

Inactivation of Cdc48p/p97 triggers formation of a complex that contains the 26S proteasome, Cdc48p/p97, ubiquitinated substrates, select components of the Hrd1 complex, and the lumenal recognition factor, Yos9p. A model is proposed in which the Hrd1 complex links substrate recognition and degradation on opposite sides of the ER membrane.
Abstract
During endoplasmic reticulum–associated degradation (ERAD), misfolded lumenal and membrane proteins in the ER are recognized by the transmembrane Hrd1 ubiquitin ligase complex and retrotranslocated to the cytosol for ubiquitination and degradation. Although substrates are believed to be delivered to the proteasome only after the ATPase Cdc48p/p97 acts, there is limited knowledge about how the Hrd1 complex coordinates with Cdc48p/p97 and the proteasome to orchestrate substrate recognition and degradation. Here we provide evidence that inactivation of Cdc48p/p97 stalls retrotranslocation and triggers formation of a complex that contains the 26S proteasome, Cdc48p/p97, ubiquitinated substrates, select components of the Hrd1 complex, and the lumenal recognition factor, Yos9p. We propose that the actions of Cdc48p/p97 and the proteasome are tightly coupled during ERAD. Our data also support a model in which the Hrd1 complex links substrate recognition and degradation on opposite sides of the ER membrane.
INTRODUCTION
The endoplasmic reticulum (ER) maintains an optimized environment for the folding and maturation of secreted and membrane proteins. However, the folding process may fail because of intracellular and external stresses and genetic mutations. These insults can result in the production of irrevocably misfolded proteins, which may trigger numerous diseases such as diabetes and neurodegeneration. Therefore ER quality control (ERQC), which detects and eliminates faulty proteins, is crucial to maintain cellular homeostasis.
One system that regulates ERQC is the unfolded protein response (UPR; Mori, 2009; Walter and Ron, 2011). In Saccharomyces cerevisiae, the accumulation of misfolded proteins elicits the UPR by activating the transmembrane kinase/nuclease Ire1p, which initiates the splicing of HAC1 mRNA. This leads to the production of Hac1p, which in turn facilitates the transcription of genes that enable the cell to cope with and fold aberrant proteins. In contrast, an inherent component of ERQC is a process known as ER-associated degradation (ERAD). Here misfolded proteins in the ER are retrotranslocated into the cytosol, polyubiquitinated, and degraded by the proteasome (Vembar and Brodsky, 2008; Xie and Ng, 2010; Bagola et al., 2011; Hampton and Sommer, 2012; Wolf and Stolz, 2012). Perhaps not surprisingly, some components of the ERAD machinery are up-regulated by the UPR, and simultaneous loss of the UPR and ERAD compromises cell survival (Friedlander et al., 2000; Travers et al., 2000; Jonikas et al., 2009). Together the UPR and ERAD alleviate ER stress by their coordinated disposal of potentially damaged proteins.
In yeast, the transmembrane Hrd1p/Der3p E3 ubiquitin ligase forms a core oligomeric complex with Hrd3p, Usa1p, and Der1p and specifically recognizes misfolded lumenal and membrane substrates for ubiquitination (Vembar and Brodsky, 2008; Xie and Ng, 2010; Bagola et al., 2011; Hampton and Sommer, 2012; Wolf and Stolz, 2012). Evidence suggests that Hrd1p directly recognizes both lumenal (Carvalho et al., 2010) and membrane substrates (Sato et al., 2009), but the precise selection of at least lumenal substrates relies on Hrd3p's lumenal domain, as well as on the lectin Yos9p (Kim et al., 2005; Szathmary et al., 2005; Denic et al., 2006; Gauss et al., 2006a) and Der1p, which associates with the Hrd1 core complex via Usa1p (Knop et al., 1996; Gauss et al., 2006b; Carvalho et al., 2006; Horn et al., 2009; Stanley et al., 2011). Once recognized, these ERAD substrates are retrotranslocated across the membrane to the cytosol and ubiquitinated by Hrd1p with the aid of the ubiquitin-conjugating enzyme Ubc7p (Bordallo et al., 1998; Bays et al., 2001).
In contrast to our understanding of the early recruitment events, later events during and after retrotranslocation are less clear. The existence of proteinous retrotranslocation channels has been proposed, and candidates include Hrd1p, Sec61p, and Der1p (Derlins in mammals). There is limited direct evidence in support of candidates (for reviews see Bagola et al., 2011; Hampton and Sommer, 2012), although studies indicate a specific interaction of lumenal substrates with Hrd1p (Carvalho et al., 2010). After ubiquitination, it is generally accepted that substrates are extracted and/or segregated from the ER by the Ubx2p-mediated recruitment of the Cdc48p/p97 AAA-ATPase (Ye et al., 2001; Jarosch et al., 2002; Neuber et al., 2005; Schuberth and Buchberger, 2005; Nakatsukasa et al., 2008; Garza et al., 2009), which, together with other ubiquitin-binding proteins, escort substrates to the proteasome (Medicherla et al., 2004; Richly et al., 2005).
Components of the Hrd1 core complex are transcriptionally up-regulated by the UPR, but the consequences of the UPR on Hrd1 complex integrity have not been assessed. In addition, because of technical obstacles to detecting the transient interactions between the Hrd1 core complex, Cdc48p/p97, the proteasome, and ubiquitinated substrates, a direct analysis of how these enzyme complexes coordinately link substrate recognition, retrotranslocation, and degradation across the ER membrane is lacking. To address these issues, we searched for a condition in which dynamic remodeling of the Hrd1p complex and its interaction partners could be monitored. We demonstrate that the oligomeric state of Hrd1p is maintained during the UPR; however, the inactivation of Cdc48p/p97 triggers the formation of a stalled retrotranslocation complex that contains Cdc48p/p97, the proteasome, ubiquitinated substrates, and Hrd1p. Because the stalled complex encapsulates Hrd3p, Usa1p, and the lumenal recognition factor, Yos9p, we propose that Hrd1p physically links substrate recognition and degradation. Moreover, our data indicate that factors required for recognition, retrotranslocation, and degradation can assemble onto an ERAD substrate in the cytoplasm in a nonordered manner.
RESULTS
The oligomeric state of Hrd1p is maintained upon UPR induction
To identify a condition in which the oligomeric state and/or the interaction partners of the Hrd1 complex could be monitored, we resolved digitonin-solubilized membranes by sucrose density gradient centrifugation and analyzed the distribution pattern of the Hrd1 core complex by SDS–PAGE and Western blotting (Figure 1, A–D, wt). Consistent with previous studies (Carvalho et al., 2006; Horn et al., 2009), Hrd1p, Hrd3p, Usa1p, and Der1p comigrated in fractions 9 and 10, which correspond to the oligomeric Hrd1 core complex. The Sec61p translocation channel showed a different distribution pattern (Figure 1E). When Hrd3p was absent, the Hrd1p peak shifted to a lighter fraction (fraction 7), suggesting the stoichiometric interaction between Hrd1p and Hrd3p (Gardner et al., 2000). Under this condition, Usa1p and Der1p also appeared in significantly lighter fractions (fractions 5 and 6). This most likely arises from depleted amounts of Hrd1p due to its degradation in hrd3Δ cells (Gardner et al., 2000), which in turn liberates Usa1p and Der1p from Hrd1p. In addition, when Usa1p was depleted, the Hrd1p and Hrd3p peak shifted to a lighter fraction (fraction 7), and Der1p no longer associated with Hrd1p, as the protein was now most abundant in fraction 5. These results are consistent with data indicating that Usa1p facilitates the oligomerization of Hrd1p and recruits Der1p to the Hrd1 core complex (Carvalho et al., 2006; Horn et al., 2009).
