

Abstract
Hepatitis B virus (HBV) vaccination is recommended for individuals with hepatitis C virus (HCV) infection given their shared risk factors and increased liver-related morbidity and mortality upon super-infection. Vaccine responses in this setting are often blunted, with poor response rates to HBV vaccinations in chronically HCV-infected individuals compared to healthy subjects. In this study, we investigated the role of T cell immunoglobulin mucin domain-3 (Tim-3)-mediated immune regulation in HBV vaccine responses during HCV infection. We found that Tim-3, a marker for T cell exhaustion, was over-expressed on monocytes, leading to a differential regulation of IL-12/IL-23 production with in turn TH17 cell accumulation, in HCV-infected HBV vaccine non-responders compared to HCV-infected HBV vaccine responders or healthy subjects (HS). Importantly, ex vivo blockade of Tim-3 signaling corrected the imbalance of IL-12/IL-23 as well as the IL-17 bias observed in HBV vaccine non-responders during HCV infection. These results suggest that Tim-3-mediated dysregulation of innate to adaptive immune responses is involved in HBV vaccine failure in individuals with chronic HCV infection, raising the possibility that blocking this negative signaling pathway might improve the success rate of HBV immunization in the setting of chronic viral infection.
Keywords: Tim-3, IL-12, IL-23, IL-17, hepatitis B vaccine, hepatitis C infection
Introduction
HCV infection is a common cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma, and is the leading indication for liver transplantation in the United States. Given the shared risk factors for transmission, co-infection of HBV with HCV is quite common and may lead to more significant liver disease, including higher rates and more rapid progression to liver cirrhosis and liver cancer1-5. As such, HBV vaccine is recommended as the primary means to prevent HBV super-infection and its associated increase in morbidity and mortality in HCV-infected individuals. However, vaccine response (seroconversion with a hepatitis B surface antibody titer >10 IU/L) in this setting is often blunted, with poor response rates to a standard course of HBV vaccinations in chronically HCV-infected individuals when compared to healthy populations (40~60% versus 90~95%); this is especially noted in the setting of advanced fibrosis and liver cirrhosis6-8. Our recent data suggests that even in the setting of relatively preserved hepatic function, seroconversion rates in HCV-infected individuals are still much lower than age-matched healthy subjects (53% versus 94%)9. To improve seroconversion rates following HBV immunization, approaches have included different administration routes (intradermal), higher doses of HBV vaccine (40 μg), or adding adjuvants (CPG 7909, levamisole, GM-CSF). These approaches have led to varying degrees of improvement in healthy subjects, but have had limited success in virally-infected individuals, in part due to a lack of information regarding cellular and molecular mechanisms that inhibit immune responses in this setting. Additionally, a poor vaccine response in HCV-infected subjects is also observed following other adult immunizations, including hepatitis A vaccine, influenza vaccine, and pneumococcal vaccine; this phenomenon is also observed in the setting of human immunodeficiency virus (HIV) infection as well as in other immunosuppressed settings, such as organ transplantation, cancer chemotherapy, and hemodialysis, suggesting a possible shared mechanism of vaccine non-response in immunocompromised hosts10-11.
The mechanism for vaccine-induced immune response involves activation of antigen presenting cells (APCs) and expansion of antigen-specific T and B lymphocytes upon encountering antigen. Given the fact that these HBV vaccine non-responders also have poor recall responses to tetanus toxoid or Candida, it has been suggested that HBV vaccine failure may be due to defects in APCs12-13, in HBsAg-reactive T cells14-17, or in accumulation of regulatory T cells18, indicating that broad immune modulation may be occurring in vaccine responses during chronic viral infection. It is well-established that the immune system is precisely regulated by an intricate balance between positive and negative signals to ensure adequate responses against pathogens, and yet prevent over-activation of lymphocytes and thus autoimmunity. Programmed death-1 (PD-1) and T cell immunoglobulin mucin domain-3 (Tim-3) represent such inhibitory pathways to regulate T cell responses and serve as markers for T cell exhaustion during chronic viral infection19-23. We have previously shown that PD-1 is up-regulated on CD4+ T cells, leading to impaired activation of T helper cells with decreased IL-2 production during chronic HCV infection, and this is particularly significant in HCV-infected HBV vaccine non-responders when compared to HBV vaccine responders9. The role of Tim-3-mediated regulation of innate to adaptive immunity during the HBV vaccine response in the setting of HCV infection has yet to be determined.
IL-12, a cytokine composed of p40 and p35 subunits, is essential for linking innate to adaptive immune responses24-26. How IL-12 produced by monocytes endows T and B lymphocyte responses is unclear, but the discovery of IL-23--a heterodimer of IL-12p40 and a unique IL-23p19 subunit with functions that differ from IL-12p35--offers new insights into the role of their balance in the immune responses during infection27-29. More specifically, IL-12 and IL-23 share a common chain, p40, but the production of p35 and p19 in response to infection is differentially regulated and their functions are distinct and often antithetical. IL-12 is involved in the induction or amplification of the TH1 response, whereas IL-23 has been associated with the generation of IL-17-producing T helper cells (TH17), which have been associated with persistent infection and autoimmune disorders24-29.
In this report, we focus on the role of Tim-3-mediated regulation on IL-12/IL-23/IL-17 production in HBV vaccine responses during chronic HCV infection, and demonstrate the involvement of Tim-3-mediated innate to adaptive immune dysregulation in HBV vaccine failure during chronic HCV infection.
Materials and Methods
Subjects
The study protocol was approved by the institutional review board at East Tennessee State University and James H. Quillen VA Medical Center (ETSU/VA IRB, Johnson City, TN). The work described herein was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). A total of 47 HCV-infected subjects with defined responses to HBV vaccine and 16 healthy subjects were recruited for this study. All subjects gave written informed consent for this study. The subjects were divided into three groups of populations: 1) HCV-infected HBV vaccine non-responders (HBV-NR, defined as hepatitis B surface antibody titer <10 IU/L at 1-6 months following a standard course of HBV vaccination, n=22); 2) HCV-infected HBV vaccine responders (HBV-R, defined as hepatitis B surface antibody titer >10 IU/L at 1-6 months following a standard course of HBV vaccination, n=25); 3) healthy subjects who were negative for HBV, HCV, and HIV infection (HS, n=16). All HCV patients have relatively preserved hepatic function and there are no significant difference in ALT and AST levels between HBV-NR and HBV-R; those with end-stage liver diseases or cirrhosis were excluded from this study. HCV genotype and viral load were performed by Lexington VAMC, with no significant differences between HCV-infected, HBV-NR and HBV-R. The majority of the study subjects were male. The mean age of the HCV-infected subjects was comparable to HS.
