

Abstract
Tubulin dynamics is a promising target for new chemotherapeutic agents. The colchicine binding site is one of the most important pockets for potential tubulin polymerization destabilizers. Colchicine binding site inhibitors (CBSI) exert their biological effects by inhibiting tubulin assembly and suppressing microtubule formation. A large number of molecules interacting with the colchicine binding site have been designed and synthesized with significant structural diversity. CBSIs have been modified as to chemical structure as well as pharmacokinetic properties, and tested in order to find a highly potent, low toxicity agent for treatment of cancers. CBSIs are believed to act by a common mechanism via binding to the colchicine site on tubulin. The present review is a synopsis of compounds that have been reported in the past decade that have provided an increase in our understanding of the actions of CBSIs.
Keywords: antimitotic, cancer, colchicine, multidrug resistance, tubulin polymerization inhibitor
INTRODUCTION
Drugs that disrupt microtubule/tubulin dynamics are used widely in cancer chemotherapy. The vast majority of these molecules act by binding to the protein tubulin, an α, β-heterodimer that forms the core of the microtubule. Microtubule targeting agents (MTA) are also named antimitotic agents which perturb not only mitosis but also arrest cells during interphase. MTAs are known to interact with tubulin through at least four binding sites: the laulimalide, taxane/epothilone, vinca alkaloid, and colchicine sites (Fig. 1). Similar to paclitaxel, Laulimalide can promote the tubulin-microtubule assembly, but binds to a different site on the microtubules (1). Taxanes, including paclitaxel and docetaxel, bind to polymerized microtubules at the inner surface of the β subunit, and are widely used in the treatment of lung, breast, ovarian and bladder cancers. Taxanes promote tubulin stabilization, thereby interfering with tubulin dynamics. Vinca alkaloids, including vinblastine, vincristine, and vinorelbine, promote depolymerization of microtubules. They generally bind with high affinity to one or a few tubulin molecules at the tip of microtubules but do not copolymerize into microtubules. Indeed, vinblastine prevents self-association of tubulin by interacting at the interface between two αβ–tubulin heterodimers (2). The fourth group of microtubule interfering agents is represented by colchicine, which also induces microtubule depolymerization. In contrast to agents binding to the other three sites, colchicine binds with high affinity to tubulin that can become copolymerized into microtubules. Colchicine binding to β-tubulin results in curved tubulin dimer and prevents it to adopt a straight structure, due to a steric clash between colchicine and α-tubulin, which inhibits microtubule assembly (3).
Fig. 1.
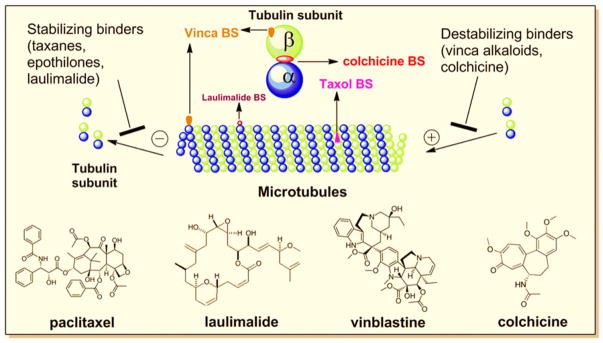
Tubulin binding sites (BS) of microtubule targeting agents.
Given the success of the taxanes and vinca alkaloids, which have established tubulin as a valid target in cancer therapy, research efforts have been focused on developing colchicine-like compounds for cancer treatment.
COLCHICINE AS A MEDICINE
Colchicine was extracted from the poisonous meadow saffron Colchicum autumnale L. and was the first tubulin destabilizing agent. It has been used for many years as an unapproved drug to treat gout, familial mediterranean fever, pericarditis and Behçet’s disease. In 2009, U.S. Food and Drug Administration (FDA) approved colchicine as a monotherapy drug to treat familial mediterranean fever and acute gout flares. Colchicine can effectively inhibits mitosis. Since cancer cells undergo mitosis at a significantly increased rate, this means that cancer cells are more susceptible to colchicine poisoning than are normal cells. Therefore colchicine is also being investigated as an anti-cancer drug. However, the therapeutic value of colchicine against cancer is restrained by its low therapeutic index. Its toxicity includes neutropenia, gastrointestinal upset, bone marrow damage and anemia.
Although colchicine is not used as an anticancer agent, there have been multiple efforts to clinically develop colchicine binding site agents (CBSI). As microtubules are important regulators of endothelial cell biology, one advantage of mechanism of actions of CBSIs is targeting the tumor vasculature. CBSIs can prevent new blood vessels formation by outgrowth from preexisting ones (angiogenesis inhibitors) or destroy the existing tumor vasculature (vascular disrupting agents, VDA). The targeting of tumor blood vessels introduces a therapeutically promising application for these compounds. Another favorable factor is that most of these drugs have no multidrug resistance (MDR) issues. The major limitation of using microtubule-targeting agents clinically is innate and acquired drug resistance. The most common form of clinical resistance is overexpression of the MDR1 gene, which encodes the P-glycoprotein (Pgp) drug efflux pump. This membrane-associated ATP-binding cassette (ABC) transporter is overexpressed in many tumor cell lines, including tissues of the liver, kidney, and gastrointestinal tract. Over-expression of Pgp decreases intracellular drug levels, consequently limiting drug cytotoxicity. In addition, over-expression of Pgp is associated with poor response to microtubule-targeted agents including taxanes and vinca alkaloids and subsequent treatment failure.
Besides over-expression of ABC transporters, other significant mechanism of resistance including mutantions in tubulin and overexpression of βIII -tubulin isoform. Among eight identified β-tubulin isotypes in human, overexpression of class III β-tubulin is an indicator of resistance to tubulin targeting agents such as paclitaxel and vinorelbine. While the efficacies of some CBSIs such as colchicines and 2-methoxyestradiol were not affected by the expression pattern of β-tubulin (4,5). The clinical development of a microtubule-targeting agent that circumvents both of these drug resistance mechanisms could have advantages for patients with drug resistant tumors.
To overcome these resistance problems, many research efforts have concentrated on developing CBSIs, although to date no agent that binds within the colchicine binding site is approved for use against cancer. Many CBSIs have been identified as potential anticancer agents for clinical studies due to their ability to overcome Pgp/β-III tubulin mediated drug resistance and their antiangiogenic or antivascular actions. Most CBSIs have small molecular weight with chemically modifiable structures, thus they afford adequate space for chemical modification to improve pharmacokinetic (PK) properties, efficacy and reduce toxicity.
The current review searched the PubMed using key words “colchicine, tubulin and cancer”. A total of 441 references were found from 1971 to 2010 and showed the trend of increasing interests every decade (Fig. 2) in CBSIs for treatment of cancer. In this article, we focus on CBSI agents in current clinical trials and recently published research papers after 2000 (Fig. 2, total 296 publications) with high citation rates.
Fig. 2.
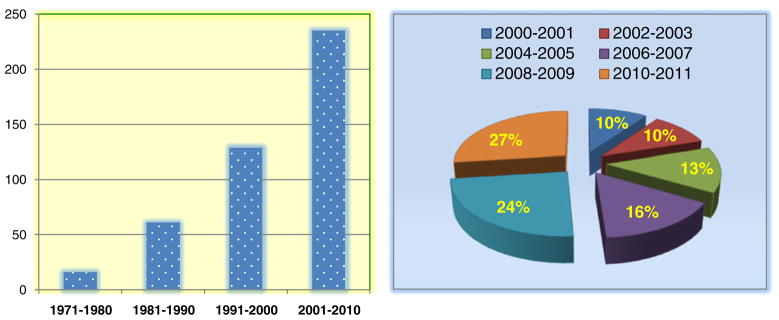
Publications related to CBSIs over last decades from PubMed (data entry implementation date until Dec 30th, 2011).
NATURAL PRODUCTS AND CBSI IN CLINICAL TRIALS
Many of the CBSIs are based on natural products such as colchicinoids and combretastatins, while others are synthetic compounds such as ABT-751. In this section, we will discuss the well-known CBSIs and recent progress of CBSIs in clinical trials (Fig. 3).
Fig. 3.

Chemical structures of established drugs bound to colchicine binding site and CBSIs in clinical trials (TMP: 3, 4, 5-trimethoxyphenyl).
Colchicine (1) and ZD6126 (2)
A number of clinical trials have been done on colchicine (1) for treatment of various diseases including cancer. However, the clinical use in treatment of cancer was hampered by its significant toxicity. ZD6126 (2) is a water-soluble phosphate prodrug of N-acetylcolchinol structurally very similar to colchicine with potential antiangiogenesis and antineoplastic activities (6,7). ZD6126 was developed by AstraZeneca for the treatment of metastatic colorectal cancer. However the study was terminated at phase II due to apparent cardiotoxicity at pharmacological doses.
CA-4 and its Analogs (3–7)
Combretastatins are a class of stilbenoid phenols isolated from Combretum caffrum. Combretastatin A-4 (CA-4, 3) is the most potent naturally occurring combretastatin known in regards to both tubulin binding ability and cytotoxicity. CA-4P (Zybrestat, fosbretabulin, and its salt fosbretabulin disodium, 3P) is the prodrug of CA-4 developed by OxiGene. Currently it is being evaluated in clinical trials as a treatment for solid tumors. In vivo, it is dephosphorylated to its active metabolite CA-4. Several phase II studies using CA-4P have finished or are ongoing for different type of cancers including anaplastic thyroid cancer, non-small cell lung cancer, relapsed ovarian cancer, etc. (8). Oxi4503 (4) is combretastatin A-1 diphosphate (CA-1P) targeting tumor vasculature. It is a phosphorylated CA-4 analog developed by OxiGene for the treatment of solid tumors. A phase I study is currently recruiting participants to determine the safety and maximum tolerated dose of OXi4503 in patients with relapsed and refractory acute myelogenous leukemia and myelodis-plastic syndrome.
AVE8062 (ombrabulin, 5) is another CA-4 analog which exerts its anticancer activity through disrupting the blood vessel formation in tumors. Compared with CA-4, it has improved water solubility and is orally available. AVE8062 has enhanced antitumor activity and decreased toxicity in a murine Colon 26 carcinoma model. It is also effective against a number of cancer cells that are resistant to taxanes (9). In a phase I study, the combination of AVE8062 with docetaxel was well tolerated. A phase III study is currently ongoing for advanced cancer treatment (10).
Phenstatin (6) is also a CA-4 analog with the double bond of CA-4 being replaced by a carbonyl group. Phenstatin showed strong cytotoxicity and antitubulin activity similar to CA-4, but it is more stable compare with CA-4 which is known to be unstable In vivo due to the transformation from the active cis-configuration to the more stable but inactive trans-configuration (11). CC-5079 (7) belongs to 1,1-diarylethenes analogs of CA-4, which is called isocombretastatins A. CC-5079 is a dual inhibitor of tubulin polymerization and phosphodiesterase-4 (PDE4) activity. It showed antiangiogenic and antitumor activities. CC-5079 can arrest cell cycle in G2/M phase, increase phosphorylation of G2/M checkpoint proteins, and induce apoptosis (12).
Podophyllotoxin (8), otherwise known as podofilox, is a non-alkaloid toxic lignan extracted from the roots and rhizomes of Podophyllum species. In 1890, Kiirsten isolated crystalline podophyllotoxin (13). Podophyllotoxin competitively inhibits the binding of colchicine. It binds to tubulin more rapidly than does colchicine. The utilization of 8 as a lead in anticancer drug design has resulted in useful cancer fighting drugs such asetoposide, teniposide, and etoposide phosphate (13).
Steganacin (9), a new lignan lactone from the alcoholic extract of Steganataenia araliacea Hochest, has significant anti-tumor activity in vivo against P388 leukemia in mice and in vitro against cells derived from a human carcinoma of the nasopharynx (KB) (14). It was found that 9 prevented the formation of the spindle that forms prior to the first cleavage. This suggested that steganacin, like other spindle poisons such as colchicine and podophyllotoxin, exerts its antimitotic activity through an effect on spindle microtubules (14).
Nocodazole (10) is a natural product which has been shown to have antimitotic and antitumor activity. The action of this agent is readily reversible and relatively rapid. Like 8 and 9, this agent exerts its effect in cells by interfering with the polymerization of microtubules. However, the full therapeutic efficacy of this agent is limited owing to the development of various side effects in patients, including bone marrow suppression, neutropenia, leukopenia and anemia (15). This agent is now often used as a lead compound to discover novel CBSIs or as a reference compound to study cell mitosis.
Curacin A (11), originally purified as a major lipid component from a strain of the cyano bacterium Lyngbya majuscula isolated in Curaçao, is a potent inhibitor of cell growth and mitosis. It binds rapidly and tightly at the colchicine site of tubulin. A recurring structural theme in the colchicine binding site agents has been at least one and generally two aromatic domains (16), while 11, as a potent colchicine binding site antimitotic agent, is a major exception to this structural generalization in that it has no aromatic residue. Poor water-solubility and lack of chemical stability prevent the clinical development of curacin A, but synthetic analogs with improved bioavailability may provide new promises.
2-Methoxyestradiol (2-ME, 12) is an endogenous estrogen metabolite, formed by hepatic cytochrome P450 2-hydroxylation of β-estradiol and 2-O-methylation via catechol O-methyltranseferase. This metabolite has attracted interest because of its potent inhibition of tumor vasculature and tumor cell growth. Because solid tumor growth is dependent on angiogenesis, the potent antiangiogenic activity and tubulin polymerization inhibition of 2-ME in vivo are of potential therapeutic value and have warranted further investigation in clinical trials. A recent clinical study indicated that the main adverse effects of 2-ME included fatigue, nausea, diarrhea, neuropathy, edema, and dyspnea (17). Studies have shown that 2-ME is metabolized by conjugation at positions 3 and 17 and oxidation at position 17. The conjugated forms of 2-ME are inactive, and oxidation to 2-methoxyestrone results in 10- to 100-fold loss in activity in vitro (18). In order to make metabolically stable analogs with improved anti-tubulin properties, ENMD-1198 (13) was generated via chemical modification at 3 and 17 position. This agent also binds to the colchicine binding site in tubulin, induces G2/M cell cycle arrest and apoptosis, and reduces hypoxia-inducible factor (HIF)-1α levels. Studies also showed that ENMD-1198 was very potent at inhibiting endothelial cell proliferation, motility, migration, and morphogenesis. In addition, ENMD-1198 induced a significant decrease in vascular endothelial growth factor receptor (VEGFR)-2 protein expression in endothelial cells. Furthermore, ENMD-1198 is able to disrupt vascular structures very quickly (19).
ABT-751 (E7010, 14) is an orally bioavailable tubulin-binding agent that is currently in a phase II clinical trial for cancer treatment. It is a novel sulfonamide antimitotic that binds to the colchicine site on β-tubulin that leads to a block in the cell cycle at the G2/M phase, resulting in cellular apoptosis. ABT-751 was investigated in a recent phase I clinical trial to assess its PK profile and safety (20). The maximum tolerated dose for the daily schedule was 250 mg/day. Dose-limiting toxicities included abdominal pain, constipation, and fatigue. ABT-751 was absorbed after oral administration with an overall mean Tmax of about 2 h. The PK properties of ABT-751 were dose-proportional and time independent. ABT-751 metabolism occurred primarily by glucuronidation and sulfation.
T138067 (15) was first reported by Shan et al. in 1999 as a novel antimitotic agent (21). This compound has been shown to covalently bind to Cys239 on β-tubulin isoforms 1, 2, and 4 by way of a nucleophilic aromatic substitution reaction (21). The covalent modification of β-tubulin prevents the polymerization of the α, β-tubulin dimers into microtubules. This leads to cell cycle arrest at the G2/M phase followed by apoptosis (21). T138067 is effective against a variety of tumors, including those that express the MDR phenotype (IC50=11–165 nM) (21). A phase II clinical trial showed that treatment with T138067 was tolerable with moderate hematologic and gastrointestinal toxicity. Neurotoxicity, an expected side effect, was minimal.
BNC-105P (16) was developed by Bionomics (Australia) as a low-molecular-weight VDA for treatment of cancers. BNC-105P is a phosphorylated prodrug which rapidly transforms to the active form BNC-105 by nonspecific endogenous phosphatases in plasma and on endothelial cells (22). BNC-105 exhibits selectivity (81 fold) for growth factor activated endothelial cells compared to quiescent human umbilical vein endothelial cells (HUVECs). A phase I study has been completed and the drug was shown to be generally well tolerated. A phase II study for BNC105P in combination with Everolimus for progressive metastatic clear cell renal cell carcinoma is currently recruiting participants (23).
Indibulin (D-24851, ZIO-301, 17) is an orally active anti-mitotic drug that is effective against various human tumor cell lines and xenografts, including taxane resistant tumors. In preclinical studies indibulin lacks neurotoxicity which is largely associated with other tubulin binding drugs. The antitumor activity against MDR cancers, the lack of neurotoxicity, and the oral dosing make indibulin a promising candidate for further development as an anticancer drug. Indibulin was reported not to overlap with the colchicine site, and it was shown to partially compete for binding with “colchicine” site binders (40% inhibition) (24). In vivo, oral application of indibulin showed a remarkable efficacy in the Yoshida AH13 rat sarcoma model without systemic toxicity being observed. Indibulin not only inhibits growth of tumor cell lines with different resistance phenotypes including MDR1 and multi-drug resistance-associated protein (MRP), but also retains its antitumor activity against cancer cell lines with resistance to cisplatin, the topoisomerase-I-inhibitor SN-38, and the thymidylate synthase inhibitors 5-FU and raltitrexed. Although indibulin also alters microtubule function, no neurotoxic effects on rats was seen at curative doses compared to paclitaxel and vincristine treatment groups.
