

Proteins play a fundamental role in establishing the diversity of cellular processes in health or disease systems. This diversity is accomplished by a vast array of protein functions. In fact, a protein rarely has a single function. The majority of proteins are involved in numerous cellular processes, and these multiple functions are made possible by interactions with other molecules. The complexity of interactions is substantially increased by the spatial and temporal diversity of proteins. For example, proteins can be part of distinct complexes within different subcellular compartments or at different stages of the cell cycle. Posttranslational modifications can regulate and further expand the ability of proteins to establish localization- or temporal-dependent interactions. This complexity and functional divergence of interactions is further increased by the simultaneous presence of stable, transient, direct, and indirect protein interactions. Thus, an understanding of protein functions cannot be fully accomplished without knowledge of its interactions. Characterizing these interactions is therefore critical to understanding the biology of health and disease systems.
Methodologies for studying protein interactions have become a core component of the proteomics field, which aims to define protein abundances, modifications, interactions, and functions. The study of protein interactions is not without difficulty. Protein interactions are inherently dynamic, and complex mixtures of stable and transient interactions routinely co-exist. Immunoaffinity purification (IP) approaches coupled to mass spectrometry (MS) can be used to study a range of these functionally relevant protein associations. Recent years have seen a significant improvement in these affinity-based methods. These improvements include advances in affinity tools, methods for sample preparation, mass spectrometry configurations offering increased sensitivity and accuracy, and bioinformatics approaches allowing a thorough analysis of large-scale or targeted interactome datasets. Therefore, affinity-based methods for identifying protein interactions have grown to encompass a wide variety of techniques with the ability to study diverse biological systems.
This review summarizes recent developments in IP-MS strategies for studying protein-protein interactions. Important considerations and optimization techniques for IP workflows are introduced, providing practical suggestions, as well as concrete examples of studies and applications. One of the greatest challenges in protein interaction studies is recognizing from the multitude of identified interactions those that are specifically associated with the isolated protein of interest. Therefore, a chapter of this review is focused on describing the types of non-specific associations and the current methods used for assessing specificity of interactions. IP-MS strategies have also improved in tackling the challenge of identifying stable and transient interactions, partly by using a combination of cross-linking, MS, and bioinformatics approaches. This review describes some of these approaches, as well as their application to understanding the structure of protein complexes. Critical to characterizing protein interactions is the ongoing development of appropriate bioinformatics tools for mapping interaction networks. An overview of the resources available for analyzing, constructing, and visualizing protein networks is provided, together with a discussion of their advantages and representative literature. While highlighting initial methods and selected developments, for each of these critical aspects of protein interaction studies, this review mainly focuses on current approaches and applications.
CONSIDERATIONS WHEN ISOLATING PROTEIN COMPLEXES USING AFFINITY-BASED METHODS
General workflow
The characterization of protein-protein interactions requires the successful isolation of protein complexes close to their physiological states. Maintaining and purifying protein associations close to their native form is challenging, and requires the ability to identify interactions of both stable and transient nature. Affinity-based approaches coupled to mass spectrometry analysis can be powerful tools in studying protein-protein interactions due to their simple and fast execution, selectivity, and sensitivity. Additionally, the enrichment of proteins of interest by immunoaffinity purification can provide insights into posttranslational modifications that may regulate protein interactions and functions1-4. In recent years, affinity-based methods have taken multiple shapes and forms due to the development of diverse workflows that can be integrated within a broad range of biological contexts. Yet, despite this diversity, there are several universal steps (Fig. 1), including: 1) generation/preparation of cell or tissue samples expressing the endogenous or affinity-tagged protein of interest, 2) cell or tissue lysis and solubilization of protein complexes under optimized lysis buffer conditions, 3) isolation of the protein of interest with its interaction partners by affinity purification, 4) elution of purified protein complexes, and 5) analysis of co-isolated proteins by mass spectrometry, leading to the identification and quantification of protein-protein interactions. This general outline is open to multiple modifications at every step to ensure efficient protein isolation, and application to various biological systems, research conditions, and experimental goals. The protein's abundance, stability, localization, and physicochemical properties are factors that have to be considered; thus, much thought must be placed into the design of the workflow for isolation and characterization of protein complexes. This section outlines critical aspects of affinity-based methods for studying protein interactions.
Figure 1. Immunoaffinity purification and mass spectrometry analysis of protein complexes.

The first step in immunoaffinity purification (IP) of protein complexes involves the generation of cell or tissue samples expressing the endogenous or affinity-tagged bait protein. Subsequent cell disruption and lysis in optimized buffers facilitates the release and solubilization of bait protein complexes, ensuring efficient protein isolation. Purified protein complexes can be eluted under native or denaturing conditions, and characterized using mass spectrometry and bioinformatics.
Affinity purification of endogenous or tagged proteins
Affinity-based approaches involve the isolation of a target protein (bait) and its interactions (preys) by affinity binding to a capture molecule immobilized on a solid support (resin). Thus, the protein of interest gets captured on the resin together with its interaction partners and, after different washing steps aimed at eliminating non-specific interactions, the isolated protein complex is eluted and analyzed by mass spectrometry. While this approach is frequently applied to small-scale studies focused on one or several proteins of interest5, 6, recent reports have also tackled large-scale studies where the interactomes of an entire system were characterized.7-10 Proteins can be purified in their endogenous form or through an affinity epitope tag (Fig. 1). The advantage of isolating the endogenous protein is the ability of capturing its physiological state, abundance, and interactions within a multitude of systems (i.e., cells, tissues, animal models), without the need for cloning or tagging. This method was successfully implemented in studies of individual endogenous proteins, as well as in large-scale interaction studies. For example, Malovannaya et al. performed a large-scale study of endogenous human regulatory protein complexes, focusing on nuclear transcriptional and signaling proteins.11 Drawbacks of isolating endogenous proteins include the dependence on availability, specificity, and affinity of antibodies recognizing the proteins of interest. Cross-reactivity and cost of the utilized antibodies are often concerns, as well as the possible interference of posttranslational modifications in epitope binding. Additionally, it is not uncommon for an antibody to work well for immunofluorescence or Western blotting, but not for IP experiments.
As an alternative to isolating endogenous proteins, a routinely utilized workflow involves the isolation of epitope tagged proteins. In this approach an epitope tag is fused to the target protein and IP is performed using an antibody specific to the tag. As the method is independent of the protein's binding properties, it is a universally applicable approach, useful for both low- and high-abundance proteins. This approach is suitable for large-scale and high-throughput experiments, as it can be applied to multiple proteins using a single tag. The workflow can be robust and reproducible, as the experimental conditions can be optimized for specific epitope tag(s). One drawback is the time requirement, since the fusion protein has to be generated and introduced into the biological system of choice. Most importantly, the fusion protein has to be assessed to confirm that the tag is not interfering with the protein's endogenous function, localization and properties.1, 12-14 This is an important consideration when studying cellular proteins from various systems (e.g., mammalian1, 14-16, bacterial17, 18, yeast13, 19), as well as proteins introduced by pathogens12, 20. Additionally, when the protein is expressed under an exogenous promoter, functional assays have to confirm that its overexpression does not alter its physiological properties and roles. Retrovirus1, 21 or lentivirus10, 22 transductions have been successfully utilized to control the level of overexpression of tagged proteins. Alternatively, expression vectors with inducible promoters (e.g., tetracycline-inducible) can be used to turn on or off the expression of the fusion protein, and avoid side effects or toxicity due to overexpression.2 A recent study showed that by titrating the amounts of tetracycline, the level of the tagged protein can be adjusted to mimic the endogenous levels prior to immunoaffinity purifications23. Other advancements in expressing tagged proteins at or near endogenous levels include the introduction of tag-encoding DNA into endogenous loci via homologous recombination24, 25, as well as the incorporation of bacterial artificial chromosomes (BACs) for BAC transgene generation allowing the expression of tagged fusion proteins upon stable transfection into mammalian or other biological systems.26-28
A variety of epitope tags have been utilized in immunoaffinity purification experiments (as reviewed in 29, 30). Commonly used single tags include small epitope tags, such as FLAG, six histidines (His)6, c-Myc, and hemaglutinin (HA), or larger tags, such as green fluorescent protein (GFP), glutathione-S-transferase (GST), and protein A (PrA). FLAG, available in 1X or 3X versions, is a frequently used tag due to its small size (8 amino acids per 1X) that limits its interference with protein function.31 3xFLAG tends to be preferred due to its higher affinity than the 1XFLAG and the corresponding increased efficiency of isolation.5, 23, 32 In a recent study, Law et al. purified 3xFLAG fusion proteins involved in RNA-directed DNA methylation, demonstrating a role for these complexes in polymerase V-dependent transcript production in plants.33 In another study, 3xFLAG tags were utilized to study virus-host protein interactions during Sindbis virus infection, unraveling host factors targeted by the virus RNA-dependent RNA polymerase and important for virus replication5. FLAG tags have been also applied to global interactome studies, including a large-scale mapping of human protein-protein interactions involved in diverse biological processes, such as proteasome function, translation, and progression through mitosis.34 GFP is another commonly used tag that has emerged as an effective tool for integrating knowledge about protein localization and interactions.35, 36 The increased use of GFP for protein isolations reflects the current emphasis on understanding the dynamics of protein interactions, as it allows placing protein interactions in a specific spatial and temporal cellular context. Cristea et al.35 have demonstrated the effectiveness of using single-step isolations via the GFP tag for characterizing protein complexes. Yeast and mammalian proteins were visualized by direct fluorescence and isolated by rapid immunoaffinity purifications via the GFP tag. As this study illustrated, the library of GFP-tagged yeast strains generated by O'Shea and Weissman using homologous recombination is a valuable resource for studying localization and interactions of GFP-tagged proteins expressed at endogenous levels in yeast.24 Nevertheless, affinity purifications via the GFP tag have been widely applied to multiple biological systems, including yeast35, 37, metazoans36, bacteria17, 18, mammalian cells1, 38 or tissue28, 39, 40. For example, Cheeseman et al. studied kinetochore proteins in C. elegans, referring to this combined visualization and purification as a Localization and Affinity Purification (LAP) tag strategy.36, 41 Goldberg et al. characterized interactions specific to distinct histone H3 isoforms in mouse embryonic stem cells, identifying H3.3 unique interactions and their roles in localizing H3.3 to telomeres.38 Additionally, these approaches were powerful for characterizing virus-host and virus-virus protein interactions that proved critical during infections with diverse viruses, including Sindbis5, 42, Human cytomegalovirus 12, 20, 43, Herpes simplex virus2, West Nile virus44, and Pseudorabies virus.45 A variation of the GFP isolation approach was designed based on single-chain antibodies raised against a GFP fragment in alpaca, which reduces contamination from antibody fragments.46, 47 The molecular weight of GFP, or other similar large tags, is a concern in IP studies, and the validation of the function and localization of the tagged protein is critical, as shown in 1.
While single-step affinity purification methods have proven effective for capturing low abundant and weak protein interactions, tandem affinity purifications (TAP) were initially introduced to preserve stable interactions and reduce non-specific associations.48 The original TAP method is based on a double TAP tag composed of a Calmodulin binding peptide (CBP) and an IgG-binding unit of Protein A from Staphylococcus aureus (PrA), allowing for a two-step affinity protein purification strategy. Although first applied in yeast, this TAP strategy has been further developed and optimized for isolations in other biological systems, including mammalian14, 49, 50, plants51, 52, viruses53, 54, and bacteria55. In recent years, multiple variations of the TAP tags have been designed for proteomic studies, as reviewed in56-58. Tags used for tandem affinity purification ranged from GFP41 and PrA to smaller tags, such as FLAG. An interesting modification was developed by Gloeckner et al. 59 based on a Strep/FLAG (SF)-TAP tag to reduce the tag size and interference with protein folding. This method has recently undergone further variations through a triple-tag approach that combines StrepII, FLAG, and yellow fluorescent protein (YFP) for parallel affinity capture (iPAC) and screening of protein interactions, localization, and expression.60 In addition, an interesting spin to the TAP methodology, with a concept similar to Förster resonance energy transfer (FRET), has been developed by Maine et al. introducing the bimolecular affinity purification system (BAP).61 This approach incorporates two affinity tags, but unlike traditional TAP procedures, each tag is fused to a different protein from a common multi-subunit complex. Thus, the method is applicable to targeted studies of a distinct molecular complex when two (or more) of its components are already known.
As discussed thus far, immunoaffinity purification methods most commonly involve the utilization of antibodies against endogenous proteins or tags for the isolation of protein complexes. In recent years alternative tools have been developed to complement the use of antibodies. For example, aptamers (nucleic acids), molecular imprinted polymers (MIPs), and engineered binding proteins (protein scaffolds) have been implemented as substitute affinity molecules, as reviewed by Ruigrok et al.62 Among the advantages of these tools are their low production costs, efficient selection, and in some cases greater tolerance to stringent affinity isolation conditions. In a representative study DeGrasse63 utilized a single-stranded DNA aptamer to specifically bind and isolate Staphylococcus aureus Enterotoxin B protein. In another example, Wiens et al.64 generated a silicatein-α fusion protein carrying a Glu tag for binding to hydroxyapatite solid matrix, and analyzed the protein interactions by mass spectrometry. Additionally, small molecules have been successfully used in quantitative chemoproteomics approaches to validate targets of drugs and identify complexes that may be preferentially bound.65, 66 Overall, these tools promise to expand the range of methods for studying interactions. Remaining challenges for their widespread use range from experimental considerations (e.g., aptamers are highly sensitive to nucleases present in lysis buffers, small molecules have to be engineered to contain a binding linker to resins) to more practical obstacles (e.g., patent-protected methodologies are oftentimes strictly licensed to a selected number of biotechnology companies).
