

Abstract
We present a method for deriving natural killer (NK) cells from undifferentiated hESCs and iPSCs using a feeder-free approach. This method gives rise to high levels of NK cells after 4 weeks culture and can undergo further 2-log expansion with artificial antigen presenting cells. hESC- and iPSC-derived NK cells developed in this system have a mature phenotype and function. The production of large numbers of genetically modifiable NK cells is applicable for both basic mechanistic as well as anti-tumor studies. Expression of firefly luciferase in hESC-derived NK cells allows a non-invasive approach to follow NK cell engraftment, distribution, and function. We also describe a dual-imaging scheme that allows separate monitoring of two different cell populations to more distinctly characterize their interactions in vivo. This method of derivation, expansion, and dual in vivo imaging provides a reliable approach for producing NK cells and their evaluation which is necessary to improve current NK cell adoptive therapies.
Keywords: Stem Cell Biology, Issue 74, Bioengineering, Biomedical Engineering, Medicine, Physiology, Anatomy, Cellular Biology, Molecular Biology, Biochemistry, Hematology, Embryonic Stem Cells, ESCs, ES Cells, Hematopoietic Stem Cells, HSC, Pluripotent Stem Cells, Induced Pluripotent Stem Cells, iPSCs, Luciferases, Firefly, Immunotherapy, Immunotherapy, Adoptive, stem cells, differentiation, NK cells, in vivo imaging, fluorescent imaging, turboFP650, FACS, cell culture
Introduction
Human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) are undifferentiated, pluripotent cells capable of unlimited self-renewal and multi-lineage differentiation. hESCs have been successfully differentiated into mature and functional subsets of each germ layer, including cells of the hematopoietic system1-3. Natural killer (NK) cells are lymphocytes of the innate immune system that can be derived from hESCs by formation of embryoid bodies (EBs)4,5 or co-culture with stromal cell lines1,2,6-8. NK cells possess anti-viral and anti-tumor capabilities and have the potential to be effective against a broad range of malignancies, as they do not require prior antigen stimulation to perform their effector functions. Thus, hESC-derived NK cells are an attractive source of cells for immunotherapy. Additionally, the derivation of NK cells from hESCs provides a genetically amenable system to study normal development in vitro.
Because they provide a genetically tractable system, hESC-derived NK cells can be experimentally modified to express fluorescent and bioluminescent reporters providing an optimal model to study NK cell effector functions in vitro and in vivo. hESC-derived NK cells possess activity against a range of targets including HIV 9, leukemia (K562) and other cancer types 7,8. However, the ability to efficiently derive enough NK cells capable of treating patients remains an important barrier for clinical translation and is, to a lesser degree, a limitation for extensive pre-clinical in vivo studies of NK cell development and anti-cancer functions. Here, we use a spin EB approach to derive hematopoietic progenitors from hESCs 4,5,10. Following 11 days the spin EBs are transferred to NK cell culture with or without feeders for 28 days. After 4 weeks in NK cell culture medium, NK cells are transferred to co-culture with K562 cells modified to express membrane-bound interleukin 21 (IL-21), which serve as artificial antigen presenting cells (aAPCs). Adapting a protocol for the expansion of peripheral blood NK cells using these artificial APCs 11,12, we are able to expand NK cells 2-logs while retaining a mature phenotype and cytotoxic capabilities.
This process of development and expansion provides sufficient hESC-derived NK cells for extensive in vivo characterization. For in vivo studies, we are able to non-invasively monitor long term engraftment and kinetics of injected firefly luciferase expressing (Fluc+), hESC-derived NK cells using bioluminescence imaging. Furthermore, we are able to follow NK cell interactions with tumor cells using a dual, bioluminescent or fluorescent imaging scheme. An earlier study by our group used bioluminescence imaging in an anti-tumor model to follow tumor progression and clearance of Fluc+ K562 cells in vivo 7. Now, by engineering our hESCs to express firefly luciferase 13,14 we can follow the biodistribution and trafficking of NK cells to K562 tumor cells that express the recently characterized fluorescent protein, turboFP650 15. We have chosen this dual reporter system in order to simultaneously follow the two cell populations in vivo (Figure 1). Most dual imaging models have been dual-luciferase systems, but these systems can be technically challenging due to the delivery requirements of coelenterazine, the substrate required for expression of most Renilla and Gaussia luciferase reporters 16-18. Fluorescent reporters have allowed easy monitoring of many cell lines and constructs in vitro, but has had limited success for in vivo imaging due to the overlap between tissue and fur autofluorescence and the emission spectra of many commonly used fluorescent reporters including GFP, DsRed, and TdTomato 15,19. This concern has encouraged the development of far-red fluorescent proteins, which allow for better tissue penetrance and higher specific signal compared to background 15,19. TurboFP650, the fluorescent protein shown in this system, is far-red shifted and overcomes many of the issues involved with imaging fluorescent proteins in living animals.