EPC2407 (Crolibulin, 18), MPI-0441138 (19), and MPC-6827 (Azixa, Verubulin, 20)
The 4-aryl-4H-chromenes, which were developed by EpiCept Corp. in California, inhibit tubulin polymerization and induce apoptosis. Through structure-activity relationship (SAR) studies of the 4-aryl-4H-chromenes, the anticancer drug candidate EPC2407 with potent vascular disrupting activity and in vivo efficacy has been identified (25) and is currently in phase II clinical trial for the treatment of anaplastic thyroid cancer. MPI-0441138 (19) is the lead compound for MPC-6827 (20) discovered by EpiCept and identified as a highly active apoptosis inducer (EC50 for caspase activation of 2 nM) and as a potent inhibitor of cell proliferation (GI50 of 2 nM) in T47D cells (26). This compound inhibits tubulin polymerization and growth of Pgp overexpressing cells, and shows efficacy in the MX-1 human breast and PC-3 prostate cancer mouse models (26). A recent phase I study indicated that MPC-6827 was well tolerated at the recommended dose. The most common adverse events were nausea, fatigue, flushing, and hyperglycemia (27). However, recent news (http://www.biopharmcatalyst.com/2011/09/myrxhalts-phase-2b-azixa-pphm-pipeline-update-scmp/) released in Sep. 2011 reported that Myrexis, which has exclusive rights to MPC-6827 from EpiCept discontinued development of MPC-6827 due to “disproportionate investment of time and resources relative to its likelihood of technical and regulatory success.”
CYT997 (21) is originally discovered as a structurally novel, orally active microtubule targeting agent. It is now in phase II clinical trials for the treatment of selected cancers. CYT997 inhibits tubulin polymerization by binding at the colchicine binding site of tubulin. CYT997 blocks the cell cycle at the G2/M phase, and western blot analysis indicates an increase in phosphorylated Bcl-2, along with increased expression of cyclin B1 (28). This compound also possesses favorable PK properties and is orally active in different tumor models, including paclitaxel resistant cancer (28). CYT997 exhibits vascular disrupting activity in vitro by effects on the permeability of human umbilical vein endothelial cell monolayers, as well as in vivo on tumor blood flow (28).
MN-029 (denibulin, 22) is a novel benzoimidazole carbamate that reversibly inhibits microtubule assembly, resulting in disruption of the cytoskeleton of tumor vascular endothelial cells. MN-029 was found to demonstrate striking antivascular effects in tumors, leading to the induction of necrosis and a consequential rapid loss of clonogenic neoplastic cells. This VDA also was successfully incorporated into conventional cisplatin or radiation therapy treatments (29). A recent phase I clinical study of MN-029 in patients with advanced solid tumors showed that MN-029 was generally well tolerated and showed decrease in tumor vascular parameters (30). The most common toxicities of MN-029 included nausea, dose related vomiting, diarrhea, fatigue, headache, and anorexia. No significant myelotoxicity, stomatitis or alopecia was observed in clinical (30).
CI-980 ((S)-(−)-NSC 613862, 23) is one of a novel class of 1, 2-dihydropyrido [3, 4-b] pyrazines that inhibits tubulin polymerization presumably by interacting with the colchicine binding site of tubulin. The (R)-(+)-isomer NSC 613863 showed potency in several biological assays. However, the S-isomer is the more potent inhibitor on tubulin polymerization and cell proliferation (31). CI-980 treated cells accumulate in the M-phase of the cell cycle and subsequently die. In sensitive tumor models, the potency for this agent is similar to that of vincristine, but the spectrum of antitumor activity is wider. CI-980 shows activity against a variety of cancer cells in vitro, including leukemia, melanoma, sarcoma, mammary adenocarcinoma, and colon adenocarcinomas. CI-980 is currently in a phase II clinical trial (32). Neurotoxicity is the biggest problem for this agent. It can cause a significant but reversible decline in recent memory functioning. So careful monitoring of cognitive function in patients receiving this agent should be performed if dose or schedule parameters are changed.
CP248 (24) and CP461 (25) are derivatives of Exisulind (Aptosyn, inhibitor of enzyme cyclic guanosine monophosphate phosphodiesterase (cGMP-PDE)). Tubulin polymerization is believed to be their target. Both CP248 and CP461 cause growth inhibition and apoptosis in several cancer cell lines. There are at least two modes of inhibiting tumor cells identified for CP248. One is its inhibition of the cGMP-specific PDE2 and PDE5 and activate a protein kinase G mediated signaling pathway that triggers apoptosis. The other is its ability to bind to tubulin, inhibit its polymerization, and cause cells to be arrested in mitosis (33). CP461 is a member of a class of novel proapoptotic drugs that inhibit cyclic GMP phosphodiesterases specifically but not cyclooxygenase-1 or -2. It was in a phase I study for the treatment of patients with advanced melanoma. CP-461 inhibits the growth of a broad range of human tumor cell lines in vitro at micromolar concentrations. It selectively induces apoptosis in cancer cells but not normal cells (34).
TN16 (26) is a tenuazonic acid derivative exhibiting anti-tumor effects in vitro and in vivo by inhibiting microtubule assembly and produces M phase arrest. TN16 has a structure distinct from the representative microtubule inhibitor colchicine, and yet it inhibits microtubule assembly, and prevents the stabilization of microtubules (35).
COMPUTER MODELING STUDIES OF CBSI
Computer-aided drug design methodologies have been increasingly applied to drug development and have already provided some useful directions in the design and discovery of anticancer drugs. Along with the increased publications of crystal structures recently, an increasing number of molecular modeling studies on tubulin have been reported.
A study published in 2000 by Hamel et al. (36) is considered the first report of structure-based approach for the colchicine binding site agents. The authors tried to identify two potential colchicinoids binding sites on tubulin with the aids of biochemical and molecular modeling techniques. The colchicine binding site was identified by Ravelli et al. in 2004 by the determination of a 3.5 Å X-ray structure of α, β-tubulin complexed with N-deacetyl-N-(2-mercaptoacetyl) colchicine (DAMA-colchicine) (3). Experimental data showed that colchicine binds to β-tubulin at its interface with α-tubulin, resulting in inhibition of tubulin polymerization. Colchicine and podophyllotoxin bind to β-tubulin at its interface with α-tubulin with a similar orientation. An X-ray diffraction study demonstrated that the trimethoxyphenyl (TMP) groups of both DAMA-colchicine and podophyllotoxin are located in the β-tubulin structure in the vicinity of the amino acid residue Cysβ241 (note: In some publications (36) this residue is numbered as Cysβ239). The width of the colchicine binding site is approximately 4–5 Å, and the volume of this site is confined in β-tubulin by helix 7 (H7) containing Cysβ241, loop 7 (T7) and helix 8 (H8).
In recent years, several molecules structurally distinct from colchicine have been crystallized in the colchicine binding site. These X-ray structures show a new and interesting binding mode of tubulin in complex with CBSIs (Table I). ABT-751 (E7010, 14) interacts with the colchicine binding site (37). The methoxy and pyridine groups of ABT-751 superimpose with the colchicine C and A rings, respectively; the sulfonamide bridge overlaps with the B ring. ABT-751 interacts with β–strand S6 via a hydrogen bond between Tyrβ202 and the phenolic group. ABT-751 is more deeply buried than colchicine in β-tubulin pocket and does not interact with the α-subunit. TN16 (26, Fig. 3) (37) competes with colchicine for tubulin binding. It is even more deeply buried in the β monomer than ABT-751. The tubulin residues involved in binding belong to β strands S4, S5, and S6 (van der Waals contact with Thrβ239, Valβ238, Tyrβ202, Gluβ200, and Pheβ169). T138067 (15, Fig. 3) is a bi-aryl molecule sharing with ABT-751 a sulfonamide linker. T138067-tubulin complex showed a dual binding mode with a covalent component (37). T138067 occupies a site largely overlapping with that of colchicine while another mode is covalently linked to Cysβ241. In the latter case, only pentafluorophenyl ring A bound covalently to residue Cysβ241 of β-tubulin. When T138067 is not covalently linked to the protein, it interacts primarily with strand S9 (Leuβ313– Argβ320) and loop T7 (Alaβ250–Pheβ244), that border the colchicine binding site on the α-tubulin side. CI-980 (23, Fig. 3) and NSC-613863 are enantiomers, which are also buried deeply into β-tubulin than colchicine (38). They show no interactions with the α subunit. The molecular partially overlapped with the A ring of colchicine. Gluβ200 has a hydrogen bonding interaction with the amino group of the pyridine. The carbamate substituent is embedded in a hydrophobic pocket though van der Waals contacting with residues of Thrβ239, Tyrβ202, Asnβ167, Glnβ136, and Ileβ4.
Table I.
Reported X-ray Structures of Tubulin in Complex with CBSIs
| CBSIs, Compd ID | PDB code | Res (Å) | Year(Ref) |
|---|---|---|---|
| Colchicine (1) | 1SA0 | 3.58 | 2004 (57) |
| Podophyllotoxin (8) | 1SA1 | 4.20 | 2004 (4) |
| ABT-751 (14) | 3HKC | 3.80 | 2009 (58) |
| T138067 (15) | 3HKE | 3.60 | 2009 (58) |
| TN16 (26) | 3HKD | 3.70 | 2009 (58) |
| CI-980 (23, S-isomer) | 3N2K | 4.00 | 2010 (59) |
| NSC613863 (R-isomer of 23) | 3N2G | 4.00 | 2010 (59) |
In order to rationalize their key common interactions at the colchicines binding site, many pharmacophore models have been reported. Gussio et al. (39) employed docking studies and molecular dynamics simulations to construct binding models for a set of structurally diverse CBSIs, using the α, β-tubulin: DAMA-colchicine X-ray structure as the template. Examination of the binding models of a set of structurally diverse CBSIs revealed common pharmacophore groups for the CBSIs and extended the understanding of interactions at the colchicine binding site. According to the report, the common pharmacophore of ligands of the colchicine binding site contains the following seven pharmacophoric points (Fig. 4): three hydrogen bond acceptors (A1, A2, and A3), one hydrogen bond donor (D1), two hydrophobic centers (H1 and H2), and one planar group (R1). Hydrogen bond acceptor A1 is in contact with Valα179, A2 is in contact with Cysβ239, and A3 establishes one contact mainly with Alaβ248, Aspβ249, and Leuβ250. Hydrogen bond donor D1 interacts with Thrα177. Drug activity requires one hydrogen bond acceptor, two hydrophobic centers, and a planar group.
Fig. 4.
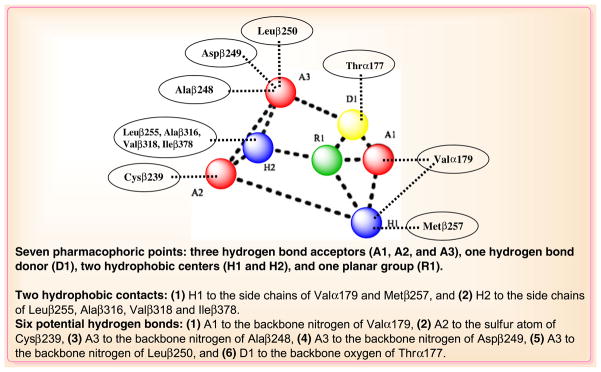
Interactions between the pharmacophoric points and the tubulin structure (based on Ref. (60)).
The above docking results provide explanations for many known and new CBSIs on their structure-binding interactions for the colchicine binding site. The fact that none of the known structures of CBSIs contains all seven pharmacophore groups suggests that the binding affinity of each chemotype can be improved by appropriate chemical modifications. The binding models and pharmacophore may provide useful insights for rational structure-based drug design.
The accuracy of computer models reported are growing, especially when relatively rapid calculations such as molecular docking and pharmacophore queries are connected with more advanced methodologies such as molecular dynamics. But there are still many challenges for researchers in this research field. The fact that all the available crystal structures of tubulin in complex with a CBSI have low resolution (3.5~4.0 Å) should not be overlooked (Table I). This can explain the poor performance of the scoring system and provide justification for why molecular dynamics is necessary to improve accuracy of model. Another point to keep in mind is that the various isoforms of tubulin and their specific roles. The tubulin superfamily includes α-, β-, γ-, δ-, ε-, ζ-, and η-tubulin (40). Different isotypes for both α and β subunits are present in human cells. β-tubulin represents the main binding domain for CBSI and includes at least eight isotypes that are expressed in different human tissue/organs. The discovery of novel CBSIs that target specific isoforms selectively could have a remarkable impact and molecular modeling could prove to be a very helpful tool in this research area.
REPORTED CBSI IN PRECLINICAL STUDIES
Along with the rapid development of colchicine binding site inhibitors in the last decade, especially with the elucidations of several tubulin colchicine pocket-ligand binding crystal structures, more structures of CBSIs were reported and many of them showed excellent potency and drug-like properties for their preclinical applications. We now summarize several classes of CBSIs with diverse chemical structures.
Combretastatin A-4 and its Analogs
Combretastatin A-4 was shown to exhibit potent antiangiogenic and antitumor activities. However, poor solubility of the drug impinged its clinical development and required the preparation of more soluble derivatives such as CA-4P phosphate sodium salt (3P, Fig. 3) and the amino acid hydrochloride salt (5). In addition, the activity of CA-4 is hampered by a short biological half-life (41,42) and isomerization of the active cis-olefinic conformation into the corresponding inactive trans-analogs under the influence of heat, light, and protic media (43,44). It could therefore be hypothesized that analogs that retain the potency and efficacy of CA-4, but that have a different pharmacokinetic profile might be useful. To overcome these problems, new analogs of CA-4 have been synthesized and developed in the recent years.
The Ethenyl Bridge of the Stilbene Moiety (Fig. 5)
Fig. 5.

Chemical structures of chemical modified CA-4 analogs.
The olefinic bridge of CA-4 represents a weak point for metabolic stability. Sulfonate analogs of CA-4 have been prepared by Gwaltney et al. (45) (i.e., 27). Compound 27 competitively binds with colchicine and CA-4 for the colchicine binding site in tubulin and is potent inhibitor of tubulin polymerization and cell proliferation. Importantly, this compound also inhibits the proliferation of Pgp over-expressing cancer cells, which are resistant to many other antitumor agents. Recently, Fortin et al. also reported that sulfonate and sulfonamide moieties are bioisosteres of ethenyl bridge (PIB-SO: 28, PIB-SA: 29) (46,47). Quantitative structure-activity relationships (QSAR) of 28 and 29 derivatives were established using comparative molecular similarity indices and comparative molecular field analyses (CoMSIA and CoMFA). Chick chorioallantoic membrane tumor assays show that active PIB-SO and PIB-SA analogs efficiently block angiogenesis and tumor growth at similar levels as CA-4 and exhibit low toxicity on the chick embryos. Interestingly, the SAR studies of PIB-SO suggest that the phenylimidazolidin-2-one moiety was utilized to mimic the TMP (A ring) moiety in CA-4, which is commonly found in the design of potent antimicrotubule agents and described as a key structural element for the binding of antimitotics to the colchicine binding site. Also, the other report from Simoni et al. (48) demonstrated that benzo[b]thiophene (30, 31) or benzofuran (32) could replace TMP in CA-4 structure and keep antiproliferative and inhibition of tubulin polymerization activity. Compounds 30 and 31 have a binding affinity to colchicine site five times stronger than CA-4.
A number of heterocyclic ring bridging CA-4 analogs have been prepared to restrict the cis-configuration and provide optimal conformational geometry for interaction with the colchicine binding site. Introducing the heterocycles in place of the double bond can prevent the isomerization of the double bond from cis- to trans-, and may improve the drug-like properties. In 1998, Ohsumi et al. reported using five-membered heterocycle rings such as pyrazole, tetrazole, and thiazole as cis-restricted bridges in combretastatin analogs which showed potent antitubulin activity and cytotoxicity (49). Inspired by this strategy, more CA-4 analogs were prepared and their activity was evaluated. Since 2000, imidazole (33-35) (50), pyrazole (36) (50), 1,3-oxazole (37) (50), 2(5H)-furanone (38) (51), cyclopentenone (39, 40) (52), oxazolone (41, 42) (53), 4-arylcoumarin (43) (54,55), furazan (44) (56), triazoles (45) (57), 4,5-dihydroisoxazole (46) (58), 2,3-dihydrothiophene (47) (59), azetidinone (48-52) (35,60), 2-aminothiazole (53-55) (61), and tetrazole (56-58) (62) have been synthesized and appeared to elicit their tumor cytoxicity in a fashion similar to combretastatin. Some compounds were found to be slightly more potent than combretastatin itself (for example, 2-aminothiazole and tetrazole bridged compounds). Incorporation of an N-methyl group into the bridging imidazole ring (35) improved PK profiles (larger oral AUC, longer half-life, and higher bioavailability). It is also the first CA-4 analog showing potent antitumor activity in vivo orally (50). Another series of rigid analogs of CA-4, which contain the 2-amino thiazole ring system in place of the ethylene bridge present in CA-4 were reported recently (61). Compounds (53-55) with different A rings displayed antiproliferative activity at picomolar concentrations against all tested cancer cell lines as well as different drug resistant cell lines. Compound 53 was the most potent inhibitor of tubulin polymerization and one of the most potent inhibitors of colchicine binding (IC50=0.44 μM for assembly, 88% inhibition of the binding of [3H]-colchicine). Compound 54 induced apoptosis and this was partially dependent on caspase activation. In 2012, a new series of tetrazole analogs with 3, 5-dihalophenyl rings (57, 58) (62) were reported and that it appearts that a dihalogen substitution can consistently increase potency by up to 5-fold when compared to the TMP ring compound 56 on HUVECs and a range of cancer cells. Similar studies show that a halogen substituted phenyl A ring could replace TMP and gave a new vision for further modifying CA-4 (3) aryl moiety (i.e. single halogen substituted compounds 59 and 60 are more active on CA-4 resistant HT-29 cells at picomolar range inhibition) (63). Tron et al. (64) synthesized rigid analogs of CA-4 (61-64) in two steps exploiting a regioselective Suzuki coupling. Compound 63 displayed low nanomolar cytotoxicity (IC50=9.4 nM) and proved to have no cis-trans isomerization and a slower phase II glucuronidation compared to CA-4.
