

Abstract
Interactions between the nucleosome and the non-histone chromosomal proteins (HMGN1 and HMGN2) were studied by circular dichroism (CD) spectroscopy to elucidate structural changes in the nucleosome induced by HMGN binding. Unlike previous studies that used a nucleosome extracted from living cells, in this study we utilized a nucleosome reconstituted from unmodified recombinant histones synthesized in Escherichia coli and a 189-bp synthetic DNA fragment harboring a nucleosome positioning sequence. This DNA fragment consists of 5′-TATAAACGCC-3′ repeats that has a high affinity to the histone octamer. A nucleosome containing a unique octamer-binding sequence at a specific location on the DNA was produced at sufficiently high yield for spectroscopic analysis. CD data have indicated that both HMGN1 and HMGN2 can increase the winding angle of the nucleosome DNA, but the extent of the structural changes induced by these proteins differs significantly. This suggests HMGN1 and HMGN2 would have different abilities to facilitate nucleosome remodeling.
Keywords: CD, Nucleosome, HMGN, Unmodified recombinant histones, Reconstitution
Abbreviations: HMG, high mobility group; HMGN1 HMGN2, non-histone chromosomal proteins; CD, circular dichroism; PCR, polymerase chain reaction; phH2A, phH2B, phH3, and phH4, vectors for the gene expression of all four recombinant human core histones H2A, H2B, H3, and H4, respectively; LB, Luria–Bertani; IPTG, isopropyl-β-d-galactopyranoside; RP-HPLC, reverse phase high performance liquid chromatography; SDS–PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; MNase, micrococcal nuclease; NMR, nuclear magnetic resonance
Highlights
-
•
A nucleosome was reconstituted from recombinant histones and a synthetic DNA.
-
•
Nucleosomes were produced at sufficiently high yield for spectroscopic analysis.
-
•
A nucleosome with and without HMGN proteins was analyzed using CD spectroscopy.
-
•
CD data indicate that HMGN proteins increase the winding angle of the nucleosome DNA.
-
•
HMGN1 and HMGN2 may have different abilities to facilitate nucleosome remodeling.
1. Introduction
An octameric protein consisting of four core histones, H2A, H2B, H3, and H4 (two copies each) is a reel around which 147-base pairs (bp) of DNA is wrapped in an 1.7 left-handed superhelical turn [1,2]. This histone–DNA complex, which is referred to as the nucleosome, serves as the basic repeating unit to effectively package whole genomes into the cell nucleus of eukaryotic cell in the following steps. First, the form of nucleosomes in a column causes a structure like beads on a string, namely, the 10-nm fiber because the nucleosomes are 10-nm in diameter. In a gene, the formation of this chromatin fiber causes repressing processing, preventing nuclear proteins such as transcriptional factors access to DNA [1,2]. Second, this fiber coils or is folded into a thicker fiber, the so-called solenoid that is 30-nm in diameter, in the DNA packaging. Finally, the solenoid is packaged in a highly compacted state of chromatin and in chromosome. Recent studies have revealed that there are several types of protein complexes that can alter the repressive nature of the chromatin fiber [3]. One such family of factors functions by covalently modifying the histone tails through phosphorylation, acetylation, and methylation [4,5]. Second group of factors is comprised of multi-protein complexes that mobilize and/or alter the structure of the nucleosome [6]. In addition to these chromatin remodeling factors, the high mobility group (HMG) proteins have been identified as a heterogeneous class of non-histone proteins that modulate the structure of the DNA in the chromatin fiber [7,8]. The HMGN subgroup of the HMG proteins includes the only known class of nuclear proteins that bind specifically to the nucleosome core particle.
Non-histone chromosomal HMGN proteins are abundant and ubiquitous in all higher eukaryotes [7,9–11]. Two structurally similar proteins, HMGN1 and HMGN2, are well known members of the HMGN protein family. They were formerly referred to as HMG-14 and HMG-17, respectively [12]. These HMGN proteins bind specifically to the nucleosome core particles without any specificity for the underlying DNA sequence [13]. These two proteins bind cooperatively to nucleosomes to form complexes containing homodimers of either HMGN1 or HMGN2 [14]. In the chromatin fiber, nucleosomes form clusters containing HMGN2 [15]. It has been suggested that the highly compacted structure of chromatin is unfolded in the HMGN2-containing regions, thus relaxing its suppressive architecture. Actually, multiple distinct foci containing either HMGN1 or HMGN2 are found in the nucleus [15], and the intranuclear distribution of the foci is shown to be similar to that of active transcription sites in cells [16,17]. Thus, the binding of HMGN proteins to nucleosomes unfolds the higher-order chromatin structure to enhance DNA-dependent activities such as transcription [18–20] and replication [21]. Chromatin unfolding is facilitated by specific interactions between the C-terminal part of HMGN1 protein and the N-terminal tail of histone H3, and also between the N-terminal part of HMGN1 and histone H2B [22]. Recent investigations have also revealed that the transcriptional co-activators such as p300 and PCAF acetylate HMGN1 and HMGN2 proteins to reduce the nucleosome binding activities of these HMGN proteins [23–25]. Modifications carried out by these co-activators apparently serve to regulate some gene expressions by altering the higher-order structure of chromatin through the control of HMGN protein binding [23–28].
Several earlier studies regarding the interactions of HMGN proteins with nucleosomes (HMGN–nucleosome interactions) have been carried out [29–32]. These works have proposed the following insights into the effect of HMGN binding to nucleosomes: HMGN binding stabilizes the nucleosome core particle against thermal denaturing [30,31] and increases the winding angle of the underlying DNA [29,32] by disrupting the highly ordered chromatin structure. However, in these previous studies, histone proteins were extracted from living cells. In living cells, the histones are subjected to a variety of post-translational modifications including acetylation, phosphorylation, methylation, ubiquitination, and ADP-ribosylation, all of which are tightly related to cell functions [1,33]. These modifications are known to affect the biophysical properties of chromatin [34–37]. Actually, as noted by Levenstein and Kadonaga [38] and Luger and co-workers [39], the histones from living cells give multiple and/or broad mass spectra peaks showing masses that are larger than the values calculated from their amino acid sequences. Therefore, the possibility that the use of nucleosomes consisting of heterogeneously modified histones causes unpredicted effects on the HMGN–nucleosome interactions has remained. In the present work, we intended to obtain definitive results on the HMGN–nucleosome interactions using properly prepared samples for spectroscopic experiments. We therefore started with reconstitution of the nucleosome from the unmodified human histones synthesized in Escherichia coli, as well as from 189-bp PCR-amplified double stranded DNA fragments harboring the nucleosome positioning sequence. The sequence consisting of the repeating motif: 5′-TATAAACGCC-3′, which is known as a high-affinity sequence for the octameric protein, was used to construct a nucleosome having a uniquely positioned histone octamer along its DNA, without the aid of nucleosome remodeling factors [40–44]. The reconstituted nucleosome allows us to perform a fine analysis of the HMGN–nucleosome interactions by use of circular dichroism (CD) spectroscopy. Consequently, it is our interest here to explore whether the binding of the HMGN proteins to the nucleosome could intrinsically provide an increase of the winding angle of the underlying DNA: the DNA is wrapped tightly on the histone octamer. This result enables us to definitively determine the difference between the structural change of nucleosome induced by HMGN1 and that of HMGN2 in the organization or formation of the HMGN–nucleosome complexes.