This method for developing and expanding NK cells derived from hESCs has allowed us to further characterize hESC-derived NK cells in vitro and in vivo, which is necessary to better understand NK cell function and clinically important to improve current NK cell adoptive therapies. It is also amenable to the derivation and expansion of iPSC-derived NK cells. The dual fluorescent and bioluminescent imaging scheme is broadly applicable to systems other than the anti-tumor model we have shown here.
Protocol
1. Adapting hESCs or iPSCs in TrypLE for Spin EB Cultures
The initial dissociation with TrypLE works best if the ES/iPS colonies from the collagenase-passaged cultures are relatively small. The starting ES/iPS populations should be cells passed no longer than 4-5 days previous. 4-5 days before initiation of TrypLE passage, pass ES cells at a density that will allow the cells to be ~70% confluent in 4 days time. Use regular ES media for culture of TrypLE-passaged ES cells. Here, we are using hESCs stably modified with a luciferase reporter construct (Materials Table). The protocol also works with iPSCs, using unmodified iPSCs for comparison of hematopoietic progenitor cell and NK cell development.
Four days before initiation of TrypLE passage, pass ES/iPS cells with collagenase IV, following a normal passage protocol. We typically pass 1:1 in the first passage with TrypLE. Expand ES/iPS cells accordingly beforehand.
One day prior to initiation of TrypLE passage, plate MEFs in 6-well plates at 0.9-1 x 105 cells/well. Pass 1:1 (or 2:1 for hard-to-adapt cell lines or for less dense cultures) in the first passage with TrypLE. Therefore, prepare 1 MEF plate for every ES/iPS plate you plan on putting into TrypLE culture.
Carefully pick off differentiated colonies (those which lack a circumscribed border or have fibroblastic/epithelial characteristics) before proceeding to TrypLE dissociation. Differentiated cells can take over the culture relatively easily with TrypLE passage, so it is important to make sure your starting population is very clean.
Aspirate media from wells and add 1.0 ml pre-warmed TrypLE Select per well.
Incubate ES/iPS cells in TrypLE for 5 min in a 37 °C incubator. After 5 min, gently pipette ES cells off the bottom of the well.
Collect the TrypLE cell suspension from wells and transfer to a conical tube. Pipette up and down gently 2-3 times to encourage the largest clumps to break apart. Dilute TrypLE with at least 1 equal volume ES media and 1 equal volume DPBS. TrypLE is not quenched by culture media, so it is important to dilute the TrypLE with media and DPBS after removing cells from the plate. We use DPBS + media for washes to save ES media.
Spin down cells at 1,500 rpm for 5 min in refrigerated centrifuge (8 °C).
Aspirate off as much of the supernatant as possible to remove TrypLE.
Resuspend cells in 4 ml ES media plus 4 ml DPBS and repeat spin to get rid of remaining traces of TrypLE.
Aspirate off supernatant and resuspend in appropriate volume ES media for plating.
Plate ES cells 1:1 (or 2:1 if need be) onto low-density MEFs in ES media.
After 24 hr, feed cells with fresh ES media. There will most likely be many single cells that did not attach. Feed ES cells daily with ES media.
Pass with TrypLE onto fresh MEFs (0.9-1 x 105 cells/well) every 3-4 days (e.g. Tuesday and Friday) using the same basic procedure as above. Normally, you should not have to pick cells passed with TrypLE.
For the first several passages (up to passage 5-10), the cells will require passing at 1:1. This makes expansion of cells difficult. In our experience, plating efficiency of the ES cells drops dramatically around passages 4-5 and then comes back up within a couple of passages. After passages 5-7, you may be able to begin passing the cells at 1:2, and eventually, 1:3. Beyond passage 20, you may need to start passing the cells at 1:5 or 1:6. Make sure to plate the ES cells at a high density the first 5-10 passages. Once you get to a point where the cells plate down efficiently enough that they can reach ~70% confluence within two days after passage, you can try to start passing at 1:2, and eventually should be able to pass at 1:3 or 1:4. iPSCs, in particular, if not passaged densely enough before fully adapted, tend to differentiate.
2. Prepare Stock Solutions for Setting Up Spin EB Cultures
10% BSA solution in IMDM: Suspend 4 g of BSA in 35 ml IMDM (in a 50 ml conical). Allow solution to sit at room temperature for 1-2 hr to allow BSA to fully dissolve. Adjust total volume to 40 ml with IMDM to get final concentration of 10%. Commercially available BSA can be cytotoxic to ES cells. Deionizing the solution can reduce the potential for cytotoxicity. To deionize, add ~1.3 g resin beads to 40 ml of BSA solution and shake well to mix.
Place BSA solution (with resin beads) at 4 °C until beads change color from blue-green to yellow (indicating that the exchange capacity has been exhausted).
Shake solution every 20-30 min to facilitate exchange
Each exchange may take up to 2 hr. When exchange is complete, centrifuge solution for 2 min at 1,000 rpm to pellet beads at bottom of tube.
