins to each other and formation of an active site for dephosphorylation. The latter involves interaction of phosphorylated CheY with the small highly conserved C-terminal helix of CheZ (CheZC), an indispensable structural component of the functional CheZ protein. To understand how the CheZC helix, representing less than 1% of the full-length protein, ascertains molecular specificity of binding to CheY, we have determined crystal structures of CheY in complex with a synthetic peptide corresponding to 15 C-terminal residues of CheZ (CheZ200-214) at resolutions ranging from 2.0 Å to 2.3 Å. These structures provide a detailed view of the CheZC peptide interaction both in the presence and absence of the phosphoryl analog, BeF3−. Our studies reveal that two different modes of binding the CheZ200-214 peptide are dictated by the conformational state of CheY in the complex. Our structures suggest that the CheZC helix binds to a “meta-active” conformation of inactive CheY and it does so in an orientation that is distinct from the one in which it binds activated CheY. Our dual binding mode hypothesis provides implications for reverse information flow in CheY and extends previous observations on inherent resilience in CheY-like signaling domains.
Keywords: CheY, CheZ peptide, crystal structure, beryllium fluoride, dual binding mode
Introduction
Chemotaxis enables motile eubacteria to regulate their swimming behavior in response to chemical gradients (reviewed in Bren et al.1, Bourret and Stock2 and Wadhams and Armitage3). In enteric species such as Salmonella enterica serovar Typhimurium and Escherichia coli, the key step in chemotaxis is the regulation of phosphorylation of the response regulator CheY in response to chemical signals in the environment. The steady-state level of phosphorylated CheY (P~CheY) determines the direction of flagellar rotation, which regulates the specific swimming behavior of the cell.
CheY is a switch molecule with multiple conformations subject to the phosphorylation state of a conserved active-site residue, 57Asp (reviewed in Silversmith and Bourret4, Robinson et al.5 and Cho et al.6). The phosphorylated form of CheY is short-lived (T1/2 ~ 10−1 sec), owing to both auto-dephosphorylation by CheY as well as dephosphorylation by CheZ, a P~CheY-specific phosphatase. Dephosphorylation of CheY by CheZ requires two interactions: tethering of the two proteins to each other and formation of an active site for dephosphorylation. The former involves interaction of P~CheY with the small C-terminal helix of CheZ (CheZC helix).
Blat and Eisenbach7, first identified the C-terminal region of CheZ as the locus of this tethering interaction. Schuster et al.8 showed that addition of the CheZC peptide (CheZ196-214) in solution enhances the phosphorylation kinetics of CheY but has no effect on its dephosphorylation. Subsequently, Zhou et al.9 reported the 2.9 Å crystal structure of CheY, activated with the phosphoryl analog, BeF3−, and bound to CheZ1-214, which presented a view of the CheZ-bound active site and addressed the structural explanation for the mechanism of CheZ-mediated dephosphorylation of CheY. The structure also provided a description of the CheY-CheZ tethering interaction, albeit limited due to apparent disorder of the region in the crystal.
The CheZC helix represents less than 1% of full-length (FL) CheZ (CheZ1-214) and is an indispensable structural component of the functional CheZ protein. Its binding to P~CheY is essential for CheZ-mediated dephosphorylation of CheY.7 The high degree of sequence conservation of the CheZC region is consistent with this role (Figure 1). An examination of 39 CheZ proteins revealed that seven of the sixteen strictly conserved residues in the FL protein are localized within the 21 C-terminal residues. Six additional residues of the C-terminus are also highly conserved (Figure 1).
Figure 1.

Sequence conservation in CheZC. A ClustalW72 sequence alignment of the C-terminal 21 residues of CheZ from 39 bacterial species is shown. Accession codes are indicated in parentheses. Sequence numbering, indicated on the first line, corresponds to S. enterica serovar Typhimurium CheZ. Hydrophobic residues are colored red, polar residues green, and acidic residues blue. The yellow box corresponds to completely conserved residues 202, 206 to 209 and 212 to 213. Strongly conserved residues are highlighted by gray boxes corresponding to hydrophobic residues, residues 198, 199, 205 and 214, a magenta box to the polar and charged residue, residue 204, and a cyan box to the less bulky residue, residue 211.
In order to understand how the small CheZC helix specifically binds to CheY and to gain insight into the significance of the CheY-CheZC interaction in chemotaxis, we solved crystal structures of wild-type (WT) S. enterica CheY, in the presence of the phosphoryl analog, BeF3− complex10-12 (CheY activated) as well as in the absence of BeF 3− (CheY inactive), each in complex with a synthetic peptide corresponding to 15 residues of the C-terminus of S. enterica CheZ (CheZ200-214). We determined the structures from two different crystal forms, F432 and P21212, at resolutions ranging between 2.0 Å and 2.3 Å.
Comparisons with previously determined structures of inactive CheY (metal-free13 and metal-bound14) and BeF3−-activated CheY, free15 and bound to CheZ1-2149, revealed distinct differences in peptide binding correlating with differences in CheY conformation. Our crystal structures suggest that the mode of binding of the CheZ200-214 peptide to activated CheY is different from its binding to inactive CheY. The conformation of CheY in the inactive CheY-CheZ200-214 complex exhibits both inactive and active features. From these structural studies, we cannot completely rule out that lattice-packing interactions have led to the trapping of an intermediate state in this complex. However, the observed conformation is similar to the “meta-active” sub-state of CheY, first documented by Simonovic and Volz.13 Therefore, the structures of the inactive CheY-CheZ200-214 complex, reported here, suggest a possible functional role for the “meta-active” intermediate of CheY. These structures also argue against the previously postulated two-state model in which long-range conformational changes in CheY are obligatorily coupled.16
The different CheY-CheZ200-214 structures illustrate the malleable nature of CheY-like signaling domains. They also suggest that the CheZ200-214 peptide induces a greater change in CheY conformation upon binding to inactive CheY than upon binding to the activated form of CheY. This might provide an explanation for the phenomenon of peptide-induced acceleration of CheY phosphorylation.8 Our structural and mutational studies suggest that this coupling of CheY phosphorylation rate and peptide binding cannot be explained by differences in conformation, bulk and charge of side chains of individual conserved residues and instead involve global changes in the backbone conformation. Based on our observations, we have proposed dual modes of CheZC-CheY binding that may be relevant to the modulation of signaling in bacterial chemotaxis.
Results and Discussion
Crystal structures of BeF3−-free and BeF3−-bound CheY-CheZ200-214 complexes from two different crystal forms
Co-crystals of S. enterica WT CheY and a synthetic peptide corresponding to S. enterica CheZ200-214 were grown by the hanging drop vapor diffusion method, as described in Materials and Methods. Crystals of both inactive and activated CheY-CheZ200-214 complexes were generated in the presence of the divalent cation, Mg2+, which plays a catalytic role at the active site of CheY.14 BeF3−, a phosphoryl analog that forms a non-covalent complex with the active-site 57Asp, was used to stabilize the active conformation of CheY.10-12 The BeF3− species was obtained by using appropriate ratios of BeCl2 and NaF, as previously described.10,15 The N-terminus of the synthetic CheZ200-214 peptide was protected by acetylation to ensure stability in solution, eliminating the charge on the α-amino group and reducing reactivity of the free N-terminus, which is prone to modification.
The CheY-CheZ200-214 complex crystallized in two different space groups: the F-centered cubic F432 and the primitive orthorhombic P21212. The F432 crystals were generated both in the presence as well as in the absence of BeCl2 and NaF in three different buffer and pH conditions: Hepes (pH 7.5), Tris (pH 8.4) and CAPS (pH 10.5). To obtain the BeF3−-bound F432 crystals, BeCl2 and NaF were soaked into inactive F432 crystals of the CheY-CheZ200-214 complex, as detailed in Materials and Methods. These crystals exhibited a range of crystal morphologies with well-defined edges including the most common cubic form (Figure 2(a)). Data from these crystals processed with one molecule per asymmetric unit and a Matthews coefficient of 5.1 Å3/Da, yielding a solvent content of 76%. The P21212 crystals were grown in MES (pH 6.0), in the presence of BeCl2 and NaF. Crystals of this form grew as needles with rough edges and with fissures running across a few (Figure 2(a)). These data processed with one molecule per asymmetric unit with a Matthews coefficient of 1.94 Å3/Da and a solvent content of only 36.6%, much lower than that of the F432 crystal form.
Figure 2.

Crystals and structures of CheY-CheZ200-214 complex. (a) Crystal morphologies of F432YZ200-214 (top two panels) and P2(1)2(1)2YZ200-214 (bottom two panels). (b) Structure of BeF3−-free F432YZ200-214 solved from a crystal grown in Tris (pH 8.4). (c) Structure of BeF3−-bound P2(1)2(1)2YZ200-214. (d) Structure of BeF3−-bound F432YZ200-214 solved from a crystal grown in Tris (pH 8.4). CheY molecules in the F432YZ200-214 structures ((b) and (d)) are shown in gray and that in the P2(1)2(1)2YZ200-214 structure ((c)) is shown in wheat. The CheZ200-214 peptide molecules in BeF3−-free F432YZ200-214 are shown in cyan ((b)), in BeF3−-bound P2(1)2(1)2YZ200-214, in orange ((c)) and in BeF3−-bound F432YZ200-214, in deep blue ((d)). Active-site water molecules are shown in deep red, Mg2+ ions are shown in magenta and BeF3− complexes are shown in yellow and the side chains of key active-site and switch residues are depicted in ball and stick models.
The molecular replacement program Phaser17 was used to obtain phases for the structures, using as a search molecule, a poly-alanine model of the BeF3−-activated CheY structure15 (PDB Accession code – 1FQW) lacking the β4-α4 loop (residues 88-92). In most response regulator receiver domains, as is the case in CheY18, the β4-α4 loop is flexible19 and undergoes substantial change upon phosphorylation. Six structures in the F432 crystal form, corresponding to different crystallization conditions, and one structure in the P21212 crystal form were solved. The statistics for data collection and those for refinement for all seven structures are summarized in Table 1. The structures were refined to resolutions ranging from 2.0 Å to 2.3 Å. All seven final models include one molecule of CheY with residues 2 to 129 and one molecule of the CheZ200-214 peptide. In case of some of the structures in the F432 crystal form, one to five N-terminal residues of the CheZ200-214 peptide could not be included because of disorder, possibly arising from the solvent-exposed nature of the peptide in this crystal form. The structures were refined to R-factors ranging between 0.186 and 0.220 and Rfree ranging between 0.207 and 0.246 with good chemical geometries, as listed in Table 1.
Table 1.
Summary of data collection and refinement statistics
| Lattice | F432 | F432 | F432 | F432 | F432 | F432 | P21212 |
| BeF3−(−/+) | + | + | + | − | − | − | + |
| pH | 10.5 | 8.4 | 7.5 | 10.5 | 8.4 | 7.5 | 6.0 |
| Data collection | |||||||
| Wavelength (Å) | 0.979 | 1.072 | 1.072 | 0.979 | 0.979 | 0.979 | 1.072 |
| Cell a= | 198.7 | 197.8 | 197.9 | 198.8 | 198.3 | 197.1 | 54.2 |
| (Å) b= | 198.7 | 197.8 | 197.9 | 198.8 | 198.3 | 197.1 | 54.2 |
| c= | 198.7 | 197.8 | 197.9 | 198.8 | 198.3 | 197.1 | 54.2 |
| α=β=γ = | 90° | 90° | 90° | 90° | 90° | 90° | 90° |
| Resolutiona (Å) | 2.0 | 2.0 | 2.0 | 2.1 | 2.2 | 2.0 | 2.0 |
| Rsym b | 0.058/ | 0.068/ | 0.071/ | 0.100/ | 0.097/ | 0.078/ | 0.052/ |
| (0.236) | (0.377) | (0.261) | (0.358) | (0.331) | (0.251) | (0.080) | |
| Completeness (%) | 99.9 / | 99.9/ | 96.9 / | 99.9 / | 99.7/ | 97.1/ | 99.4 |
| (99.5) | (100.0) | (98.5) | (98.7) | (100.0) | (97.0) | (99.3) | |
| I/σI | 34.9/ | 35.7/ | 35.5/ | 21.3/ | 22.8/ | 22.0/ | 38.6/ |
| (7.1) | (7.0) | (8.2) | (6.3) | (8.0) | (6.6) | (21.5) | |
| Refinement | |||||||
| Resolutiona (Å) | 2.0 | 2.0 | 2.0 | 2.1 | 2.3 | 2.0 | 2.0 |
| Rworking c | 0.210 | 0.220 | 0.212 | 0.197 | 0.193 | 0.200 | 0.186 |
| Rfree d | 0.224 | 0.246 | 0.228 | 0.207 | 0.220 | 0.213 | 0.232 |
| No. of reflections | 20,589 | 20,593 | 20,015 | 17,947 | 13,445 | 19,511 | 7,864 |
| No. of protein atoms | 1058 | 1067 | 1090 | 1083 | 1090 | 1078 | 1097 |
| Small moleculese | |||||||
| Mg2+ | 1 | 1 | 1 | − | − | − | 1 |
| BeF3− | 1 | 1 | 1 | − | − | − | 1 |
| Water | 81 | 53 | 89 | 80 | 87 | 86 | 90 |
| CAPS | 2 | − | − | 2 | − | − | − |
| Tris | − | 1 | − | − | 1 | − | − |
| Sulfate | 1 | 1 | 1 | 1 | 1 | 1 | − |
| Glycerol | − | 1 | − | − | 1 | − | − |
| Acetyl group | − | − | − | − | − | − | 1 |
| r.m.s.d. values from ideality | |||||||
| Bond lengths (Å) | 0.010 | 0.013 | 0.011 | 0.011 | 0.013 | 0.011 | 0.008 |
| Bond angles (°) | 1.250 | 1.281 | 1.220 | 1.184 | 1.217 | 1.409 | 1.116 |
| B-valuef (Å2) | 25.60 | 28.76 | 24.13 | 20.04 | 21.28 | 27.45 | 10.92 |
Values corresponding to highest resolution shells are shown in parentheses.