2. Materials and methods
2.1. Plasmid construction for the synthesis of human core histones in E. coli
The four core histone genes comprising a gene cluster [45,46] were amplified by the polymerase chain reaction (PCR) using the genomic DNA extracted from HeLa cells. In order to amplify each histone gene, two primers were prepared. In one primer that has the 5′ end sequence of gene, an NdeI restriction site was incorporated with the initiating Met codon, as summarized in Table 1A. In the other primer that has the complementary sequence of the 3′ end of the gene, an XhoI site was added to the sequence, except for an H3 primer, as summarized in Table 1B. In the H3 primer, a BamHI site was added to the sequence, as summarized in Table 1C. The amplified DNA fragments of H2A, H2B and H4 were digested with NdeI and XhoI enzymes, whereas the fragment of H3 was digested with NdeI and BamHI enzymes. The products were inserted into the NdeI/XhoI and NdeI/BamHI sites of pET-22b (Novagen), respectively, to create vectors (phH2A, phH2B, phH3, and phH4) for the gene expression of all four recombinant human core histones. The histone genes in each of the resulting plasmids were sequenced to confirm their integrity.
Table 1.
PCR primer sequences.
| name | DNA sequence |
|---|---|
| A | |
| H2A-NdeI | agatagccatatgtctggtcgcggcaaac |
| H2B-NdeI | agatagccatatgccagagccagcgaag |
| H3-NdeI | agatagcatatggctcgtactaaacagac |
| H4-NdeI | agatagccatatgtccggcagaggaaag |
| B | |
| H2A-XhoI | agatagctcgagtcactttcccttggccttatg |
| H2B-XhoI | agatagctcgagttacttagcgctggtgtac |
| H4-XhoI | agatagctcgagctagcctccgaagccg |
| C | |
| H3-BamHI | atataggatccttacgctctttctccgcg |
| D | |
| Tetrad-EcoRI | ggaattcagatcttcctgggaaaaccctggcgaggtc |
| Tetrad-BamHI | taggtgggatcccgagctgtttcctgtgtgaagac |
| Tetrad-1 | aaaaccctggcgaggtctataagcgtctataagcgtctatgaacgtctataaacgtctataaacgcctataaacgcctataaacgcctatacaag |
| Tetrad-2 | ctgtttcctgtgtgaagacgtgtatagtcgtgcatagacgtgtataggcgtttataggcttgtataggcgtttatagg |
| E | |
| N1-NdeI | agatagcatatgcccaagaggaaggtc |
| N1-BamHI | atataggatccttattaatcagacttggcttc |
| N2-NdeI | agatagcatatgcccaagagaaaggc |
| N2-BamHI | atataggatcctcacttggcatctccag |
2.2. Expression and purification of human core histones
Each of the expression vectors was introduced into the BL21(DE3) strain of E. coli. The BL21 cells containing the recombinant plasmid were grown at 37 °C until OD600 reached 0.6–1.0 in Luria–Bertani (LB) medium. Induction was carried out for 5 h at 37 °C with 0.4 mM IPTG. Cells were harvested by centrifugation and frozen with liquid nitrogen. The frozen cells were resuspended in a wash buffer (300 mM NaCl, 50 mM sodium phosphate, pH 7.8). The suspension was kept on ice for 5 min and then subjected to repeated vigorous shaking and sonication with full output for 60 s. The membrane-containing pellet was collected by centrifugation (30,000g at 4 °C for 30 min), and the supernatant was discarded. The pellet was resuspended in ice-cold desalting buffer (50 mM sodium phosphate, pH 7.8), and the supernatant was removed by centrifugation (20,000g at 4 °C for 20 min). The pellet containing inclusion bodies of histone protein was resuspended in a freshly prepared unfolding buffer (8 M urea, 50 mM sodium phosphate, pH 7.8) with the help of a short application of sonication for disrupting the pellet at room temperature. High-grade urea from ICN Pharmaceuticals, Inc. was used. The insoluble material in the urea solution was removed by centrifugation (20,000g at 25 °C for 10 min). The supernatant was applied onto an SP-Sepharose (GE Healthcare) ion-exchange column pre-equilibrated in the unfolding buffer, 8 M urea in 50 mM sodium phosphate buffer, pH 7.8. The histone proteins were eluted with a salt gradient from 0 to 1 M NaCl. The collected histones were further purified by reverse phase chromatography with the POROS R1/M column (4.6 × 100 mm, Applied Biosystems). Elution was performed using linear gradients of acetonitrile (0–100% in 10 min) in water containing 0.1% trifluoroacetic acid. The elution profile was monitored at 215 nm absorbance. The collected histone solution was extensively dialyzed against H2O then lyophilized for long term storage. Protein purification was checked by 20% SDS–PAGE gels [47,48].
2.3. Preparation of human histone octamer
The preparation of refolded histone octamer was accomplished according to the method by Luger et al., with slight modification [39,49]. The lyophilized histone proteins were dissolved in 20 mM sodium acetate buffer, pH 5.2, containing 7 M urea, 0.2 M NaCl, 5 mM 2-mercaptoethanol, and 1 mM EDTA. The dissolved histone solution was kept at room temperature for 30 min. The concentration of each histone protein was adjusted to 1.0 × 10−4 M (see also Section 2.11). This histone mixture was dialyzed against a refolding buffer, 10 mM Tris–HCl, pH 7.4, containing 2 M NaCl, 5 mM 2-mercaptoethanol, 1 mM EDTA at 4 °C. The dialyzed protein mixture was concentrated to 5.0 × 10−4 M using a 10-kDa cutoff Centriprep device (Millipore). Insoluble material was removed by centrifugation. The refolded octamer was separated from the misfolded components by size-exclusion chromatography on a Superdex-200 column (15 × 750 mm, GE Healthcare) equilibrated with refolding buffer. The collected fractions were subjected to SDS–PAGE to identify the desired fractions.
2.4. Preparation of the high-affinity DNA fragment
A 189-bp synthetic DNA fragment having a nucleosome-positioning sequence consisting of the consensus repeat sequence TATAAACGCC [44] was constructed by using the primer overlap extension method. In the method, PCR primers were as summarized in Table 1D. The synthetic fragment was generated from an equimolar mixture (2.5 μM for each histone) of the four overlapping oligonucleotides shown above plus dNTPs (250 μM) in a buffer optimized for KOD DNA polymerase (Toyobo). After the addition of the DNA polymerase, the following amplification protocol was applied: heating at 94 °C for 30 s, annealing at 52 °C for 30 s and elongation at 72 °C for 60 s, and 30 cycles. The resulting DNA fragment was digested with EcoRI and BamHI enzymes, and then inserted into the pUC18 vector for a large-scale preparation. This vector was designated as the pUC18-tetrads. The inserted DNA sequence was confirmed using the forward and reverse pUC/M13 universal primers. In order to perform CD experiments, the TATAAACGCC repeat fragment in the pUC18-tetrads was amplified by the KOD polymerase under the following conditions: heating at 94 °C for 30 s, annealing at 62 °C for 30 s and elongation at 72 °C for 60 s, and 33 cycles using the primers Tetrad-EcoRI and Tetrad-BamHI. The PCR product was collected by isopropanol precipitation and was further purified by ion-exchange chromatography using a POROS HQ/20 HPLC column (Applied Biosystems).