Pipette BSA solution into a fresh 50 ml conical, leaving beads to be discarded.
Repeat steps 2.2-2.5 at least two additional times for a total of three or more exchanges. During the last exchange, the capacity of the beads may not fully be exhausted even after two hour incubation with the beads.
Sterile filter the BSA solution and remove any remaining beads.
The BSA solution should be stable at 4 °C for at least 2 months.
5% PVA solution: Measure 100 ml Millipore H2O into 125 ml glass bottle.
Add 5 g PVA to the water. It is important to add the PVA to the water so that the PVA gets fully "wet." If you add the water to the PVA, it will take a very long time for the PVA to go into solution.
PVA will NOT dissolve immediately. Place PVA/water solution at 4 °C overnight.
Next morning, place PVA/water solution in 37 °C waterbath for 4-8 hr. PVA should be mostly in solution by the end of the incubation.
Place bottle back in 4 °C for an additional 24-48 hr, or until fully dissolved. Solution might be slightly viscous.
PVA solutions should be stable at 4 °C for at least 6 weeks.
Linoleic and Linolenic Acids (10,000X solutions): Dilute to 1 mg/ml in 200-proof ethanol. Add 10 ml pure oil to 10 ml ethanol. Aliquot and store at -20 °C.
α-MTG stock solution: Dilute 13 μl α-MTG in 1 ml IMDM.
Ascorbic Acid 2-Phosphate solution (100X): Make a 5 mg/ml solution. 250 mg ascorbic acid 2-phosphate in 50 ml sterile, tissue-culture grade H2O. Dissolves readily.
3. Assembly of BPEL Media (for ~200 ml Total Volume)
Make a PVA-lipids mixture (this step prevents the PVA from forming insoluble droplets in the medium that would get filtered out).
Add 20 ml IMDM and 20 ml F-12 nutrient mixture to 50 ml conical tube.
Add 10 ml of 5% PVA stock, 0.4 ml synthechol, 20 μl linolenic acid, and 20 μl linoleic acid for 200 ml BPEL media. Shake tube well to mix. Set aside while other media components are assembled.
Add all remaining media components to top of 250 ml Stericup filtration unit. Add the balance of required IMDM and F-12 volumes (66 ml each), BSA, a-MTG, 100X ITS, Glutamax I, Pen/Strep, Protein-free hybridoma mixture II, and ascorbic acid. Add the PVA-lipids mixture from step 1 to the top of the Stericup. Filter the medium. Medium should be stable at 4 °C for at least 2 weeks. Use old medium for wash steps.
4. Setting up TrypLE-passaged ES Cells into Stage I Spin EBs
Use ES/iPS cells that have been adapted to TrypLE passage on lower-density MEFs(i.e. ES/iPS cells that have been passaged with TrypLE at least 5-7 times). If cells can be passed at ~1:3 and become 70-80% confluent in 3-4 days, the cells should be good to use.
Two days before setting up Spin EB differentiation (day -2): Pass TrypLE-adapted ES/iPS cells onto fresh MEFs at a density allowing them to be 70-80% confluent on the day of differentiation setup. We typically pass 48 hr prior to Spin EB setup.
Day of Spin EB Setup into Stage I Differentiation (day 0): To prepare for Spin EB plating, pipette 150 μl sterile water into the 36 outer wells of each 96-well plate to minimize evaporation. Use round-bottom, low attachment 96 well plates only.
Aspirate culture media off of ES/iPS cells and add 1.0 ml pre-warmed TrypLE Select to each well.
Place plates in incubator (37 °C) for 5 min. Gently pipette cells off of the plate.
Collect dissociated cells in a conical tube and pipette up and down to break apart clumps. Dilute TrypLE with 1 equal volume BPEL media and at least 1 equal volume DPBS.
Remove supernatant and resuspend the cells in 5 ml BPEL media plus 5 ml DPBS. Repeat spin.
Remove supernatant and resuspend cells in 5-10 ml BPEL media, depending on anticipated cell number. Pass cells through 70 μm filter (BD Falcon #352350) into a fresh 50 ml conical in order to remove clumps (which can interfere with EB formation).
Count filtered cells. Aliquot cells to be used for plating into a 50 ml conical and spin. For plating: 3,000 ES cells/well, 60 wells/plate = 1.8x105 ES cells/plate.
After centrifugation, cells will need to be resuspended to 3x104 cells/ml (100 μl volume/well, 3,000 ES cells/well).
Prepare a volume of BPEL media + cytokines sufficient for resuspending the cells (this will be 6 ml/plate). For stage I differentiation, we use SCF (40 ng/ml), BMP4 (20 ng/ml), and VEGF (20 ng/ml) for derivation of hematopoietic progenitor cells.
Plate 100 μl cells/well (=3,000 cells/well) into each of the inner 60 wells of the prepared 96-well plates. This is easiest when using a multichannel pipettor and sterile trough. Be sure to mix cells by pipetting before putting in trough and work quickly so that cells do not have a chance to settle out.
