

Abstract
Background
More than half of the cells in the brain are glia and yet the impact of general anaesthetics on these cells is largely unexamined. We hypothesized that astroglia, which are strongly implicated in neuronal well-being and synapse formation and function, are vulnerable to adverse effects of isoflurane.
Methods
Cultured rat astrocytes were treated with 1.4% isoflurane in air or air alone for 4 h. Viability, proliferation, and cytoskeleton were assessed by colorimetric assay, immunocytochemistry, or a migration assay at the end of treatment or 2 days later. Also, primary rat cortical neurones were treated for 4 days with conditioned medium from control [astrocyte-conditioned media (ACM)], or isoflurane-exposed astrocytes (Iso-ACM) and synaptic puncta were assessed by synapsin 1 and PSD-95 immunostaining.
Results
By several measures, isoflurane did not kill astrocytes. Nor, based on incorporation of a thymidine analogue, did it inhibit proliferation. Isoflurane had no effect on F-actin but reduced expression of α-tubulin and glial fibrillary acidic protein both during exposure (P<0.05 and P<0.001, respectively) and 2 days later (P<0.01), but did not impair astrocyte motility. ACM increased formation of PSD-95 but not synapsin 1 positive puncta in neuronal cultures, and Iso-ACM was equally effective.
Conclusions
Isoflurane decreased expression of microtubule and intermediate filament proteins in astrocytes in vitro, but did not affect their viability, proliferation, motility, and ability to support synapses.
Keywords: astrocytes, isoflurane
Editor's key points.
General anaesthetics can have toxic effects on neurones, but their effects on glia are unclear.
Isoflurane reduced astrocyte cytoskeletal protein expression, but did not reduce their survival, proliferation, or motility.
The neurotoxic effects of isoflurane are unlikely due to disruption of astrocytes, which are not seriously impaired despite their many shared signalling mechanisms with neurones.
There is growing evidence from rodents and primates that exposure to commonly used general anaesthetics can lead to structural and functional changes in the brain. During key periods of brain development these effects include apoptotic neurodegeneration, synapse loss or gain, decreased neurogenesis, and cognitive and behavioural deficits that last into adulthood.1–8 Likewise, in adult or senescent animals general anaesthetic exposure can result in altered mitochondrial energetics, activation of pro-apoptotic caspases, and long-lasting learning deficits.9–11
In these situations, the target of the anaesthetics is presumed to be neurones. However, glia outnumber neurones in the brain and are vital for its integrity and function.12,13 Astroglia in particular play major metabolic, structural, and functional roles in supporting neurones and synapses and protecting them from injury12,14,15 and astrocyte dysfunction is implicated in a variety of neurologic and neurodevelopmental disorders, including those marked by learning and memory defects.16,17 Nonetheless, the impact of general anaesthetics on astrocytes is largely unexamined. Most general anaesthetics are γ-amminobutyric acid (GABA) or N-methyl-d-aspartate (NMDA) receptor modulators or both,18 and astrocytes express abundant GABA and NMDA receptors, which have properties similar to those of neurones.16,19 Indeed, isoflurane produces more profound suppression of astrocyte than neural activity in the adult rat brain in vivo.20 Accordingly, we hypothesized that adverse effects of these agents on the central nervous system (CNS) might reflect astrocyte dysfunction. We tested this possibility in vitro by investigating the impact of the commonly used general anaesthetic agent isoflurane on astrocyte viability and proliferation, motility and cytoskeleton, and ability to support synapses.
Methods
The experimental protocol was approved by the Harvard Medical Area Standing Committee on Animals. Astrocytes were exposed in vitro to 1.4% isoflurane in air or air alone for 4 h and assayed at the conclusion of exposure or 2 days later. We have described most of the procedures and methods elsewhere.21
Astrocyte harvest and culture
Pregnant female rats (Harlan Sprague Dawley, Indianapolis, IN, USA) were killed on embryonic day 18 by CO2 intoxication, the embryos harvested and placed in ice-cold phosphate-buffered saline plus penicillin-streptomycin [(PBS+), Invitrogen, Carlsbad, CA, USA] and the cerebral cortex removed. The cortices were washed three times by centrifugation in PBS+ and then incubated in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) with 4% papain (Worthington Biochemical Corporation, Lakewood, NJ, USA), Dispase II (Roche Diagnostics, Indianapolis, IN, USA) and Dnase 1 (Recominbant Rnase-free Dnase 1, Roche Diagnostics). Samples were triturated, washed and then suspended in DMEM/fetal bovine serum (FBS) [Advanced DMEM with glutamine and penicillin-streptomycin (Invitrogen)]. Five million cells were plated in tissue culture flasks (BD Biosciences, Bedford, MA, USA) containing DMEM/FBS and placed in a humidified cell culture incubator at 37°C with 5% CO2. Five millilitres of fresh media were added to each flask three times per week. When cells reached 75–80% confluence, they were passaged, subjected to a partial media exchange, and re-plated at a density 5×106 cells. At Passage 2 [day in vitro (DIV) 14], cells were plated into 96-well tissue culture plates at 103 cells per well. Matched plates were prepared for each assay from the same batch of cells, and then were placed in the incubator for 2 days before randomization to isoflurane or control treatment. To reduce variability, matched plates were treated, fixed, stained, and imaged at the same time. Compared with the in vivo state, the age of cells cultured under these conditions is indeterminate. However, the cells stain prominently with vimentin, a marker of immature astroglia (data not shown), suggesting they maintain an immature phenotype.