High resolution limits for data collection and refinement are indicated.
, where Iobs = observed integrated intensity and Iavg = average integrated intensity from multiple measurements.
, where Fobs and Fcalc are the observed and calculated structure factor amplitudes for hkl indices, respectively.
Rfree is identical to Rworking but is calculated from 10% of the reflections set aside as a disjoint set prior to refinement.
Number of small molecules in each case are indicated.
Overall B-value includes all atoms in a given model.
The FO-FC difference maps contoured at +3σ for all activated models showed the presence of a BeF3− species and a Mg2+ ion exhibiting octahedral coordination geometry at the active site. Following refinement, BeF3− refined with 100% occupancy in all activated structures, except one. In the structure solved from an F432 crystal grown in Hepes (pH 7.5), which was soaked in BeCl2 and NaF prior to data collection (see Materials and Methods), the occupancy for BeF3− refined to only 65%. The BeF3−-free or inactive CheY-CheZ200-214 complex was crystallized only in the F432 crystal form. At the active site of CheY in all these structures, a water molecule with tetrahedral geometry occupies the position of Mg2+. The lack of metal binding in this case, in spite of the presence of 7 mM MgCl2 during crystallization, likely results from the high ionic strength of the crystallization solutions (see Materials and Methods). Low metal occupancy in the presence of high concentrations of ammonium sulfate has been previously observed.14 The adverse effect of high ionic strength on metal binding in the F432 crystal form is overcome in the presence of BeF3−, which provides additional ligands for coordination, as observed in the BeF3−-bound CheY-CheZ200-214 structures solved from the F432 crystals (BeF3−-bound F432YZ200-214). Additional small molecules included in the final models are summarized in Table 1.
CheY is a 129-residue doubly-wound α/β protein with a central sheet of five beta strands, surrounded by five alpha helices. The CheZ200-214 peptide, in helical configuration, is bound to the α4-β5-α5 face of the CheY molecule in all seven structures, reported in this manuscript, as is the CheZC helix in the CheY-CheZ1-214 structure.9 The overall structures of all six models in the F432 crystal form are similar. Superpositions of the CheY molecules from the BeF3−-bound and BeF3−-free F432YZ200-214 complexes yield main chain root mean square deviations (r.m.s.d.) of 0.12 Å to 0.27 Å for CheY and 0.22 Å to 0.28 Å for CheZ200-214. The CheY-CheZ200-214 structures in the F432 crystal form (F432YZ200-214) differ from that in the P21212 crystal form (P2(1)2(1)2YZ200-214) by main chain r.m.s.d. values ranging between 0.70 Å and 0.83 Å for CheY and between 0.46 Å and 0.60 Å for CheZ200-214. The BeF3−-free and BeF3−-bound F432YZ200-214 and the BeF3−-bound P2(1)2(1)2YZ200-214 structures are shown in Figure 2(b), Figure 2(d) and Figure 2(c), respectively.
Comparison of CheY-CheZC contacts – dual binding modes
An examination of the CheY-CheZ200-214 interface in the F432YZ200-214 and the P2(1)2(1)2YZ200-214 structures revealed that the CheZ200-214 helix in the two crystal forms is positioned on the α4-β5-α5 face of CheY in two distinct orientations, as illustrated in Figure 3(a) and Figure 3(b). For ease of discussion, the orientation of the CheZ200-214 helix observed in the F432YZ200-214 structures will be referred to as the F432mode and that observed in the P2(1)2(1)2YZ200-214 structure, as the P2(1)2(1)2mode.
Figure 3.

The CheZ200-214 peptide-CheY interface. Ribbon representation of (a) the F432YZ200-214 interface and (b) the P2(1)2(1)2YZ200-214 interface. The side chains of key contacting residues are illustrated as ball and stick models and hydrophobic contacts are shown as light green patches. (c) Relative B-factors of CheZ200-214 in the CheY-CheZ200-214 structures. The relative B-factor versus CheZ residue number plot in BeF3−-free F432YZ200-214 is shown in cyan, that in BeF3−-bound F432YZ200-214 in deep blue and that in BeF3−-bound P2(1)2(1)2YZ200-214 in orange. Bresidue is the overall B-factor for each residue and BCheZ is the overall B-factor for all atoms of CheZ200-214 included in the final model. (d) Schematic representation of the CheY-CheZ200-214 contacts. The F432Z200-214 primary sequence in cyan and the P2(1)2(1)2Z200-214 primary sequence in orange are shown on either side of the C-terminal half of CheY, represented in secondary structural elements. Participating residues are highlighted. Hydrophobic contacts are illustrated as solid grey lines, salt bridges as dashed black lines and hydrogen bonds as solid black lines. The BeF3−-free and BeF3−-bound F432YZ200-214 structures solved from crystals grown in Tris (pH 8.4) are used as representatives of F432YZ200-214 structures in this figure.
In both modes, there is some disorder to the termini of the CheZ200-214 helix, but disorder occurs at opposite ends in the two modes. In the F432mode, the CheZ200-214 helix (F432Z200-214) is parallel to the α4 helix of CheY and the N-terminal region of the F432Z200-214 helix is highly disordered and is exposed to solvent (Figure 3(a)). Undefined density in the FO-FC difference maps, precluding the placement of one to five N-terminal residues in the final models, and relatively higher B-factors of the N-terminal region of CheZ200-214 compared to the rest of CheZ200-214 in the F432YZ200-214 structures (Figure 3(c)) are consistent with this observation. At the C-terminus, the F432Z200-214 peptide is anchored by the phenyl ring of the terminal residue, 214Phe that hooks into a hydrophobic pocket on the α4-β5-α5 face of the CheY protein. This pocket is created by the solvent-buried conformation of the side chain of 106Tyr on the β5 strand of CheY. 106Tyr is one of the switch residues involved in conformational change that accompanies phosphorylation of CheY (reviewed in Robinson et al.5).
In the P2(1)2(1)2mode, the CheZ200-214 helix (P2(1)2(1)2Z200-214) is parallel to the α5 helix of CheY (Figure 3(b)). The N-terminus of the P2(1)2(1)2Z200-214 helix is ordered and is buried within the α4-β5-α5 face while the C-terminus is disordered, consistent with the relatively higher B-factors of the C-terminal region of CheZ200-214 compared to those of the rest of the peptide in the P2(1)2(1)2YZ200-214 structure (Figure 3(c)).
The high-resolution crystal structures of the CheY-CheZ200-214 complexes provided the opportunity to analyze details of the protein-protein interactions between CheY and the CheZ200-214 peptide. The two different interfaces involve similar types of interactions. However, different residues mediate the interactions in the two binding modes (Figure 3(d) and Table 2). Both interfaces are predominantly hydrophobic. In the two modes of binding, the hydrophobic interface is overlapping but it is flanked by distinct hydrogen bond and salt bridge contacts. Buried surface analyses revealed that the C-terminal residues, 212Leu, 213Gly and 214Phe of the F432Z200-214 peptide contribute to 30% of the surface buried at the F432YZ200-214 interface. In contrast, contribution to buried surface at the P2(1)2(1)2YZ200-214 interface is substantially higher from the P2(1)2(1)2Z200-214 N-terminus than the C-terminus. The C-terminal residues, 212Leu and 214Phe contribute to only 17% of the buried surface at the P2(1)2(1)2YZ200-214 interface.
Table 2.
Comparison of CheZC peptide contacts with CheY
| Hydrophobic interactionsa | ||||
|---|---|---|---|---|
| CheY | CheZC | F432YZ200-214c | P2(1)2(1)2YZ200-214 | CheY-CheZ1-2149 |
| 95Ile | 200Ala | − | +d | − d |
| 95Ile | 208Leu, 209Leu | + | + | + |
| 95Ile | 212Leu | + | − | − |
| 95Ile | 205Val | + | + | + |
| 96Ile | 208Leu | + | − | − |
| 99Ala | 209Leu | − | + | + |
| 99Ala | 212Leu | + | − | − |
| 99Ala | 214Phe | − | + | − e |
| 106Tyr | 205Val | − | + | + |
| 106Tyr | 209Leu | + | − | − |
| 106Tyr | 214Phe | + | − | − |
| 108Val | 205Val | − | + | + |
| Hydrogen bonds b | ||||||
|---|---|---|---|---|---|---|
| CheY | CheZC | F432YZ200-214c | P2(1)2(1)2YZ200-214 | CheY-CheZ1-2149 | ||
| 90Ala | O | 200Ala | N | − | 2.80d | − d |
| 106Tyr | O | 202Gln | Nε2 | − | 2.99 | 2.85 |
| 108Val | N | 202Gln | Oε1 | − | 3.02 | 2.42 |
| 119Lys | Nζ | 214Phe | O | 2.78f | − | − e |
| Salt bridges b | ||||||
|---|---|---|---|---|---|---|
| CheY | CheZC | F432YZ200-214c | P2(1)2(1)2YZ200-214 | CheY-CheZ1-2149 | ||
| 119Lys | Nζ | 206Asp | Oσ1 | − | 3.16 | 2.73 |
| 119Lys | Nζ | 206Asp | Oσ2 | − | 2.78 | 2.91 |
| 119Lys | Nζ | 214Phe | OXT | 2.78f | − | − |
Carbon-carbon distances in the hydrophobic interactions are within 4.0 Å.
Hydrogen bonds and salt bridge contact distances are between 2.6 Å and 3.2 Å.
Values are given for the BeF3−-free F432YZ200-214 structure solved from a crystal grown in Tris (pH 8.4). Contact distances differ marginally in the rest of the F432YZ200-214 models.
It is not clear if the contacts involving 200Ala of CheZ, observed in the P2(1)2(1)2YZ200-214 structure, is physiological or an artifact of acetylating the N-terminus of CheZ200-214 peptide. The backbone NH group of 200Ala should still be available for hydrogen bond interaction if the chain is not truncated at this residue, as in CheZ1-214. However, in contrast to the other contacting residues in both CheY and CheZ, only 200Ala is not conserved (Figure 1). The absence of this contact in the CheY-CheZ1-214 structure is due to the absence of 200Ala from the final CheY-CheZ1-214 model9.
214Phe is not included in the final CheY-CheZ1-214 model due to high disorder9.
The lone electron pair on the C-terminal carboxylate group of CheZ200-214 should exist in resonance with the carbonyl group of the C-terminal residue 214Phe. Hence, in all the F432YZ200-214 structures, the salt bridge between the positively-charged 119Lys sidechain and the negatively-charged C-terminus of the peptide would predominate over the hydrogen bond between the 119Lys sidechain and the backbone carbonyl of 214Phe of the CheZ200-214 peptide.
Comparison of binding modes of the CheZC helices in the CheY-CheZ structures was initiated by structural superposition of the CheY molecules in the CheY complexes. Figure 4 depicts such a structural alignment of the three CheY-CheZC models (F432YZ200-214, P2(1)2(1)2YZ200-214 and CheY-CheZ1-2149). In the CheY-CheZ1-214 structure, the CheY active site contacts the coiled coil region of CheZ and the α4-β5-α5 face binds to the CheZ C-terminal tail, located at the end of a long flexible linker (CheZ169-200) that is disordered and absent from the final model. Thus, the region of the CheY-CheZ1-214 model with the C-terminus of CheZ contacting CheY is analogous to the models of CheY bound to a synthetic peptide corresponding to the 15 C-terminal residues of CheZ, described in this work.
Figure 4.
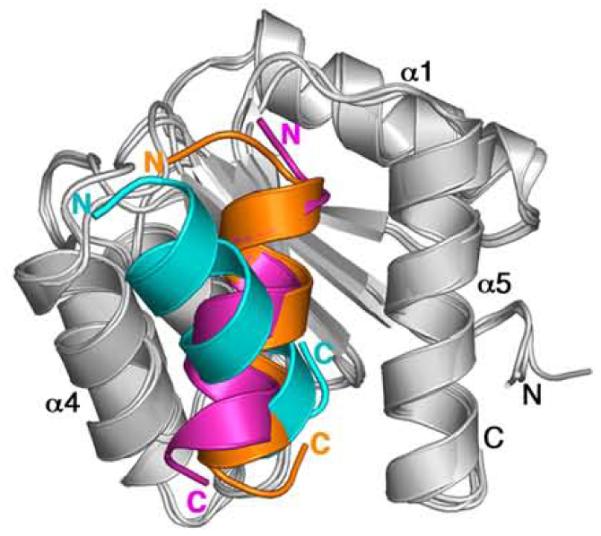
Ribbon diagrams of CheY-CheZC structures upon superposition of CheY showing different orientations of CheZC. CheY molecules in F432YZ200-214, P2(1)2(1)2YZ200-214 and CheY-CheZ1-2149 structures are shown in light gray and the respective CheZC helices are shown in cyan, orange and magenta, respectively. The BeF3−-free F432YZ200-214 structure solved from a crystal grown in Tris (pH 8.4) is used in this figure as a representative of all six F432YZ200-214 structures.