FIGURE 1:

Sucrose density gradient analysis of the Hrd1 core complex in retrotranslocation-defective cells. (A–E) Membrane fractions were solubilized with 1% digitonin and subjected to sucrose density gradient analysis. Fractions were collected and analyzed by Western blotting with anti-Hrd1p (A), -Hrd3p (B), -Usa1p (C), -Der1p (D), and -Sec61p (E) antibodies. Cells were cultured at 30°C, except that cdc48-3 cells were cultured at 25°C and shifted to 37°C for 1 h. Where indicated, cells were treated with 5 mM DTT or 2 μg/ml tunicamycin for 1 h. The proteasome was inactivated by treatment with 50 μM of MG132 for 1 h. The expression of an active form of Hac1p (Hac1(i)) was induced under the control of the GAL1 promoter from a low-copy plasmid for 4 h in galactose-containing media. Distribution patterns of the Hrd1 core complex and Sec61p were unchanged when wild-type cells were treated with dimethyl sulfoxide (the solvent for MG132), when wild-type cells with an empty vector were cultured in galactose-containing media for 4 h (vector only control for Hac1(i)) or when wild-type cells were shifted to 37°C for 1 h (as a control for cdc48-3 cells; data not shown). Dashed lines depict fractions that correspond to the oligomeric Hrd1 core complex, and arrows denote the migrations of components mentioned in the text. The asterisk in B indicates unglycosylated Hrd3p that was generated by the treatment of cells with tunicamycin. The migrations of molecular mass markers are also indicated.
When cells were exposed to ER stress by dithiothreitol (DTT), which prevents disulfide bond formation, the migration of oligomeric Hrd1p was barely changed (Figure 1A). However, the levels of Usa1p and Der1p were up-regulated twofold to threefold in a Hac1p- dependent manner (Figure 2, A and B), indicating that up-regulation occurs downstream of UPR signaling. In addition, these proteins were most abundant in fractions 5–7 (Figure 1, C and D), suggesting that excess Usa1p and Der1p were not incorporated into the Hrd1 core complex. Further, the level of Hrd3p was moderately decreased (∼0.8-fold; Figure 2A), and nearly 60% of Hrd3p distributed in higher–molecular weight fractions (Figure 1B, fractions 11–17 and asterisk), whereas a portion of Hrd3p remained in the core complex. These data suggest that at least a part of Hrd3p was inactive. A similar distribution pattern of the core complex was observed when ER stress was induced by tunicamycin, which inhibits N-linked glycosylation. Such imbalanced and altered distribution patterns most likely occur because the damaged Hrd3p species inefficiently binds to and stabilizes Hrd1p (Supplemental Figure S1). Pleiotropic effects caused by these compounds might also generally affect the architecture of the Hrd1 complex, which could arise from misfolding of other lumenal and membrane proteins, a change of the redox status of membrane lipids (especially by DTT), and/or UPR-independent membrane expansion (Schuck et al., 2009).
FIGURE 2:
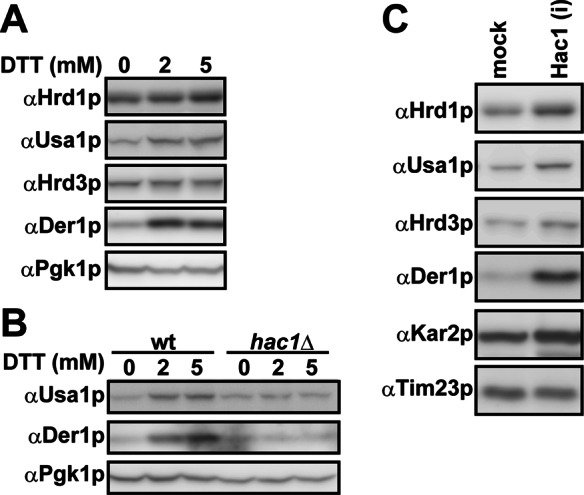
Balanced vs. imbalanced up-regulation of the Hrd1 complex upon UPR activation. (A, B) Wild-type or hac1Δ cells were treated with 0, 2, or 5 mM DTT for 1 h at 30°C. The levels of components in the Hrd1 core complex were assessed by Western blotting. Pgk1p, a cytosolic protein, served as a loading control. (C) Overexpression of Hac1(i) up-regulates the Hrd1 core complex. The expression of Hac1(i) was induced as described. The levels of components in the Hrd1 core complex were assessed by Western blotting. Kar2p (BiP) and Tim23p served as a positive and a loading control, respectively. Note that DTT treatment causes a portion of Hrd3 to aggregate (Figure 1) and is thus unable to enter the gel; thus the amount of this protein does not increase in A.
To more directly analyze the consequence of the UPR on complex integrity, we activated the UPR by overexpressing a constitutively active form of Hac1p (designated here as Hac1(i)). In this case, there was controlled up-regulation and maintenance of the oligomeric Hrd1 core complex (Figure 1, A–D, Hac1(i)). In addition, we found that the levels of each component increased: 1.6-fold (Hrd1p), 1.8-fold (Hrd3p), 1.8-fold (Usa1p), and 4.1-fold (Der1p) (Figure 2C). Therefore the UPR induces the expression of each of the components that constitute the Hrd1 core complex and preserves its functional oligomeric state. In contrast, chemical perturbation of lumenal protein homeostasis and possible damage to the degradation machinery cause defects in core complex formation.
The Hrd1 complex is remodeled when retrotranslocation stalls
We next asked whether the Hrd1 core complex was remodeled when degradation was blocked. On thermal inactivation of a Cdc48p mutant (cdc48-3 allele), the Hrd1p peak shifted slightly but reproducibly to higher–molecular weight fractions (Figure 1A, cdc48-3, fractions 10 and 11). The Hrd3p, Usa1p, and Der1p peaks also shifted to higher–molecular weight fractions (Figure 1, B–D, cdc48-3, fractions 10 and 11). A similar result was obtained when solubilized membranes from ubx2Δ cells were resolved (Figure 1, A–D, ubx2Δ). In contrast, when the activity of the proteasome was blocked, no significant change was observed in the distribution patterns of the Hrd1 core complex (MG132). These results indicate that the Hrd1 core complex may be remodeled when Cdc48p is inactivated.
Sucrose gradient analysis is a relatively harsh method by which weakly and/or transiently associated proteins might be dissociated. To better probe for the presence of other components as well as for ubiquitinated proteins, we immunoprecipitated a functional tagged form of Hrd1p-3FLAG (Supplemental Figure S2) from digitonin or Triton X-100–solubilized membranes. In Cdc48p-defective cells, ubiquitinated proteins coprecipitated with Hrd1p-3FLAG (Figure 3, A, lanes 10 and 24; B, lanes 6 and 12) in a Ubc7p-dependent manner (Figure 3A, lanes 12 and 26). Of importance, nearly quantitative dissociation of ubiquitinated proteins was observed under denaturing conditions (i.e., solubilization with SDS/urea; Figure 3B, lane 18). Therefore most but not all ubiquitinated proteins coprecipitated with Hrd1p represent ubiquitinated substrates. When Usa1p was absent, the binding of ubiquitinated proteins to the Hrd1 core complex was maintained in digitonin-solubilized lysate (Figure 3A, lane 14), but this interaction was Triton X-100 sensitive (lane 28). Thus Usa1p may help to maintain ubiquitinated substrates on the Hrd1 core complex.