Isoflurane exposure
Matched 96-well plates were placed in identical air-tight, humidified chambers (Billups-Rothenberg, Del Mar, CA, USA) flushed with 21% oxygen—5% CO2—74% nitrogen (control) or the control gas mixed with 1.4% isoflurane. The entire experiment was performed in a room maintained at 37°C and the chambers and gas content-certified canisters (Airgas, Hingham, MA, USA) were temperature equilibrated in the room overnight. Isoflurane, oxygen, and carbon dioxide concentrations were measured every 30 min with an agent analyzer (Ohmeda 5250 RGM, Louisville, CO, USA). Treatment was terminated when the plates were removed from the chambers.
Cell viability
Cell viability and death were assessed at the end of isoflurane exposure or 48 h later by propidium iodide (PI) staining, lactate dehydrogenase (LDH) release, and fluorescence immunocytochemistry for cleaved caspase 3 and cytochrome C.21 Hoechst 33 342, a nuclear stain, was used to determine cell number. PI is a fluorescent molecule that binds DNA; as it is membrane impermeant, PI labels only non-viable cells. LDH is released into the culture medium as cells die and is a measure of necrotic cell death. Translocation of cyctochrome C from the cytoplasm to the nucleus and cleavage and activation of caspase 3 are indices of apoptotic cell death. Cells treated with staurosporine (3 μM), a caspase 3 activator, served as a positive control. LDH was measured in the supernatant with a commercially available colorimetric cytotoxicity detection kit (Roche Applied Science, Mannheim, Germany) and a plate reader (SpectraMax M2, Molecular Devices, Sunnyvale, CA, USA) according to the manufacturer's instructions.21 For immunocytochemistry, cells were fixed with 4% paraformaldehyde in PBS and then incubated overnight with primary antibodies for the apoptotic markers activated caspase 3 (Abcam, Inc., Cambridge, MA, USA; 1:500 dilution) or cytochrome C (Abcam, Inc., Cambridge, MA, USA; 1:650 dilution). After application of a secondary antibody (as appropriate) and washing, Hoechst 33 342 was applied. Cells were washed again and stored in the dark at 4°C until image acquisition and cell number and protein expression were measured with a high-throughput fluorescence microscope, as outlined below.
Cell division
Cell proliferation was assessed during and 48 h after exposure to isoflurane by incorporation of 5-ethynyl-2′-deoxyuridine (EdU) using a commercially available kit [Click-iT™ EdU Alexa Fluor® High-Throughput Imaging (HCS) Assay, Invitrogen] according to the manufacturer's instructions.21 EdU is a thymidine analogue that is incorporated into cells only during S-phase of cell division and hence is used to assess proliferation.22 Briefly, EdU (final concentration 10 μM) was added to 20 wells of a 96-well plate containing cells in DMEM/FBS media either immediately before treatment or 44 h later. Cells were incubated with EdU for 4 h and then fixed with 4% paraformaldehyde in PBS and treated with 0.5% Triton® X-100. After washing, cells were incubated with Click-iT™ reaction cocktail, washed, and blocked with bovine serum albumin in PBS. To identify astrocytes, cells were incubated with an anti-glial fibrillary acidic protein (GFAP) antibody (Millipore, Billerica, MA, USA) and labelled with Hoechst 33 342 to determine cell number. Specimens were stored in the dark at 4°C until image acquisition.
Cytoskeletal protein expression
Control and isoflurane-exposed cells were processed at the end of treatment or 48 h later for immunocytochemistry for the intermediate filament protein GFAP (Millipore; 1:500 dilution); the microtubule protein α-tubulin (Abcam, Inc.; 1:1000 dilution); and Alexa Fluor® 555 phalloidin (Invitrogen, Molecular Probes, Eugene, OR, USA) for F-actin. Cells were fixed and processed as described above and stored in the dark at 4°C until image acquisition. Changes in GFAP were confirmed by reverse transcription polymerase chain reaction (RT-PCR) in three plates (106 cells per plate) per treatment condition using a Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA, USA) for RNA isolation and a NanoDrop ND-1000 (NanoDrop Products, Wilmington, DE, USA) for measurement, as described previously.23 Because of the differences noted in some of the cytoskeletal proteins, post hoc analysis was performed to evaluate the size of the astrocytes and the distribution of F-actin and α-tubulin both at the end and 48 h after treatment with isoflurane.