It is apparent from Figure 4 that the orientation of the CheZC helix in CheY-CheZ1-214 is similar to that in P2(1)2(1)2YZ200-214. In both these cases, the interacting CheZC helix is positioned at the α4-β5-α5 face in the P2(1)2(1)2mode and is parallel to the α5 helix of CheY, tilted by ~30° with respect to the axis of the CheZC helix in the F432mode. Comparison of the details of the interacting residues in the three CheY-CheZC models (F432YZ200-214, P2(1)2(1)2YZ200-214 and CheY-CheZ1-214) reveal that the specific hydrophobic interactions, hydrogen bonds and salt bridges observed for the P2(1)2(1)2mode in both the CheY-CheZ1-214 and the P2(1)2(1)2YZ200-214 models are almost identical, but distinct from those observed for the F432mode (Table 2).
Comparison of the crystal structures of CheY bound to CheZ200-214 with previously published crystal structures of CheY bound to the N-terminal peptide of FliM20,21, a component of the flagellar motor and to the P2 domain of the histidine kinase CheA22-24 reveal that the binding interfaces of CheY with its binding partners are overlapping but not identical, a paradigm that was formerly pointed out in mutation25 and NMR26 studies. Additionally, the residues involved in the CheY-CheZ200-214 binding interface, as revealed in the CheY-CheZ200-214 structures, reported here, were also implicated in previous studies.25,26 Two different protein interfaces have been previously observed in crystal structures of CheY bound to the P2 domain of CheA.22 This study provides the first evidence for dual modes of binding of CheY to CheZ.
Comparison of binding interfaces, model quality, experimental conditions and crystal lattice influences
An understanding of the functional significance of dual binding modes of CheY and CheZ, the F432mode and the P2(1)2(1)2mode, necessitates a thorough comparison of all available crystal structures of CheY-CheZC complex (F432YZ200-214, P2(1)2(1)2YZ200-214 and CheY-CheZ1-2149). A detailed analysis of interface characteristics, structural model features, crystallization conditions and influences of the crystal lattice in these structures is presented in Table 3. For simplicity, the BeF3−-free F432YZ200-214 structure solved from a crystal grown in Tris (pH 8.4) will be used as a representative of all six F432YZ200-214 models.
Table 3.
Comparison of interface characteristics, model quality, experimental conditions and crystal lattice characteristics
| Models | F432YZ200-214a | P2(1)2(1)2YZ200-214 | CheY-CheZ1-2149 |
| Binding modes | F432Mode | P2(1)2(1)2Mode | P2(1)2(1)2Mode |
| Interface characteristics | |||
| Buried surface area (Å2) | 789.6 (762 – 821) | 1044 | 677 |
| H-bondsb | 1 | 2 | 1 |
| Salt bridgesb | 1 | 2 | 2 |
| Hydrophobic contactsb | 8 | 8 | 5 |
| Gap volume indexc | 2.14 (1.39 – 2.16) | 1.25 | 2.70 |
| Model quality | |||
| Resolution (Å) | 2.3 (2.0 – 2.3) | 2.0 | 2.9 |
| Rworking | 0.193 (0.193 – 0.220) | 0.186 | 0.279 |
| Rfree | 0.220 (0.207 – 0.246) | 0.232 | 0.298 |
| CheZC B-valuesd (Å2) | 38.8 (29.5 – 46.0) | 17.67 | 164.8 |
| No. of CheZC residuese | 14 (10 – 14) | 15 | 13 |
| Experimental conditions | |||
| CheZ FL/peptidef | peptide | peptide | FL |
| Crystallization conditions | |||
| [Protein] (mM)g | 0.9/ 3.6 | 0.8/ 1.2 | 0.264/ 0.264 |
| Chemical conditions | |||
| [BeCl2] (mM) | 5.0 | 35.0 | 3.6 |
| [NaF] (mM) | 30.0 | 35.0 | 10.0 |
| [MgCl2] (mM) | 7.0 | 7.0 | 10.0 |
| Buffer (0.1 M) | Hepes (pH 7.5)/ Tris (pH 8.4)/ CAPS (pH 10.5) |
MES (pH 6.0) | Bicine (pH 8.5) |
| Precipitant | 2 M ammonium | 0.05 M sodium | 0.2 M ammonium |
| sulfate | phosphate | acetate | |
| (1.8 M – 2.3 M) | (monobasic) | 30% isopropanol | |
| 0.2M lithium sulfate | 37.5% PEG-8000 | ||
| Cryoprotectanth | 20% glycerol/ | 18% PEG-400 2% sucrose |
50% sucrose |
| Temperature | 25°C | 25°C | 4°C |
| Crystal lattice | F432 | P21212 | P43212 |
Values are given for the BeF3−-free F432YZ200-214 structure solved from a crystal at Tris (pH 8.4). Range of values within parenthesis pertains to all six F432YZ200-214 structures.
Hydrogen bonds and salt bridges are enumerated as atom-atom contacts. Hydrophobic contacts are enumerated as residue-residue contacts.
Gap volume index is defined by the volume of gaps between two interacting molecules per Å2 of interface accessible surface area.
Average B-value indicates the averaged B-value for all modeled backbone and side-chain atoms of the CheZC region in a given model.
Number of CheZC residues included in the models is given in each case.
CheZ full-length (FL) protein or peptide is indicated in each case.
Concentrations of CheY/ CheZ200-214 peptide are given for the CheY-CheZ200-214 structures. Concentrations of CheY/ CheZ1-214 protein are given for the CheY-CheZ1-214 structure.
Final concentrations of cryoprotectant solutions are given.
Analyses of buried surface at the CheY-CheZC interfaces revealed that the specific interactions in the F432mode provide ~790 Å2 of buried surface area while those in P2(1)2(1)2mode contribute 1044 Å2 and 680 Å2 at the interfaces in the P2(1)2(1)2YZ200-214 and CheY-CheZ1-214 structures, respectively. The difference in buried surface between P2(1)2(1)2YZ200-214 and CheY-CheZ1-214, in spite of an identical mode of binding, is due to the fact that two residues, 200Ala and 214Phe in the CheZC region are absent from the CheY-CheZ1-214 model due to disorder but are well-ordered in the P2(1)2(1)2YZ200-214 model. The surface of CheY buried by the peptide in the two binding modes is largely overlapping but not identical. This contrast arises from the differences in the specific contacts, as discussed in the previous section and as detailed in Table 2. Together these moderately distinct interactions contribute to comparable buried surface areas.
The solvent-exposed face of the CheZC helix is similar in both binding modes (data not shown). Difference in gap volume index, a measure of complementarity of interacting surfaces27, is only moderate and this parameter does not strongly favor one binding mode over the other. Notably, all the residues in CheY that contact CheZ in either binding mode are strongly conserved among the proteobacters that synthesize CheZ (data not shown). Similarly, in the C-terminus of CheZ, most residues that contact CheY (Table 2) are completely conserved (Figure 1). This conservation supports the biological significance of the protein-protein interactions observed in both modes of binding.
The quality of the models of the CheY-CheZ200-214 and CheY-CheZ1-214 complexes differs substantially. The Fo-Fc difference maps contoured at similar sigma levels reveal complete backbone and discernible side chain density for the CheY-CheZ200-214 peptide models determined at ~2.0 Å from both crystal forms, F432 and P21212, in contrast to limited backbone and a complete absence of side chain density for the CheY-CheZ1-214 model determined at 2.9 Å resolution (data not shown). Both the F432YZ200-214 and P2(1)2(1)2YZ200-214 structures have substantially lower Rfree values than the CheY-CheZ1-214 structure, indicating better agreement of the CheY-CheZ200-214 models to the measured data (Table 3). The average crystallographic B-factors of all the CheZC atoms in the F432YZ200-214 and P2(1)2(1)2YZ200-214 structures are 38.8 Å2 and 17.7 Å2, respectively, while that in the CheY-CheZ1-214 structure is 164.8 Å2. The lower B-factors indicate a more defined spread of electron density, less dynamic mobility and less errors in model building for the structural models of the CheY-CheZ200-214 peptide complex (Table 3). However, while the poor model quality of the CheY-CheZ1-214 model raises questions regarding details of the CheY-CheZC interface in this structure, the Fo-Fc maps suggest no ambiguity in the orientation of the CheZC helix axis relative to the α4-β5-α5 face of CheY.9 This is further strengthened by the fact that contact analyses show that both the orientation and the specific interactions at the CheY-CheZC interface in the CheY-CheZ1-214 structure are similar to those in P2(1)2(1)2YZ200-214, which exhibits better model characteristics.
The comparative analyses presented above indicate that neither of the two modes of binding of the CheZ200-214 peptide to CheY appears to be an obviously better interface. Differences in experimental conditions (Table 3) are likely to have influenced the conformations observed, though none provides a readily discernable explanation for the different modes of binding. Crystals of the CheY-CheZ1-214 complex were generated by co-crystallization of E. coli CheY and FL E. coli CheZ (CheZ1-214) whereas crystals of CheY-CheZ200-214 peptide complex, reported in this work, were generated by co-crystallization of S. enterica CheY and a synthetic peptide corresponding to the 15 C-terminal residues of S. enterica CheZ (CheZ200-214). The C-terminal residues 200 to 214 in E. coli and S. enterica CheZ are identical (Figure 1) and the three minor amino acid differences between E. coli and S. enterica CheY proteins map outside the α4-β5-α5 binding face. Likewise, contacts of CheY with the N-terminal regions of CheZ1-214 occur near the CheY active site and do not appear to impart long-range perturbations to the α4-β5-α5 binding face that interacts with the CheZ C-terminal peptide. In the F432YZ200-214 structures solved from crystals grown in CAPS (pH 10.5), a CAPS buffer molecule interacts with both CheY and CheZ200-214. However, the absence of this molecule in the structures of the complex solved from F432 crystals grown at different buffer and pH conditions suggests that the orientation of the CheZ200-214 peptide in the F432mode is not influenced by the specific crystallization buffer.
Contributions of lattice contacts are more difficult to assess. In the F432 crystal lattice, each asymmetric unit is subject to three symmetry operations, one three-fold and two two-fold operators (Figure 5(a) and Figure 5(b)). The CheZ200-214 peptide in this lattice is part of the 3-fold interface (Figure 5(a)). The CheZ200-214 peptide makes nonspecific van der Waals interactions with parts of the α2 helix of a symmetry-related CheY molecule with contact distances >3.5 Å, but there are no specific hydrogen bonds or salt bridges that appear to constrain the position of the peptide. On the other hand, the P21212 crystal lattice is characterized by three mutually perpendicular two-fold symmetry operators, two of which are associated with 21 translational symmetry functions (Figure 5(c)). The CheZ200-214 peptide in the P21212 lattice is involved in a few specific electrostatic contacts in addition to nonspecific van der Waals interactions with the α1 and α2 helices of a CheY molecule in the neighbouring asymmetric unit.
Figure 5.
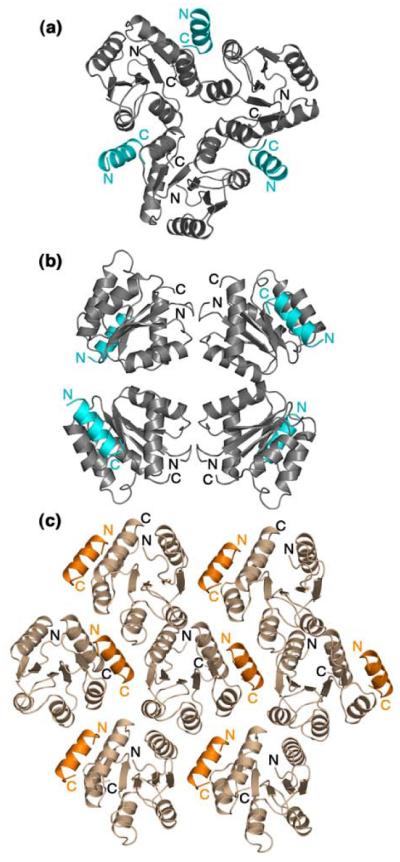
Symmetry-related molecules of CheY-CheZ200-214 in the F432 lattice shown at the 3-fold axis (a) and at the 4-fold axis (b) and in the P21212 crystal lattice (c). CheY molecules in the F432 lattice are shown in gray and those in the P21212 lattice are shown in wheat. The CheZ200-214 peptide molecules in the F432 lattice are shown in cyan and those in the P21212 lattice are shown in orange. The BeF3−-free F432YZ200-214 structure solved from a crystal grown in Tris (pH 8.4) is used as a representative of all six F432YZ200-214 structures in (a) and (b).