Cell isolation and culture
Human peripheral blood mononuclear cells (PBMC) were isolated from the peripheral blood of study subjects by Ficoll-density centrifugation with lympho-H (Atlanta biological, Lawrenceville, GA), which were then viably cryopreserved in freezing medium in liquid nitrogen. If indicated, CD14+ T cells were further purified from isolated PBMC by incubation with a magnetic beads-conjugated with anti-CD14 antibody, followed by positive selection per the manufacturer’s instruction (purity > 95%; Miltenyi Biotec., Auburn, CA). The purified cells were cultured as described19.
Flow cytometry
Purified CD14+ monocytes were stimulated by 1 μg/ml of Toll-like receptor (TLR) 4 ligand - lipopolysaccharide (LPS) (BD Pharmingen) and 2.5 μg/ml of TLR 7/8 ligand - R848 (InvivoGen, San Diego, CA) for 6 h, Brefeldin A (BioLegend, San Diego, CA) was added 5h prior to harvest the cells forbidding cytokine secretion. PBMCs were stimulated by 100 ng/ml of phorbol 12-myristate 13-acetate (PMA) and 1 μg/ml of Ionomycin (InvivoGen, San Diego, CA) for 6h, with Brefeldin A added 5h prior to harvest the cells. Specific antibody direct conjugates for cell surface staining were carried out using Tim-3-APC (R&D, Minneapolis, MN), CD4-APC or CD14-FITC (Miltenyi Biotec Inc, Auburn CA), followed by intracellular staining for IL-12p35-APC (R&D), IL-23p19-PE (eBioscience), or IL-17A-PE (eBioscience). The intracellular cytokine staining was carried out using the Inside Stain kit (Miltenyi Biotec) per manufacturer’s instructions. Isotype-matched control antibodies (eBioscience) and fluorescence minus one (FMO) controls were used to determine background levels of staining and adjust multicolor compensation as a gating strategy. The cells were analyzed on a FACSCalibur flow cytometer (BD, Franklin Lakes, NJ) using CELLQuest or FlowJo software.
Tim-3 blockade
Purified CD14+ cells or PBMCs were incubated with LEAF™ anti-human Tim-3 antibody (10 μg/ml, BioLegend) or control IgG for 72h, followed by stimulation with LPS/R848 for 6h or PMA/Ionomycin for 6h, then subjected for flow cytometric analysis of IL-12p35, IL-23p19, or IL-17A as described above.
Statistical analysis
Study results for each group are expressed as the mean ± standard deviation (SD). Comparison between two groups is performed by SPSS-18 software. Pair wise t-test is used to compare the significance of changes in Tim-3 blocking experiments. Correlation between Tim-3 expression on monocytes and IL-12p35 or IL-23p19 as well as IL-17A expressions were analyzed using a Pearson Correlation program. Values of P< 0.05 (*), P< 0.01(**), or P<0.001 (***) were considered significant. NS denoted no significance.
Results
Differential expression of IL-12/IL-23 by monocytes in HCV-infected HBV-NR and HBV-R or HS
To determine whether the blunted HBV vaccine response is associated with an impaired innate immunity during HCV infection, we first compared IL-12/IL23 production by monocytes in HCV-infected subjects with defined HBV vaccine responses. To this end, CD14+ monocytes isolated from HCV-infected, HBV-NR and HBV-R, or HS, were ex vivo stimulated with LPS/R848 for 6h; intracellular IL-12p35 and IL-23p19 expressions by CD14+ monocytes were examined by flow cytometry. As shown in Fig. 1A (left panel), the percentage of IL-12p35 expressing monocytes was found to be significantly lower in the group of individuals with chronic HCV infection (n=45) when compared to HS (n=16). Within the group of HCV-infected individuals, however, IL-12p35 expression by monocytes from HBV-NR (n=20) was found to be significantly lower than those in HBV-R (n=25). Notably, the mean fluorescence intensity (MFI) of IL-12p35 expression level by monocytes was also lower in HBV-NR compared with HBV-R of HCV-infected individuals versus HS, although there were no significant differences observed between HBV-NR and HBV-R or between HBV-R and HS; the MFI between HBV-NR and HS was significant (Fig. 1A right panel).
Fig. 1. Differential regulation of IL-12p35 and IL-23p19 productions by monocytes in HCV-infected HBV-NR versus HBV-R or HS.
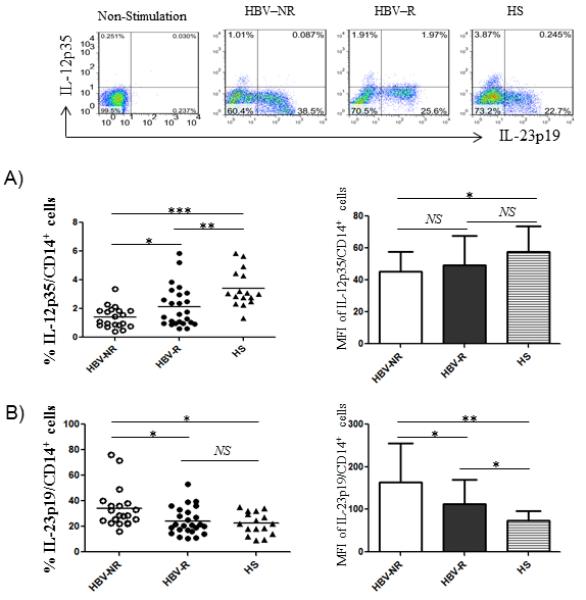
Purified CD14+ monocytes from chronically HCV-infected HBV-NR (n=20) and HBV-R (n=25) or HS (n=16) were stimulated with TLR ligands LPS and R848 for 6h, immune stained with conjugated antibodies against IL-12p35 and IL-23p19, followed by flow cytometric analysis. Isotype-matched control antibodies and fluorescence minus one (FMO) controls were used to determine background levels of staining and adjust multicolor compensation as gating strategy. A) The representative dot plots of IL-12p35 versus IL-23p19 expressions in CD14+ monocytes without stimulation and TLR-stimulated monocytes from HCV-infected HBV-NR and HBV-R or HS is shown above. Summary data of the positive cell frequency and the MFI of IL-12p35 expression in gated CD14+ cells in different group of subjects. Each symbol represents an individual subject, and the horizontal bars represent median values. *P<0.05; **P<0.01; ***P<0.001; NS=no significance. B) Summary data of the percentage of IL-23p19+ cells and the MFI of IL-23 expression level in CD14+ monocytes of HCV-infected HBV-NR versus HBV-R or HS. *P<0.05; **P<0.01; NS=no significance.