Lysis approaches
In order to effectively isolate a protein of interest, the tissue or cells are first subjected to lysis. This lysis has to be performed in a manner that ensures exposure of cellular contents for immunoaffinity purification, without disruption of protein interactions. Several types of lysis are routinely performed in IP experiments. The most common methods involve physical cell (or tissue) disruption, preferably done cryogenically, or direct lysis in optimized detergent-containing buffers. Cryogenic cell lysis is recommended as an optimal choice in numerous IP-MS workflows. The immediate freezing of the sample (cell or tissue) helps to preserve protein complexes close to their cellular state. Subsequent mechanical grinding of the frozen sample provides an effective mean to shatter through cell wall, cytoskeletal networks, membranes and vesicles, providing access to various baits of interest and reducing non-specific associations35, 67-69. Cryogenic lysis can be performed using mortar and pestle if large amounts of starting material are available, or within grinding jars or plastic tubes if starting with smaller amounts of frozen cells and for more consistent grinding. This type of lysis has been extensively utilized by many groups when working with tissue or cells samples, and is usually followed by suspension of the frozen sample powder in an optimized lysis buffer. For example, we successfully applied this workflow to recent studies of protein complexes in bacteria17, yeast18, mammalian cells1, 21, as well as during viral infection.5, 12, 20 Mechanical disruption can be also performed by shearing the cellular sample by passing through a needle, or by applying temperature shifts using freeze/thaw cycles (for review see70). This type of lysis may not be the appropriate choice when IPs of the bait protein aim to isolate an intact large structure (e.g., postsynaptic density from brain tissue samples) or organelle (e.g., mitochondria). In such cases, direct lysis in a detergent- and salt-containing buffer is more suitable. One caveat of whole cell lysis is that proteins with different sub-cellular localizations can mix during the lysis procedure, becoming available for non-specific binding. Therefore, if localization of the bait protein is known, fractionation can be incorporated into an IP workflow to achieve efficient and clean isolation. Fractionation strategies have been utilized in various biological systems and cell types, especially in large-scale studies aimed at characterizing proteomes of organelles (e.g., mitochondria, Golgi, nucleus).71 However, numerous proteins have multiple sub-cellular localizations, in which case IPs can be either performed from whole cell lysates or as parallel isolations from the fractionated sub-cellular compartments (e.g., parallel nuclear and cytoplasmic isolations). In the latter case, localization-dependent unique and shared interactions can be compared by quantitative mass spectrometry. For example, Trinkle-Mulcahy et al. characterized nuclear- and cytoplasmic-specific interactions of the Survival of Motor Neuron (SMN) complex, known to function in both compartments.72 If the mechanisms regulating protein localization are known (e.g., phosphorylation sites), then mutants can be used to trigger the localization of a protein to a certain sub-cellular compartment, and isolations of mutants can be used to define localization-dependent interactions. For example, Greco et al. utilized phospho-mutants to control the nuclear-cytoplasmic shuttling of histone deacetylase 5 (HDAC5) and determine its localization-dependent interactions.1
Regardless of the procedure utilized for sample preparation (i.e., cryogenic lysis or fractionation), the optimization of lysis buffer conditions is one of the most important steps in IP experiments, providing a balance between efficient protein solubilization, while preserving stable and weak interactions. In addition, this step is critical for reducing non-specific interactions, as discussed in section “Determining specificity of interactions”. Lysis buffers have to be tailored to the chemical properties and localization of the bait protein. Most lysis buffers include protease inhibitors to avoid protein degradation, as well as DNases and/or RNases to decrease sample viscosity and facilitate protein isolation. Sonication can also be applied to mechanically shear DNA and RNA molecules. DN/RNases should be avoided when investigating protein interactions that are facilitated by DNA or RNA molecules. Different types and concentrations of salts and detergents, as well as the pH and ionic strength of the solution should be considered when optimizing the lysis buffer composition, as this determines the accessibility of the bait for isolation and, therefore, the efficiency of extraction. A list of commonly used detergents and their properties have been described.39 Among these, milder detergents that can be used to study more sensitive protein complexes and protein-lipid interactions include Triton X-100 and NP-40 (depending on the selected concentrations). More stringent detergents (e.g., Sodium Deoxycholate, Digitonin) solubilize lipid molecules, making these suitable for studying interactions of membrane-bound proteins. Optimization of lysis buffer detergents is also necessary to ensure the isolation of protein complexes close to their native state. For example, Everberg et al. used a combination of milder detergents (i.e., Zwittergent 3-10, Triton X-114) followed by a polymer two-phase partitioning system to enrich for solubilized membrane protein complexes in their native state.73 Additional considerations include the impact of detergents on the selected mass spectrometry analysis workflow. Norris et al. utilized cleavable detergents for the isolation and analysis of intracellular and membrane proteins, as these proved to be less detrimental for matrix-assisted laser desorption/ionization (MALDI) mass spectrometry and could be eliminated prior to MS analyses.74 Further information on the selection of lysis buffer conditions when studying proteins in different biological contexts is provided in several recent studies and reviews.39, 70, 75
In addition to lysis conditions, other factors critical for efficient isolation of protein complexes include the choice of antibodies and tags (as discussed above), as well as the selection of affinity resin and duration of protein purification (Fig 1). Several types of affinity resins have been commonly used in IP experiments, including natural (e.g., agarose and sepharose beads), organic (e.g., glass), and synthetic (e.g., acrylamide-based supports) resins, as reviewed in76. Magnetic beads are a more recent and continuously improving addition to affinity resins. Important advantages of the latter include the available varied chemistry for binding and their inherent property of surface binding that allows the isolation of a broad range of protein complex sizes. Additionally, since magnetic beads do not require centrifugation, but instead use a magnet for their collection, their easy handling reduces non-specific binding. The impact of the resin type and duration of isolation on the accumulation of non-specific associations is discussed in the “Determining specificity of interactions” section.
Protein elution
The strategies for eluting the isolated protein complexes depend on the purpose of the subsequent analysis, and whether the isolated proteins will be analyzed in their denatured or native forms (Fig. 1). The most common workflows utilize the elution of isolated complexes in a denatured manner, such as with buffers containing sodium dodecyl sulfate (SDS) or lithium dodecyl sulfate (LDS). The advantage of such buffers is the resulting high efficiency of elution, and LDS-based elutions have been recently adapted for sample preparation by in-solution digestion prior to mass spectrometry (e.g., filter-aided sample preparation (FASP77)). However, this type of elution can also trigger the release of non-specific molecules attached to the affinity resin, as well as immunoglobulin (IgG). In a recent study Antrobus and Borner attempt to reduce the amounts of contaminant IgG by modifying the elution conditions for isolated endogenous proteins.78 In this study, “soft” elution conditions using lower detergent concentrations and a lower temperature for elution decreased the amounts of contaminant IgG without dramatically compromising the efficiency of protein isolation. The type of isolated protein complex may, however, play a role in this efficiency of elution. Another common strategy for achieving a denaturing elution is the use of solutions with acidic or basic pH. Citric acid and trifluoroacetic acid (TFA) are commonly used for acidic elutions21, while ammonium hydroxide in combination with ethylenediaminetetraacetic acid (EDTA) is often preferred for basic pH conditions.35 In addition, protein elution can be performed over a pH gradient providing a comprehensive view of the nature of the isolated protein-protein interactions, as multiple fractions can be collected and analyzed. This strategy, while being labor intensive and less practical for high-throughput proteomic studies, can help reveal the stability of interactions (i.e., retained at acidic or basic pH conditions).
As an alternative to denaturing strategies, protein complexes can be eluted in a non-denatured way to preserve intact isolated assemblies, possibly for further functional studies. These types of elution are well integrated with recent mass spectrometry technology developments that allow the analysis of native proteins and complexes.79-85 In such studies, elution can be performed by utilizing reagents for competitive binding to the resin. For example, FLAG (1X or 3X) peptides are used for eluting complexes isolated via anti-FLAG antibodies.86, 87 Additionally, a cyclic peptide has been developed for the native elution of complexes isolated via PrA tag.88 Competitive elutions have also been utilized when purified proteins, rather than antibodies, have been conjugated to resin and used for isolating interacting proteins. For example, Dubois et al. used competition with a synthetic phosphopeptide to elute phosphorylated substrates of sepharose-immobilized 14-3-3 proteins.89 In another example, phenyl phosphate was utilized to elute phosphotyrosine-containing protein complexes involved in receptor tyrosine kinases signaling.90
Upon isolation, a comprehensive characterization of the isolated protein complex(es) can be achieved by taking advantage of the multitude of available mass spectrometry-based workflows (Fig. 1). Depending on the complexity of the isolated assembly, the proteins can be separated by SDS-PAGE electrophoresis (1-D or 2-D), in-solution isoelectric focusing (at the protein or peptide level)85, or directly prepared for mass spectrometry analysis. Protein complexes are routinely analyzed using bottom-up or middle-down approaches, in which the eluted proteins are digested with proteases.82 Bottom-up approaches utilize enzymes that digest the proteins into numerous small to medium length peptides (e.g, trypsin, GluC). Middle-down approaches utilized digestions with proteases that aim to preserve larger parts of the protein intact (e.g., LysC), being frequently integrated in studies of cross-talk between posttranslational modifications. A combination of enzymes can be used to increase the observed protein sequence coverage.2 Isolated protein complexes can also be analyzed using top-down approaches for studying intact proteins.91 Additionally, intact protein complexes, eluted using non-denaturing methods, can be separated by blue native gel electrophoresis (BN-PAGE).90 This type of gel separation, in combination with mass spectrometry, allows for analysis of structure and function of protein complexes.92, 93 Western Blot analyses frequently accompany mass spectrometry analyses for validation or targeted follow-up studies. While not the focus of this review, further information on sample preparation and analyses by mass spectrometry is available in several reviews.94-96
DETERMINING SPECIFICITY OF PROTEIN INTERACTIONS
Proteins can have numerous direct and indirect interactions that are spatially and temporally regulated and critical for their diverse functions. In view of this complexity of interactions, a great challenge lies in distinguishing specific interactions from numerous potentially non-specific associations. Therefore, substantial effort has been recently devoted toward developing approaches for assessing the specificity of protein interactions. This section presents an overview of the sources of non-specific binding and the strategies utilized to address them.
Sources of non-specific interactions
Common contaminants in IP experiments include proteins that interact with the resin (e.g., magnetic beads, agarose), immunoglobulin molecules (e.g., heavy or light chains of antibodies), and tags (e.g., FLAG, GFP) (Fig.2A). The choice of resin for IP experiments can aid in reducing non-specific contaminants. For example, Trinkle-Mulcahy et al. indicated that sepharose beads isolated less non-specific associations from cytoplasmic extracts, while magnetic beads performed better with nuclear extracts.28,70 On the other hand, magnetic beads do not require centrifugation, but instead use a magnet for their collection after IP. This type of surface binding and easy washing may partly contribute to the observation of reduced non-specific binding when comparing magnetic to agarose beads.23 The type of antibodies selected for IP can also impact the level of non-specific associations. Polyclonal antibodies tend to have high affinities for binding, providing higher efficiency of isolation when compared to monoclonal antibodies. However, the use of monoclonal antibodies leads to fewer non-specific associations than polyclonal antibodies. Also, it is common that commercially available antibodies against a protein of interest produce higher levels of non-specific binders than custom in-house generated and purified antibodies.
Figure 2. Specificity of interactions in IP experiments.

A) Sources of non-specific interactions include associations with resin, and antibody, as well as isolated proteins. B) A balance between stringency of lysis buffer and incubation period with the antibody can aid in isolating specific interactions, while reducing the presence of non-specific interactions.
Even more challenging are non-specific associations that bind to the isolated proteins of interest, which may be referred to as non-specific interactions that are particular to the studied complex(es). Once affinity purified on the resin, the co-isolated proteins can act as interacting sites for numerous readily adherent (“sticky”), abundant, or domain-recognizing proteins that are present in the cell lysate. Non-specific binding is, in part, driven by the fact that proteins in solution do not retain the subcellular localization they possess in vivo and, therefore, may have previously non-existent opportunities to bind upon lysis (Fig. 2A). Table 1 lists commonly used methods for identification of non-specific interactions from these different sources of non-specific binding, indicating the advantages and disadvantages of individual methods.
Table 1.
Examples of common methods used for identification of non-specific protein interactions.
| Name | Advantages | Disadvantages | Examples |
|---|---|---|---|
| LABEL-FREE METHODS | |||
| Biochemical approaches | |||
| Control IP | Removes non-specific associations, including proteins interacting with beads, antibody, or tag | Can exclude highly abundant proteins that may also specifically interact with bait | 1, 2, 12, 21, 35, 45, 98, 99, 101, 102 |
| Reciprocal IP | If bait protein is present in prey IP, these proteins are likely to associate specifically | Can produce false-negatives if prey is not abundant. Does not distinguish direct versus indirect interactions | 6, 34, 35, 99, 101-103, 106, 109, 113 |
| Bioinformatics approach based on mass spectrometry data | |||
| SAI | Accounts for high-abundance proteins that also specifically associate with the bait | Labor-intensive, requiring several rounds of IPs and reciprocal IPs | 105, 220 |
| PE score | Uses combined datasets from independent studies | Might be limited for organisms that do not have large-scale datasets | 10, 107, 221, 222 |
| IR score | Accounts for interactions between bait and prey in reciprocal IPs, as well as in IPs of other baits | If the threshold is not set properly, false-positives or false-negatives could be enriched | 109, 223, 224 |
| NSAF | NSAF normalizes spectral counts to protein length | Cannot accurately reflect proteins of low abundance with low spectral counts | 6, 112, 113 |
| PAX | PAX can increase the confidence in interactions with low abundance proteins | Data not yet available for all cell/tissue types or different conditions (e.g. virus-infected cells) | 6, 114 |
| CompPASS | Combines multiple score systems to identify high-confidence interacting partners that are found in multiple runs, as well as rarely found proteins | Some utilized scores were derived empirically and lack clear probability model for assigning threshold values to them | 116-119, 225 |
| SAINT | Likelihood of a true interaction is estimated from spectral counts probability distribution model. The method is applicable to datasets of all sizes | False-positives can be assigned to proteins non-specifically associated with the isolated complex; Low abundance or small proteins may be missed because of low spectral counts. | 7, 120, 226, 227 |
| MasterMap | Bona fide interactions are identified due to change in their abundance (MS1 signals) in sequential dilutions of control and specific IP samples | Increased number of samples due to sequential dilutions makes experiments labor-intensive for large-scale studies | 111 |
| MiST | Composite score is derived from multiple types of data and applicable to host-pathogen interactions | Relevant but weak interactions can be missed if the score cutoff is set too high | 121, 228 |
| LABELING METHODS | |||
| Metabolic labeling | |||
| I-DIRT | Allows differentiation of non-specific associations to purified complexes after cell lysis | Can assign fast-exchanging interactions as non-specific | 6, 124, 229 |
| QUICK | Contaminants in IPs of endogenous proteins are distinguished by comparison to knock-down conditions | Same limits as for SILAC approach, plus availability of shRNA with high knock-down efficiency | 126-128 |
| SILAC | Combination of (Tc) PAM-SILAC and MAP-SILAC allows accurate identification of dynamic (transient) interactions | Expensive and requires cell culture, like most metabolic labeling approaches. Both PAM and MAP are required to differentiate stable and transient interactions. | 123, 130, 131 |
The presence of non-specific interactions from these different sources is influenced by a number of factors that can be experimentally controlled. One of the critical aspects and primary steps in reducing non-specific interactions is the careful optimization of lysis conditions. Lysis buffers can be adjusted to retain specific strong or weak interactions, while reducing non-specific interactions. For example, more stringent lysis buffer compositions (e.g., high salt or detergent concentrations) may be used to primarily preserve strong interactions, while milder conditions can be optimized to retain weaker interactions. While stringent lysis conditions may also be necessary for accessing proteins within membranes or vesicles, one caveat of using high detergent concentrations is the possible denaturing of proteins. Protein denaturation can trigger an additional accumulation of non-specific associations (e.g., heat shock proteins that bind to unfolded proteins97). On the other hand, mild lysis buffer conditions can lead to an increase in not only specific and weak interactions, but also in non-specific binding39. Another critical factor influencing the presence of non-specific associations is the duration of the steps involved in immunoaffinity purification. Cristea, et al. demonstrated that the time frame utilized for cell lysis and subsequent incubation of cell lysate with antibody (conjugated to the resin) can significantly impact the cleanliness of the isolation.35 Incubations of a few minutes to a maximum of one hour can help reduce the number and abundance of non-specific associations. A proper balance between the stringency and incubation time can help maintain the majority of relevant interactions, while reducing background (Fig. 2B).
Although optimal lysis and isolation conditions can help reduce the background, contaminants that interact with resins, tags, IgG, or isolated complexes will still be present, even if at lower levels. Therefore, a requirement for all IPs is to design and incorporate appropriate controls. The experimental conditions for the control isolation should be consistent with the entire workflow (i.e., the conditions for isolating the proteins of interest). Most approaches for determining interaction specificity address non-specific associations with the resin, tag, and antibody, as discussed below. Several approaches have also been designed to tackle the issue of non-specific associations with the isolated proteins themselves. These methods are discussed in the “Assessing interaction specificity using metabolic labeling.”
Label-free biochemical approaches
The most frequently used controls in IPs of tagged bait proteins involve the generation of control cell lines expressing only the tag under the same promoter as the bait. This method has been successfully used by our laboratory to control for non-specific association of proteins to GFP or FLAG tags in studies characterizing functions of protein complexes in mammalian cells, as well as in cells infected with viruses.1, 2, 12, 21, 35, 98 For example, GFP control cell lines were used in a study aimed at determining localization- and phosphorylation-dependent interactions of histone deacetylase 5 (HDAC5)1. In another study, the possibility of temporal changes in non-specific associations was addressed using control isolations at different time points after infection with Sindbis virus42. Liu et al. used TAP tagging and compared the results to control TAP-only IPs to characterize the interactome of the hepatitis B (HBV) viral protein, HBx.99 This study identified a factor involved in apoptosis inhibition during HBV infection. A novel HaloTag technology was developed by Daniels et al. to characterize interactions of RNA polymerases I, II, and III, in which control HaloTag isolations were performed to determine non-specific interactions.100 Yang et al. utilized a GST control to specifically identify ribosomal protein 1 as a novel interacting partner of GST-tagged MSMEG_2731 protein of M. smegmatis (the model organism used to study M. tuberculosis), indicating a role for MSMEG_2731 in the processes of transcription and translation.101 When isolating endogenous proteins, a similar concept is used in designing controls. Resin conjugated with IgG is frequently used to capture proteins that bind non-specifically to the antibody rather than the bait. Malovannaya et al. incorporated additional steps into their IP protocol for elimination of common sources of non-specific associations when isolating an endogenous protein.102 For instance, ultracentrifugation at 100,000xg prior to bead incubation allowed for removal of proteins that had precipitated out of solution during antibody incubation.
