

Abstract
Background
Sexually deceptive orchids of the genus Ophrys mimic the mating signals of their pollinator females to attract males as pollinators. This mode of pollination is highly specific and leads to strong reproductive isolation between species. This study aims to identify candidate genes responsible for pollinator attraction and reproductive isolation between three closely related species, O. exaltata, O. sphegodes and O. garganica. Floral traits such as odour, colour and morphology are necessary for successful pollinator attraction. In particular, different odour hydrocarbon profiles have been linked to differences in specific pollinator attraction among these species. Therefore, the identification of genes involved in these traits is important for understanding the molecular basis of pollinator attraction by sexually deceptive orchids.
Results
We have created floral reference transcriptomes and proteomes for these three Ophrys species using a combination of next-generation sequencing (454 and Solexa), Sanger sequencing, and shotgun proteomics (tandem mass spectrometry). In total, 121 917 unique transcripts and 3531 proteins were identified. This represents the first orchid proteome and transcriptome from the orchid subfamily Orchidoideae. Proteome data revealed proteins corresponding to 2644 transcripts and 887 proteins not observed in the transcriptome. Candidate genes for hydrocarbon and anthocyanin biosynthesis were represented by 156 and 61 unique transcripts in 20 and 7 genes classes, respectively. Moreover, transcription factors putatively involved in the regulation of flower odour, colour and morphology were annotated, including Myb, MADS and TCP factors.
Conclusion
Our comprehensive data set generated by combining transcriptome and proteome technologies allowed identification of candidate genes for pollinator attraction and reproductive isolation among sexually deceptive orchids. This includes genes for hydrocarbon and anthocyanin biosynthesis and regulation, and the development of floral morphology. These data will serve as an invaluable resource for research in orchid floral biology, enabling studies into the molecular mechanisms of pollinator attraction and speciation.
Introduction
The orchids (Orchidaceae) are one of the most species-rich plant families, and their remarkable floral diversity and pollination biology have long fascinated evolutionary biologists [1], [2]. It has been estimated that about one third of orchids are pollinated by deception, i.e. without rewarding their pollinators [2], [3]. For example, Ophrys L., a Euro-Mediterranean genus of sexually deceptive orchids, is mostly pollinated by male insects, primarily solitary bees [4]. These orchids mimic the visual, tactile, and olfactory signals of the females of their pollinators, so that male insects are attracted and try to copulate with the flower labellum (a modified petal). During these so-called ‘pseudo-copulations’ the pollinia (pollen packets) become attached to the bees and are transferred during subsequent visits of the males to other flowers [5], [6], [7]. Numerous behavioural studies have shown that the Ophrys-pollinator relationship is highly specific: each Ophrys species is usually pollinated by only one (or very few) insect species [4], [5], [6], [8], [9], [10]. It has also been shown that floral odour is the key factor in attracting specific pollinators and eliciting male mating behaviour [11], [12], [13]. In addition to odour, flower colour (including UV) and morphology (shape, size and texture) including epidermal structure (e.g. trichomes) also contribute to successful pollination [4], [14], [15], [16], [17]. Nonetheless, colour signals are of less importance than floral odour in a group of solitary bee-pollinated species [18] similar to those analysed in the present study.
In Ophrys orchids, floral odour mimics the sex pheromone produced by the female of the pollinators [11], [12], [19]. This pseudo-pheromone is a mixture of cuticular alkane and alkene hydrocarbons produced by the flower labellum: specifically alkanes (saturated straight-chain hydrocarbons) with different carbon chain length (C21-C31) and alkenes (monounsaturated hydrocarbons) that can additionally vary in their cis-double-bond positions (e.g. 7-, 9-, or 12-) [17], [20]. The relative amounts of alkanes and alkenes differ significantly among Ophrys species, thereby producing different pseudo-pheromone odour bouquets that attract different species of male bees as their pollinators [13], [21], [22]. These hydrocarbons are therefore crucial for pollinator-mediated reproductive isolation among Ophrys species [22], and thus play an important role in pollinator-mediated speciation in these orchids [20], [21], [23], [24].
Because of their strong pollinator-mediated reproductive isolation and the relatively well-understood chemical ecology of their highly specific pollination, Ophrys orchids provide an excellent system for studying pollinator-driven speciation and for identifying reproductive ‘barrier genes’ [17], [25], that is, genes directly involved in reproductive isolation [26]. Three closely related and sympatric Ophrys species, O. exaltata, O. sphegodes and O. garganica (Fig. S1) are investigated in this study. They are genetically compatible and crossable, but are strongly isolated from each other by pollinator-mediated, odour-based reproductive isolation, whereas post-pollination reproductive barriers are weak [22]. These species produce different odour bouquets: O. exaltata produces high levels of 7-alkenes, whereas O. sphegodes and O. garganica produce high levels of 9- and 12-alkenes in different proportions and carbon chain lengths [22]. Therefore, genes underlying these floral odour differences are candidate barrier genes, or possibly even speciation genes, among the study species. Alkanes and alkenes are expected to be derived from very-long-chain fatty acid (VLCFA) biosynthesis in epidermal cells of the flower labellum [17], [20], [27], [28]. Although acyl-ACP (acyl carrier protein) desaturases that introduce a double-bond into alkene precursors have previously been identified as barrier genes among O. sphegodes and O. exaltata [20], [24], other genes responsible for odour differences, such as hydrocarbon chain length differences among O. sphegodes and O. garganica, are still unknown.
The study of candidate genes in Ophrys orchids has so far been hindered by the lack of sequence resources. Currently, there is no genome sequence publically available for any orchid, and there are no comprehensive genome, transcriptome, or proteome resources for sexually deceptive orchids. A small number of orchid expressed sequence tags (ESTs) obtained by Sanger sequencing are available [29], [30], [31], [32], including 277 ESTs from Ophrys [33]. Although transcriptomes of the Phalaenopsis and Oncidium ‘Gower Ramsey’ orchids have recently been released [34], [35], these orchids are from the subfamily Epidendroideae and are only distantly related to Ophrys (subfamily Orchidoideae). Recently, next-generation sequencing such as 454 pyrosequencing has been widely used for de novo sequencing and EST analyses. These technologies have proven effective for expanding the available sequence information not only for model species [36], [37] but also for non-model species [38], [39] such as Ophrys, the large genome size of which (∼10 Gbp [40]) makes transcriptome sequencing a good choice for gene discovery. Moreover, shotgun proteomics by tandem mass spectroscopy (MS/MS) has recently been successfully used for the discovery of the protein components of various biological systems for which no prior information was available [41].
The current study aims to aid progress in orchid biology by (1) uncovering candidate genes for specific pollinator attraction and pollinator-mediated reproductive isolation among three Ophrys species, (2) providing a benchmark reference transcriptome from the orchid subfamily Orchidoideae, and (3) providing the first proteomic data for orchids. We address these questions by use of a systems biology approach, combining high throughput next-generation sequencing technologies (454 pyrosequencing, together with Solexa and Sanger sequencing) and MS/MS shotgun proteomics, in three sexually deceptive Ophrys species.
Results and Discussion
Sequencing and Assembly
Three normalised cDNA libraries were constructed from three different Ophrys species, O. exaltata, O. garganica, and O. sphegodes. For all three libraries, different tissues (mostly of floral origin) were pooled (see Materials and Methods; Fig. 1). These libraries were 454 pyrosequenced, resulting in a total of 71.3 Mbp of sequence data after processing (Table S1), with approximately 80% of the reads between 100 and 450 bp in length (Fig. 2A). All the high quality reads generated in this study are available in the Sequence Read Archive (SRA) of the National Centre for Biotechnology Information (NCBI) with the accession number SRA060767. Additional sequencing of O. sphegodes flower labella yielded 1.7 Mbp of Sanger (dbEST library LIBEST_028084; dbEST IDs 77978749-77979571; GenBank accessions JZ163765-JZ164587) and 2.5 Gbp of Illumina Solexa (SRA060767) data (Table S1).
Figure 1. Flow chart of transcriptome and proteome analysis.
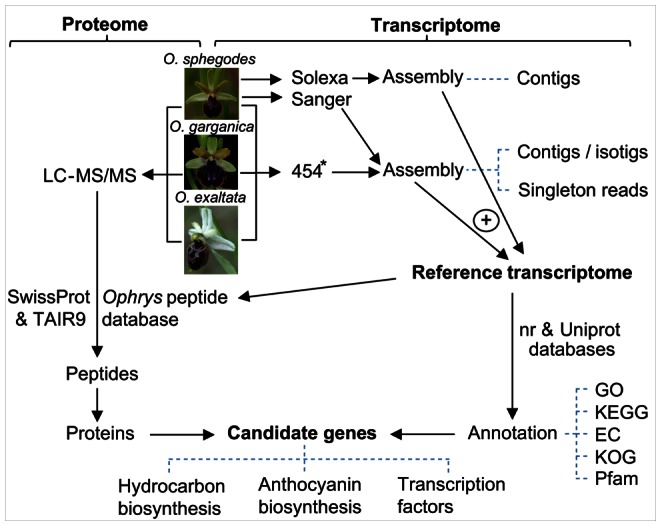
Labellum tissue from mature, unpollinated flowers was used, except where marked by an asterisk (*), indicating that additional material from sepals, petals, leaves, bracts, and flower buds was included.
Figure 2. Sequence length distributions.
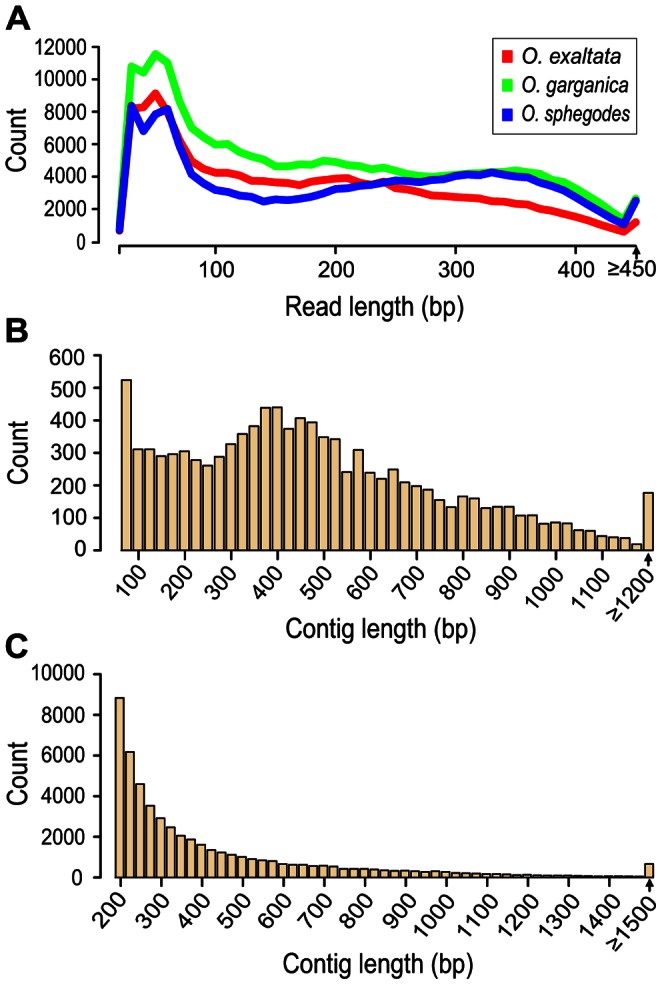
(A) Distribution of 454 read lengths after filtering and adapter removal for the three Ophrys species. (B) Contig length distribution (20 bp windows) for the pooled 454 assembly of the three Ophrys species. (C) Distribution of contig/isotig lengths in the Ophrys reference transcriptome (20 bp windows).