IL-12 and IL-23 are both mainly produced by activated antigen-presenting cells, including monocytes/macrophages and dendritic cells (DCs), upon encountering pathogens. They have distinct and often contradictory roles in promoting antimicrobial immune-responses and diseases in vivo. IL-12 is important in stimulating TH1 responses that are essential for host defense and pathogen clearance24-26. IL-23 is a pro-inflammatory cytokine that plays an important role in chronic infection and autoimmune diseases27-29. In addition to determining IL-12 expression, we examined IL-23 level in our cohort. As shown in Fig. 1B, in contrast to IL-12p35, IL-23p19 expression was found to be significantly higher in monocytes from HCV-infected, HBV-NR than HBV-R, or HS; and this held true in measuring the MFI of IL-23p19 expression levels among the different groups of subjects. Taken together, these results suggest a differential regulation of IL-12/IL-23 expressions by monocytes from HCV-infected, HBV-NR versus HBV-R, or HS.
Tim-3 expression is associated with an imbalance of IL-12/IL-23 production by monocytes of HCV-infected HBV-NR versus HBV-R or HS
We have previously observed that Tim-3 is involved in the differential regulation of IL-12/IL-23 expressions during HCV infection (Wang JM et al. J Virol. in press). To determine the role of this inhibitory pathway in regulation of innate immune responses to HBV vaccination, we examined Tim-3 expression on CD14+ monocytes from chronically HCV-infected, HBV-NR versus HBV-R, or HS. As shown in Fig. 2A, Tim-3 expression on monocytes was found significantly higher in HCV-infected, HBV-NR (n=22) than HBV-R (n=25), or HS (n=16), not only in the percentage of Tim-3 positive cell frequency, but also in the MFI of Tim-3 expression level. With Pearson correlation analysis, Tim-3 expression was found to be closely associated with the ratio of IL-23/IL-12 production by monocytes, not only in HCV-infected, HBV-NR and HBV-R, but also in HS (Fig. 2B). These results suggest that Tim-3 may function as a regulator for control of IL-12/IL-23 expressions, and its expression level might serve as a marker to predict HBV vaccine response in the setting of HCV infection.
Fig. 2. Tim-3 is over-expressed on CD14+ monocytes, which positively correlates with up-regulation of IL-23p19 and negatively correlates with IL-12p35 expression in HCV-infected HBV-NR and HBV-R or HS.
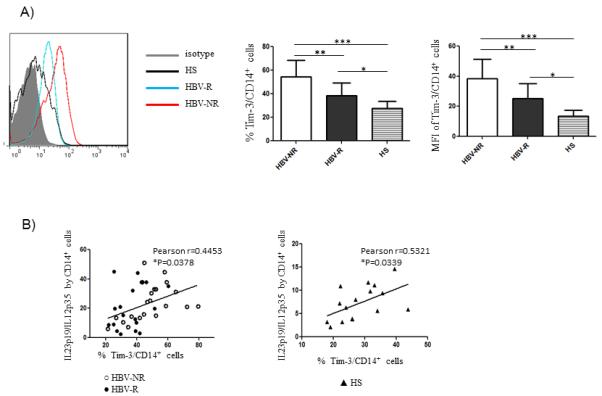
Purified CD14+ monocytes from chronically HCV-infected HBV-NR (n=22) and HBV-R (n=25) or HS (n=16) were stimulated with or without TLR ligands LPS and R848 for 6h, then immune stained with conjugated antibodies against Tim-3, IL-12p35 and IL-23p19, followed by flow cytometric analysis. A) The representative overlay histogram of Tim-3 expression on CD14+ monocytes from HCV-infected HBV-NR (red line) and HBV-R (blue line) versus HS (black line) against isotype control (grey filled area) is shown on the left. The percentage of positive cell frequency and the MFI of Tim-3 expression level on gated CD14+ cells in HCV-infected HBV-NR versus HBV-R or HS are shown on the right. Each symbol represents an individual subject, and the horizontal bars represent median values. *P<0.05; **P<0.01; ***P<0.001. B) The correlation between Tim-3 expression and the ratio of IL-23p19/IL-12p35 production by CD14+ monocytes from HBV-NR and HBV-R of HCV-infected individuals as well as HS, analysis by Pearson Correlation with 2-tailed significance. *P<0.05.
Tim-3-mediated differential regulation of IL-12/IL-23 expressions by monocytes is associated with an increase in TH17 cells in HCV infected HBV-NR versus HBV-R or HS
Given the pivotal role of IL-12/IL-23 balance in TH17 cell differentiation, we next sought to determine whether the observed differential regulation of IL-12/IL-23 expression may lead to generation of TH17 cells, and whether there is any difference in TH17 cell development between HBV-NR and HBV-R during HCV infection. To this end, PBMC isolated from chronically HCV-infected individuals with defined HBV vaccine responses and HS were stimulated with PMA/ionomycin, and the prevalence of IL-17+ CD4+ T cells was assessed by flow cytometric analysis. As shown in Fig. 3A, chronically HCV-infected HBV-NR (n=14) exhibited significantly higher frequencies of IL-17+ cells in CD4+ T populations than HBV-R (n=14) or HS (n=16). Additionally, the IL-17 expression level (MFI) in CD4+ T cells was also observed to be higher in HCV-infected, HBV-NR versus HBV-R, or HS. Importantly, the increased frequency of IL-17+ CD4+ T cells was found to positively correlate with Tim-3 expression on CD14+ monocytes as well as IL-23/IL-12 productions by CD14+ monocytes (Fig. 3B), suggesting that Tim-3-mediated differential regulation of IL-12/IL-23 production contributes to the development of TH17 cells, which might in turn contribute to the HBV vaccine non-respons frequently observed during chronic HCV infection.