Motility assay
Astrocytes were plated into 10 wells of two identical 24-well tissue culture treated plates at Passage 2 at a density of 106 cells per well. Once the cells reached confluence a longitudinal scratch was made in the centre of each well using a 10 μm plastic pipette tip24 and the plates randomly assigned to control and isoflurane treatment groups as described above. Scratch closure was examined over the next 24 h using phase contrast microscopy and IN Cell Analyzer 2000 (GE Healthcare, Piscataway, NJ, USA).21
Imaging and analysis
All images, except those acquired for the motility assay, were acquired and analysed using an IN Cell Analyzer 1000 and associated software (GE Healthcare), as described previously.21 For the motility assay, two phase contrast images were acquired per well at 20× at time 0, 4, 12, 16, and 24 h after treatment using the IN Cell Analyzer 2000. Positioning of the images was standardized for each well such that serial images captured identical areas. Images were stitched together using the ImageJ software (National Institutes of Health, Bethesda, MD, USA) and the unfilled area of the scratch was determined at each time point and normalized to the percentage of the scratch area at time zero. For fluorescence imaging, parameters were set based on a control plate stained with the fluorophor or antibody of interest and the parameters used to image an entire set of matched control and isoflurane-treated plates. Once parameters were set, images were acquired automatically, meaning there was no cell selection bias. In these experiments, 12 images were acquired per well at a 20× objective from 20 wells per treatment condition per assay per time (n=20) in matched control and isoflurane-treated plates. Based on control experiments with staurosporine, a PI or CC3 positive cell was defined as one with a nuclear/cellular intensity ratio >2 or 3, respectively. The nuclear intensity threshold for an EdU positive cell was defined by the intensity of EdU positive staining in matched control cells. Cytochrome C translocation was defined by the ratio of its nuclear to cellular intensity in the control cells, with an increase in this ratio taken as evidence for activation of the apoptotic cascade. For the remaining markers, expression intensity was measured and expressed as a percentage of control. Experiments were completed in duplicate and matched control and isoflurane-treated plates were processed at the same time.
Impact of astrocyte-conditioned media on synapses
Astrocytes were harvested, cultured, and exposed to isoflurane 1.4% or control conditions for 4 h as described above. They were then returned to the incubator and left undisturbed for 2 days, at which time the astrocyte-conditioned media (ACM) was removed and concentrated 10-fold using Vivaspin concentrating columns (Sartorius Stedium Biotech, Göttingen, Germany). In parallel, primary rat cortical neurones (Invitrogen) were grown for 10 days on poly-d-lysine/laminin coated coverslips in 24-well tissue culture plates containing Neurobasal media supplemented with penicillin-streptomycin, B27 with vitamin A, l-glutamine, and BDNF (Invitrogen). A half media exchange was performed every other day until DIV 10, at which time coverslips were randomly assigned to incubation with conditioned media from astrocytes exposed to control conditions (ACM), media from astrocytes exposed to isoflurane (Iso-ACM), or Neurobasal media alone (control). This was accomplished by replacing half of the existing culture media with ACM, Iso-ACM, or control media. Neurones were cultured under these conditions until DIV 14 and were then fixed and stained for the presynaptic protein synapsin 1 (Abcam) and the postsynaptic protein PSD-95 (Abcam). A blinded observer imaged three neurones per cover slip (10 coverslips per treatment condition) using a Zeiss Axio Observer Inverted fluorescence microscope under a 63× oil emersion lens. The average numbers of synapsin 1 and PSD-95 puncta per cell per coverslip were counted using ImageJ with the Puncta Analyzer plug in.
Statistics
Data obtained from each assay were analysed using a Student's t-test that compared matched control and isoflurane-treated cells (plates) at each time point. For the motility assay, data from each time point from matched control and isoflurane-treated wells were analysed using a two-way analysis of variance (anova), with treatment and time being the two conditions. In all cases, P<0.05 was considered statistically significant.