Solexa reads were assembled into contigs, whereas 454 and Sanger reads of the three species were separately assembled into isotigs (transcripts) (Table 1; Fig. 2B), thereby retaining putative gene/transcript relationships. This process left unassembled high-quality singleton reads, which can be considered to be either rare transcripts or artefacts. However, cross-validation of singleton reads (Table 2) by mapping of Solexa data suggested that the majority of singleton reads are not artefacts but represent real transcripts. Pooled 454 and Sanger assembly of all species together increased the effectiveness of the assembly (Table 1). Also, hybrid assemblies combining short (e.g. Solexa) and long (454 or Sanger) reads can improve de novo assembly of genomes and transcriptomes (for average read lengths, see Table S1) [42], [43], and short reads can improve coverage and the mining for lowly expressed genes [42]. Therefore, our pooled 454 and Sanger assembly was further merged with the O. sphegodes Solexa assembly to produce a final assembly, which we refer to as the Ophrys reference transcriptome (Table 1). Overall, this assembly contained 51 795 contigs and isotigs, with an average contig/isotig length of 441 bp, and 70 122 singleton reads with an average length of 285 bp (Table 1, Fig. 2C). The proportion of transcripts shared among species was determined by mapping the 454 reads of each species to the assembled contigs and isotigs present in the reference transcriptome (Fig. 3A). The majority of transcripts were shared among three or at least two species (Fig. 3B), the highest number of transcripts being shared among O. garganica and O. sphegodes (Fig. 3A), suggesting a closer overall transcriptome similarity among these species, which is consistent with overall floral similarities (see Fig. S1).
Table 1. Sequence assembly summary.
| O. exa. 454 | O. gar. 454 | O. sph. 454+ Sanger | O. sph. Solexa | O. sph. 454+ Sanger+Solexa | Pooled 454+ Sanger | Reference transcriptome (Pooled 454+ Sanger+Solexa) | |
| Number of contigs/isotigs | 2205 | 4172 | 3815 | 50 230 | 51 465 | 9375 | 51 795 |
| Bases in assembly (nt) | 1 022 978 | 2 334 132 | 1 925 958 | 19 166 533 | 20 501 805 | 5 777 134 | 22 837 772 |
| Average contig/isotig length (nt) | 463 | 559 | 504 | 382 | 398 | 616 | 441 |
| N50 length (nt) | 498 | 610 | 524 | 394 | 303 | 692 | 322 |
| Longest contig/isotig length (nt) | 1860 | 2777 | 1811 | 4273 | 4273 | 4771 | 4780 |
| Number of singleton reads1 | 35 567 | 42 261 | 43 2282 | N/A | 43 228 | 70 122 | 70 122 |
| Average singleton read length (nt) | 273 | 289 | 290 | N/A | 290 | 285 | 285 |
.Individual assembly statistics are shown for data from different sequencing technologies (454, Sanger, Solexa) and different species (O. exaltata, O. garganica, and O. sphegodes), where ‘Pooled 454’ refers to 454 data pooled from all three species. N50 length denotes the length-weighted median contig length of a given assembly.
Singleton reads from 454 and Sanger data only.
Out of these, 92 are Sanger reads.
Table 2. Cross-validation of NGS data sets.
| Reference data | Nref | Mapped by | Nmap | %age (mapping) |
| Solexa-only contigs (O. sph.) | 42 493 | 454 reads (3 spp.) | 16 255 | 38.3% |
| 454 reads (3 spp.) and Sanger singleton reads (O. sph.)1 | 25 287 (92) | Solexa reads (O. sph.) | 15 316 (52) | 60.6% (56.5%) |
| 454 singleton reads (O. exa.) | 18 664 | Solexa reads (O. sph.) | 9752 | 52.3% |
| 454 singleton reads (O. gar.) | 26 171 | Solexa reads (O. sph.) | 16 225 | 62.0% |
Summary of the proportion of a given sequence data set to which reads from another NGS data set can be mapped. Nref: number of sequences in the reference data set; Nmap: number of sequences in the reference data set that is mapped by the query data set; %age (mapping): Nmap expressed as a percentage. The term “3 spp.” refers to data from all three orchid species.
In this row, values in brackets are for Sanger reads.
Figure 3. Overlap of transcriptome and proteome data among three orchid species.
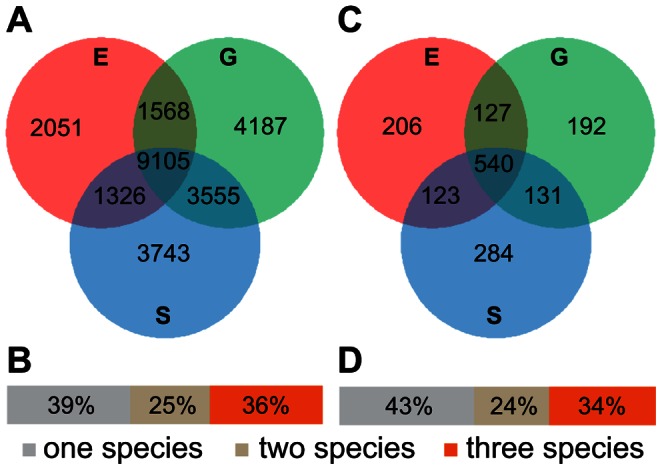
(A) Venn diagram showing the species overlap in 454 reads mapped back onto the reference transcriptome. (B) Bar graph indicating the extent of read sharing. (C) Venn diagram showing the overlap of Ophrys proteomes (HQ data of proteins with corresponding transcripts). (D) Bar graph indicating the extent of proteome overlap among species. (A,C) E, O. exaltata; G, O. garganica; S, O. sphegodes.
The combination of the 454 transcriptomes of the three species with the EST Sanger sequencing and Solexa data into a reference transcriptome represents the maximum amount of genetic information available to date for Ophrys. In total, 121 917 unique putative Ophrys transcripts were obtained and used for annotation and subsequent analysis. This is considerably more than the published Phalaenopsis orchid transcriptome (42 863 transcripts) [35], and comparable to that of Oncidium [34] in terms of contigs (50 908), although the latter transcriptome had a higher number of singleton reads (120 219). We note that our Ophrys reference transcriptome (assembled sequences and annotation provided in Supplementary Files S1 and S2, respectively) was generated mostly from floral tissues. This is the first comprehensive sequence resource both from orchids of the Orchidoideae subfamily, and for sexually deceptive orchids.
Proteomics Results
To obtain a more comprehensive understanding of Ophrys flowers and to further corroborate the authenticity of Ophrys transcripts, we performed large-scale shotgun proteomic analysis using MS/MS. Proteins were extracted from labellum tissue of each study species, digested and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) to obtain three proteomics data sets. Mass spectra were searched against SwissProt and Arabidopsis TAIR9 protein databases to identify peptides. Additionally, spectra were searched against protein databases created from the Ophrys reference transcriptome obtained in this study. Stringent criteria were used for the assignment of spectra to peptides (95% peptide identification probability) in Scaffold 3.3 (Proteome Software Inc., USA). In order to maximise the utility of proteomics data for uncovering proteins predicted by the orchid transcriptome, a minimum of one unique peptide was used for protein identification, while using two different stringency levels for the probabilistic assignment of peptides to proteins (99% for highest quality, HQ; 90% to maximise protein discovery, PD, in the absence of a fully sequenced genome). The proteomics data generated in this study are available from the PRIDE database [44] under accession numbers 27721–27914 and the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) under accession number PXD000069 (doi: 10.6019/PXD000069).
A total of 5496 contaminant-free, HQ unique peptides (PD: 7487) were found in the Ophrys proteome (Tables 3, S2 and S3). Out of these data, 93.8% of HQ spectra matched 1603 HQ (2644 PD) proteins predicted from Ophrys transcriptome data. This demonstrates the high quality of the transcriptome assembly and the necessity of its use for Ophrys protein identification. Out of these HQ proteins, 1328 were only found in Ophrys transcriptome data (PD: 1880), but not in SwissProt or TAIR9 databases, whereas 16.6% of HQ spectra (275 HQ, 764 PD proteins) also matched proteins in those databases. An additional 204 putative HQ proteins (PD: 887) from 1222 HQ spectra not present in Ophrys transcriptome data (Table 3) could be identified using TAIR and SwissProt databases. Using the FunCat classification scheme [45] for spectra with a hit in the TAIR database, no significant difference (p>0.01; Fisher’s exact test) in any functional category could be found among the entire protein set and proteins present in the Ophrys transcriptome data. No orchid peptides matched translations from different strands of the same transcript (Table 3), further indicating the overall high quality of the assembly.
Table 3. Summary of proteomics data.
| Protein discovery (PD) | Highest quality (HQ) | |||||||
| Statistic | O. exa. | O. gar. | O. sph. | Total | O. exa. | O. gar. | O. sph. | Total |
| N raw spectra | 9676 | 8419 | 7840 | 25 935 | 7433 | 6648 | 6344 | 20 425 |
| N clean spectra | 9357 | 8181 | 7588 | 25 126 | 7127 | 6426 | 6102 | 19 655 |
| N unique peptides | 4072 | 3835 | 4137 | 7487 | 2932 | 2878 | 3164 | 5496 |
| N proteins (transcriptome)1 | 1748 | 1672 | 1610 | 2644 | 1201 | 1222 | 1206 | 1603 |
| N proteins (non-transcriptome)2 | 403 | 405 | 439 | 887 | 130 | 134 | 143 | 204 |
| Total N proteins | 2110 | 1943 | 2031 | 3531 | 1331 | 1356 | 1349 | 1807 |
Data are presented for species individually and for the total of all three species combined. The protein discovery (PD) analysis was performed at a protein identification threshold of 90%, the highest quality (HQ) data set was compiled at a threshold of 99%. For spectra, numbers are given before (‘raw’) and after (‘clean’) removal of known contaminants. All other numbers were obtained from cleaned spectra only.
Proteins matching a sequence in the Ophrys reference transcriptome. In the HQ data set, 115 were 454 singleton reads, 4 Sanger singleton reads, 388 Solexa contigs, 343 pyrosequencing isotigs and 753 combined 454/Solexa contigs. Among these 1603 HQ orchid proteins, no peptide matched translations from different strands, and only 11 proteins (0.7%) had peptides matching to two reading frames of the same transcript strand (in each case due to a single frame shift).
Proteins not matching any Ophrys transcript, but with SwissProt and/or TAIR9 database hit.
Like in other published studies [42], [46], [47], [48], far fewer unique proteins (whether HQ or PD) could be identified than transcripts. Possible reasons include the complexity of the proteome and lower protein coverage as compared to transcript sequence data. The overlap among orchid-specific proteins for the three study species (Fig. 3C,D) was similar to the proportions of shared transcripts among species. All in all, proteomic corroboration of sequence data supports the good quality of our reference transcriptome. Moreover, proteomic data allowed the identification of up to 887 (PD) proteins for which no transcripts were observed, possibly due to a short half-life of the corresponding transcripts.
Functional Annotation
All unique sequences were annotated using BLASTX based on sequence similarity searches against public NCBI non-redundant (nr) and UniProt databases [49], [50]. Among all sequences, 44 034 (36.1%; 53.0% of contigs/isotigs and 23.6% of singleton reads) had at least one significant hit to existing genes in the databases. This is nearly twice the number of annotated Phalaenopsis (only 22 234 transcripts; 51.9%) [35] and Oncidium sequences (22 810, or 44.8% of contigs; 23 591, or 19.6% of singleton reads) [34]. The remaining 77 883 sequences did not match any known sequences, and may be considered novel transcripts. Alternatively, these sequences may be too short to match known sequences in the databases, or they may be derived from untranslated or nonconserved regions with low homology to known protein sequences, as has been reported in several studies [51], [52], [53]. We observed that annotation success was positively correlated (R2 = 0.41, p<0.001; Fig. S2) with sequence length, similar to data shown by Hoffman [54]. Therefore, long transcripts without BLAST hits are most likely to represent novel genes in Ophrys (Fig. S2).