Once protein interactions are identified as unique to bait isolations and absent in the control isolations, the interactions of interest must be further validated. Validation of interactions is frequently carried out by performing reciprocal isolations, i.e. isolation of the newly identified protein as a bait and confirmation of the co-isolation of the initial protein of interest as a prey. However, this strategy is limited by the availability and cost of antibodies recognizing the proteins of interest or the requirement of tagging novel proteins. For example, reciprocal IPs were used to confirm putative interactions identified in a study of mitotic protein complexes in cells expressing LAP-tagged bait proteins.103 The large-scale nature of this study allowed the comparison of numerous samples, and proteins found in multiple isolations or in control purifications were considered contaminants. This combination of control parameters led to relatively low rates of false-positive identifications. While reciprocal isolations are valuable in confirming interactions, these approaches do not distinguish between direct and indirect interactions. Additionally, reciprocal isolations can be challenging depending on the abundances of the bait or prey proteins. If the analyzed prey is abundant and involved in numerous complexes, it may prove difficult to isolate the prey protein at a sufficient level for confirmation of an interaction with a low abundance protein. Alternatively, if the prey protein is of low cellular abundance, its isolation may be difficult and confirmation experiments will depend on the achievable efficiency of isolation. Other common approaches for validation of interactions include assessing co-localization of bait and prey proteins6, 103, as well as determining whether the interaction is direct by using binary interaction assays104 (e.g., yeast two-hybrid).
Label-free bioinformatics approaches based on mass spectrometry data
Following optimization of IP conditions to reduce non-specific contaminants, further analysis based on mass spectrometry data can be applied to recognize the remaining non-specific associations. This is commonly done by statistical analysis of label-free qualitative or quantitative data. The qualitative approach compares the presence or absence of a protein in bait and control isolations, and is most commonly applied to large-scale studies that provide sufficient data for statistical analyses. Due to the continuous recent advancements in quantitative mass spectrometry, the quantitative approach is routinely implemented in IP studies of small or large scale, as described below.
Highly abundant proteins are frequently assigned as common contaminants in IP studies. However, interpreting the specificity of such proteins can be challenging. While their presence in isolated complexes can be derived from non-specific associations, true interactions may also be excluded as a side-effect of their abundances (e.g., tubulin, actin, heat shock proteins). Several large-scale studies of yeast proteome aimed to address this and other challenges in identifying specific interactions by developing statistical analysis approaches based on the presence or absence of interactions between multiple isolations.105-107 For example, Gavin and colleagues introduced “socio-affinity” index (SAI) as a tool to assign interaction specificity.105 SAI describes the tendency of two proteins to be present in reciprocal isolations or to associate in isolations of other tagged proteins, allowing to retain highly abundant proteins that would otherwise be removed.105 In another large-scale study, Krogan and colleagues applied two mass spectrometry methods, MALDI-TOF and LC-MS/MS, to identify interactomes of TAP-tagged yeast proteins with high coverage and confidence.106 Using two rounds of machine learning algorithms trained on the curated MIPS database108 of protein complexes, probabilities were assigned to each pairwise interaction. The combined dataset from the above mentioned studies105, 106 was later analyzed by purification enrichment (PE) scoring system that takes into account positive and negative evidences for and against interactions with the goal of decreasing the presence of false-positives.107 Recent study by Babu et al. utilized the PE score to analyze membrane-protein complexes in yeast, providing insights into organization of eukaryotic membranes.10 Similar and alternative computational approaches have been also employed in IP studies in mammalian systems. For example, Jeronimo et al. studied human protein complexes involved in transcription and RNA processing using several computational approaches.109 First, the Mascot110 scores for proteins in control isolations were used to account for non-specific binding of proteins to the resin and abundance of proteins in the isolation. Second, an interaction reliability (IR) score was assigned to each interaction, which was based on both the Mascot score and its network connections. Another human interactome study conducted by Ewing et al. used FLAG-tagged bait proteins with diverse biological functions for IP and high-throughput mass spectrometry analysis.34 To remove non-specific interactions, in addition to removing frequently associated proteins, a partial least squares-based regression model was built to assign interaction confidence scores to observed prey proteins.
In more recent IP-MS studies, quantitative information from mass spectrometry has become a valuable tool for the analysis of interaction specificity. For example, Rinner et al. used a MasterMap integrating MS1 signals from several LC-MS runs to identify specific interaction partners of FoxO3A.111 To increase specificity and sensitivity of analysis, proteins isolated in HA-FoxO3A IP were sequentially added in increasing amounts into control isolations. As a result, protein profiles extracted from LC-MS analyses showed progressive enrichment of specific interacting partners, while non-specific interactions remained identified at constant levels. Information on the number of peptides observed per protein (spectral counts) derived from IP-MS analysis is becoming increasingly popular for label-free quantification. To account for protein length and variation between separate MS analyses, an approach utilizing normalized spectral abundance factors (NSAF) was developed.112 NSAF values are calculated from the total number of spectra for each protein, normalized to its length and the total number of spectra for all proteins in the sample.112 Therefore, NSAF values can provide a view of the relative abundance for each protein among co-isolated proteins, highlighting proteins that may be prominent interactions. Sardiu et al. utilized NSAF values for building protein networks of chromatin remodeling complexes.113 To determine probable contaminants, an NSAF ratio was calculated for each protein by comparing IPs of FLAG-tagged bait and FLAG control.113 In a recent modification of this strategy, Tsai et al. integrated NSAF values with the estimated proteome abundance (PAX) values from PaxDb to build protein interaction networks for proteins with limited known functions.6 PAX values are reflective of the approximate total cellular abundance of proteins.114 Therefore, normalization of NSAF to PAX values6 provides a means for assessing the relative enrichment of proteins within an isolated complex, while correcting for the possible bias resulting from their total cellular abundances. This approach led to the identification of SIRT7 interactions with nucleolar chromatin remodeling complexes, providing insight into its role in rDNA transcription.6 While we expect PAX values to continue providing useful information in interaction studies, several considerations must be taken into account. Proteins are well known to have differential expression levels in distinct cell types and tissues. While PAX values have already been derived for proteins in multiple tissue types, including brain, heart, liver, and lung, information is still unavailable for many cell types and many proteins. Nevertheless, this concern is partly addressed by the fact that many highly abundant proteins, such as cytoskeletal and heat shock proteins, tend to be consistently abundant across different cell types. Therefore, even if the current PAX values may not reflect the precise levels of many medium or low abundance proteins, these values may still allow for correction of the more highly-abundant proteins. One additional concern that remains more difficult to address is the fact that proteomes can be significantly altered by environmental stimuli or pathogens. As an example, viruses can trigger substantial overexpression of certain proteins (e.g, 10-20-fold increase in p53 levels in cytomegalovirus-infected cells115). Therefore, future analyses of PAX values in the presence of different environmental and pathogenic stressors would provide a mean for increasingly accurate normalizations.
Other computational algorithms utilizing spectral counting for assessing specificity of interactions were developed to expand application to different experimental workflows (e.g., use of control and/or reciprocal IPs in the experimental setup). In a global proteomic analysis of deubiquitinating (Dub) enzymes, Sowa et al. developed the CompPass software that uses two scoring metrics to identify high-confidence candidate interacting proteins from parallel, non-reciprocal datasets obtained from FLAG-HA-Dub IPs.116 A Z-score was applied to weigh the presence of unique interactions in multiple immune complexes, and a D-score was used to take into account the abundance of the interactions (total spectral counts, TSC) and their reproducibility. The effectiveness of this approach for generating the Dub interactome was demonstrated by validation analyses using reciprocal IPs, parallel IPs in different cell lines, and comparisons to previously reported interactions. Interestingly, when CompPass was compared to the SAI approach developed by Gavin, et al.105, only 47% of interactions were found to overlap. This observation could be due to different experimental workflows (e.g., cell lysis conditions, sample preparation) or the fact that reciprocal IPs were not included in the initial CompPass assessment. Additionally, when CompPass was compared to the NSAF method used by Sardiu et al.113, and D-scores were calculated based on NSAF or TSC values, the overlap of identified bona fide interactions increased to 87%. One experimental workflow difference between these studies was that Sardiu et al. utilized several control IPs in their analysis, while the CompPass study relied on multiple parallel, non-reciprocal IPs. The CompPass algorithm was successfully applied to studies of E7 human papillomavirus protein interactions, bromodomain protein 4 associations that are important for its transcription regulatory functions, and others.117-119
In 2010, the Nesvizhskii laboratory introduced the SAINT (significance analysis of interactome) algorithm that allows not only generation of confidence scores for protein-protein interactions based on mass spectrometry data, but also creation of a probability model to distribute true and false interactions.7,120 This approach was applied to construction of global kinase and phosphatase interaction networks in yeast, and further optimized for easy implementation in the analysis of various datasets.120 The SAINT algorithm combines experimental data for interactions between bait and prey proteins with values derived from negative controls (e.g., control tag isolations) through a semi-supervised approach. The requirement for negative controls can be avoided if sufficient numbers of distantly connected baits are used for IPs (unsupervised approach).120 Overall, SAINT proved as a useful tool for analysis of both large-and small-scale datasets, as even single-bait IPs performed with sufficient numbers of control IPs can be analyzed by this method.
As an alternative to spectral counting, a specialized scoring system termed MiST (mass spectrometry interaction statistics) was devised by Jager et al. for identification of HIV-1-host protein interactions following transfection of HIV-1 proteins into HEK293 or Jurkat T cells.121 The MiST algorithm was designed to overcome some of the limitations of aforementioned spectral counting-based approaches (e.g., their reliance on the abundance of interactions that can vary depending on intracellular abundances of bait and prey, efficiency of IP experiments, and detection by MS). MiST analysis accounted for protein abundance as indicated by peak intensities, reproducibility of abundance across replicates, and specificity of interactions across all purifications.
Overall, these label-free techniques for assessing interaction specificity can be easily incorporated into small- or large-scale experiments, and do not require expensive reagents when compared to labeling approaches (see section below). However, such methods may not reliably detect small changes in protein abundances and may not comprehensively address non-specific binding to the isolated complexes (Fig. 2A).
Assessing interaction specificity using metabolic labeling
Techniques incorporating metabolic labeling were introduced into proteomics workflows to address the need for accurate relative quantification. Incorporation of stable isotopes into proteins in cell culture provides global labeling of all expressed proteins and a means for simultaneous processing of samples for comparison, thus minimizing experimental variations and improving accuracy in relative quantification. Metabolic labeling was first utilized by Oda et al. in the form of 15N labeling in yeast.122 Mann and colleagues developed the SILAC (stable isotope labeling in cell culture) approach in which heavy amino acids containing 2H, 13C, and/or 15N were introduced into growing mammalian cells.123 This method simplified quantification because the expected mass differences between heavy and light peptides were known.
More recently, metabolic labeling has been used to distinguish specific from non-specific interactions in IP experiments. The I-DIRT (Isotopic Differentiation of Interactions as Random or Targeted) method was developed by Chait and colleagues to overcome the challenge of determining contaminants that bind non-specifically to the isolated protein complex (Fig. 2A) of the yeast DNA polymerase ε complex.124 In this approach, cells expressing an affinity-tagged protein are grown in light isotopic medium, while wild-type cells are cultured in heavy medium. As the IP is performed from a 1:1 mixture of light- and heavy-cultured cells, specific associations to the tagged bait are detected by MS analysis to have light peptides only, while non-specific interactions have light and heavy peptides. As in this approach the complexity of the mixed sample is increased especially for non-specific components, Tsai et al. demonstrated that a targeted “SRM-like” I-DIRT approach can be used to increase identification of specific and low abundance interactions of interest in mammalian cells.6 One limitation of the I-DIRT technique is that fast-exchanging interactions can be falsely assigned as non-specific. To differentiate between specific stable, specific transient, and non-specific interactions, a transient I-DIRT strategy using cross-linking in cell culture was developed and applied to study the multi-subunit complex NuA3.125 In the MS analysis, stable interactions produced 100% light peptides, non-specific interactions produced 50% light, while transient interactions generated peptides in an intermediate range (between 50% and 100% light).
While I-DIRT was developed for experiments with tagged proteins, a separate approach was designed for the study of endogenous protein complexes. The QUICK (quantitative immunoprecipitation combined with knockdown) approach integrates metabolic labeling with RNAi to assess interaction specificity, as demonstrated for β-catenin and Cbl interactions.126 By RNAi-mediated knockdown of a protein of interest in light-labeled cell culture, while preserving expression of the target protein in heavy-labeled culture, specific interactions can be distinguished from non-specific interaction through comparison of light and heavy peptide intensities. This approach was applied in several studies, including the assessment of Stat3 interactions and the involvement of human leucine-rich repeat kinase 2 (Lrrk2) in cytoskeleton function.127, 128 Retention of specific versus non-specific interactions also depends on whether purification is done before mixing (MAP, mixing after purification) or after mixing (PAM, purification after mixing) of cell lysates when using SILAC.129 The PAM-SILAC approach is useful in identifying stable interactions from background interactions, but can miss dynamic interactions. To overcome this challenge, Huang and colleagues proposed a combination of time-controlled (Tc) PAMSILAC and MAP-SILAC.130 As shown, dynamic interactions had significantly increased SILAC ratios with the MAP-SILAC approach, while behaving like background proteins (1:1 SILAC ratio) in the PAM-SILAC approach. This method was used for studying the human 26S proteasome, the tumor suppressor PTEN, and others.130, 131
Assessing interaction specificity using chemical labeling
As metabolic labeling is not applicable to analyzing all sample types (e.g., tissues) and requires culturing cells for several passages in heavy media to ensure stable isotope incorporation, chemical labeling methods provide valuable alternatives. Chemical labeling is typically done after IP, either at the protein or peptide level. Therefore, this approach cannot account for variations in sample processing prior to the point of labeling. Chemical labeling has been utilized for the analysis of specificity of interactions. ICAT (isotope-coded affinity tag) was developed by Aebersold and co-workers for use in quantification in MS analyses and was applied to the characterization of protein complexes.132, 133 In this approach, chemical labeling of cysteine residues on intact proteins was performed with either heavy or light ICAT reagents in bait and control purifications, respectively. Specific interactions were distinguished from background proteins by comparison of heavy and light ICAT-labeled peptides. However, the limitations of this method include loss of information about non-cysteine containing peptides, and a reduced ability to characterize differences in post-translational modifications. Chemical labeling with isobaric tags has also been implemented in studies of interaction specificity. iTRAQ, a multiplex isotopic labeling method for amine-containing N-termini and lysine residues of peptides134, was utilized for distinguishing specific interactions in IP experiments.135 As the methodology for multiplexed chemical labeling continues to advance, we envisage that these approaches will become more extensively incorporated into studies of protein complexes and determination of specificity of interactions.
CONSTRUCTION AND BIOLOGICAL INTERPRETATION OF PROTEIN INTERACTION NETWORKS
Following the isolation of protein complexes, mass spectrometry analysis, and determination of interaction specificity, the frequently large number of identified putative interactions presents a challenge for determining biologically significant targets, even in small-scale studies with a single bait protein. Therefore, detailed computational analysis is required, involving determination of ontological protein relationships and construction of functional protein networks, to deduce the biological significance of identified protein complexes (Fig. 3). This section discusses the availability and advantages of recently developed computational tools. These include resources for generation and visualization of protein network maps, and deduction of interaction clusters based on functional enrichment and co-localization patterns. Due to the high number and variety of such tools, Table 2 and 3 aim to provide a representative list of commonly used software tools and database resources to date.