FIGURE 3:

Ubiquitinated substrates associate with the Hrd1 core complex upon Cdc48p inactivation. (A, B) Digitonin (1%)- or Triton X-100 (0.75%)-solubilized membranes were prepared from the indicated strains, and 3xFLAG-tagged Hrd1p was immunoprecipitated with anti-FLAG antibody. Coprecipitated proteins were analyzed by Western blotting with specific antibodies. Cells were cultured at 25°C and shifted to 37°C for 1 h to inactivate Cdc48-3p before membrane fractions were prepared. Where indicated, membranes were solubilized with SDS (1%)/urea (4 M). The concentration of SDS/urea was diluted 10-fold with 1% Triton X-100 before immunoprecipitation. The asterisk shows a nonspecific band that cross-reacted with anti-Sec61p antibody.
The amount of Usa1p and Der1p coprecipitated with Hrd1p-3FLAG was unaltered by Cdc48p inactivation, irrespective of the detergent. However, we observed an ∼2-fold increase in the amount of Hrd3p and Yos9p that coprecipitated with Hrd1p-3FLAG in Triton X-100–solubilized lysates (Figure 3, A, compare lanes 23 and 24; B, lanes 11 and 12). Increased association was Ubc7p (Figure 3A, lane 26) and Usa1p (Figure 3A, lane 28) independent. The nature of this phenomenon is unclear, but it implies Cdc48p-dependent remodeling between Hrd1p and the Hrd3p–Yos9p surveillance complex. Cdc48p-dependent but Usa1p-independent remodeling of the Hrd1 core complex was also suggested by sucrose density gradient analysis when the integrity of the Hrd1 core complex was examined in cdc48-3, usa1Δ, and cdc48-3usa1Δ cells (Supplemental Figure S3). Of interest, the mutant form of Cdc48p (Cdc48-3p) interacted more avidly with Hrd1p (Figure 3, A, lanes 10 and 24; B, lanes 6 and 12). This was probably because Cdc48-3p, which has two point mutations in the first AAA ATPase domain (P257L and R387K; Verma et al., 2011; see also Materials and Methods), failed to dissociate completely from the Hrd1 core complex and/or from ubiquitinated substrates. Overall, a block in retrotranslocation rearranged Hrd1 core complex interactions and led to the accumulation of ubiquitinated substrates on the complex. As a consequence of these multiple events, the Hrd1 core complex was remolded so that it migrated at an apparent greater molecular mass.
Ubiquitinated substrates on the Hrd1 core complex bridge interaction with Ubx2p
The fact that Hrd1p associated with ubiquitinated proteins in cdc48-3 yeast implied that incompletely extracted and ubiquitinated substrates also associated with downstream components. One candidate for a downstream component was Ubx2p, which helps recruit Cdc48p to the Hrd1 core complex (Neuber et al., 2005; Schuberth and Buchberger, 2005). To test this hypothesis, we immunoprecipitated a functional tagged form of Ubx2p (Supplemental Figure S2) from cdc48-3 cells. As shown in Figure 4A, a significant amount of ubiquitinated proteins coimmunoprecipitated with Ubx2p from cdc48-3 cells in a Ubc7p-dependent manner (lanes 8 and 10). Moreover, when Cdc48p was inactivated, more Ubx2p was recovered from denser fractions in the sucrose gradient (Figure 4B, top, fractions 11–17), and ubiquitinated proteins bound to Ubx2p were preferentially recovered (bottom). Note that Ubx2p itself was not ubiquitinated because the majority of the ubiquitinated species that coprecipitated with Ubx2p dissociated under denaturing conditions; moreover, Ubx2p is a relatively stable protein (Supplemental Figure S4).
FIGURE 4:

Ubx2p-associated ubiquitinated substrates bridge Ubx2p to the Hrd1 core complex. (A) Digitonin-solubilized membrane fractions were prepared from the indicated cells, and Ubx2p-3FLAG was immunoprecipitated with anti-FLAG antibody. Ubiquitinated proteins that coprecipitated with Ubx2p-3FLAG were detected with anti-ubiquitin antibody. (B) After sucrose density gradient ultracentrifugation, performed as in Figure 1, Ubx2p-3FLAG was immunoprecipitated with anti-FLAG antibody under native conditions. Ubiquitinated proteins were detected as in A. Top, arrow indicates the Ubx2p-3FLAG peak in the gradient. Bottom, arrow indicates ubiquitinated proteins that coprecipitated with Ubx2p-3FLAG from cdc48-3 cells. Note that a 4% step gradient (4–32%) was used instead of a 5% step gradient to facilitate the immunoprecipitation of Ubx2p-3FLAG, particularly from denser sucrose fractions. Under this condition, the peak of Hrd1p appeared in fractions 14 and 16 for the CDC48 and cdc48-3 lysates, respectively. (C) Digitonin (1%)- or Triton X-100 (0.75%)-solubilized membrane fractions were prepared from the indicated strains. Ubx2p-3FLAG was immunoprecipitated with anti-FLAG antibody, and coprecipitated proteins were analyzed with specific antibodies. The amount of the Hrd1 core complex that coprecipitated with Ubx2p was quantified based on quantification of immunoprecipitated Ubx2p.
We next analyzed the interaction of Ubx2p with the Hrd1 core complex in cdc48-3 cells. As anticipated, 1.5- to 2.0-fold more Hrd1p, Usa1p, Hrd3p, and Der1p coprecipitated with Ubx2p from Cdc48p-defective yeast than from wild-type cells in the presence of digitonin (Figure 4C, compare lanes 7 and 8). The interaction between Ubx2p and the Hrd1 core complex in cdc48-3 cells was maintained regardless of whether digitonin or Triton X-100 was used (lanes 17 and 18). When Ubc7p was depleted from cdc48-3 cells, the binding of Ubx2p to the Hrd1 core complex was also maintained in digitonin-solubilized lysate (lane 10); however the interaction was Triton X-100 sensitive (lane 20). These data suggest that the interaction between Ubx2p and the Hrd1 core complex is weaker in the absence of ubiquitinated proteins.