In contrast, some cis-restricted bridging CA-4 analogs were observed with reduced activity (Fig. 6). Three-membered cyclopropyl ring (65) (65) and anti-epoxide (66) (66) retained a certain antiproliferative activity. Substituted five membered rings such as 3-aminopyrazole (67) (67), 1,2,3-triazole (68) (68), imidazol-2-one (69) (69) showed decreased activity. Six-membered pyridine (70) (58), cyclohexenone (71) (70), 1,2,3,4-tetrahydro-2-thioxopyrimidine (72) (71) were also used to replace the olefinic moiety. A 3,5-pyrazoline (73) (72) analog was also prepared, but it showed reduced cytotoxicity.
Fig. 6.
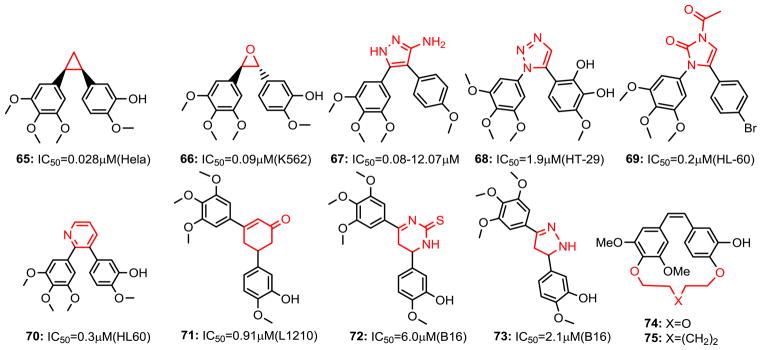
Chemical structures of chemical modified CA-4 analogs (continued).
Overall, it can be concluded that certain five-membered ring systems seem to be the best option for chemical modifications of CA-4 analogs. It is difficult to say if the decrease in activity, observed in some cases, is due to steric interactions or an incorrect orientation of the two phenyl rings in the binding site. In some cases, the two phenyl rings should have 1, 2-substituents to maximize the potency; while 1, 3- relationships (73) give a strong reduction in potency. Moreover, the presence of an aromatic character does not seem to be necessary (27-29, 46-52 and 64). An interesting investigation to rigidify the cis-conformation of CA-4 is to synthesize a para-cyclophane derivative. Although the idea of a macrocyclic analog of CA-4 is quite attractive, the resulting compounds (e.g., 74, 75) did not lead to biological activity on tubulin, precluding their anticancer applications (73).
Indole, Quinolone and Thiophene-Based CBSIs
Many natural products such as alkaloids contain indole group showed a variety of biological activities. The indole ring is a structural component of a large amount of antimitotic compounds. A series of microtubule inhibitors and anti-cancer drugs bearing indole nucleus are reviewed with their enhanced cytotoxic activity.
Arylthioindole (ATI, 76-81, Fig. 7) analogs, which possess a 3-(3,4,5-trimethoxyphenyl)thio moiety at the 2-position of the indole ring were effective tubulin assembly inhibitors (74,75). Sulfur bridging linker ATI along with the corresponding methylene (77) and ketone (78) compounds were potent tubulin assembly inhibitors reported by Silvestri et al. (76). Sulfur derivatives were superior or equivalent to the ketones for growth inhibition of MCF-7 breast cancer cells, while the methylene derivatives were substantially less effective. These compounds inhibited the growth of MCF-7, HEK, M14, and U937 cells with IC50 values in the 13–220 nM range. All three linked (S 76, CH2 77, CO 78) analogs bearing either 5-Br or 5-OMe and a 2-COOMe of the indole were found to be potent inhibitors in both tubulin polymerization and MCF-7 cell growth assays, with potencies comparable to that of CA-4.
Fig. 7.

Chemical structures of indole, quinolone and thiophene-based CBSIs.
Further investigation of new ATIs by replacing the 2-ester in 76 with 5-membered heterocyclic rings (79-81) improved their biological profile. New ATI agents were active in the Pgp-overexpressing and human transformed cell lines with improved solubility. They triggered caspase-3 activation and induced p53-independent apoptosis, differently from the classical apoptotic response induced by DNA damage that requires functional p53. The sulfur bridging ATI 79 showed satisfactory metabolic stability and PK properties (77). Molecular modeling simulation from the most active to the least active analogs in ATI series confirmed the importance of TMP by its interaction with Cysβ241. The key role of H-bond between the indole nitrogen atom and Thrα179 amide group was also confirmed by molecular dynamics simulation (77).
2-Aroylindoles with 5-methoxy-1H-2-indolyl-phenylmethanone (D-64131, 82) were discovered as tubulin inhibitors by high throughput screening from synthetic 2-aroylindole derivatives (78). D-64131 arrests tumor cells in G2/M phase, interferes with the colchicine binding site of tubulin, but does not affect β-tubulin GTPase activity. It is also cytotoxic toward MDR/MRP resistant cell lines, depicts antiangiogenic activity and shows oral bioavailability with marked in vivo antitumor activity in the human MEXF 989 melanoma xenograft model.
BPR0L075 (83) is a 3-indolyl-phenylmethanone with antimitotic activity in human cancer cells. It exerts potent antitumor and antimitotic activities through the inhibition of tubulin polymerization by binding to tubulin at the colchicine binding site. BPR0L075 showed in vitro anticancer activity against a variety of human tumor cell lines including glioblastoma, breast, gastric, leukemia, liver, and colorectal cancer cells. Furthermore, phosphorylated Bcl-2, perturbed mitochondrial membrane potential, and activation of the caspase-3 cascade may be involved in BPR0L075-induced apoptosis. Notably, BPR0L075 can overcome Pgp170/MDR and MRP mediated multidrug-resistant to vincristine, paclitaxel, and colchicine. Moreover, BPR0L075 shows potent activity against the growth of xenograft tumors at i.v. doses of 50 mg/kg in nude mice (79).
N-Heterocyclic indolyl glyoxylamide BPR0C261 (84) is an analog of indibulin (17) and possesses in vitro/in vivo anticancer activities (80,81). BPR0C261 destabilizes microtubules and blocks cell cycle transition specifically at G2/M phase. Moreover, apoptosis induction in the cancer cells is another underlying mechanism for the anticancer effects of BPR0C261. Colchicine binding assay indicated BPR0C261 at both 5 and 20 μM competitively binds to tubulin and strongly interferes with the colchicine binding to tubulin (80). In addition, BPR0C261 concentration-dependently inhibited the proliferation and migration of HUVECs with an IC50 value of 1.6 nM and disrupted the endothelial capillary-like 2D tube formations of HUVEC. Given orally, BPR0C261 suppressed angiogenesis in a mouse model. It was found orally absorbable in mice and showed a good oral bioavailability (F=43%) in dogs. Moreover, the combination of BPR0C261 plus cisplatin synergistically prolonged the lifespans of mice inoculated with murine leukemia cells.
1-Aroylindole, 1-aroylindoline, 1-aroyl-1,2,3,4-tetrahydroquinoline, and 2-, 3-, 4-, 5-, 6-, 7-, 8-aroylquinolines were synthesized and evaluated for anticancer activity as CA-4 derivatives (82,83). Among these substituents, 1-aroylindoles with C4-amino (85) and C4-hydroxy (86) substituents exhibited antitubulin activity superior or comparable to that of colchicine and CA-4 with IC50 values of 0.9 and 0.6 μM, respectively. They also showed antiproliferative activity with an IC50 range of 0.3–5.4 nM in a set of human cancer cell lines. In an aroylquinoline series, only 2- and 6-aroylquinoline showed potency against five cancer cell lines. 5-Amino-6-methoxy-2-aroylquinoline 87 has the most potent antiproliferative activity (IC50=0.2 to 0.4 nM) against various human cancer cell lines including a MDR-resistant cancer cell line KB-vin10. Compound 87 exhibited more potent inhibition of tubulin polymerization (IC50=1.6 μM) than CA-4 (IC50= 2.1 μM) and showed strong binding property to the colchicine binding site on microtubules. A continued modification of the linkers (-O-, -NH-, -S-, -SO2-, and direct bond) between TMP and 5-amino-6-methoxyquinoline identified active oxygen and sulfur atom linked analogs (88-90) (84). Although less active than parent compound 87, the most potent compounds, sulfide 89 and sulfone 90, still showed low nanomolar inhibition against cancer growth and revealed that the linkage has a capacity for various bridging groups. Moreover, Compound 87 demonstrated the ability to overcome the efflux protein (MDR/Pgp or MRP) mediated drug-resistance in human cancer cell lines (KB-Vin 10, KB-S15, and KB-7D) with IC50 values ranging from 2.4 to 2.8 nM.
Florent’s group explored the possibility of replacing the usual TMP ring present in a large majority of CA-4 analogs with a trimethoxyindole ring (91) to obtain substituted 2-aroylindoles through a palladium-catalyzed domino reaction (85). Compound 91 only displayed fair activity (IC50= 4 μM). However, Liou et al. (86,87) successfully identified potent 1-benzyl-4, 5, 6-trimethoxyindole (92 and 93, mean IC50=26 and 27 nM, respectively), 1-indolylindole (94) and 1-quinolinylindole (95) as a new class of colchicine binding site microtubule destabilizing agents. 4, 6- or 5, 6-Dimethoxyindole analogs showed dramatically reduced bio-activity indicating that the trimethoxyindole moiety in the 1-benzylindoles series is critical for activity. Compounds 94 and 95 exhibited anti-proliferative activity with IC50 values ranging from 11 to 49 nM in a diverse set of human cancer cell lines, including MDR1-expressing cervical carcinoma cell line KB-VIN10. These two compounds also demonstrated potent tubulin polymerization inhibitory activity with IC50 values of 1.7 and 2.7 μM, respectively. Molecular modeling and docking studies showed A-ring of 94 and CA-4 are located in a similar area and have hydrophobic interactions with Cys241, Leu248, Ala250 and Leu255. The Brings of both compounds also occupied the same pocket and have hydrophobic interactions with Asn258, Met259, Lys352 and Val181.
Hu et al. (88) discovered 2-(2-amino-5-(1-ethyl-1H-indol-5-yl)pyrimidin-4-yl)phenol (97) from 2-(2-amino-5-(4-(ethyl (methyl)amino)phenyl)pyrimidin-4-yl)phenol (96, IC50 range from 90 to 550 nM) by forming a 5-indolyl substitution via cyclization of N-methyl to phenyl ring. Compound 97 displays activity as an inhibitor of tubulin polymerization (IC50 =0.79 μM, 3.39 fold more active than colchicine IC50=2.68 μM), and it possesses the ability to arrest cells at the G2/M phase of the cell cycle and antiproliferative activities against several tumor cell lines with IC50 values ranging from 16 to 62 nM.
El-Nakkady et al. (89) reported introduction of hydrazides at the 3 position of 2-phenylindole. The most potent compound 98 exhibited an IC50 value of 1.6 nM, being more active than vincristine (IC50=2.0 nM). Structures of compound 98 docked in the colchicine binding site of tubulin showed a hydrogen bond between the indole NH and Asnα101 in the colchicine binding site of tubulin, suggesting that these phenolic indoles might act through inhibition of tubulin.
T115 (99) is a recently reported N-substituted 1, 2, 4-triazole based colchicine binding site tubulin inhibitor. It has potent and selective inhibitory effects against several cancer cell lines and their corresponding drug resistance cells. T115 is devoid of the instability issue of stilbene-like CA-4 analogs by introducing the triazole ring system as a bridge to retain the cis-configuration which is the biologically active form. Acute toxicity studies showed T115 was well-tolerated in vivo with a maximum tolerated dose of 400 mg/kg and showed no cytotoxicity against normal fibroblasts cell lines. T115 significantly inhibited tumor growth (i.p.) in mouse xenograft HT-29 and PC-3 models (90).
Compound 100 was originally designed by Kelly’s group (91) for a nuclear hormone receptor program but it exhibited potent inhibition of mitosis at the G2/M stage and proved to be a colchicine binding site tubulin polymerization inhibitor. The 4-indolyl group at the 1-position of phenyl was found to be critical for potency. Compounds with NH/C=O linkers or without the linker at the 3-position all showed good activities against multiple cell lines (as low as 0.01 μM). Replacement of the phenolic hydroxyl group in 100, which suffered from rapid glucuronidation, with a sulfonylamide gave a potent compound LP-261 (101) with significantly improved oral bioavailability in rat PK studies (F=24% vs. 80%).
NSC 664171 (102) is a quinolinone derivative that has demonstrated strong cytotoxic effects with GI50 values in the nanomolar or subnanomolar range in many different tumor cell lines such as lung, ovary, prostate, breast cancers. It is also a potent inhibitor of tubulin polymerization with activity comparable to those of the other well-known antimitotic natural products such as colchicine, podophyllotoxin, and CA-4 (92).
CHM-1 (103) was discovered via SAR studies of a series of 2-phenyl-4-quinolones as a new class of antimitotic antitumor agents. It showed potent cytotoxicity with an average log GI50 value of −6.47 (log of the concentration that reduced cell growth by 50%) in the National Cancer Institute (NCI)’s 60 human tumor cell line (93). This compound was also a potent inhibitor of tubulin polymerization with an IC50 value of 0.85 μM. Most importantly, it demonstrated good in vivo activity against the OVCAR-3 ovarian cell line, prolonging the life span of mice bearing the tumor by 130%.
S9 (104) (94), a hybrid molecule of α-methylene-γ-lactones and 2-phenyl indoles derived from PI3K inhibitor wortmannin, is a multi-inhibitor simultaneously targeting both PI3K-Akt-mTOR pathway and the microtubule cytoskeleton. S9 down-regulated phosphorylation of Akt, mTOR, p70S6K and 4EBP1 through stimulation of EGF in Rh30 cells. It inhibited the polymerization of tubulin by binding to the colchicine binding site and cause M phase arrest. Dual mechanism of PI3k-Akt-mTOR signaling and tubulin inhibiting contributes to cytotoxicity observed in vitro against a panel of tumor cells including MDR tumor cells and in vivo antitumor activity in human tumor xenografted mice models.
A series of thiophene derivatives (105, 106) have been synthesized and were found to be potent inhibitors of tubulin polymerization (61,95). Compounds 105 and 106 inhibit cancer cell growth at subnanomolar concentrations and interact strongly with tubulin by binding to the colchicine site. Flow cytometry of these compounds had cellular effects typical of agents that bind to tubulin, causing accumulation of cells in the G2/M phase of the cell cycle and a substantial increase in the number of apoptotic cells.
Dodd’s group reported a series of fused indole ring compounds (107-109) (96,97), which contain a seven-member amide ring. It showed similar effect with colchicine in potency and inhibiting tubulin polymerization, by the interruption of cancer cell growth at the G2/M stage. The ability of these compounds to promote apoptosis in the cancer cells studied was also clearly demonstrated. Compound 108 was more effective than colchicine in causing tumor volume regression after 7 days of treatment in a chick chorioallantoic membrane model of transplanted human glioma (U87) tumor growth. The research group also synthesized rigid analogs of these compounds by fusing another five-member ring between the phenyl and seven member ring, but this led to a loss of activity (97).
Chalcone Compounds
Antitubulin activity was found in chalcone compounds which bears an aromatic ketone and an enone as the central core. Chalcones are precursors of flavonoids and scaffolds for a variety of important biological compounds. They are abundant in edible plants and display biological activities, including anti-cancer, anti-inflammatory, anti-tubercular, and anti-fungal, etc. Their biological properties are largely due to the α, β-unsaturated ketone moiety. Modifications on the two aromatic rings remains an area of pharmacological interest in the screening of active chalcones, such as MDL-27048 (110, Fig. 8) and JAI-51 (111) (98). The effects of MDL-27048 on microtubules are similar to those of colchicine or combretastatin analogs. It represents a new type of antitubulin agent, which could prove to be valuable as an experimental inhibitor in the study of microtubules and microtubule-mediated functions.
Fig. 8.
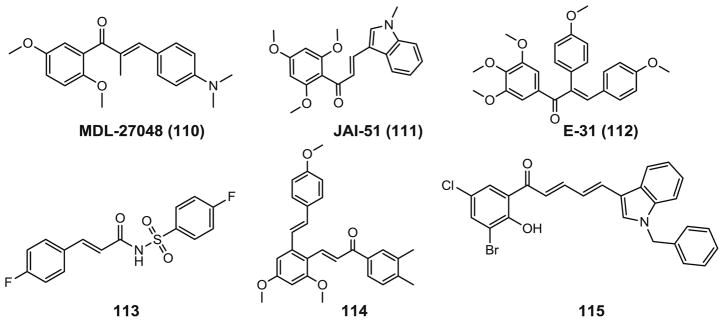
Chemical structures of chalcone analogs.
A series of aryl- and aroyl-substituted chalcone analogs of the tubulin binding agent CA-4 (3) were prepared by Flynn’s group (99). All compounds were assessed as inhibitors of tubulin polymerization, but only E-31 (112) had activity similar to that of CA-4 (2.5 vs. 2.0 μM). However, compound E-31 did not exhibit antiproliferative activity against the MCF-7 cell line.
Zhu’s group recently reported a series of novel antitubulin polymerization inhibitors containing the chalcone skeleton and a sulfonamide moiety (113) (100) or containing the resveratrol skeleton and chalcone moiety (114) (101). Compound 113 showed the most potent inhibitory activity with an IC50 value of 0.8 μg/mL and antitubulin polymerization activity with an IC50 of 2.4 μg/mL in the cinnamic acyl sulfonamide derivatives. Among the resveratrol derivatives, compound 114 showed the most potent inhibitory activity. It inhibited the growth of a number of cancer cell lines with IC50 values ranging from 0.1 to 1.4 μg/mL. It also inhibited the polymerization of tubulin with an IC50 of 2.6 μg/mL. Computational docking analysis of the binding conformation of compound 113 and 114 in the colchicine binding site demonstrated that interactions with the protein residues in tubulin led to the antiproliferative activity.
A series of dihalogenated chalcones and structurally related dienones have been synthesized and showed fair cytotoxic activities (IC50 in low micromolar range) toward individual cancer cell lines. Most of this series of compounds are tubulin polymerization inhibitors. However, one dienone derivative (115) was found unexpectedly to stabilize tubulin similar to docetaxel. This is the first reported chalcone derivative with microtubule-stabilizing activity.