Possible functions of transcripts with significant BLAST hits were classified for the three main Gene Ontology (GO) functional categories: biological process, molecular function, and cellular component (GO level 2, Fig. 4; levels 1–12, Table S4). The largest number of transcripts (21 138 transcripts) was annotated by molecular function, followed by biological process (19 960) and cellular component (19 272). In the molecular function category, most transcripts were assigned to ‘binding’ (46.1%) and ‘catalytic activity’ (42.1%). Within biological process, the most abundant categories were ‘metabolic process’ (42.0%) and ‘cellular process’ (35.9%). For cellular components, ‘cell’ and ‘organelle’ had the highest number of transcripts (55.0% and 37.1%, respectively).
Figure 4. Functional annotation.

(A–C) Pie charts showing the composition of 2nd level GO terms of the Ophrys reference transcriptome, broken up into the three major GO categories: (A) biological process, (B) cellular components, and (C) molecular function. (D) Summary of KOG functional annotation of Ophrys transcripts. The KOG terms are: D: Cell cycle control, cell division, chromosome partitioning; M: Cell wall, membrane, envelope biogenesis; N: Cell motility; O: Posttranslational modification, protein turnover, chaperones; T: Signal transduction mechanisms; U: Intracellular trafficking, secretion, and vesicular transport; V: Defence mechanisms; W: Extracellular structures; Y: Nuclear structure; Z: Cytoskeleton; A: RNA processing and modification; B: Chromatin structure and dynamics; J: Translation, ribosomal structure and biogenesis; K: Transcription; L: Replication, recombination and repair; C: Energy production and conversion; E: Amino acid transport and metabolism; F: Nucleotide transport and metabolism; G: Carbohydrate transport and metabolism; H: Coenzyme transport and metabolism; I: Lipid transport and metabolism; P: Inorganic ion transport and metabolism; Q: Secondary metabolites biosynthesis, transport and catabolism; R: General function prediction only; S: Unknown function.
To obtain additional annotation information for Ophrys transcripts and to identify putative orthologues and paralogues, all sequences were compared to the eukaryotic clusters of orthologous groups of proteins (KOG) database [55]. In total, 24 412 transcripts (20.0%) were assigned to different eukaryotic orthologous groups (Fig. 4D). The two major functional groups assigned are ‘general function prediction only’ (2893 transcripts, 11.9%) and ‘posttranslational modification, protein turnover, chaperones’ (2527 transcripts, 10.4%).
KEGG Pathways
The Kyoto Encyclopaedia of Genes and Genomes (KEGG) classification system provides an alternative functional annotation for genes according to their association with biochemical pathways [56]. To evaluate the completeness of the Ophrys reference transcriptome, transcripts were assigned to KEGG pathways and to enzyme commission (EC) numbers. A total of 7394 transcripts were assigned to KEGG pathways (Table 4) and the presence of Ophrys sequences for the majority of enzymes involved in essential biochemical pathways provides further evidence that the Ophrys transcriptome is relatively complete.
Table 4. Summary of KEGG pathway analysis.
| KEGG pathways | KEGG sub-pathways | N transcripts | N enzymes |
| Metabolism | Amino Acid Metabolism | 892 | 172 |
| Biosynthesis of Other Secondary Metabolites | 252 | 37 | |
| Carbohydrate Metabolism | 1300 | 206 | |
| Energy Metabolism | 707 | 81 | |
| Glycan Biosynthesis and Metabolism | 173 | 35 | |
| Lipid Metabolism | 393 | 82 | |
| Metabolism of Cofactors and Vitamins | 444 | 63 | |
| Metabolism of Other Amino Acids | 195 | 35 | |
| Metabolism of Terpenoids and Polyketides | 87 | 28 | |
| Nucleotide Metabolism | 657 | 55 | |
| Overview | 1934 | 351 | |
| Xenobiotics Biodegradation and Metabolism | 170 | 30 | |
| Genetic Information Processing | Translation | 84 | 20 |
| Environmental Information Processing | Signal Transduction | 66 | 9 |
| Organismal Systems | Immune System | 40 | 1 |
The number of Ophrys transcripts in a given sub-pathway, as well as the corresponding number of distinct enzymes in the KEGG database are shown.
Protein Domains
A total of 20 858 unique Ophrys transcripts matched 4274 protein domains in the Pfam conserved domain database (Tables 5 and S5), with Pkinase, RRM_1 and PPR being the most highly represented classes. Among these, Pkinase represents a conserved protein domain containing the catalytic function of protein kinases, which play roles in various cellular processes including division, proliferation, apoptosis, and differentiation [57]. RRM proteins are the largest group of single strand RNA-binding proteins in eukaryotes that play important roles in RNA processing and protein synthesis regulation [58]. PPR repeat proteins represent the biggest multigene family in plants, and are involved in almost all stages of gene expression, including mRNA transcription, processing, splicing, editing, translation and stability [59]. Among the protein domains of the enzymes putatively involved in hydrocarbon and anthocyanin biosynthesis (Table 5), the AMP-binding and 2OG-FeII_Oxy domains were most highly represented.
Table 5. Summary of Pfam domains.
| Accession | ID | Description | Occurrence |
| Protein domains in the reference transcriptome | |||
| PF00069.19 | Pkinase | Protein kinase domain | 507 |
| PF00076.16 | RRM_1 | RNA recognition motif. (a.k.a. RRM, RBD, or RNP domain) | 237 |
| PF01535.14 | PPR | pentatricopeptide repeat | 220 |
| PF00400.26 | WD40 | WD domain, G-beta repeat | 203 |
| PF07714.1 | Pkinase_Tyr | Protein tyrosine kinase | 199 |
| PF07727.8 | RVT_2 | Reverse transcriptase (RNA-dependent DNA polymerase) | 190 |
| PF00067.16 | p450 | Cytochrome P450 | 148 |
| PF12854.1 | PPR_1 | Pentatricopeptide repeat | 142 |
| PF00665.20 | rve | Integrase core domain | 93 |
| PF00004.23 | AAA | ATPase family associated with various cellular activities (AAA) | 90 |
| PF00270.23 | DEAD | DEAD/DEAH box helicase | 90 |
| PF00271.25 | Helicase_C | Helicase conserved C-terminal domain | 80 |
| PF00153.21 | Mito_carr | Mitochondrial carrier protein | 77 |
| PF00481.15 | PP2C | Protein phosphatase 2C | 75 |
| PF00036.26 | efhand | EF hand | 74 |
| Other domains | 18 055 | ||
| Protein domains in candidate genes for hydrocarbon biosynthesis | |||
| PF00501 | AMP-binding | AMP-binding enzyme | 62 |
| PF01061 | ABC2_membrane | ABC-2 type transporter | 43 |
| PF00106 | adh_short | short chain dehydrogenase | 35 |
| PF00378 | ECH | Enoyl-CoA hydratase/isomerase family | 23 |
| PF01553 | Acyltransferase | Acyltransferase | 13 |
| PF03405.8 | FA_desaturase_2 | Fatty acid desaturase | 13 |
| PF08392.1 | FAE1_CUT1_RppA | FAE1/Type III polyketide synthase-like protein | 13 |
| PF07993 | NAD_binding_4 | Male sterility protein | 13 |
| Other domains | 60 | ||
| Protein domains in candidate genes for anthocyanin biosynthesis | |||
| PF03171 | 2OG-FeII_Oxy | 2OG-Fe(II) oxygenase superfamily | 63 |
| PF00201 | UDPGT | UDP-glucoronosyl and UDP-glucosyl transferase | 19 |
| PF02797 | Chal_sti_synt_C | Chalcone and stilbene synthases, C-terminal domain | 10 |
| PF00195 | Chal_sti_synt_N | Chalcone and stilbene synthases, N-terminal domain | 7 |
| PF02431 | Chalcone | Chalcone-flavanone isomerase | 4 |
Highly abundant protein domains in the Ophrys reference transcriptome and among candidate genes for hydrocarbon and anthocyanin biosynthesis. ‘Occurrence’ lists the number of transcripts matching a given domain.
Candidate Genes for Pollinator Attraction
Genes involved in VLCFA and hydrocarbon biosynthesis
We combined transcriptome and proteome data sets to create a comprehensive resource for gene discovery in Ophrys orchids, with the aim of identifying candidate genes for pollinator attraction and reproductive isolation among Ophrys species. An important group of such candidate genes is putatively involved in hydrocarbon (and thus VLCFA) biosynthesis [17] (Fig. 5A). While all biosynthetic enzymes in the pathway are potential candidate genes for species-specific pollinator attraction, stearoyl-ACP desaturase (SAD) and β-ketoacyl-CoA synthase (KCS) enzymes are obvious a priori candidates for changes in hydrocarbon double-bonds and chain lengths, respectively [17], [60], which constitute the main odour differences among our study species. Homologues of all enzymes putatively involved in hydrocarbon biosynthesis were found in the Ophrys reference transcriptome, with 156 unique transcripts (132 putative genes; numbers include singleton reads) representing 20 candidate gene classes (Fig. 5A, Table S5). Seventy per cent (14/20) of these enzymes were confirmed by peptides (Table S6), including SAD and KCS. Proteins that were not confirmed by proteomics data were mostly membrane-associated (e.g., FAD or transporters) or very small (ferredoxin or ACP), and as such are less likely to be detected by shotgun proteomics.
Figure 5. Candidate genes in biosynthetic pathways.

Schematic diagrams of (A) hydrocarbon and (B) core anthocyanin biosynthesis, indicating candidate protein classes. The number of transcripts for a given candidate gene class is indicated in blue numbers, where an asterisk (*) indicates that a given candidate gene was confirmed at the protein level. Fatty acids in (A) are shown using C:D notation, where C is the number of carbon atoms and D is the number of double-bonds. Greyed, dotted arrows in panel (B) indicate metabolic reactions hypothesised to be absent. Protein abbreviations and further details on the listed candidates are provided in Table S6.
Plant SAD proteins are a class of ubiquitous soluble enzymes that catalyse the insertion of a double-bond into acyl-ACP. Differential expression and/or activity of different orchid SADs is responsible for alkene double-bond differences among O. sphegodes and O. exaltata [20], [24]. Homologous to seven known Arabidopsis SAD genes [61], we identified 13 different SAD transcripts in 7 gene models (isogroups) from transcriptome assembly that likely correspond to 6 known Ophrys SAD genes [20], [24], [33]. This suggests that our knowledge of the identity of Ophrys SAD genes is relatively complete. KCS, a critical component of the fatty acid (FA) elongase (FAE) complex, catalyses the condensation of malonyl-CoA and fatty acyl-CoA to extend the FA by two carbon units [62]. KCS is a good candidate for the differences in alkene chain length between O. sphegodes and O. garganica. Twenty-one KCS-like genes have been annotated in the Arabidopsis genome [63], [64], [65], and we identified 25 KCS transcripts (24 gene models) in Ophrys. This high number of transcripts indicates ample potential for evolutionary change in KCS-like genes. However, experimental studies are required to test the role of KCS in changing hydrocarbon chain length in Ophrys.