Results
Isoflurane produces structural changes in astrocytes
Cultured astrocytes have an intricate, lattice-like cytoskeletal protein network (Fig. 1). To determine whether isoflurane affected astrocyte morphology, we examined expression and localization of the cytoskeletal proteins F-actin, α-tubulin, and GFAP. Based on phalloidin immunostaining, isoflurane did not change expression of F-actin either during or 48 h after exposure (Fig. 1a and b). In contrast, α-tubulin expression decreased 16% during exposure (P<0.001; Fig. 1c) and remained low for 48 h (93% of control, P≤0.01; Fig. 1d–f). Isoflurane likewise decreased expression of GFAP during exposure (84% of control, P<0.001; Fig. 2a) and 48 h later (96% of control, P<0.01; Fig. 2b–d). This was confirmed by RT-PCR, which revealed a 50% decrease in transcription of GFAP during exposure to isoflurane, with recovery to control 48 h later (Fig. 2e and f). To determine if isoflurane caused redistribution of these proteins we performed a post hoc analysis using the intensity spreading function, where a higher number reflects distribution of expression intensity towards the periphery of the cell. By this measure, there was a small but statistically significant redistribution of F-actin towards the periphery both at the end of isoflurane exposure [1 (0.02) vs 1.02 (0.01) in control and isoflurane-treated cells, respectively, P≤0.01] and 48 h later [1 (0.01) vs 1.01 (0.01) in control and isoflurane-treated cells, respectively, P≤0.05] but no such effect on α-tubulin. Because cytoskeletal proteins are important for astrocyte migration, we used a standard scratch assay to examine astrocyte motility. Prior exposure to isoflurane did not hinder scratch closure (Fig. 3), indicating abnormalities in cytoskeletal gene and protein expression did not grossly impair astrocyte mobility.
Fig 1.

Isoflurane had no effect on phalloidin binding, but decreases α-tubulin expression, both during and 48 h after isoflurane treatment. (a) Isoflurane had no effect on phalloidin binding at the end of exposure or (b) 48 h after exposure. In contrast, isoflurane decreased expression of α-tubulin both at the end of exposure (c) and 48 h later (d). (e) Control and (f) isoflurane-treated cells fixed at 48 h after exposure and stained immunohistochemically for α-tubulin [Alexa Fluor® 488 (green) and Hoechst (blue)]. Data expressed as a percentage of control values [mean (sd), n=20 wells per treatment condition], **P<0.01, ***P<0.001.
Fig 2.

Isoflurane altered GFAP expression both during and 48 h after treatment. GFAP, a marker of astrocytes, is strongly expressed in control astrocyte cultures. (a) Isoflurane led to a decrease in GFAP expression both during treatment and (b) 48 h later. (c) GFAP image 48 h after exposure from control plate. (d) GFAP image 48 h after anaesthesia from isoflurane-treated plate. Data expressed as a percentage of control values [mean (sd), n=20 wells per treatment condition]; GFAP Alexa Fluor® 488 (green), Hoechst (blue). Isoflurane also led to a decrease in GFAP gene expression as measured by RT-PCR at the end of treatment (e) but this had resolved by 48 h later (f) (n=3, per treatment condition), *P<0.05, **P<0.01, ***P<0.001.
Fig 3.

Isoflurane had no effect on the wound assay. Astrocytes were exposed to control conditions or 1.4% isoflurane for 4 h after a scratch was placed with a 10 μl plastic pipette tip. The area of the wound was measured at the end of treatment (T=0, Fig. 5b), 4 h after treatment (T=4), 12 h after treatment (T=12, Fig. 5c), 16 h after treatment (T=16), and 24 h after treatment (T=24, Fig. 5d), and normalized to T=0 controls. There was no effect of isoflurane on the migration of astrocytes into the wound. Data were analysed with a two-way anova with treatment condition and time being the two variables. There was a significant effect of time P<0.001 but no effect of treatment condition (P=0.80).
Isoflurane has no effect on astrocyte viability or proliferation
Virtually all of the cells [98 (2%)] in our cultures were astrocytes as determined by immunopositivity for GFAP. Less than 1% of cells stained positive for PI or cleaved caspase 3 under control conditions, indicating the cultures were healthy. Staurosporin killed cells as revealed by more PI [1.9 (3.0) %] and cleaved caspase positive cells [46.7 (13.1) %]. Isoflurane had no effect on PI staining at either time point (Fig. 4a and b). LDH release decreased during exposure to isoflurane but not 48 h after withdrawal (Fig. 4c and d). Similiarly, whereas staurosporine increased translocation of cytochrome C by 216 (16) % compared with control, isoflurane had little or no effect (Fig. 4e and f). Likewise, there were no differences in the percentage of cleaved caspase 3 positive cells at either timepoint in cells exposed to isoflurane compared with control cultures (Fig. 4g and h). To determine the effect of isoflurane on astrocyte proliferation, we performed the EdU proliferation assay. In control cultures, 14.1 (1.5) % and 11 (0.7) % of cells were dividing based on EdU incorporation during treatment and 48 h later, respectively, with the slightly lower rate at 48 h likely reflecting contact inhibition as cells continued to proliferate and plating density increased over time. Compared with the corresponding control, exposure to isoflurane did not affect EdU incorporation either during exposure or 48 h after withdrawal (Fig. 5). Together, these data indicate a clinically relevant dose of isoflurane attenuated natural necrotic cell death but otherwise had no effect on viability or proliferation of cultured astrocytes.
Fig 4.