2.5. Reconstitution of nucleosome core particles
The reconstitution of nucleosome from the constructed histone octamer, as well as from the DNA fragment containing the high-affinity sequence was accomplished by using the exponentially gradient dilution method, according to a previously described [39,49] with slight modifications. Recombinant histone octamer was mixed with equimolar amounts of the synthetic TATAAACGCC repeat fragment in ice-cold TE buffer (10 mM Tris–HCl (pH 7.5), 0.1 mM EDTA) supplemented with 2 M NaCl (see also Section 2.11). The mixture was kept on ice for 10 min and then transferred into dialysis bags with a molecular weight cutoff 3.5-kDa. The sample mixture was dialyzed against a 100-fold excess of TE buffer supplemented with 0.4 M NaCl. The dialysis buffer was changed twice every 3 h at 4 °C. The sample mixture was then further dialyzed against a 100-fold excess of TE buffer with 16 mM NaCl for another 10 h. The dialyzed nucleosome core particle solution was concentrated to the desired concentration using a 50-kDa cutoff filtercup.
2.6. Plasmid construction for the synthesis of human HMGN1 and HMGN2 proteins
HeLa cells were cultivated in Dulbecco's modified minimum essential medium supplemented with 10% fetal calf serum in order to isolate HMGN RNAs. Cultures were maintained at 37 °C in a 5% CO2 atmosphere. Total RNA was extracted from 1 to 5 × 106 frozen HeLa cells, then the mRNA was purified using a Quick Prep Micro mRNA Purification Kit (GE Healthcare). The cDNA was synthesized using a First-Strand cDNA Synthesis Kit (GE Healthcare) with the extracted mRNA as template. The HMGN1 and HMGN2 genes were amplified from the cDNA by PCR. In the primers, an NdeI restriction site was incorporated with the initiating Met codon of the gene, as well as a BamHI site is added to the complementary sequence of the 3′ end of the gene, as summarized in Table 1E. The PCR products were digested with NdeI and BamHI enzymes. Vectors for the gene expression of HMGN were created by the insertion of each fragment into pET-22b (Novagen) that was previously digested with NdeI and BamHI. The HMGN genes in the resulting plasmids (phHMGN1 and phHMGN2) were sequenced to confirm their integrity.
2.7. Expression and purification of the human HMGN1 and HMGN2 proteins
Each of the vectors for expressing HMGN genes was introduced into the BL21(DE3) strain of E. coli, and then the gene expression was induced by 0.4 mM IPTG when OD600 reached 1.0 after shaking at 37 °C in LB medium. After the induction, the culture was continued for another 7 h at 37 °C to produce the protein in cells. Cells were harvested by centrifugation and frozen with liquid nitrogen. Purification of the protein was performed according to essentially the same method described before [50]. The culture broth was centrifuged at 5000g for 20 min and the obtained pellet was resuspended and lysed by vigorous vortexing in a phosphate-buffered saline solution containing 0.1% Triton X-100 and 1 mM PMSF. An equal volume of cold 10% perchloric acid was added and the mixture was incubated on ice for 15 min with occasional vortexing. The materials precipitated by the acid were removed by centrifugation and the remaining soluble proteins were further precipitated by adding 100% (w/v) trichloroacetic acid to 25% final concentration. The recombinant HMGN1 was purified from the crude precipitate by anion exchange chromatography with a Q-Sepharose (GE Healthcare) column employing a NaCl gradient, of 10 mM to 1 M NaCl in 20 mM Tris–HCl buffer pH 8.2. On the other hand, HMGN2 protein was purified by cation exchange chromatography using an SP-Sepharose column with 20 mM to 1 M NaCl in sodium bicine buffer, pH 8.0. The HMGN proteins were further purified by reverse phase chromatography using a POROS R1/M perfusion chromatography column (4.6 × 100 mm, Applied Biosystems). Purification was performed using a linear gradient of acetonitrile (0–100% in 10 min) in 0.1% trifluoroacetic acid. The sample was subjected to extensive dialysis against H2O and then frozen and lyophilized. The purified proteins were analyzed by electrospray ionization (ESI) MS. Protein purification was monitored by 15% SDS–PAGE [35] and 15% acid–urea PAGE [48].
2.8. Electrophoretic mobility shift assay
The DNA and nucleosome were analyzed on nondenaturing gels containing 4% acrylamide, 0.1% bisacrylamide, 0.5% glycerol, and TAE buffer (40 mM Tris–acetate, 1mM EDTA). Samples were mixed with the sample buffer containing TAE at the same concentration as in the gel plus 25% glycerol. Gels were stained with ethidium bromide and placed on the surface of a Vilber Lourmat TFX-20MC ultraviolet transilluminator (France) to photograph the results.
2.9. Micrococcal nuclease digestion
The micrococcal nuclease (MNase, Affymetrix) digestion was performed by adding five units of MNase/μl of the reconstituted nucleosome solution containing 3 mM CaCl2. The reaction time was set to 2, 5 and 10 min at 25 °C and stopped by the addition of SDS, and then the samples were subjected to ethanol precipitation. The samples were dissolved in H2O and were mixed with 6 x loading buffer (30% Glycerol, 30 mM EDTA, 0.03% bromophenol blue, 0.03% xylene cyanol) with 1/5 volume of DNA solution. The resulting DNA fragments were resolved by 6% native polyacrylamide (20:1 acrylamide/bisacrylamide) gel electrophoresis at 110 V with 0.5 × TBE (45 mM Tris–HCl pH 7.4, 45 mM boric acid, 1 mM EDTA) as elution buffer. After elution, the gel was submerged in staining solution (0.5 μg/ml ethidium bromide in 1 × TBE) for 20 min. Gel photographs were taken with the Vilber Lourmat TFX-20MC ultraviolet transilluminator and analyzed with EDAS290FF (Kodak).
2.10. Circular dichroism spectra measurement for nucleosome and HMGN–nucleosome complexes
CD spectra were recorded with a Jasco J-720 spectrophotometer. The spectra were measured for the octamer sample at the concentration of 10 μM in TE buffer, pH 7.5, 2M NaCl. For the nucleosome and the HMGN–nucleosome complexes, the nucleosome concentration was 1.0 μM in TE buffer, pH 7.5, 16 mM NaCl. All measurements were done at 25 °C using a 0.1-cm path length quartz cuvette. Each spectrum was obtained from eight scans in wavelength ranges from 190 nm to 320 nm, keeping the scanning rate at 100 nm/min.
2.11. Determination of protein and DNA concentrations
The extinction coefficient of the histone octamer was assumed to be ɛ280 = 48,900 cm−1 M−1 (0.45 cm2 mg−1) [51] to calculate the ellipticity for circular dichroism analysis. The extinction coefficient at 276 nm of each unfolded histone protein was calculated according to the method of Gill and von Hippel [52], yielding H2A, 3870; H2B, 5950; H3, 4720; and H4, 5180 M−1 cm−1. The extinction coefficients at 220 nm for HMGN proteins were determined by the quantitative amino acid analysis, yielding HMGN1, 6550; and HMGN2, 5900 M−1 cm−1. The experimental procedure for this purpose is described here briefly. First, the absorbance at 276 or 220 nm for the sample to be analyzed was measured. Then 30 μl of sample solution was transferred to a vacuum hydrolysis tube (Pierce) together with 100 μl of constantly boiling HCl. The tube was sealed under vacuum and placed at 110 °C for 24 h. The hydrolysis mixture was dried under vacuum. In addition to running standard amino acids to calibrate the elution profile, three control experiments were performed to correct the profile for background. The amino acid content of each sample was used to calculate its concentration based on the amino acid composition of histone proteins, and this, along with the absorbance of the original sample at 220 nm, was used to calculate the extinction coefficients. The molar extinction coefficient (ɛ260) of the DNA fragment was calculated from the sequence using the following constants: dA, 15,300; dC, 7400; dG, 11,500; and dT, 8700 M−1 cm−1. The ɛ260 value of the 189-bp double strand DNA was thus 4,190,400 M−1cm−1. The circular dichroism data were expressed in terms of mean residue ellipticity, [θ].