Genes involved in anthocyanin biosynthesis
Anthocyanidin pigments are formed as part of the flavonoid pathway, accumulate in the vacuole of epidermal cells, and can be responsible for red, orange, purple, and blue colours in flowers [66], [67]. These pigments play important ecological functions, such as providing visual signals to attract pollinators from a distance. Because of their ubiquity in flowering plants, the biosynthesis and regulation of anthocyanins are well understood, and genes involved in these processes have been characterised in several orchid species (e.g. [68], [69]). While pollinator specificity in Ophrys is mostly due to hydrocarbon differences, floral coloration is involved in the mimicry of the pollinator female’s body colour [17]. For instance, O. garganica has darker flowers than O. sphegodes (see Fig. S1), corresponding to the darker body colour of its pollinator, the black Andrena pilipes. With one exception, homologues of all enzymes of the core anthocyanin biosynthesis pathway were found in the Ophrys reference transcriptome, with 61 unique transcripts (59 putative genes), representing 7 candidate enzyme classes (Fig. 5B, Table S6). Seventy-one per cent (5/7) of these enzymes were confirmed by peptides (Table S6). Interestingly, the one exception was flavonoid 3′,5′-hydroxylase (F3′5′H), for which no transcripts (or peptides) were found. It is possible that this absence of F3′5′H is not due to limited transcriptome coverage, but reflects the biology of (rather reddish) Ophrys flowers, because F3′5′H is required for the formation of (often bluish) delphinidin pigments [66], [70]. Similar situations are known in several other plants without delphinidin pigments (e.g., Ipomoea, Rosa, Dianthus, and Chrysanthemum) [71], in which F3′5′H either is not expressed or was lost from the genome (e.g. in Arabidopsis thaliana) [72]. Given that delphinidin pigments may be present in the distantly related O. speculum [73], this loss of F3′5′H transcript and/or gene may only have occurred relatively recently.
Transcription factors
Transcription factors (TFs) are important regulators of gene expression in response to plant developmental processes and environmental factors [74]. Since they are potential candidates for species differences in pollinator attraction, TFs in the Ophrys transcriptome were identified by a comparison to Arabidopsis thaliana and Oryza sativa transcription factor databases [75] and by KOG annotation. Overall, 3319 unique transcripts (2.7% of the Ophrys transcriptome), encoding members of 56 putative TF families, were identified (Fig. 6; Table S7), which is higher than the number of TFs identified in Phalaenopsis (786 transcripts, 1.83%). The most abundant TF families were WRKY, NF-YA and NAC factors (Fig. 6). Moreover, LFY, M-type, STAT, VOZ and WOX factors, which had not been found in the Phalaenopsis transcriptome [35], were detected in the Ophrys transcriptome. Other abundantly represented TF families in our dataset include Myb, bHLH and MADS factors.
Figure 6. Transcription factors.

Bar plot showing the number of transcripts in different transcription factor classes; additional TFs identified by KOG annotation are plotted as a separate column.
MADS-domain (M-type and MIKC) TFs are involved in controlling all major aspects of plant development [76] and have been shown to regulate anthocyanin biosynthesis in pigmented tubers of sweet potato [77]. MADS-domain proteins are important regulators of floral development [78], [79], and duplications of B-class MADS-box genes have been implicated in the evolution of complex orchid flowers [80]. For instance, the combinatorial expression of several paralogues is thought to specify flower labellum identity, which may also depend on the action of TCP TFs [80]. In the Ophrys transcriptome, MADS-domain factors were represented by 56 transcripts. These included 4 transcripts of GLOBOSA/PISTILLATA and 3 transcripts of DEFICIENS/APETALA3 (clade 2, 3 and 4 [80], [81]; Table S8) B-class gene homologues. Moreover, 6 TCP TFs were found in the transcriptome. These genes represent candidate genes for labellum development.
The Myb and basic helix-loop-helix (bHLH) families are the largest and second largest classes of TFs in plants, respectively [82]. In rice and Arabidopsis there are 339 and 230 Myb factors, and 162 and 111 bHLH TFs, respectively [82]. Myb (Myb and MYB-related) factors account for 197 transcripts in the Ophrys transcriptome, whereas the bHLH family is represented by 111 transcripts. Myb TFs are especially interesting candidate genes, because they are implicated in a range of functions, including the regulation of secondary metabolism [83], [84], [85], [86], cell morphogenesis [87], [88], [89], control of the cell cycle [90], [91], floral and seed development [92], [93], [94], [95], responses to biotic and abiotic stresses [96], [97], [98], [99], and light and hormone signalling pathways [100], [101], [102]. Particularly, Myb factors controlling cell shape as well as VLCFA and anthocyanin biosynthesis may be important candidates for changes in floral traits like odour, colour and epidermal cell shape, all of which are involved in specific pollinator attraction in Ophrys [17].
One interesting Myb gene, MYB30, has been found to be involved in VLCFA biosynthesis by controlling the expression of four enzymes forming the FAE complex in Arabidopsis [103]. Two transcripts homologous to MYB30 were identified in Ophrys (Table S7). A specific group of Myb factors, R2R3 MYBs, interacts with bHLH factors and WD-repeat (WDR) proteins to form the MYB–bHLH–WDR (MBW) complexes that regulate anthocyanin biosynthesis [70]; R2R3 MYB subgroup 6 (SG6) TFs such as AtMYB113, AtMYB114, AtMYB75/PAP1 and AtMYB90/PAP2 activate anthocyanin biosynthesis in many different species [83], [104], [105]. Four transcripts represent SG6 genes in the Ophrys transcriptome (Table S7).
Conical cells may enhance the colour intensity and brightness of petal surface and, thereby, increase flower attractiveness to the pollinators [106], [107], [108], [109]. MIXTA, a Myb-related TF, drives the formation of conical epidermal cells from the flat epidermal cells of the snapdragon, Antirrhinum majus [109], [110], and AmMYBML1 encodes a similar R2R3 MYB TF that has a role in controlling trichome, conical cell and mesophyll cell morphogenesis in the ventral petal of Antirrhinum flowers [111]. One AmMYBML1-like and three MIXTA-like genes were identified in Ophrys (Table S7).
Taken together, our Ophrys reference transcriptome with its focus on flowers allowed us to identify several putative transcription factors that may be relevant to our understanding of the molecular basis of pollinator attraction. This includes MADS, TCP and Myb factors that may be involved in the orchid’s mimicry of pollinator females by regulating odour, colour and floral morphological traits. Our data thus provide a starting point for investigating the molecular mechanisms of pollinator attraction and adaptation.
Evolutionary Implications
Evolution of sexual deception
Sexual deception in Ophrys may have evolved from food deception [112], and alkene production (and possibly labellum size) may have served as pre-adaptations for this mode of pollination [112], [113]. Floral morphological development, hydrocarbon and anthocyanin biosynthesis play important roles in facilitating sexually deceptive pollination [17]. These pathways, however, are likely to be similar in Ophrys and related genera, which share many of its floral features and (pre-)adaptive traits, although with quantitative differences (e.g. in alkene levels [113]). This implies that sexual deception likely arose by modification of existing pathways, possibly by regulatory changes or changes associated with gene duplication (and subsequent neo- and subfunctionalisation). Therefore, comparative genomic studies incorporating other orchid species may lead to the identification of the genetic changes that allowed the evolution of sexual deception.
Molecular mechanisms of reproductive isolation
All biosynthetic and regulatory candidate genes uncovered in the transcriptome were present in all species, with the exception of F3′H and some transcription factors, all of which were only detected in O. sphegodes (Tables S6 and S7). However, this may be due to lower sequence coverage in the other two species, which might have prevented detection of rare transcripts. The widespread presence of candidate gene transcripts in all species therefore suggests that species differences in pollinator attraction, and consequently reproductive isolation, are unlikely to be caused by the simple presence or absence of expression in any candidate gene class. Rather, it may be expected that subtler changes, such as paralogue-, isoform-, or allele-specific sequence and/or expression changes, or even epigenetic phenomena [114], [115], underlie reproductive isolation. This holds true at least for SAD genes specifying alkene double-bond positions [20], [24] and appears similarly plausible for KCS and hydrocarbon chain length, where the presence of 25 transcript sequences presages complexity. More detailed studies that tease apart the effects of different gene copies, alleles and quantitative expression changes are required to understand the exact molecular architecture of pollinator-mediated reproductive isolation among Ophrys species.
Conclusions
Here, we employed high throughput next-generation sequencing technologies combined with shotgun proteomics to provide the first floral reference transcriptome and proteome data for sexually deceptive Ophrys from the Orchidoideae subfamily of orchids, for which proteome data were previously absent and sequence information scarce. We thereby provide significant resources for gene discovery and systems biology in orchids in general, by enabling sequence comparisons among disparate lineages of orchids. Likewise, our data set considerably expands the resources available for sexually deceptive plants, and provides an opportunity to advance our understanding of the molecular basis of plant-pollinator interactions, as well as pollinator-mediated selection and speciation. Our data are relevant for the characterisation of candidate genes for these and other processes, as demonstrated here by the identification of genes potentially involved in plant pseudo-pheromone biosynthesis and regulation. Based upon a priori knowledge and the abundance of these gene classes in the Ophrys transcriptome, especially KCS biosynthetic genes and Myb transcription factors warrant further attention as candidate genes for differences in specific pollinator attraction. Subtle changes in such genes may be responsible for reproductive isolation among sexually deceptive orchids.
Materials and Methods
Plant Materials
Plant material of O. exaltata subsp. archipelagi (Gölz & Reinhard) Del Prete, O. garganica Nelson, and O. sphegodes Miller for 454 sequencing was grown in a greenhouse at the Botanic Garden of the University of Zurich, Switzerland. Additional flower labellum samples of these three species were used for proteome analysis and two further O. sphegodes flower labella for EST library and Illumina Solexa sequencing, all of these samples being from different plants. All flowers used in this study were unpollinated. Plant tissues were collected, flash-frozen in liquid nitrogen and stored at −80°C until RNA or protein extraction. For 454 sequencing, the following tissues were used: leaves, bracts, labella, sepals, petals and columns, both from open flowers and flower buds from all developmental stages available on the sampled plants, the smallest available bud being five positions from the latest open flower on the inflorescence.
cDNA Normalisation and 454 Sequencing
Total RNA was extracted separately from different tissues (see above) collected from three different individual plants per species (21 RNA samples/species) using TRIzol® reagent (Invitrogen) and the supplier’s protocol. Extracts were further purified by using RNeasy MinElute Cleanup columns (Qiagen) according to the manufacturer’s protocol. The extracted RNAs were analysed for potential degradation by gel electrophoresis and on a Bioanalyzer 2100 (Agilent), and RNA concentration was quantified using the fluorometric Qubit Quantitation Platform (Invitrogen). Equal amounts of RNA from each biological individual were pooled to yield one RNA sample for each Ophrys species. To avoid genomic DNA contamination, RNA was treated with RNase-free DNase I (Qiagen). Full-length double-stranded cDNA was synthesised from 0.5 µg pooled total RNA using SMARTer PCR cDNA Synthesis Kit (Clontech, Palo Alto, CA, USA) according to the manufacturer’s instructions. To enhance gene discovery, the contribution of highly abundant transcripts was reduced before sequencing. To do so, 1 µg of each cDNA was normalised using the Trimmer cDNA Normalization Kit (Evrogen, Moscow, Russia), according to the manufacturer’s instructions.
Approximately 500 ng of normalised cDNA of each sample were used to generate a single strand cDNA transcriptome library for the Roche/454 Life Sciences GS-FLX Titanium platform (Roche, Basel, Switzerland) following the Rapid Library Preparation Method Manual. Briefly, cDNA of each sample was sheared by nebulisation to produce fragments of approximately 300–400 bp, and oligonucleotide adapters were ligated to the fragmented cDNA. One adapter contained a barcode sequence that was used to discriminate the samples (i.e. species) from each other after sequencing, as all libraries were combined in a single pool. Transcriptome library sequencing was then performed according to the Roche GS-FLX XLR70 Titanium emPCR and sequencing manuals. The pooled sample was sequenced on a full picotitre plate on a Genome Sequencer FLX Instrument, according to the manufacturer’s instructions, at the Functional Genomics Centre Zurich, Switzerland.