Figure 3. Constructing and analyzing protein interaction networks.

Following IP and mass spectrometry analysis, a combination of computational analysis approaches can be utilized to obtain insights into the biological significance of identified protein associations. These include computational tools for construction and visualization of protein clusters and large protein networks, as well as resources for functional enrichment analysis to determine ontological protein relationships.
Table 2.
Protein interaction databases.
| PROTEIN INTERACTION DATABASES | ||
|---|---|---|
| Name / Ref. | Description / Website | Range of Species |
|
STRING158
IntAct 140, 141 BIND 149 DIP 146 MINT 139 SCOPPI 230 Predictome 143, 144 IMEx 152 MatrixDB 153 iHOP 231 |
Search Tool for the Retrieval of Interacting Genes/Proteins
http://string.embl.de/ Protein Interaction Database and Analysis System www.ebi.ac.uk/intact/ Biomolecular Interaction Network Database http://bind.ca Database of Interacting Proteins http://dip.doe-mbi.ucla.edu Molecular Interactions Database http://mint.bio.uniroma2.it/mint/ Structural Classification of Protein-protein Interfaces http://www.scoppi.org Database of Putative Functional Links between Proteins (VisANT) http://visant.bu.edu/ International Molecular Exchange Consortium http://www.imexconsortium.org/ Extracellular Matrix Interaction Database http://matrixdb.ibcp.fr Information Hyperlinked over Proteins http://www.ihop-net.org/UniPub/iHOP/ |
Theoretically Unrestricted |
|
BioGRID
148
CORUM232 MITOP2 151 I2D 233 InnateDB 234 |
Biological General Repository for Interaction Datasets
http://www.thebiogrid.org Comprehensive Resource of Mammalian Protein Complexes http://mips.helmholtz-muenchen.de/genre/proj/corum Database of mitochondrial proteins http://www.mitop.de Interlogous Interaction Database http://ophid.utoronto.ca/ophidv2.201/ Innate Immunity Interactome Database http://www.innatedb.com |
Major model organisms Mammalian organisms Major model organisms Major model organisms Human and mouse |
|
HPRD
145
HPID 150 HomoMINT 235 HIV-1 154 MPact 142 MPIDB 147 Bacteriome 159 Hybrigenics 236 DroID 237 |
Human Protein Reference Database
http://www.hprd.org Human Protein Interaction Database http://wilab.inha.ac.kr/hpid/ Human Molecular Interactions Database http://mint.bio.uniroma2.it/mint/ HIV-1 - Human Protein Interaction Database http://www.ncbi.nlm.nih.gov/RefSeq/HIVInteractions/ Yeast Protein-Protein Interaction Dataset by the Munich Information Center for Protein Sequences http://mips.gsf.de/genre/proj/mpact Microbial Protein Interaction Database http://www.jcvi.org/mpidb/about.php http://www.bacteriome.org https://pimr.hybrigenics.com/ Drosophila Interactions Database http://www.droidb.org/Index.jsp |
Human Human Human Human in HIV-1 infection Yeast (S. cerevisiae) Microbial species Bacteria Helicobacter Drosophila |
Table 3.
Protein interaction visualization and network mapping resources.
| Name/Ref. | Description / Website |
|---|---|
| NETWORK MAPPING AND VISUALIZATION TOOLS | |
| Cytoscape 160 |
http://www.cytoscape.org/ Tool for analysis and visualization of biological networks; used for genetic and molecular interaction datasets |
|
Pathway
Palette 161 |
http://blais.dfci.harvard.edu/index.php?id=61 Web-based application for representation of proteomics data in the context of biological pathways and protein networks. Based on peptide rather than protein data input |
| Medusa 238 |
https://sites.google.com/site/medusa3visualization/ Data Java application for analysis and visualization of large-scale biological networks in 2D. Aims to integrate heterogeneous data from different sources into a single network |
| PINA 239 |
http://cbg.garvan.unsw.edu.au/pina/ Protein Interaction Network Analysis Platform integrating data from six protein-protein interaction databases (IntAct, MINT, BioGRID, DIP, HPRD, and Mpact) to provide tools for network construction |
| Arena3D 162 |
http://arena3d.org/ Visualization tool specifically developed for high level relationship and analysis of large networks incorporating three dimensional representation |
| Bio Layout Express 3D240 |
http://www.biolayout.org/ Analysis and visualization tool developed for network graphs, offering 2D and 3D visualization of data from multiple inputs |
| VisANT 144 |
http://visant.bu.edu/ Computational system for visualization of networks and pathways, built on interaction data from MIPS, BIND, and the HPRD databases. Provides navigation for disease/ drug hierarchy and visual construction of disease therapy networks |
| GEOMI 241, 242 |
http://www.systemsbiology.org.au/downloads_geomi.html 3D and 4D network visualization platform for protein associations utilizing various parameters, such as sub-cellular localization, protein abundance, post-translational modifications, and gene ontology classification |
| GENE ONTOLOGY DATABASES AND ANALYSIS TOOLS | |
| COG 243 |
Clusters of Orthologous Groups of proteins http://www.ncbi.nlm.nih.gov/COG/ Provides phylogenetic protein classification and tools for evolutionary and functional genome analysis |
| GOA 164 |
Gene Ontology Annotation project
http://www.ebi.ac.uk/goa |
| DAVID 169 |
Database for Annotation, Visualization and Integrated Discovery http://david.abcc.ncifcrf.gov/ Provides tools for building functional pathways based on gene functional classifications, annotations, and clustering. Well suited for high-throughput genomic experiments |
| ClueGO 167 |
http://www.ici.upmc.fr/cluego/cluegoDownload.shtml Cytoscape plug-in developed specifically for functional analysis and Gene Ontology/pathway term networks. Incorporates GO terms and KEGG/BioCarta pathways |
| FatiGO244 |
http://bioinfo.cipf.es/babelomicswiki/tool:fatigo Can be used to compile two lists of GO terms and highlight overlapping members. Provides also search tools for signaling pathways, protein motifs, keywords, and others |
| Gene MANIA 245 |
Gene Multiple Association Network Integration Algorithm http://www.genemania.org/plugin/ Cytoscape plug-in created to provide fast prediction of gene function. Uses over 800 networks from multiple organisms. |
| BiNGO168 |
A Biological Network Gene Ontology Tool http://www.psb.ugent.be/cbd/papers/BiNGO/Home.html Cytoscape plug-in developed to outline statistically enriched GO functional groups |
| GO::Term Finder 166 |
Gene Ontology (GO) Term Finder http://search.cpan.org/dist/GO-TermFinder/ Designed to assess GO information and annotate correctly gene lists to GO terms |
| GOMiner 165 |
http://discover.nci.nih.gov/gominer/index.jsp Biological interpretation of genomic microarray and proteomic data by incorporating statistical analysis, visualization platform, and query output. |
Resources for assembling protein interaction networks
As a first step in interactions data analysis, all proteins must have an associated identifier, which allows retrieval of known attributes (e.g., chemical and biological feature sets) from relational databases. While the selection of identifiers is not trivial, the choice is primarily between two distinct classes, gene-centric or protein/organism-specific identifiers. For the former, mapping of protein groups back to gene symbols often provides greatest coverage across databases and does not a priori restrict attribute retrieval by organism. In contrast, selection of database-specific (UniProt accessions, NCBI gi numbers) or species-specific (SGD, MGD, FlyBase) identifiers is often advantageous for retrieving the most accurate functional attributes for the intended target protein/protein group. Unless the software tool provides user control of organism selection, the identifier system is recommended, particularly since many well-developed knowledgebases, such as UniProt, allow facile conversion of accessions to other identifiers. As a second step, gathering general information for a list of putative protein interactions (e.g., protein length, amino acid sequence, phylogenetic data, protein abundance) is often beneficial for subsequent interaction network analysis, as this information may be used to generate a richer picture of the topology of individual protein clusters or larger protein networks. For example, NSAF and PAX abundance values, discussed earlier in this review, reveal valuable information on the extent of enrichment of individual proteins within a protein cluster and could point to a housekeeping role for highly represented interactions. The calculation of such values depends on the availability of general information on protein properties, in particular protein length and global protein abundance. Great resources for obtaining such protein characteristics are protein interaction databases, discussed in great detail below, but primarily the major protein knowledgebases, such as UniProt (Universal Protein Resource Database)136, PIR (Protein Information Resource)137, and RefSeq (NCBI Reference Sequence collection)138, are used for this purpose.
Having gathered initial details on all protein interactions to be analyzed, a common approach used for reconstructing protein complexes and performing network analysis involves the utilization of protein interaction databases (PIDs). These PIDs often contain both direct and indirect protein functional relationships, which have been collected from published literature, experimental data, text-mined abstracts, and computationally-based predictions representing the likelihood of interaction. Table 2 presents a detailed list of universally applicable PIDs. Information about proteins and protein interactions is collected by the process of curation (e.g. MINT139, IntAct140, 141, MPact142) or by employment of prediction softwares that analyze previously published data by text-mining (e.g. Predictome143, 144). Many databases are built on both approaches and are complemented by data deposition from scientists in the appropriate field in an effort to generate the most comprehensive database possible (e.g. HPRD145, DIP146, MPIDB147). Generally, the curation approach, which relies primarily on experimental data is preferred to prediction-based computational tools, as it is based on manual gathering of information from published literature performed by recruited expert scientists. This is why many databases are compiled solely on the basis of manual data curation, with some being even more stringent in their data collection and including only information from experimental data (e.g. MINT139, DIP146). Since variation in the interpretation of data due to human error is possible, it is useful to perform data analysis based on information recruited from multiple PIDs. This strategy was recently applied by Meixner et al.128, utilizing two independent databases (HPRD145 and BioGRID148) for characterizing interactions of the Leucine-rich Repeat Kinase 2.
In addition to the variation in the method of data collection, PIDs also differ in other aspects, including the range of covered species, how comprehensive a given database is, and the specificity of input data (e.g., protein-protein, protein-polysaccharides, protein-small molecule interactions). Thus, it is important to take into account such differences when selecting the databases that suit best a particular study. Multiple databases cover a broad range of species, without particular limitations, including IntAct140, 141, BIND149, DIP146, and MINT139, while others are more specialized and focus on specific species or a subset of commonly studied ones. The MPact142 and the HPID150 databases, for example, include only data from the yeast specie Saccharomyces cerevisiae and humans, respectively, while the MITOP2151 database is built on data exclusively from yeast, mouse, Arabidopsis thaliana, neurospora and humans. There are also databases, such as HPID, that integrate information from other resources like BIND, DIP, and HPRD. Such “secondary” databases collect information from multiple primary databases, offering a comprehensive set of information about putative protein interactions (e.g. IMEx152). At times, more specialized databases can allow for more specific analyses, especially when a particular type of proteins or system is the focus of investigation. One such case is the MatrixDB153 database that aims to report data primarily from extracellular protein interactions and protein-polysaccharides associations. Another is the HIV-1 Human Protein Interaction Database154 that, as implied, recruits data specifically on HIV-1 viral proteins and known protein interactions.
One concern with the utilization of a wide variety of interaction databases over the years has been the fact that with such a vast array of data available, reported and analyzed in many different formats and on multiple platforms, a universal system is required to standardize all molecular interaction data. The initiative to solve this problem was launched by the Human Proteome Organization Proteomics Standards Initiative (HUPO-PSI)155 with the objective to establish community standards for proteomics data representation and to facilitate exchange, verification, and comparison of interactions data. As a result, the International Molecular Exchange (IMEx) consortium (Table 2) to standardize protein interaction data curation was created152, and data providers (e.g., DIP, IntAct, MINT, BioGRID, MatrixDB) have agreed to coordinate literature curation efforts and share information to decrease data redundancy. Another step forward was the introduction of standard data formats, such as Molecular Interaction (MI) extensible markup language (XML), MITAB156, and mzIdentML157.
Network mapping and visualization tools
The next step in data analysis involves the generation of network maps and their visualization. Some interaction databases have incorporated tools for these purposes (e.g., STRING158, VisANT144, Bacteriome159 in Table 2). STRING, for example, creates interactive protein networks with customizable view options of interaction clusters built on the basis of fusion, co-expression, experimental, neighborhood, and text-mining data, among others methods. However, protein interaction databases are best utilized when complemented by other tools, specifically developed for visualization and representation of protein networks and maps. Table 3 provides a list of some of these commonly utilized computational tools. Of note, Cytoscape160 is a widely used software supporting genetic and molecular interaction datasets in multiple formats, and allowing for advanced data analysis and modeling. This effective tool is commonly used for functional protein annotation and visual mapping of interactions from small- and large-scale studies. In addition, numerous Cytoscape plug-ins are available to integrate external functional attributes to the network of interest, including tools for protein clustering by gene ontology annotations, as discussed below. Other examples of useful resources include Pathway Palette161 and Arena3D162. The first is practical for representing proteomics data in the context of biological pathways or protein networks on the basis of peptide rather than protein input data. Arena3D has been specifically developed for three dimensional representations of complex data sets and bigger protein networks. Such platforms have been incorporated in recent analyses of proteomics data from affinity isolation-based studies.128, 163
Functional analysis of protein interactions
Interpretation of large datasets of protein interactions can be a challenging task but it is facilitated greatly by analysis of clustered protein associations based on gene ontology (GO) annotations. Functional analysis based on GO annotations can be utilized before or after constructing the protein network maps. This piece of the puzzle provides valuable functional information about the putative biological roles of identified protein complexes and can point researchers in the right direction for further follow-up studies. The three main GO categories that are particularly informative for interpretation of protein interactions are biological process, cellular component, and molecular function (Fig. 3). Biological process clusters reveal a general process or pathway that protein complexes may be involved in, for example cell cycle or transcription; cellular component clusters provide information about the putative localization of identified proteins within cellular compartments or established complexes; and molecular function points to the role of protein in the cell with regard to its activity, for example, kinase or demethylase activity. If the functional analysis based on GO annotations is performed prior to constructing protein networks, this analysis can help narrow down the list of putative interaction partners to specific categories of interest (e.g., protein localization or biological role). Therefore, protein interactions from each category can be further subjected to network analysis to highlight any well-defined protein complexes. Additionally, in cases where the localization of the bait is strictly distinct (e.g., nucleus, mitochondria), contaminating protein interactions restricted to other compartments can be easily filtered if the interaction data are first clustered by GO localization.
There are numerous resources for GO annotation databases. Table 3 provides a list of key features for some of these resources, such as GOA164, GOMiner165, GO::TermFinder166. Particularly useful are multiple plug-ins developed for Cytoscape.160 For instance, ClueGO167, BiNGO168 and DAVID169 were specifically developed to provide GO classification, statistical enrichment of GO terms, and subsequent visualization. These are commonly used in large- and small-scale proteomics studies. For example, Echeverria et al. integrated ClueGO in an analysis of the Heat shock protein Hsp90 molecular chaperone machinery to build a functional map of protein clusters linked to biological processes.170 In another study, Doolittle and Gomez utilized DAVID as a tool to analyze the biological role of predicted protein interactions formed during Dengue virus infection in human and insect host cells.171
Overall, these tools can aid in analyzing large datasets obtained from immunoaffinity purification studies by constructing protein interactome networks. Identification of novel complexes and/or functional relationships is often complemented by orthogonal validation of selected protein associations of interest, as described in the section on “Determining specificity of interactions”. The resulting functional networks can serve as a foundation for understanding the biological roles of the observed associations.
CROSS-LINKING IN PROTEIN INTERACTION STUDIES
Cross-linking strategies coupled to mass spectrometry analyses have become fundamental tools for studying protein interactions and complexes. This section describes some of the main cross-linking methods in use today and their application to protein interaction studies. In particular, approaches to gain either structural insights into protein complex or to identify specific stable and transient interactions in vitro and in vivo will be discussed.