3. Results
3.1. Purification of human histone proteins and formation of octameric protein
The human histone genes used in the present work were obtained from a gene cluster [45,46] containing at least one copy of each histone gene in HeLa cells. Each of the expression vectors was introduced into E. coli cells. The purification procedures for bacterially synthesized histone proteins from Xenopus laevis [39], Saccharomyces cerevisiae [53,54], and Drosophila melanogaster [38] have been reported. Our purification process was essentially the same as described in these works, with a slight modification, to completely remove contamination by reverse phase (RP) HPLC. The human histone proteins were purified by ion-exchange chromatography in the presence of urea followed by RP-HPLC. Typically, 3.0–15 mg of each histone protein was obtained from 1 L culture of LB medium. To examine the sample homogeneity, each purified sample was subjected to mass analysis with ESI mass spectroscopy. The observed molecular weight of each protein was consistent with the value deduced from the amino acid sequence within 10 mass units, confirming the absence of any chemical modification to the recombinant histones. The N-terminal methionine added due to the plasmid construction was not present in the final polypeptides.
The histone proteins were refolded using a stepwise dilution of urea in the dialysis buffer to create the octameric protein, as reported in a earlier paper [49]. The resultant histone octamer mixture was further purified by size-exclusion chromatography. The properly folded histone octamer was separated from the misfolded histone aggregates. Fig. 1A shows the elution profile of size-exclusion chromatography, in which two distinct peaks can be seen. The SDS–PAGE analysis of each fraction using a 20% polyacrylamide gel shows the faster eluant included all four kinds of histone proteins, while the slower one contained only the H2A–H2B dimer, as shown in Fig. 1B. The yield of the refolded histone octamer was approximately 50% of the input material, resulting in a few mg histone octamer.
Fig. 1.
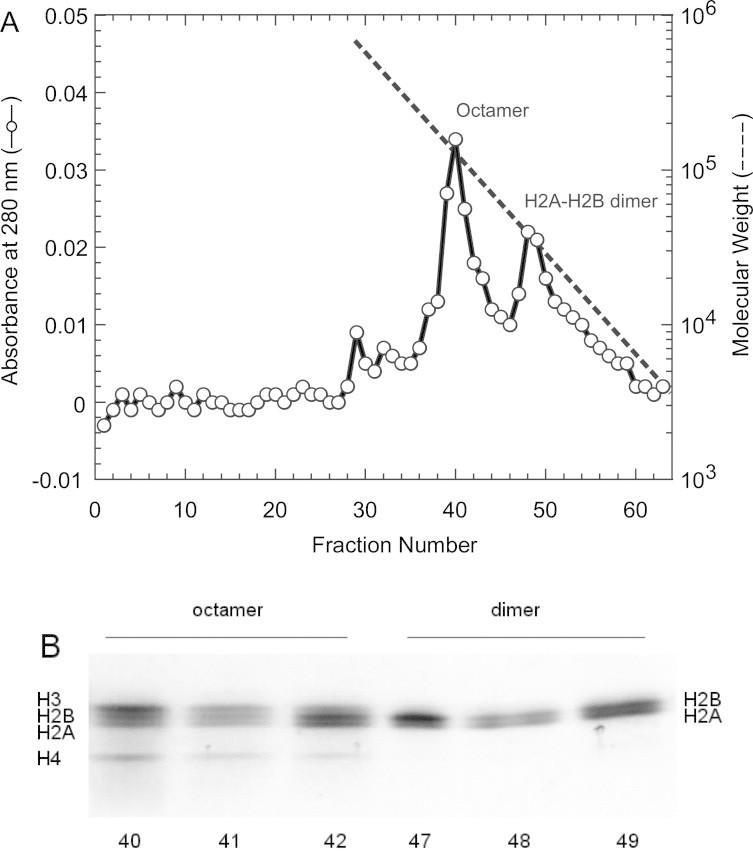
Purification of recombinant human histone complexes. (A) Gel filtration on a Superdex-200 column (15 × 750 mm). An aliquot of the concentrate (1 ml) was applied to a gel-filtration column. The column was developed at the flow rate of 0.3 ml/min, and 1 ml fractions were collected. The peak fractions (40–42, 47–49) were analyzed by SDS–PAGE. (B) Analysis of proteins by SDS–PAGE under reducing conditions on a 20% gel. The protein was stained with Coomassie Brilliant Blue R-250.
Fig. 2 shows the circular dichroism spectrum of the histone octamer in a solution having a high salt concentration, 2 M NaCl. Under such condition, the histone octamer exists as a stable entity [55,56]. The histone octamer shows a strong negative band around 209 nm and 222 nm, which indicates a high α-helix content of histones. These results show that the histone proteins were properly folded into octamers. The CD spectrum obtained for the recombinant histone octamer was essentially identical to those previously reported for the native histone octamer extracted from chicken erythrocytes [57] and HeLa cells [51].
Fig. 2.
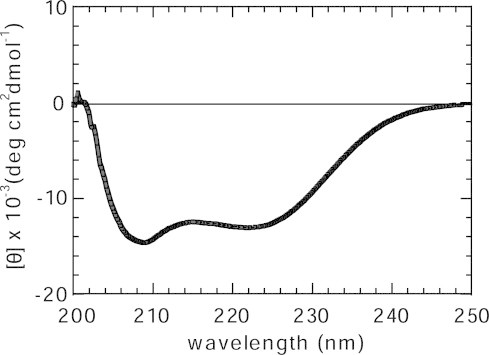
CD spectrum of the human histone octamer. The human histone octamer in 2 M NaCl, 0.1 mM EDTA, 10 mM Tris–HCl (pH 7.5).
3.2. Reconstitution of the recombinant nucleosome core particle
In order to construct the nucleosome core particle harboring uniquely located core histones on the DNA without using nucleosome remodeling factors, a 189-bp DNA fragment with a sequence consisting of the repeated 10-bp “TATA-tetrad” sequence of 5′-TATAAACGCC-3′ [44] was used. This repeated sequence was originally identified from a mouse genomic DNA library. It was found to position a nucleosome core particle with very high affinity, ∼350-fold higher than that of a random sequence [42,43]. This repeat sequence is known to function as a nucleosome positioning signal [41–43]. In the present work, the DNA fragment was obtained by PCR amplification from a plasmid that included the pre-cloned desired DNA sequence. Typically, 10−9 mol of DNA fragment was obtained from a large-scale preparation (30 ml) by PCR. The nucleosome was reconstituted from the mixture of the high affinity DNA sequence and the reconstituted histone octamer. The buffer exchange from high to low ionic strength is achieved by an exponential gradient for 2 days. The gel mobility shift assay in Fig. 3 shows the nucleosome was formed.