EST Library Preparation and Sequencing
Poly (A)+ RNA was purified from 2 µg O. sphegodes flower labellum total RNA using the Oligotex mRNA isolation kit (Qiagen AG, Hombrechtikon, Switzerland) according to the manufacturer’s instructions. One standard cDNA library was prepared using the Creator SMART cDNA Library Construction Kit and the protocol provided, except that insert size selection was performed using Zymoclean Gel DNA Recovery Kit (Zymo Research, Orange, CA, USA). Oligo-dT-primed cDNA inserts larger than 1 kb were directionally cloned in pDNR-LIB vector (Clontech, Palo Alto, CA, USA) and transformed into XL-10 Gold Kan+ ultracompetent Escherichia coli (Stratagene, LaJolla, CA, USA). Colony PCR reactions were performed to test the library efficiency and insert size range. The library was stored by the addition of glycerol (20% v/v final concentration) and sent for Sanger sequencing (Applied Biosystems) at the Purdue University sequencing platform, West Lafayette, Indiana (USA).
Illumina Solexa Sequencing
Total RNA of one O. sphegodes flower labellum was used for an RNA-Seq experiment by Illumina Solexa sequencing at BGI Shenzhen (China), following Illumina’s sample preparation guidelines. Briefly, poly(A)+ was purified from total RNA and fragmented, cDNA synthesised and adapters ligated. After size selection, cDNA was subjected to Solexa paired-end sequencing on an Illumina Genome Analyzer II generating 75 nt long reads.
Processing of Sequence Reads
For Sanger reads, base calling, masking of vector sequences and low quality ends were done with phred (version 071220) via the phredPhrap script (version 080818) in consed (version 20.0) [116], and trimming of poly-A/T tails with seqclean (http://compbio.dfci.harvard.edu/tgi/software/). Raw Solexa reads were filtered by quality using the manufacturer’s software and default parameters. Raw 454 sequencing data was obtained with the GS Run Processor 2.5.3 (Roche) using shotgun quality filtering and trimming as defined by the default settings. Raw 454 reads were first trimmed of adapter sequences used in cDNA library preparation and normalisation with newbler in the Roche 454 Software Suite 2.5.3 (454 Life Science, Branford, CT). MEGABLAST 2.2.21 [117] was run to ascertain that trimmed reads were clean of any adapter and primer sequences. Trimmed 454 reads were further processed with seqclean to remove low complexity and low quality reads, and to remove any left-over poly-A/T tails.
Transcriptome Assembly
O. sphegodes Solexa reads were first assembled into contigs using SOAPdenovo [118], using paired-end information to link contigs into scaffolds, and where possible, to fill gaps with reads. Scaffolds were clustered using TGICL [119], each cluster producing one or more consensus sequences; unclustered scaffolds were termed singleton contigs. O. sphegodes Sanger reads and 454 reads from all three species were both (1) assembled separately for each species and (2) pooled and assembled with 454 newbler 2.5.3 (454 Life Science, Branford, CT). The pooled assembly was further merged with the O. sphegodes Solexa assembly using minimus2 under the criteria of minimum 40 bases overlap with at least 94% identity [120]. The merged assembly from Sanger, 454 and Solexa data is referred to as the Ophrys reference transcriptome.
Transcriptome Annotation and Analysis
Assembled sequences and singleton reads were compared to the NCBI non-redundant (nr) database using BLASTX [121]. Based on the search results, Gene Ontology (GO) term annotation was performed to predict the function of the sequences using BLAST2GO software [122]. Enzyme commission (EC) number and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway [56], [123] were inferred from GO annotations using the same software. Moreover, GO, EC and KEGG annotation was also done with annot8r based on BLASTX results from searches against the UniProt database. Orthologous genes (KOG annotation) were identified by searching against the NCBI KOG database using RPSTBLASTN 2.2.26 [121]. Coding regions were predicted using ESTScan 3.0.3 [124] and then compared to the Pfam protein domain database [125] using pfam_scan 1.3 [126].
Read Mapping
Read mapping was performed for purposes of data set cross-validation. Solexa reads were mapped onto 454/Sanger singleton reads using Bowtie 0.12.7 [127] (parameters: ‘-n 1 -l 35 -e 100 -m 5–best –strata’) as independent evidence for singleton reads. 454 reads were mapped back on the reference transcriptome using the BWA-SW algorithm in BWA 0.5.9 [128] (with parameter ‘-s 5′).
Identification of Candidate Genes
A priori candidate genes were identified by homology to genes of known or putative function in model organisms. To identify hydrocarbon and anthocyanin biosynthetic genes, a BLASTN search of the Ophrys reference transcriptome against Arabidopsis thaliana TAIR10 coding sequences was performed (at e-value of<E-03), retaining information of the best BLAST hit for each orchid transcript. Homologues of Arabidopsis genes for selected gene classes were considered candidate genes. In addition, text/term searches of BLAST/NR best hits, GO annotations and EC numbers were used to identify candidate orchid genes. For transcription factor identification, KOG searches for transcription terms were performed alongside BLAST searches against A. thaliana and Oryza sativa TF databases publically available from PlantTFDB 2.0 (which currently does not contain sequences from Asparagales) [75]. TFs of special interest, putatively involved in the regulation of VLCFA, anthocyanin biosynthesis and flower morphology, were identified from the literature.
Shotgun Proteomics
Frozen labellum tissue of an unpollinated flower from each study species was ground to powder without allowing it to thaw, and resuspended in 150 µL urea protein extraction buffer (65 mM Tris-HCl, 8 M urea, 10% glycerol, 5% β-mercaptoethanol, 2% SDS, 0.025% bromophenol blue), denatured and separated by SDS-PAGE, sliced (13 gel slices per sample) and trypsinised as described previously [129]. Proteins were subjected to electrospray ionisation-based LC-MS/MS analysis with a 2D linear ion trap Finnigan LTQ (Thermo Electron Corporation), equipped with an Ultimate Nano HPLC System (Dionex Corporation) exactly as described by Grobei et al. [129].
Proteome Analysis
MS/MS-derived spectra were searched against different peptide databases using Mascot Search Engine version 2.3 (Matrix Science Ltd., UK). The databases used were SwissProt and TAIR9 [130], [131], including decoys and known contaminants (as in Grobei et al. 2009 [129]), and (1) orchid peptides predicted by ESTScan and (2) a 6-frame translation of the orchid transcriptome. Mascot searches, with a peptide mass tolerance of 3 Da, allowed for one trypsin miscleavage, for Met oxidation and Cys carbamidomethylation, and were further analysed and validated in R 2.14.2 [132] and Scaffold 3.3 (Proteome Software Inc., USA), which uses the Peptide-Prophet and Protein-Prophet algorithms [133], [134]. Spectra from known contaminants were removed from the data set for final analysis.
Supporting Information
Comparison of Ophrys flowers. Five flowers each from different individuals of (A) O. exaltata subsp. archipelagi, (B) O. sphegodes and (C) O. garganica, showing inter-species and intra-species variation (plants all from Gargano, Southern Italy). Images were scaled for comparison, the white bar indicating 1 cm. O. sphgodes tends to have comparatively small flowers with a brown labellum and a greenish perigon, whereas O. garganica flowers are usually larger, with a darker labellum and sometimes coloured petals. O. exaltata tends to have comparatively large flowers with a slightly elongated brown labellum, typically with a protrusion at its apex, and usually a white perigon. The speculum (brighter, more reflective part of the labellum) can be quite variable in all species, and they all have longer trichomes at the sides of the labellum (‘hairy margin’) as compared to its centre. Micromorphological features of Ophrys flowers are described elsewhere [15].
(PDF)
Transcript length/annotation relationship. Plot showing the percentage of hits with annotation information from NCBI nr and UniProt databases versus sequence length.
(PDF)
Summary of sequencing data. This table lists details of the 454, Sanger and Solexa sequence data sets obtained in this study.
(DOCX)
List of orchid proteins. This table lists all proteins matching sequences in the Ophrys transcriptome by their transcriptome sequence ID. In the column HQ, an asterisk (*) denotes proteins in the HQ data set (no asterisk indicates PD data set). The ‘Species’ column indicates in which orchid species a given protein was found, where: E, O. exaltata; G, O. garganica; S, O. sphegodes. In the column ‘Orchid-only’, y/n (yes/no) indicate if a given protein was found only in the Ophrys transcriptome (y) or if it also matched proteins in the TAIR and/or SwissProt databases (n). The ‘Description’ column refers to the term provided in the transcript ID’s best BLASTX/nr database hit.
(XLSX)
List of proteins without transcript. This table lists all proteins that were identified in the SwissProt/TAIR9 databases, but did not match any sequence in the Ophrys transcriptome, listing the protein description, source database and associated accession numbers. In the column HQ, an asterisk (*) denotes proteins in the HQ data set (no asterisk indicates PD data set). The ‘Species’ column indicates in which orchid species a given protein was found, where: E, O. exaltata; G, O. garganica; S, O. sphegodes.
(XLSX)
GO classification. This table contains the GO classification of the assembled sequences into three main categories (biological function, cellular component and molecular function) at all levels (1–12). ‘All species’ lists the number of transcripts in a given category in the Ophrys reference transcriptome, and additional columns list the corresponding number of transcripts for the individual species separately.
(XLS)
Pfam protein domains. This table lists the Pfam annotations in the Ophrys reference transcriptome by transcript ID.
(XLSX)
List of candidate genes. Details of transcripts encoding candidate biosynthetic proteins putatively involved in hydrocarbon and anthocyanin biosynthesis, sorted alphabetically per category. TAIR ID lists the Arabidopsis gene for which homologues were found; EC is the enzyme commission number searched for a given candidate protein; ‘N transcripts’ lists the number of unique transcripts matching a candidate protein category in the Ophrys reference transcriptome (counting singleton reads as transcripts); ‘N gene models’ is composed of the number of isogroups from sequence assembly plus transcripts that were not assigned to isogroups (so that for instance, singleton reads would be counted as a new gene model); ‘Unique transcripts’ lists the unique transcriptome sequence IDs. Species with transcripts and peptides list in which Ophrys species transcripts or peptides were found, respectively, where: E, O. exaltata; G, O. garganica; S, O. sphegodes. In the column ‘Species with peptides’, an asterisk indicates that a protein was part of the HQ data set (no asterisk, PD data set). The ordering of elements in the two rightmost columns corresponds to the ordering in the ‘Unique transcripts’ column.
(DOCX)
List of transcription factors. This table lists the identification method (TFDB, transcription factor database; KOG, KOG annotation) for TF identification, along with the unique transcript ID and annotation details (accession number or KOG term). The column ‘Transcription Factor’ lists the TF class identified from the database or the KOG description term, as appropriate. Species with transcripts or peptides for a given TF are listed, where: E, O. exaltata; G, O. garganica; S, O. sphegodes. Proteins identified in the HQ data set are marked with an asterisk. The Comments column provides further information on TFs of special interest. In addition to the TFs listed in this table, one additional transcript (transcript ID 3673) was found to have significant BLAST homology (e-value 6E–12) to a TF of interest, namely Antirrhinum majus AmMYBML1-like (AY661653.1).
(XLSX)
BLAST results for DEF/AP3 and GLO/PI MADS-box gene homologues. Table of BLASTN results, showing top 10 hits against the NCBI nr database (sorted by e-value) for each putative Ophrys B-class MADS-box gene homologue identified in the Ophrys reference transcriptome. The column ‘Lineage’ lists the gene lineage (DEF clades 1, 2, 3 and 4; Orchidoideae GLO1 and GLO2) assigned to the accession number of a BLAST hit, as defined by Mondragón-Palomino & Theißen (2008; [81]) for DEF, and Kim & al. (2007; [135]) and Cantone & al. (2011; [136]) for GLO. The best lineage assignment is highlighted in bold for each transcript. Accession numbers with defined lineage are as follows: DEF clade 1: AY378149, DQ119838, DQ683575, FJ804097, FJ804106, FJ804115; clade 2: AY196350, AY378148, FJ804098, FJ804105, FJ804111; clade 3: AY378150, AB232663, DQ119839, FJ804099, FJ804107, FJ804110, FJ804117; clade 4: AY378147, FJ804108, FJ804112, FJ804116; GLO1: AB232665, AB450305, AB450307, AB450310, AB450302, AB450308, AB450309, AB450299, AB450303, AB450306; GLO2: AB232664, AB537512, AB537507, AB537511, AB537513, AB537509, AB537508, AB537506, AB537504, AB537510.