Cross-linking is the process of joining molecules through formation of covalent bonds. Reagents for protein cross-linking include moderately-reactive reagents for chemical cross-linking and highly-reactive intermediates generated during photo-cross-linking.172 Currently, chemical cross-linking is more frequently used in MS studies than photo-cross-linking. The specificity of cross-linking reagents determines the targeted protein groups to be linked (e.g., amines, sulfhydryls, carboxyls), while the size of the cross-linker determines the distance between captured proteins (Fig. 4). To optimize cross-linking strategies for MS analysis (i.e., to reduce the complexity of cross-linking samples and improve detection of cross-linked peptides), cross-linking reagents with several new features were developed. These features include affinity tags, chemically- or MS-reversible cross-linkers, and isotope labels for ease of detection and identification by MS. Several comprehensive reviews describe the structures and properties of available cross-linking reagents.172-174 Figure 4 illustrates how different cross-linking strategies can be utilized to study protein interactions within existing mass spectrometry workflows.
Figure 4. Common steps in integrated cross-linking and mass spectrometry workflows.
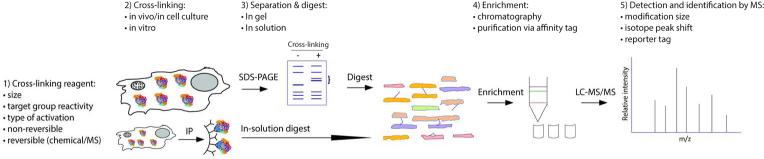
Cross-linking reagents, chosen based on experimental design and goals, can be utilized in vivo, in cell culture, or in vitro. The resulting protein mixture is digested in solution or after SDS-PAGE separation to reduce sample complexity. Cross-linked peptides can be enriched by chromatography or affinity isolation of tagged cross-links. Samples are analyzed by mass spectrometry, where different specialized algorithms can be utilized for detection and subsequent identification of cross-linked peptides and proteins.
Elucidation of protein complex structures
Protein cross-linking coupled to MS analysis is a useful tool for structural characterization of heterogeneous protein complexes and serves as an alternative to crystallography- and NMR-based approaches. In earlier studies that utilized cross-linking coupled to mass spectrometry analysis, several challenges had to be addressed.175-177 Cross-linking reagents with different properties had to be tested to maximize product diversity for in-depth structural analysis. In addition, the amount of available starting material and the instrument sensitivity had a critical impact on the detection of cross-linked sites. Due to these limitations, early approaches were applicable to the analysis of smaller protein complexes. In recent years, cross-linking methodologies have improved through enrichment for and labeling of cross-linked peptides for easier detection, improved sensitivity in MS analysis, and development of automated algorithms for database searching.172, 173, 178
As a result of improved MS instrument sensitivity, cross-linking combined with MS analysis has been successfully used in a variety of structural studies, including analysis of yeast 19S proteasome lid, yeast RNA Pol II, human protein phosphatase 2A (PP2A) complexes, and phage DNA packaging machinery.179-183 Sharon et al. cross-linked the endogenous yeast 19S proteasome lid complex by bis(sulfosuccinimidyl)suberate (BS3) amine-reactive reagent and analyzed gel bands corresponding to cross-linked proteins by MALDI-TOF/TOF. As a result, this analysis was able to identify additional interactions within the complex.179 To improve detection of cross-linked peptides, Chen et al. applied charge-based enrichment of BS3-cross-linked peptides and high-resolution MS analysis for elucidation of a 15-subunit Pol II complex structure.180 Cross-linking using disuccinimidyl tartrate (DST) has been integrated with mutagenesis analysis in studies of phage packaging mechanisms.181 More recently, Aebersold and colleagues used isotopically light- or heavy-labeled disuccinimidyl suberate (DSS) cross-linkers to study the topology of affinity-purified human protein phosphatase 2A (PP2A) in complex with its adaptor proteins.182 To identify isotopically coded cross-linked, the xQuest search engine was employed.178 The algorithm works by first analyzing MS spectra for isotopic peptide pairs, which are separately sequenced by MS/MS and analyzed for the presence of common ions or isotopically shifted, cross-linked ions. Caveats of this strategy include requirements for efficient labeling with isotopically light or heavy cross-linkers and labor-intensive scoring procedure.174 An alternative workflow that does not make use of isotopically labeled cross-linkers was developed by Goodlett and co-workers.184 This strategy utilizes the Popitam software185, and the cross-linked peptides are considered to be modified at unknown residues with unknown modifications. The resulting cross-linked pair of peptides would have matching modifications, in which one peptide from the pair is modified with a mass equivalent to the mass of the other peptide in the pair plus the cross-linker, and vice versa. Limitations of this approach include the ability of Popitam to effectively match the theoretical spectrum for a single peptide to the more complex spectrum of a cross-linked peptide pair, as well as the requirement for manual verification of cross-linked peptide spectra.186 To address these challenges, a database containing every possible cross-linked product was generated for use with a SEQUEST-style search.186 Additionally, xComb, a publicly available database processing tool, was introduced for use with standard proteome search engine to simplify the identification of cross-linked peptides.187 Other bioinformatics tools were also developed to aid the interpretation of complex cross-linking peptides spectra.188-193
Although the development of search algorithms for cross-linked peptide spectra contributed significantly to the optimization of cross-linking and MS workflows, interpretation of spectra remains labor-intensive, partly due to the low abundance of cross-linked peptides. Several strategies that address this challenge have been proposed, including enrichment of cross-linked peptides using affinity tags194-197, detection by isotope-labeling182, 198-201, fluorescence-labeling,202 or reporter tag labeling,194, 203 as well as cleavable cross-linkers194, 200, 201, 204-207. For example, Chowdhury et al. developed the CLIP (click-enabled linker for interacting proteins) reagent that contains an alkyne tag for enrichment of cross-linked peptides and a detection tag (NO2) for identification of cross-linked peptides in MS/MS spectra.196 Zelter et al. labeled the C-termini of cross-linked peptides with stable isotopes during digestion in the presence of H218O, leading to peptide identification based on signature isotopic peak distributions in MS spectra.208 This method overcomes the need for isotope-coded cross-linkers, detection of peptide mass shifts, and manual inspection. To characterize the yeast 20S proteasome complex, Kao et al. utilized a disuccinimidyl sulfoxide (DSSO) cross-linker with collision-induced dissociation (CID)-cleavable sites that cause separation of cross-linked peptides at the MS/MS level, allowing the identification of peptide chain fragment ions at the MS3 level.201 Overall, the development of new cross-linking reagents in conjunction with careful mass spectrometry analysis has contributed significantly to generation of structural views of protein complexes. In combination with computational modeling approaches, cross-linking enables reconstruction of the architectures of heterogeneous protein complexes at high levels of resolution182, and is expected to continue to impact this field of study.
Capturing stable and transient protein interactions
Understanding the composition and assembly of macromolecular complexes can provide valuable insight into their functions. Additionally, transient interactions, such as enzyme-substrate interactions, are known to contribute significantly to the dynamic regulation of protein functions. However, these transient interactions are particularly challenging to be captured by traditional IP experiments. For example, in the previously mentioned study by Malovannaya et al.102, variability between the abundances (i.e., spectral counts) of co-isolated proteins was considered indicative of non-specific binding. However, exclusion of proteins that demonstrated significant variation in their spectral counts between isolations may simultaneously eliminate transient interactions (false negatives). Cross-linking methods have provided effective tools for studying both stable and transient interactions in vitro and in vivo. As an example of an in vitro study, Lambert et al. utilized BS3 cross-linking to identify both the transient interaction itself and the specific residues involved in binding of small heat-shock protein (HSp21) to its substrate, malate dehydrogenase (MDH).209 Cross-linking was successfully incorporated into IP-MS/MS workflows for identification of transient or weak interactions under stringent purification conditions. One major challenge is performing in vivo cross-linking to capture stable, as well as spatially and temporally transient specific interactions.210 Guerrero et al. developed the QTAX (quantitative analysis of tandem affinity purified cross-linked protein complexes) approach to determine the composition of the yeast 26S proteasome.211 This approach employs a similar concept to metabolic labeling methodologies (i.e., I-DIRT124 and SILAC123, see section on “Determining specificity of interactions”), integrating it with formaldehyde cross-linking in cell culture and tandem affinity purification. Informatively, proteins at the core of the 25S proteasome were represented by smaller numbers of peptides, consistent with their likelihood of low accessibility by trypsin. Reversible cross-linking may be used to resolve this issue. One such method, ReCLIP (reversible cross-link immune-precipitation) was recently proposed by Smith et al.212 Cross-linking via DSP (dithiobis succinimidyl propionate) and DTME (Dithio-bismaleimidoethane) cell-permeable reagents that are thiol-cleavable can be reversed using a reducing agent such as dithiothreitol (DTT). Another example of cross-linking in cell culture for identifying transient interactions is the study of the dynamic NuA3 histone acetyltransferase complex.213 Through integration of metabolic labeling using I-DIRT (see section on “Determining specificity of interactions”) with cross-linking and IPMS/MS, transient interactions were identified and placed in the context of interaction specificity.125
While numerous studies have shown the effective use of cross-linking in cell culture, its use in animal models was recently demonstrated. Bai et al. utilized in vivo time-controlled transcardiac perfusion cross-linking214 with 4% formaldehyde to study amyloid precursor protein (APP) interactions in mice.215 This method allowed for limited cross-linking to occur in tissue, followed by purification of protein complexes in the presence of high salt and detergent concentrations that are particularly useful for studying membrane-bound proteins. As a control for non-specific interactions, parallel immunoaffinity purifications of APP paralogues were performed. While the utility of this approach was apparent by the identification of both known and novel APP interactions, several well-characterized APP-binding proteins were absent. These results highlight some of the remaining difficulties in this approach, including the precise selection of the time for in vivo cross-linking, the accessibility of the interacting surfaces for cross-linking, and the optimization of lysis buffer conditions.
A novel cross-linking reagent called PIR (protein interaction reporter), which contains labile bonds with a reporter group and an affinity tag, was developed in the Bruce laboratory with the goal of reducing sample and data output complexity in the MS/MS workflow.216 The labile bonds of PIR can be selectively cleaved following either LC separation by photo-activation or during the mass spectrometry analysis by any dissociation mechanism, thus releasing peptides that can be identified by a standard MS/MS workflow. A reporter group marks the spectra containing these cleaved products. PIR technology was shown to be effective in cell culture, as well as for cross-linking of virions.205, 217, 218
One approach that has not been yet integrated with mass spectrometry, but has the potential to be used in IP-MS/MS workflows, is the metabolic incorporation of modified amino acids that can generate cross-links in growing eukaryotic cells.219 Photo-inducible amino acids that resemble leucine and methionine were metabolically incorporated into protein chains and activated by UV light to initiate cross-linking reactions. This approach addresses the limitation of currently available cross-linking reagents that cannot efficiently penetrate cell membranes to achieve high levels of cross-linking.
In summary, recent developments in cross-linking approaches with utility in cell culture or in vivo supply new pipelines for distinguishing stable from transient interactions. Therefore, it is expected that these approaches will further contribute to elucidating the spatial and temporal regulation of cellular pathways involved in the proper maintenance of cellular functions, as well as pathways crucial in disease-associated mechanisms.
Summary Statement
As summarized in this review, developments in IP-MS experimental workflows have significantly aided the identification and characterization of protein interactions in different biological contexts. Given the increasing sensitivity of mass spectrometry, the interpretation of the resulting large datasets of protein interactions was made possible by developments in computational tools for constructing and visualizing interaction networks. These functional protein networks can be powerful in guiding follow-up biological studies for elucidating the functions of identified interactions. Furthermore, advances in label-free and labeling quantitative approaches for assessing specificity of interactions have contributed to improving the outcome and reliability of IP-MS experiments. With the current expansion of the proteomics field, a better understanding of protein interactions within their temporal and spatial biological context is timely and necessary. Distinguishing stable from transient interactions within a cellular pathway can provide important insights into its normal functions, as well as disregulation in disease. This is made possible by the continuous integration of multidisciplinary methods within the proteomics arsenal of tools. For example, cross-linking has become an integral part of protein interaction studies using mass spectrometry-based methods. Similarly, the combination of microscopy and proteomics has placed protein expression, interactions and posttranslational modifications in the context of localization. Therefore, these are exciting years in advancing our understanding of protein function. We envisage that in coming years, the methodologies will continue to expand and add to our ability to study the temporal and spatial dynamics of protein interactions. Furthermore, knowledge of interactions within different cell types, tissues, or following different environmental stimuli or pathogen infection will be necessary for creating a more complete view of protein regulation. Identifying protein interactions involved in the initiation or progression of disease can provide an array of condition-specific targets for development of new therapeutics.
Acknowledgments
We are grateful for funding from NIH/NIDA grant DP1DA026192, NIH grant R21AI102187, and HFSPO award RGY0079/2009-C to IMC, and an NSF graduate fellowship to HGB. We also thank Amanda J. Guise and Todd M. Greco in the Cristea lab for critical reading of the manuscript.
Biography
Ileana M. Cristea has performed her graduate research at the Michael Barber Center for Mass Spectrometry, University of Manchester, U.K., under the supervision of Simon Gaskell. She then pursued her postdoctoral work in the mass spectrometry laboratory of Brian Chait at The Rockefeller University. She is currently an Assistant Professor in the Department of Molecular Biology at Princeton University, where her laboratory focuses on understanding mechanisms that control the fate of cells under invasion by pathogens, with particular emphasis on the viral modulation of chromatin remodeling machineries. Her research employs a multi-disciplinary approach, integrating modern mass spectrometry and proteomics with molecular biology, biochemistry, and virology. She is the recipient of the Bordoli Prize from the British Mass Spectrometry Society (2001), NIDA Avant-Garde Award for HIV/AIDS Research (2008), Human Frontiers Science Program Young Investigator Award (2009), Early Career Award in Mass Spectrometry from the American Chemical Society NJ Section (2011), and the American Society for Mass Spectrometry Research Award (2012).
Yana V. Miteva completed her B.A. degree in Biochemistry and Molecular Biology at the Pennsylvania State University, during which she performed research in the Robert Schlegel lab and completed a one-year science exchange program at The University Louis Pasteur, France. She then held a research specialist position in the Physiology Department at the University of Pennsylvania in the lab of Todd Lamitina. In 2008 she joined Princeton University and is currently finalizing her Ph.D. thesis work in the Cristea lab, investigating the biological roles of deacetylating complexes during viral infection.
Hanna G. Budayeva completed her B.S. degree in Biochemistry at Rider University, NJ. Her undergraduate research in laboratory of Bryan Spiegelberg focused on in vitro binding assays for characterizing protein interactions involved in transcription regulation. She is presently a Ph.D. student in the Cristea laboratory at Princeton University. In her research, she utilizes systems biology approaches to study changes in sirtuin interactions and downstream pathways during environmental stresses. She is the recipient of a National Science Foundation Graduate Research fellowship.