Fig. 3.
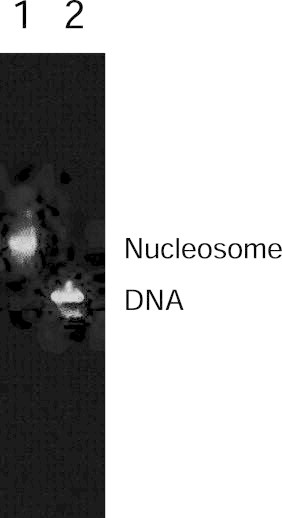
Nucleosome reconstitution on the TATAAACGCC repeat sequence. Lane 1, salt-induced nucleosome reconstitution mixture; Lane 2, free probe DNA. The reconstituted products were subjected to 4% native PAGE.
Rotational positioning, which refers to the orientation of the DNA relative to the surface of the histone octamer, is generally characterized by nuclease digestion or hydroxyl radical footprinting. The rotational positioning of the high affinity repeat sequence to the histone octamer from living cells has been previously characterized by hydroxyl radical footprinting and MNase digestion [44]. These experiments have shown that the repeat sequence is capable of positioning a nucleosome core particle with a unique translational position. Fig. 4 shows the result of MNase footprinting of our reconstituted nucleosome. The periodic MNase cleavage of the DNA can be seen every 10-bp within the central 147-bp of the nucleosome core particle. This periodic cleavage was not observed outside of the central 147-bp. In this footprinting result, the nucleosome was confirmed to be properly reconstituted with unique rotational positioning.
Fig. 4.
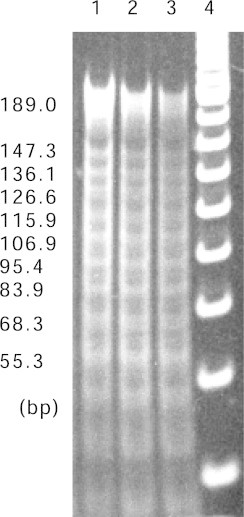
MNase cleavage analysis of the nucleosome core DNA. Aliquots of the reconstituted nucleosome sample used in the native PAGE analysis (Fig. 3) were characterized by MNase cleavage analysis, in which each nucleosome sample was partially digested with three different incubation times. Lane 1, 2 min; Lane 2, 5 min; Lane 3, 10 min. Protein was removed from the reaction products, and the resulting DNA fragments were resolved by 6% native PAGE and visualized by staining with ethidium bromide.
The reconstituted nucleosome was further characterized by CD spectroscopy. Fig. 5A compares the CD spectra of the nucleosome core particle and the free high-affinity sequence DNA. The CD spectrum of DNA has a characteristic positive band above the 260 nm region. The intensity of this positive band has been well characterized as being sensitive to the winding angle of the duplex [58,59]. As shown in the expanded spectra above 260 nm, Fig. 5B, the intensity of the positive DNA band decreased upon its wrapping around the octamer, meaning that the winding angle in the DNA duplex was increased in the process of nucleosome formation. This result was consistent with reports on the nucleosome harboring the histones from chicken erythrocytes [57] and calf thymus nucleus [30,31]. From these characterizations, we concluded that large-scale preparation of nucleosomes in mg quantity could provide properly folded particles to be used in subsequent experiments.
Fig. 5.
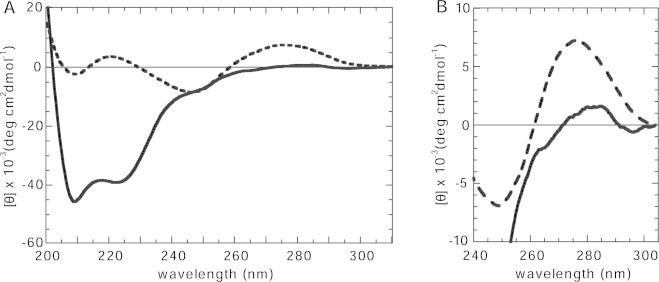
CD spectra of human recombinant nucleosome core particles. (A) The human recombinant nucleosome core particle (thick line) and the naked DNA (thick dashed line). (B) An enlarged view of the near UV region portion of A.
3.3. Purification of human HMGN proteins
The human HMGN genes used in the present work were obtained from a cDNA, synthesized with the extracted mRNA from HeLa cells. Each of the expression vectors was introduced into E. coli cells. Our purification process was essentially the same as describied in the works [50], with a slight modification, to completely remove contamination by ion-exchange chromatography followed by RP–HPLC. The SDS–PAGE analysis using 15% polyacrylamide gel shows a single band, as shown in Fig. 6. Typically, 2.0–3.9 mg of each HMG protein was obtained from 1 L culture of LB medium.
Fig. 6.
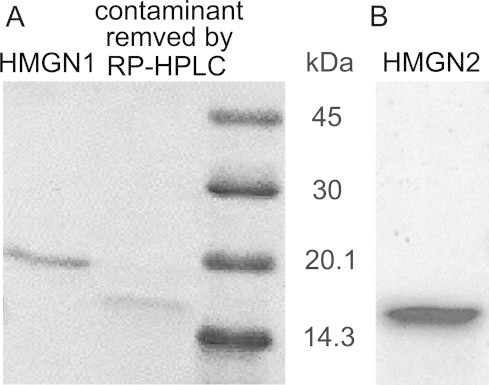
SDS–PAGE analysis of recombinant human HMGN proteins. (A) HMGN1 and (B) HMGN2 under reducing conditions on a 15% gel. The proteins were stained with Coomassie Brilliant Blue R-250.
3.4. CD analysis of structural changes induced by HMGN proteins
We compared the observed spectra for the HMGN/nucleosome mixture with the mathematical sum of the spectra for the pure HMGN proteins at the tested concentration plus the pure nucleosomes (HMGN+nucleosome). Fig. 7A and B shows the CD spectral changes caused by binding of HMGN1 and HMGN2, respectively, to the reconstituted nucleosome. The nucleosomes were mixed with either HMGN1 or HMGN2 (up to the molar ratio for HMGN/nucleosome = 3) in TE buffer (pH 7.5) with 16 mM NaCl. In Fig. 7A and B, the binding of both HMGN1 and HMGN2 decreased the intensity of the positive CD band in the region between 265 and 295 nm. The observed ellipticities [θ] at 280 nm ([θ]280) were 2.9 × 103 deg cm2 dmol−1 (HMGN1/nucleosome = 3), 3.2 × 103 deg cm2 dmol−1 (HMGN2/nucleosome = 3). Fig. 7C and D shows the dependencies of [θ]280 value on the molar ratio HMGN1/nucleosome and on the molar ratio HMGN2/nucleosome, respectively. The plots for the molar ratio (HMGN1/nucleosome) from 2 to 3 showed relatively fixed values, thus the ellipticities for the fully bound forms of the complexes were safely obtained in the tested region. This result appears to be consistent with the observation that the HMGN protein forms homodimer on a nucleosome [14]. On the other hand, the reduction of the [θ]280 value was smaller for the nucleosome–HMGN2 complex, compared to that seen upon HMGN1 binding. Because of the limited sensitivity for the CD signal in the region, the plotted signal intensities would contain noises. Thus, such ellipticities for the fully bound forms of the complexes were not confirmed.
Fig. 7.