(XLSX)
This zip-compressed file contains (1) the assembled Ophrys reference transcriptome (FASTA), along with (2) an MD5 check-sum, (3) an FAI index file, (4) 454 isotig to isogroup (transcript to gene) mapping (tab-delimited text), and (5) a text file describing the naming scheme for the transcript identifiers used.
(ZIP)
This spreadsheet document contains the annotation information for the Ophrys reference transcriptome in several tables.
(XLSX)
Acknowledgments
The authors thank Natalia Dudareva and the Purdue Sanger sequencing platform, Bernd Roschitzki and Simon Barkow for help with proteome bioinformatics, Wen-Chieh Tsai for providing their Phalaenopsis transcriptome data, Salvatore Cozzolino for discussion, and Stephen Schauer and two reviewers for discussion and comments.
Funding Statement
This work was funded by the Swiss National Science Foundation (Project 31003A_130796 to PMS), and the University Research Priority Programme Functional Genomics/Systems Biology of the University of Zurich (to UG). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Darwin CR (1877) The various contrivances by which orchids are fertilised by insects. London: John Murray, 2nd ed.
- 2.Dressler RL (1981) The orchids: natural history and classification: Harvard University Press. 332 p.
- 3. Cozzolino S, Widmer A (2005) Orchid diversity: an evolutionary consequence of deception? Trends Ecol Evol 20: 487–494. [DOI] [PubMed] [Google Scholar]
- 4. Gaskett AC (2011) Orchid pollination by sexual deception: pollinator perspectives. Biol Rev 86: 33–75. [DOI] [PubMed] [Google Scholar]
- 5. Kullenberg B (1961) Studies in Ophrys pollination. Zool Bidr Upps 34: 1–340. [Google Scholar]
- 6. Paulus HF, Gack C (1990) Pollinators as prepollinating isolation factors: evolution and speciation in Ophrys (Orchidaceae). Israel J Bot 39: 43–79. [Google Scholar]
- 7. Borg-Karlson A-K (1990) Chemical and ethological studies of pollination in the genus Ophrys (Orchidaceae). Phytochem 29: 1359–1387. [Google Scholar]
- 8.Ayasse M, Gögler J, Stökl J (2010) Pollinator-driven speciation in sexually deceptive orchids of the genus Ophrys. In: Glaubrecht M, editor. Evolution in action: case studies in adaptive radiation, speciation and the origin of biodiversity. 101–118.
- 9. Schiestl FP (2005) On the success of a swindle: pollination by deception in orchids. Naturwissenschaften 92: 255–264. [DOI] [PubMed] [Google Scholar]
- 10.Vereecken NJ (2009) Deceptive behavior in plants: I. Pollination by sexual deception in orchids: a host-parasite perspective. In: Baluska F, editor. Plant-environment interactions. 203–222.
- 11. Schiestl FP, Ayasse M, Paulus HF, Löfstedt C, Hansson BS, et al. (1999) Orchid pollination by sexual swindle. Nature 399: 421–421. [Google Scholar]
- 12. Mant J, Brändli C, Vereecken NJ, Schulz CM, Francke W, et al. (2005) Cuticular hydrocarbons as sex pheromone of the bee Colletes cunicularius and the key to its mimicry by the sexually deceptive orchid, Ophrys exaltata . J Chem Ecol 31: 1765–1787. [DOI] [PubMed] [Google Scholar]
- 13. Mant J, Peakall R, Schiestl FP (2005) Does selection on floral odor promote differentiation among populations and species of the sexually deceptive orchid genus Ophrys? Evolution 59: 1449–1463. [PubMed] [Google Scholar]
- 14. Spaethe J, Moser WH, Paulus HF (2007) Increase of pollinator attraction by means of a visual signal in the sexually deceptive orchid, Ophrys heldreichii (Orchidaceae). Plant Syst Evol 264: 31–40. [Google Scholar]
- 15. Bradshaw E, Rudall PJ, Devey DS, Thomas MM, Glover BJ, et al. (2010) Comparative labellum micromorphology of the sexually deceptive temperate orchid genus Ophrys: diverse epidermal cell types and multiple origins of structural colour. Bot J Linn Soc 162: 504–540. [Google Scholar]
- 16. Ågren L, Kullenberg B, Sensenbaugh T (1984) Congruences in pilosity between three species of Ophrys (Orchidaceae) and their hymenopteran pollinators. Nova Acta Reg Soc Sci 3: 15–25. [Google Scholar]
- 17. Schlüter PM, Schiestl FP (2008) Molecular mechanisms of floral mimicry in orchids. Trends Plant Sci 13: 228–235. [DOI] [PubMed] [Google Scholar]
- 18. Vereecken NJ, Schiestl FP (2009) On the roles of colour and scent in a specialized floral mimicry system. Ann Bot 104: 1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schiestl FP, Ayasse M, Paulus HF, Löfstedt C, Hansson BS, et al. (2000) Sex pheromone mimicry in the early spider orchid (Ophrys sphegodes): patterns of hydrocarbons as the key mechanism for pollination by sexual deception. J Comp Physiol A 186: 567–574. [DOI] [PubMed] [Google Scholar]
- 20. Schlüter PM, Xu S, Gagliardini V, Whittle E, Shanklin J, et al. (2011) Stearoyl-acyl carrier protein desaturases are associated with floral isolation in sexually deceptive orchids. Proc Natl Acad Sci USA 108: 5696–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schiestl FP, Ayasse M (2002) Do changes in floral odor cause speciation in sexually deceptive orchids? Plant Syst Evol 234: 111–119. [Google Scholar]
- 22. Xu S, Schlüter PM, Scopece G, Breitkopf H, Gross K, et al. (2011) Floral isolation is the main reproductive barrier among closely related sexually deceptive orchids. Evolution 65: 2606–2620. [DOI] [PubMed] [Google Scholar]
- 23.Xu S, Schlüter PM, Schiestl FP (2012) Pollinator-driven speciation in sexually deceptive orchids. Int J Ecol vol. 2012, Article ID 285081.
- 24. Xu S, Schlüter PM, Grossniklaus U, Schiestl FP (2012) The genetic basis of pollinator adaptation in a sexually deceptive orchid. PLoS Genet 8: e1002889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schiestl FP, Schlüter PM (2009) Floral isolation, specialized pollination, and pollinator behavior in orchids. Annu Rev Entomol 54: 425–446. [DOI] [PubMed] [Google Scholar]
- 26. Noor MAF, Feder JL (2006) Speciation genetics: evolving approaches. Nat Rev Genet 7: 851–861. [DOI] [PubMed] [Google Scholar]
- 27. Kunst L, Samuels AL (2003) Biosynthesis and secretion of plant cuticular wax. Prog Lipid Res 42: 51–80. [DOI] [PubMed] [Google Scholar]
- 28. Perera MADN, Qin W, Yandeau-Nelson M, Fan L, Dixon P, et al. (2010) Biological origins of normal-chain hydrocarbons: a pathway model based on cuticular wax analyses of maize silks. Plant J 64: 618–632. [DOI] [PubMed] [Google Scholar]
- 29. Tan J, Wang HL, Yeh KW (2005) Analysis of organ-specific, expressed genes in Oncidium orchid by subtractive expressed sequence tags library. Biotechnol Lett 27: 1517–1528. [DOI] [PubMed] [Google Scholar]
- 30. Hsiao YY, Tsai WC, Kuoh CS, Huang TH, Wang HC, et al. (2006) Comparison of transcripts in Phalaenopsis bellina and Phalaenopsis equestris (Orchidaceae) flowers to deduce monoterpene biosynthesis pathway. BMC Plant Biol 6: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsai WC, Hsiao YY, Lee SH, Tung CW, Wang DP, et al. (2006) Expression analysis of the ESTs derived from the flower buds of Phalaenopsis equestris . Plant Sci 170: 426–432. [Google Scholar]
- 32. Teh SL, Chan WS, Abdullah JO, Namasivayam P (2011) Development of expressed sequence tag resources for Vanda Mimi Palmer and data mining for EST-SSR. Mol Biol Rep 38: 3903–3909. [DOI] [PubMed] [Google Scholar]
- 33. Monteiro F, Sebastiana M, Figueiredo A, Sousa L, Cotrim HC, et al. (2012) Labellum transcriptome reveals alkene biosynthetic genes involved in orchid sexual deception and pollination-induced senescence. Funct Integr Genomics 12: 693–703. [DOI] [PubMed] [Google Scholar]
- 34. Chang Y-Y, Chu Y-W, Chen C-W, Leu W-M, Hsu H-F, et al. (2011) Characterization of Oncidium ‘Gower Ramsey’ transcriptomes using 454 GS-FLX pyrosequencing and their application to the identification of genes associated with flowering time. Plant Cell Physiol 52: 1532–1545. [DOI] [PubMed] [Google Scholar]
- 35. Hsiao Y-Y, Chen Y-W, Huang S-C, Pan Z-J, Fu C-H, et al. (2011) Gene discovery using next-generation pyrosequencing to develop ESTs for Phalaenopsis orchids. BMC Genomics 12: 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shin H, Hirst M, Bainbridge MN, Magrini V, Mardis E, et al. (2008) Transcriptome analysis for Caenorhabditis elegans based on novel expressed sequence tags. BMC Biol 6: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Toth AL, Varala K, Newman TC, Miguez FE, Hutchison SK, et al. (2007) Wasp gene expression supports an evolutionary link between maternal behavior and eusociality. Science 318: 441–444. [DOI] [PubMed] [Google Scholar]
- 38. Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, et al. (2008) Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol Ecol 17: 1636–1647. [DOI] [PubMed] [Google Scholar]
- 39. Wheat CW (2010) Rapidly developing functional genomics in ecological model systems via 454 transcriptome sequencing. Genetica 138: 433–451. [DOI] [PubMed] [Google Scholar]
- 40. Leitch IJ, Kahandawala I, Suda J, Hanson L, Ingrouille MJ, et al. (2009) Genome size diversity in orchids: consequences and evolution. Ann Bot 104: 469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sabidó E, Selevsek N, Aebersold R (2011) Mass spectrometry-based proteomics for systems biology. Curr Opin Biotechnol 23: 1–7. [DOI] [PubMed] [Google Scholar]
- 42. He R, Kim M-J, Nelson W, Balbuena TS, Kim R, et al. (2012) Next-generation sequencing-based transcriptomic and proteomic analysis of the common reed, Phragmites australis (Poaceae), reveals genes involved in invasiveness and rhizome specificity. Am J Bot 99: 232–247. [DOI] [PubMed] [Google Scholar]
- 43. Xu X, Pan SK, Cheng SF, Zhang B, Mu DS, et al. (2011) Genome sequence and analysis of the tuber crop potato. Nature 475: 189–194. [DOI] [PubMed] [Google Scholar]
- 44. Vizcaino JA, Cote R, Reisinger F, Barsnes H, Foster JM, et al. (2010) The proteomics identifications database: 2010 update. Nucleic Acids Res 38: D736–D742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ruepp A, Zollner A, Maier D, Albermann K, Hani J, et al. (2004) The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res 32: 5539–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Desgagné-Penix I, Khan MF, Schriemer DC, Cram D, Nowak J, et al. (2010) Integration of deep transcriptome and proteome analyses reveals the components of alkaloid metabolism in opium poppy cell cultures. BMC Plant Biol 10: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adamidi C, Wang Y, Gruen D, Mastrobuoni G, You X, et al. (2011) De novo assembly and validation of planaria transcriptome by massive parallel sequencing and shotgun proteomics. Genome Res 21: 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang H, Zhang H, Wong YH, Voolstra C, Ravasi T, et al. (2010) Rapid transcriptome and proteome profiling of a non-model marine invertebrate, Bugula neritina . Proteomics 10: 2972–2981. [DOI] [PubMed] [Google Scholar]
- 49. Pruitt KD, Tatusova T, Maglott DR (2007) NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35: D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. UniProt Consortium (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res 40: D71–D75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mittapalli O, Bai X, Mamidala P, Rajarapu SP, Bonello P, et al. (2010) Tissue-specific transcriptomics of the exotic invasive insect pest emerald ash borer (Agrilus planipennis). PLoS ONE 5: e13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang H, Carlson JE, Leebens-Mack JH, Wall P, Mueller L, et al. (2008) An EST database for Liriodendron tulipifera L. floral buds: the first EST resource for functional and comparative genomics in Liriodendron . Tree Genet Genomes 4: 419–433. [Google Scholar]