Notably, the CheZ200-214 peptide in both the F432 and the P21212 lattices is positioned at interfaces of symmetry elements and contributes to 40% of the total buried surface due to lattice contacts (data not shown). Nonetheless, it is difficult to argue that the binding modes are solely a consequence of crystal packing. Although it cannot be ruled out that the peptide orientation in the F432mode is influenced by the van der Waals interactions imposed by the cubic F432 lattice, analyses of the CheY-CheZ200-214 interfaces in these structures suggest that the contact displays all the characteristics of bonafide biological interfaces. Furthermore, any assumption of bias imposed on the orientation of the peptide in the P21212 lattice is undermined by the fact that the peptide orientation in the P21212 lattice is identical to that in the CheY-CheZ1-214 P43212 lattice, in which the CheZC helix is not part of any crystal contacts.9
Comparison of CheY conformational states
CheY is a switch protein with different conformations dictating distinct signaling states (reviewed in Robinson et al.5 and Cho et al.6). The ability of CheY to signal in bacterial chemotaxis is regulated by its phosphorylation state. Phosphorylation at the active site stabilizes an active conformation, in which the α4-β5-α5 signaling face of CheY, distant from the active site, is altered relative to the inactive conformation. Structural changes promoted by phosphorylation are conserved in most bacterial response regulator receiver domains. Structural studies of N-terminal receiver domains activated by phosphorylation or BeF3− are the basis of the present understanding of these conserved changes and have defined signatures for the active conformation.15,18,28-35
In activated CheY, one of the phosphoryl oxygens (or fluorines of BeF3−) serves as one of the six conserved ligands for the octahedrally coordinated divalent cation at the active site. Based on previous mutational and structural evidence, CheY activation is associated with three major changes: 1) displacement of the protein backbone of the conserved residue 87Thr at the tip of the β4 strand towards the active site, allowing a strong hydrogen bond interaction with the phosphoryl group (or BeF3−), 2) repositioning of the conserved residue 109Lys on the β5-α5 loop, enabling a salt bridge interaction with the phosphoryl group (or BeF3−) and 12Asp, one of the three conserved aspartate residues at the active site, and 3) rotameric conversion of the conserved aromatic switch residue 106Tyr on the β5 strand from a solvent-exposed to a solvent-buried conformation.15,16,18,36-39 According to previous dynamic studies and NMR analysis of inactive CheY-Mg2+, the β4-α4 loop in the inactive protein is flexible and the active-site interactions involving 87Thr and 109Lys, promoted by phosphorylation or binding of the BeF3− ligand, influence the protein backbone in the β4-α4 and the β5-α5 loop regions, respectively, thereby stabilizing the active conformation.15,18,40 Whether these structural changes occur in conjunction with, or as a consequence of, one another is not clear. Although a mechanism of activation in FixJN was suggested based on a molecular dynamics study, further experimental evidence for such a mechanism is lacking.19
The conformational states of CheY in the CheZ200-214 peptide-bound complexes (F432YZ200-214 and P2(1)2(1)2YZ200-214) were compared with previously determined structures of the Mg2+-bound inactive state14 (PDB Accession code – 2CHE) and the BeF3−-activated state15 (PDB Accession code – 1FQW). Least-squares superpositions of the main chain atoms of CheY molecules in the F432YZ200-214 and P2(1)2(1)2YZ200-214 structures with those of Mg2+-bound inactive and BeF3−-activated CheY structures were performed. Side chain conformations of the conserved active-site (Figure 6(a) and Figure 6(c)) and switch residues (Figure 6(b) and Figure 6(d)) are illustrated in superpositions with inactive Mg2+-bound CheY (Figure 6(a) and Figure 6(b)) and with BeF3−-activated CheY (Figure 6(c) and Figure 6(d)).
Figure 6.

Comparison of active-site and switch residues in the CheY-CheZ200-214 complexes with those in inactive and active CheY. CheY molecules in BeF3−-free F432YZ200-214 (key features shown in cyan), BeF3−-bound F432YZ200-214 (key features shown in deep blue) and BeF3−-bound P2(1)2(1)2YZ200-214 (key features shown in orange) are superposed on inactive Mg2+-bound CheY (2CHE)14 (key features shown in red) in (a) and (b) and on BeF3−-activated CheY (1FQW)15 (key features shown in green) in (c) and (d), focusing on the active site in (a) and (c), and on the activation-sensitive α4β4α5 region in (b) and (d). The side chains of key active-site and switch residues are illustrated as ball and stick models. The Mg2+ ions (magenta), coordinating water molecules (deep red) and the BeF3−-complexes (yellow) at the active site in BeF3−-bound F432YZ200-214 are shown in (a) and those in BeF3−-bound P2(1)2(1)2YZ200-214 are shown in (c). The BeF3−-free F432YZ200-214 and the “metaactive” conformer of the BeF3−-bound F432YZ200-214 structures solved from crystals grown in Tris (pH 8.4) are used as representatives of F432YZ200-214 structures in this figure.
Following a least-squares superposition procedure, analysis was performed to fully assess the extent of differences in the tertiary structures of the CheY-CheZ200-214 complexes and the previously determined structures of CheY (see Materials and Methods). reflects the magnitude of a main chain displacement vector that is a measure of individual atomic displacement vectors with summations carried out over a short peptide of three contiguous residues in the protein.41 Concerted structural changes result in constructive addition of these vectors while uncorrelated differences lower the magnitude of these vectors. The magnitudes of displacement vectors versus CheY residue number upon least-squares superposition of backbone atoms of CheY molecules in the CheY-CheZ200-214 peptide structures with those of inactive Mg2+-bound CheY and BeF3−-activated CheY are shown in Figure 7. Using low r.m.s.d. values and small magnitudes of the displacement vectors as indications of structural similarity, both the overall r.m.s.d. and analyses suggest that CheY molecules in the BeF3−-free F432YZ200-214 structures are similar to inactive Mg2+-CheY while the CheY molecule in the BeF3−-bound P2(1)2(1)2YZ200-214 structure is similar to BeF3−-activated CheY. Intriguingly, CheY molecules in the BeF3−-bound F432YZ200-214 structures are similar to inactive Mg2+-CheY, rather than to BeF3−-activated CheY.
Figure 7.
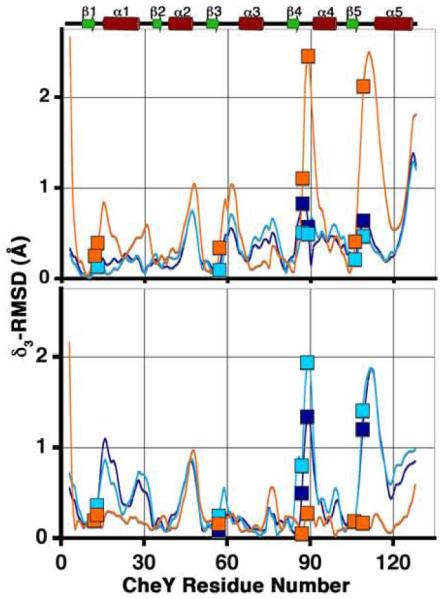
Comparison of the CheY backbone conformations in the CheY-CheZ200-214 complexes with those in inactive and active CheY. plots are shown for main chain atoms of CheY in BeF3−-free F432YZ200-214 (cyan), BeF3−-bound F432YZ200-214 (deep blue) and BeF3−-bound P2(1)2(1)2YZ200-214 (orange) following “sieve-fit” superposition with inactive Mg2+-bound CheY (2CHE)14 (upper panel), resulting in overall r.m.s.d. values of 0.50 Å, 0.48 Å and 0.98 Å, respectively and with BeF3−-activated CheY (1FQW)15 (lower panel), resulting in overall r.m.s.d. values of 0.80 Å, 0.77 Å and 0.35 Å, respectively. The BeF3−-free F432YZ200-214 and the “meta-active” conformer of the BeF3−-bound F432YZ200-214 structures solved from crystals grown in Tris (pH 8.4) were used for and r.m.s.d. calculations, shown here. Key active-site and switch residues are highlighted on the plots by square symbols. The secondary structure of CheY is plotted for reference.
Partial occupancy of BeF3− in BeF3−-bound F432YZ200-214 could likely explain the absence of CheY activation in these structures, as is the case in the BeF3−-bound F432YZ200-214 structure solved from a crystal grown in Hepes (pH 7.5), where the BeF3− occupancy refined to only 65%. However, in the remaining two BeF3−-bound F432YZ200-214 structures, one solved from a crystal grown in CAPS (pH 10.5) and the other solved from a crystal grown in Tris (pH 8.4), the BeF3− species is fully occupied. To resolve this paradox, structural features at the active site and at the activation-sensitive α4-β5-α5 face of CheY were compared in the CheY-CheZ200-214 structures and the structures of the inactive metal-free apoCheY13 (PDB Accession code – 1JBE), inactive Mg2+-bound and BeF3−-activated CheY (Table 4).
Table 4.
Comparison of CheY conformational signatures and pockets on signaling surface
| Models | F432YZC(−)a | F432YZC(+)b | P2(1)2(1)2YZC(+)c | Actived | Inactivee | Apof |
| BeF3−(+/−) | − | + | + | + | − | − |
| M2+/H2O at active siteg | H2O | M2+ | M2+ | M2+ | M2+ | |
| H2O | ||||||
| Active-site features | ||||||
| Coordination tetrahedral |
tetrahedral | octahedral | octahedral | octahedral | octahedral | |
| No. of ligands 4 |
4 | 6 | 6 | 6 | 6 | |
| M2+/H2O contact distancesg (Å) | ||||||
| 57Asp Oσ | 2.6 | 2.0 | 2.0 | 2.2 | 2.1 | |
| 3.0 | ||||||
| 59Asn CO | 2.8 | 2.2 | 2.1 | 2.3 | 2.2 | |
| 2.8 | ||||||
| 13Asp Oσ | 2.6 | 2.0 | 2.2 | 2.3 | 2.1 | |
| 2.8 | ||||||
| H2O # (range)h | 1 (>3.0) | 2 (2.1 – 2.3) | 2 (2.1 – 2.2) | 2 (2.4) | 3 (2.1 – 2.2) | |
| SO4 (2.8) | ||||||
| BeF3− | n/a | 2.2 | 2.0 | 2.1 | n/a | |
| n/a | ||||||
| BeF3− contact distances (Å) | ||||||
| Be – 57Asp Oσ | n/a | 1.6 | 1.7 | 1.5 | n/a | |
| n/a | ||||||
| F− – 59Asn NH | n/a | 2.9 | 2.9 | 3.0 | n/a | |
| n/a | ||||||
| F− – 58Trp NH | n/a | 3.2 | 3.2 | 2.9 | n/a | |
| n/a | ||||||
| F− – 88Ala NH | n/a | 4.2/ 3.2i | 2.9 | 2.9 | n/a | |
| n/a | ||||||
| F− – 109Lys Nζ | n/a | 2.5 | 2.9 | 2.9 | n/a | |
| n/a | ||||||
| 12Asp Oσ – H2O (Å) | 2.9 | 2.7 | 2.6 | 2.6 | 2.2 | |
| 2.8 | ||||||
| 109Lys Nζ – 12Asp Oσ (Å) | 4.5 | 3.1 | 2.8 | 2.8 | 6.2 | |
| 4.5 | ||||||
| Naturej | indirect | direct | direct | direct | none | |
| indirect | ||||||
| 109Lys Nζ – 57Asp Oσ (Å) | 2.8 | 3.3 | 3.3 | 3.3 | 5.0 | |
| 2.7 | ||||||
| 57Asp χ1 (°) | −157 | −177 | −175 | 178 | 165 | |
| −151 | ||||||
| 109Lys χ4 (°) | −177 | −176 | 173 | 172 | 37 | |
| −165 | ||||||
| 87Thr position | ||||||
| 57Asp Oσ – 87Thr Oγ (Å) | 6.9 | 6.9/ 4.2i | 4.2 | 4.1 | 6.5 | 6.0 |
| F− – 87Thr Oγ | n/a | 5.2/ 2.6i | 2.6 | 2.5 | n/a | n/a |
| 106Tyr features | ||||||
| 106Tyr OH | 87Thr Oγ/ | 87Thr Oγ/ | 89Glu NH | 89Glu NH | n/a | 87Thr |
| Oγ/ | ||||||
| 94Asn Nσ | 94Asn Nσ | 94Asn | ||||
| Nσ | ||||||
| Naturej | indirect | indirect | direct | direct | n/a | |
| indirect | ||||||
| 106Tyr conformation | buried | buried | buried | buried | exposed | |
| dual | ||||||
| ϕ (°) | −147 | −152 | −155 | −145 | −136 | |
| −145 | ||||||
| Ψ (°) | −52 | −54 | −60 | −57 | −31 | |
| −52 | ||||||
| χ1 (°) | −178 | −178 | −155 | −164 | 31 | |
| 179/ 72k | ||||||
| χ2 (°) | −94 | 99 | 105 | 110 | 35 | |
| −71 | ||||||
| Volume of α4-β4-α5 surface pockets (Å3) | ||||||
| α4-β5 | − | − | 12.0 | 15.7 | 285.0 | −/ |
| 170.0k | ||||||
| β5-α5 | 55.5 | 61.5 | 119.1 | 96.0 | − | |
| 35.0/ 40.0k | ||||||
Values are given for the BeF3−-free F432YZ200-214 structure solved from a crystal grown in Tris (pH 8.4).