Fig. 3. TH17 cells are significantly up-regulated in HCV-infected HBV-NR and are associated with Tim-3 expression and differential regulation of IL-23/IL-12 production by monocytes.
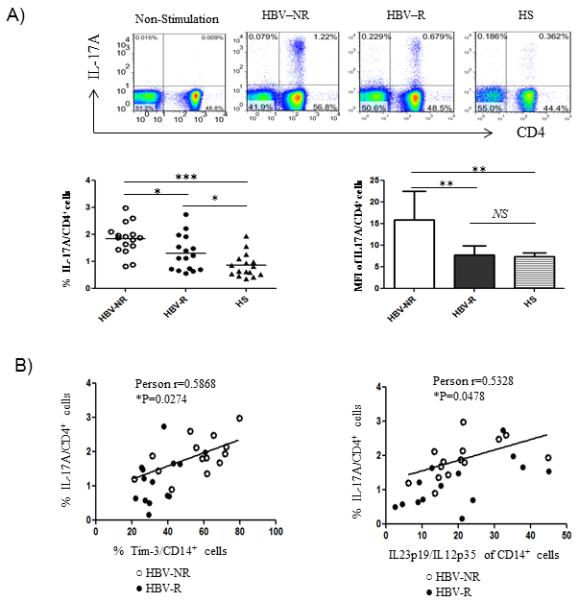
PBMC from HCV-infected HBV-NR and HBV-R or HS were stimulated with PMA/ionomycin for 6h, immune stained with CD4 and IL-17A, followed by flow cytometry analysis. A) Representative dot plots of IL-17A expression in CD4+ T cells without stimulation and stimulated CD4+ T cells from HCV-infected HBV-NR versus HBV-R or HS are shown on the left; and summary data of percentages of the positive cell frequency and MFI of IL-17A expression level in CD4+ cells from each group is shown on the right. *P<0.05; **P<0.01; ***P<0.001; NS=no significance. B) IL-17 expression by CD4+ cells is positively correlated to the Tim-3 expression on CD14+ cells. *P<0.05. IL-17 expression by CD4+ cells is also positively associated with the ratio of IL-23/IL-12 produced by CD14+ cells. *P<0.05.
Tim-3 blockade on monocytes corrects the balance of IL-12/IL-23 and reverses IL-17 expressions in HCV-infected HBV-NR
The observed Tim-3 expression and differential regulation of IL-12/IL-23/IL-17 productions might be a concurrent but unrelated phenomenon during HCV infection. To determine the role of Tim-3 on IL-12, IL-23, and IL-17 expressions, we isolated CD14+ monocytes or PBMCs from HCV-infected individuals (HBV-NR n=22, HBV-R 25) and HS (n=16), incubated ex vivo with anti-Tim-3 (αTim-3) or a control antibody (IgG) for 72h, and then stimulated with LPS/R848 or PMA/ionomycin for 6h, followed by detecting IL-12/IL-23 and IL-17 expressions by flow cytometry. As shown in Fig. 4A, representative dot plots and summary data of IL-12p35 versus IL-23p19 expressions in CD14+ monocytes, blocking Tim-3 signaling significantly improved IL-12p35 production in HCV-infected, HBV-NR as well as HBV-R, but not in HS. In contrast, blocking Tim-3 pathway significantly inhibited IL-23p19 in HCV-infected HBV-NR, but not in HBV-R or HS.
Fig. 4. Tim-3 blockade improves IL-12p35 and inhibits IL-23p19 productions by CD14+ monocytes, leading to reduction of TH17 cells in HCV-infected HBV-NR.
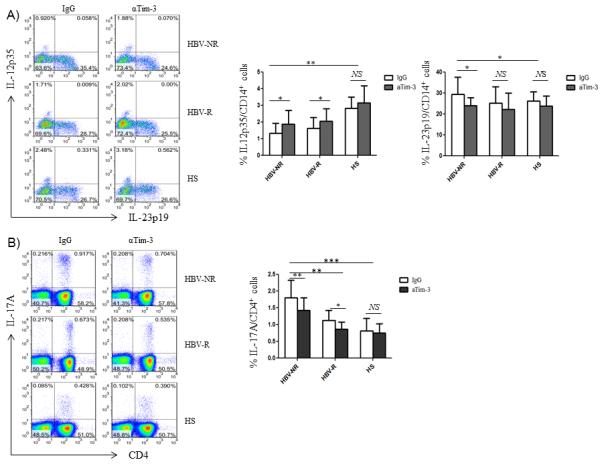
A) Purified CD14+ monocytes from HCV-infected HBV-NR and HBV-R or HS were incubated with αTim-3 and control IgG antibody for 72h; then stimulated with LPS/R848 for 6h, followed by flow cytometry analysis of IL-12p35 and IL-23p19 expressions. Representative dot plots and summary data measuring IL-12p35 and IL-23p19 productions by CD14+ monocytes in HCV-infected HBV-NR versus HBV-R or HS with the blockade of Tim-3 or IgG antibody are shown. *P<0.05; **P<0.01; NS = no significance. B) PBMCs from HCV-infected HBV-NR and HBV-R or HS were incubated with αTim-3 and control IgG antibody for 72h, and then stimulated with PMA/ionomycin for 6h, followed by flow cytometry analysis of IL-17A expression in CD4+ T cells. Representative dot plots and summary data measuring IL-17 production by CD4+ T cells in HCV-infected HBV-NR versus HBV-R or HS with the blockade of Tim-3 or IgG antibody are shown. *P<0.05; **P<0.01; *** P<0.001; NS = no significance.