- 53. Wang J-PZ, Lindsay BG, Leebens-Mack J, Cui L, Wall K, et al. (2004) EST clustering error evaluation and correction. Bioinformatics 20: 2973–2984. [DOI] [PubMed] [Google Scholar]
- 54. Hoffman JI (2011) Gene discovery in the Antarctic fur seal (Arctocephalus gazella) skin transcriptome. Mol Ecol Resour 11: 703–710. [DOI] [PubMed] [Google Scholar]
- 55. Tatusov R, Fedorova N, Jackson J, Jacobs A, Kiryutin B, et al. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Manning G, Plowman GD, Hunter T, Sudarsanam S (2002) Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci 27: 514–520. [DOI] [PubMed] [Google Scholar]
- 58. Wang L-L, Zhou Z-Y (2009) RNA recognition motif (RRM)-containing proteins in Bombyx mori . Afr J Biotechnol 8: 1121–1126. [Google Scholar]
- 59. Andrés C, Lurin C, Small ID (2007) The multifarious roles of PPR proteins in plant mitochondrial gene expression. Physiol Plant 129: 14–22. [Google Scholar]
- 60. Schlüter PM, Ruas PM, Kohl G, Ruas CF, Stuessy TF, et al. (2009) Genetic patterns and pollination in Ophrys iricolor and O. mesaritica (Orchidaceae): sympatric evolution by pollinator shift. Bot J Linn Soc 159: 583–598. [Google Scholar]
- 61. Kachroo A, Shanklin J, Whittle E, Lapchyk L, Hildebrand D, et al. (2007) The Arabidopsis stearoyl-acyl carrier protein-desaturase family and the contribution of leaf isoforms to oleic acid synthesis. Plant Mol Biol 63: 257–271. [DOI] [PubMed] [Google Scholar]
- 62. Kunst L, Samuels L (2009) Plant cuticles shine: advances in wax biosynthesis and export. Curr Opin Plant Biol 12: 721–727. [DOI] [PubMed] [Google Scholar]
- 63. Blacklock BJ, Jaworski JG (2006) Substrate specificity of Arabidopsis 3-ketoacyl-CoA synthases. Biochem Biophys Res Commun 346: 583–590. [DOI] [PubMed] [Google Scholar]
- 64. Dunn TM, Lynch DV, Michaelson LV, Napier JA (2004) A post-genomic approach to understanding sphingolipid metabolism in Arabidopsis thaliana . Ann Bot 93: 483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lechelt-Kunze C, Meissner RC, Drewes M, Tietjen K (2003) Flufenacet herbicide treatment phenocopies the fiddlehead mutant in Arabidopsis thaliana . Pest Manag Sci 59: 847–856. [DOI] [PubMed] [Google Scholar]
- 66. Holton TA, Cornish EC (1995) Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell 7: 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Winkel-Shirley B (2001) Flavonoid biosynthesis. a colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol 126: 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kuehnle AR, Lewis DH, Markham KR, Mitchell KA, Davies KM, et al. (1997) Floral flavonoids and pH in Dendrobium orchid species and hybrids. Euphytica 95: 187–194. [Google Scholar]
- 69. Hieber AD, Mudalige-Jayawickrama RG, Kuehnle AR (2006) Color genes in the orchid Oncidium Gower Ramsey: identification, expression, and potential genetic instability in an interspecific cross. Planta 223: 521–531. [DOI] [PubMed] [Google Scholar]
- 70. Grotewold E (2006) The genetics and biochemistry of floral pigments. Annu Rev Plant Biol 57: 761–780. [DOI] [PubMed] [Google Scholar]
- 71.Rausher MD (2006) The evolution of flavonoids and their genes. In: Grotewold E, editor. The science of flavonoids. New York, USA: Springer. 175–212.
- 72. Falginella L, Castellarin SD, Testolin R, Gambetta GA, Morgante M, et al. (2010) Expansion and subfunctionalisation of flavonoid 3′,5′-hydroxylases in the grapevine lineage. BMC Genomics 11: 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vignolini S, Davey MP, Bateman RM, Rudall PJ, Moyroud E, et al. (2012) The mirror crack’d: both pigment and structure contribute to the glossy blue appearance of the mirror orchid, Ophrys speculum . New Phytol 196: 1038–1047. [DOI] [PubMed] [Google Scholar]
- 74. Singh KB, Foley RC, Oñate-Sánchez L (2002) Transcription factors in plant defense and stress responses. Curr Opin Plant Biol 5: 430–436. [DOI] [PubMed] [Google Scholar]
- 75. Zhang H, Jin J, Tang L, Zhao Y, Gu X, et al. (2011) PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res 39: D1114–D1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gramzow L, Theißen G (2010) A hitchhiker’s guide to the MADS world of plants. Genome Biol 11: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lalusin AG, Nishita K, Kim S-H, Ohta M, Fujimura T (2006) A new MADS-box gene (IbMADS10) from sweet potato (Ipomoea batatas (L.) Lam) is involved in the accumulation of anthocyanin. Mol Genet Genomics 275: 44–54. [DOI] [PubMed] [Google Scholar]
- 78. Schwarz-Sommer Z, Huijser P, Nacken W, Saedler H, Sommer H (1990) Genetic control of flower development by homeotic genes in Antirrhinum majus . Science 250: 931–936. [DOI] [PubMed] [Google Scholar]
- 79. Theißen G, Saedler H (2001) Plant biology: Floral quartets. Nature 409: 469–471. [DOI] [PubMed] [Google Scholar]
- 80. Mondragón-Palomino M, Theißen G (2009) Why are orchid flowers so diverse? Reduction of evolutionary constraints by paralogues of class B floral homeotic genes. Ann Bot 104: 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mondragón-Palomino M, Theißen G (2008) MADS about the evolution of orchid flowers. Trends Plant Sci 13: 51–59. [DOI] [PubMed] [Google Scholar]
- 82. Feller A, Machemer K, Braun EL, Grotewold E (2011) Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J 66: 94–116. [DOI] [PubMed] [Google Scholar]
- 83. Borevitz JO, Xia Y, Blount J, Dixon RA, Lamb C (2000) Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell 12: 2383–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jin H, Cominelli E, Bailey P, Parr A, Mehrtens F, et al. (2000) Transcriptional repression by AtMYB4 controls production of UV-protecting sunscreens in Arabidopsis . EMBO J 19: 6150–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nesi N, Jond C, Debeaujon I, Caboche M, Lepiniec L (2001) The Arabidopsis TT2 gene encodes an R2R3 MYB domain protein that acts as a key determinant for proanthocyanidin accumulation in developing seed. Plant Cell 13: 2099–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Baudry A, Heim MA, Dubreucq B, Caboche M, Weisshaar B, et al. (2004) TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana . Plant J 39: 366–380. [DOI] [PubMed] [Google Scholar]
- 87. Lee MM (1999) Schiefelbein J (1999) WEREWOLF, a MYB-related protein in Arabidopsis, is a position-dependent regulator of epidermal cell patterning. Cell 99: 473–483. [DOI] [PubMed] [Google Scholar]
- 88. Lee MM (2001) Schiefelbein J (2001) Developmentally distinct MYB genes encode functionally equivalent proteins in Arabidopsis . Development 128: 1539–1546. [DOI] [PubMed] [Google Scholar]
- 89. Higginson T, Li SF, Parish RW (2003) AtMYB103 regulates tapetum and trichome development in Arabidopsis thaliana . Plant J 35: 177–192. [DOI] [PubMed] [Google Scholar]
- 90. Ito M, Araki S, Matsunaga S, Itoh T, Nishihama R, et al. (2001) G2/M-phase-specific transcription during the plant cell cycle is mediated by c-Myb-like transcription factors. Plant Cell 13: 1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Araki S, Ito M, Soyano T, Nishihama R, Machida Y (2004) Mitotic cyclins stimulate the activity of c-Myb-like factors for transactivation of G2/M phase-specific genes in tobacco. J Biol Chem 279: 32979–32988. [DOI] [PubMed] [Google Scholar]
- 92. Penfield S, Meissner RC, Shoue DA, Carpita NC, Bevan MW (2001) MYB61 is required for mucilage deposition and extrusion in the Arabidopsis seed coat. Plant Cell 13: 2777–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schmitz G, Tillmann E, Carriero F, Fiore C, Cellini F, et al. (2002) The tomato Blind gene encodes a MYB transcription factor that controls the formation of lateral meristems. Proc Natl Acad Sci USA 99: 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shin B, Choi G, Yi H, Yang S, Cho I, et al. (2002) AtMYB21, a gene encoding a flower-specific transcription factor, is regulated by COP1. Plant J 30: 23–32. [DOI] [PubMed] [Google Scholar]
- 95. Steiner-Lange S, Unte US, Eckstein L, Yang C, Wilson ZA, et al. (2003) Disruption of Arabidopsis thaliana MYB26 results in male sterility due to non-dehiscent anthers. Plant J 34: 519–528. [DOI] [PubMed] [Google Scholar]
- 96. Yang Y, Klessig DF (1996) Isolation and characterization of a tobacco mosaic virus-inducible myb oncogene homolog from tobacco. Proc Natl Acad Sci USA 93: 14972–14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Abe H, Yamaguchi-Shinozaki K, Urao T, Iwasaki T, Hosokawa D, et al. (1997) Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid-regulated gene expression. Plant Cell 9: 1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Geri C, Cecchini E, Giannakou ME, Covey SN, Milner JJ (1999) Altered patterns of gene expression in Arabidopsis elicited by cauliflower mosaic virus (CaMV) infection and by a CaMV gene VI transgene. Mol Plant Microbe Interact 12: 377–384. [DOI] [PubMed] [Google Scholar]
- 99. Sugimoto K, Takeda S, Hirochika H (2000) MYB-related transcription factor NtMYB2 induced by wounding and elicitors is a regulator of the tobacco retrotransposon Tto1 and defense-related genes. Plant Cell 12: 2511–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gocal GFW, Sheldon CC, Gubler F, Moritz T, Bagnall DJ, et al. (2001) GAMYB-like genes, flowering, and gibberellin signaling in Arabidopsis . Plant Physiol 127: 1682–1693. [PMC free article] [PubMed] [Google Scholar]
- 101. Seo HS, Yang J-Y, Ishikawa M, Bolle C, Ballesteros ML, et al. (2003) LAF1 ubiquitination by COP1 controls photomorphogenesis and is stimulated by SPA1. Nature 423: 995–999. [DOI] [PubMed] [Google Scholar]
- 102. Newman LJ, Perazza DE, Juda L, Campbell MM (2004) Involvement of the R2R3-MYB, AtMYB61, in the ectopic lignification and dark-photomorphogenic components of the det3 mutant phenotype. Plant J 37: 239–250. [DOI] [PubMed] [Google Scholar]
- 103. Raffaele S, Vailleau F, Leger A, Joubes J, Miersch O, et al. (2008) A MYB transcription factor regulates very-long-chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis . Plant Cell 20: 752–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Davies KM, Albert NW, Schwinn KE (2012) From landing lights to mimicry: the molecular regulation of flower colouration and mechanisms for pigmentation patterning. Funct Plant Biol 39: 619–638. [DOI] [PubMed] [Google Scholar]
- 105. Gonzalez A, Zhao M, Leavitt JM, Lloyd AM (2008) Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J 53: 814–827. [DOI] [PubMed] [Google Scholar]
- 106. Comba L, Corbet SA, Hunt H, Outram S, Parker JS, et al. (2000) The role of genes influencing the corolla in pollination of Antirrhinum majus . Plant Cell Environ 23: 639–647. [Google Scholar]
- 107. Glover BJ, Martin C (1998) The role of petal cell shape and pigmentation in pollination success in Antirrhinum majus . Heredity 80: 778–784. [Google Scholar]
- 108. Kay QON, Daoud HS, Stirton CH (1981) Pigment distribution, light reflection and cell structure in petals. Bot J Linn Soc 83: 57–84. [Google Scholar]