Isoflurane did not alter astrocyte viability. Astrocytes were exposed to control conditions or 1.4% isoflurane for 4 h. (a, b) Very few cells stained positive for PI under control conditions and isoflurane had no effect on the percentage of astrocytes stained by PI at the end of exposure 48 h later. (c) Isoflurane decreased LDH release in astrocytes during exposure but there were no effects on LDH activity 48 h later (d). There was a small but statistically significant decrease in nuclear translocation of cytochrome C during isoflurane exposure (e) but no effect (f) 48 h later. Similarly, there were very few control or isoflurane-treated cells that stained positive for cleaved caspase 3 either at the end of exposure (g) or 48 h later (h). Taken together these data suggest that isoflurane prevents endogenous astrocyte death during exposure but has no effect on necrotic cell death 48 h later. Data expressed as mean (sd), n=12–20 wells per treatment condition, *P<0.05, **P<0.001.
Fig 5.
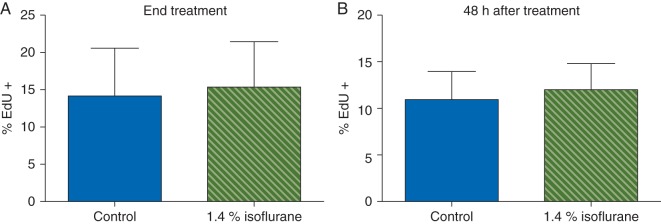
Isoflurane had no effect on EdU incorporation in astrocytes. Isoflurane had no effect on EdU incorporation into astrocytes either at end of exposure (a) or 48 h later (b), suggesting that isoflurane has no effect on the S-phase of cell division in rat astrocytes. Data expressed as a percentage of control values [mean (sd), n=20 wells per treatment condition].
Isoflurane-treated astrocytes support synaptogenesis in vitro
Astrocytes are potent regulators of CNS synaptogenesis via release of synaptogenic factors.13 To address whether isoflurane impairs the synaptogenic capacity of astrocytes, we treated cortical neurones with conditioned medium (ACM) obtained from astrocytes exposed to isoflurane or control conditions, and subsequently assayed the number of synaptic puncta per neurone. As expected from previous studies in retinal ganglion cells,25 cortical neurones treated with control ACM had more PSD-95 positive puncta than those cultured without ACM (P<0.01, Fig. 6a). Neurones treated with ACM from astrocytes that were exposed to isoflurane also had more PSD-95 labelled puncta than those cultured without ACM (P<0.01, Fig. 6a), but ACM whether from control or isoflurane-treated astrocytes had no effect on the number of synapsin 1 puncta (Fig. 6b). This suggests that in this in vitro model isoflurane exposure did not impair the ability of astrocytes to support development and maintenance of synapses and that ACM increased the number of PSD-95 but not synapsin 1 puncta.
Fig 6.
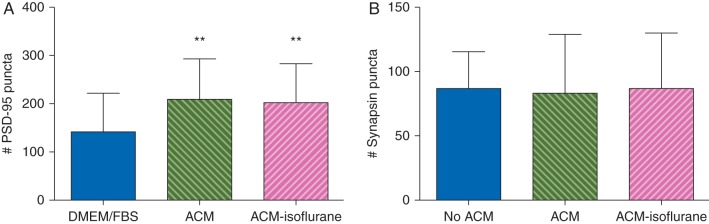
ACM increased the number of PSD-95 but not synapsin 1 positive puncta in cultured astrocytes. Neurones treated with ACM from control astrocytes had more PSD-95 puncta (a) but not synapsin 1 puncta (b) than cells treated with media alone. Isoflurane-ACM was equally effective. n=10 coverslips per treatment condition, three cells imaged per coverslip, **P<0.01.
Discussion
Our findings show that astrocytes in culture are remarkably resistant to toxic effects of isoflurane. At a clinically relevant concentration and duration of exposure, isoflurane neither killed astrocytes nor slowed their division. Although it decreased expression of the microtubule protein α-tubulin and the intermediate filament protein GFAP for at least 2 days, it spared F-actin and did not alter astrocyte motility. Moreover, media collected from isoflurane-exposed astrocytes was as effective as media from control astrocytes in supporting and maintaining synapses as judged by PSD-95 labelling, suggesting the trophic function of astrocytes is intact after isoflurane exposure. If the same is true in vivo, astrocyte elimination or dysfunction is an unlikely mechanism or mediator of the adverse effects of isoflurane on neurones.
Apoptotic neurodegeneration is a prominent feature of isoflurane toxicity in cultured neurones and the developing brain1,8 and astrocytes have many of the same receptors and signalling systems as neurones.19 Thus, we were surprised that isoflurane did not kill astroglia. Similarly, based on studies showing that isoflurane decreases division of neural progenitors in vitro and reduces neurogenesis in the neonatal brain in vivo,6,7,21,26 we expected it to inhibit proliferation of astrocytes. Neither was true under our experimental conditions. Using harsher experimental conditions (isoflurane 3.4% for 24 h) and exposure at an earlier time in vitro (DIV4), others have observed negligible effects of isoflurane on astrocyte viability and growth but reduced proliferation,27 whereas in vivo exposure on postnatal day 14 produced gliosis in adulthood.7 Together, these data suggest responsiveness of astrocytes to isoflurane depends upon developmental phenotype and age, with the agent promoting astrocyte differentiation and proliferation in the intact animal.