Sulfonanilides Compounds
The sulfonamides have been in clinical use for yearsdue to their biological activities such as antibacterial, antidiabetic, antithyroid, antihypertensive, or antiviral activities. Recently, many structurally novel sulfonamide derivatives have shown substantial antitumor activity. Several CBSIs containing the sulfonamide group were used in clinical studies such as ABT-751 (14, Fig. 3) and T138067 (15). Sulfonamide analog ELR510444 (116, Fig. 9) (102) has potent microtubule disrupting activity via direct interaction with tubulin at the colchicine binding site. ELR510444 potently inhibited cell proliferation with an IC50 value of 9.0 nM in MDA-MB-435 cells and did not serve as a substrate for the Pgp drug transporter and it retains activity in class III βtubulin overexpressing cell lines, suggesting that it circumvents both clinically relevant mechanisms of drug resistance to this class of agents. ELR510444 also shows efficacy in the MDA-MB-231 xenograft model with at least a 2-fold therapeutic window. Studies in tumor endothelial cells show that ELR510444 has potential antivascular effects. ELR510444 also leads to caspase-3/7 activation and subsequent apoptosis with cellular EC50 values of 50–100 nM. The compound induces an initial cellular arrest in G2/M and a significant tubulin depolymerizing effect, followed by an increase in a sub-G1 (apoptotic) population after 24 h.
Fig. 9.
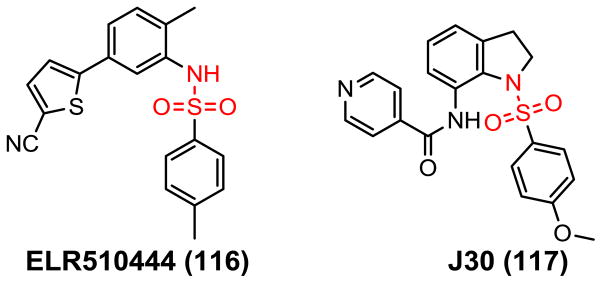
Chemical structures of sulfonamide CBSIs.
J30 (117) is an orally active sulfonamide CBSI discovered by Liou’s group (103,104) and shows strong antiproliferative activity with IC50 values ranging from 8.6 to 11.1 nM against human tumor cell lines, as well as the ability to overcome drug resistance. J30 depolymerizes microtubules in the KB cell line, resulting in an accumulation of G2/M phase cells. Further studies indicate that J30 causes cell cycle arrest, as assessed by flow analyses and the appearance of MPM-2 (a specific mitotic marker), and is associated with up-regulation of cyclin B1, phosphorylation of Cdc25C, and dephosphorylation of Cdc2. J30 also causes Bcl-2 phosphorylation, cytochrome C translocation, and activation of the caspase-9 and caspase-3 cascades, suggesting it mediated apoptotic signaling pathway that depends on caspases and mitochondria. J30 given orally inhibits tumor growth in NOD/scid mice bearing human oral, gastric, and drug-resistant tumor xenografts.
2-Methoxyestradiol and its Derivatives
2-Methoxyestradiol (2-ME, 12, Fig. 3) is the main metabolite of the hormone β–estradiol and is a weak competitive inhibitor of colchicine binding to tubulin. 2-ME and its analog ENMD-1198 (13, Fig. 3) are under clinical studies as promising anticancer agents with dual activity against cancer cell proliferation and angiogenesis (17,19). Cushman and colleagues have focused on changing substituents at the 2- and 6-positions (105) of 2-methoxyestradiol to increase cytotoxicity and tubulin polymerization inhibition, and their efforts have produced 2-ethoxyestradiol (118, Fig. 10), which was 17-fold more cytotoxic than 2-ME in the whole panel of tumor cell lines in MGM value (Mean graph midpoint for growth inhibition of all human cancer cell lines tested, 76 nM vs. 1.3 μM). Furthermore, molecules carrying an NOH moiety in the 6-position with either CH3CH2O or CF3CH2O in the 2-position (119) were also superior to 2-ME in all three parameters (MGM=79 and 66 nM). A 2-ethoxyestradiol derivative with an additional ketone moiety in position 6 (120) was still highly potent (MGM=130 nM). Later modifications at the 2-position by the same research group resulted in some other potent compounds, although not superior to 2-ME, such as 2-(E-3′-hydroxy-1′-propenyl) estradiol (121, MGM=1.1 μM) and 2-(1′-propynyl) estradiol (122, MGM=1.7 μM) (106). Additionally, the 17-position contributes significantly to the anticancer activity and pharmacokinetic profile of 2-ME. Oxidation of the 17-OH of estradiol to estrone is one of the main routes for metabolic deactivation and steroid clearance. 2-Ethoxy-17-methylene analog of 2-ME (123, MGM=0.79 μM) showed greater tubulin polymerization inhibition and cytotoxicity than 2-ME and contained moieties that are expected to inhibit deactivating metabolic processes (107). Agoston and colleagues investigated whether substituents at the neighboring 16-position may inhibit this process (108). Larger substituents tended to result in lower cytotoxicity, but a few 16-position modified compounds including 124 were comparable to 2-ME in assays with HUVEC and MDA-MD-231 cells. However, all these agents did not address the major problem of low oral bioavailability. If sulfamate is introduced on the hydroxyl groups of the 3 and 17 positions (125 and 126), (109,110) the sulfamoylated compounds retained the ability to bind to the colchicine site of tubulin, conferred superior biological activity and demonstrated resistance to metabolism with a high bioavailability (85%). Most importantly, superior biological activity in vivo was found after oral application of bis-sulfamate (125) compared to 2-ME at a daily dose of 20 mg/kg in mouse xenograft models of MDA-MB-435 and MCF-7 cancer cells. Similar results were obtained in the MCF-7 model for a 17-cyanomethyl analog (126). Given orally, 125 and 126 were also superior to 2-ME in an assay testing the inhibition of blood vessel growth, and demonstrated a superior ability to inhibit neovascularization. These agents work as inhibitors of tubulin polymerization and inhibit the binding of radiolabeled colchicine to tubulin. The potential hydrogen bonding of the terminal NH2 group of the sulfamate is not essential based on SAR studies of this series of compounds. As illustrated by compounds 127 and 128 (111), when the NH2 is replaced by a CH3 along with a 2-ethyl substituents, A 4–11 fold enhancement of anti-proliferative activity was observed.
Fig. 10.
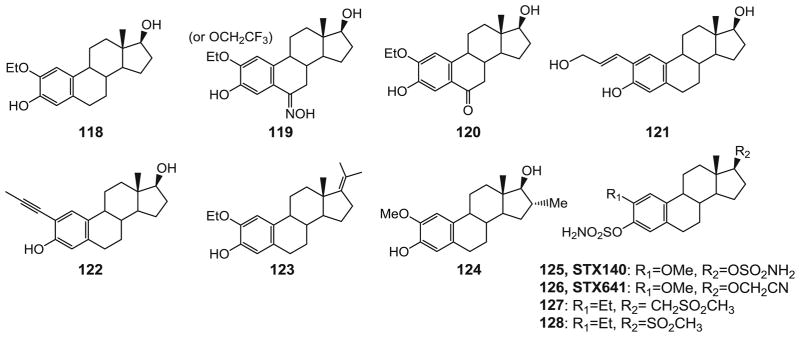
Chemical structures of 2-ME analogs.
Analogs of Natural Products
Natural products are excellent sources for drug discovery and development. A large amount of anticancer drugs are either natural products or derivatives from natural sources including plants, animals and microorganisms. Colchicinoids and combretastatins are isolated from plants and are well-studied tubulin binding agents. Structure modification of natural products which bind to the colchicine site are continually being performed and have successfully generated plenty of synthesized derivatives.
Desmosdumotins and Analogs
Desmosdumotins AC (129-131, Fig. 11) are isolated from the root bark of Desmos dumosus. These natural flavonoid and chalcone scaffolds represent promising new lead structures for further new analog development as potential antitumor agents. Lee’s group synthesized derivatives of desmosdumotin B and C with improved anticancer activity and selectivity against Pgp overexpressing multidrug resistant cell lines (112). Triethyl analogs with 4′-alkyl desmosdumotin B (130) derivatives 132 and 133 showed significant selectivity with ED50 values of 0.03 and 0.025 μg/mL, respectively, against KB-VIN cells (MDR) with ratios of >460- and 320-fold compared with that of KB (non-MDR) cells (113). Further modifications on B-ring systems with bicyclic or tricyclic aromatic rings identified derivative 134 with a benzo[b]thiophenyl B-ring, which was highly active with GI50 values of 0.06–0.16 μM over a panel of cancer cell lines including Pgp expressed MDR cells. Furthermore, 134 inhibited tubulin assembly in vitro with an IC50 value of 2.0 μM and colchicine binding by 78% as well as cellular microtubule polymerization and spindle formation, which confirmed they are a new class of CBSIs (114). Modifications of desmosdumotin C (131) led to 3,5,5-tripropyl-4′-bromo analog 135, which possessed the most potent activity against A549, HCT-8, 1A9, PC-3, KB and KB-VIN cells with ED50 values of 0.87–2.25 μg/mL (1.8–2.6 μM). A MeO- or PrO- group at C-4 was generally preferred over other alkyl ether groups. Oligonucleotide microarray studies showed that 135 may modulate spindle assembly checkpoint and chromosome separation and arrest cells mainly in the G2/M phase (112,115).
Fig. 11.

Chemical structures of CBSIs derivatives of natural products.
Centaureidin (136, Fig. 11) is an O-methylated flavonol isolated from plants like tanacetum microphyllum, brickellia veronicaefolia, bidens pilosa and polymnia fruticosa. Centaureidin inhibits tubulin polymerization and competes with the binding of [3H]-colchicine to tubulin. It can induce mitotic figure formation in whole cells at cytotoxic concentrations. Centaureidin is the first known example of a flavone with anti-mitotic activity (116).
Noscapine (137, Fig. 11), an anti-cough opium alkaloid, binds to tubulin, alters its conformation, affects microtubule assembly, and causes apoptosis in many cell types. Oral administration of noscapine has potent antitumor activity against solid murine lymphoid tumors and human breast and bladder tumors implanted in nude mice. Amino-noscapine was designed and chemically synthesized following the guidance of a linear interaction energy (LIE) method with a surface generalized Born (SGB) continuum solvation model. The amino noscapine derivative 138 has higher tubulin binding activity with the binding pocket of tubulin involved in three hydrogen bonds and they are distinct compared to noscapine which involved only one hydrogen bond. The LIE–SGB model constitutes the first evidence that this class of compounds binds to the colchicine binding site. Amino noscapine has overall much stronger anti-tumor activity than noscapine against the NCI-60 cancer cells panel (117).
Polygamain (139, Fig. 11)
Lignan polygamain was recently isolated as the microtubule-active constituent from the crude extract of the Mountain torchwood, Amyris madrensis (118). Polygamain has structural similarities to podophyllotoxin (8, Fig. 3) and has potent inhibition against a wide panel of cancer cell lines (average IC50 is 52.7 nM). It inhibits the tubulin assembly and interacts within the colchicine binding site. Molecular modeling suggests that the benzodioxole group of polygamain occupies the same pocket as the TMP group of podophyllotoxin, but has distinct interactions within the hydrophobic pocket. Polygamain circumvents two common mechanisms of drug resistance for microtubule targeting agents, the expression of Pgp pump and the class βIII isotype of tubulin.
Diketopiperazine
Cyclopeptides are known to exhibit biological activities ranging from cell-cycle inhibition to specific enzyme-activity modulation. The smallest cyclopeptides studied for their potential therapeutic effects are diketopiperazines. Tryprostatin A and B (140 and 141, Fig. 11), diketopiperazines isolated from the fermentation broth of Aspergillus fumigatus by Osada and co-workers in 1995, hold great anti-cancer promise (119). Tryprostatin A reverses resistance against mitoxantrone in various breast cancer resistance protein-expressing human cancer cells accompanied by a selective inhibition of the ATP-dependent drug transport activity of breast cancer resistance protein (120). Phenylahistin (NPI-2350, 142), a metabolite isolated from Aspergillus ustus, is another fungal isoprenylated diketopiperazine, composed of the phenylalanine and dehydrohistidine. The fungus produces phenylahistin as a racemic mixture, but only the (-)-enantiomer proved to be cytotoxic (121). Interestingly, (Z)-dehydrophenylahistin (143), in which chirality is lost by dehydrogenation of the phenylalanine moiety, has also been reported as an antimitotic agent being 1,000 times more active than (-)-phenylahistin toward the first cleavage of sea urchin embryo (122). 143 also showed significant activity against human cancer cells lines, with IC50 values in the nanomolar range. The isoprenyl group attached to the imidazole ring was also indicated to be important for activity. A series of analogs of dehydrophenylahistin was synthesized, resulting in the identification of plinabulin (NPI-2358, 144). Plinabulin is a tert-butyl analog of phenylahistin with a colchicine-like tubulin depolymerization activity and a potent microtubule-targeting diketopiperazine derivative with IC50 values in the low nanomolar range. Compound 144 is equally active against MDR tumor cell lines and is now under phase II clinical trials as a vascular disrupting anti-cancer drug. To develop more potent anti-microtubule and cytotoxic derivatives based on the dehydrophenylahistin skeleton, Hayashi’s group performed further SAR study on the tert-butyl and the phenyl groups of 144, and evaluated their cytotoxic and tubulin-binding activities (123). Compounds 145 with a 2, 5-difluorophenyl and 146 with a benzophenone in place of the phenyl group had both vascular disrupting and cytotoxic activities (5- and 10-times more potent than that of CA-4, respectively). Compounds 145 and 146 exhibited a lowest effective concentration of 2 nM and 1 nM for microtubule depolymerization, respectively.
Curacin A
The marine natural product curacin A (11, Fig. 3) is a potent inhibitor of cellmitosis, binding quickly at the colchicine binding site of tubulin. The lipid structure of curacin A differs greatly from that of colchicine and other CBSIs. Analog studies of curacin A were made to simplify the structure and increase water solubility and chemical stability. However, minor changes in the lipid chain or fragments of curacin A can lead to inactive derivatives: opening of the thiazoline ring and longer chains or acyl groups as oxygen substituents on C13 are not tolerated. The C7-C10 diene segment of curacin A is sensitive to modification. Replacement of cyclopropyl ring with a tert-butyl group (147-150, Fig. 11) leads to a>2-fold decrease in activity. The oxazoline and oxazole analogs 147 and 148 lack any appreciable biological efficacy (124). Compound 151, with an ethylenedioxy bridge at C13 causing loss of chirality, is equivalent to curacin A in potency. Classic TMP group introductions provide effective replacements for the labile cyclopropyl thiazoline moiety in analog 152. Further introducing the oxime ether linker to replace the (Z)-C3-C4 alkene moiety lead to discovery of analog 153, which is found to be more potent than curacin A in inhibiting the assembly of purified tubulin and shows more chemical stability in the presence of plasma (125).
Agents Covalently Binding to the Tubulin Colchicine Binding Site (Fig. 12)
Fig. 12.
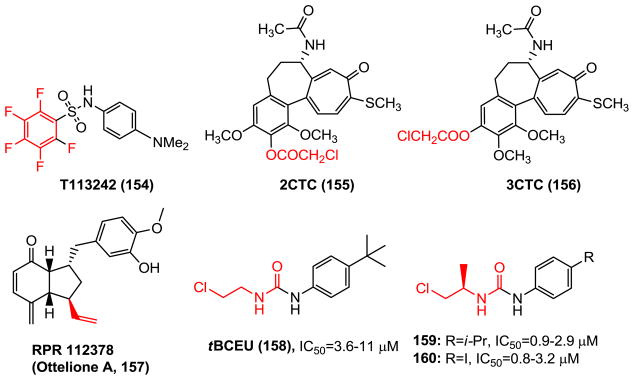
Chemical structures of CBSIs covalently binding to tubulin colchicine binding site.
While most of CBSIs reversibly bind to the tubulin pocket, design and synthesis of irreversible CBSIs has aroused interest since covalent bond formation could circumvent the resistance caused by the mutations of tubulin residues and trigger microtubule disruption. A subset of irreversible CBSIs was reported, which formed covalent bonds with tubulin amino acid residues, generally cysteines. Alkylation of Cysβ239 caused loss of tubulin’s ability to polymerize. As previously mentioned aryl-pentafluorosulfonamaide T138067 (15, Fig. 3), as well as its analog T113242 (154, Fig. 12) (126), covalently alkylate tubulin Cysβ239 and induce microtubule depolymerization. Colchicinoids 2-chloroacetyl-2-demethylthiocolchicine (2CTC, 155) and 3-chloroacetyl-3-demethylthiocolchicine (3CTC, 156) resemble colchicine in binding to tubulin and react covalently with β–tubulin, forming adducts with Cysβ239 and Cysβ354 (127).
A screening program aimed at the discovery of new antimicrotubule agents yielded RPR112378 (ottelione A, 157), a natural inhibitor of tubulin polymerization, first isolated from the fresh water plant Ottelia alismoides. RPR112378 is an efficient inhibitor of tubulin polymerization (IC50 =1.2 μM) and a highly cytotoxic compound with an IC50 of 0.02 nM against KB cell growth. The cytotoxicity of RPR112378 is probably caused by an addition reaction of ethylenic bond with sulfhydryl of cysteine residues (128).
tBCEU (158), a derivative of N-aryl-N′-2-chloroethylurea (CEU), was reported to react exclusively with Cysβ239. Further modification of CEU by introducing a branched R-alkyl chain (159, 160) led to enhanced cytotoxicity (including cell line with mutations in tubulin) and rapid alkylation of β-tubulin in cells. It is speculated that the active site of the β-tubulin is stereo selective and the R-isomer of the branched chloroethyl group allowed a sterically favored orientation of the alkylating moiety, promoting the approach of the chlorine atom toward the sulfhydryl of Cysβ239 residue. However, subsequent work using mass spectrometry has identified that the Gluβ198, which is adjacent to the colchicine binding site behind the two potent nucleophilic residues, Cysβ239 and Cysβ354, has been shown to covalently react with CEU (129,130). None of the cysteine residues of β-tubulin was linked to the alkylating agent. This result is contrary to the previous hypothesis that the reacting amino acids in tubulins would be mainly the cysteine residues.