Spin the 96-well plates in refrigerated centrifuge for 4 min at ~1,500 rpm, 8 °C.
Carefully transfer plates from centrifuge to 37 °C incubator. Avoid disrupting the plates as much as possible. Check plates to make sure that the cells formed uniform pellets in the bottom of the wells.
Stage I spin EBs should not be fed or otherwise disturbed through days 3-4 differentiation until the EBs have formed a more durable outer layer. If EBs will be left in stage I beyond 10-12 days, we sometimes feed EBs with 50 μl fresh BPEL media with cytokines to each well (first take out 50 μl of old media from each well). Within 24 hr, the cells should start to form a 3-D EB structure, and by day 3-5 should have a distinct outer layer, and start developing cystic structures.
5. Dissociating Stage I EBs for FACS Analysis
Collect around18-36 spin EB for FACS analysis. If analyzing cells before day 6, more wells are needed to obtain sufficient cell numbers.
Prepare necessary volume of trypsin with 2% chicken serum. Place in 37 °C waterbath until use.
Transfer spin EBs and media from 96-well plates to 15 ml conical tubes.
Allow EBs to settle to bottom of conical tubes. With a 5 ml pipette, carefully pipette out the supernatant and transfer to a separate tube (you can pool all of the supernatant into one 15 ml or 50 ml conical tube). As some hematopoietic cells have already been released from the spin EB between days 9 and 11, separating the supernatant containing these cells prevents excessive trypsinization.
Resuspend EBs in trypsin + 2% chicken serum: 3-4 ml to each 15 ml conical tube. Place EBs with trypsin in 37 °C waterbath for 3-7 min. Shake or gently vortex every 1-2 min. At earlier timepoints (before day 8), EBs will break apart more readily and can take as little as 3 min. At later timepoints (day 8+) the EBs may take longer to dissociate. We usually don't let them trypsinize for more than 5 min at 37 °C. Note, these times are a guide only. For any particular cell line or EB differentiation, it may take more or less time to dissociate the EBs. Closely monitor the dissociation. It is important not to leave cells in trypsin for too long. It is better to stop the trypsin dissociation when only a few small clumps remain visible rather than try to eliminate all clumps.
Quench trypsin with at least 1 equal volume of FBS-containing media. Spin down cells (1,500 rpm, 5 min, 8 °C).
Resuspend cells in an appropriate volume of media for counting (usually 3-5 ml).
Filter cells through a 100 μm cell strainer. Remove an aliquot of cells for counting. Proceed with cells to standard lab FACS protocols.
6. Development, Expansion, and Characterization of NK Cells from hPSC-derived Progenitor Cells
Because the spin EBs contain a high percentage of hematopoietic progenitor cells, they can be directly transferred to NK cell culture on day 11. The spin EBs may be transferred to 24 well plates with or without feeders. We have shown no difference in phenotype or function between NK cells derived in each condition. Therefore, we generally transfer to cultures without feeders to have a completely defined system for NK cell development. If using an hESC/iPSC line with suboptimal hematopoietic differentiation the use of feeder layers is recommended 7,8.
Using a multichannel pipette set to 100 μl, transfer 6 spin EBs to each well of a 24 well plate.
With Feeders: transfer spin EBs to 24-well plates containing 100,000 irradiated (3,000 cGY) EL08-1D2 (a murine embryonic liver cell line) cells per well. Without feeders: transfer EBs directly to uncoated, 24 well plates.
Add 400 μl NK differentiation media containing cytokines (IL-3, IL-15, IL-7, SCF, Flt3L, all from Peprotech) so the total volume in each well is ~1 ml.
Place cells in incubator and feed every 5-7 days with NK differentiation media containing all cytokines in step 6.3 except IL-3.
NK cells will express a mature phenotype at day 28 following transfer to NK cell differentiation culture (Figure 2).
To test NK cell cytotoxicity, a 4 hr chromium release assay can be performed 8.
If large numbers of cells are needed for in vivo studies, co-culture with artificial antigen presenting cells (aAPCs) can yield a 2-log or greater expansion. For a detailed protocol, visit http://www.jove.com/video/2540/expansion-purification-functional-assessment-human-peripheral-blood 11,12.
7. In vivo Fluorescent and Bioluminescent Imaging to Monitor hESC-derived NK Cells and Tumor Cells in Immunodeficient Mice
We use nonobese diabetic/severe combined immunodeficiency with gamma-chain knockout (NOD/SCID/ γC-/-) mice that are 6 to 8 weeks old in all of our experiments. We use a dual imaging model to simultaneously track tumor progression (turboFP650 reporter) and hESC-NK cells kinetics (Fluc reporter expressed in parent ES cells) in vivo. This imaging scheme is broadly applicable to systems other than the anti-tumor model shown here. The IVIS Spectrum (Caliper Life Sciences) is optimal for simultaneous fluorescent and bioluminescent in vivo imaging.
24 hr prior to tumor cell injection, irradiate mice (225-250 cGY).