References
- 1.Greco TM, Yu F, Guise AJ, Cristea IM. Molecular & cellular proteomics: MCP. 2011;10:M110.004317–M004110.004317. doi: 10.1074/mcp.M110.004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li T, Diner BA, Chen J, Cristea IM. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10558–10563. doi: 10.1073/pnas.1203447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Old WM, Shabb JB, Houel S, Wang H, Couts KL, Yen CY, Litman ES, Croy CH, Meyer-Arendt K, Miranda JG, Brown RA, Witze ES, Schweppe RE, Resing KA, Ahn NG. Molecular cell. 2009;34:115–131. doi: 10.1016/j.molcel.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farley AR, Powell DW, Weaver CM, Jennings JL, Link AJ. J Proteome Res. 2011;10:1481–1494. doi: 10.1021/pr100877m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cristea IM, Rozjabek H, Molloy KR, Karki S, White LL, Rice CM, Rout MP, Chait BT, MacDonald MR. Journal of virology. 2010;84:6720–6732. doi: 10.1128/JVI.01983-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai YC, Greco TM, Boonmee A, Miteva Y, Cristea IM. Molecular & cellular proteomics : MCP. 2012;11:60–76. doi: 10.1074/mcp.A111.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin Z-Y, Breitkreutz B-J, Stark C, Liu G, Ahn J, Dewar-Darch D, Reguly T, Tang X, Almeida R, Qin ZS, Pawson T, Gingras A-C, Nesvizhskii AI, Tyers M. Science (New York, N.Y.) 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Sali A, Rout MP. Nature. 2007;450:695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- 9.Hutchins JRA, Toyoda Y, Hegemann B, Poser I, Hériché J-K, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, Conrad C, Comartin D, Schleiffer A, Sarov M, Pozniakovsky A, Slabicki MM, Schloissnig S, Steinmacher I, Leuschner M, Ssykor A, Lawo S, Pelletier L, Stark H, Nasmyth K, Ellenberg J, Durbin R, Buchholz F, Mechtler K, Hyman AA, Peters J-M. Science (New York, N.Y.) 2010;328:593–599. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babu M, Vlasblom J, Pu S, Guo X, Graham C, Bean BD, Burston HE, Vizeacoumar FJ, Snider J, Phanse S, Fong V, Tam YY, Davey M, Hnatshak O, Bajaj N, Chandran S, Punna T, Christopolous C, Wong V, Yu A, Zhong G, Li J, Stagljar I, Conibear E, Wodak SJ, Emili A, Greenblatt JF. Nature. 2012;489:585–589. doi: 10.1038/nature11354. [DOI] [PubMed] [Google Scholar]
- 11.Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, Ding C, Shi Y, Yucer N, Krenciute G, Kim BJ, Li C, Chen R, Li W, Wang Y, O'Malley BW, Qin J. Cell. 2011;145:787–799. doi: 10.1016/j.cell.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moorman NJ, Sharon-Friling R, Shenk T, Cristea IM. Molecular & cellular proteomics: MCP. 2010;9:851–860. doi: 10.1074/mcp.M900485-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez-Martinez J, Phillips J, Sekedat MD, Diaz-Avalos R, Velazquez-Muriel J, Franke JD, Williams R, Stokes DL, Chait BT, Sali A, Rout MP. The Journal of cell biology. 2012;196:419–434. doi: 10.1083/jcb.201109008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao J, Yu H, Lin L, Tu J, Cai L, Chen Y, Zhong F, Lin C, He F, Yang P. Journal of proteomics. 2011;75:588–602. doi: 10.1016/j.jprot.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 15.Singh VK, Munro K, Jia Z. Cellular signalling. 2012;24:1790–1796. doi: 10.1016/j.cellsig.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Liu D, Qian D, Dai J, An Y, Jiang S, Stanley B, Yang J, Wang B, Liu DX. The Journal of biological chemistry. 2012;287:19599–19609. doi: 10.1074/jbc.M112.363622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carabetta VJ, Silhavy TJ, Cristea IM. Journal of bacteriology. 2010;192:3713–3721. doi: 10.1128/JB.00300-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carabetta VJ, Li T, Shakya A, Greco TM, Cristea IM. J Am Soc Mass Spectrom. 2010;21:1050–1060. doi: 10.1016/j.jasms.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dokudovskaya S, Waharte F, Schlessinger A, Pieper U, Devos DP, Cristea IM, Williams R, Salamero J, Chait BT, Sali A, Field MC, Rout MP, Dargemont C. Molecular & cellular proteomics: MCP. 2011;10:M110.006478–M006110.006478. doi: 10.1074/mcp.M110.006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cristea IM, Moorman NJ, Terhune SS, Cuevas CD, O'Keefe ES, Rout MP, Chait BT, Shenk T. Journal of virology. 2010;84:7803–7814. doi: 10.1128/JVI.00139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guise AJ, Greco TM, Zhang IY, Yu F, Cristea IM. Molecular & cellular proteomics: MCP. 2012 doi: 10.1074/mcp.M112.021030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni Z, Olsen JB, Emili A, Greenblatt JF. Methods Mol Biol. 2011;781:31–45. doi: 10.1007/978-1-61779-276-2_2. [DOI] [PubMed] [Google Scholar]
- 23.Domanski M, Molloy K, Jiang H, Chait BT, Rout MP, Jensen TH, Lacava J. BioTechniques. 2012;0:1–6. doi: 10.2144/000113864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Guo C, Chen Y, Shulha HP, Schnetz MP, LaFramboise T, Bartels CF, Markowitz S, Weng Z, Scacheri PC, Wang Z. Nature methods. 2008;5:163–165. doi: 10.1038/nmeth1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poser I, Sarov M, Hutchins JR, Heriche JK, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW, Pelletier L, Kittler R, Hua S, Naumann R, Augsburg M, Sykora MM, Hofemeister H, Zhang Y, Nasmyth K, White KP, Dietzel S, Mechtler K, Durbin R, Stewart AF, Peters JM, Buchholz F, Hyman AA. Nature methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciotta G, Hofemeister H, Maresca M, Fu J, Sarov M, Anastassiadis K, Stewart AF. Methods. 2011;53:113–119. doi: 10.1016/j.ymeth.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Selimi F, Cristea IM, Heller E, Chait BT, Heintz N. PLoS biology. 2009;7:e83. doi: 10.1371/journal.pbio.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brizzard B. BioTechniques. 2008;44:693–695. doi: 10.2144/000112841. [DOI] [PubMed] [Google Scholar]
- 30.Trinkle-Mulcahy L. Proteomics. 2012;12:1623–1638. doi: 10.1002/pmic.201100438. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z. Methods in molecular biology (Clifton, N.J.) 2009;567:87–98. doi: 10.1007/978-1-60327-414-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang L, Hernan R, Brizzard B. Molecular biotechnology. 2001;19:313–321. doi: 10.1385/MB:19:3:313. [DOI] [PubMed] [Google Scholar]
- 33.Law JA, Ausin I, Johnson LM, Vashisht AA, Zhu JK, Wohlschlegel JA, Jacobsen SE. Current biology : CB. 2010;20:951–956. doi: 10.1016/j.cub.2010.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O'Connor L, Li M, Taylor R, Dharsee M, Ho Y, Heilbut A, Moore L, Zhang S, Ornatsky O, Bukhman YV, Ethier M, Sheng Y, Vasilescu J, Abu-Farha M, Lambert JP, Duewel HS, Stewart II, Kuehl B, Hogue K, Colwill K, Gladwish K, Muskat B, Kinach R, Adams SL, Moran MF, Morin GB, Topaloglou T, Figeys D. Molecular systems biology. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cristea IM, Williams R, Chait BT, Rout MP. Molecular & cellular proteomics: MCP. 2005;4:1933–1941. doi: 10.1074/mcp.M500227-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Cheeseman IM, Niessen S, Anderson S, Hyndman F, Yates JR, 3rd, Oegema K, Desai A. Genes & development. 2004;18:2255–2268. doi: 10.1101/gad.1234104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bloom J, Cristea IM, Procko AL, Lubkov V, Chait BT, Snyder M, Cross FR. The Journal of biological chemistry. 2011;286:5434–5445. doi: 10.1074/jbc.M110.205054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver RC, Miller JC, Lee YL, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D, Allis CD. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conlon FL, Miteva Y, Kaltenbrun E, Waldron L, Greco TM, Cristea IM. Methods in molecular biology (Clifton, N.J.) 2012;917:369–390. doi: 10.1007/978-1-61779-992-1_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heller EA, Zhang W, Selimi F, Earnheart JC, Slimak MA, Santos-Torres J, Ibanez-Tallon I, Aoki C, Chait BT, Heintz N. PloS one. 2012;7:e39572. doi: 10.1371/journal.pone.0039572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheeseman IM, Desai A. Science's STKE : signal transduction knowledge environment. 2005;2005:pl1. doi: 10.1126/stke.2662005pl1. [DOI] [PubMed] [Google Scholar]
- 42.Cristea IM, Carroll J-WN, Rout MP, Rice CM, Chait BT, MacDonald MR. The Journal of biological chemistry. 2006;281:30269–30278. doi: 10.1074/jbc.M603980200. [DOI] [PubMed] [Google Scholar]
- 43.Terhune SS, Moorman NJ, Cristea IM, Savaryn JP, Cuevas-Bennett C, Rout MP, Chait BT, Shenk T. PLoS pathogens. 2010;6:e1000965. doi: 10.1371/journal.ppat.1000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. J Virol. 2012;86:7360–7371. doi: 10.1128/JVI.00157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kramer T, Greco TM, Enquist LW, Cristea IM. Journal of virology. 2011;85:6427–6441. doi: 10.1128/JVI.02253-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H. Molecular & cellular proteomics : MCP. 2008;7:282–289. doi: 10.1074/mcp.M700342-MCP200. [DOI] [PubMed] [Google Scholar]
- 47.Pichler G, Leonhardt H, Rothbauer U. Methods in molecular biology (Clifton, N.J.) 2012;911:475–483. doi: 10.1007/978-1-61779-968-6_29. [DOI] [PubMed] [Google Scholar]
- 48.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B. Nature biotechnology. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 49.Gloeckner CJ, Boldt K, Schumacher A, Ueffing M. Methods in molecular biology (Clifton, N.J.) 2009;564:359–372. doi: 10.1007/978-1-60761-157-8_21. [DOI] [PubMed] [Google Scholar]
- 50.Burckstummer T, Bennett KL, Preradovic A, Schutze G, Hantschel O, Superti-Furga G, Bauch A. Nature methods. 2006;3:1013–1019. doi: 10.1038/nmeth968. [DOI] [PubMed] [Google Scholar]
- 51.Andres C, Agne B, Kessler F. Methods Mol Biol. 2011;775:31–49. doi: 10.1007/978-1-61779-237-3_3. [DOI] [PubMed] [Google Scholar]
- 52.Pflieger D, Bigeard J, Hirt H. Proteomics. 2011;11:1824–1833. doi: 10.1002/pmic.201000635. [DOI] [PubMed] [Google Scholar]
- 53.Lester JT, DeLuca NA. J Virol. 2011;85:5733–5744. doi: 10.1128/JVI.00385-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robb NC, Chase G, Bier K, Vreede FT, Shaw PC, Naffakh N, Schwemmle M, Fodor E. J Virol. 2011;85:5228–5231. doi: 10.1128/JVI.02562-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu R, Zhou M, Wu H. Applied and environmental microbiology. 2010;76:7966–7971. doi: 10.1128/AEM.01434-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Y. Biotechnology and applied biochemistry. 2010;55:73–83. doi: 10.1042/BA20090273. [DOI] [PubMed] [Google Scholar]
- 57.Xu X, Song Y, Li Y, Chang J, Zhang H, An L. Protein expression and purification. 2010;72:149–156. doi: 10.1016/j.pep.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 58.Gavin A-C, Maeda K, Kühner S. Current opinion in biotechnology. 2011;22:42–49. doi: 10.1016/j.copbio.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 59.Gloeckner CJ, Boldt K, Schumacher A, Roepman R, Ueffing M. Proteomics. 2007;7:4228–4234. doi: 10.1002/pmic.200700038. [DOI] [PubMed] [Google Scholar]
- 60.Rees JS, Lowe N, Armean IM, Roote J, Johnson G, Drummond E, Spriggs H, Ryder E, Russell S, St Johnston D, Lilley KS. Molecular & cellular proteomics : MCP. 2011;10:M110 002386. doi: 10.1074/mcp.M110.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maine GN, Gluck N, Zaidi IW, Burstein E. Cold Spring Harb Protoc. 2009;2009:pdb prot5318. doi: 10.1101/pdb.prot5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruigrok VJB, Levisson M, Eppink MHM, Smidt H, van der Oost J. The Biochemical journal. 2011;436:1–13. doi: 10.1042/BJ20101860. [DOI] [PubMed] [Google Scholar]
- 63.DeGrasse JA. PloS one. 2012;7:e33410. doi: 10.1371/journal.pone.0033410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wiens M, Schroder HC, Wang X, Link T, Steindorf D, Muller WE. Biochemistry. 2011;50:1981–1990. doi: 10.1021/bi101429x. [DOI] [PubMed] [Google Scholar]
- 65.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G. Nat Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 66.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Nat Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 67.Westblade LF, Minakhin L, Kuznedelov K, Tackett AJ, Chang EJ, Mooney RA, Vvedenskaya I, Wang QJ, Fenyo D, Rout MP, Landick R, Chait BT, Severinov K, Darst SA. J Proteome Res. 2008;7:1244–1250. doi: 10.1021/pr070451j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yaciuk P. Methods in molecular medicine. 2007;131:103–111. doi: 10.1007/978-1-59745-277-9_8. [DOI] [PubMed] [Google Scholar]
- 69.Archambault V, Li CX, Tackett AJ, Wasch R, Chait BT, Rout MP, Cross FR. Molecular biology of the cell. 2003;14:4592–4604. doi: 10.1091/mbc.E03-06-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grabski AC. Methods in enzymology. 2009;463:285–303. doi: 10.1016/S0076-6879(09)63018-4. [DOI] [PubMed] [Google Scholar]
- 71.Altelaar AF, Heck AJ. Current opinion in chemical biology. 2012;16:206–213. doi: 10.1016/j.cbpa.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 72.Trinkle-Mulcahy L, Boulon S, Lam YW, Urcia R, Boisvert FM, Vandermoere F, Morrice NA, Swift S, Rothbauer U, Leonhardt H, Lamond A. The Journal of cell biology. 2008;183:223–239. doi: 10.1083/jcb.200805092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Everberg H, Leiding T, Schioth A, Tjerneld F, Gustavsson N. J Chromatogr A. 2006;1122:35–46. doi: 10.1016/j.chroma.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 74.Norris JL, Porter NA, Caprioli RM. Analytical chemistry. 2003;75:6642–6647. doi: 10.1021/ac034802z. [DOI] [PubMed] [Google Scholar]
- 75.Peach M, Marsh N, Macphee DJ. Methods Mol Biol. 2012;869:37–47. doi: 10.1007/978-1-61779-821-4_4. [DOI] [PubMed] [Google Scholar]
- 76.Ayyar BV, Arora S, Murphy C, O'Kennedy R. Methods (San Diego, Calif.) 2012;56:116–129. doi: 10.1016/j.ymeth.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 77.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Nature methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 78.Antrobus R, Borner GH. PloS one. 2011;6:e18218. doi: 10.1371/journal.pone.0018218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van den Heuvel RH, Heck AJ. Current opinion in chemical biology. 2004;8:519–526. doi: 10.1016/j.cbpa.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 80.Barrera NP, Robinson CV. Annual review of biochemistry. 2011;80:247–271. doi: 10.1146/annurev-biochem-062309-093307. [DOI] [PubMed] [Google Scholar]
- 81.Breuker K, McLafferty FW. Proceedings of the National Academy of Sciences. 2008;105:18145–18152. doi: 10.1073/pnas.0807005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chait BT. Annual review of biochemistry. 2011;80:239–246. doi: 10.1146/annurev-biochem-110810-095744. [DOI] [PubMed] [Google Scholar]