Other CBSI: Screened and Synthesized
Anthracenone
Antimitotic anthracenone-based molecules (161-168, Fig. 13) that inhibit tubulin polymerization have been reported (131). The majority of these compounds possess an unsaturated bond between the anthracenone and the terminal aromatic ring. The modifications of the linker between the anthracenone and the terminal phenyl ring were performed with the benzylidene C=C double bond (161, 162), chalcone (165), oxime (166) and a C=N bond (167, 168). The active anthracenone analogs inhibit the growth of various tumor cell lines in the G2/M phase with a cell cycle dependent manner by interacting with tubulin at the colchicine binding site.
Fig. 13.

Chemical structures of screened and synthesized CBSIs.
Chromene
4-Aryl-4H-chromenes (169, Fig. 13) are identified as apoptosis-inducing agents, possessing vascular disrupting activity through a cell-based apoptosis screening assay (25,132). One chromene lead compound EPC2407 (crolibulin, 18, Fig. 3) has been used in clinical trials. The 4-aryl-4H-chromenes inhibit tubulin polymerization and bind at or close to the binding site of colchicine. The cells treated with these agents undergo a G2/M arrest prior to caspase activation. They are also active in the multidrug resistant MES-SA/DX5 tumor cells and are highly active as single agents (subnanomolar range potency) and in combination with other anticancer agents such as cisplatin in several tumor xenograft models. SAR studies of the 4-aryl group with 5-, 6-, 7-, 8-substituents and fused 7,8-chromene rings were carried out and led to the discovery of a 7-methyl-4H-pyrrolo[2,3-H]chromene analog with low nanomolar potency (GI50=0.3 nM against T47D colon cancer cells) (133).
The small-molecule fluorenone UA62784 (170, Fig. 13) was first reported by Henderson et al. in 2009 (134) from a high throughput cytotoxicity screening that selectively targeted DPC4-deleted pancreatic cancer cells. UA62784 shows cytotoxic in the nanomolar range, causes G2/M arrest, induces apoptosis, and prevents the formation of a functional bipolar spindle during mitosis by inhibiting the microtubule-associated ATPase activity of the CENP-E kinesin-like protein, but it shows no effect on tubulin polymerization. Continued research by Abrieu et al. (135) on this molecule revealed an alternative mechanism of antitumor action that showed UA62784 interacts with tubulin at or near the colchicine binding site, not via inhibiting ATPase activity of kinesin CENP-E. Immunofluorescence and live cell imaging indicate that UA62784 perturbs the mitotic spindles. It also shows additive effects with some known microtubule-depolymerizing drugs including vinca alkaloids, colchicine, or nocodazole, but not paclitaxel.
JG-03-14 (171, Fig. 13) (136,137), a tetra-substituted brominated pyrrole, has been shown to have broad cytotoxic and antiproliferative effects against cancer cells in vitro (IC50=36–80 nM, including drug-resistant cell line) and in vivo activity due to its ability to potently bind at or near the colchicine binding site on tubulin. JG-03-14 can cause dose-dependent loss of cellular microtubules and it can phosphorylate Bcl-2, arrest cells in the G2/M phase and it is a poor substrate of Pgp (137). JG-03-14 has direct effects on endothelial cells that could be indicative of therapeutically useful anti-vascular actions. Molecular modeling studies have indicated that while the dimethoxyphenyl group of JG-03-14 occupies a space similar to that of the TMP group of colchicine and interacts with Cysβ241, the tetra-substituted pyrrole group interacted with both α- and β-tubulin in space not shared with colchicine. The side chain ethoxy oxygen forms a bifurcated H-bond interaction with the NH2 of Asnα101. This may suggest significant differences as compared to colchicine in the mechanism of binding to tubulin.
MT119 (172, Fig. 13) is a planar-structured compound, which was optimized as a new CBSI from a combinatorial library (138). MT119 inhibit tubulin polymerization significantly both in tumor cells and in cell-free systems, which is followed by the disruption of mitotic spindle assembly. It arrests different tumor cells at the G2/M phase, and inhibits the proliferation of ten tested tumor cells with IC50s ranging from 0.06 μM to 0.53 μM. MT119 is also cytotoxic to cancer cells resistant to vincristine, adriamycin or mitoxantrone.
NSC 676693 (173, Fig. 13) is a novel antimitotic compound based on the arylthienopyrrolizinone molecular skeleton. It has strong anticancer activity in human cancer cells with IC50 in the nanomolar range and it interacts with tubulin in the micromolar range. Based on the structure of NSC 676693, a series of it analogs have been synthesized and the best compound shows 10-fold improvement of potency compared with NSC 676693 (139).
Indanocine (174, Fig. 13) is a synthetic indanone that was first identified by the NCI’s Developmental Therapeutics Program as an antiproliferative agent. It is a tubulin inhibitor as well as an apoptosis inducer in certain cancers. The activity of indanocine on multidrug-resistant cancer cells indicates that indanocine could be a potential lead compound for the development of chemotherapeutic strategies for drug-resistant cancers (140).
IRC-083927 (175, Fig. 13) is an orally available synthetic imidazole derivative, which was developed by ISPEN, and inhibits tubulin polymerization by binding to the colchicine site. It shows highly potent antiproliferative activity on human tumor cell lines including taxane, vinca alkaloid, or epothilone resistant cells due to the presence of efflux pumps (Pgp, MRP) and/or mutated tubulin (141). IRC-083927 displayed cell cycle arrest in G2/M phase in tumor cells and inhibited endothelial cell proliferation in vitro and vessel formation in the low nanomolar range supporting an antiangiogenic action. Furthermore, the oral administration of IRC-083927 in athymic mouse models showed a significant in vivo antitumor activity without apparent toxicity (141). The antitumor effect induced by IRC-083927 supports its potential for the treatment of advanced cancers, particularly those resistant to current clinically available drugs.
XRP44X (176, Fig. 13) is a pyrazole derivative reported by Wasylyk et al. Itintergrates two anticancer mechanisms: antimitotic and inhibition of MAPK/ERK signaling pathway. It binds to the colchicine binding site of tubulinand depolymerizes the microtubules, thus affects the morphology of the actin cytoskeleton. It also inhibits fibroblast growth factor 2 (FGF-2)–and phosphorylation by the Ras-Erk signaling upstream from Ras (142). XRP44X also inhibits the growth of transformed cells in culture and angiogenesis in an ex vivo assay of endothelial cell sprouting (142).
KRIBB3 (177, Fig. 13) is a novel microtubule inhibitor with an isoxazole moiety in the structure. It can induce mitotic arrest and apoptosis in human cancer cells. It was first developed by Korea Research Institute of Bioscience and Biotechnology. KRIBB3 exerts significant antitumor activity against a variety of cancer cells such as colon, prostate, breast, and lung by inhibiting tubulin polymerization. KRIBB3 selectively arrests cell cycle at the mitotic phase. In vivo, KRIBB3 decreased tumor volume by 49.5% (50 mg/kg) and 70.3% (100 mg/kg) compared to control mice (143).
A-105972 (178) and A-204197 (179, Fig. 13) are both oxadiazoline analogs identified by Abbott Laboratories from a high throughput screening of 60,000 compounds. The two drugs have the exact same scaffold and the only difference is a functional group in the aromatic ring. They are potential anticancer agents and were undergoing preclinical development by Abbott Laboratories (144). A-105972 inhibited the growth of several tumor cell lines, including breast, central nervous system, colon, liver, lung, and prostate cancer cell lines, as well as multidrug-resistant cells with IC50 in between 20 and 200 nM depending on specific cell types, but its utility in vivo was limited by a short half-life. A-105972 and A-204197 interact with tubulin and induce apoptosis and Bcl-2 phosphorylation. A-204197 has shown high potency in vitro (IC50 36–48 nM) and is especially effective against MDR cancer cell lines. These results indicate A-204197 is a promising antimitotic agent that has potential for treating neoplastic diseases with great efficacy (145).
A-289099 (180, Fig. 13) is an indolyloxazoline derivative with antimitotic activity developed also by Abbott Laboratories. It was discovered as a result of structural optimizations of the lead compound A-105972 (146). A-289099 exerts its anticancer activity by inhibition of tubulin polymerization and by binding at the colchicine binding site. A-289099 has high anti-proliferative activity in a number of cancer cells with EC50 values ranging from 5.1 to 12.8 nM. A-289099 is orally active in a syngeneic M5076 murine reticulum sarcoma flank tumor model. Pharmacokinetic study showed that the bioavailability of A-289099 in monkey is 18.6%. The half-life ranges from 1.1 h to 3.3 h depending on species (146,147).
2-Indolinones (181-183, Fig. 13), a series of methylene-bridged heterocycles discovered by Andreani et al. (148,149) have similar inhibitory effects on colchicine binding site and tubulin polymerization. These compounds arrest the cells in the G2/M phase of the cell cycle, and are associated with activation of apoptosis, disrupt the mitochondrial membrane potential, and increase ROS production and Bax translocation into mitochondria. The apoptosis in the HT-29 cells is accompanied by caspase activation and phosphatidylserine externalization. Interestingly, the most potent compounds 181 and 183, which show more potent mean GI50 (40–70 nM in NCI screening) than vincristine (GI50=200 nM), strongly inhibit the activation of the kinase Akt associated with cell survival and proliferation.
CBSI with Improved Solubility and PK Profiles
A major problem with CBSIs is their limited aqueous solubility that substantially reduces absorption of a drug. Problems with the delivery of drugs to the tumor occur also when the active agent has a high molecular weight which limits tissue penetration. Various strategies and structural modifying approaches have been investigated to solve this problem.
Prodrugs have been successfully used to increase aqueous solubility. For example, phosphate prodrugs are typically converted to the parent molecule via non-specific alkaline or acidic phosphatases. This approach has allowed intravenous administration of phosphate prodrugs to overcome otherwise bleak pharmacokinetic profiles. In particular, CA-4 has recently benefited from a phosphate prodrug administration (3P). Similarly, Cushman’s group also addressed the issue of low water solubility by producing water soluble 2-ME derivatives by coupling phosphate to the hydroxyl groups in positions 3 and 17 (150). The 3-phosphate (184, Fig. 14) exhibited similar properties as compared with the parental drug 2-ME. As expected, in rats the 3- phosphate was metabolized rapidly (within one hour) to the active drug 2-ME. However, the oral administration of the 3-phosphate did not enhance 2-ME plasma levels in rats relative to 2-ME feeding. But the enhanced water solubility facilitates intravenous application. Similarly, hydrophilic monosodium phosphate prodrug (185) of CHM-1 (103, Fig. 7) was prepared, and possesses improved pharmacological effects over CHM-1, and readily converts to its parent molecule during both i.v. and p.o. administration, and shows antitumor activity in the SKOV-3 xenograft mouse model.
Fig. 14.

Chemical structures of prodrugs of CBSIs.
Another strategy to increase aqueous solubility of CA-4 is replacing the C-3 hydroxyl group in ring B with an amine substituent as an HCl salt (AC-7739, 186, Fig. 14). The L-serine amide HCl salt (AC-7700, 187) has also been prepared. Both compounds showed enhanced aqueous solubility over CA-4 and were efficacious in vivo (49). On the basis of these results, a variety of amide prodrugs of irreversible tubulin inhibitor T138067 (15) were prepared. T138067 has the ability to penetrate the blood brain barrier (BBB). While compounds that have the ability to penetrate the BBB are promising candidates for treating brain tumors, it brought the main dose-limiting side effect of central nervous system (CNS) toxicity. Amides 188-191 showed no detectable or extremely small amounts of crossing the BBB. The in vivo efficacy of amide 191 approached that of T138067 and was better tolerated when administered to athymic nude mice bearing MX-1 human mammary tumor xenografts (151).
A PEG-based polymeric nanomedicine prodrug (192, Fig. 14) of colchicine was synthesized to increase its aqueous solubility, reduce systemic toxicity and to enhance its therapeutic window in a recent study (152). Cell viability studies with human umbilical vein endothelial cells demonstrated the degradation kinetics of the prodrug accordingly. It was observed that distinct vascular disruption and consequent tumor necrosisin the prodrug treatment but not for free colchicine at an equivalent dose by i.v. treatment in the B16F10 melanoma-bearing mouse model. Furthermore, a five-times-higher dose of the prodrug is well tolerated with reduced toxicity. The polymeric conjugated prodrug is another potential strategy to improve solubility/efficacy/toxicity compared with the parent drug, and it appears to be a promising approach for the application of CBSIs in cancer therapy.
Liposome based drug delivery possesses advantages including their prolonged circulation kinetics, passive targeting properties, and the ability to encapsulate both hydrophobic and hydrophilic drugs. However, colchicinoids are difficult to retain in liposomes due to their physicochemical properties. Two hydrolysable PEGylated derivatives (153) of colchicine were developed for encapsulation into the aqueous core of long-circulating liposomes with glycolic acid linker (193), and a lactic acid linker (194, Fig. 14). Hydrolysis studies at 37°C and pH 7.4 showed that 193 possessed a relatively rapid half-life (5.4 h) whereas 194 hydrolyzed much slower (T1/2 =217 h). The parent drug, colchicine, was released rapidly after encapsulation into liposomes, whereas both PEGylated colchicine prodrugs were efficiently retained and released only after cleavage of the PEG-linker. Unlike colchicine, these PEGylated colchicine-derived prodrugs are retained within the aqueous interior after encapsulation into liposomes, and the release of colchicine can be controlled by using different biodegradable linkers.
Chemical Structure Modifications
Our groups (154) discovered a series of 4-substituted methoxybenzoyl-aryl-thiazoles (SMART, 195, Fig. 15) as a result of structural modifications of the lead compounds 2-arylthiazolidine-4-carboxylic acid amides (ATCAA). The antiproliferative activity of the SMART agents against melanoma and prostate cancer cells has been improved from micromolar to low nanomolar range as compared to the early ATCAA series. Preliminary mechanism of action studies indicated that the SMART compounds exert their anticancer activity through inhibition of tubulin polymerization via the colchicine binding site, overcome the Pgp mediated MDR and show lower neurotoxicity as compared to vinblastine (155). In order to improve the poor aqueous solubility of SMART, it was formulated in polyethylene-b-poly(D,L-lactide) (PEG-PLA) micelles. The solubility of SMART was increased by more than 1.1×105 fold (156). In a continuing effort at chemical structure modifications to improve the poor aqueous solubility and bioavailability, an imidazole B ring analog (ABI, 196) and a phenyl-aminothiazole (PAT, 197) template have been designed in which an amino linkage was inserted between the “A” and “B” rings of compound 195. The most potent ABI analogs possess low nanomolar activity and have substantially improved aqueous solubility by 50-fold (157). The PAT analogs maintained nanomolar range potency against cancer cell lines via inhibiting tubulin polymerization and markedly improved solubility and bioavailability (R=Fluoro, F=21%) compared with the SMART template (F=3.3%).
Fig. 15.

Chemical structures of CBSIs with improved solubility.
Lee’s group (158,159) developed a series of acetylated (198) and methylpyrazoline (199, Fig. 15) analogs of CA-4 with the intention to improve solubility of the CA-4 analogs. Compound 199 has aqueous solubility of 372 μM, which is slightly higher than that for CA-4 (350 μM), but they also lost the potency.
Gangjee et al. (160) reported water soluble colchicine binding site inhibitor and microtubule depolymerizing agents (200-201, Fig. 15) that inhibited the growth of cancer cells with GI50 values in the nanomolar range. This molecule contains a chiral center and both the R and S enantiomers cause a G2/M cell cycle arrest and circumvent tumor resistance due to overexpression of Pgp and βIII tubulin. The S isomer (200) is a single digit nanomolar inhibitor in 51 cancer cell lines and is 10- to 88-fold more potent against most of the cell lines than either the racemate or the R isomer (201).
CONCLUSION
Tubulin targeting agents have played a key role in cancer treatment since the approval of vincristine by FDA in 1963, and research in this field maintains active. Among the different types of anti-tubulin agents, only CBSIs have not yet reached the commercial phase. Literature searching shows hundreds of potential CBSIs have been synthesized and tested with the hope to find a better clinical drug for cancer therapy. A large number of structurally diverse CBSI molecules display their anticancer activity through their abilities to arrest cell cycle and to kill cancer cells via both mitotic and apoptotic pathways, thus hold great promise for new family of anticancer drugs. Over the last decade, the potential of CBSIs to act as selective VDAs and overcome efflux pump/mutant tubulin/βIII tubulin overexpression mediated multidrug resistance has been recognized. A number of agents have been examined in clinical studies, and their capacity to inhibit growth of the tumor vasculature and resistant tumor has been confirmed. The current drawbacks in developing this series of agents as anticancer drugs exist in several areas including narrow therapeutic windows and lack of oral bioavailability. The side effects such as neural toxicity, cardiovascular and thromboembolic events remain major concerns and their therapeutic efficacy as single agents has been disappointing to this point. However, combination strategies of established anticancer drugs with CBSIs suggest that targeting the vascular tissue may be a profitablet direction for the further development of CBSIs as anti-cancer drugs. Furthermore, many CBSIs have limited clinical application due to their poor aqueous solubility, inadequate metabolic stability, and unsatisfactory pharmacokinetic profiles. Future new CBSIs will concentrate on the design of metabolically stable analogs and pharmacokinetic optimization will focus on how to improve their oral bioavailability as well as to increase their potency. With the new insights into the design of new agents, there is hope that CBSIs will move from basic research to clinical practice.
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA148706.