To test this hypothesis, we uncoupled Ubx2p binding from Cdc48p by deleting the UBX domain at its C-terminus. As a result, more ubiquitinated proteins and 2.0- to 2.4-fold more Hrd1p, Usa1p, Hrd3p, and Der1p bound to Ubx2pΔUBX (Figure 5A, lane 9). However, the added deletion of the UBA domain, which is located at the N-terminus and may bind to ubiquitin, reduced the extent of the interaction between Ubx2pΔUBXΔUBA and the complex (lane 10). Note that the deletion of UBA domain of Ubx2p did not slow the degradation of CPY*, a soluble ERAD substrate (Wang and Lee, 2012; also see Supplemental Figure S5); in contrast, insertion of an epitope tag at the N-terminus of Ubx2p inhibits ERAD (Neuber et al., 2005). Although the precise role of the N-terminal UBA domain of Ubx2p during ERAD is unknown, ubiquitinated substrates may be recognized redundantly by several UBA-containing proteins and can be delivered to other pathway(s) en route to the proteasome when Ubx2p's UBA domain is deleted. To confirm the specific nature of the identified interactions, we also demonstrated that polyubiquitin chain cleavage on microsomal proteins decreased interaction of Ubx2p with Hrd1p, even when Cdc48p was inactivated (Figure 5B, lane 7 vs. lane 8). Therefore Cdc48p inactivation leads to the accumulation of ubiquitinated proteins, which bridge the interaction between Ubx2p and the Hrd1 core complex (Figure 5C).
FIGURE 5:

The interaction between Ubx2p and the Hrd1 complex in cdc48-3 cells is ubiquitin chain dependent. (A) Digitonin (1%)-solubilized membranes were prepared from ubx2Δ cells expressing 3xFLAG-tagged Ubx2p or the indicated mutants lacking the UBA and/or UBX domains from low-copy plasmids under the control of the endogenous promoter. Proteins were immunoprecipitated with anti-FLAG antibody, and bound proteins were analyzed by Western blotting with specific antibodies. Note that ubiquitinated proteins bound to wild-type Ubx2p (lane 7) were more abundant than those bound to chromosomally expressed Ubx2p (e.g., Figure 4A, lane 7). This was probably due to different levels of Ubx2p expression from the plasmid (this figure) compared with the chromosome-integrated gene in Figure 4A. The asterisks show nonspecific bands that cross-reacted with anti-FLAG antibody (αFLAG) and anti-Sec61p antibody (αSec61), respectively. (B) Membranes were prepared from CDC48 and cdc48-3 cells expressing chromosomally integrated Ubx2p-3FLAG that were shifted to 37°C for 1 h. The membranes were then incubated with His6-USP2CD (UBP41 catalytic domain; DUB +) or an identical amount of BSA (DUB –) at 37°C for 1 h. After the reaction was quenched with 10 mM NEM, the membranes were solubilized with 1% digitonin and subjected to immunoprecipitation with anti-FLAG antibody as in A. Note that 10 mM NEM, which prevents the nonspecific degradation of ubiquitin chains by deubiquitinating enzymes, was omitted in this experiment. Therefore the extent of the polyubiquitin chains in lysates and Ubx2p-3FLAG immunoprecipitates was less than that in Figure 4A. (C) Cdc48p inactivation leads to the accumulation of ubiquitinated proteins, which bridge the interaction between Ubx2p and the Hrd1 core complex. Note that only a portion of the ubiquitinated protein substrate is drawn as a dotted line, because it may loosely associate with Hrd1p.
Proteasome accumulation on the Hrd1 complex upon Cdc48p inactivation
The widely accepted view of Cdc48p/p97’s function is that it actively extracts (and/or segregates) ubiquitinated substrates from an upstream macromolecular component and recruits processing factors such as deubiquitinating enzymes and peptide N-glycanase to substrates before proteasome entry (Elsasser and Finley, 2005; Richly et al., 2005). Based on this model, extraction and targeting to the proteasome occur in a sequential order, but one can imagine that the proteasome might be excluded from the observed Hrd1p-containing stalled complex that is generated upon Cdc48p inactivation. Alternatively, it is possible that the proteasome accesses the Hrd1 core complex, possibly via ubiquitinated substrates, before Cdc48p-mediated extraction is completed. In this case, degradation could start during extraction or after extraction is completed. A related mechanism was proposed for the Cdc48p-dependent degradation of Rpb1, the largest RNA pol II subunit, after ultraviolet irradiation (Verma et al., 2011).
To test this model, we analyzed the interaction of Rpt5p, which resides in the 19S regulatory particle of the proteasome, with the Hrd1 core complex. Because of the transient nature of interactions among the proteasome, the Hrd1/E3 ligase complex, and ubiquitinated substrates, we developed a liquid nitrogen lysis–cross-linking method (see Materials and Methods). After lysis, the extract was immediately treated with a chemical cross-linker (dithio-bis(succinimidyl propionate) [DSP]) to fix protein interactions, and Rpt5-3FLAG was immunoprecipitated. Next the bound proteins were detected by immunoblot analysis. As shown in Figure 6A, Rpt5p coprecipitated with the 20S proteasome and Cdc48p from both wild-type and cdc48-3 cells. The 26S proteasome integrity was previously shown to remain intact upon Cdc48p inactivation (Verma et al., 2011), and our results are consistent with this observation. However, significantly more Hrd1p, Usa1p, Hrd3p, and even the lumenal recognition factor Yos9p coprecipitated with Rpt5p from cdc48-3 cells. In contrast, Der1p and Sec61p were barely detected in the immunoprecipitates. The faint amount of Der1p in the Rpt5p-immunocomplex is consistent with the observation that Der1p is a peripherally associated protein that can be dissociated from the Hrd1p immunocomplex by a strong nonionic detergent (Figure 3). When the gene encoding Usa1p was deleted in cdc48-3 cells, we observed a decrease in Hrd1p-Rpt5p association, consistent with a suggested role for Usa1p in maintaining ubiquitinated substrates at the Hrd1 core complex (Figures 6B, lane 8, and 3A). Depletion of Ubc7p from cdc48-3 cells reduced binding of Hrd1p to Rpt5p (Figure 6B, lane 5). Taken together, these results strongly suggest that ubiquitinated substrates bound to Hrd1p facilitate proteasome–Hrd1p interaction and that the proteasome and Cdc48p bind these species noncompetitively.
FIGURE 6:

Increased proteasome association with the Hrd1 core complex when Cdc48p is inactivated. (A, B) The indicated yeast strains were lysed in liquid nitrogen, and proteins were cross-linked with DSP. The membranes were collected and solubilized, and 3xFLAG-tagged Rpt5p was immunoprecipitated. Bound proteins were detected with specific antibodies after DTT treatment. The asterisk shows a nonspecific band that that cross-reacted with anti-FLAG antibody after the immunoprecipitation. (C) A model for the formation of the stalled retrotranslocation complex (see the text for details). As in Figure 5C, note that only a part of the ubiquitinated protein is drawn as a dotted line, because it may loosely associate with Hrd1p.
DISCUSSION
In this study, we analyzed the architecture of the Hrd1 core complex under a variety of conditions and by using a combination of genetic and biochemical tools. We found that a stalled retrotranslocation complex contains Cdc48p/p97, the proteasome, ubiquitinated substrates, and Hrd1p (Figure 6A). Because the proteasome gains access to Hrd1p—possibly via ubiquitinated substrates even in the absence of active Cdc48p—our results indicate that the actions of Cdc48p and the proteasome are tightly coupled rather than sequential. Moreover, because the stalled complex contains select components of the Hrd1 core complex (Hrd1p, Hrd3p, and Usa1p) and the lumenal recognition factor, Yos9p, we suggest that Hrd1p links substrate recognition and proteasomal degradation on opposite sides of the ER membrane (Figure 6C). Normally, Cdc48p prevents the accumulation of this stalled complex by repeated cycles of extraction and/or disassembly of ubiquitinated substrates from the Hrd1 core complex, thereby facilitating ERAD.