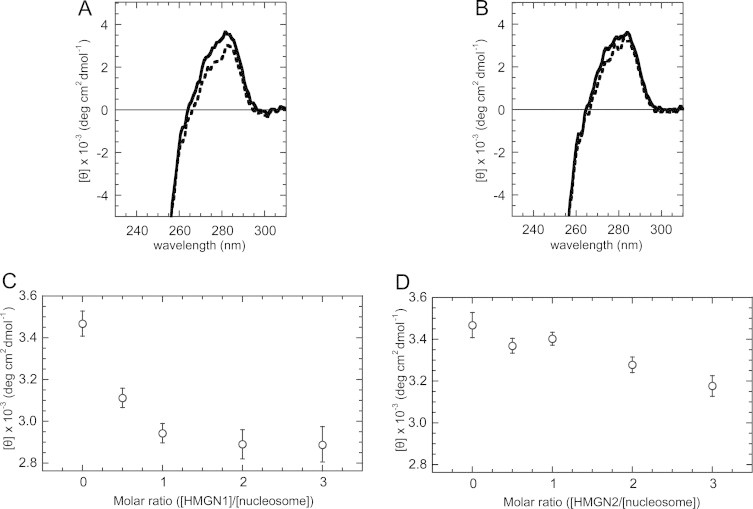
CD spectra of HMGN–nucleosome core complexes containing recombinant histones and HMGN. (A) HMGN1-nucleosome; (B) HMGN2–nucleosome. Stoichiometry is 3 mol of HMGN proteins per mol of nucleosomes. Samples are HMGN/nucleosome mixture with (thick discontinuous line) and the mathematical sum of the spectra for the pure HMGN proteins at the tested concentration plus the pure nucleosomes (thick line). Plot of the mean residue ellipticity [θ] at 280 nm according to the titration of HMGN1 (C), and HMGN2 (D), proteins.
4. Discussion
4.1. Conformational change of the reconstituted nucleosome by binding HMGN proteins
In the present work, conformational changes of the nucleosome DNA that are induced by the binding of HMGN1 and HMGN2 proteins were studied. The changes were investigated by using nucleosomes that were devoid of any covalent modifications to detect the intrinsic effect of the HMGN protein binding to nucleosomes. Our reconstituted nucleosome has the unmodified octameric protein and the unique DNA sequence that has the potency for the specific positioning of histone core along the DNA [40–44]. Its affinity to the octameric protein is approximately 350-fold higher than random sequence [40–44]. Therefore, the highly attractive interaction between unmodified octameric protein and DNA in the reconstituted nucleosome is thought to cause tightly stabilizing the structure of the nucleosome, implying that its structural change is suppressed more than that of the heterogeneously native nucleosome. Considering the binding of HMG protein to such reconstituted nucleosome, the attractive interaction is expected to somewhat suppress the structural change of DNA induced by binding of HMGN proteins, compared to the native nucleosome in the previous experiment [29,32].
Conformational changes could be detected by circular dichroism (CD) spectroscopy in the present work. Here, we found that the binding of HMGN protein to the unmodified nucleosome induced the reduction in the positive CD band in the region between 265 and 295 nm. In fact, the presently observed CD spectral changes by binding of HMGN proteins is smaller than those reported in previous works [29,32]. The change of the CD band in this region is known to be related to the DNA conformational change [58,59]. Especially, the reduction of the CD band in this region indicates an increase in winding angles of the DNA duplex [58,59]. Such conformational change of DNA appears to stabilize the nucleosome structure. This is thought to be consistent with the increased thermal stability of the complexes [29].
4.2. Difference between HMGN1 and HMGN2 in Complexes
The binding of two types of HMGN protein to nucleosomes induced different degree from each other. Considering that the magnitude of the positive band in the region between 265 and 295 nm decreases in a linear manner as the duplex winding angle increases [59], the observed difference in the CD band reduction in Fig. 7A and B implies that HMGN1 induces a tighter winding of the DNA than HMGN2 does. This in turn suggests that different conformational states of the nucleosome DNA are induced by binding of either HMGN1 or HMGN2. Thus, the observed CD spectral changes in Fig. 7A and B indicate that HMGN1 and HMGN2 have different abilities to induce DNA deformation in nucleosomes.
The affinities of HMGN1 and HMGN2 to the extracted nucleosomes from native cells were determined by using gel retardation assays: the dissociation constants are 1.05 × 10−7 M and 0.44 × 10−7 M for HMGN1 and HMGN2, respectively [14]. Because HMGN2 is expected to have a higher affinity to the unmodified nucleosome than HMGN1 as well, the lesser reduction of its CD band would not explained by the difference in their affinities. Therefore, the different structural changes induced by HMGN1 and HMGN2 should be ascribed to their different potencies for DNA deformation in the nucleosome. The different potency for affecting the structural changes that accompany binding of HMGN1 and HMGN2 might be related to the facts that (1) these proteins bind to nucleosomes to form complexes containing exclusively homodimers of either HMGN1 or HMGN2 [14] and (2) the proteins are organized into multiple distinct foci containing either HMGN1 or HMGN2 in cells [15]. Also, our results are consistent with the approaches to functionally distinguish between the two Xenopus laevis HMGN variants [60]. To explain the formation of exclusively homodimers on nucleosomes, we consider that binding of the first HMGN protein may cause a specific structural change in the nucleosome, leading to preferential recruitment of the second HMGN protein to form the homodimer. In the present CD analysis, specific details cannot be deduced regarding changes in the nucleosome structure facilitated by HMGN binding. However, the differences in the induced DNA conformations indicated by the present CD data may support a model of the structural alterations mediated by specific dimer formation. To gain further insight into the binding modes of HMGN1 and HMGN2, we will next analyze interactions between HMGN proteins and the reconstituted nucleosome, which is devoid of any covalent modification, by NMR spectroscopy [61].
In the present work done with the intention of constructing the uniquely formed nucleosome particle that includes an attractive interaction between unmodified octameric protein and DNA, we can detect the induced spectral changes by HMGN binding. Thus, we conclude that the binding of HMGN to the nucleosome can definitively alter its structure. The CD data obtained have indicated that binding of HMGN1 or HMGN2 increase the winding angle of the DNA in nucleosomes, and then the HMGN1 protein might induce tighter winding of the DNA, compared to the HMGN2.