Values are given for the BeF3−-bound F432YZ200-214 structure solved from a crystal grown in Tris (pH 8.4).
Values are given for the BeF3−-bound P2(1)2(1)2YZ200-214 structure.
Values are given for BeF3−-activated CheY15.
Values are given for inactive Mg2+-bound CheY14
Values are given for apoCheY13.
M2+ indicates a divalent cation
Number of water molecules at the active site are indicated and the range of contact distances are given in parentheses.
Values correspond to “A” conformer/ “B” conformer of BeF3−-bound F432YZ200-214 (pH 8.4).
Indirect contacts are water or solvent-mediated hydrogen bonds; direct contacts are direct hydrogen bonds.
Comparison of active sites
Comparison of the active-site coordination geometry revealed that in both the BeF3−-bound F432YZ200-214 and the BeF3−-bound P2(1)2(1)2YZ200-214 structures, the active site of CheY is occupied by an octahedrally coordinated Mg2+ with coordination geometry similar to that in BeF3−-activated CheY (Figure 6(c) and Table 4). In the BeF3−-free F432YZ200-214 structure, the CheY active site is occupied by a tetrahedrally coordinated water molecule in place of the metal ion, similar to the active site of CheY in the inactive metal-free apoCheY structure (Table 4).
Examination of the contact geometries of the conserved residue 109Lys showed that in the BeF3−-bound F432YZ200-214 structures, in which the BeF3− ligand is fully occupied, the side chain of the conserved residue 109Lys on the β5-α5 loop of CheY extends towards the active site and makes ionic interactions with BeF3− and 12Asp. This is similar to the conformation of 109Lys in BeF3−-activated CheY as well as in BeF3−-bound P2(1)2(1)2YZ200-214 (Figure 6(c)). In the BeF3−-free F432YZ200-214 structure, while the 109Lys Cα position is similar to that in BeF3−-bound F432YZ200-214 (Figure 7) and the 109Lys side chain also extends into the active site, 109Lys is involved in a salt bridge interaction with the active-site 57Asp residue, similar to that in inactive metal-free apoCheY (Table 4).
Comparison of 87Thr and β4-α4 loops
Interpretation of differences in the conserved residue 87Thr and the β4-α4 loop is complex. In BeF3−-free F432YZ200-214, the position of 87Thr is similar to that in the inactive Mg2+-bound structure, but the β4-α4 loop is slightly displaced from the inactive position (Figure 6(a), Figure 6(b) and Table 4) while in the BeF3−-bound P2(1)2(1)2YZ200-214 structure, 87Thr and the β4-α4 loop are in the active conformation, as in BeF3−-activated CheY (Figure 6(c), Figure 6(d) and Table 4). The differences in the corresponding plots are consistent with these observations (Figure 7).
In the three different BeF3−-bound F432YZ200-214 structures, 87Thr and the β4-α4 loop are found in different positions. In the structure solved from a crystal grown in Hepes (pH 7.5), in which BeF3− occupancy is partial, 87Thr is in the inactive position, as in inactive Mg2+-CheY, while the β4-α4 loop is slightly displaced, as in BeF3−-free F432YZ200-214. In the BeF3−-bound F432YZ200-214 structure solved from a crystal grown in CAPS (pH 10.5), 87Thr is displaced towards the active site by only 0.3 Å and the β4-α4 loop is positioned intermediate to that in Mg2+-bound inactive and BeF3−-activated CheY.
In the BeF3−-bound F432YZ200-214 structure solved from a crystal grown in Tris (pH 8.4), 87Thr and the β4-α4 loop show two distinct positions, each with 50% occupancy. In one conformation, 87Thr is similar to that in inactive Mg2+-CheY and the β4-α4 loop is slightly displaced, as in the BeF3−-free F432YZ200-214 structures and the BeF3−-bound F432YZ200-214 structure solved from a crystal grown in Hepes (pH 7.5). In the alternate conformation, the position of 87Thr is identical to that in BeF3−-activated CheY but the β4-α4 loop is placed intermediate to that in inactive Mg2+-bound CheY and BeF3−-activated CheY (Figures 6(a), Figure 6(b) and Table 4).
Differences in the positions of 87Thr and the β4-α4 loop observed in the BeF3−-bound F432YZ200-214 structures are not likely to be related to differences in buffer and pH conditions during crystallization but rather to non-specific crystal to crystal variation in characteristics such as size and morphology that might influence diffusion rates of the BeF3− species, resulting in varying degrees of propagative change in 87Thr and the β4-α4 loop.
Comparison of 106Tyr
106Tyr, the key aromatic switch residue in CheY is located on the β5 strand that is part of the signaling surface.16 The rotameric conformation of 106Tyr is highly correlated with the signaling state of the protein. The two rotamers have large differences in solvent accessibility of the bulky aromatic phenol group with the solvent-exposed position of 106Tyr corresponding to the inactive signaling surface and the solvent-buried conformer to the active α4-β5-α5 face. In all seven structures of the CheY-CheZ200-214 complex from the two crystal forms, 106Tyr is in the solvent-buried or active conformation irrespective of the presence of BeF3− and Mg2+ at the active site (Figure 6(b) and Figure 6(d) and Table 4). The solvent-exposed conformation of 106Tyr is not competent to bind to the CheZC helix in either of the two orientations observed because of steric clashes. This has also been noted in the FliMN-bound CheY structures.20,21
Y-T coupling
Previous structural studies of CheY activation established that the solvent-buried conformation of 106Tyr and the movement of 87Thr towards the active site and the consequent repositioning of the β4-α4 loop are obligatorily coupled, a phenomenon termed “Y-T coupling”.16 A molecular dynamics study of the FixJ receiver domain further suggested that during the mechanism of activation, the movement of the β4-α4 loop occurs prior to rotation of the aromatic switch residue (101Phe in FixJ), and the rearranged β4-α4 loop is stabilized by a hydrogen bond between the conserved Thr (82Thr in FixJ) and the phosphoryl oxygen at the active site.19 Based on previous studies supporting “Y-T coupling”, the solvent-buried “active” conformation of 106Tyr is not compatible with the inactive conformation of 87Thr and the β4-α4 loop because of steric hindrance imposed by the short distance (1.5 Å) between the hydroxyl oxygen of the solvent-buried 106Tyr side chain and the C atom of the 87Thr side chain.16
A direct consequence of “Y-T coupling” during CheY activation is the perturbation of the backbone of the β4-α4 loop. This is the case in the BeF3−-bound P2(1)2(1)2YZ200-214 structure as well as the previously determined BeF3−-CheY structure.15 In this conformation, the solvent-buried rotamer of 106Tyr is stabilized by a hydrogen bond between its side chain hydroxyl and the backbone amide NH of the repositioned Glu89 of the β4-α4 loop (Table 4).
However, in all six F432YZ200-214 structures, the solvent-buried conformation of 106Tyr is not associated with the complete repositioning of the β4-α4 loop and 87Thr, as detailed above. In this intermediate state, the side chain hydroxyl of 106Tyr is stabilized by a water-mediated contact with the side chain hydroxyl of 87Thr and the side chain amide NH of 94Asn (Table 4). Two additional structures of CheY have been previously reported in which 106Tyr is buried from solvent but 87Thr and the β4-α4 loop are not repositioned.13,28 In all these cases as well as in the F432YZ200-214 structures, reported here, the solvent-buried conformation of 106Tyr does not impose steric clashes with 87Thr because of moderate shifts in Cα positions (~0.3Å), ~10-20° rotation of the 87Thr C-Cα bond as well as minor changes in 106Tyr torsion angles (Table 4). These subtle changes position the 106Tyr side chain OH group ~3.0 Å away from the side chain Cγ of 87Thr, allowing such a conformation to exist. These observations provide evidence for plasticity and are contrary to a two-state hypothesis in which long-range conformational changes are obligatorily coupled.
Summary of CheY conformations
The comparisons described above allow categorizations of the CheY conformations in the different CheY-CheZ200-214 structures. CheY in BeF3−-bound P2(1)2(1)2YZ200-214 has all the characteristic signatures of a fully activated receiver domain. CheY in the F432YZ200-214 structures is not as easily categorized. In the BeF3−-free F432YZ200-214 structures, 87Thr is in an inactive position, 106Tyr is in an active conformation, albeit with small differences in χ1 from that of active CheY, while the β4-α4 and the β5-α5 loops are slightly different from those in inactive CheY. The analysis shows that the difference between the CheY conformations in the peptide-bound and peptide-free structures in the absence of BeF3− activation, i.e., between F432YZ200-214 and Mg2+-CheY, is greater than the difference in CheY conformations in the peptide-bound and peptide-free structures in the presence of BeF3− activation, i.e., between P2(1)2(1)2YZ200-214 and BeF3−-CheY (Figure 7), suggesting that CheZC peptide binding to inactive CheY is associated with a greater change in CheY conformation than that associated with CheZC binding to fully activated CheY. The greater similarity of conformation signatures of CheY in the F432YZ200-214 complex to those in the active state relative to those in the inactive state provides the basis for designating this CheY conformation as the “meta-active” state. Although not identical, this conformation of CheY closely resembles the “meta-active” state, first introduced by Simonovic and Volz13 (Figure 8).
Figure 8.
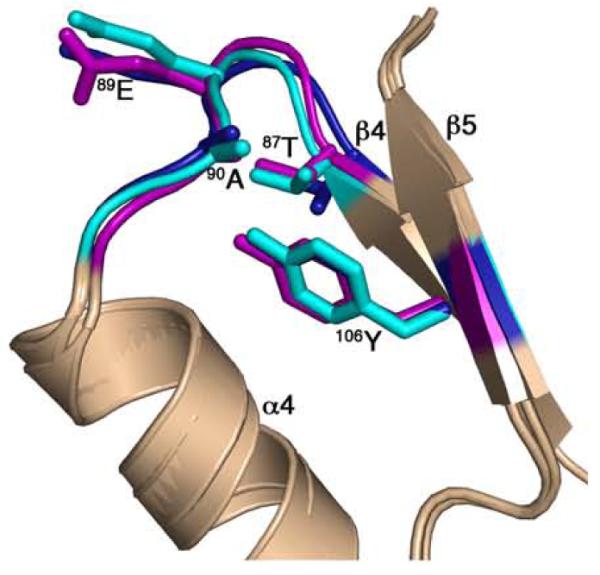
Comparison of the CheY conformation in F432YZ200-214 with “meta-active” apoCheY. The activation-sensitive α4β4α5 region of CheY molecules in BeF3−-free F432YZ200-214 (key features shown in cyan) and BeF3−-bound F432YZ200-214 (key features shown in deep blue) are superposed on “meta-active” apoCheY (1JBE)13 (key features shown in magenta) with side chains of key switch residues depicted in ball and stick models.
In the BeF3−-bound F432YZ200-214 structures, 87Thr and the β4-α4 loop are in multiple positions while 106Tyr is similar to the active conformation, albeit with small differences in χ1 (Table 4). Conformational analysis revealed that CheY molecules in these structures have the signatures of a partially activated structure. The multiple conformations of CheY in the BeF3−-bound F432YZ200-214 structures likely represent intermediates in the pathway from a “meta-active” to a fully active conformation. The lack of complete activation in these structures might result from restrictions imposed by lattice contacts although the regions of the CheY protein that define its conformational state are not directly associated with symmetry interfaces. Modeling the BeF3−-bound P2(1)2(1)2YZ200-214 structure in the F432 cell showed that the P2(1)2(1)2mode cannot be accommodated in the F432 cell because of steric clashes with a symmetry-related CheY molecule (Figure 9). The idea that lattice contacts constrain the conformation of BeF3−-bound CheY is further supported by relatively higher disorder in the CheZ200-214 peptide (Figure 3(c)) as well as a higher degree of variation in the “meta-active” conformation of CheY in the BeF3−-bound F432YZ200-214 structures compared to those in the BeF3−-free F432YZ200-214 structures.
Figure 9.
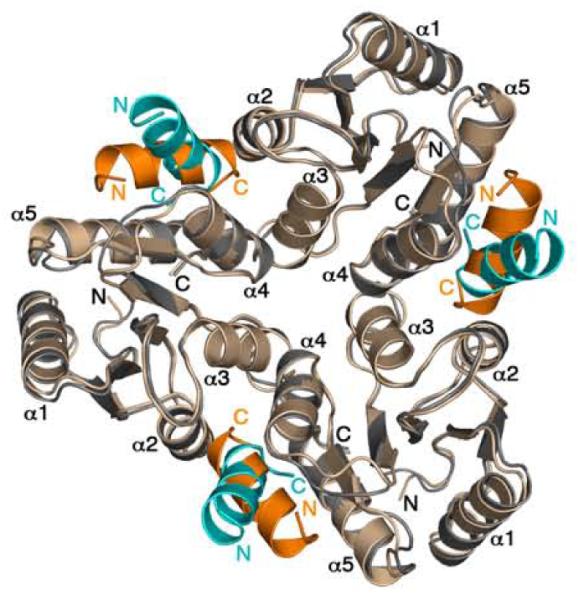
Incompatibility of the P2(1)2(1)2mode of CheZ200-214 peptide binding in the F432 lattice. CheY molecules in P2(1)2(1)2YZ200-214, (CheY shown in wheat and CheZ200-214 shown in orange) are superimposed on the symmetry-related CheY molecules in F432YZ200-214 (CheY shown in gray and CheZ200-214 shown in cyan) in the F432 lattice. The BeF3−-free F432YZ200-214 structure solved from a crystal grown in Tris (pH 8.4) is used as a representative of all F432YZ200-214 structures in this figure.