Although previous studies indicated that Cdc48p provides the energy for retrotranslocation, its precise point of action has not been definitively established. During ERAD, Cdc48p might actively pull substrates through a putative retrotranslocon and/or extract hydrophobic transmembrane region of polytopic substrates (Rouiller et al., 2000; Ye et al., 2004; Zhong et al., 2004). Alternatively, Cdc48p might segregate ubiquitinated substrates that have already been retrotranslocated and remain bound to membrane-associated components (Rape et al., 2001). In some cases, retrotranslocation might instead require the ATPase activity of the 19S proteasome cap, thus subverting the Cdc48p requirement (Mayer et al., 1998; Lee et al., 2004; Kothe et al., 2005; Carlson et al., 2006; Wahlman et al., 2007; Lipson et al., 2008). Moreover, it has been suggested that p97/Cdc48p-associated deubiquitination activity is a prerequisite for retrotranslocation in mammalian cells (Ernst et al., 2009, 2011). These diverse models have been illustrated through the use of model substrates and by analyzing degradation mediated by the activities of enzymes in the ubiquitin-proteasome system. In the present study, we assessed the interactions of relevant macromolecular complexes and found that the catalytic cycle of Cdc48p regulates the spatial recruitment of the proteasome to the cytosolic surface of the Hrd1 complex. Moreover, the architecture within the complex (e.g., the Hrd1p–Hrd3p interaction) appeared to be altered when defects in Cdc48p function led to the formation of a stalled complex. Therefore we propose that the assembly of the Hrd1 complex is regulated by Cdc48p, a hypothesis that is being investigated in our laboratories.
On the basis of our model, we propose that the extraction of ERAD substrates from the ER membrane and their targeting to the proteasome are coupled and may not necessarily take place in a fixed order. Because extraction/segregation is likely to be much slower than targeting, we envision that the proteasome and Cdc48p simultaneously bind to emerging ubiquitinated substrates on Hrd1p; the proteasome then stands by for the completion of Cdc48p-mediated extraction/segregation of substrates. This state is probably transient under normal conditions. However, in Cdc48p-defective cells, the Hrd1p-proteasome association is maintained because the rate of extraction/segregation of ubiquitinated substrates slows. It is this state that we detected by chemical cross-linking.
Our data support the idea that ubiquitinated substrates associating with Hrd1p bridge the proteasome–Hrd1p interaction, because the majority of ubiquitinated proteins that were in complex with Hrd1p dissociated upon treatment with SDS/urea (Figure 3B). However, we do not rule out the possibility that other factors also facilitate this bridge. One such factor may be the polyubiquitin chain on Hrd1p that arises from autoubiquitination, which is known to occur in the absence of Hrd3p (Bays et al., 2001). Another possibility is that ubiquitin-binding proteins such as Ufd1p bridge the interaction between ubiquitinated substrates, Cdc48p, and the proteasome (see also Supplemental Figure S6). A detailed description of the interactions within the stalled complex awaits future studies.
Although the importance of the transmembrane domain of Hrd1p as a channel component remains to be established, especially for polytopic, integral membrane substrates (Garza et al., 2009), Hrd1p is considered a prime retrotranslocon candidate for lumenal substrates (Gauss et al., 2006b; Carvalho et al., 2010). Our study is in line with but significantly extends this notion. First, we discovered that the proteasome–Hrd1p complex also harbors a lumenal recognition factor; therefore substrates in the ER lumen are recognized and may be transferred to the proteasome through the Hrd1 core complex. Second, we found that other retrotranslocon candidates, including Der1p and Sec61p, were barely detectable in proteasome-containing immunoprecipitates (Figure 6A). Der1p in yeast supports the degradation of only lumenal soluble substrates (ERAD-L; Carvalho et al., 2006). Moreover, the binding of Der1p to Hrd1p is believed to be peripheral and is mediated by Usa1p (Carvalho et al., 2006; Horn et al., 2009). Indeed, a small amount of Der1p could be recovered in the Hrd1 immunocomplex when membranes were solubilized with a nonionic detergent Triton X-100 (Figure 3) or NP-40 (Gauss et al., 2006b), whereas a greater amount could be recovered in the presence of a milder detergent, digitonin (Figure 3). In contrast, Derlin-1 in mammals supports the degradation of many substrates that reside both in the ER lumen and membrane and is a central component of the membrane-associated retrotranslocation complex (Lilley and Ploegh, 2004; Ye et al., 2004). Therefore we suggest that Der1p may have an ancillary function or act upstream during the transfer of substrates from Hrd1p to the proteasome in yeast, whereas Derlin-1 in mammals is a key member of the ERAD machinery that facilitates retrotranslocation (Greenblatt et al., 2011).
Finally, our methods may be applicable to resolving the dynamic interactions within cellular quality control compartments, such as the juxtanuclear quality control compartment, which concentrates disaggregating chaperones and 26S proteasomes (Kaganovich et al., 2008). The residence and participation of the Hrd1 core complex in a specific compartment are unknown, but the methods developed in this study can be used to better characterize this and perhaps related complexes.
MATERIALS AND METHODS
Strains
Strains used in this study are listed in Supplemental Table S1. The gene insertion or disruption cassettes were amplified by PCR from the following plasmids: pFA6a-3FLAG-KanMX (a generous gift from Y. Tamura and H. Sesaki, Johns Hopkins University, Baltimore, MD) and pRS305, pUC18-CgTRP1, and pUC19-CgHIS3 (provided by the National Bio-Resource Project of the Ministry of Education, Culture, Sports, Science and Technology of Japan). The pdr5::HPH cassette was a generous gift from S. Mimura (Osaka University, Osaka, Japan). The pep4::LEU2 cassette was amplified by PCR from pRS305 using primers OKN113 (5′-ATG TTC AGC TTG AAA GCA TTA TTG CCA TTG GCC TTG TTG TTG GTC AGC GCA GAT TGT ACT GAG AGT GCA C-3′) and OKN114 (5′-TCA AAT TGC TTT GGC CAA ACC AAC CGC ATT GTT GCC CAA ATC GTA AAT AGC TGT GCG GTA TTT CAC ACC G-3′). The resulting cassettes were introduced into yeast cells, and correct integration was confirmed by PCR.
KNY208 (cdc48-3) was constructed as follows. We first amplified and sequenced the cdc48-3 open reading frame (ORF) by PCR from genomic DNA of RSY1181 (a generous gift from R. Schekman, University of California, Berkeley, CA). We found that this mutant allele has two mutations, P257L (CCT>CTT) and R387K (AGA>AAA). The same mutation points in the cdc48-3 allele were reported in a recent article (Verma et al., 2011). Subsequently, a part of the cdc48-3 ORF that contains two mutations and its 3′-untranslated region was amplified from RSY1181 using 5′-phosphorylated primers OKN144 (5′-ATG GGT GAA GAA CAT AAA CCA CTT TTG GAC-3′) and OKN145 (5′-TAA TAC CGA GTA ACG TTT ATG GTA TAG AGA-3′). The resulting fragment was blunt ligated into the PvuII site in pRS306, which was then cut with PvuII (located inside the cdc48-3 ORF but downstream of the two mutations). The yeast strain KNY140 was transformed with this linearized plasmid, and the temperature-sensitive clones were selected after 5-fluoroorotic acid selection. The cdc48-3 allele was again amplified by PCR from the resulting clone and was confirmed to have the P257L and R387K mutations.