The fact that isoflurane decreased α-tubulin and GFAP and redistributed F-actin, albeit weakly, indicates it affects the astrocyte cytoskeleton. This is generally, if not universally,28 concordant with studies in multiple cell types including neurones, where isoflurane produces sustained changes in F-actin rich dendritic spines,5,29,30 and immature astroglia, where it reduces F-actin derived stress fibre formation and expression of RhoA, a small GTPase that regulates the actin and microtubule cytoskeletons.27,31,32 However, we found no change in F-actin expression or astrocyte motility, which is principally an F-actin dependent process, differences we attribute to dissimilar dosing protocols (1.4% for 2 h vs 3.4% for 24 h), measurement methods, and age of the astrocytes (DIV 14–15 vs 4). The changes in α-tubulin and GFAP are intriguing because microtubules and intermediate filaments perform numerous intracellular functions including acting as the scaffold for glutamate transporters and the conduits along which signal transduction molecules, vesicles, and trophic factors move.33,34 Indeed, other studies show that isoflurane and similar compounds can disaggregate microtubules, interfere with intermediate filament assembly and microtubule associated protein binding, and impair glutamate transport,28,35–37 which means isoflurane may alter astroglial functions not tested here.
Astrocytes secrete trophic factors that enhance synaptogenesis and are vital to ensuring the integrity of existing synapses.12,13,25 Our data indicate isoflurane does not interfere with this critical astrocyte function; more PSD-95 labelled synapses were formed in the presence of astrocyte-conditioned medium, as expected, and ACM from astrocytes pretreated with isoflurane was equally effective. Thus, it appears that isoflurane-exposed astrocytes are capable of supporting synapse formation in otherwise normal neurones. This does not necessarily mean that astrocytes protect isoflurane-threatened neurones against synaptotoxicity, as in our model the neurones were not exposed to the anaesthetic.2,30 In fact, in vivo studies confirm that isoflurane produces synapse loss in the neonatal brain,2 suggesting that astrocytes are not enough to prevent the toxicity of this agent.
This work is limited in some important respects. First, in vitro systems do not faithfully replicate in vivo conditions. Plating density affects the concentration of astrocyte secreted trophic substances and designating cells as immature or mature based on days in vitro is not entirely valid. Secondly, astrocytes are morphologically and functionally heterogeneous; astrocytes in culture are likely to differ from those in the brain, just as astrocytes in one brain region differ from those in another. As such, extrapolation of our in vitro data to the brain must be made cautiously. Thirdly, we have examined only one anaesthetic agent at a single concentration. This restricts generalizability of the results, but the conditions closely approximate the clinical situation in terms of dosage and duration of exposure. Fourthly, considering that a single astrocyte typically ensheaths as many as 100 000 synapses in vivo and performs numerous subtle and complex tasks,12,38 our measures of astrocyte function are limited. In particular, the scratch assay cannot detect the minute but biologically important changes in astrocytic process motility that occur in response to synaptic activity. Finally, even minor contamination of the neurone culture with astrocytes could have compensated for deficiencies in ACM from isoflurane-treated astrocytes and rescued the effect.
To understand and mitigate the adverse neural and behavioural effects of general anaesthetics it seems wise to look beyond neurones and synapses to the neuroglia that support and modulate them. Based on recent data and our results, we conclude that in contrast to neurones and neural stem cells, astrocytes are comparatively resistant to isoflurane. This does not preclude the possibility that isoflurane has subtle but meaningful undesirable effects on these cells in vivo but it does suggest that astrocyte dysfunction is not a major factor in the overt adverse effects of this agent on the brain.
Authors’ contributions
D.J.C.: experimental design and development, data analysis, oversight, and manuscript preparation. E.K.C.: experimental design and development, experiment execution, and manuscript preparation. E.K.: experimental execution and manuscript preparation. A.P.: experimental design and development, data analysis, and manuscript preparation. J.D.B.: experimental execution, data analysis, and manuscript preparation. Z.X.: experimental design and development. G.C.: experimental design and development, data analysis, and manuscript preparation.
Funding
This study was supported by National Institutes of Health grants K08 GM077057 (D.J.C.), R01 GM088817 (G.C.), the Milton Fund of Harvard University (D.J.C.), and the Department of Anesthesiology, Perioperative and Pain Medicine, Brigham and Women's Hospital, Boston, MA, USA.