Binding model
Two inferences can be drawn from analyses of the modes of CheZ200-214 peptide binding and the conformational states of CheY in the structures of the CheY-CheZ200-214 peptide complexes: 1) in the BeF3−-bound P2(1)2(1)2YZ200-214 complex, in which CheY is in the fully activated conformation, the CheZ200-214 peptide binds in the P2(1)2(1)2mode, and 2) in the F432YZ200-214 complexes, in which CheY exists in the “meta-active” state, the CheZ200-214 peptide binds in the F432mode. Based on the correlation between dual CheZC binding modes and the CheY conformational state, we propose that the CheZ200-214 peptide binds to CheY in two different modes that are reflective of the activation state of the CheY protein. The 20-fold lower binding affinity of the CheZC peptide to inactive versus active CheY is likely to be reflective of the different modes of binding.26 Whether a similar difference in Kd’s for binding of the FliMN peptide to active and inactive CheY might correspond to dual modes of binding is unknown as FliMN-bound inactive CheY structures have not been reported.
Superpositions of CheY molecules bound to the CheZC peptide in the two distinct orientations did not reveal any steric clashes that might explain why one conformation binds in one mode and not the other. Two surface pockets on the α4-β5-α5 signaling face of CheY, one between α4 and β5 (α4-β5) and the other between β5 and α5 (β5-α5), were compared (see Materials and Methods). The volume (Table 4), hydrophobicity and electrostatic characteristics (data not shown) of the two pockets differ in different conformations of CheY.
In the fully activated protein, the α4-β5 pocket is smaller in volume and more hydrophobic than the β5-α5 pocket, which has a higher concentration of positively-charged residues, as in the BeF3−-activated CheY and the BeF3−-bound P2(1)2(1)2YZ200-214 structures. In the inactive protein, the β5-α5 pocket is absent while the α4-β5 pocket is ~20-fold greater in volume, because of the solvent-exposed conformation of 106Tyr coupled with the positions of the β4-α4 and β5-α5 loops, as in the CheY-Mg2+ structure. However, when 106Tyr is buried with only small changes in the β4-α4 and β5-α5 loops, as in the “meta-active” state, the α4-β5 pocket disappears while part of the β5-α5 pocket exhibits a hydrophobic characteristic because of slightly altered torsions of the solvent-buried 106Tyr residue, as in “meta-active” apoCheY and CheY in the F432YZ200-214 structures. It is possible that these conformation-dependent differences in surface profiles of the α4-β5-α5 face favor binding of the CheZC helix in different orientations.
Implications of CheZ binding to inactive CheY
CheZ binding to inactive CheY offers interesting scenarios for discussion although its physiological significance is yet to be established. Binding to inactive CheY might allow CheZ to present inactive CheY to CheA for phosphotransfer. Although the binding affinity of inactive CheY to CheA-P2 is two orders of magnitude higher than that for CheZ26,42, the relative intracellular concentrations of chemotaxis proteins might allow for such a situation. A comprehensive quantitative determination of chemotaxis components in bacterial cells, reported by Li and Hazelbauer43, suggests that 70% of unphosphorylated CheY molecules could exist in complex with P2 domains of CheA. It follows that the remaining 30% of unphosphorylated CheY might exist as soluble cytoplasmic protein, potentially available for binding to CheZ. Based on the calculations of Li and Hazelbauer, such a scenario would be possible only within the 44% of receptor signaling complexes that contain both CheAL, proficient in phosphorylating CheY, and CheAS, a shorter form of CheA that cannot transfer phosphoryl groups to CheY but localizes CheZ to the receptor complex.44,45
Localization of the source (kinase CheA) and the sink (phosphatase CheZ) of phosphoryl groups for CheY to the same site in the cell (polar chemoreceptor patches) creates a uniform distribution of P~CheY throughout the cytoplasm, ensuring rapid responses to stimuli.46,47 It can be speculated that binding of unphosphorylated CheY to CheZ at the core signaling complex would result in an increase in the rate or number of futile cycles of phosphorylation and dephosphorylation of CheY, competing with the productive transfer of phosphoryl groups from CheA to CheY and CheB.
In a number of different response regulators, it has been reported that mutations or ligands that stabilize the active conformation increase rates of phosphorylation, suggesting that phosphorylation occurs preferentially in a sub-population that exists in an active state.13,18,30,48-50 A “meta-active” conformation of CheY in the BeF3−-free F432YZ200-214 structures suggests that the CheZ200-214 peptide binds preferentially to the “meta-active” sub-population of inactive CheY. This provides the evidence for the functional nature of the proposed “meta-active” state of the CheY protein. This might provide the structural explanation for the coupling of CheY phosphorylation and peptide binding8 and suggest a functional role for the “meta-active” state of CheY. It was shown that the CheZC and FliMN peptides cause a 10-fold increase and CheA-P2 causes a 6-fold decrease in the rate of phosphoryl transfer to CheY from small molecule phosphodonors.8 The observation that CheY exists in a “meta-active” state in the F432YZ200-214 complex, provides evidence that the increased rate phosphorylation upon peptide binding results from a shift in equilibrium towards a more active conformation.
It was previously shown that 87Thr, 109Lys and 106Tyr, key conserved residues involved in the conformational switch are not important in the coupling of peptide binding to increased rates of phosphorylation.8 Two additional residues near the active site, 14Phe and 59Asn, display completely different rotamer conformations in the CheZ200-214 peptide-bound structures as compared to inactive and active CheY structures not bound to peptide (Figure 10(a) and Figure 10(b)). 59Asn is highly conserved in all species within the proteobacter group and 14Phe is highly conserved in γ-proteobacteriaceae, Enterobacteriaceae and β-proteobacteriaceae families (data not shown). 14Phe has been previously implicated in regulating the solvent accessibility of the active-site pocket in structures of CheY bound to CheA-P2.22,24 It has been reasoned that when CheY is bound to P2, the phenyl group of 14Phe is rotated away from the active site, increasing accessibility of the phospho-histidine. A similar configuration of 14Phe is observed in the CheZC peptide-bound structure but not in the FliMN-bound structure. The change in rotamer conformation of 59Asn is also observed in the FliMN-bound20,21 as well as CheA-P2-bound22-24 structures of CheY proteins.
Figure 10.

Role of key residues in peptide-induced acceleration of CheY phoshorylation. (a) Active site features of CheY in BeF3−-free F432YZ200-214 superimposed on inactive Mg2+-bound CheY. (b) Active site features of CheY in BeF3−-bound P2(1)2(1)2YZ200-214 superimposed on BeF3−-activated CheY. Key features in BeF3−-free F432YZ200-214 are shown in cyan, in BeF3−-bound P2(1)2(1)2YZ200-214 in orange, in inactive Mg2+-bound CheY in red and in BeF3−-activated CheY in green. The Mg2+ ion (magenta), water molecules (deep red) and the BeF3−species (yellow) at the active site are shown. The BeF3−-free F432YZ200-214 structure solved from a crystal grown in Tris (pH 8.4) is used as a representative of all F432YZ200-214 structures in (a). (c) Role of 14Phe and 59Asn. Rates of phosphotransfer to WT CheY, CheY14F→A and CheY59N→A proteins from ammonium phosphoramidate in the presence (+) and absence (−) of the CheZ200-214 peptide are shown as bars (see Materials and Methods). Each rate corresponds to an average from three independent experiments with standard errors indicated.
In order to assess the significance of the different rotameric conformations of 14Phe and 59Asn in the acceleration of CheY phosphorylation upon CheZC binding, phosphorylation kinetics of CheY proteins containing alanine substitutions at 14Phe and 59Asn (CheY14F→A and CheY59N→A) were measured in the presence and absence of the CheZ200-214 peptide by following changes in 58Trp fluorescence (see Materials and Methods). Extending previous observations, we found that changes in charge, bulk and rotameric conformations of key residues have little effect on the enhancement of phosphorylation upon peptide binding (Figure 10(c)). Consistent with earlier results, the presence of the CheZ200-214 peptide increased the phosphorylation rate of the WT protein by 10-fold.8 While both mutant proteins alone have phosphorylation rates 1.5- to 2-fold less than the WT protein, both mutant proteins exhibit 10-fold greater rates in the presence of CheZ200-214 peptide, exactly similar to the WT protein.
Implications of dual binding modes
Structures of the CheY-CheZ200-214 complexes indicate that CheY is a flexible molecule capable of accommodating dual modes of binding to the CheZ200-214 peptide. Previous evidence of such flexibility in the α4-β5-α5 face of CheY was found in crystal structures displaying two distinct modes of binding of this region to the P2 domain of the histidine kinase CheA.22 The physiological significance of each of the CheZ peptide binding modes remains to be determined. However, a distinct mode of binding of inactive CheY to CheZ offers interesting implications.
The consequence of CheY activation is the elevated binding affinity for CheZ and FliM and reduced binding affinity for CheA.26,42 Peptide-induced enhancement of CheY phosphorylation suggests that relay of information in the reverse direction is also possible.8 Reverse flow of signaling information has also been reported between effector and receiver domains of OmpR upon DNA binding.51 The different binding modes observed in the CheY-CheZ200-214 structures may provide different mechanisms of information flow in the forward and reverse directions, allowing bacteria to fine-tune signaling along the two-way pathway of signal transduction to CheY.
Although the CheZ protein is not a canonical phosphatase, analogies can be made with 4-amino-5-hydroxymethyl-2-methylpyrimidine (HMP) phosphate kinase, which catalyzes two independent ATP-dependent phosphotransfer reactions of the substrate, HMP, in which the product of the first reaction, HMP phosphate acts as the substrate for the second reaction.52 The HMP phosphate kinase is able to catalyze the two reactions by allowing two different binding modes for HMP phosphate, corresponding to its role as product or substrate. This mechanism is in some ways analogous to the two binding modes of the CheZC helix observed in the CheY-CheZ200-214 structures where one mode corresponds to binding of the primary substrate of the CheZ protein, the activated form of CheY, and the other mode corresponds to binding of the secondary substrate for CheZ phosphatase for reverse flow of signaling information, the inactive or “meta-active” forms of CheY.
Conclusions
The high-resolution models of the CheZ200-214 peptide bound to inactive and active CheY provide evidence for dual binding modes subject to the conformational state of the CheY protein. Our studies suggest that the CheZ200-214 peptide binds to the “meta-active” sub-population of inactive CheY molecules. This provides a potential structural explanation for the reverse information flow from peptide binding to increased phosphotransfer at the active site. Our studies also provide evidence against the proposed obligatory “Y-T coupling”, and indicate that repositioning of 87Thr and the β4-α4 loop and the rotameric conversion of 106Tyr need not be obligatorily coupled. The dual modes of binding displayed by our CheY-CheZ200-214 complexes extend previous conclusions about the inherent malleability of CheY. Such plasticity allows for different binding modes of CheY partners, which may be relevant to the fine-tuning of signaling responses.
Materials and Methods
Materials
The CheZ200-214 peptide, corresponding to the C-terminal 15 residues of S. enterica serovar Typhimurium CheZ, bearing the sequence ASQDQVDDLLDSLGF with the N-terminus protected by acetylation, was obtained from the peptide synthesis core facility of Massachusetts General Hospital (Boston, MA) as lyophilized powder. Oligonucleotides for site-directed mutagenesis, bearing sequences 5′-TTT TTG GTT GTG GAT GAC GCT TCG ACC ATG CGT CGT ATC-3′ and 5′-GAT ACG ACG CAT GGT CGA AGC GTC ATC CAC AAC CAA AAA-3′ for CheY14Phe→Ala mutagenesis and 5′-ATT ATC TCC GAC TGG GCC ATG CCG AAC CTG-3′ and 5′-CAT GTT CGG CAT GGC CCA GTC GGA GAT AAT-3′ for CheY59Asn→Ala mutagenesis, were obtained from the DNA core facility of the University of Medicine and Dentistry of New Jersey (Piscataway, NJ). The ammonium salt of phosphoramidate was synthesized by the method of Sheridan et al.53
Proteins
The S. enterica serovar Typhimurium WT cheY gene was previously cloned within a 0.8 kilobase SnaI-SmaI fragment in a pUC12-based CheY expression vector (pME124).54,55 Plasmids, pEF41 and pJG1, carrying the 14Phe→Ala and 59Asn→Ala mutations, respectively, were constructed using the plasmid pME124 as template and respective oligonucleotides using the site-directed mutagenesis kit (Stratagene).