KNY269 strain expressing USA1 under the control of the GPD promoter was constructed as follows. HIS3MX6-PGPD cassette was amplified by PCR from a plasmid pKN41 using primers OKN466 (5′-CTT AAG CGG CTA TAT AAA GTG TCA TAT ACA CCC TTC ACC AAA TAC GCA ACA CAG CTA TGA CCA TGA TTA CGC C-3′) and OKN467 (5′-CTT GAC CAT ATA GTA AAT TTG CAG GGC GTC TGA GCT AGA TAT TCA GAC ATA TCC GTC GAA ACT AAG TTC TGG T-3′). The resulting fragment was introduced into KNY140 to give KNY269.
Construction of plasmids
Plasmids used in this study are listed in Supplemental Table S2. pKN41 encoding the HIS3MX6-PGPD cassette was constructed as follows. The GPD promoter was removed from p426GPD with SacI/EcoRI and ligated into the same sites of pUC119 to give pKN40. HIS3MX6 was amplified by PCR from pFA6a-PA-GFP-His3MX6 using primers OKN111 (5′-GCG AAG CTT AGA TCT GTT TAG CTT GCC T-3′) and OKN112 (5′-GCG GAG CTC TCG TTT AAA CTG GAT GGC-3′). The resulting fragment was cut with HindIII/SacI and ligated into the same sites of pKN40 to give pKN41.
pKN107 encoding PGAL1-HAC1(i)-TCYC1 was constructed as follows. The ORF of HAC1(i) was amplified from pTYE493 (a generous gift from T. Yoshihisa, Nagoya University, Nagoya, Japan) using primers OKN474 (5′-GCG TCT AGA ATG GAA ATG ACT GAT TTT GAA CTA ACT-3′) and OKN475 (5′-GCG AAG CTT TCA TCA TGA AGT GAT GAA GAA ATC ATT C-3′). The resulting fragment was cut with XbaI and HindIII and inserted into the SpeI-HindIII sites of pKN16. pKN16 is a pRS316-based plasmid with PGAL1 at the SacI–XbaI sites and TCYC1 is at the XhoI–KpnI sites in pRS316.
pKN72 encoding PUBX2-UBX2-3FLAG was constructed as follows. The promoter and ORF of UBX2 gene (PUBX2-UBX2) were amplified by PCR from genomic DNA using primers OKN412 (5′-GCG CTC GAG GGT GAT CAG GCA CGT TCT ATC GAA-3′) and OKN413 (5′-AAG GAA AAA AGC GGC CGC TTG TTC TTC ATT TTC CTC ATC TTC TTC ATC TT-3′). The resulting fragment was cut with XhoI/NotI and ligated into the same sites of pSM293 (a pRS314-based plasmid with a 3xFLAG tag and a stop codon between the NotI–SacI sites, a generous gift from S. Mimura) to give pKN72.
pKN73 encoding PUBX2-UBX2 (UBAΔ)-3FLAG (lacking amino acids [aa] 9–61) was constructed as follows. Two overlapping fragments of PUBX2-UBX2 (UBAΔ) were amplified by PCR from genomic DNA using primers OKN412/OKN415 (5′-CCA TCT GGT CTG GTT CGC CTT TAT CCT CAT GAT TGA CTA CTG GCA T-3′) and OKN416 (5′-ATG CCA GTA GTC AAT CAT GAG GAT AAA GGC GAA CCA GAC CAG ATG G-3′)/OKN413. Using these two fragments as templates, we amplified full-length PUBX2-UBX2 (UBAΔ) by PCR using OKN412 and OKN413. The resulting fragment was ligated into pSM293 as before to give pKN73.
pKN75 encoding PUBX2-UBX2 (UBAΔUBXΔ)-3FLAG (lacking both aa 9–61 and 421–584) was constructed as follows. PUBX2-UBX2 (UBAΔUBXΔ) was amplified by PCR from pKN73 using primers OKN412 and OKN414 (5′-AAG GAA AAA AGC GGC CGC ATC GAT GCA AGC CTT TAA CCA TTT TAA TT-3′). The resulting fragment was ligated into pSM293 as before to give pKN75.
pKN81 encoding PUBX2-UBX2 (UBXΔ)-3FLAG (lacking aa 418–584) was constructed as follows. PUBX2-UBX2 (UBXΔ) was amplified by PCR from pKN72 using OKN412 and OKN419 (5′-AAG GAA AAA AGC GGC CGC AGC CTT TAA CCA TTT TAA TTG CCG A-3′). The resulting fragment was ligated into pSM293 as before to give pKN81.
Antibodies and immunoblotting
Anti-Usa1p antiserum was a generous gift from P. Carvalho and T. Rapoport (Harvard Medical School, Boston, MA). In some experiments, anti-Usa1p antiserum that was raised by our laboratory in rabbits against KLH-conjugated CTATGAQPHLYIPDED peptide was used. Anti-Hrd3p antiserum was a generous gift from T. Sommer and J. Ernst (Max-Delbrück-Center for Molecular Medicine, Berlin, Germany). Anti-Hrd1p antiserum and anti-Der1p antiserum were raised in rabbits against histidine-tagged fragments of Hrd1p (amino acids 348–551) and Der1p (amino acids 112–211) expressed from pRSET-B (Invitrogen, Carlsbad, CA) in Escherichia coli. Anti-Yos9p and anti-Tim23p antibodies were generous gifts from T. Endo (Nagoya University). Anti-Cdc48p antibody was a generous gift from M. Esaki (Kumamoto University, Kumamoto, Japan). Anti-Kar2p antibody was a generous gift from R. Schekman. Anti-Ufd1p antibody was a generous gift from C. Moore (Tufts University, Medford, MA; del Olmo et al., 1997). Anti-FLAG (M2) monoclonal antibody and anti-FLAG M2 affinity gel were purchased from Sigma-Aldrich (St. Louis, MO). Anti-Sec61p polyclonal antiserum was raised in rabbits against a C-terminal peptide. One lot of anti-Sec61p antiserum cross-reacts with a higher–molecular weight cytosolic protein, which is indicated by asterisks in some figures. Anti-20S proteasome antibodies were purchased from Enzo. Anti-mouse immunoglobulin G (IgG)–horseradish peroxidase (HRP) and anti-rabbit IgG-HRP (Sigma-Aldrich) were used as secondary antibodies. The extent of the polyubiquitin chains was detected as described previously (Nakatsukasa et al., 2008), with minor modifications. In brief, proteins were separated by SDS–PAGE and transferred to a polyvinylidene fluoride membrane, which was then incubated with monoubiquitinated and polyubiquitinated conjugated/mouse monoclonal antibody HRP conjugate (FK2H; PW0150; Biomol International, Enzo Life Sciences, Plymouth, PA) in Tris-buffered saline (TBS)–Tween 20.