- 83.Robinson CV. Cold Spring Harb Protoc. 2009;2009:pdb prot5180. doi: 10.1101/pdb.prot5180. [DOI] [PubMed] [Google Scholar]
- 84.Jia W, Shaffer JF, Harris SP, Leary JA. J Proteome Res. 2010;9:1843–1853. doi: 10.1021/pr901006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fonslow BR, Kang SA, Gestaut DR, Graczyk B, Davis TN, Sabatini DM, Yates JR., 3rd Analytical chemistry. 2010;82:6643–6651. doi: 10.1021/ac101235k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Prasad R, Williams JG, Hou EW, Wilson SH. Nucleic acids research. 2012 doi: 10.1093/nar/gks898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li BZ, Huang Z, Cui QY, Song XH, Du L, Jeltsch A, Chen P, Li G, Li E, Xu GL. Cell research. 2011;21:1172–1181. doi: 10.1038/cr.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Strambio-de-Castillia C, Tetenbaum-Novatt J, Imai BS, Chait BT, Rout MP. J Proteome Res. 2005;4:2250–2256. doi: 10.1021/pr0501517. [DOI] [PubMed] [Google Scholar]
- 89.Dubois F, Vandermoere F, Gernez A, Murphy J, Toth R, Chen S, Geraghty KM, Morrice NA, MacKintosh C. Molecular & cellular proteomics : MCP. 2009;8:2487–2499. doi: 10.1074/mcp.M800544-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Darie CC, Deinhardt K, Zhang G, Cardasis HS, Chao MV, Neubert TA. Proteomics. 2011;11:4514–4528. doi: 10.1002/pmic.201000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Nature. 2011;480:254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Camacho-Carvajal MM, Wollscheid B, Aebersold R, Steimle V, Schamel WW. Molecular & cellular proteomics : MCP. 2004;3:176–182. doi: 10.1074/mcp.T300010-MCP200. [DOI] [PubMed] [Google Scholar]
- 93.Huang R, Yu M, Li CY, Zhan YQ, Xu WX, Xu F, Ge CH, Li W, Yang XM. Proteomics. Clinical applications. 2012;6:467–475. doi: 10.1002/prca.201200009. [DOI] [PubMed] [Google Scholar]
- 94.Domon B, Aebersold R. Science. 2006;312:212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 95.Walther TC, Mann M. The Journal of cell biology. 2010;190:491–500. doi: 10.1083/jcb.201004052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mallick P, Kuster B. Nat Biotechnol. 2010;28:695–709. doi: 10.1038/nbt.1658. [DOI] [PubMed] [Google Scholar]
- 97.Wu Z, Moghaddas Gholami A, Kuster B. Molecular & cellular proteomics : MCP. 2012;11:M111 016675. doi: 10.1074/mcp.M111.016675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luo Y, Li T, Yu F, Kramer T, Cristea IM. Journal of the American Society for Mass Spectrometry. 2010;21:34–46. doi: 10.1016/j.jasms.2009.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu H, Yuan Y, Guo H, Mitchelson K, Zhang K, Xie L, Qin W, Lu Y, Wang J, Guo Y, Zhou Y, He F. J Proteome Res. 2012 doi: 10.1021/pr2012297. [DOI] [PubMed] [Google Scholar]
- 100.Daniels DL, Mendez J, Mosley AL, Ramisetty SR, Murphy N, Benink H, Wood KV, Urh M, Washburn MP. J Proteome Res. 2012;11:564–575. doi: 10.1021/pr200459c. [DOI] [PubMed] [Google Scholar]
- 101.Yang M, Chen Y, Zhou Y, Wang L, Zhang H, Bi LJ, Zhang XE. PloS one. 2012;7:e36666. doi: 10.1371/journal.pone.0036666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Malovannaya A, Li Y, Bulynko Y, Jung SY, Wang Y, Lanz RB, O'Malley BW, Qin J. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:2431–2436. doi: 10.1073/pnas.0912599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, Conrad C, Comartin D, Schleiffer A, Sarov M, Pozniakovsky A, Slabicki MM, Schloissnig S, Steinmacher I, Leuschner M, Ssykor A, Lawo S, Pelletier L, Stark H, Nasmyth K, Ellenberg J, Durbin R, Buchholz F, Mechtler K, Hyman AA, Peters JM. Science. 2010;328:593–599. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Braun P. Proteomics. 2012;12:1499–1518. doi: 10.1002/pmic.201100598. [DOI] [PubMed] [Google Scholar]
- 105.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, Edelmann A, Heurtier MA, Hoffman V, Hoefert C, Klein K, Hudak M, Michon AM, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick JM, Kuster B, Bork P, Russell RB, Superti-Furga G. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 106.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MH, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O'Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 107.Collins SR, Kemmeren P, Zhao XC, Greenblatt JF, Spencer F, Holstege FC, Weissman JS, Krogan NJ. Molecular & cellular proteomics : MCP. 2007;6:439–450. doi: 10.1074/mcp.M600381-MCP200. [DOI] [PubMed] [Google Scholar]
- 108.Mewes HW, Amid C, Arnold R, Frishman D, Guldener U, Mannhaupt G, Munsterkotter M, Pagel P, Strack N, Stumpflen V, Warfsmann J, Ruepp A. Nucleic acids research. 2004;32:D41–44. doi: 10.1093/nar/gkh092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Therien C, Bergeron D, Bourassa S, Greenblatt J, Chabot B, Poirier GG, Hughes TR, Blanchette M, Price DH, Coulombe B. Molecular cell. 2007;27:262–274. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. ELECTROPHORESIS. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 111.Rinner O, Mueller LN, Hubalek M, Muller M, Gstaiger M, Aebersold R. Nat Biotechnol. 2007;25:345–352. doi: 10.1038/nbt1289. [DOI] [PubMed] [Google Scholar]
- 112.Paoletti AC, Parmely TJ, Tomomori-Sato C, Sato S, Zhu D, Conaway RC, Conaway JW, Florens L, Washburn MP. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18928–18933. doi: 10.1073/pnas.0606379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sardiu ME, Cai Y, Jin J, Swanson SK, Conaway RC, Conaway JW, Florens L, Washburn MP. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1454–1459. doi: 10.1073/pnas.0706983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, von Mering C. Molecular & cellular proteomics : MCP. 2012;11:492–500. doi: 10.1074/mcp.O111.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Muganda P, Mendoza O, Hernandez J, Qian Q. J Virol. 1994;68:8028–8034. doi: 10.1128/jvi.68.12.8028-8034.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR. Nature cell biology. 2012;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, Howley PM. Molecular and cellular biology. 2011;31:2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.White EA, Sowa ME, Tan MJ, Jeudy S, Hayes SD, Santha S, Munger K, Harper JW, Howley PM. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E260–267. doi: 10.1073/pnas.1116776109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras AC, Nesvizhskii AI. Nature methods. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang GM, Roth SL, Akiva E, Marlett J, Stephens M, D'Orso I, Fernandes J, Fahey M, Mahon C, O'Donoghue AJ, Todorovic A, Morris JH, Maltby DA, Alber T, Cagney G, Bushman FD, Young JA, Chanda SK, Sundquist WI, Kortemme T, Hernandez RD, Craik CS, Burlingame A, Sali A, Frankel AD, Krogan NJ. Nature. 2012;481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6591–6596. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Molecular & cellular proteomics : MCP. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 124.Tackett AJ, DeGrasse JA, Sekedat MD, Oeffinger M, Rout MP, Chait BT. J Proteome Res. 2005;4:1752–1756. doi: 10.1021/pr050225e. [DOI] [PubMed] [Google Scholar]
- 125.Byrum S, Smart SK, Larson S, Tackett AJ. Methods Mol Biol. 2012;833:143–152. doi: 10.1007/978-1-61779-477-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Selbach M, Mann M. Nature methods. 2006;3:981–983. doi: 10.1038/nmeth972. [DOI] [PubMed] [Google Scholar]
- 127.Zheng P, Zhong Q, Xiong Q, Yang M, Zhang J, Li C, Bi LJ, Ge F. Journal of proteomics. 2012;75:1055–1066. doi: 10.1016/j.jprot.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 128.Meixner A, Boldt K, Van Troys M, Askenazi M, Gloeckner CJ, Bauer M, Marto JA, Ampe C, Kinkl N, Ueffing M. Molecular & cellular proteomics : MCP. 2011;10:M110 001172. doi: 10.1074/mcp.M110.001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kaake RM, Wang X, Huang L. Molecular & cellular proteomics : MCP. 2010;9:1650–1665. doi: 10.1074/mcp.R110.000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang X, Huang L. Molecular & cellular proteomics : MCP. 2008;7:46–57. doi: 10.1074/mcp.M700261-MCP200. [DOI] [PubMed] [Google Scholar]
- 131.Gunaratne J, Goh MX, Swa HL, Lee FY, Sanford E, Wong LM, Hogue KA, Blackstock WP, Okumura K. The Journal of biological chemistry. 2011;286:18093–18103. doi: 10.1074/jbc.M111.221184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 133.Ranish JA, Brand M, Aebersold R. Methods Mol Biol. 2007;359:17–35. doi: 10.1007/978-1-59745-255-7_2. [DOI] [PubMed] [Google Scholar]
- 134.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Molecular & cellular proteomics : MCP. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 135.Zieske LR. Journal of experimental botany. 2006;57:1501–1508. doi: 10.1093/jxb/erj168. [DOI] [PubMed] [Google Scholar]
- 136.UniProt C. Nucleic acids research. 2011;39:D214–219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wu CH, Yeh LS, Huang H, Arminski L, Castro-Alvear J, Chen Y, Hu Z, Kourtesis P, Ledley RS, Suzek BE, Vinayaka CR, Zhang J, Barker WC. Nucleic acids research. 2003;31:345–347. doi: 10.1093/nar/gkg040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pruitt KD, Tatusova T, Brown GR, Maglott DR. Nucleic acids research. 2012;40:D130–135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G. Nucleic acids research. 2007;35:D572–D574. doi: 10.1093/nar/gkl950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hermjakob H, Montecchi-Palazzi L, Lewington C, Mudali S, Kerrien S, Orchard S, Vingron M, Roechert B, Roepstorff P, Valencia A, Margalit H, Armstrong J, Bairoch A, Cesareni G, Sherman D, Apweiler R. Nucleic acids research. 2004;32:D452–455. doi: 10.1093/nar/gkh052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, Duesbury M, Dumousseau M, Feuermann M, Hinz U, Jandrasits C, Jimenez RC, Khadake J, Mahadevan U, Masson P, Pedruzzi I, Pfeiffenberger E, Porras P, Raghunath A, Roechert B, Orchard S, Hermjakob H. Nucleic acids research. 2012;40:D841–846. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Güldener U, Münsterkötter M, Oesterheld M, Pagel P, Ruepp A, Mewes H-W, Stümpflen V. Nucleic acids research. 2006;34:D436–441. doi: 10.1093/nar/gkj003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mellor JC, Yanai I, Clodfelter KH, Mintseris J, DeLisi C. Nucleic acids research. 2002;30:306–309. doi: 10.1093/nar/30.1.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hu Z, Hung J-H, Wang Y, Chang Y-C, Huang C-L, Huyck M, DeLisi C. Nucleic acids research. 2009;37:W115–W121. doi: 10.1093/nar/gkp406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, Surendranath V, Niranjan V, Muthusamy B, Gandhi TKB, Gronborg M, Ibarrola N, Deshpande N, Shanker K, Shivashankar HN, Rashmi BP, Ramya MA, Zhao Z, Chandrika KN, Padma N, Harsha HC, Yatish AJ, Kavitha MP, Menezes M, Choudhury DR, Suresh S, Ghosh N, Saravana R, Chandran S, Krishna S, Joy M, Anand SK, Madavan V, Joseph A, Wong GW, Schiemann WP, Constantinescu SN, Huang L, Khosravi-Far R, Steen H, Tewari M, Ghaffari S, Blobe GC, Dang CV, Garcia JGN, Pevsner J, Jensen ON, Roepstorff P, Deshpande KS, Chinnaiyan AM, Hamosh A, Chakravarti A, Pandey A. Genome research. 2003;13:2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. Nucleic acids research. 2004;32:D449–451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Goll J, Rajagopala SV, Shiau SC, Wu H, Lamb BT, Uetz P. Bioinformatics. 2008;24:1743–1744. doi: 10.1093/bioinformatics/btn285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stark C, Breitkreutz B-J, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, Reguly T, Rust JM, Winter A, Dolinski K, Tyers M. Nucleic acids research. 2011;39:D698–704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Alfarano C, Andrade CE, Anthony K, Bahroos N, Bajec M, Bantoft K, Betel D, Bobechko B, Boutilier K, Burgess E, Buzadzija K, Cavero R, D'Abreo C, Donaldson I, Dorairajoo D, Dumontier MJ, Dumontier MR, Earles V, Farrall R, Feldman H, Garderman E, Gong Y, Gonzaga R, Grytsan V, Gryz E, Gu V, Haldorsen E, Halupa A, Haw R, Hrvojic A, Hurrell L, Isserlin R, Jack F, Juma F, Khan A, Kon T, Konopinsky S, Le V, Lee E, Ling S, Magidin M, Moniakis J, Montojo J, Moore S, Muskat B, Ng I, Paraiso JP, Parker B, Pintilie G, Pirone R, Salama JJ, Sgro S, Shan T, Shu Y, Siew J, Skinner D, Snyder K, Stasiuk R, Strumpf D, Tuekam B, Tao S, Wang Z, White M, Willis R, Wolting C, Wong S, Wrong A, Xin C, Yao R, Yates B, Zhang S, Zheng K, Pawson T, Ouellette BFF, Hogue CWV. Nucleic acids research. 2005;33:D418–424. doi: 10.1093/nar/gki051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Han K, Park B, Kim H, Hong J, Park J. Bioinformatics. 2004;20:2466–2470. doi: 10.1093/bioinformatics/bth253. [DOI] [PubMed] [Google Scholar]
- 151.Prokisch H, Andreoli C, Ahting U, Heiss K, Ruepp A, Scharfe C, Meitinger T. Nucleic acids research. 2006;34:D705–711. doi: 10.1093/nar/gkj127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Orchard S, Kerrien S, Abbani S, Aranda B, Bhate J, Bidwell S, Bridge A, Briganti L, Brinkman FS, Cesareni G, Chatr-aryamontri A, Chautard E, Chen C, Dumousseau M, Goll J, Hancock RE, Hannick LI, Jurisica I, Khadake J, Lynn DJ, Mahadevan U, Perfetto L, Raghunath A, Ricard-Blum S, Roechert B, Salwinski L, Stumpflen V, Tyers M, Uetz P, Xenarios I, Hermjakob H. Nature methods. 2012;9:345–350. doi: 10.1038/nmeth.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chautard E, Fatoux-Ardore M, Ballut L, Thierry-Mieg N, Ricard-Blum S. Nucleic acids research. 2011;39:D235–240. doi: 10.1093/nar/gkq830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fu W, Sanders-Beer BE, Katz KS, Maglott DR, Pruitt KD, Ptak RG. Nucleic acids research. 2009;37:D417–422. doi: 10.1093/nar/gkn708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hermjakob H, Montecchi-Palazzi L, Bader G, Wojcik J, Salwinski L, Ceol A, Moore S, Orchard S, Sarkans U, von Mering C, Roechert B, Poux S, Jung E, Mersch H, Kersey P, Lappe M, Li Y, Zeng R, Rana D, Nikolski M, Husi H, Brun C, Shanker K, Grant SGN, Sander C, Bork P, Zhu W, Pandey A, Brazma A, Jacq B, Vidal M, Sherman D, Legrain P, Cesareni G, Xenarios I, Eisenberg D, Steipe B, Hogue C, Apweiler R. Nature biotechnology. 2004;22:177–183. doi: 10.1038/nbt926. [DOI] [PubMed] [Google Scholar]