ABBREVIATIONS
- 2-ME
2-methoxyestradiol
- ABC
ATP binding cassette
- BS
binding site
- CA-4
combretastatin A-4
- CBSI
colchicine binding site inhibitors
- cGMP
cyclic guanosine monophosphate
- CNS
central nervous system
- CoMFA
comparative molecular field analyses
- CoMSIA
comparative molecular similarity indices analyses
- DAMA-colchicine
N-deacetyl-N-(2-mercaptoacetyl) colchicine
- FDA
Food and Drug Administration
- FGF
fibroblast growth factor
- HIF
hypoxia-inducible factor
- HUVEC
human umbilical vein endothelial cell
- LIE
linear interaction energy
- MDR
multidrug resistance
- MRP
multidrug resistance-associated protein
- MTA
microtubule targeting agent
- NCI
National Cancer Institute
- PDE
phosphodiesterase
- Pgp
P-glycoprotein
- PK
pharmacokinetic
- QSAR
quantitative structure-activity relationships
- SAR
structure-activity relationship
- SGB
surface-generalized Born
- TMP
trimethoxyphenyl
- TNF-α
tumor necrosis factor-α
- VDA
vascular-disrupting agent
- VEGFR
vascular endothelial growth factor receptor
Footnotes
DISCLOSURES
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional supports were provided by the Van Vleet Endowed Professorship.
References
- 1.Pryor DE, O’Brate A, Bilcer G, Diaz JF, Wang Y, Kabaki M, et al. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry. 2002;41:9109–15. doi: 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]
- 2.Gigant B, Wang C, Ravelli RB, Roussi F, Steinmetz MO, Curmi PA, et al. Structural basis for the regulation of tubulin by vinblastine. Nature. 2005;435:519–22. doi: 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- 3.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, et al. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 4.Seveand P, Dumontet C. Is class III beta-tubulin a predictive factor in patients receiving tubulin-binding agents? The Lancet Oncology. 2008;9:168–75. doi: 10.1016/S1470-2045(08)70029-9. [DOI] [PubMed] [Google Scholar]
- 5.Stengel C, Newman SP, Leese MP, Potter BV, Reed MJ, Purohit A. Class III beta-tubulin expression and in vitro resistance to microtubule targeting agents. Br J Cancer. 2010;102:316–24. doi: 10.1038/sj.bjc.6605489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goto H, Yano S, Zhang H, Matsumori Y, Ogawa H, Blakey DC, et al. Activity of a new vascular targeting agent, ZD6126, in pulmonary metastases by human lung adenocarcinoma in nude mice. Cancer Res. 2002;62:3711–5. [PubMed] [Google Scholar]
- 7.Lippert JW., 3rd Vascular disrupting agents. Bioorg Med Chem. 2007;15:605–15. doi: 10.1016/j.bmc.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 8.Rustin GJ, Shreeves G, Nathan PD, Gaya A, Ganesan TS, Wang D, et al. A Phase Ib trial of CA4P (combretastatin A-4 phosphate), carboplatin, and paclitaxel in patients with advanced cancer. Br J Cancer. 2010;102:1355–60. doi: 10.1038/sj.bjc.6605650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim TJ, Ravoori M, Landen CN, Kamat AA, Han LY, Lu C, et al. Antitumor and antivascular effects of AVE8062 in ovarian carcinoma. Cancer Res. 2007;67:9337–45. doi: 10.1158/0008-5472.CAN-06-4018. [DOI] [PubMed] [Google Scholar]
- 10.http://clinicaltrials.gov/ct2/results?term=AVE8062.
- 11.Pettit GR, Toki B, Herald DL, Verdier-Pinard P, Boyd MR, Hamel E, et al. Antineoplastic agents. 379. Synthesis of phenstatin phosphate. J Med Chem. 1998;41:1688–95. doi: 10.1021/jm970644q. [DOI] [PubMed] [Google Scholar]
- 12.Zhang LH, Wu L, Raymon HK, Chen RS, Corral L, Shirley MA, et al. The synthetic compound CC-5079 is a potent inhibitor of tubulin polymerization and tumor necrosis factor-alpha production with antitumor activity. Cancer Res. 2006;66:951–9. doi: 10.1158/0008-5472.CAN-05-2083. [DOI] [PubMed] [Google Scholar]
- 13.Bohlinand L, Rosen B. Podophyllotoxin derivatives: drug discovery and development. Drug Discov Today. 1996;1:343–51. [Google Scholar]
- 14.Kupchan SM, Britton RW, Ziegler MF, Gilmore CJ, Restivo RJ, Bryan RF. Steganacin and steganangin, novel antileukemic lignan lactones from Steganotaenia araliacea. J Am Chem Soc. 1973;95:1335–6. doi: 10.1021/ja00785a054. [DOI] [PubMed] [Google Scholar]
- 15.Attia SM. Molecular cytogenetic evaluation of the mechanism of genotoxic potential of amsacrine and nocodazole in mouse bone marrow cells. Journal of Applied Toxicology: JAT. 2011 doi: 10.1002/jat.1753. [DOI] [PubMed] [Google Scholar]
- 16.Hamel E. Antimitotic natural products and their interactions with tubulin. Medicinal Research Reviews. 1996;16:207–31. doi: 10.1002/(SICI)1098-1128(199603)16:2<207::AID-MED4>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Matei D, Schilder J, Sutton G, Perkins S, Breen T, Quon C, et al. Activity of 2 methoxyestradiol (Panzem NCD) in advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: a Hoosier Oncology Group trial. Gynecol Oncol. 2009;115:90–6. doi: 10.1016/j.ygyno.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 18.LaVallee TM, Burke PA, Swartz GM, Hamel E, Agoston GE, Shah J, et al. Significant antitumor activity in vivo following treatment with the microtubule agent ENMD-1198. Mol Cancer Ther. 2008;7:1472–82. doi: 10.1158/1535-7163.MCT-08-0107. [DOI] [PubMed] [Google Scholar]
- 19.Pasquier E, Sinnappan S, Munoz MA, Kavallaris M. ENMD-1198, a new analogue of 2-methoxyestradiol, displays both anti-angiogenic and vascular-disrupting properties. Mol Cancer Ther. 2010;9:1408–18. doi: 10.1158/1535-7163.MCT-09-0894. [DOI] [PubMed] [Google Scholar]
- 20.Hande KR, Hagey A, Berlin J, Cai Y, Meek K, Kobayashi H, et al. The pharmacokinetics and safety of ABT-751, a novel, orally bioavailable sulfonamide antimitotic agent: results of a phase 1 study. Clin Cancer Res. 2006;12:2834–40. doi: 10.1158/1078-0432.CCR-05-2159. [DOI] [PubMed] [Google Scholar]
- 21.Shan B, Medina JC, Santha E, Frankmoelle WP, Chou TC, Learned RM, et al. Selective, covalent modification of beta-tubulin residue Cys-239 by T138067, an antitumor agent with in vivo efficacy against multidrug-resistant tumors. Proc Natl Acad Sci U S A. 1999;96:5686–91. doi: 10.1073/pnas.96.10.5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patterson DM, Rustin GJS, Serradell N, Rosa E, Bolos J. Combretastatin A-4 phosphate. Drugs of the Future. 2007;32:1025–32. [Google Scholar]
- 23.Rischin D, Bibby DC, Chong G, Kremmidiotis G, Leske AF, Matthews CA, et al. Clinical, pharmacodynamic, and pharmacokinetic evaluation of BNC105P: a phase I trial of a novel vascular disrupting agent and inhibitor of cancer cell proliferation. Clin Cancer Res. 2011;17:5152–60. doi: 10.1158/1078-0432.CCR-11-0937. [DOI] [PubMed] [Google Scholar]
- 24.Bacher G, Nickel B, Emig P, Vanhoefer U, Seeber S, Shandra A, et al. D-24851, a novel synthetic microtubule inhibitor, exerts curative antitumoral activity in vivo, shows efficacy toward multidrug-resistant tumor cells, and lacks neurotoxicity. Cancer Res. 2001;61:392–9. [PubMed] [Google Scholar]
- 25.Gourdeau H, Leblond L, Hamelin B, Desputeau C, Dong K, Kianicka I, et al. Antivascular and antitumor evaluation of 2-amino-4-(3-bromo-4,5-dimethoxy-phenyl)-3-cyano-4H-chromenes, a novel series of anticancer agents. Mol Cancer Ther. 2004;3:1375–84. [PubMed] [Google Scholar]
- 26.Sirisoma N, Kasibhatla S, Pervin A, Zhang H, Jiang S, Willardsen JA, et al. Discovery of 2-chloro-N-(4-methoxy-phenyl)-N-methylquinazolin-4-amine (EP128265, MPI-0441138) as a potent inducer of apoptosis with high in vivo activity. J Med Chem. 2008;51:4771–9. doi: 10.1021/jm8003653. [DOI] [PubMed] [Google Scholar]
- 27.Tsimberidou AM, Akerley W, Schabel MC, Hong DS, Uehara C, Chhabra A, et al. Phase I clinical trial of MPC-6827 (Azixa), a microtubule destabilizing agent, in patients with advanced cancer. Mol Cancer Ther. 2010;9:3410–9. doi: 10.1158/1535-7163.MCT-10-0516. [DOI] [PubMed] [Google Scholar]
- 28.Burns CJ, Fantino E, Phillips ID, Su S, Harte MF, Bukczynska PE, et al. CYT997: a novel orally active tubulin polymerization inhibitor with potent cytotoxic and vascular disrupting activity in vitro and in vivo. Mol Cancer Ther. 2009;8:3036–45. doi: 10.1158/1535-7163.MCT-09-0076. [DOI] [PubMed] [Google Scholar]
- 29.Shiand W, Siemann DW. Preclinical studies of the novel vascular disrupting agent MN-029. Anticancer Res. 2005;25:3899–904. [PubMed] [Google Scholar]
- 30.Ricart AD, Ashton EA, Cooney MM, Sarantopoulos J, Brell JM, Feldman MA, et al. A phase I study of MN-029 (denibulin), a novel vascular-disrupting agent, in patients with advanced solid tumors. Cancer Chemotherapy and Pharmacology. 2011;68:959–70. doi: 10.1007/s00280-011-1565-4. [DOI] [PubMed] [Google Scholar]
- 31.de Ines C, Leynadier D, Barasoain I, Peyrot V, Garcia P, Briand C, et al. Inhibition of microtubules and cell cycle arrest by a new 1-deaza-7,8-dihydropteridine antitumor drug, CI 980, and by its chiral isomer, NSC 613863. Cancer Res. 1994;54:75–84. [PubMed] [Google Scholar]
- 32.Thomas JP, Moore T, Kraut EH, Balcerzak SP, Galloway S, Vandre DD. A phase II study of CI-980 in previously untreated extensive small cell lung cancer: an Ohio State University phase II research consortium study. Cancer Investigation. 2002;20:192–8. doi: 10.1081/cnv-120001146. [DOI] [PubMed] [Google Scholar]
- 33.Yoon JT, Palazzo AF, Xiao D, Delohery TM, Warburton PE, Bruce JN, et al. CP248, a derivative of exisulind, causes growth inhibition, mitotic arrest, and abnormalities in microtubule polymerization in glioma cells. Mol Cancer Ther. 2002;1:393–404. [PubMed] [Google Scholar]
- 34.Sun W, Stevenson JP, Gallo JM, Redlinger M, Haller D, Algazy K, et al. Phase I and pharmacokinetic trial of the proapoptotic sulindac analog CP-461 in patients with advanced cancer. Clin Cancer Res. 2002;8:3100–4. [PubMed] [Google Scholar]
- 35.Tripodi F, Pagliarin R, Fumagalli G, Bigi A, Fusi P, Orsini F, et al. Synthesis and biological evaluation of 1,4-Diaryl-2-azetidinones as specific anticancer agents: activation of adenosine monophosphate activated protein kinase and induction of apoptosis. J Med Chem. 2012;55:2112–24. doi: 10.1021/jm201344a. [DOI] [PubMed] [Google Scholar]
- 36.Bai R, Covell DG, Pei XF, Ewell JB, Nguyen NY, Brossi A, et al. Mapping the binding site of colchicinoids on beta -tubulin. 2-Chloroacetyl-2-demethylthiocolchicine covalently reacts predominantly with cysteine 239 and secondarily with cysteine 354. J Biol Chem. 2000;275:40443–52. doi: 10.1074/jbc.M005299200. [DOI] [PubMed] [Google Scholar]
- 37.Dorleans A, Gigant B, Ravelli RB, Mailliet P, Mikol V, Knossow M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc Natl Acad Sci U S A. 2009;106:13775–9. doi: 10.1073/pnas.0904223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbier P, Dorleans A, Devred F, Sanz L, Allegro D, Alfonso C, et al. Stathmin and interfacial microtubule inhibitors recognize a naturally curved conformation of tubulin dimers. J Biol Chem. 2010;285:31672–81. doi: 10.1074/jbc.M110.141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen TL, McGrath C, Hermone AR, Burnett JC, Zaharevitz DW, Day BW, et al. A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach. J Med Chem. 2005;48:6107–16. doi: 10.1021/jm050502t. [DOI] [PubMed] [Google Scholar]
- 40.Dutcher SK. The tubulin fraternity: alpha to eta. Curr Opin Cell Biol. 2001;13:49–54. doi: 10.1016/s0955-0674(00)00173-3. [DOI] [PubMed] [Google Scholar]
- 41.Simoni D, Romagnoli R, Baruchello R, Rondanin R, Rizzi M, Pavani MG, et al. Novel combretastatin analogues endowed with antitumor activity. J Med Chem. 2006;49:3143–52. doi: 10.1021/jm0510732. [DOI] [PubMed] [Google Scholar]
- 42.Stevenson JP, Rosen M, Sun W, Gallagher M, Haller DG, Vaughn D, et al. Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: magnetic resonance imaging evidence for altered tumor blood flow. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2003;21:4428–38. doi: 10.1200/JCO.2003.12.986. [DOI] [PubMed] [Google Scholar]
- 43.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinal chemistry of combretastatin A4: present and future directions. J Med Chem. 2006;49:3033–44. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 44.Aprile S, Del Grosso E, Tron GC, Grosa G. In vitro metabolism study of combretastatin A-4 in rat and human liver microsomes. Drug Metabolism and Disposition: the Biological Fate of Chemicals. 2007;35:2252–61. doi: 10.1124/dmd.107.016998. [DOI] [PubMed] [Google Scholar]
- 45.Gwaltney SL, 2nd, Imade HM, Barr KJ, Li Q, Gehrke L, Credo RB, et al. Novel sulfonate analogues of combretastatin A-4: potent antimitotic agents. Bioorganic & Medicinal Chemistry Letters. 2001;11:871–4. doi: 10.1016/s0960-894x(01)00098-1. [DOI] [PubMed] [Google Scholar]
- 46.Fortin S, Wei L, Moreau E, Lacroix J, Cote MF, Petitclerc E, et al. synthesis, biological evaluation, and structure-activity relationships of substituted phenyl 4-(2-oxoimidazolidin-1-yl)benzenesulfonates as new tubulin inhibitors mimicking combretastatin A-4. J Med Chem. 2011;54:4559–80. doi: 10.1021/jm200488a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fortin S, Wei L, Moreau E, Lacroix J, Cote MF, Petitclerc E, et al. Substituted phenyl 4-(2-oxoimidazolidin-1-yl)benzenesulfonamides as antimitotics. Antiproliferative, antiangiogenic and antitumoral activity, and quantitative structure-activity relationships. European Journal of Medicinal Chemistry. 2011;46:5327–42. doi: 10.1016/j.ejmech.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 48.Simoni D, Romagnoli R, Baruchello R, Rondanin R, Grisolia G, Eleopra M, et al. Novel A-ring and B-ring modified combretastatin A-4 (CA-4) analogues endowed with interesting cytotoxic activity. J Med Chem. 2008;51:6211–5. doi: 10.1021/jm8005004. [DOI] [PubMed] [Google Scholar]
- 49.Ohsumi K, Hatanaka T, Fujita K, Nakagawa R, Fukuda Y, Nihei Y, et al. Syntheses and antitumor activity of cis-restricted combretastatins: 5-membered heterocyclic analogues. Bioorganic & Medicinal Chemistry Letters. 1998;8:3153–8. doi: 10.1016/s0960-894x(98)00579-4. [DOI] [PubMed] [Google Scholar]
- 50.Wang L, Woods KW, Li Q, Barr KJ, McCroskey RW, Hannick SM, et al. Potent, orally active heterocycle-based combretastatin A-4 analogues: synthesis, structure-activity relationship, pharmacokinetics, and in vivo antitumor activity evaluation. J Med Chem. 2002;45:1697–711. doi: 10.1021/jm010523x. [DOI] [PubMed] [Google Scholar]
- 51.Kim Y, Nam NH, You YJ, Ahn BZ. Synthesis and cytotoxicity of 3,4-diaryl-2(5H)-furanones. Bioorganic & Medicinal Chemistry Letters. 2002;12:719–22. doi: 10.1016/s0960-894x(01)00831-9. [DOI] [PubMed] [Google Scholar]
- 52.Nam NH, Kim Y, You YJ, Hong DH, Kim HM, Ahn BZ. Synthesis and anti-tumor activity of novel combretastatins: combretocyclopentenones and related analogues. Bioorganic & Medicinal Chemistry Letters. 2002;12:1955–8. doi: 10.1016/s0960-894x(02)00321-9. [DOI] [PubMed] [Google Scholar]
- 53.Nam NH, Kim Y, You YJ, Hong DH, Kim HM, Ahn BZ. Combretoxazolones: synthesis, cytotoxicity and antitumor activity. Bioorganic & Medicinal Chemistry Letters. 2001;11:3073–6. doi: 10.1016/s0960-894x(01)00622-9. [DOI] [PubMed] [Google Scholar]
- 54.Bailly C, Bal C, Barbier P, Combes S, Finet JP, Hildebrand MP, et al. Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. J Med Chem. 2003;46:5437–44. doi: 10.1021/jm030903d. [DOI] [PubMed] [Google Scholar]