Sucrose density gradient centrifugation
Cells were grown to log phase (OD600 < 1.0), and 30–50 OD600 equivalents of cells were harvested, washed with ice-cold distilled water, and stored at −80°C. Temperature-sensitive strains and their isogenic wild-type strains were shifted to 37°C for 1 h before cells were collected. To overexpress Hac1p(i) under the control of the GAL1 promoter, cells were first grown in a synthetic complete medium with 2% glucose and appropriate amino acids, washed with yeast extract/peptone (YP)-galactose twice, and grown in YP-galactose for 4 h. The frozen cells were disrupted with glass beads in lysis buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, 50 mM KOAc, 2 mM EDTA, 0.1 M sorbitol, 1 mM DTT, 20 μM MG132, and complete protease inhibitor cocktail [Roche, Indianapolis, IN]) by agitation on a Vortex mixer eight times for 30 s with 30-s intervals on ice between each cycle. The homogenate was collected and pooled with rinses of the beads with buffer 88, pH 7.4 (20 mM HEPES, pH 7.4, 150 mM KOAc, 250 mM sorbitol, 5 mM MgOAc). After unbroken cells were removed by centrifugation at 300 × g for 5 min at 4°C, the supernatant was centrifuged at 20,000 × g for 20 min at 4°C. The pelleted membranes were solubilized in solubilization buffer (30 mM Tris-Cl, pH 7.6, 150 mM NaCl, 2 mM MgCl2, 5% glycerol, and complete protease inhibitor cocktail) plus 1% digitonin (Wako, Osaka, Japan) on ice for 30 min. The lysates were cleared by centrifugation at 20,000 × g for 10 min at 4°C. The supernatant (420 μl) was layered onto a 5–40% sucrose step gradient (420 μl of 5, 10, 15, 20, 25, 30, 35, 40% sucrose in solubilization buffer containing 0.5% digitonin). Centrifugation was performed at 45,000 rpm for 3.5 h at 4°C in an SW60Ti rotor (Beckman Coulter, Brea, CA). Fractions (210 μl) were collected from the top. Proteins were precipitated with trichloroacetic acid (TCA), washed with ice-cold acetone, and dissolved in KN sample buffer (80 mM Tris, pH 8.0, 8 mM EDTA, 0.5 M DTT, 3.5% SDS, 15% glycerol, 0.08% Tris-base, and 0.01% bromophenol blue) by incubation at 42°C, followed by SDS–PAGE and immunoblotting.
Coimmunoprecipitation
Membrane fractions were isolated as described, and the lysates were prepared in solubilization buffer supplemented with 1% digitonin or 0.75% Triton X-100. In experiments in which the extent of the polyubiquitin chains was assessed, 10 mM N-ethylmaleimide (NEM) was included in all buffers throughout the procedure. Next the lysate was clarified by centrifugation at 20,000 × g for 10 min at 4°C. For an input control, a 100-μl aliquot was removed, precipitated with TCA, washed with ice-cold acetone, and solubilized in KN sample buffer at 42°C for 20 min. The remaining supernatant was added to anti-FLAG antibody–conjugated Dynabeads Protein G (Invitrogen) or anti-FLAG M2 affinity gel (Sigma-Aldrich). When digitonin was used, its concentration was lowered to 0.66% before antibody addition. After nutating at 4°C for 2–4 h, the beads were washed four times with the solubilization buffer supplemented with 0.5% digitonin or 0.5% Triton X-100 before proteins were eluted with KN sample buffer.
For the denaturing immunoprecipitation performed in Figure 3, membranes were solubilized in denaturing solubilization buffer (1% SDS, 4 M urea, 10 mM NEM, 10 mM EDTA in 1× TBS) with a complete protease inhibitor cocktail by heating at 42°C for 30 min. The lysate was cleared by centrifugation at 13,000 rpm for 2 min at room temperature. The resulting supernatant was diluted with a 10-fold volume of TTS buffer (1% Triton X-100, 0.1% SDS, complete protease inhibitor cocktail in 1× TBS). For the input control, a 100-μl aliquot was removed, precipitated with TCA, washed with ice-cold acetone, and solubilized in KN sample buffer at 42°C for 20 min. The remaining supernatant was added to anti-FLAG M2 affinity gel and nutated as before. The agarose was washed four times with TTS buffer supplemented with 10 mM NEM before proteins were eluted with KN sample buffer.
The interaction between the Hrd1 complex and the proteasome is analyzed in Figure 6 by the liquid nitrogen lysis-DSP cross-linking method developed in this study. Cells were grown to log phase, and 400 OD600 equivalents of cells were harvested. Temperature-sensitive strains and their isogenic wild-type strains were shifted to 37°C for 1 h before harvesting. Subsequently, cells were washed with distilled water and resuspended in 1 ml of buffer 88, pH 7.4, plus 15% glycerol. The cell slurry was frozen by dribbling a thin stream into liquid nitrogen. The frozen “popcorn”-like cells were stored at −80°C. The frozen cells were then broken with a coffee mill (MK-61M-G Panasonic, Osaka, Japan) for 2 min by adding a small amount of liquid nitrogen every 15–20 s. The resulting fine “flour”-like powder was stored at −80°C or immediately thawed by adding 2 ml of PM buffer (buffer 88, pH 7.4, supplemented with 15% glycerol, 5 mM ATP, and 50 μM MG132) for 1–2 min. Unbroken cells were removed by centrifugation at 500 × g for 5 min at 4°C, and the supernatant was centrifuged again at 400 × g for 5 min at 4°C. The supernatant (2 ml) containing cytosol and membranes was immediately added to 200 μg/ml of DSP cross-linker (Pierce, Rockford, IL) and incubated at 4°C for 90 min. Membranes were collected by centrifugation at 20,000 × g for 15 min at 4°C, washed in 900 μl of PM buffer supplemented with 100 μl of 1 M Tris-Cl, pH 7.5, to quench the reaction, and incubated in 1 ml of the same buffer for 15 min at 4°C. Membranes were reisolated, rinsed quickly with 1 ml of ice-cold 1× TBS, and solubilized in 1100 μl of TTS buffer supplemented with 50 μM MG132 by nutating at 4°C for 30 min. The lysate was cleared by centrifugation at 20,000 × g for 10 min at 4°C. For the input control, 100 μl was removed, TCA precipitated, washed with ice-cold acetone, and solubilized in KN sample buffer-2 (80 mM Tris, pH 8.0, 8 mM EDTA, 8 M urea, 4% SDS, 15% glycerol, 0.08% Tris-base, and 0.01% bromophenol blue) supplemented with 200 mM DTT at 42°C for 20 min. The remaining supernatant was incubated with 40 μl of 60% (vol/vol) 1× TBS-equilibrated anti-FLAG M2 affinity gel at 4°C for 90 min to precipitate 3xFLAG-tagged Rpt5p. The agarose was washed four times with 1 ml of TTS buffer, and proteins were eluted with KN sample buffer-2 at 42°C for 20 min, followed by addition of 200 mM DTT and incubation at 42°C for 30 min to cleave the cross-linker.