Protein expression and purification
The pME124 plasmids bearing WT and mutant cheY genes were transformed into HB101 cells.14,56 The WT and mutant CheY proteins were purified by a modification of previously described procedures.57 The previously used ion-exchange and gel filtration columns were substituted with a HiTrap Q Fast Flow column (GE Healthcare) and a Superdex 26/60 column (GE Healthcare) in fast performance liquid chromatography using an AKTA system (GE Healthcare). The purified protein was quantitated by measuring UV absorbance at 280 nm, using extinction coefficients calculated from amino acid composition.58 The ε280 value in ml mg−1 cm−1 280 calculated for WT CheY is 0.493, for CheY14F→A is 0.496 and for CheY59N→A is 0.495.
Crystallization
Purified WT CheY protein was dialyzed into a buffer containing 50 mM Hepes and 7 mM MgCl2 (pH 7.0) (buffer A) and concentrated to 25 mg/ml (1.8 mM) using a Centriprep-10 (Millipore). A 20 mM stock solution of CheZ200-214 peptide was prepared in buffer A. Both purified protein and dissolved peptide were filtered through 0.22 μm-poresize cellulose acetate filters (Corning Costar).
For crystallization of the BeF3−-free F432YZ200-214 complex, WT CheY in buffer A was mixed with the CheZ200-214 peptide to a final concentration of 0.9 mM CheY and 3.6 mM CheZ200-214. Crystals were grown by the hanging drop vapor diffusion method at room temperature by mixing equal volumes of the protein/peptide mixture and reservoir solutions. The BeF3−-free F432YZ200-214 crystals grew to ~100 μm × 100 μm × 50 μm in ~3 days in the presence of reservoir solutions containing 1.8 M to 2.3 M ammonium sulfate and 0.2 M lithium sulfate (precipitant A) in three different buffer and pH conditions: 0.1 M Hepes (pH 7.5) (buffer B), 0.1 M Tris (pH 8.4) (buffer C), and 0.1 M CAPS (pH 10.5) (buffer D). A few BeF3−-free F432YZ200-214 crystals from each buffer and pH condition were incubated in gradually increasing concentrations of BeCl2 and NaF in precipitant A to a final concentration of 5 mM BeCl2 and 30 mM NaF, at least 4-5 hours prior to data collection to obtain BeF3−-bound F432YZ200-214 crystals. Cryoprotection was achieved by serially transferring the F432YZ200-214 crystals into their respective reservoir solutions, supplemented with glycerol at a final concentration of 20% (v/v) at 5% steps, soaking for 1 min at every step. Crystals were placed in a 100 K nitrogen cryostream for data collection.
For crystallization of the BeF3−-bound P2(1)2(1)2YZ200-214 complex, WT CheY in buffer C was activated with 5 mM BeCl2 and 30 mM NaF, prior to mixing with the CheZ200-214 peptide, dissolved in buffer C to a final concentration of 0.8 mM CheY and 1.2 mM CheZ200-214. These crystals were also grown by the hanging drop vapor diffusion method at room temperature by mixing equal volumes of the protein/peptide mixture and the reservoir solution, containing 0.05 M sodium phosphate (monobasic) and 37.5% PEG-8000 (precipitant B) in 0.1 M MES (pH 6.0) (buffer E). These crystals at first grew as fuzzy needles in the absence of BeCl2 and NaF at the high throughput crystallization lab at the Hauptman Woodward Medical Research Institute, Buffalo, NY and were subsequently optimized to grow in the presence of BeCl2 and NaF to ~100 μm × 10 μm × 10 μm by repeated microseeding procedures. For crypotection, the BeF3−-bound P2(1)2(1)2YZ200-214 crystals were serially transferred into reservoir solution, containing PEG-400 and sucrose to a final concentration of 18% (w/v) PEG-400 at 4.5% steps and 2% (w/v) sucrose at 0.5% steps, soaked for 1 min at each step and frozen in a 100 K nitrogen cryostream for data collection.
Data collection and processing
Data were collected at beamline X4A at the National Synchrotron Light Source at Brookhaven National Laboratory, Upton, NY. For the F432YZ200-214 crystals, 100° of data with 0.5° oscillations and for the P2(1)2(1)2YZ200-214 crystals, 200° of data with 1° oscillations were collected, processed with DENZO and SCALEPACK.59 Data collection and processing statistics are listed in Table 1.
Structure determination and refinement
All seven structures were solved using the molecular replacement program, Phaser.17 Following rigid-body refinement using Refmac 5.1.2460, as part of the CCP4 suite of programs61, interative cycles of maximum likelihood and isotropic temperature factor and model building using O62 were performed, in which the data were extended gradually from 2.4 Å to the highest resolution shell until convergence. Water molecules were initially modeled using the ARP-WARP routine63 and subsequently only waters and other small molecules with Fourier difference peaks greater than 3σ were included in the final models. Refinement statistics of the seven final models are listed in Table 1.
Quality of structural models
In Ramachandran plots, generated by the PROCHECK program64, 91.3% to 94.4% of the residues occupy the most favored regions. Similar to all previously solved CheY structures, only 62Asn is disallowed because of its location within a γ-turn.65 The average B-factors for all atoms of the structures range between 10.9 Å2 and 27.4 Å2.
Structural analyses
Superpositions of atomic models were accomplished by a previously described least-squares based iterative superposition procedure41 that is similar to the “sievefit” program66 to define a “static core” of main chain atoms for CheY molecules, achieved by omitting residues with atomic displacements greater than twice the r.m.s.d. value. “Sievefit” superpositions of CheY coordinates in the F432YZ200-214 structures with those in apoCheY13, Mg2+-bound CheY14 and BeF3−-activated CheY15 structures converged within 9 to 12, 3 to 6 and 20 to 35 cycles, respectively, yielding r.m.s. residuals of 0.33 to 0.36, 0.31 to 0.37 and 0.37 to 0.39 Å, respectively, defining a static core of 392 to 412, 456 to 476 and 364 to 400 main chain atoms, respectively. “Sievefit” superpositions of CheY coordinates in the P2(1)2(1)2YZ200-214 structures with those in Mg2+-bound CheY and BeF3−-activated CheY structures converged within 25 and 6 cycles, yielding r.m.s. residuals of 0.29 and 0.26 Å, and defining a static core of 364 and 464 main chain atoms, respectively. Overall r.m.s.d. values were obtained using the r.m.s.d. calculation algorithm within CNS 1.1.67
To estimate the extent of difference in tertiary structure in the superimposed CheY molecules, the magnitude of the main chain displacement vector, was plotted versus residue number as in Kavanaugh et al.41 is defined as,
where summation is over n contiguous residues and denotes the atomic displacement vector corresponding to the jth main chain atom of the n-residue peptide. Summations were carried out over 3 contiguous residues.
CNS 1.167, iMOLTALK ver. 2.068 and the protein-protein interaction server (http://www.biochem.ucl.ac.uk/bsm/PP/server/) were used for the peptide-CheY contact analyses and the WHATIF69 web interface (http://swift.cmbi.kun.nl/WIWWWI/) was used for the lattice contact analyses. Buried surface areas were calculated using CNS 1.167 and cavity/pocket searches and analyses were carried out using the CASTp v 1.170 server. Graphical representations were generated using Microsoft Excel 2004 version 11.1.1 (Microsoft Corporation). All structural figures were generated using Pymol.71
CheY phosphorylation kinetics
To investigate the role of the residues, 14Phe and 59Asn on peptide-induced acceleration of phosphorylation, time courses of pre steady-state phosphotransfer from the small molecule phosphodonor ammonium phosphoramidate to WT CheY versus mutant CheY proteins (CheY14F→A and CheY59N→A) were recorded by following the quenching of intrinsic fluorescence of 58Trp as a probe of phosphorylation. Fluoresence measurements were carried out using an SLM-Aminco Series 2 luminescence spectrometer with a stopped-flow attachment, with an excitation wavelength of 295 nm and emission wavelength of 345 nm and 4 nm band passes for both. The dead time on the stopped-flow attachment was 4 msec. The pre steady-state CheY phosphorylation data were collected and analyzed as previously detailed8 using SIGMAPLOT 8.0 (Systat Software, Inc.).
Protein data bank accession numbers
The coordinates and structure factors have been deposited in the RCSB Protein Data Bank. The PDB ID codes for the BeF3−-free F432YZ200-214 structures solved from crystals grown in CAPS (pH 10.5), Tris (pH 8.4) and Hepes (pH 7.5) are 2FLK, 2FMI and 2FMF, respectively. The corresponding PDB ID codes for the BeF3−-bound F432YZ200-214 structures solved from crystals grown in CAPS (pH 10.5), Tris (pH 8.4) and Hepes (pH 7.5) are 2FKA, 2FMH and 2FLW, respectively. The PDB ID code for the BeF3−-bound P2(1)2(1)2YZ200-214 structure is 2FMK.
Acknowledgments
We thank E. Fox and T. Wu for assistance with construction of expression vectors and protein purification, the beamline staff at X4A at the National Synchrotron Light Source at Brookhaven National Laboratory for technical assistance, Dr. L. Falzon for technical help in using the luminescence spectrometer and Dr. J. Kavanaugh for the sievefit and delta r.m.s.d. fortran codes. This work was supported by NIH grant 2R37GM47958. A.M.S. is an investigator of the Howard Hughes Medical Institute.
Abbreviations used
- P~CheY
phosphorylated CheY
- CheZC
C-terminal region of CheZ
- FL
full-length
- WT
wild-type
- F432YZ200-214
the CheY-CheZ200-214 complex in the F432 crystal form
- P2(1)2(1)2YZ200-214
the CheY-CheZ200-214 complex in the P21212 crystal form
- r.m.s.d.