Resuspend desired number of tumor cells (1 x 106 K562 cells are shown) in 200 μl Iscove modified Dulbecco medium (IMDM) supplemented with 20% FBS. Tumor cell dose can be varied depending on the model used. TurboFP650 could also be expressed in non-tumor types. It is best imaged with cells localized to a single anatomic location.
Inject tumor cells subcutaneously into the upper left thorax of the mice using a 27 or 28 gauge needle and allow to engraft for 4 days. Cells should form a bubble under the skin of the mouse. Mice can be shaved in this portion of the body to better visualize injection. It also exposes the area for better in vivo fluorescent imaging. Injection around the thorax of the mice was used in this model because the ip injected NK cells are best visualized while the mouse is supine. Alternative methods of subcutaneous delivery, including the back or flank, are less stressful to the animal and should be considered.
Resuspend the desired number of hESC-derived NK cells (10 x 106 NK cells are shown) in 300 μl Iscove modified Dulbecco medium (IMDM) supplemented with 20% FBS without antibiotics.
Inject hESC-derived NK cells into each mouse intraperitoneally (IP) using a 27 or 28 gauge needle. For all experiments, include mice receiving no tumor or NK cell infusion as a negative control.
To sustain NK cell populations in vivo, give mice a 250 μl IP injection of IL-2 (1x104 U/mouse) and IL-15 (10 ng/mouse) in sterile DPBS every day for the first 7 days followed by IL-2 only every 2 to 3 days until the time of sacrifice.
Monitor engraftment of hESC-derived NK cells by bioluminescent imaging. Inject each mouse IP with 120 μl D-luciferin (150 mg/kg). Image the mice 10 min after D-luciferin injection.
Anesthetize mice using a chamber allowing isoflurane entry into the box at 0.8 l/min. Make sure there is also oxygen flow into the chamber.
Place anesthetized mice in IVIS Spectrum and secure mice on their backs using tape or Velcro and allow isoflurane entry into the box at 0.5 l/min to maintain anesthesia.
Set IVIS machine to correct imaging platform (D for 4-5 mice, C for 3 or fewer mice), set binning to medium, f/stop to 1 and acquire image for 1 min.
To perform fluorescent imaging for turboFP650+ cells, use the imaging wizard to set up an emission scan measuring the probe of interest as well as background signal. From the list of probes choose Input Em/Ex, and enter in correct excitation and emission. For turboFP650, use 605 excitation and 660-720 emission to measure tumor signal and 570 excitation and 640-720 emission to measure background signal. Use auto exposure settings and select the desired imaging platform. The entire imaging sequence will generally take 3-5 min to acquire.
Allow mice to fully recover from anesthesia before transporting from the imaging system. Analyze images using the Living Image software package version 4.2 (Caliper Life Sciences).
Quantify bioluminescent signal using a region of interest (ROI) set to total flux (photons/sec) or average radiance (photons/sec/cm2/sr) and set all images to the same scale (Figure 3). Here, representative images are used to demonstrate both cell populations in each mouse (Figure 3). Other studies from our lab have demonstrated colocalization using this technique20. Colocalization timing will need to be optimized based on the experimental model used.
To quantify fluorescent signal from the emission scan sequence use the "Spectral Unmixing" feature in Living Image. Select the images to include in the analysis and draw a mask over the area of the mouse containing the signal of interest, in this case, the upper thorax. Select tissue autofluorescense and unknown probe to be unmixed. If mice are given food containing alfalfa, food signal can also be unmixed from the probe of interest and tissue autofluorescence.
If the resulting images do not show an even distribution of tissue autofluorescence (including the area where the signal is located) constraints can be set to get consistent distribution and better separation of the components being unmixed. This is typical and often necessary for probes that are not in the software and when specific signal is low, or close to background signal. Set a high pass filter, found under the update tab, for 640 nm, which corresponds to the lower end of the emission curve for turboFP650. A low pass filter constraint can also be set, but it was unnecessary for the imaging analysis shown here.
Quantify fluorescent signal from the unmixed image using a region of interest (ROI) set to radiant efficiency (photons/sec/cm2/sr)/μW) and set all images to the same scale (Figure 3).
At a desired time point, mice can be sacrificed and analyzed for engraftment of hESC-derived cells by flow cytometry. For intraperitoneally injected NK cells we typically analyze the peripheral blood (facial vein bleed), spleen, and peritoneum. Peritoneal washes can be performed by exposing just the peritoneal membrane and making a medial incision just wide enough to allow a glass Pasteur pipette to access the peritoneal cavity. Using sterile PBS, perform several washes of the peritoneal cavity making sure to mix solution thoroughly by rotating the mouse. Collect enough fluid until you have a visible pellet following centrifugation (typically 5-10 ml of wash).