Acknowledgements
We thank Drs. Yoshiaki Onishi and Ryoichi Kiyama for technical suggestions. We also thank Dr. Hitoshi Shirakawa for helpful comments on the manuscript. This work was supported in part by a Grand-in-Aid for Scientific Research from the Ministry of Education, Science, Sport and Culture of Japan (Nos. 13780482 and 12680608). Dr. Sailaja S. Seeram was supported by the Japan Society for the Promotion of Science (JSPS) postdoctoral fellowship for foreign researchers (ID: P99374).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Wolffe A.P. third ed. Academic press; London: 1998. Chromatin: structure and function. [Google Scholar]
- 2.Wolffe A.P., Kurumizaka H., Kivie M. The nucleosome: a powerful regulator of transcription. Prog. Nucleic Acid Res. Mol. Biol. 1998;61:379–422. doi: 10.1016/s0079-6603(08)60832-6. [DOI] [PubMed] [Google Scholar]
- 3.Wolffe A.P., Guschin D. Review: chromatin structural features and targets that regulate transcription. J. Struct. Biol. 2000;129:102–122. doi: 10.1006/jsbi.2000.4217. [DOI] [PubMed] [Google Scholar]
- 4.Cheung P., Allis C.D., Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103:263–271. doi: 10.1016/s0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 5.Roth S.Y., Denu J.M., Allis C.D. Histone acetyl transferases. Annu. Rev. Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 6.Vignali M., Hassan A.H., Neely K.E., Workman J.L. ATP-dependent chromatin-remodeling complexes. Mol. Cell. Biol. 2000;20:1899–1910. doi: 10.1128/mcb.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bustin M., Reeves R., Waldo E.C., Moldave K. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog. Nucleic Acid Res. Mol. Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 8.Rattner B.P., Yusufzai T., Kadonaga J.T. HMGN proteins act in opposition to ATP-dependent chromatin remodeling factors to restrict nucleosome mobility. Mol. Cell. 2009;34:620–626. doi: 10.1016/j.molcel.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bustin M., Trieschmann L., Postnikov Y. The HMG-14/-17 chromosomal protein family: architectural elements that enhance transcription from chromatin templates. Semin. Cell Biol. 1995;6:247–255. doi: 10.1006/scel.1995.0033. [DOI] [PubMed] [Google Scholar]
- 10.Bustin M. Regulation of DNA-dependent activities by the functional motifs of the High-Mobility-Group chromosomal proteins. Mol. Cell. Biol. 1999;19:5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bustin M. Chromatin unfolding and activation by HMGN chromosomal proteins. Trends Biochem. Sci. 2001;26:431–437. doi: 10.1016/s0968-0004(01)01855-2. [DOI] [PubMed] [Google Scholar]
- 12.Bustin M. Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem. Sci. 2001;26:152–153. doi: 10.1016/s0968-0004(00)01777-1. [DOI] [PubMed] [Google Scholar]
- 13.Shirakawa H., Herrera J.E., Bustin M., Postnikov Y. Targeting of high mobility group-14/-17 proteins in chromatin is independent of DNA sequence. J. Biol. Chem. 2000;275:37937–37944. doi: 10.1074/jbc.M000989200. [DOI] [PubMed] [Google Scholar]
- 14.Postnikov Y.V., Trieschmann L., Rickers A., Bustin M. Homodimers of chromosomal proteins HMG-14 and HMG-17 in nucleosome cores. J. Mol. Biol. 1995;252:423–432. doi: 10.1006/jmbi.1995.0508. [DOI] [PubMed] [Google Scholar]
- 15.Postnikov Y.V., Herrera J.E., Hock R., Scheer U., Bustin M. Clusters of nucleosomes containing chromosomal protein HMG-17 in chromatin. J. Mol. Biol. 1997;274:454–465. doi: 10.1006/jmbi.1997.1391. [DOI] [PubMed] [Google Scholar]
- 16.Grande M.A., van der Kraan I., de Jong L., van Driel R. Nuclear distribution of transcription factors in relation to sites of transcription and RNA polymerase II. J. Cell Sci. 1997;110:1781–1791. doi: 10.1242/jcs.110.15.1781. [DOI] [PubMed] [Google Scholar]
- 17.Zeng C., Kim E., Warren S.L., Berget S.M. Dynamic relocation of transcription and splicing factors dependent upon transcriptional activity. EMBO J. 1997;16:1401–1412. doi: 10.1093/emboj/16.6.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crippa M.P., Trieschmann L., Alfonso P.J., Wolffe A.P., Bustin M. Deposition of chromosomal protein HMG-17 during replication affects the nucleosomal ladder and transcriptional potential of nascent chromatin. EMBO J. 1993;12:3855–3864. doi: 10.1002/j.1460-2075.1993.tb06064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tremethick D.J., Hyman L. High mobility group proteins 14 and 17 can prevent the close packing of nucleosomes by increasing the strength of protein contacts in the linker DNA. J. Biol. Chem. 1996;271:12009–12016. doi: 10.1074/jbc.271.20.12009. [DOI] [PubMed] [Google Scholar]
- 20.Trieschmann L., Alfonso P.J., Crippa M.P., Wolffe A.P., Bustin M. Incorporation of chromosomal proteins HMG-14/HMG-17 into nascent nucleosomes induces an extended chromatin conformation and enhances the utilization of active transcription complexes. EMBO J. 1995;14:1478–1489. doi: 10.1002/j.1460-2075.1995.tb07134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vestner B., Bustin M., Gruss C. Stimulation of replication efficiency of a chromatin template by chromosomal protein HMG-17. J. Biol. Chem. 1998;273:9409–9414. doi: 10.1074/jbc.273.16.9409. [DOI] [PubMed] [Google Scholar]
- 22.Trieschmann L., Martin B., Bustin M. The chromatin unfolding domain of chromosomal protein HMG-14 targets the N-terminal tail of histone H3 in nucleosomes. Proc. Natl. Acad. Sci. 1998;95:5468–5473. doi: 10.1073/pnas.95.10.5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergel M., Herrera J.E., Thatcher B.J., Prymakowska-Bosak M., Vassilev A., Nakatani Y., Martin B., Bustin M. Acetylation of Novel Sites in the Nucleosomal Binding Domain of Chromosomal Protein HMG-14 by p300 Alters Its Interaction with Nucleosomes. J. Biol. Chem. 2000;275:11514–11520. doi: 10.1074/jbc.275.15.11514. [DOI] [PubMed] [Google Scholar]
- 24.Herrera J.E., Sakaguchi K., Bergel M., Trieschmann L., Nakatani Y., Bustin M. Specific acetylation of chromosomal protein HMG-17 by PCAF alters its interaction with nucleosomes. Mol. Cell. Biol. 1999;19:3466–3473. doi: 10.1128/mcb.19.5.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pogna E.A., Clayton A.L., Mahadevan L.C. Signalling to chromatin through post-translational modifications of HMGN. Biochim. Biophys. Acta. 2010;1799:93–100. doi: 10.1016/j.bbagrm.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 26.Abuhatzira L., Shamir A., Schones D.E., Schaffer A.A., Bustin M. The chromatin-binding protein HMGN1 regulates the expression of methyl CpG-binding protein 2 (MECP2) and affects the behavior of mice. J. Biol. Chem. 2011;286:42051–42062. doi: 10.1074/jbc.M111.300541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuddapah S. Genomic profiling of HMGN1 reveals an association with chromatin at regulatory regions. Mol. Cell. Biol. 2011;31:700–709. doi: 10.1128/MCB.00740-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Postnikov Y.V., Kurahashi T., Zhou M., Bustin M. The nucleosome binding protein HMGN1 interacts with PCNA and nacilitates Its binding to chromatin. Mol. Cell. Biol. 2012;32:1844–1854. doi: 10.1128/MCB.06429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paton A.E., Wilkinson-Singley E., Olins D.E. Nonhistone nuclear high mobility group proteins 14 and 17 stabilize nucleosome core particles. J. Biol. Chem. 1983;258:13221–13229. [PubMed] [Google Scholar]