Since Tim-3 is also up-regulated on T cells and other types of immune cells, such as natural killer cells, B lymphocytes, and regulatory T cells, in HCV-infected patients, we examined the role of Tim-3 on lymphocyte IL-17 expression. Interestingly, blocking Tim-3 in purified CD4+ T cells or monocyte-depleted PBMCs increases the Th17 cell population, suggesting a direct inhibitory role of Tim-3 on IL-17-secreting CD4+ T (Th17) cells. However, Th17 cell differentiation is inhibited in CD4+ T cells incubated with monocytes pre-treated with Tim-3 blocking antibody, which may correct IL-12/IL-23 secretions (Wang JM et al. J Virol. in press). Additionally, we observed inhibition of Tim-3 signaling in whole PBMCs to determine its overall effect on IL-17 production by TH cells. As shown in Fig. 4B, representative dot plots and summary data of IL-17A expression in CD4+ T cells, blocking Tim-3 signaling in bulk PBMCs ex vivo for 72h followed by PMA/ionomycin stimulation for 6h significantly reduced the number of TH17 cells up-regulated in HCV-infected HBV-NR, but not in HBV-R or HS. Taken together, these results suggest that Tim-3 signaling on monocytes overrides its effect on other types of cells in PBMCs, resulting in an overall stimulatory effect on IL-17 production or TH17 cell development, and indicating that Tim-3 regulation on monocytes IL-12/IL-23 secretion alters IL-17 production or TH17 cell development. This may play a critical role in HBV vaccine response during chronic HCV infection.
Discussion
Vaccine responses in the setting of chronic HCV infection are blunted, with poor response rates to a standard course of HBV vaccination in chronically HCV-infected individuals when compared to healthy populations (40~60% versus 90~95%)6-9. This study again confirms this discrepancy and raises the possibility that responses to vaccination may be impaired at least in part by up-regulated expression of the negative immunomodulator, Tim-3. Our data consistently demonstrate that HBV vaccine non-responders have up-regulation of Tim-3 expression that correlates with an imbalance of IL-12/IL-23 production by monocytes, and accumulation of TH17 cells, an immune dysregulation that can be corrected by Tim-3 blockade.
Tim-3 is an immunoinhibitory receptor predominantly expressed on activated T cells and functioning through interaction with its natural ligand, galectin-9, expressed on both haematopoetic and parenchymal cells. Up-regulation of Tim-3 appears to be a marker for T cell anergy, with selective up-regulation on “exhausted” T cells; inhibition of its signaling via anti-Tim-3 antibody has led to improved T cell function and viral clearance in several models30-33. While Tim-3 has been identified as an inhibitory receptor expressed on dysfunctional T cells, its role in human innate immune responses during viral infection remains less-well studied. We have recently studied the trans- versus cis-association of Tim-3 and Gal-9 on monocytes stimulated with TLR ligands, and found that trans-association of Tim-3/Gal-9 leads to an inhibition, whereas cis-association of Tim-3/Gal-9 causes activation, of monocytes through TLR signaling (Ma CJ et al. manuscript submitted). Additionally, Tim-3 in viral clearance during HCV and/or HIV infections has been extensively studied30-33, but its role in vaccine responses during chronic viral infections remains unknown; it may serve as a novel marker or readout of immune exhaustion and vaccine non-response during chronic infection. Our preliminary results presented here add to a compendium of data by suggesting a role for Tim-3 in vaccine responses in immunocompromised hosts. We aim to generate more data to establish a cut-off level of Tim-3 and IL-12/IL-23 expressions that is predictive of HBV vaccine responses in chronic viral infections. We are also working towards establishing new guidelines or policies to improve the vaccine success rate by blocking Tim-3 signaling while re-immunizing in HBV-NR as a clinical application.
IL-12 and IL-23 are paradoxically regulated, exhibiting contradictory functions in disease conditions; the precise mechanism for their differential expression remains unclear. IL-12 suppresses IL-23 and IL-17 production and, vice versa, IL-23 inhibits IL-12 and IFN-γ production, indicating cross-regulation between the IL-23/Th17 and IL-12/Th1 pathways. We have recently shown that HCV-induced Tim-3 expression inhibits IL-12 production through delivering a negative signal to TLR-mediated STAT-1 activation in monocytes34-35. We further discovered that HCV-induced Tim-3 expression up-regulates IL-23 expression by stimulating TLR-mediated STAT-3 phosphorylation (Wang JM et al. J Virol. in press). Therefore, we propose a theoretical model in which HCV-induced Tim-3 controls the balance of IL-12/IL-23 through differential regulation of STAT-1/STAT-3 in innate immunity, leading towards differentiation or expansion of TH17 cells, a process that is more significantly evident in HBV-NR during chronic HCV infection. This novel model, as depicted in Fig. 5, is plausible by providing an insight into the understanding of immune modulation of vaccine failure as well as the pathogenesis of HCV persistence.
Fig. 5. Proposed model for Tim-3-mediated differential regulation of IL-12 and IL-23 production by monocytes.
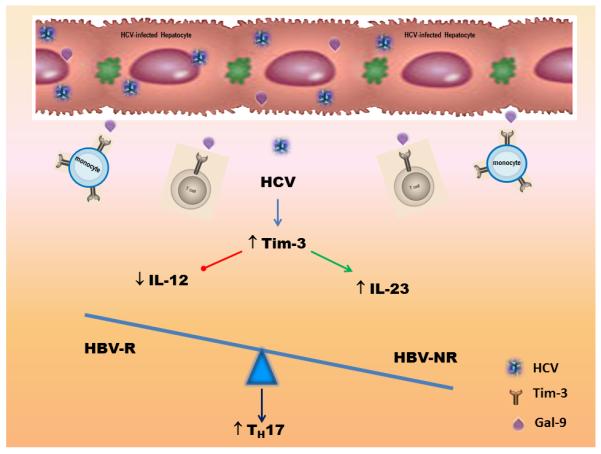
HCV-driven up-regulation of Tim-3 expression on monocytes inhibits IL-12 and enhances IL-23 productions, respectively. This Tim-3-mediated IL-12/IL-23 alteration in innate immunity would lead to a milieu conducive for accumulation of TH17 cells in the adaptive immune response, which might contribute to vaccine non-response during chronic HCV infection.