root mean square deviation
- F432mode
binding mode observed in the F432 crystal form
- P2(1)2(1)2mode
binding mode observed in the P21212 crystal form
- F432Z200-214
the CheZ200-214 helix in the F432 crystal form
- P2(1)2(1)2Z200-214
the CheZ200-214 helix in the P21212 crystal form
- HMP
4-amino-5-hydroxymethyl-2-methylpyrimidine
References
- 1.Bren A, Eisenbach M. How signals are heard during bacterial chemotaxis: protein-protein interactions in sensory signal propagation. J. Bacteriol. 2000;182:6865–6873. doi: 10.1128/jb.182.24.6865-6873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bourret RB, Stock AM. Molecular information processing: lessons from bacterial chemotaxis. J. Biol. Chem. 2002;277:9625–9628. doi: 10.1074/jbc.R100066200. [DOI] [PubMed] [Google Scholar]
- 3.Wadhams GH, Armitage JP. Making sense of it all: bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 2004;5:1024–1037. doi: 10.1038/nrm1524. [DOI] [PubMed] [Google Scholar]
- 4.Silversmith RE, Bourret RB. Throwing the switch in bacterial chemotaxis. Trends Microbiol. 1999;7:16–22. doi: 10.1016/s0966-842x(98)01409-7. [DOI] [PubMed] [Google Scholar]
- 5.Robinson VL, Buckler DR, Stock AM. A tale of two components: a novel kinase and a regulatory switch. Nat. Struct. Biol. 2000;7:628–633. doi: 10.1038/77915. [DOI] [PubMed] [Google Scholar]
- 6.Cho HS, Pelton JG, Yan D, Kustu S, Wemmer DE. Phosphoaspartates in bacterial signal transduction. Curr. Opin. Struct. Biol. 2001;11:679–684. doi: 10.1016/s0959-440x(01)00271-8. [DOI] [PubMed] [Google Scholar]
- 7.Blat Y, Eisenbach M. Conserved C-terminus of the phosphatase CheZ is a binding domain for the chemotactic response regulator CheY. Biochemistry. 1996;35:5679–5683. doi: 10.1021/bi9530447. [DOI] [PubMed] [Google Scholar]
- 8.Schuster M, Silversmith RE, Bourret RB. Conformational coupling in the chemotaxis response regulator CheY. Proc. Natl. Acad. Sci. USA. 2001;98:6003–6008. doi: 10.1073/pnas.101571298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao R, Collins EJ, Bourret RB, Silversmith RE. Structure and catalytic mechanism of the E. coli chemotaxis phosphatase CheZ. Nat. Struct. Biol. 2002;9:570–575. doi: 10.1038/nsb816. [DOI] [PubMed] [Google Scholar]
- 10.Yan D, Cho HS, Hastings CA, Igo MM, Lee SY, Pelton JG, Stewart V, Wemmer DE, Kustu S. Beryllofluoride mimics phosphorylation of NtrC and other bacterial response regulators. Proc. Natl. Acad. Sci. USA. 1999;96:14789–14794. doi: 10.1073/pnas.96.26.14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho H, Wang W, Kim R, Yokota H, Damo S, Kim S-H, Wemmer DE, Kustu S, Yan D. BeF3− acts as a phosphate analog in proteins phosphorylated on aspartate: structure of a BeF3− complex with phosphoserine phosphatase. Proc. Natl. Acad. Sci. USA. 2001;98:8525–8530. doi: 10.1073/pnas.131213698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chabre M. Aluminofluoride and beryllofluoride complexes: a new phosphate analogues in enzymology. Trends Biochem. Sci. 1990;15:6–10. doi: 10.1016/0968-0004(90)90117-t. [DOI] [PubMed] [Google Scholar]
- 13.Simonovic M, Volz K. A distinct meta-active conformation in the 1.1-Å resolution structure of wild-type apoCheY. J. Biol. Chem. 2001;276:28637–28640. doi: 10.1074/jbc.C100295200. [DOI] [PubMed] [Google Scholar]
- 14.Stock AM, Martinez-Hackert E, Rasmussen BF, West AH, Stock JB, Ringe D, Petsko GA. Structure of the Mg2+-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry. 1993;32:13375–13380. doi: 10.1021/bi00212a001. [DOI] [PubMed] [Google Scholar]
- 15.Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, Wemmer DE. Crystal structure of activated CheY. J. Biol. Chem. 2001;276:16425–16431. doi: 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 16.Zhu X, Rebello J, Matsumura P, Volz K. Crystal structures of CheY mutants Y106W and T87I/Y106W: CheY activation correlates with movement of residue 106. J. Biol. Chem. 1997;272:5000–5006. doi: 10.1074/jbc.272.8.5000. [DOI] [PubMed] [Google Scholar]
- 17.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 18.Cho HS, Lee SY, Yan D, Pan X, Parkinson JS, Kustu S, Wemmer DE, Pelton JG. NMR structure of activated CheY. J. Mol. Biol. 2000;297:543–551. doi: 10.1006/jmbi.2000.3595. [DOI] [PubMed] [Google Scholar]
- 19.Roche P, Mouawad L, Perahia D, Samama JP, Kahn D. Molecular dynamics of the FixJ receiver domain: movement of the β4-α4 loop correlates with the in and out flip of Phe101. Protein Sci. 2002;11:2622–2630. doi: 10.1110/ps.0218802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S-Y, Cho HS, Pelton JG, Yan D, Henderson RK, King DS, Huang L-S, Kustu S, Berry EA, Wemmer DE. Crystal structure of an activated response regulator bound to its target. Nat. Struct. Biol. 2001;8:52–56. doi: 10.1038/83053. [DOI] [PubMed] [Google Scholar]
- 21.Dyer CM, Quillin ML, Campos A, Lu J, McEvoy MM, Hausrath AC, Westbrook EM, Matsumura P, Matthews BW, Dahlquist FW. Structure of the constitutively active double mutant CheYD13K Y106W alone and in complex with a FliM peptide. J. Mol. Biol. 2004;342:1325–1335. doi: 10.1016/j.jmb.2004.07.084. [DOI] [PubMed] [Google Scholar]
- 22.McEvoy MM, Hausrath AC, Randolph GB, Remington SJ, Dahlquist FW. Two binding modes reveal flexibility in kinase/response regulator interactions in the bacterial chemotaxis pathway. Proc. Natl. Acad. Sci. USA. 1998;95:7333–7338. doi: 10.1073/pnas.95.13.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welch M, Chinardet N, Mourey L, Birck C, Samama J-P. Structure of the CheY-binding domain of histidine kinase CheA in complex with CheY. Nat. Struct. Biol. 1998;5:25–29. doi: 10.1038/nsb0198-25. [DOI] [PubMed] [Google Scholar]
- 24.Gouet P, Chinardet N, Welch M, Guillet V, Cabantous S, Birck C, Mourey L, Samama JP. Further insights into the mechanism of function of the response regulator CheY from crystallographic studies of the CheY-CheA124-257 complex. Acta Crystallogr. D. 2001;57:44–51. doi: 10.1107/s090744490001492x. [DOI] [PubMed] [Google Scholar]
- 25.Zhu X, Volz K, Matsumura P. The CheZ-binding surface of CheY overlaps the CheA- and FliM-binding surfaces. J. Biol. Chem. 1997;272:23758–23764. doi: 10.1074/jbc.272.38.23758. [DOI] [PubMed] [Google Scholar]
- 26.McEvoy MM, Bren A, Eisenbach M, Dahlquist FW. Identification of the binding interfaces on CheY for two of its targets, the phosphatase CheZ and the flagellar switch protein FliM. J. Mol. Biol. 1999;289:1423–1433. doi: 10.1006/jmbi.1999.2830. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski RA. SURFNET: a program for visualizing molecular surfaces, cavities, and intermolecular interactions. J. Mol. Graph. 1995;13:323–330. doi: 10.1016/0263-7855(95)00073-9. [DOI] [PubMed] [Google Scholar]
- 28.Halkides CJ, McEvoy MM, Casper E, Matsumura P, Volz K, Dahlquist FW. The 1.9 Å resolution crystal structure of phosphono-CheY, an analogue of the active form of the response regulator, CheY. Biochemistry. 2000;39:5280–5286. doi: 10.1021/bi9925524. [DOI] [PubMed] [Google Scholar]
- 29.Birck C, Mourey L, Gouet P, Fabry B, Schumacher J, Rousseau P, Kahn D, Samama J-P. Conformational changes induced by phosphorylation of the FixJ receiver domain. Structure Fold. Des. 1999;7:1505–1515. doi: 10.1016/s0969-2126(00)88341-0. [DOI] [PubMed] [Google Scholar]
- 30.Kern D, Volkman BF, Luginbuhl P, Nohaile MJ, Kustu S, Wemmer DE. Structure of a transiently phosphorylated switch in bacterial signal transduction. Nature. 1999;40:894–898. doi: 10.1038/47273. [DOI] [PubMed] [Google Scholar]
- 31.Lewis RJ, Brannigan JA, Muchová K, Barák I, Wilkinson AJ. Phosphorylated aspartate in the structure of a response regulator protein. J. Mol. Biol. 1999;294:9–15. doi: 10.1006/jmbi.1999.3261. [DOI] [PubMed] [Google Scholar]
- 32.Bachhawat P, Swapna GV, Montelione GT, Stock AM. Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure. 2005;13:1353–1363. doi: 10.1016/j.str.2005.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toro-Roman A, Mack TR, Stock AM. Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: a symmetric dimer mediated by the α4-β5-α5 face. J. Mol. Biol. 2005;349:11–26. doi: 10.1016/j.jmb.2005.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nixon BT, Yennawar HP, Doucleff M, Pelton JG, Wemmer DE, Krueger S, Kondrashkina E. SAS solution structures of the apo and Mg2+/BeF3−-bound receiver domain of DctD from Sinorhizobium meliloti. Biochemistry. 2005;44:13962–13969. doi: 10.1021/bi051129u. [DOI] [PubMed] [Google Scholar]
- 35.Toro-Roman A, Wu T, Stock AM. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci. 2005;14:3077–3088. doi: 10.1110/ps.051722805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lowry DF, Roth AF, Rupert PB, Dahlquist FW, Moy FJ, Domaille PJ, Matsumura P. Signal transduction in chemotaxis: a propagating conformation change upon phosphorylation of CheY. J. Biol. Chem. 1994;269:26358–26362. [PubMed] [Google Scholar]
- 37.Ganguli S, Wang H, Matsumura P, Volz K. Uncoupled phosphorylation and activation in bacterial chemotaxis: the 2.1-Å structure of a threonine to isoleucine mutant at position 87 of CheY. J. Biol. Chem. 1995;270:1–8. [PubMed] [Google Scholar]
- 38.Bellsolell L, Cronet P, Majolero M, Serrano L, Coll M. The three-dimensional structure of two mutants of the signal transduction protein CheY suggest its molecular activation mechanism. J. Mol. Biol. 1996;257:116–128. doi: 10.1006/jmbi.1996.0151. [DOI] [PubMed] [Google Scholar]
- 39.Appleby JL, Bourret RB. Proposed signal transduction role for conserved CheY residue Thr87, a member of the response regulator active-site quintet. J. Bacteriol. 1998;180:3563–3569. doi: 10.1128/jb.180.14.3563-3569.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moy FJ, Lowry DF, Matsumura P, Dahlquist FW, Krywko JE, Domaille PJ. Assignments, secondary structure, global fold, and dynamics of chemotaxis Y protein using three- and four-dimensional heteronuclear (13C, 15N) NMR spectroscopy. Biochemistry. 1994;33:10731–10742. doi: 10.1021/bi00201a022. [DOI] [PubMed] [Google Scholar]
- 41.Kavanaugh JS, Weydert JA, Rogers PH, Arnone A. High-resolution crystal structures of human hemoglobin with mutations at tryptophan 37β: structural basis for a high-affinity T-state. Biochemistry. 1998;37:4358–4373. doi: 10.1021/bi9708702. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Swanson RV, Simon MI, Weis RM. The response regulators CheB and CheY exhibit competitive binding to the kinase CheA. Biochemistry. 1995;34:14626–14636. doi: 10.1021/bi00045a003. [DOI] [PubMed] [Google Scholar]
- 43.Li M, Hazelbauer GL. Cellular stoichiometry of the components of the chemotaxis signaling complex. J. Bacteriol. 2004;186:3687–3694. doi: 10.1128/JB.186.12.3687-3694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Matsumura P. Characterization of the CheAs/CheZ complex: a specific interaction resulting in enhanced dephosphorylating activity on CheY-phosphate. Mol. Microbiol. 1996;19:695–703. doi: 10.1046/j.1365-2958.1996.393934.x. [DOI] [PubMed] [Google Scholar]
- 45.Cantwell BJ, Draheim RR, Weart RB, Nguyen C, Stewart RC, Manson MD. CheZ phosphatase localizes to chemoreceptor patches via CheA-short. J. Bacteriol. 2003;185:2354–2361. doi: 10.1128/JB.185.7.2354-2361.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaknin A, Berg HC. Single-cell FRET imaging of phosphatase activity in the Escherichia coli chemotaxis system. Proc. Natl. Acad. Sci. USA. 2004;101:17072–17077. doi: 10.1073/pnas.0407812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lipkow K, Andrews SS, Bray D. Simulated diffusion of phosphorylated CheY through the cytoplasm of Escherichia coli. J. Bacteriol. 2005;187:45–53. doi: 10.1128/JB.187.1.45-53.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang M, Bourret RB, Simon MI, Volz K. Uncoupled phosphorylation and activation in bacterial chemotaxis: The 2.3 Å structure of an aspartate to lysine mutant at position 13 of CheY. J. Biol. Chem. 1997;272:11850–11855. doi: 10.1074/jbc.272.18.11850. [DOI] [PubMed] [Google Scholar]
- 49.Feher VA, Cavanagh J. Millisecond-timescale motions contribute to the function of the bacterial response regulator protein Spo0F. Nature. 1999;400:289–293. doi: 10.1038/22357. [DOI] [PubMed] [Google Scholar]
- 50.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 51.Ames SK, Frankema N, Kenney LJ. C-terminal DNA binding stimulates N-terminal phosphorylation of the outer membrane protein regulator OmpR from Escherichia coli. Proc. Natl. Acad. Sci. USA. 1999;96:11792–11797. doi: 10.1073/pnas.96.21.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng G, Bennett EM, Begley TP, Ealick SE. Crystal structure of 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate kinase from Salmonella typhimurium at 2.3 A resolution. Structure. 2002;10:225–235. doi: 10.1016/s0969-2126(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 53.Sheridan RC, McCullough JF, Wakefield ZT. Phosphoramidic acid and its salts. Inorg. Synth. 1971;13:23–26. [Google Scholar]
- 54.Messing J. New M13 vectors for cloning. Methods Enzymol. 1983;101:20–78. doi: 10.1016/0076-6879(83)01005-8. [DOI] [PubMed] [Google Scholar]
- 55.Stock AM, Mottonen JM, Stock JB, Schutt CE. Three-dimensional structure of CheY, the response regulator of bacterial chemotaxis. Nature. 1989;337:745–749. doi: 10.1038/337745a0. [DOI] [PubMed] [Google Scholar]
- 56.Bolivar F, Backman K. Plasmids of Escherichia coli as cloning vectors. Methods Enzymol. 1979;68:245–267. doi: 10.1016/0076-6879(79)68018-7. [DOI] [PubMed] [Google Scholar]
- 57.Stock A, Koshland DE, Jr., Stock J. Homologies between the Salmonella typhimurium CheY protein and proteins involved in the regulation of chemotaxis, membrane protein synthesis, and sporulation. Proc. Natl. Acad. Sci. USA. 1985;82:7989–7993. doi: 10.1073/pnas.82.23.7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 59.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 60.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 61.Collaborative Computational Project, N The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 62.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 63.Lamzin VS, Wilson KS. Automated refinement of protein models. Acta Crystallogr. D. 1993;49:129–147. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- 64.Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:282–291. [Google Scholar]
- 65.Volz K, Matsumura P. Crystal structure of Escherichia coli CheY refined at 1.7 Å resolution. J. Biol. Chem. 1991;266:15511–15519. doi: 10.2210/pdb3chy/pdb. [DOI] [PubMed] [Google Scholar]
- 66.Gerstein M, Chothia C. Analysis of protein loop closure: two types of hinges produce one motion in lactate dehydrogenase. J. Mol. Biol. 1991;220:133–149. doi: 10.1016/0022-2836(91)90387-l. [DOI] [PubMed] [Google Scholar]
- 67.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 68.Diemand AV, Scheib H. iMolTalk: an interactive, internet-based protein structure analysis server. Nucl. Acids Res. 2004;32:W512–W516. doi: 10.1093/nar/gkh403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56. 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 70.Binkowski TA, Naghibzadeh S, Liang J. CASTp: Computed Atlas of Surface Topography of proteins. Nucl. Acids Res. 2003;31:3352–3355. doi: 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Delano WL. The Pymol Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 72.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl. Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]