Declaration of interest
E.K. has previously worked for AstraZeneca in Sweden as an assistant in a Quality Assurance department and currently works as a consultant in medical information with AstraZeneca in Sweden, Norway, Finland and Denmark. E.K. has previously worked on product overview for Actavis in Sweden, Norway and Denmark. E.K. has previously worked on product overview for Janssen in Sweden, Norway, Denmark, Finland.
Acknowledgements
The authors thank Beth Stevens, PhD and Allison M. Rosen, PhD candidate, Department of Neurology, Children's Hospital, Harvard Medical School, for their assistance in the development of astrocyte and neurone co-cultures.
References
- 1.Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci, 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Head BP, Patel HH, Niesman IR, Drummond JC, Roth DM, Patel PM. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology. 2009;110:813–25. doi: 10.1097/ALN.0b013e31819b602b. doi:10.1097/ALN.0b013e31819b602b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lunardi N, Ori C, Erisir A, Jevtovic-Todorovic V. General anesthesia causes long-lasting disturbances in the ultrastructural properties of developing synapses in young rats. Neurotox Res. 2010;17:179–88. doi: 10.1007/s12640-009-9088-z. doi:10.1007/s12640-009-9088-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Roo M, Klauser P, Briner A, et al. Anesthetics rapidly promote synaptogenesis during a critical period of brain development. PLoS ONE. 2009;4:e7043. doi: 10.1371/journal.pone.0007043. doi:10.1371/journal.pone.0007043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briner A, De Roo M, Dayer A, Muller D, Habre W, Vutskits L. Volatile anesthetics rapidly increase dendritic spine density in the rat medial prefrontal cortex during synaptogenesis. Anesthesiology. 2010;112:546–56. doi: 10.1097/ALN.0b013e3181cd7942. doi:10.1097/ALN.0b013e3181cd7942. [DOI] [PubMed] [Google Scholar]
- 6.Stratmann G, Sall JW, May LD, et al. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology. 2009;110:834–48. doi: 10.1097/ALN.0b013e31819c463d. doi:10.1097/ALN.0b013e31819c463d. [DOI] [PubMed] [Google Scholar]
- 7.Zhu C, Gao J, Karlsson N, et al. Isoflurane anesthesia induced persistent, progressive memory impairment, caused a loss of neural stem cells, and reduced neurogenesis in young, but not adult, rodents. J Cereb Blood Flow Metab. 2010;30:1017–30. doi: 10.1038/jcbfm.2009.274. doi:10.1038/jcbfm.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brambrink AM, Evers AS, Avidan MS, et al. isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:8. doi: 10.1097/ALN.0b013e3181d049cd. doi:10.1097/ALN.0b013e3181c62a1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Z, Dong Y, Maeda U, et al. The inhalation anesthetic isoflurane induces a vicious cycle of apoptosis and amyloid beta-protein accumulation. J Neurosci. 2007;27:1247–54. doi: 10.1523/JNEUROSCI.5320-06.2007. doi:10.1523/JNEUROSCI.5320-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Culley DJ, Baxter MG, Yukhananov R, Crosby G. Long-term impairment of acquisition of a spatial memory task following isoflurane-nitrous oxide anesthesia in rats. Anesthesiology. 2004;100:309–14. doi: 10.1097/00000542-200402000-00020. doi:10.1097/00000542-200402000-00020. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Xu Z, Wang H, et al. Anesthetics isoflurane and desflurane differently affect mitochondrial function, learning, and memory. Ann Neurol. 2012;71:687–98. doi: 10.1002/ana.23536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron, 2008;60:430–40. doi: 10.1016/j.neuron.2008.10.013. doi:10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 13.Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–31. doi: 10.1038/nature09612. doi:10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen NJ, Barres BA. Neuroscience: glia—more than just brain glue. Nature. 2009;457:675–7. doi: 10.1038/457675a. doi:10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- 15.Wang DD, Bordey A. The astrocyte odyssey. Prog Neurobiol. 2008;86:342–67. doi: 10.1016/j.pneurobio.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–55. doi: 10.1146/annurev-physiol-021909-135843. doi:10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molofsky AV, Krenick R, Ullian E, et al. Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 2012;26:891–907. doi: 10.1101/gad.188326.112. doi:10.1101/gad.188326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemmings H, Jr, Akabas M, Goldstein P, Trudell J, Orser B, Harrison N. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–10. doi: 10.1016/j.tips.2005.08.006. doi:10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Lee M, Schwab C, McGeer PL. Astrocytes are GABAergic cells that modulate microglial activity. Glia. 2011;59:152–65. doi: 10.1002/glia.21087. doi:10.1002/glia.21087. [DOI] [PubMed] [Google Scholar]