The adaptive immune system is crucial for the elimination of viral infections, but dysregulation of adaptive immune responses can also lead to the development of inflammatory and autoimmune diseases. TH17 cells are the newest member of the TH cell family that is characterized by their ability to produce specific cytokines such as IL-17A, IL-17F, IL-22, and CCL2036. The conditions for the differentiation of TH17 cells in human system have not been fully elucidated, but the cytokine ratio of IL-12/IL-23 produced by monocytes seems to play a pivotal role in several disease models37-39. Functionally, TH17 cells contribute to host defense as a new effector TH cell subset with a role in protection against pathogens through activities on immune and non-immune epithelial cells. Their activities, however, are also pivotal in the development of autoimmune diseases under pathologic conditions40-44. In this study, we provide pilot evidence indicating that TH17 cells accumulate in the peripheral blood of HCV-infected individuals, especially noted in HBV vaccine non-responders. Tim-3 blocking ex vivo reverses TH17 cell development, suggesting that HCV-induced, Tim-3-mediated differential regulation of IL-12/IL-23 expressions by monocytes is critical in TH17 cell development. It thereby might be evident that the STAT-3/IL-23/IL-17 pathway, rather than the STAT-1/IL-12/IFN-γ axis, may be crucial for viral persistence and vaccine non-response during HCV infection.
HBV vaccine responders and non-responders are usually classified by antibody response and are generally believed to result from differentiation of TH1/TH2 polarization induced by the vaccine in the setting of chronic viral infection. We and others have found that the mechanisms underlying viral persistence and vaccine non-response during HCV infection are likely multiple and that HCV-mediated expressions of inhibitory molecules regulating innate to adaptive immune responses may play a pivotal role. Specifically, we and others have shown that HCV-induced negative signaling molecules, including program death-1 (PD-1), suppressor of cytokine signaling-1 (SOCS-1), killer cell lectin-like receptor subfamily G member 1 (KLRG-1), and Tim-3, play key roles in inhibiting several intracellular signaling pathways, including MAPK, Jak/STAT, and Akt/PI3K, and lead to inhibition of monocyte IL-12 production, suppression of T cell responses, promotion of B cell activation and proliferation, and induction of CD4+CD25+Foxp3+ regulatory T cell accumulation during HCV infection9,34-35,45-56. It should be noted that activated B cells during HCV infection are often antigen non-specific and produce “non-sense” IgG/IgM or autoimmune antibodies, whereas their ability to produce antigen-specific antibodies, such as anti-HBs or HCV neutralizing antibody, is jeopardized during the course of HCV infection46-48. Nevertheless, this study provides pilot evidence that HCV-induced Tim-3 expression can differentially regulate IL-12/IL-23 expression by monocytes, leading to TH17 cell development. At this point, it is still unclear why Th17 correlates with HBV vaccine response, particularly whether it is a cause or result of HBV vaccine non-response. We do know that, in addition to TH1 response being switched to TH2 response, anti-inflammatory cytokine IL-10/TGF-β expressions are up-regulated, leading to a more significant increase of CD4+CD25+Foxp3+ regulatory T cells (Tregs)52-54 in HBV vaccine non-responders versus responders18. Our data is in line with a report that tumor-associated Tregs express the IL-23 receptor, which activates STAT3 in this cell type and leads to up-regulation of the Treg-specific transcription factor, Foxp3, and the regulatory cytokine, IL-1057. These findings add new insight to the overall picture of HCV pathogenesis, underscoring the importance of immunotherapy in conjunction with antiviral treatment in the management of chronic HCV infection and its associated vaccine non-response56.
Acknowledgements
This work was supported by an NIH NIDDK grant to ZQY/JPM (R01DK093526) and by a Wake Forrest University and East Tennessee State University Collaborative Research Pilot Award. This publication is the result of work supported with resources and the use of facilities at the James H. Quillen Veterans Affairs Medical Center. The contents in this publication do not represent the views of the Department of Veterans Affairs or the United States Government.
Abbreviations
- ALT
alanine transaminase
- AST
aspartate transaminase
- HBV
hepatitis B virus
- HBV-NR
hepatitis B vaccine non-responder
- HBV-R
hepatitis B vaccine responder
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- HS
healthy subject
- IL-12
interleukin-12
- IL-17
interleukin-17
- IL-23
interleukin-23
- KLRG-1
killer cell lectin-like receptor subfamily G member 1
- PBMC
peripheral blood mononuclear cells
- PD-1
programmed death-1
- SD
standard deviation
- STAT
signal transducer and activator of transcription
- Tim-3
T cell immunoglobulin mucin domain-3
Footnotes
Authorship Contributions and Disclosure of Conflicts of Interest: Yao ZQ, Moorman JP, and High KP, PI and co-PIs of this study, conceived the idea and wrote the paper; Wang JM, and Ma CJ carried out the experiments and summarized the data; Wu XY and Li GY provided technical support and laboratory management. Thayer P, Greer P, and Smith MA facilitated collection of clinical samples. The authors disclose no conflicts of interest.