- 55.Combes S, Barbier P, Douillard S, McLeer-Florin A, Bourgarel-Rey V, Pierson JT, et al. Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. Part 2. J Med Chem. 2011;54:3153–62. doi: 10.1021/jm901826e. [DOI] [PubMed] [Google Scholar]
- 56.Tron GC, Pagliai F, Del Grosso E, Genazzani AA, Sorba G. Synthesis and cytotoxic evaluation of combretafurazans. J Med Chem. 2005;48:3260–8. doi: 10.1021/jm049096o. [DOI] [PubMed] [Google Scholar]
- 57.Pati HN, Wicks M, Holt HL, Leblanc R, Weisbruch P, Forrest L, et al. Synthesis and biological evaluation of cis-combretastatin analogs and their novel 1,2,3-triazole derivatives. Heterocyclic Commun. 2005;11:117–20. [Google Scholar]
- 58.Simoni D, Grisolia G, Giannini G, Roberti M, Rondanin R, Piccagli L, et al. Heterocyclic and phenyl double-bond-locked combretastatin analogues possessing potent apoptosis-inducing activity in HL60 and in MDR cell lines. J Med Chem. 2005;48:723–36. doi: 10.1021/jm049622b. [DOI] [PubMed] [Google Scholar]
- 59.Flynn BL, Flynn GP, Hamel E, Jung MK. The synthesis and tubulin binding activity of thiophene-based analogues of combretastatin A-4. Bioorganic & Medicinal Chemistry Letters. 2001;11:2341–3. doi: 10.1016/s0960-894x(01)00436-x. [DOI] [PubMed] [Google Scholar]
- 60.O’Boyle NM, Carr M, Greene LM, Bergin O, Nathwani SM, McCabe T, et al. Synthesis and evaluation of azetidinone analogues of combretastatin A-4 as tubulin targeting agents. J Med Chem. 2010;53:8569–84. doi: 10.1021/jm101115u. [DOI] [PubMed] [Google Scholar]
- 61.Romagnoli R, Baraldi PG, Brancale A, Ricci A, Hamel E, Bortolozzi R, et al. Convergent synthesis and biological evaluation of 2-amino-4-(3′,4′,5′-trimethoxyphenyl)-5-aryl thiazoles as microtubule targeting agents. J Med Chem. 2011;54:5144–53. doi: 10.1021/jm200392p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beale TM, Allwood DM, Bender A, Bond PJ, Brenton JD, Charnock-Jones DS, et al. Xian A-ring dihalogenation increases the cellular activity of combretastatin-templated tetrazoles. ACS Medicinal Chemistry Letters: Ahead of Print. 2012 doi: 10.1021/ml200149g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schobert R, Biersack B, Dietrich A, Effenberger K, Knauer S, Mueller T. 4-(3-Halo/amino-4,5-dimethoxyphenyl)-5-aryloxazoles and -N-methylimidazoles that are cytotoxic against combretastatin A resistant tumor cells and vascular disrupting in a cisplatin resistant germ cell tumor model. J Med Chem. 2010;53:6595–602. doi: 10.1021/jm100345r. [DOI] [PubMed] [Google Scholar]
- 64.Theeramunkong S, Caldarelli A, Massarotti A, Aprile S, Caprioglio D, Zaninetti R, et al. Regioselective Suzuki coupling of dihaloheteroaromatic compounds as a rapid strategy to synthesize potent rigid combretastatin analogues. J Med Chem. 2011;54:4977–86. doi: 10.1021/jm200555r. [DOI] [PubMed] [Google Scholar]
- 65.Furst R, Zupko I, Berenyi A, Ecker GF, Rinner U. Synthesis and antitumor-evaluation of cyclopropyl-containing combretastatin analogs. Bioorganic & Medicinal Chemistry Letters. 2009;19:6948–51. doi: 10.1016/j.bmcl.2009.10.064. [DOI] [PubMed] [Google Scholar]
- 66.Hadfield JA, Gaukroger K, Hirst N, Weston AP, Lawrence NJ, McGown AT. Synthesis and evaluation of double bond substituted combretastatins. European Journal of Medicinal Chemistry. 2005;40:529–41. doi: 10.1016/j.ejmech.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 67.Liu T, Cui R, Chen J, Zhang J, He Q, Yang B, et al. 4,5-Diaryl-3-aminopyrazole derivatives as analogs of Combretastatin A-4: synthesis and biological evaluation. Arch Pharm. 2011;344:279–86. doi: 10.1002/ardp.201000069. [DOI] [PubMed] [Google Scholar]
- 68.Akselsen OW, Odlo K, Cheng JJ, Maccari G, Botta M, Hansen TV. Synthesis, biological evaluation and molecular modeling of 1,2,3-triazole analogs of combretastatin A-1. Bioorg Med Chem. 2012;20:234–42. doi: 10.1016/j.bmc.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 69.Xue N, Yang X, Wu R, Chen J, He Q, Yang B, et al. Synthesis and biological evaluation of imidazol-2-one derivatives as potential antitumor agents. Bioorg Med Chem. 2008;16:2550–7. doi: 10.1016/j.bmc.2007.11.048. [DOI] [PubMed] [Google Scholar]
- 70.Ruprich J, Prout A, Dickson J, Younglove B, Nolan L, Baxi K, et al. Design, synthesis and biological testing of cyclohexenone derivatives of combretastatin-A4. Letters in Drug Design & Discovery. 2007;4:144–8. [Google Scholar]
- 71.Lee L, Davis R, Vanderham J, Hills P, Mackay H, Brown T, et al. 1,2,3,4-tetrahydro-2-thioxopyrimidine analogs of combretastatin-A4. European Journal of Medicinal Chemistry. 2008;43:2011–5. doi: 10.1016/j.ejmech.2007.11.030. [DOI] [PubMed] [Google Scholar]
- 72.Johnson M, Younglove B, Lee L, LeBlanc R, Holt H, Jr, Hills P, et al. Design, synthesis, and biological testing of pyrazoline derivatives of combretastatin-A4. Bioorganic & Medicinal Chemistry Letters. 2007;17:5897–901. doi: 10.1016/j.bmcl.2007.07.105. [DOI] [PubMed] [Google Scholar]
- 73.Mateo C, Perez-Melero C, Pelaez R, Medarde M. Stilbenophane analogues of deoxycombretastatin A-4. J Org Chem. 2005;70:6544–7. doi: 10.1021/jo0508393. [DOI] [PubMed] [Google Scholar]
- 74.De Martino G, Edler MC, La Regina G, Coluccia A, Barbera MC, Barrow D, et al. New arylthioindoles: potent inhibitors of tubulin polymerization. 2. Structure-activity relationships and molecular modeling studies. J Med Chem. 2006;49:947–54. doi: 10.1021/jm050809s. [DOI] [PubMed] [Google Scholar]
- 75.De Martino G, La Regina G, Coluccia A, Edler MC, Barbera MC, Brancale A, et al. Arylthioindoles, potent inhibitors of tubulin polymerization. J Med Chem. 2004;47:6120–3. doi: 10.1021/jm049360d. [DOI] [PubMed] [Google Scholar]
- 76.La Regina G, Sarkar T, Bai R, Edler MC, Saletti R, Coluccia A, et al. New arylthioindoles and related bioisosteres at the sulfur bridging group. 4. Synthesis, tubulin polymerization, cell growth inhibition, and molecular modeling studies. J Med Chem. 2009;52:7512–27. doi: 10.1021/jm900016t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.La Regina G, Bai R, Rensen W, Coluccia A, Piscitelli F, Gatti V, et al. Design and synthesis of 2-Heterocyclyl-3-arylthio-1H-indoles as potent tubulin polymerization and cell growth inhibitors with improved metabolic stability. J Med Chem. 2011;54:8394–406. doi: 10.1021/jm2012886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mahboobi S, Pongratz H, Hufsky H, Hockemeyer J, Frieser M, Lyssenko A, et al. Synthetic 2-aroylindole derivatives as a new class of potent tubulin-inhibitory, antimitotic agents. J Med Chem. 2001;44:4535–53. doi: 10.1021/jm010940+. [DOI] [PubMed] [Google Scholar]
- 79.Kuo CC, Hsieh HP, Pan WY, Chen CP, Liou JP, Lee SJ, et al. BPR0L075, a novel synthetic indole compound with antimitotic activity in human cancer cells, exerts effective antitumoral activity in vivo. Cancer Res. 2004;64:4621–8. doi: 10.1158/0008-5472.CAN-03-3474. [DOI] [PubMed] [Google Scholar]
- 80.Hu CB, Chen CP, Yeh TK, Song JS, Chang CY, Chuu JJ, et al. BPR0C261 is a novel orally active antitumor agent with antimitotic and anti-angiogenic activities. Cancer Science. 2011;102:182–91. doi: 10.1111/j.1349-7006.2010.01744.x. [DOI] [PubMed] [Google Scholar]
- 81.Li WT, Hwang DR, Chen CP, Shen CW, Huang CL, Chen TW, et al. Synthesis and biological evaluation of N-heterocyclic indolyl glyoxylamides as orally active anticancer agents. J Med Chem. 2003;46:1706–15. doi: 10.1021/jm020471r. [DOI] [PubMed] [Google Scholar]
- 82.Nien CY, Chen YC, Kuo CC, Hsieh HP, Chang CY, Wu JS, et al. 5-Amino-2-aroylquinolines as highly potent tubulin polymerization inhibitors. J Med Chem. 2010;53:2309–13. doi: 10.1021/jm900685y. [DOI] [PubMed] [Google Scholar]
- 83.Liou JP, Wu ZY, Kuo CC, Chang CY, Lu PY, Chen CM, et al. Discovery of 4-amino and 4-hydroxy-1-aroylindoles as potent tubulin polymerization inhibitors. J Med Chem. 2008;51:4351–5. doi: 10.1021/jm800150d. [DOI] [PubMed] [Google Scholar]
- 84.Lee HY, Chang JY, Nien CY, Kuo CC, Shih KH, Wu CH, et al. 5-Amino-2-aroylquinolines as highly potent tubulin polymerization inhibitors. Part 2. The impact of bridging groups at position C-2. J Med Chem. 2011;54:8517–25. doi: 10.1021/jm201031f. [DOI] [PubMed] [Google Scholar]
- 85.Arthuis M, Pontikis R, Chabot GG, Quentin L, Scherman D, Florent JC. Domino approach to 2-aroyltrimethoxyindoles as novel heterocyclic combretastatin A4 analogues. European Journal of Medicinal Chemistry. 2011;46:95–100. doi: 10.1016/j.ejmech.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 86.Lai MJ, Chang JY, Lee HY, Kuo CC, Lin MH, Hsieh HP, et al. Synthesis and biological evaluation of 1-(4′-Indolyl and 6′-Quinolinyl) indoles as a new class of potent anticancer agents. European Journal of Medicinal Chemistry. 2011;46:3623–9. doi: 10.1016/j.ejmech.2011.04.065. [DOI] [PubMed] [Google Scholar]
- 87.Lai MJ, Kuo CC, Yeh TK, Hsieh HP, Chen LT, Pan WY, et al. Synthesis and structure-activity relationships of 1-benzyl-4,5,6-trimethoxyindoles as a novel class of potent antimitotic agents. ChemMedChem. 2009;4:588–93. doi: 10.1002/cmdc.200800405. [DOI] [PubMed] [Google Scholar]
- 88.Xie F, Zhao H, Li D, Chen H, Quan H, Shi X, et al. Synthesis and biological evaluation of 2,4,5-substituted pyrimidines as a new class of tubulin polymerization inhibitors. J Med Chem. 2011;54:3200–5. doi: 10.1021/jm101388d. [DOI] [PubMed] [Google Scholar]
- 89.El-Nakkady SS, Hanna MM, Roaiah HM, Ghannam IA. Synthesis, molecular docking study and antitumor activity of novel 2-phenylindole derivatives. European Journal of Medicinal Chemistry. 2012;47:387–98. doi: 10.1016/j.ejmech.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 90.Arora S, Wang XI, Keenan SM, Andaya C, Zhang Q, Peng Y, et al. Novel microtubule polymerization inhibitor with potent anti-proliferative and antitumor activity. Cancer Res. 2009;69:1910–5. doi: 10.1158/0008-5472.CAN-08-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shetty RS, Lee Y, Liu B, Husain A, Joseph RW, Lu Y, et al. Synthesis and pharmacological evaluation of N-(3-(1H-indol-4-yl)-5-(2-methoxyisonicotinoyl)phenyl)methanesulfonamide (LP-261), a potent antimitotic agent. J Med Chem. 2011;54:179–200. doi: 10.1021/jm100659v. [DOI] [PubMed] [Google Scholar]
- 92.Li L, Wang HK, Kuo SC, Wu TS, Mauger A, Lin CM, et al. Antitumor agents. 155. Synthesis and biological evaluation of 3′,6,7-substituted 2-phenyl-4-quinolones as antimicrotubule agents. J Med Chem. 1994;37:3400–7. doi: 10.1021/jm00046a025. [DOI] [PubMed] [Google Scholar]
- 93.Li L, Wang HK, Kuo SC, Wu TS, Lednicer D, Lin CM, et al. Antitumor agents. 150. 2′,3′,4′,5′,5,6,7-substituted 2-phenyl-4-quinolones and related compounds: their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J Med Chem. 1994;37:1126–35. doi: 10.1021/jm00034a010. [DOI] [PubMed] [Google Scholar]
- 94.Zhang C, Yang N, Yang CH, Ding HS, Luo C, Zhang Y, et al. S9, a novel anticancer agent, exerts its anti-proliferative activity by interfering with both PI3K-Akt-mTOR signaling and microtubule cytoskeleton. PLoS One. 2009;4:e4881. doi: 10.1371/journal.pone.0004881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Romagnoli R, Baraldi PG, Carrion MD, Lopez Cara C, Preti D, Fruttarolo F, et al. Synthesis and biological evaluation of 2- and 3-aminobenzo[b]thiophene derivatives as antimitotic agents and inhibitors of tubulin polymerization. J Med Chem. 2007;50:2273–7. doi: 10.1021/jm070050f. [DOI] [PubMed] [Google Scholar]
- 96.Keller L, Beaumont S, Liu JM, Thoret S, Bignon JS, Wdzieczak-Bakala J, et al. New C5-alkylated indolobenzazepinones acting as inhibitors of tubulin polymerization: cytotoxic and antitumor activities. J Med Chem. 2008;51:3414–21. doi: 10.1021/jm701466p. [DOI] [PubMed] [Google Scholar]
- 97.Pons V, Beaumont S, Dau METH, Iorga BI, Dodd RH. Rigid analogues of antimitotic indolobenzazepinones: new insights into tubulin binding via molecular modeling. ACS Medicinal Chemistry Letters. 2011;2:565–570. doi: 10.1021/ml200024y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boumendjel A, McLeer-Florin A, Champelovier P, Allegro D, Muhammad D, Souard F, et al. A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivo glioblastoma models. BMC Cancer. 2009;9:242. doi: 10.1186/1471-2407-9-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kerr DJ, Hamel E, Jung MK, Flynn BL. The concise synthesis of chalcone, indanone and indenone analogues of combretastatin A4. Bioorg Med Chem. 2007;15:3290–8. doi: 10.1016/j.bmc.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 100.Luo Y, Qiu KM, Lu X, Liu K, Fu J, Zhu HL. Synthesis, biological evaluation, and molecular modeling of cinnamic acyl sulfonamide derivatives as novel antitubulin agents. Bioorg Med Chem. 2011;19:4730–8. doi: 10.1016/j.bmc.2011.06.088. [DOI] [PubMed] [Google Scholar]
- 101.Ruan BF, Lu X, Tang JF, Wei Y, Wang XL, Zhang YB, et al. Synthesis, biological evaluation, and molecular docking studies of resveratrol derivatives possessing chalcone moiety as potential antitubulin agents. Bioorg Med Chem. 2011;19:2688–95. doi: 10.1016/j.bmc.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 102.Risinger AL, Westbrook CD, Encinas A, Mulbaier M, Schultes CM, Wawro S, et al. ELR510444, a novel microtubule disruptor with multiple mechanisms of action. J Pharmacol Exp Ther. 2011;336:652–60. doi: 10.1124/jpet.110.175331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chang JY, Hsieh HP, Chang CY, Hsu KS, Chiang YF, Chen CM, et al. 7-Aroyl-aminoindoline-1-sulfonamides as a novel class of potent antitubulin agents. J Med Chem. 2006;49:6656–9. doi: 10.1021/jm061076u. [DOI] [PubMed] [Google Scholar]
- 104.Liou JP, Hsu KS, Kuo CC, Chang CY, Chang JY. A novel oral indoline-sulfonamide agent, N-[1-(4-methoxybenzenesulfonyl)-2,3-dihydro-1H-indol-7-yl]-isonicotinamide (J30), exhibits potent activity against human cancer cells in vitro and in vivo through the disruption of microtubule. J Pharmacol Exp Ther. 2007;323:398–405. doi: 10.1124/jpet.107.126680. [DOI] [PubMed] [Google Scholar]
- 105.Cushman M, He HM, Katzenellenbogen JA, Varma RK, Hamel E, Lin CM, et al. Synthesis of analogs of 2-methoxyestradiol with enhanced inhibitory effects on tubulin polymerization and cancer cell growth. J Med Chem. 1997;40:2323–34. doi: 10.1021/jm9700833. [DOI] [PubMed] [Google Scholar]
- 106.Cushman M, Mohanakrishnan AK, Hollingshead M, Hamel E. The effect of exchanging various substituents at the 2-position of 2-methoxyestradiol on cytotoxicity in human cancer cell cultures and inhibition of tubulin polymerization. J Med Chem. 2002;45:4748–54. doi: 10.1021/jm020218r. [DOI] [PubMed] [Google Scholar]
- 107.Edsall AB, Mohanakrishnan AK, Yang D, Fanwick PE, Hamel E, Hanson AD, et al. Effects of altering the electronics of 2-methoxyestradiol on cell proliferation, on cytotoxicity in human cancer cell cultures, and on tubulin polymerization. J Med Chem. 2004;47:5126–39. doi: 10.1021/jm049647a. [DOI] [PubMed] [Google Scholar]