In vitro deubiquitination
Membrane fractions were prepared by the same method described for sucrose density gradients. Membranes (prepared from ∼30 OD600 equivalent of cells) were resuspended in 100 μl of 50 mM Tris-Cl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 2 mM DTT, and 0.2 M sorbitol. To digest polyubiquitin chains attached to the membrane-associated proteins, 10 μg of hexahistidine-USP2CD (UBP41 catalytic domain; Boston Biochem, Cambridge, MA) was mixed with the membranes and incubated at 37°C for 1 h. The addition of the same amount of bovine serum albumin (BSA) served as a negative control. The reaction was quenched by adding 1 ml of buffer 88 (20 mM HEPES, pH 6.8, 150 mM KOAc, 250 mM sorbitol, and 5 mM MgOAc) supplemented with 10 mM NEM. Membranes were collected by centrifugation, solubilized with 1% digitonin, and subjected to the immunoprecipitation protocol described earlier.
Supplementary Material
Acknowledgments
We thank P. Carvalho, T. Endo, J. Ernst, C. Moore, S. Nishikawa, T. A. Rapoport, R. Schekman, and T. Sommer for antibodies and K. A. Arndt, T. Endo, T. Izawa, S. Mimura, S. Nishikawa, D. T. Ng, R. Schekman, H. Sesaki, Y. Tamura, J. S. Weissman, T. Yoshihisa, and the National Bio-Resource Project of the Ministry of Education, Culture, Sports, Science and Technology of Japan for strains and plasmids. This work was supported in part by a grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan to K.N and T.K., by the Sumitomo Foundation and the Toyoaki Foundation to K.N., and by National Institutes of Health Grant GM75061 to J.L.B.
Abbreviations used:
- ERAD
endoplasmic reticulum–associated degradation
- UPR
unfolded protein response
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-12-0907) on March 27, 2013.
REFERENCES
- Bagola K, Mehnert M, Jarosch E, Sommer T. Protein dislocation from the ER. Biochim Biophys Acta. 2011;1808:925–936. doi: 10.1016/j.bbamem.2010.06.025. [DOI] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001;3:24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, Wolf DH. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell. 1998;9:209–222. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson EJ, Pitonzo D, Skach WR. p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J. 2006;25:4557–4566. doi: 10.1038/sj.emboj.7601307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Olmo M, Mizrahi N, Gross S, Moore CL. The Uba2 and Ufd1 proteins of Saccharomyces cerevisiae interact with poly(A) polymerase and affect the polyadenylation activity of cell extracts. Mol Gen Genet. 1997;255:209–218. doi: 10.1007/s004380050491. [DOI] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Elsasser S, Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol. 2005;7:742–749. doi: 10.1038/ncb0805-742. [DOI] [PubMed] [Google Scholar]
- Ernst R, Claessen JH, Mueller B, Sanyal S, Spooner E, van der Veen AG, Kirak O, Schlieker CD, Weihofen WA, Ploegh HL. Enzymatic blockade of the ubiquitin-proteasome pathway. PLoS Biol. 2011;8:e1000605. doi: 10.1371/journal.pbio.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Mueller B, Ploegh HL, Schlieker C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell. 2009;36:28–38. doi: 10.1016/j.molcel.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2:379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Sato BK, Hampton RY. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J Biol Chem. 2009;284:14710–14722. doi: 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss R, Jarosch E, Sommer T, Hirsch C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol. 2006a;8:849–854. doi: 10.1038/ncb1445. [DOI] [PubMed] [Google Scholar]
- Gauss R, Sommer T, Jarosch E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006b;25:1827–1835. doi: 10.1038/sj.emboj.7601088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt EJ, Olzmann JA, Kopito RR. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat Struct Mol Biol. 2011;18:1147–1152. doi: 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Sommer T. Finding the will and the way of ERAD substrate retrotranslocation. Curr Opin Cell Biol. 2012;24:460–466. doi: 10.1016/j.ceb.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, Heinemann U, Sommer T, Jarosch E. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell. 2009;36:782–793. doi: 10.1016/j.molcel.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- Jonikas MC, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–1697. doi: 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Spear ED, Ng DT. Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol Cell. 2005;19:753–764. doi: 10.1016/j.molcel.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Kothe M, Ye Y, Wagner JS, De Luca HE, Kern E, Rapoport TA, Lencer WI. Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J Biol Chem. 2005;280:28127–28132. doi: 10.1074/jbc.M503138200. [DOI] [PubMed] [Google Scholar]
- Lee RJ, Liu CW, Harty C, McCracken AA, Latterich M, Romisch K, DeMartino GN, Thomas PJ, Brodsky JL. Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. EMBO J. 2004;23:2206–2215. doi: 10.1038/sj.emboj.7600232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- Lipson C, Alalouf G, Bajorek M, Rabinovich E, Atir-Lande A, Glickman M, Bar-Nun S. A proteasomal ATPase contributes to dislocation of endoplasmic reticulum-associated degradation (ERAD) substrates. J Biol Chem. 2008;283:7166–7175. doi: 10.1074/jbc.M705893200. [DOI] [PubMed] [Google Scholar]
- Mayer TU, Braun T, Jentsch S. Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. EMBO J. 1998;17:3251–3257. doi: 10.1093/emboj/17.12.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicherla B, Kostova Z, Schaefer A, Wolf DH. A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 2004;5:692–697. doi: 10.1038/sj.embor.7400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146:743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat Cell Biol. 2005;7:993–998. doi: 10.1038/ncb1298. [DOI] [PubMed] [Google Scholar]
- Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S. Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell. 2001;107:667–677. doi: 10.1016/s0092-8674(01)00595-5. [DOI] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Rouiller I, Butel VM, Latterich M, Milligan RA, Wilson-Kubalek EM. A major conformational change in p97 AAA ATPase upon ATP binding. Mol Cell. 2000;6:1485–90. doi: 10.1016/s1097-2765(00)00144-1. [DOI] [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell. 2009;34:212–222. doi: 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C, Buchberger A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol. 2005;7:999–1006. doi: 10.1038/ncb1299. [DOI] [PubMed] [Google Scholar]
- Schuck S, Prinz WA, Thorn KS, Voss C, Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. 2009;187:525–536. doi: 10.1083/jcb.200907074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley AM, Carvalho P, Rapoport T. Recognition of an ERAD-L substrate analyzed by site-specific in vivo photocrosslinking. FEBS Lett. 2011;585:1281–1286. doi: 10.1016/j.febslet.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szathmary R, Bielmann R, Nita-Lazar M, Burda P, Jakob CA. Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol Cell. 2005;19:765–775. doi: 10.1016/j.molcel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Oania R, Fang R, Smith GT, Deshaies RJ. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol Cell. 2011;41:82–92. doi: 10.1016/j.molcel.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlman J, DeMartino GN, Skach WR, Bulleid NJ, Brodsky JL, Johnson AE. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell. 2007;129:943–955. doi: 10.1016/j.cell.2007.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang CW, Lee SC. The ubiquitin-like (UBX)-domain-containing protein Ubx2/Ubxd8 regulates lipid droplet homeostasis. J Cell Sci. 2012;125:2930–2939. doi: 10.1242/jcs.100230. [DOI] [PubMed] [Google Scholar]
- Wolf DH, Stolz A. The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim Biophys Acta. 2012;1823:117–124. doi: 10.1016/j.bbamcr.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Xie W, Ng DT. ERAD substrate recognition in budding yeast. Semin Cell Dev Biol. 2010;21:533–539. doi: 10.1016/j.semcdb.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem. 2004;279:45676–45684. doi: 10.1074/jbc.M409034200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.