- 156.Kerrien S, Orchard S, Montecchi-Palazzi L, Aranda B, Quinn AF, Vinod N, Bader GD, Xenarios I, Wojcik J, Sherman D, Tyers M, Salama JJ, Moore S, Ceol A, Chatr-Aryamontri A, Oesterheld M, Stumpflen V, Salwinski L, Nerothin J, Cerami E, Cusick ME, Vidal M, Gilson M, Armstrong J, Woollard P, Hogue C, Eisenberg D, Cesareni G, Apweiler R, Hermjakob H. BMC biology. 2007;5:44. doi: 10.1186/1741-7007-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jones AR, Eisenacher M, Mayer G, Kohlbacher O, Siepen J, Hubbard SJ, Selley JN, Searle BC, Shofstahl J, Seymour SL, Julian R, Binz PA, Deutsch EW, Hermjakob H, Reisinger F, Griss J, Vizcaino JA, Chambers M, Pizarro A, Creasy D. Molecular & cellular proteomics : MCP. 2012;11:M111 014381. doi: 10.1074/mcp.M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. Nucleic acids research. 2003;31:258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Su C, Peregrin-Alvarez JM, Butland G, Phanse S, Fong V, Emili A, Parkinson J. Nucleic acids research. 2008;36:D632–636. doi: 10.1093/nar/gkm807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Askenazi M, Li S, Singh S, Marto JA. Proteomics. 2010;10:1880–1885. doi: 10.1002/pmic.200900723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Secrier M, Pavlopoulos G, Aerts J, Schneider R. BMC bioinformatics. 2012:13. doi: 10.1186/1471-2105-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Satagopam VP, Theodoropoulou MC, Stampolakis CK, Pavlopoulos GA, Papandreou NC, Bagos PG, Schneider R, Hamodrakas SJ. Database : the journal of biological databases and curation. 2010;2010:baq019. doi: 10.1093/database/baq019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Barrell D, Dimmer E, Huntley RP, Binns D, O'Donovan C, Apweiler R. Nucleic acids research. 2009;37:D396–403. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zeeberg B, Feng W, Wang G, Wang M, Fojo A, Sunshine M, Narasimhan S, Kane D, Reinhold W, Lababidi S, Bussey K, Riss J, Barrett J, Weinstein J. Genome Biology. 2003:4. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman W-H, Pagès F, Trajanoski Z, Galon J. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Maere S, Heymans K, Kuiper M. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 169.Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. Genome biology. 2003:4. [PubMed] [Google Scholar]
- 170.Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. PloS one. 2011;6:e26044. doi: 10.1371/journal.pone.0026044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Doolittle JM, Gomez SM. PLoS neglected tropical diseases. 2011;5:e954. doi: 10.1371/journal.pntd.0000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rappsilber J. Journal of structural biology. 2011;173:530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Leitner A, Walzthoeni T, Kahraman A, Herzog F, Rinner O, Beck M, Aebersold R. Molecular & cellular proteomics : MCP. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Singh P, Panchaud A, Goodlett DR. Analytical chemistry. 2010;82:2636–2642. doi: 10.1021/ac1000724. [DOI] [PubMed] [Google Scholar]
- 175.Chen X, Chen YH, Anderson VE. Analytical biochemistry. 1999;273:192–203. doi: 10.1006/abio.1999.4243. [DOI] [PubMed] [Google Scholar]
- 176.Rappsilber J, Siniossoglou S, Hurt EC, Mann M. Analytical chemistry. 2000;72:267–275. doi: 10.1021/ac991081o. [DOI] [PubMed] [Google Scholar]
- 177.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R. Nature methods. 2008;5:315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sharon M, Taverner T, Ambroggio XI, Deshaies RJ, Robinson CV. PLoS biology. 2006;4:e267. doi: 10.1371/journal.pbio.0040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chen ZA, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski-Wills JC, Nilges M, Cramer P, Rappsilber J. The EMBO journal. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fu CY, Uetrecht C, Kang S, Morais MC, Heck AJ, Walter MR, Prevelige PE., Jr. Molecular & cellular proteomics : MCP. 2010;9:1764–1773. doi: 10.1074/mcp.M900625-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, Leitner A, Beck M, Hartl FU, Ban N, Malmstrom L, Aebersold R. Science. 2012;337:1348–1352. doi: 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- 183.Tardiff DF, Abruzzi KC, Rosbash M. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19948–19953. doi: 10.1073/pnas.0710179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Singh P, Shaffer SA, Scherl A, Holman C, Pfuetzner RA, Larson Freeman TJ, Miller SI, Hernandez P, Appel RD, Goodlett DR. Analytical chemistry. 2008;80:8799–8806. doi: 10.1021/ac801646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hernandez P, Gras R, Frey J, Appel RD. Proteomics. 2003;3:870–878. doi: 10.1002/pmic.200300402. [DOI] [PubMed] [Google Scholar]
- 186.McIlwain S, Draghicescu P, Singh P, Goodlett DR, Noble WS. J Proteome Res. 2010;9:2488–2495. doi: 10.1021/pr901163d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Panchaud A, Singh P, Shaffer SA, Goodlett DR. J Proteome Res. 2010;9:2508–2515. doi: 10.1021/pr9011816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Maiolica A, Cittaro D, Borsotti D, Sennels L, Ciferri C, Tarricone C, Musacchio A, Rappsilber J. Molecular & cellular proteomics : MCP. 2007;6:2200–2211. doi: 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- 189.Chu F, Baker PR, Burlingame AL, Chalkley RJ. Molecular & cellular proteomics : MCP. 2010;9:25–31. doi: 10.1074/mcp.M800555-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lee YJ, Lackner LL, Nunnari JM, Phinney BS. J Proteome Res. 2007;6:3908–3917. doi: 10.1021/pr070234i. [DOI] [PubMed] [Google Scholar]
- 191.Nadeau OW, Wyckoff GJ, Paschall JE, Artigues A, Sage J, Villar MT, Carlson GM. Molecular & cellular proteomics : MCP. 2008;7:739–749. doi: 10.1074/mcp.M800020-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, Luo SK, Ding YH, Wang LH, Hao Z, Xiu LY, Chen S, Ye K, He SM, Dong MQ. Nature methods. 2012;9:904–906. doi: 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- 193.Walzthoeni T, Claassen M, Leitner A, Herzog F, Bohn S, Forster F, Beck M, Aebersold R. Nature methods. 2012;9:901–903. doi: 10.1038/nmeth.2103. [DOI] [PubMed] [Google Scholar]
- 194.Tang X, Munske GR, Siems WF, Bruce JE. Analytical chemistry. 2005;77:311–318. doi: 10.1021/ac0488762. [DOI] [PubMed] [Google Scholar]
- 195.Chu F, Mahrus S, Craik CS, Burlingame AL. J Am Chem Soc. 2006;128:10362–10363. doi: 10.1021/ja0614159. [DOI] [PubMed] [Google Scholar]
- 196.Chowdhury SM, Du X, Tolic N, Wu S, Moore RJ, Mayer MU, Smith RD, Adkins JN. Analytical chemistry. 2009;81:5524–5532. doi: 10.1021/ac900853k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nessen MA, Kramer G, Back J, Baskin JM, Smeenk LE, de Koning LJ, van Maarseveen JH, de Jong L, Bertozzi CR, Hiemstra H, de Koster CG. J Proteome Res. 2009;8:3702–3711. doi: 10.1021/pr900257z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Collins CJ, Schilling B, Young M, Dollinger G, Guy RK. Bioorganic & medicinal chemistry letters. 2003;13:4023–4026. doi: 10.1016/j.bmcl.2003.08.053. [DOI] [PubMed] [Google Scholar]
- 199.Petrotchenko EV, Olkhovik VK, Borchers CH. Molecular & cellular proteomics : MCP. 2005;4:1167–1179. doi: 10.1074/mcp.T400016-MCP200. [DOI] [PubMed] [Google Scholar]
- 200.Petrotchenko EV, Borchers CH. BMC bioinformatics. 2010;11:64. doi: 10.1186/1471-2105-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kao A, Chiu CL, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L. Molecular & cellular proteomics : MCP. 2011;10:M110 002212. doi: 10.1074/mcp.M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sinz A, Wang K. Analytical biochemistry. 2004;331:27–32. doi: 10.1016/j.ab.2004.03.075. [DOI] [PubMed] [Google Scholar]
- 203.Back JW, Hartog AF, Dekker HL, Muijsers AO, de Koning LJ, de Jong L. J Am Soc Mass Spectrom. 2001;12:222–227. doi: 10.1016/S1044-0305(00)00212-9. [DOI] [PubMed] [Google Scholar]
- 204.Yang L, Tang X, Weisbrod CR, Munske GR, Eng JK, von Haller PD, Kaiser NK, Bruce JE. Analytical chemistry. 2010;82:3556–3566. doi: 10.1021/ac902615g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhang H, Tang X, Munske GR, Tolic N, Anderson GA, Bruce JE. Molecular & cellular proteomics : MCP. 2009;8:409–420. doi: 10.1074/mcp.M800232-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Soderblom EJ, Goshe MB. Analytical chemistry. 2006;78:8059–8068. doi: 10.1021/ac0613840. [DOI] [PubMed] [Google Scholar]
- 207.Gardner MW, Vasicek LA, Shabbir S, Anslyn EV, Brodbelt JS. Analytical chemistry. 2008;80:4807–4819. doi: 10.1021/ac800625x. [DOI] [PubMed] [Google Scholar]
- 208.Zelter A, Hoopmann MR, Vernon R, Baker D, MacCoss MJ, Davis TN. J Proteome Res. 2010;9:3583–3589. doi: 10.1021/pr1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lambert W, Rutsdottir G, Hussein R, Bernfur K, Kjellstrom S, Emanuelsson C. Cell stress & chaperones. 2012 doi: 10.1007/s12192-012-0360-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bruce JE. Proteomics. 2012;12:1565–1575. doi: 10.1002/pmic.201100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Guerrero C, Tagwerker C, Kaiser P, Huang L. Molecular & cellular proteomics : MCP. 2006;5:366–378. doi: 10.1074/mcp.M500303-MCP200. [DOI] [PubMed] [Google Scholar]
- 212.Smith AL, Friedman DB, Yu H, Carnahan RH, Reynolds AB. PloS one. 2011;6:e16206. doi: 10.1371/journal.pone.0016206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Smart SK, Mackintosh SG, Edmondson RD, Taverna SD, Tackett AJ. Protein science : a publication of the Protein Society. 2009;18:1987–1997. doi: 10.1002/pro.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Schmitt-Ulms G, Hansen K, Liu J, Cowdrey C, Yang J, DeArmond SJ, Cohen FE, Prusiner SB, Baldwin MA. Nat Biotechnol. 2004;22:724–731. doi: 10.1038/nbt969. [DOI] [PubMed] [Google Scholar]
- 215.Bai Y, Markham K, Chen F, Weerasekera R, Watts J, Horne P, Wakutani Y, Bagshaw R, Mathews PM, Fraser PE, Westaway D, St George-Hyslop P, Schmitt-Ulms G. Molecular & cellular proteomics : MCP. 2008;7:15–34. doi: 10.1074/mcp.M700077-MCP200. [DOI] [PubMed] [Google Scholar]
- 216.Tang X, Bruce JE. Molecular bioSystems. 2010;6:939–947. doi: 10.1039/b920876c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Zheng C, Yang L, Hoopmann MR, Eng JK, Tang X, Weisbrod CR, Bruce JE. Molecular & cellular proteomics : MCP. 2011;10:M110 006841. doi: 10.1074/mcp.M110.006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chavez JD, Cilia M, Weisbrod CR, Ju HJ, Eng JK, Gray SM, Bruce JE. J Proteome Res. 2012;11:2968–2981. doi: 10.1021/pr300041t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Suchanek M, Radzikowska A, Thiele C. Nature methods. 2005;2:261–267. doi: 10.1038/nmeth752. [DOI] [PubMed] [Google Scholar]
- 220.Singh GP, Ganapathi M, Dash D. Proteins. 2007;66:761–765. doi: 10.1002/prot.21281. [DOI] [PubMed] [Google Scholar]
- 221.Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, Schuldiner M, Gebbia M, Recht J, Shales M, Ding H, Xu H, Han J, Ingvarsdottir K, Cheng B, Andrews B, Boone C, Berger SL, Hieter P, Zhang Z, Brown GW, Ingles CJ, Emili A, Allis CD, Toczyski DP, Weissman JS, Greenblatt JF, Krogan NJ. Nature. 2007;446:806–810. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- 222.Gupta R, Kus B, Fladd C, Wasmuth J, Tonikian R, Sidhu S, Krogan NJ, Parkinson J, Rotin D. Molecular systems biology. 2007;3:116. doi: 10.1038/msb4100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Cloutier P, Al-Khoury R, Lavallee-Adam M, Faubert D, Jiang H, Poitras C, Bouchard A, Forget D, Blanchette M, Coulombe B. Methods. 2009;48:381–386. doi: 10.1016/j.ymeth.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cojocaru M, Bouchard A, Cloutier P, Cooper JJ, Varzavand K, Price DH, Coulombe B. The Journal of biological chemistry. 2011;286:5012–5022. doi: 10.1074/jbc.M110.176628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Jackson PK. Cell. 2009;138:222–224. doi: 10.1016/j.cell.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 226.Glatter T, Schittenhelm RB, Rinner O, Roguska K, Wepf A, Junger MA, Kohler K, Jevtov I, Choi H, Schmidt A, Nesvizhskii AI, Stocker H, Hafen E, Aebersold R, Gstaiger M. Molecular systems biology. 2011;7:547. doi: 10.1038/msb.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Choi H, Glatter T, Gstaiger M, Nesvizhskii AI. J Proteome Res. 2012;11:2619–2624. doi: 10.1021/pr201185r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. Nature. 2012;481:371–375. doi: 10.1038/nature10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Lee DJ, Busby SJ, Westblade LF, Chait BT. J Bacteriol. 2008;190:1284–1289. doi: 10.1128/JB.01599-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Winter C, Henschel A, Kim WK, Schroeder M. Nucleic acids research. 2006;34:D310–314. doi: 10.1093/nar/gkj099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Hoffmann R, Valencia A. Nat Genet. 2004;36:664. doi: 10.1038/ng0704-664. [DOI] [PubMed] [Google Scholar]
- 232.Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, Mewes HW. Nucleic acids research. 2010;38:D497–501. doi: 10.1093/nar/gkp914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Brown KR, Jurisica I. Bioinformatics. 2005;21:2076–2082. doi: 10.1093/bioinformatics/bti273. [DOI] [PubMed] [Google Scholar]
- 234.Lynn DJ, Chan C, Naseer M, Yau M, Lo R, Sribnaia A, Ring G, Que J, Wee K, Winsor GL, Laird MR, Breuer K, Foroushani AK, Brinkman FSL, Hancock REW. BMC Syst Biol. 2010:4. doi: 10.1186/1752-0509-4-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Persico M, Ceol A, Gavrila C, Hoffmann R, Florio A, Cesareni G. BMC bioinformatics. 2005;6(Suppl 4) doi: 10.1186/1471-2105-6-S4-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schächter V, Chemama Y, Labigne A, Legrain P. Nature. 2001;409:211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- 237.Murali T, Pacifico S, Yu J, Guest S, Roberts GG, 3rd, Finley RL., Jr. Nucleic acids research. 2011;39:D736–743. doi: 10.1093/nar/gkq1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hooper SD, Bork P. Bioinformatics. 2005;21:4432–4433. doi: 10.1093/bioinformatics/bti696. [DOI] [PubMed] [Google Scholar]
- 239.Wu J, Vallenius T, Ovaska K, Westermarck J, Mäkelä TP, Hautaniemi S. Nature methods. 2009;6:75–77. doi: 10.1038/nmeth.1282. [DOI] [PubMed] [Google Scholar]
- 240.Theocharidis A, van Dongen S, Enright AJ, Freeman TC. Nature protocols. 2009;4:1535–1550. doi: 10.1038/nprot.2009.177. [DOI] [PubMed] [Google Scholar]
- 241.Ho E, Webber R, Wilkins MR. Journal of proteome research. 2008;7:104–112. doi: 10.1021/pr070274m. [DOI] [PubMed] [Google Scholar]
- 242.Goel A, Li SS, Wilkins MR. Proteomics. 2011;11:2672–2682. doi: 10.1002/pmic.201000546. [DOI] [PubMed] [Google Scholar]
- 243.Tatusov RL, Koonin EV, Lipman DJ. Science (New York, N.Y.) 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 244.Al-Shahrour F, Diaz-Uriarte R, Dopazo J. Bioinformatics. 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- 245.Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, Donaldson SL, Morris Q, Bader GD. Bioinformatics. 2010;26:2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]