- 30.Sasi R., Huvos P.E., Fasman G.D. A conformational study of the binding of a high mobility group protein with chromatin. J. Biol. Chem. 1982;257:11448–11454. [PubMed] [Google Scholar]
- 31.Sasi R., Fasman G.D. The effect of a high mobility group protein (HMG 17) on the structure of acetylated and control core HeLa cell chromatin. Biochim. Biophys. Acta. 1984;782:55–66. doi: 10.1016/0167-4781(84)90106-4. [DOI] [PubMed] [Google Scholar]
- 32.Zama M., Mita K., Ichimura S. Conformation of the HMG17-nucleosome complex. Biochim. Biophys. Acta. 1984;783:100–104. doi: 10.1016/0167-4781(84)90083-6. [DOI] [PubMed] [Google Scholar]
- 33.Masaoka A., Gassman N.R., Kedar P.S., Prasad R., Hou E.W., Horton J.K., Bustin M., Wilson S.H. HMGN1 protein regulates poly(ADP-ribose) polymerase-1 (PARP-1) self-PARylation in mouse fibroblasts. J. Biol. Chem. 2012;287:27648–27658. doi: 10.1074/jbc.M112.370759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Ramirez M., Rocchini C., Ausio J. Modulation of chromatin folding by histone acetylation. J. Biol. Chem. 1995;270:17923–17928. doi: 10.1074/jbc.270.30.17923. [DOI] [PubMed] [Google Scholar]
- 35.Norton V.G., Imai B.S., Yau P., Bradbury E.M. Histone acetylation reduces nucleosome core particle linking number change. Cell. 1989;57:449–457. doi: 10.1016/0092-8674(89)90920-3. [DOI] [PubMed] [Google Scholar]
- 36.Norton V.G., Marvin K.W., Yau P., Bradbury E.M. Nucleosome linking number change controlled by acetylation of histones H3 and H4. J. Biol. Chem. 1990;265:19848–19852. [PubMed] [Google Scholar]
- 37.Tse C., Sera T., Wolffe A.P., Hansen J.C. Disruption of higher-order folding by core histone acetylation rramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell. Biol. 1998;18:4629–4638. doi: 10.1128/mcb.18.8.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levenstein M.E., Kadonaga J.T. Biochemical analysis of chromatin containing recombinant drosophila core histones. J. Biol. Chem. 2002;277:8749–8754. doi: 10.1074/jbc.M111212200. [DOI] [PubMed] [Google Scholar]
- 39.Luger K., Rechsteiner T.J., Flaus A.J., Waye M.M.Y., Richmond T.J. Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 1997;272:301–311. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- 40.Roychoudhury M., Sitlani A., Lapham J., Crothers D.M. Global structure and mechanical properties of a 10-bp nucleosome positioning motif. Proc. Natl. Acad. Sci. 2000;97:13608–13613. doi: 10.1073/pnas.250476297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shrader T.E., Crothers D.M. Artificial nucleosome positioning sequences. Proc. Natl. Acad. Sci. 1989;86:7418–7422. doi: 10.1073/pnas.86.19.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thastrom A., Lowary P.T., Widlund H.R., Cao H., Kubista M., Widom J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 43.Widlund H.R. Identification and characterization of genomic nucleosome-positioning sequences. J. Mol. Biol. 1997;267:807–817. doi: 10.1006/jmbi.1997.0916. [DOI] [PubMed] [Google Scholar]
- 44.Widlund H.R., Kuduvalli P.N., Bengtsson M., Cao H., Tullius T.D., Kubista M. Nucleosome structural features and intrinsic properties of the TATAAACGCC repeat sequence. J. Biol. Chem. 1999;274:31847–31852. doi: 10.1074/jbc.274.45.31847. [DOI] [PubMed] [Google Scholar]
- 45.Albig W., Kardalinou E., Drabent B., Zimmer A., Doenecke D. Isolation and characterization of two human H1 histone genes within clusters of core histone genes. Genomics. 1991;10:940–948. doi: 10.1016/0888-7543(91)90183-f. [DOI] [PubMed] [Google Scholar]
- 46.Zhong R., Roeder R.G., Heintz N. The primary structure and expression of four cloned human histone genes. Nucleic Acids Res. 1983;11:7409–7425. doi: 10.1093/nar/11.21.7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cleveland D.W., Fischer S.G., Kirschner M.W., Laemmli U.K. Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J. Biol. Chem. 1977;252:1102–1106. [PubMed] [Google Scholar]
- 48.Panyim S., Chalkley R. High resolution acrylamide gel electrophoresis of histones. Arch. Biochem. Biophys. 1969;130:337–346. doi: 10.1016/0003-9861(69)90042-3. [DOI] [PubMed] [Google Scholar]
- 49.Luger K., Rechsteiner T.J., Richmond T.J., Paul M., Wassarman A.P.W. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 50.Bustin M., Becerra P.S., Crippa M.P., Lehn D.A., Pash J.M., Shiloach J. Recombinant human chromosomal proteins HMG-14 and HMG-17. Nucleic Acids Res. 1991;19:3115–3121. doi: 10.1093/nar/19.11.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prevelige P.E., Fasman G.D. Structural studies of acetylated and control inner core histones. Biochemistry. 1987;26:2944–2955. doi: 10.1021/bi00384a041. [DOI] [PubMed] [Google Scholar]
- 52.Gill S.C., von Hippel P.H. Calculation of protein extinction coefficients from amino acid equence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 53.Gelbart M.E., Rechsteiner T., Richmond T.J., Tsukiyama T. Interactions of Isw2 chromatin remodeling complex with nucleosomal arrays: analyses using recombinant yeast histones and Immobilized templates. Mol. Cell. Biol. 2001;21:2098–2106. doi: 10.1128/MCB.21.6.2098-2106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White C.L., Suto R.K., Luger K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001;20:5207–5218. doi: 10.1093/emboj/20.18.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karantza V., Baxevanis A.D., Freire E., Moudrianakis E.N. Thermodynamic studies of the core histones: Ionic strength and pH dependence of H2A–H2B dimer stability. Biochemistry. 1995;34:5988–5996. doi: 10.1021/bi00017a028. [DOI] [PubMed] [Google Scholar]
- 56.Karantza V., Freire E., Moudrianakis E.N. Thermodynamic studies of the core histones: pH and ionic strength effects on the stability of the (H3–H4)/(H3–H4)2 system. Biochemistry. 1996;35:2037–2046. doi: 10.1021/bi9518858. [DOI] [PubMed] [Google Scholar]
- 57.Wang X., Moore S.C., Laszckzak M., Ausio J. Acetylation increases the alpha-helical content of the histone tails of the nucleosome. J. Biol. Chem. 2000;275:35013–35020. doi: 10.1074/jbc.M004998200. [DOI] [PubMed] [Google Scholar]
- 58.Baase W.A., Johnson W.C. Circular dichroism and DNA secondary structure. Nucleic Acids Res. 1979;6:797–814. doi: 10.1093/nar/6.2.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chan A., Kilkuskie R., Hanlon S. Correlations between the duplex winding angle and the circular dichroism spectrum of calf thymus DNA. Biochemistry. 1979;18:84–91. doi: 10.1021/bi00568a013. [DOI] [PubMed] [Google Scholar]
- 60.Ong, M.S., Vasudevan, D., Davey, C.A. (2010) Divalent metal- and high mobility group N protein-dependent nucleosome stability and conformation. Journal of Nucleic Acids Article ID 143890, doi:10.4061/2010/143890. [DOI] [PMC free article] [PubMed]
- 61.Kato H., van Ingen H., Zhou B.-R., Feng H., Bustin M., Kay L.E., Bai Y. Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. Proc. Natl. Acad. Sci. 2011;108:12283–12288. doi: 10.1073/pnas.1105848108. [DOI] [PMC free article] [PubMed] [Google Scholar]