- 20.Schummers J, Yu H, Sur M. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science. 2008;320:1638–43. doi: 10.1126/science.1156120. doi:10.1126/science.1156120. [DOI] [PubMed] [Google Scholar]
- 21.Culley DJ, Boyd JD, Palanisamy A, et al. Isoflurane decreases self-renewal capacity of rat cultured neural stem cells. Anesthesiology. 2011;115:754–63. doi: 10.1097/ALN.0b013e318223b78b. doi:10.1097/ALN.0b013e318223b78b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chehrehasa F, Meedeniya AC, Dwyer P, Abrahamsen G, Mackay-Sim A. EdU, a new thymidine analogue for labelling proliferating cells in the nervous system. J Neurosci Methods. 2009;177:122–30. doi: 10.1016/j.jneumeth.2008.10.006. doi:10.1016/j.jneumeth.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 23.Xie Z, Culley DJ, Dong Y, et al. The common inhalation anesthetic isoflurane induces caspase activation and increases amyloid beta-protein level in vivo. Ann Neurol. 2008;64:618–27. doi: 10.1002/ana.21548. doi:10.1002/ana.21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brozzi F, Arcuri C, Giambanco I, Donato R. S100B protein regulates astrocyte shape and migration via Interaction with Src Kinase: implications for astrocyte development, activation, and tumor growth. J Biol Chem. 2009;284:8797–811. doi: 10.1074/jbc.M805897200. doi:10.1074/jbc.M805897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–61. doi: 10.1126/science.291.5504.657. doi:10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- 26.Sall JW, Stratmann G, Leong J, et al. Isoflurane inhibits growth but does not cause cell death in hippocampal neural precursor cells grown in culture. Anesthesiology. 2009;110:826–33. doi: 10.1097/ALN.0b013e31819b62e2. doi:10.1097/ALN.0b013e31819b62e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lunardi N, Hucklenbruch C, Latham JR, Scarpa J, Jevtovic-Todorovic V. Isoflurane impairs immature astroglia development in vitro: the role of actin cytoskeleton. J Neuropathol Exp Neurol. 2011;70:281–91. doi: 10.1097/NEN.0b013e31821284e9. doi:10.1097/NEN.0b013e31821284e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samain E, Bouillier H, Rucker-Martin C, et al. Isoflurane alters angiotensin II-induced Ca2+ mobilization in aortic smooth muscle cells from hypertensive rats: implication of cytoskeleton. Anesthesiology. 2002;97:642–51. doi: 10.1097/00000542-200209000-00019. doi:10.1097/00000542-200209000-00019. [DOI] [PubMed] [Google Scholar]
- 29.Kaech S, Brinkhaus H, Matus A. Volatile anesthetics block actin-based motility in dendritic spines. Proc Natl Acad Sci USA. 1999;96:10433–7. doi: 10.1073/pnas.96.18.10433. doi:10.1073/pnas.96.18.10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemkuil BP, Head BP, Pearn ML, Patel HH, Drummond JC, Patel PM. Isoflurane neurotoxicity is mediated by p75NTR-RhoA activation and actin depolymerization. Anesthesiology. 2011;114:49–57. doi: 10.1097/ALN.0b013e318201dcb3. doi:10.1097/ALN.0b013e318201dcb3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35. doi: 10.1038/nature01148. doi:10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 32.Tas PWL, Gambaryan S, Roewer N. Volatile anesthetics affect the morphology of rat glioma C6 cells via RhoA, ERK, and Akt activation. J Cell Biochem. 2007;102:368–76. doi: 10.1002/jcb.21294. doi:10.1002/jcb.21294. [DOI] [PubMed] [Google Scholar]
- 33.Sullivan SM, Lee A, Björkman ST, et al. Cytoskeletal anchoring of GLAST determines susceptibility to brain damage: an identified role for GFAP. J Biol Chem. 2007;282:29414–23. doi: 10.1074/jbc.M704152200. doi:10.1074/jbc.M704152200. [DOI] [PubMed] [Google Scholar]
- 34.Geraldo S, Gordon-Weeks PR. Cytoskeletal dynamics in growth-cone steering. J Cell Sci. 2009;122:3595–604. doi: 10.1242/jcs.042309. doi:10.1242/jcs.042309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vergara GA, Livingston A. Halothane modifies colchicine binding to tubulin. Pharmacology. 1981;23:264–70. doi: 10.1159/000137559. doi:10.1159/000137559. [DOI] [PubMed] [Google Scholar]
- 36.Hinkley RE. Microtubule-macrotubule transformations induced by volatile anesthetics. Mechanism of macrotubule assembly. J Ultrastruct Res. 1976;57:237–50. doi: 10.1016/s0022-5320(76)80113-x. doi:10.1016/S0022-5320(76)80113-X. [DOI] [PubMed] [Google Scholar]
- 37.Tang JX, Mardini F, Caltagarone BM, et al. Anesthesia in presymptomatic Alzheimer's disease: a study using the triple-transgenic mouse model. Alzheimers Dement. 2011;7:521–31.e1. doi: 10.1016/j.jalz.2010.10.003. doi:10.1016/j.jalz.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]