References
- 1.Cheruvu S, Marks K, Talal AH. Understanding the pathogenesis and management of hepatitis B/HIV and hepatitis B/hepatitis C virus coinfection. Clinics in liver disease. 2007;11:917–943. doi: 10.1016/j.cld.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Duberg AS, Torner A, Davidsdottir L, Aleman S, Blaxhult A, Svensson A, et al. Cause of death in individuals with chronic HBV and/or HCV infection, a nationwide community-based register study. Journal of viral hepatitis. 2008;15:538–550. doi: 10.1111/j.1365-2893.2008.00982.x. [DOI] [PubMed] [Google Scholar]
- 3.Filippini P, Coppola N, Pisapia R, Martini S, Marrocco C, Di Martino F, et al. Virological and clinical aspects of HBV-HCV coinfection in HIV positive patients. Journal of medical virology. 2007;79:1679–1685. doi: 10.1002/jmv.20992. [DOI] [PubMed] [Google Scholar]
- 4.Torbenson M, Kannangai R, Astemborski J, Strathdee SA, Vlahov D, Thomas DL. High prevalence of occult hepatitis B in Baltimore injection drug users. Hepatology (Baltimore, Md. 2004;39:51–57. doi: 10.1002/hep.20025. [DOI] [PubMed] [Google Scholar]
- 5.Zarski JP, Bohn B, Bastie A, Pawlotsky JM, Baud M, Bost-Bezeaux F, et al. Characteristics of patients with dual infection by hepatitis B and C viruses. Journal of hepatology. 1998;28:27–33. doi: 10.1016/s0168-8278(98)80198-0. [DOI] [PubMed] [Google Scholar]
- 6.Keeffe EB. Acute hepatitis A and B in patients with chronic liver disease: prevention through vaccination. The American journal of medicine. 2005;118(Suppl 10A):21S–27S. doi: 10.1016/j.amjmed.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Kramer ES, Hofmann C, Smith PG, Shiffman ML, Sterling RK. Response to hepatitis A and B vaccine alone or in combination in patients with chronic hepatitis C virus and advanced fibrosis. Digestive diseases and sciences. 2009;54:2016–2025. doi: 10.1007/s10620-009-0867-4. [DOI] [PubMed] [Google Scholar]
- 8.Tsai IJ, Chang MH, Chen HL, Ni YH, Lee PI, Chiu TY, et al. Immunogenicity and reactogenicity of the combined hepatitis A and B vaccine in young adults. Vaccine. 2000;19:437–441. doi: 10.1016/s0264-410x(00)00205-x. [DOI] [PubMed] [Google Scholar]
- 9.Moorman JP, Zhang CL, Ni L, et al. Impaired hepatitis B vaccine responses during chronic hepatitis C infection: involvement of the PD-1 pathway in regulating CD4+ T cell responses. Vaccine. 2011;29:3169–3176. doi: 10.1016/j.vaccine.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malaspina A, Moir S, Orsega SM, Vasquez J, Miller NJ, Donoghue ET, et al. Compromised B cell responses to influenza vaccination in HIV-infected individuals. The Journal of infectious diseases. 2005;191:1442–1450. doi: 10.1086/429298. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Barradas MC, Alexandraki I, Nazir T, Foltzer M, Musher DM, Brown S, et al. Response of human immunodeficiency virus-infected patients receiving highly active antiretroviral therapy to vaccination with 23-valent pneumococcal polysaccharide vaccine. Clin Infect Dis. 2003;37:438–447. doi: 10.1086/375841. [DOI] [PubMed] [Google Scholar]
- 12.Hohler T, Stradmann-Bellinghausen B, Starke R, et al. C4A deficiency and nonresponse to hepatitis B vaccination. J Hepatol. 2002;37:387–392. doi: 10.1016/s0168-8278(02)00205-2. [DOI] [PubMed] [Google Scholar]
- 13.Verkade MA, van Druningen CJ, op de Hoek CT, et al. Decreased antigen-specific T cell proliferation by moDC among hepatitis B vaccine non-responders on haemodialysis. Clin Exp Med. 2007;7:65–71. doi: 10.1007/s10238-007-0127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albarran B, Goncalves B, Salmen B, et al. Profiles of NK, NKT cell activation and cytokine production following vaccination against hepatitis B. Apmis. 2005;113:526. doi: 10.1111/j.1600-0463.2005.apm_191.x. [DOI] [PubMed] [Google Scholar]
- 15.Bauer T, Jilg W. Hepatitis B surface antigen-specific T and B cell memory in individuals who had lost protective antibodies after hepatitis B vaccination. Vaccine. 2006;24:572–577. doi: 10.1016/j.vaccine.2005.08.058. [DOI] [PubMed] [Google Scholar]
- 16.Goncalves L, Albarran B, Salmen S, et al. The nonresponse to hepatitis B vaccination is associated with impaired lymphocyte activation. Virology. 2004;326:20–28. doi: 10.1016/j.virol.2004.04.042. [DOI] [PubMed] [Google Scholar]
- 17.Salazar M, Deulofeut H, Granja C, et al. Normal HBsAg presentation and T cell defect in the immune response of nonresponders. Immunogenetics. 1995;41:366–374. doi: 10.1007/BF00163994. [DOI] [PubMed] [Google Scholar]
- 18.Bauer T, Gunther M, Bienzie I, et al. Vaccination against hepatitis B in liver transplant receipients: pilot analysis of cellular immune response shows evidence of HBsAg-specific regulatory T cells. Liver Transplantation. 2007;13:434–442. doi: 10.1002/lt.21061. [DOI] [PubMed] [Google Scholar]
- 19.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nature Rev. Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 20.Nishimura H, Honjo T. PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends in Immunol. 2001;22:265–268. doi: 10.1016/s1471-4906(01)01888-9. [DOI] [PubMed] [Google Scholar]
- 21.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 22.Hafler DA, Kuchroo V. TIMs: central regulators of immune responses. J.Exp.Med. 2008;205:2699–2701. doi: 10.1084/jem.20082429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuchroo VK, Umetsu DT, DeKruyff RH, et al. The TIM gene family: emerging roles in immunity and disease. Nat Rev Immunol. 2003;3:454–462. doi: 10.1038/nri1111. [DOI] [PubMed] [Google Scholar]
- 24.Hikens CM, Kalinski P, de Boer M, Kapsenberg ML. Human dendritic cells require exogenous interleukin-12-inducing factors to direct the development of naïve T-helper cells towards the Th1 phenotype. Blood. 1997;90:1920–1926. [PubMed] [Google Scholar]
- 25.Feili-Hariri M, Falkner DH, Morel PA. Polarization of naïve T cells into Th1 or Th2 by distinct cytokine-driven murine dendritic cell populations: implications for immunotherapy. J Leukoc Biol. 2005;78:656–664. doi: 10.1189/jlb.1104631. [DOI] [PubMed] [Google Scholar]
- 26.Vasconcellos R, Carter NA, Rosser EC, Mauri C. IL-12p35 subunit contributes to autoimmunity by limiting IL-27-driven regulatory responses. J Immunol. 2011;187:3402–3412. doi: 10.4049/jimmunol.1100224. [DOI] [PubMed] [Google Scholar]
- 27.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 28.Hunter C. A. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 2005;5:521–531. doi: 10.1038/nri1648. [DOI] [PubMed] [Google Scholar]