- 109. Noda K, Glover BJ, Linstead P, Martin C (1994) Flower colour intensity depends on specialized cell shape controlled by a Myb-related transcription factor. Nature 369: 661–664. [DOI] [PubMed] [Google Scholar]
- 110. Martin C, Bhatt K, Baumann K, Jin H, Zachgo S, et al. (2002) The mechanics of cell fate determination in petals. Philos Trans R Soc Lond B Biol Sci 357: 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Perez-Rodriguez M, Jaffe FW, Butelli E, Glover BJ, Martin C (2005) Development of three different cell types is associated with the activity of a specific MYB transcription factor in the ventral petal of Antirrhinum majus flowers. Development 132: 359–370. [DOI] [PubMed] [Google Scholar]
- 112. Inda LA, Pimentel M, Chase MW (2012) Phylogenetics of tribe Orchideae (Orchidaceae: Orchidoideae) based on combined DNA matrices: inferences regarding timing of diversification and evolution of pollination syndromes. Ann Bot 110: 71–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schiestl FP, Cozzolino S (2008) Evolution of sexual mimicry in the orchid subtribe orchidinae: the role of preadaptations in the attraction of male bees as pollinators. BMC Evol Biol 8. [DOI] [PMC free article] [PubMed]
- 114. Paun OG, Bateman RM, Fay MF, Luna JA, Moat J, et al. (2011) Altered gene expression and ecological divergence in sibling allopolyploids of Dactylorhiza (Orchidaceae). BMC Evol Biol 11: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liu X-J, Chuang Y-N, Chiou C-Y, Chin D-C, Shen F-Q, et al. (2012) Methylation effect on chalcone synthase gene expression determines anthocyanin pigmentation in floral tissues of two Oncidium orchid cultivars. Planta 236: 401–409. [DOI] [PubMed] [Google Scholar]
- 116. Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8: 186–194. [PubMed] [Google Scholar]
- 117. Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7: 203–214. [DOI] [PubMed] [Google Scholar]
- 118. Li R, Zhu H, Ruan J, Qian W, Fang X, et al. (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Pertea G, Huang X, Liang F, Antonescu V, Sultana R, et al. (2003) TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19: 651–652. [DOI] [PubMed] [Google Scholar]
- 120.Schatz MC, Phillippy AM, Sommer DD, Delcher AL, Puiu D, et al.. (2011) Hawkeye and AMOS: visualizing and assessing the quality of genome assemblies. Brief Bioinform: doi: 10.1093/bib/bbr1074. [DOI] [PMC free article] [PubMed]
- 121. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- 123. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, et al. (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Iseli C, Jongeneel CV, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol: 138–148. [PubMed]
- 125.Coggill P, Finn RD, Bateman A (2008) Identifying protein domains with the Pfam database. Curr Protoc Bioinformatics 23: Unit 2.5. [DOI] [PubMed]
- 126. Finn RD, Mistry J, Tate J, Coggill P, Heger A, et al. (2010) The Pfam protein families database. Nucleic Acids Res 38: D211–D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Langmead B, Trapnell C, Pop M, Salzberg S (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Grobei MA, Qeli E, Brunner E, Rehrauer H, Zhang R, et al. (2009) Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function. Genome Res 19: 1786–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Bairoch A, Apweiler R (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28: 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, et al. (2012) The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res 40: D1202–D1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.R Development Core Team (2008) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- 133. Keller A, Purvine S, Nesvizhskii AI, Stolyar S, Goodlett DR, et al. (2002) Experimental protein mixture for validating tandem mass spectral analysis. OMICS 6: 207–212. [DOI] [PubMed] [Google Scholar]
- 134. Nesvizhskii AI, Keller A, Kolker E, Aebersold R (2003) A Statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75: 4646–4658. [DOI] [PubMed] [Google Scholar]
- 135. Kim S-Y, Yun P-Y, Fukuda T, Ochiai T, Yokoyama J, et al. (2007) Expression of a DEFICIENS-like gene correlates with the differentiation between sepal and petal in the orchid, Habenaria radiata (Orchidaceae). Plant Sci 172: 319–326. [Google Scholar]
- 136. Cantone C, Gaudio L, Aceto S (2011) The PI/GLO-like locus in orchids: Duplication and purifying selection at synonymous sites within Orchidinae (Orchidaceae). Gene 481: 48–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of Ophrys flowers. Five flowers each from different individuals of (A) O. exaltata subsp. archipelagi, (B) O. sphegodes and (C) O. garganica, showing inter-species and intra-species variation (plants all from Gargano, Southern Italy). Images were scaled for comparison, the white bar indicating 1 cm. O. sphgodes tends to have comparatively small flowers with a brown labellum and a greenish perigon, whereas O. garganica flowers are usually larger, with a darker labellum and sometimes coloured petals. O. exaltata tends to have comparatively large flowers with a slightly elongated brown labellum, typically with a protrusion at its apex, and usually a white perigon. The speculum (brighter, more reflective part of the labellum) can be quite variable in all species, and they all have longer trichomes at the sides of the labellum (‘hairy margin’) as compared to its centre. Micromorphological features of Ophrys flowers are described elsewhere [15].
(PDF)
Transcript length/annotation relationship. Plot showing the percentage of hits with annotation information from NCBI nr and UniProt databases versus sequence length.
(PDF)
Summary of sequencing data. This table lists details of the 454, Sanger and Solexa sequence data sets obtained in this study.
(DOCX)
List of orchid proteins. This table lists all proteins matching sequences in the Ophrys transcriptome by their transcriptome sequence ID. In the column HQ, an asterisk (*) denotes proteins in the HQ data set (no asterisk indicates PD data set). The ‘Species’ column indicates in which orchid species a given protein was found, where: E, O. exaltata; G, O. garganica; S, O. sphegodes. In the column ‘Orchid-only’, y/n (yes/no) indicate if a given protein was found only in the Ophrys transcriptome (y) or if it also matched proteins in the TAIR and/or SwissProt databases (n). The ‘Description’ column refers to the term provided in the transcript ID’s best BLASTX/nr database hit.
(XLSX)
List of proteins without transcript. This table lists all proteins that were identified in the SwissProt/TAIR9 databases, but did not match any sequence in the Ophrys transcriptome, listing the protein description, source database and associated accession numbers. In the column HQ, an asterisk (*) denotes proteins in the HQ data set (no asterisk indicates PD data set). The ‘Species’ column indicates in which orchid species a given protein was found, where: E, O. exaltata; G, O. garganica; S, O. sphegodes.
(XLSX)
GO classification. This table contains the GO classification of the assembled sequences into three main categories (biological function, cellular component and molecular function) at all levels (1–12). ‘All species’ lists the number of transcripts in a given category in the Ophrys reference transcriptome, and additional columns list the corresponding number of transcripts for the individual species separately.
(XLS)
Pfam protein domains. This table lists the Pfam annotations in the Ophrys reference transcriptome by transcript ID.
(XLSX)
List of candidate genes. Details of transcripts encoding candidate biosynthetic proteins putatively involved in hydrocarbon and anthocyanin biosynthesis, sorted alphabetically per category. TAIR ID lists the Arabidopsis gene for which homologues were found; EC is the enzyme commission number searched for a given candidate protein; ‘N transcripts’ lists the number of unique transcripts matching a candidate protein category in the Ophrys reference transcriptome (counting singleton reads as transcripts); ‘N gene models’ is composed of the number of isogroups from sequence assembly plus transcripts that were not assigned to isogroups (so that for instance, singleton reads would be counted as a new gene model); ‘Unique transcripts’ lists the unique transcriptome sequence IDs. Species with transcripts and peptides list in which Ophrys species transcripts or peptides were found, respectively, where: E, O. exaltata; G, O. garganica; S, O. sphegodes. In the column ‘Species with peptides’, an asterisk indicates that a protein was part of the HQ data set (no asterisk, PD data set). The ordering of elements in the two rightmost columns corresponds to the ordering in the ‘Unique transcripts’ column.
(DOCX)
List of transcription factors. This table lists the identification method (TFDB, transcription factor database; KOG, KOG annotation) for TF identification, along with the unique transcript ID and annotation details (accession number or KOG term). The column ‘Transcription Factor’ lists the TF class identified from the database or the KOG description term, as appropriate. Species with transcripts or peptides for a given TF are listed, where: E, O. exaltata; G, O. garganica; S, O. sphegodes. Proteins identified in the HQ data set are marked with an asterisk. The Comments column provides further information on TFs of special interest. In addition to the TFs listed in this table, one additional transcript (transcript ID 3673) was found to have significant BLAST homology (e-value 6E–12) to a TF of interest, namely Antirrhinum majus AmMYBML1-like (AY661653.1).
(XLSX)
BLAST results for DEF/AP3 and GLO/PI MADS-box gene homologues. Table of BLASTN results, showing top 10 hits against the NCBI nr database (sorted by e-value) for each putative Ophrys B-class MADS-box gene homologue identified in the Ophrys reference transcriptome. The column ‘Lineage’ lists the gene lineage (DEF clades 1, 2, 3 and 4; Orchidoideae GLO1 and GLO2) assigned to the accession number of a BLAST hit, as defined by Mondragón-Palomino & Theißen (2008; [81]) for DEF, and Kim & al. (2007; [135]) and Cantone & al. (2011; [136]) for GLO. The best lineage assignment is highlighted in bold for each transcript. Accession numbers with defined lineage are as follows: DEF clade 1: AY378149, DQ119838, DQ683575, FJ804097, FJ804106, FJ804115; clade 2: AY196350, AY378148, FJ804098, FJ804105, FJ804111; clade 3: AY378150, AB232663, DQ119839, FJ804099, FJ804107, FJ804110, FJ804117; clade 4: AY378147, FJ804108, FJ804112, FJ804116; GLO1: AB232665, AB450305, AB450307, AB450310, AB450302, AB450308, AB450309, AB450299, AB450303, AB450306; GLO2: AB232664, AB537512, AB537507, AB537511, AB537513, AB537509, AB537508, AB537506, AB537504, AB537510.
(XLSX)
This zip-compressed file contains (1) the assembled Ophrys reference transcriptome (FASTA), along with (2) an MD5 check-sum, (3) an FAI index file, (4) 454 isotig to isogroup (transcript to gene) mapping (tab-delimited text), and (5) a text file describing the naming scheme for the transcript identifiers used.
(ZIP)
This spreadsheet document contains the annotation information for the Ophrys reference transcriptome in several tables.
(XLSX)