Representative Results
The generation of hematopoietic progenitor cells using the spin EB approach allows for optimal NK cell development from hESCs and iPSCs. As demonstrated in Figure 2, day 11 spin EBs contain high percentages of progenitor cells expressing CD34, CD45, CD43, and CD31. High levels of CD34 and CD45 allows direct transfer to NK conditions without need for sorting or supporting stromal cells. If there is suboptimal spin EB differentiation, it is recommended that stromal cells such as EL08-1D2 are used in the secondary NK conditions. Following 4 weeks of culture the spin EBs give rise to large numbers of NK cells (approximately 1-2 x 106 cells per well of a 24-well plate). Cultures can also be phenotyped at earlier time points to see the gradual progression of NK cell development in this system 7. NK cells maintain the expression of reporter genes that were modified into the parent ES/iPS line and therefore can then be followed in vivo. Here, we have used the IVIS Spectrum to demonstrate simultaneous imaging of both NK cells and tumor cells in the same mouse (Figure 3). Using the spectral unmixing feature in Living Image software, tissue autofluorescence can be subtracted to optimize signal from fluorescently labeled cells.
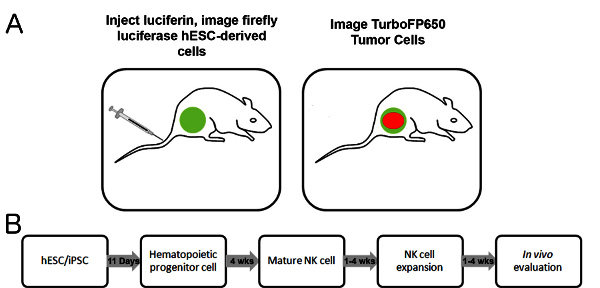 Figure 1. Dual imaging scheme and developmental timeline. A) Schematic of in vivo model. Mice are injected IP with luciferin, and following proper incubation time, firefly luciferase+ cells are imaged for bioluminescent signal. Following acquisition of the bioluminescent image, fluorescent signal from TurboFP650+ cells can be imaged. B) Timeline for the development and testing of hESC-derived NK cells.
Figure 1. Dual imaging scheme and developmental timeline. A) Schematic of in vivo model. Mice are injected IP with luciferin, and following proper incubation time, firefly luciferase+ cells are imaged for bioluminescent signal. Following acquisition of the bioluminescent image, fluorescent signal from TurboFP650+ cells can be imaged. B) Timeline for the development and testing of hESC-derived NK cells.
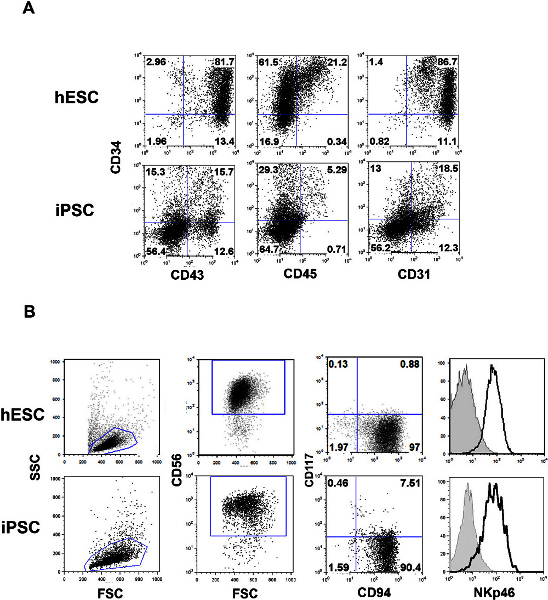 Figure 2. Phenotype of spin EBs and NK cells derived from hESCs and iPSCs. A) Following 11 days in spin EB culture, cells can be analyzed by flow cytometry. The day 11 time point gives high percentages of CD34, CD43, CD45, and CD31 expressing cells optimal for NK cell differentiation. B) NK cells can be detected using flow cytometry following 4 weeks of NK cell culture. Cells within the lymphocyte gate (FSC/SSC plot) are analyzed for the expression of CD56. CD56+ cells can be further analyzed for co-expression of markers such as CD117, CD94, NKp46, or others. Click here to view larger figure.
Figure 2. Phenotype of spin EBs and NK cells derived from hESCs and iPSCs. A) Following 11 days in spin EB culture, cells can be analyzed by flow cytometry. The day 11 time point gives high percentages of CD34, CD43, CD45, and CD31 expressing cells optimal for NK cell differentiation. B) NK cells can be detected using flow cytometry following 4 weeks of NK cell culture. Cells within the lymphocyte gate (FSC/SSC plot) are analyzed for the expression of CD56. CD56+ cells can be further analyzed for co-expression of markers such as CD117, CD94, NKp46, or others. Click here to view larger figure.