- 108.Agoston GE, Shah JH, Lavallee TM, Zhan X, Pribluda VS, Treston AM. Synthesis and structure-activity relationships of 16-modified analogs of 2-methoxyestradiol. Bioorg Med Chem. 2007;15:7524–37. doi: 10.1016/j.bmc.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 109.Chander SK, Foster PA, Leese MP, Newman SP, Potter BV, Purohit A, et al. In vivo inhibition of angiogenesis by sulphamoylated derivatives of 2-methoxyoestradiol. Br J Cancer. 2007;96:1368–76. doi: 10.1038/sj.bjc.6603727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ireson CR, Chander SK, Purohit A, Perera S, Newman SP, Parish D, et al. Pharmacokinetics and efficacy of 2-methoxyoestradiol and 2-methoxyoestradiol-bis-sulphamate in vivo in rodents. Br J Cancer. 2004;90:932–7. doi: 10.1038/sj.bjc.6601591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jourdan F, Leese MP, Dohle W, Hamel E, Ferrandis E, Newman SP, et al. Synthesis, antitubulin, and antiproliferative SAR of analogues of 2-methoxyestradiol-3,17-O,O-bis-sulfamate. J Med Chem. 2010;53:2942–51. doi: 10.1021/jm9018806. [DOI] [PubMed] [Google Scholar]
- 112.Nakagawa-Goto K, Chen TH, Peng CY, Bastow KF, Wu JH, Lee KH. Antitumor agents 259. Design, syntheses, and structure-activity relationship study of desmosdumotin C analogs. J Med Chem. 2007;50:3354–8. doi: 10.1021/jm0702534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nakagawa-Goto K, Bastow KF, Chen TH, Morris-Natschke SL, Lee KH. Antitumor agents 260. New desmosdumotin B analogues with improved in vitro anticancer activity. J Med Chem. 2008;51:3297–303. doi: 10.1021/jm701208v. [DOI] [PubMed] [Google Scholar]
- 114.Nakagawa-Goto K, Wu PC, Lai CY, Hamel E, Zhu H, Zhang L, et al. Antitumor agents. 284. New desmosdumotin B analogues with bicyclic B-ring as cytotoxic and antitubulin agents. J Med Chem. 2011;54:1244–55. doi: 10.1021/jm1011947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nakagawa-Goto K, Wu PC, Bastow KF, Yang SC, Yu SL, Chen HY, et al. Antitumor agents 283. Further elaboration of desmosdumotin C analogs as potent antitumor agents: activation of spindle assembly checkpoint as possible mode of action. Bioorg Med Chem. 2011;19:1816–22. doi: 10.1016/j.bmc.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Beutler JA, Cardellina JH, II, Lin CM, Hamel E, Cragg GM, Boyd MR. Centaureidin, a cytotoxic flavone from Polymnia fruticosa, inhibits tubulin polymerization. Bioorganic & Medicinal Chemistry Letters. 1993;3:581–4. [Google Scholar]
- 117.Naik PK, Chatterji BP, Vangapandu SN, Aneja R, Chandra R, Kanteveri S, et al. Rational design, synthesis and biological evaluations of amino-noscapine: a high affinity tubulin-binding noscapinoid. J Comput Aided Mol Des. 2011;25:443–54. doi: 10.1007/s10822-011-9430-4. [DOI] [PubMed] [Google Scholar]
- 118.Hartley RM, Peng J, Fest GA, Dakshanamurthy S, Frantz DE, Brown ML, et al. Polygamain, a new microtubule depolymerizing agent that occupies a unique pharmacophore in the colchicine site. Molecular Pharmacology. 2011 doi: 10.1124/mol.111.075838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cui CB, Kakeya H, Okada G, Onose R, Ubukata M, Takahashi I, et al. Tryprostatins A and B, novel mammalian cell cycle inhibitors produced by Aspergillus fumigatus. J Antibiot. 1995;48:1382–4. doi: 10.7164/antibiotics.48.1382. [DOI] [PubMed] [Google Scholar]
- 120.Woehlecke H, Osada H, Herrmann A, Lage H. Reversal of breast cancer resistance protein-mediated drug resistance by try-prostatin A. International Journal of Cancer Journal International Du Cancer. 2003;107:721–8. doi: 10.1002/ijc.11444. [DOI] [PubMed] [Google Scholar]
- 121.Kanoh K, Kohno S, Katada J, Takahashi J, Uno I. (-)-Phenyl-ahistin arrests cells in mitosis by inhibiting tubulin polymerization. J Antibiot. 1999;52:134–41. doi: 10.7164/antibiotics.52.134. [DOI] [PubMed] [Google Scholar]
- 122.Kanzaki H, Yanagisawa S, Kanoh K, Nitoda T. A novel potent cell cycle inhibitor dehydrophenylahistin–enzymatic synthesis and inhibitory activity toward sea urchin embryo. J Antibiot. 2002;55:1042–7. doi: 10.7164/antibiotics.55.1042. [DOI] [PubMed] [Google Scholar]
- 123.Yamazaki Y, Tanaka K, Nicholson B, Deyanat-Yazdi G, Potts B, Yoshida T, et al. Synthesis and Structure-Activity Relationship Study of Antimicrotubule Agents Phenylahistin Derivatives with a Didehydropiperazine-2,5-dione Structure. J Med Chem. 2012;55:1056–71. doi: 10.1021/jm2009088. [DOI] [PubMed] [Google Scholar]
- 124.Wipf P, Reeves JT, Balachandran R, Day BW. Synthesis and biological evaluation of structurally highly modified analogues of the antimitotic natural product curacin A. J Med Chem. 2002;45:1901–17. doi: 10.1021/jm0105171. [DOI] [PubMed] [Google Scholar]
- 125.Wipf P, Reeves JT, Balachandran R, Giuliano KA, Hamel E, Day BW. Synthesis and Biological Evaluation of a Focused Mixture Library of Analogues of the Antimitotic Marine Natural Product Curacin A. J Am Chem Soc. 2000;122:9391–5. [Google Scholar]
- 126.Ziegelbauer J, Shan B, Yager D, Larabell C, Hoffmann B, Tjian R. Transcription factor MIZ-1 is regulated via microtubule association. Molecular Cell. 2001;8:339–49. doi: 10.1016/s1097-2765(01)00313-6. [DOI] [PubMed] [Google Scholar]
- 127.Bai R, Pei XF, Boye O, Getahun Z, Grover S, Bekisz J, et al. Identification of cysteine 354 of beta-tubulin as part of the binding site for the A ring of colchicine. J Biol Chem. 1996;271:12639–45. doi: 10.1074/jbc.271.21.12639. [DOI] [PubMed] [Google Scholar]
- 128.Combeau C, Provost J, Lancelin F, Tournoux Y, Prod’homme F, Herman F, et al. RPR112378 and RPR115781: two representatives of a new family of microtubule assembly inhibitors. Mol Pharmacol. 2000;57:553–63. doi: 10.1124/mol.57.3.553. [DOI] [PubMed] [Google Scholar]
- 129.Bouchon B, Chambon C, Mounetou E, Papon J, Miot-Noirault E, Gaudreault RC, et al. Alkylation of beta-tubulin on Glu 198 by a microtubule disrupter. Mol Pharmacol. 2005;68:1415–22. doi: 10.1124/mol.105.015586. [DOI] [PubMed] [Google Scholar]
- 130.Fortin S, Bouchon B, Chambon C, Lacroix J, Moreau E, Chezal JM, et al. Characterization of the covalent binding of N-phenyl-N′-(2-chloroethyl)ureas to {beta}-tubulin: importance of Glu198 in microtubule stability. J Pharmacol Exp Ther. 2011;336:460–7. doi: 10.1124/jpet.110.171082. [DOI] [PubMed] [Google Scholar]
- 131.Prinz H, Schmidt P, Bohm KJ, Baasner S, Muller K, Gerlach M, et al. Phenylimino-10H-anthracen-9-ones as novel antimicrotubule agents-synthesis, antiproliferative activity and inhibition of tubulin polymerization. Bioorg Med Chem. 2011;19:4183–91. doi: 10.1016/j.bmc.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 132.Kasibhatla S, Gourdeau H, Meerovitch K, Drewe J, Reddy S, Qiu L, et al. Discovery and mechanism of action of a novel series of apoptosis inducers with potential vascular targeting activity. Mol Cancer Ther. 2004;3:1365–74. [PubMed] [Google Scholar]
- 133.Kemnitzer W, Drewe J, Jiang S, Zhang H, Crogan-Grundy C, Labreque D, et al. Discovery of 4-aryl-4H-chromenes as a new series of apoptosis inducers using a cell- and caspase-based high throughput screening assay. 4. Structure-activity relationships of N-alkyl substituted pyrrole fused at the 7,8-positions. J Med Chem. 2008;51:417–23. doi: 10.1021/jm7010657. [DOI] [PubMed] [Google Scholar]
- 134.Henderson MC, Shaw YJ, Wang H, Han H, Hurley LH, Flynn G, et al. UA62784, a novel inhibitor of centromere protein E kinesin-like protein. Mol Cancer Ther. 2009;8:36–44. doi: 10.1158/1535-7163.MCT-08-0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tcherniuk S, Deshayes S, Sarli V, Divita G, Abrieu A. UA62784 Is a cytotoxic inhibitor of microtubules, not CENP-E. Chem Biol. 2011;18:631–41. doi: 10.1016/j.chembiol.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 136.Dalyot-Herman N, Delgado-Lopez F, Gewirtz DA, Gupton JT, Schwartz EL. Interference with endothelial cell function by JG-03-14, an agent that binds to the colchicine site on microtubules. Biochem Pharmacol. 2009;78:1167–77. doi: 10.1016/j.bcp.2009.06.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mooberry SL, Weiderhold KN, Dakshanamurthy S, Hamel E, Banner EJ, Kharlamova A, et al. Identification and characterization of a new tubulin-binding tetrasubstituted brominated pyrrole. Mol Pharmacol. 2007;72:132–40. doi: 10.1124/mol.107.034876. [DOI] [PubMed] [Google Scholar]
- 138.Zhang Z, Meng T, Yang N, Wang W, Xiong B, Chen Y, et al. MT119, a new planar-structured compound, targets the colchicine site of tubulin arresting mitosis and inhibiting tumor cell proliferation. International Journal of Cancer Journal International Du Cancer. 2011;129:214–24. doi: 10.1002/ijc.25661. [DOI] [PubMed] [Google Scholar]
- 139.Lisowski V, Leonce S, Kraus-Berthier L, Sopkovade Oliveira Santos J, Pierre A, Atassi G, et al. Design, synthesis, and evaluation of novel thienopyrrolizinones as antitubulin agents. J Med Chem. 2004;47:1448–64. doi: 10.1021/jm030961z. [DOI] [PubMed] [Google Scholar]
- 140.Leoni LM, Hamel E, Genini D, Shih H, Carrera CJ, Cottam HB, et al. Indanocine, a microtubule-binding indanone and a selective inducer of apoptosis in multidrug-resistant cancer cells. J Natl Cancer Inst. 2000;92:217–24. doi: 10.1093/jnci/92.3.217. [DOI] [PubMed] [Google Scholar]
- 141.Liberatore AM, Coulomb H, Pons D, Dutruel O, Kasprzyk PG, Carlson M, et al. IRC-083927 is a new tubulin binder that inhibits growth of human tumor cells resistant to standard tubulin-binding agents. Mol Cancer Ther. 2008;7:2426–34. doi: 10.1158/1535-7163.MCT-08-0208. [DOI] [PubMed] [Google Scholar]
- 142.Wasylyk C, Zheng H, Castell C, Debussche L, Multon MC, Wasylyk B. Inhibition of the Ras-Net (Elk-3) pathway by a novel pyrazole that affects microtubules. Cancer Res. 2008;68:1275–83. doi: 10.1158/0008-5472.CAN-07-2674. [DOI] [PubMed] [Google Scholar]
- 143.Shin KD, Yoon YJ, Kang YR, Son KH, Kim HM, Kwon BM, et al. KRIBB3, a novel microtubule inhibitor, induces mitotic arrest and apoptosis in human cancer cells. Biochem Pharmacol. 2008;75:383–94. doi: 10.1016/j.bcp.2007.08.027. [DOI] [PubMed] [Google Scholar]
- 144.Szczepankiewicz BG, Liu G, Jae HS, Tasker AS, Gunawardana IW, von Geldern TW, et al. New antimitotic agents with activity in multi-drug-resistant cell lines and in vivo efficacy in murine tumor models. J Med Chem. 2001;44:4416–30. doi: 10.1021/jm010231w. [DOI] [PubMed] [Google Scholar]
- 145.Tahir SK, Han EK, Credo B, Jae HS, Pietenpol JA, Scatena CD, et al. A-204197, a new tubulin-binding agent with antimitotic activity in tumor cell lines resistant to known microtubule inhibitors. Cancer Res. 2001;61:5480–5. [PubMed] [Google Scholar]
- 146.Li Q, Woods KW, Claiborne A, Gwaltney SL, 2nd, Barr KJ, Liu G, et al. Synthesis and biological evaluation of 2-indolyloxazolines as a new class of tubulin polymerization inhibitors. Discovery of A-289099 as an orally active antitumor agent. Bioorganic & Medicinal Chemistry Letters. 2002;12:465–9. doi: 10.1016/s0960-894x(01)00759-4. [DOI] [PubMed] [Google Scholar]
- 147.Tahir SK, Nukkala MA, Zielinski Mozny NA, Credo RB, Warner RB, Li Q, et al. Biological activity of A-289099: an orally active tubulin-binding indolyloxazoline derivative. Mol Cancer Ther. 2003;2:227–33. [PubMed] [Google Scholar]
- 148.Andreani A, Burnelli S, Granaiola M, Leoni A, Locatelli A, Morigi R, et al. Antitumor activity of new substituted 3-(5-imidazo[2,1-b]thiazolylmethylene)-2-indolinones and 3-(5-imidazo[2,1-b]thiadiazolylmethylene)-2-indolinones: selectivity against colon tumor cells and effect on cell cycle-related events. J Med Chem. 2008;51:7508–13. doi: 10.1021/jm800827q. [DOI] [PubMed] [Google Scholar]
- 149.Andreani A, Granaiola M, Locatelli A, Morigi R, Rambaldi M, Varoli L, et al. Substituted 3-(5-Imidazo[2,1-b]thiazolyl-methylene)-2-indolinones and analogues: synthesis, cytotoxic activity, and study of the mechanism of action (1) J Med Chem. 2012 doi: 10.1021/jm2012694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Edsall AB, Agoston GE, Treston AM, Plum SM, McClanahan RH, Lu TS, et al. Synthesis and in vivo antitumor evaluation of 2-methoxyestradiol 3-phosphate, 17-phosphate, and 3,17-diphosphate. J Med Chem. 2007;50:6700–5. doi: 10.1021/jm070639e. [DOI] [PubMed] [Google Scholar]
- 151.Rubenstein SM, Baichwal V, Beckmann H, Clark DL, Frankmoelle W, Roche D, et al. Hydrophilic, prodrug analogues of T138067 are efficacious in controlling tumor growth in vivo and show a decreased ability to cross the blood brain barrier. J Med Chem. 2001;44:3599–605. doi: 10.1021/jm000478d. [DOI] [PubMed] [Google Scholar]
- 152.Crielaard BJ, van der Wal S, Lammers T, Le HT, Hennink WE, Schiffelers RM, et al. A polymeric colchicinoid prodrug with reduced toxicity and improved efficacy for vascular disruption in cancer therapy. International Journal of Nanomedicine. 2011;6:2697–703. doi: 10.2147/IJN.S24450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Crielaard BJ, van der Wal S, Le HT, Bode AT, Lammers T, Hennink WE, et al. Liposomes as carriers for colchicine-derived prodrugs: Vascular disrupting nanomedicines with tailorable drug release kinetics. European Journal of Pharmaceutical Sciences: Official Journal of the European Federation for Pharmaceutical Sciences. 2012;45:429–35. doi: 10.1016/j.ejps.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 154.Lu Y, Li CM, Wang Z, Ross CR, 2nd, Chen J, Dalton JT, et al. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as novel anticancer agents: synthesis, biological evaluation, and structure-activity relationships. J Med Chem. 2009;52:1701–11. doi: 10.1021/jm801449a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li CM, Wang Z, Lu Y, Ahn S, Narayanan R, Kearbey JD, et al. Biological activity of 4-substituted methoxybenzoyl-aryl-thiazole: an active microtubule inhibitor. Cancer Res. 2011;71:216–24. doi: 10.1158/0008-5472.CAN-10-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li F, Lu Y, Li W, Miller DD, Mahato RI. Synthesis, formulation and in vitro evaluation of a novel microtubule destabilizer, SMART-100. Journal of Controlled Release: Official Journal of the Controlled Release Society. 2010;143:151–8. doi: 10.1016/j.jconrel.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 157.Chen J, Wang Z, Li CM, Lu Y, Vaddady PK, Meibohm B, et al. Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicines binding site in tubulin as potential anticancer agents. J Med Chem. 2010;53:7414–27. doi: 10.1021/jm100884b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Babu B, Lee M, Lee L, Strobel R, Brockway O, Nickols A, et al. Acetyl analogs of combretastatin A-4: synthesis and biological studies. Bioorg Med Chem. 2011;19:2359–67. doi: 10.1016/j.bmc.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 159.Lee M, Brockway O, Dandavati A, Tzou S, Sjoholm R, Satam V, et al. A novel class of trans-methylpyrazoline analogs of combretastatins: synthesis and in-vitro biological testing. European Journal of Medicinal Chemistry. 2011;46:3099–104. doi: 10.1016/j.ejmech.2011.03.064. [DOI] [PubMed] [Google Scholar]
- 160.Gangjee A, Zhao Y, Hamel E, Westbrook C, Mooberry SL. Synthesis and biological activities of (R)- and (S)-N-(4-Methoxy-phenyl)-N,2,6-trimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimi din-4-aminium chloride as potent cytotoxic antitubulin agents. J Med Chem. 2011;54:6151–5. doi: 10.1021/jm2007722. [DOI] [PMC free article] [PubMed] [Google Scholar]