- 29.Duvallet E, Semerano L, Assier E, et al. Interleukin-23: a key cytokine in inflammatory diseases. Ann Med. 2011;43:503–511. doi: 10.3109/07853890.2011.577093. [DOI] [PubMed] [Google Scholar]
- 30.Jin HT, Anderson AC, Tan WG, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. PNAS. 2010;107:14733–14738. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones RB, Ndhlovu LC, Barbour JD, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008;205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahan RH, Golden-Mason L, Nishimura MI, et al. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J. Clin. Invest. 2010;1172:4546–2557. doi: 10.1172/JCI43127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vali B, Jones RB, Sakhdari A, et al. HCV-specific T cells in HCV/HIV co-infection show elevated frequencies of dual Tim-3/PD-1 expression that correlate with liver disease progression. Eur. J. Immunol. 2010;40:2493–2505. doi: 10.1002/eji.201040340. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Ma CJ, Wu XY, et al. Tim-3 Negatively Regulates IL-12 production by Monocytes during Chronic Hepatitis C Infection. PLoS One. 2011;6:e19664. doi: 10.1371/journal.pone.0019664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Ma CJ, Wu XY, et al. Tim-3 negatively regulates IL-12 production in human CD14+ Monocytes. J Leu Bio. 2011;91:189–196. [Google Scholar]
- 36.Louten J, Boniface K, Malefyt Rene de W. Development and function of TH17 cells in health and disease. J Allergy Clin Immunol. 2009;123:1004–1011. doi: 10.1016/j.jaci.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 37.Khayrullina T, Yen JH, Jing H, Ganea D. In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. J Immunol. 2008;181:721–735. doi: 10.4049/jimmunol.181.1.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukine-23 production and the T-helper 17 response in human. Immunol Rev. 2008;226:112–131. doi: 10.1111/j.1600-065X.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stamp LK, Easson A, Pettersson L, et al. Monocyte derived interleukin (IL)-23 is an important determinant of synovial IL-17A expression in rheumatoid arthritis. J Rheumatol. 2009;36:2403–2408. doi: 10.3899/jrheum.081304. [DOI] [PubMed] [Google Scholar]
- 40.Oosting M, ter Hofstede H, van de Veerdonk FL, et al. Role of interleukin-23 (IL-23) receptor signaling for IL-17 responses in human Lyme disease. Infect Immun. 2011;79:4681–4687. doi: 10.1128/IAI.05242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gasse P, Riteau N, Vacher R, et al. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS One. 2011;6:e23185. doi: 10.1371/journal.pone.0023185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mok MY, Wu HJ, Lo Y, Lau CS. The relation of interleukin 17 (IL-17) and IL-23 to Th1/Th2 cytokines and disease activity in systemic lupus erythematosus. J Rheumatol. 2010;37:2046–2052. doi: 10.3899/jrheum.100293. [DOI] [PubMed] [Google Scholar]
- 43.Rocha AM, Souza C, Rocha GA, et al. The levels of IL-17A and of the cytokines involved in Th17 cell commitment are increased in patients with chronic immune thrombocytope-nia. Haematologica. 2011;96:1560–1564. doi: 10.3324/haematol.2011.046417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang JY, Zhang Z, Lin F, et al. Interleukin-17-producing CD4 (+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology. 2010;51:81–91. doi: 10.1002/hep.23273. [DOI] [PubMed] [Google Scholar]
- 45.Yao ZQ, King E, Prayther D, et al. T cell dysfunction by hepatitis C virus core protein involves PD-1/PDL-1 signaling. Viral Immunol. 2007;20:276–287. doi: 10.1089/vim.2006.0096. [DOI] [PubMed] [Google Scholar]
- 46.Yao ZQ, Prayther D, Trabu C, et al. Differential regulation of SOCS-1 signaling in T and B lymphocytes by HCV core protein. Immunology. 2008;125:197–207. doi: 10.1111/j.1365-2567.2008.02829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moorman JP, Dong ZP, Ni L, et al. Abnormal B lymphocyte activation associated with TALL-1 overexpression and SOCS-1 deregulation in chronic HCV infection. Immunology. 2009;128:227–235. doi: 10.1111/j.1365-2567.2009.03106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao ZQ, Ni L, Zhang Y, et al. Differential Regulation of T and B lymphocytes by PD-1 and SOCS-1 signaling in Hepatitis C Virus-associated non-Hodgkin’s Lymphoma. Immunol Invest. 2010;40:243–264. doi: 10.3109/08820139.2010.534218. [DOI] [PubMed] [Google Scholar]
- 49.Ma CJ, Ni L, Zhang Y, et al. PD-1 negatively regulates IL-12 expression by limiting STAT-1 phosphorylation in monocytes/macrophages during chronic hepatitis C infection. Immunology. 2011;132:421–431. doi: 10.1111/j.1365-2567.2010.03382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frazier AD, Zhang CL, Ni L, et al. Program death-1 pathway affects suppressor of cytokine signaling-1 expression in T cells during hepatitis C infection. Viral Immunology. 2010;23:487–495. doi: 10.1089/vim.2010.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Ma CJ, Ni L, et al. Crosstalk between PD-1 and SOCS-1 in HCV core-mediated IL-12 suppression. J of Immunology. 2011;186:3093–3103. [Google Scholar]
- 52.Ni L, Ma CJ, Zhang Y, et al. PD-1 modulates Regulatory T cells and suppresses T cell responses in HCV-associated Lymphoma. Immunology & Cell Biology. 2010;89:535–539. doi: 10.1038/icb.2010.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moorman JP, Wang JM, Zhang Y, et al. Tim-3 controls regulatory and effector T cell balance during HCV Infection. J. Immunol. 2012;189:755–766. doi: 10.4049/jimmunol.1200162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ji XJ, Ma CJ, Wang JM, et al. Hepatitis C Virus Induces CD4+CD25+Foxp3+ Regulatory T cell Development through the Tim-3/Gal-9 Pathway. Eur. J Immunol. 2012 doi: 10.1002/eji.201242768. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Y, Zhang Y, Yao ZQ, et al. Dendritic cell-based immunity and vaccination against hepatitis C virus infection. Immunology. 2012;136:385–96. doi: 10.1111/j.1365-2567.2012.03590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao ZQ, Moorman JP. Immune exhaustion and immune senescence - two distinct pathways for HBV vaccine failure during HCV and/or HIV infection. AITE; 2013. Review. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kortylewski M, Xin H, Kujawski M, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]