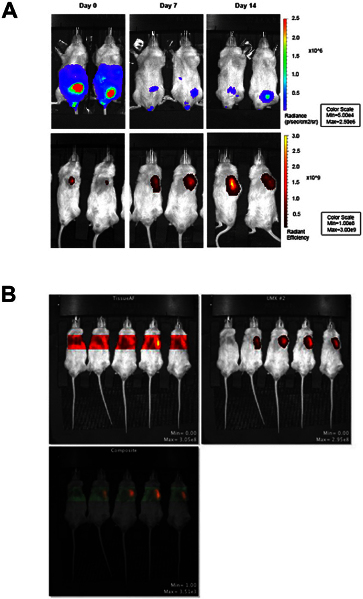 Figure 3. Fluorescent and bioluminescent imaging in vivo using the IVIS Spectrum. In vivo monitoring is the primary aim of this protocol. Therefore, mice are not followed out for tumor clearance. Our previous report (using 200,000 K562 cells) demonstrates that tumor clearance occurs at three weeks 7. A) Bioluminescent imaging of Fluc+hESC-derived NK cells on day 0, 7, and 14 after NK cell injection is shown in the top row. Fluorescent imaging of TurboFP650+ K562 cells on day 0, 7, and 14 after NK cell injection is shown in the bottom row. Fluorescent images were acquired immediately following bioluminescent image acquisition. B) Sequence showing tissue autofluorescence and fluorescent signal from turboFP650+ cells, generated using the spectral unmixing feature in Living Image software. The mouse on the far left in each image is a non-injection control and only shows the background signal. Click here to view larger figure.
Figure 3. Fluorescent and bioluminescent imaging in vivo using the IVIS Spectrum. In vivo monitoring is the primary aim of this protocol. Therefore, mice are not followed out for tumor clearance. Our previous report (using 200,000 K562 cells) demonstrates that tumor clearance occurs at three weeks 7. A) Bioluminescent imaging of Fluc+hESC-derived NK cells on day 0, 7, and 14 after NK cell injection is shown in the top row. Fluorescent imaging of TurboFP650+ K562 cells on day 0, 7, and 14 after NK cell injection is shown in the bottom row. Fluorescent images were acquired immediately following bioluminescent image acquisition. B) Sequence showing tissue autofluorescence and fluorescent signal from turboFP650+ cells, generated using the spectral unmixing feature in Living Image software. The mouse on the far left in each image is a non-injection control and only shows the background signal. Click here to view larger figure.
Discussion
hESCs are an ideal platform to study diverse cell types and hold remarkable potential for clinical translation. We use a defined, spin EB approach to differentiate hESC/iPSCs to hematopoietic progenitor cells. The spin EB approach has yielded consistent derivation of hematopoietic progenitor cells and differentiation to NK cells; yet, variation still exists in differentiation efficiency across cell lines and may need to be modified for generation of other hematopoietic cell lineages. While comparable results can be obtained from derivation in other EB or stromal culture methods, this system is serum-free and represents a more defined approach for producing hematopoietic progenitors compared to other methods. We have also standardized an efficient method of deriving NK cells from these hematopoietic progenitors in a similarly feeder-free manner, which overcomes a major concern for clinical translation. It also provides a defined system to study NK cell development in vitro. Extensive pre-clinical, in vivo characterization of hematopoietic cells, such as NK cells, is also necessary to specifically define these experimentally derived cell types and their implementation to clinical trials.
Advances in imaging and microscopy techniques have enabled long-term and non-invasive in vivo monitoring of cells expressing bioluminescent and fluorescent reporters15-17,19,21. Most notable is the use of the firefly luciferase construct, which has been used to visualize diverse cell types since its initial characterization. This is an ideal reporter for in vivo imaging, but is limited to only monitoring one cell population. The biggest advantage of a dual-reporter system is the ability to follow the interactions between two distinct cell populations. However, few reporters have been developed that are both easy to image in vivo and have sufficient and quantifiable signal above background.
TurboFP650, the fluorescent protein used in our dual-imaging scheme, is far-red shifted, which gives signal distinct from background autofluorescence. However, utilization of spectral unmixing methods in Living Image software (Caliper Life Sciences) was necessary to maximize the signal to background ratio in our system. There are many features in the Living Image software able to help with the intrinsic challenges of fluorescent protein imaging. Still, signal penetrance remains a challenge. The system needs further optimization for imaging of small cell populations or imaging of dispersed cells, as would be the case if the tumor cells were injected IP. However, the consistency and ease of fluorescent imaging makes its use an ideal platform to use in a dual imaging model. The animals are under anesthesia for less time and there is no need to deliver a second substrate as with most dual-luciferase systems.
This method of derivation, expansion, and dual-imaging in vivo provides a consistent approach for producing hESC-derived or iPSC-derived NK cells and their evaluation which is necessary to improve current NK cell adoptive therapies. The dual fluorescent and bioluminescent imaging model is broadly applicable for long-term study of two distinct cell populations other than anti-tumor model we have shown.
Disclosures
This work was supported by NIH/NHLBI R01-HL77923 (D.S.K.) and by NIH MSTP grant T32 GM008244 (D.A.K.), Stem Cell Biology Training Grant T32 (D.A.K.)(T32HD060536), Undergraduate Research Opportunities Program (UROP) Grant, University of Minnesota (A.M.B), the Leukemia Research Fund of the University of Minnesota Cancer Center, and the William L and Blanche Hughes Foundation.
Acknowledgments
The authors would like to thank Melinda Hexum for initiation of the spin EB protocol within our lab. We would like to thank other members of the lab, including Laura E. Bendzick, Michael Lepley, and Zhenya Ni for their technical assistance with this work. The authors would also like to thank Brad Taylor at Caliper Life Sciences for his expert technical advice.
References
- Keller GM. In vitro differentiation of embryonic stem cells. Current Opinion in Cell Biology. 1995;7:862–869. doi: 10.1016/0955-0674(95)80071-9. [DOI] [PubMed] [Google Scholar]
- Kaufman DS. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proceedings of the National Academy of Sciences. 2001;98:10716–10721. doi: 10.1073/pnas.191362598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman DS. Toward clinical therapies using hematopoietic cells derived from human pluripotent stem cells. Blood. 2009;114:3513–3523. doi: 10.1182/blood-2009-03-191304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng ES, Davis R, Stanley EG, Elefanty AG. A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies. Nature Protocols. 2008;3:768–776. doi: 10.1038/nprot.2008.42. [DOI] [PubMed] [Google Scholar]
- Ng ES, Davis RP, Azzola L, Stanley EG, Elefanty AG. Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood. 2005;106:1601–1603. doi: 10.1182/blood-2005-03-0987. [DOI] [PubMed] [Google Scholar]
- Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood. 2005;105:617–626. doi: 10.1182/blood-2004-04-1649. [DOI] [PubMed] [Google Scholar]
- Woll PS, et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. 2009;113:6094–6101. doi: 10.1182/blood-2008-06-165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. Journal of Immunology. 2005;175:5095–5103. doi: 10.4049/jimmunol.175.8.5095. [DOI] [PubMed] [Google Scholar]
- Ni Z, et al. Human Pluripotent Stem Cells Produce Natural Killer Cells That Mediate Anti-HIV-1 Activity by Utilizing Diverse Cellular Mechanisms. Journal of Virology. 2011;85:43–50. doi: 10.1128/JVI.01774-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng ES, Davis RP, Hatzistavrou T, Stanley EG, Elefanty AG. Directed differentiation of human embryonic stem cells as spin embryoid bodies and a description of the hematopoietic blast colony forming assay. Current Protocols in Stem Cell Biology. 2008;Chapter 1:Unit 1D.3. doi: 10.1002/9780470151808.sc01d03s4. [DOI] [PubMed] [Google Scholar]
- Denman CJ, et al. Membrane-Bound IL-21 Promotes Sustained Ex Vivo Proliferation of Human Natural Killer Cells. PLoS ONE. 2012;7:e30264. doi: 10.1371/journal.pone.0030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanchi SS, Senyukov VV, Denman CJ, Lee DA. Expansion, Purification, and Functional Assessment of Human Peripheral Blood NK Cells. J. Vis. Exp. 2011. p. e2540. [DOI] [PMC free article] [PubMed]
- Tian X, et al. Bioluminescent imaging demonstrates that transplanted human embryonic stem cell-derived CD34+ cells preferentially develop into endothelial cells. Stem Cells. 2009;27:2675–2685. doi: 10.1002/stem.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilber A, et al. Efficient and stable transgene expression in human embryonic stem cells using transposon-mediated gene transfer. Stem Cells. 2007;25:2919–2927. doi: 10.1634/stemcells.2007-0026. [DOI] [PubMed] [Google Scholar]
- Shcherbo D, et al. Near-infrared fluorescent proteins. Nature Methods. 2010;7:827–829. doi: 10.1038/nmeth.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Morange M, Bensaude O. Firefly luciferase luminescence assays using scintillation counters for quantitation in transfected mammalian cells. Analytical Biochemistry. 1988;171:404–408. doi: 10.1016/0003-2697(88)90505-2. [DOI] [PubMed] [Google Scholar]
- Sadikot RT, Blackwell TS. Bioluminescence Imaging. Proceedings of the American Thoracic Society. 2005;2:537–540. doi: 10.1513/pats.200507-067DS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos EB, et al. Sensitive in vivo imaging of T cells using a membrane-bound Gaussia princeps luciferase. Nature Medicine. 2009;15:338–344. doi: 10.1038/nm.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiological Reviews. 2010;90:1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- Knorr DA, Bock A, Brentjens RJ, Kaufman DS. Engineered Human Embryonic Stem Cell-Derived Lymphocytes to Study In Vivo Trafficking and Immunotherapy. Stem Cells Dev. 2013;28:28. doi: 10.1089/scd.2012.0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN, Robey EA, Cahalan MD. A Decade of Imaging Cellular Motility and Interaction Dynamics in the Immune System. Science. 2012;336:1676–1681. doi: 10.1126/science.1221063. [DOI] [PMC free article] [PubMed] [Google Scholar]