

Abstract
SUMMARY
Long-chain-length hydrophobic acyl residues play a vital role in a multitude of essential biological structures and processes. They build the inner hydrophobic layers of biological membranes, are converted to intracellular storage compounds, and are used to modify protein properties or function as membrane anchors, to name only a few functions. Acyl thioesters are transferred by acyltransferases or transacylases to a variety of different substrates or are polymerized to lipophilic storage compounds. Lipases represent another important enzyme class dealing with fatty acyl chains; however, they cannot be regarded as acyltransferases in the strict sense. This review provides a detailed survey of the wide spectrum of bacterial acyltransferases and compares different enzyme families in regard to their catalytic mechanisms. On the basis of their studied or assumed mechanisms, most of the acyl-transferring enzymes can be divided into two groups. The majority of enzymes discussed in this review employ a conserved acyltransferase motif with an invariant histidine residue, followed by an acidic amino acid residue, and their catalytic mechanism is characterized by a noncovalent transition state. In contrast to that, lipases rely on completely different mechanism which employs a catalytic triad and functions via the formation of covalent intermediates. This is, for example, similar to the mechanism which has been suggested for polyester synthases. Consequently, although the presented enzyme types neither share homology nor have a common three-dimensional structure, and although they deal with greatly varying molecule structures, this variety is not reflected in their mechanisms, all of which rely on a catalytically active histidine residue.
INTRODUCTION
Long-chain-length hydrophobic acyl residues play a vital role in a multitude of essential biological structures and processes. They build the inner hydrophobic layers of biological membranes, are converted to intracellular storage compounds, and are used to modify protein properties or function as membrane anchors, to name only a few important and versatile functions. (Hydroxy-)Fatty acids are usually activated for subsequent reactions by esterification of their carboxyl groups with the thiol group of coenzyme A (CoA) or of the acyl carrier protein (ACP), yielding acyl-thioesters. In general, there are two basic routes to provide long-chain fatty acids: (i) via de novo fatty acid synthesis from the central metabolite acetyl-CoA, yielding acyl-ACPs, or (ii) via uptake of exogenous fatty acids or other compounds that are converted to fatty acids such as alkanes and their conversion to acyl-CoAs by acyl-CoA synthetases (1, 2). Acyltransferases or transacylases utilize these activated acyl chains and transfer them to a variety of different substrates or polymerize them. Lipases form another important enzyme class dealing with fatty acyl chains; however, they cannot be regarded as acyltransferases in the strict sense. Since they employ a completely different enzymatic mechanism to cleave and transfer fatty acids, which is similar to the mechanism that is suggested for polyester synthases, their key features are presented and compared to those of other acyltransferases in this review.
Thus, this review aims at presenting an overview of the enzymatic processes where fatty acyl moieties are transferred from one molecule to another. The scope is not only the versatility of enzymes and mechanisms but also a comparison and generalization of these processes. Therefore, general key features as well as fundamental differences of acyl-transferring enzymes are pointed out whenever possible or known. Due to the nearly inexhaustible diversity of acyltransferases, this review will focus on several important and widespread processes, such as those catalyzed by enzymes involved in storage lipid synthesis (or degradation) and in membrane glycerolipid and lipid A synthesis, as well as enzymes that synthesize or modify polyketide (PK)-containing lipids, bacterial toxins, or antibiotics. Enzymes involved in the synthesis and elongation of fatty acids are beyond the scope of this review but have been reviewed comprehensively elsewhere (1, 3). Furthermore, the general focus is on acyl-transferring processes occurring in prokaryotes, but whenever possible, eukaryotic enzymes are described in comparison to their prokaryotic counterparts or in order to point to the distribution of a certain enzyme class.
ACYLTRANSFERASES INVOLVED IN STORAGE LIPID SYNTHESIS
In many habitats, bacteria are exposed to an unsteady and imbalanced nutrition supply. Thus, the ability to deposit intracellular carbon storage compounds conveys an advantage over competitors in the habitat when growth substrates become scarce. Consequently, nearly all prokaryotes known so far are able to accumulate at least one type of storage compound. Lipids represent ideal reserve materials, as they are highly calorific, water insoluble, and osmotically inert (4, 5). The most common lipophilic storage compounds in prokaryotes consist of esterified (hydroxy-)fatty acids and can be divided into polymeric lipids [poly(3-hydroxyalkanoic acids) {PHA}] and nonpolymeric, neutral lipids (triacylglycerols [TAG] and wax esters [WE]) (Fig. 1). While PHA are the predominant type of bacterial reserve materials, TAG are by far the most important storage lipids for eukaryotes. Wax esters are rather uncommon storage compounds but often fulfill specialized purposes (6–9).
Fig 1.
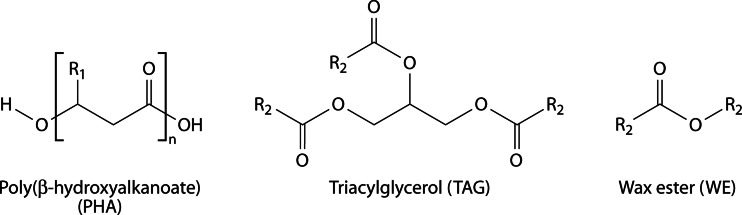
Chemical structures of common lipophilic storage compounds in prokaryotes: poly(3-hydroxyalkanoate) (PHA), triacylglycerol (TAG), and wax ester (WE). R1, alkyl chain with a length ranging from C1 to C13; R2, saturated or unsaturated long-chain-length alkyl residue.
The following section deals with a family of acyltransferases important for neutral lipid storage (TAG and WE), whereas enzymes involved in bacterial PHA storage, which employ a different mode of catalysis, will be discussed in the last section of the review.
TAG and WE in Eukaryotes
TAG consist of three long-chain fatty acids esterified with one molecule of glycerol (Fig. 1) and are, for example, deposited in large amounts in plant seeds or animal adipocytes and are also synthesized by various fungi or yeasts (8, 10, 11). WE also belong to the class of neutral lipids, as they are hydrophobic esters of long-chain fatty acids and primary long-chain fatty alcohols (Fig. 1). They are synthesized by various eukaryotes (including vertebrates), but, as mentioned above, they fulfill rather specialized purposes. For example, as one compound of cuticular wax, WE help to protect plant cells from exsiccation, UV light, and pathogens (12). Usually, WE are only a minor component of the cuticular waxes; e.g., in Arabidopsis the complex mixture contains only around 0.1 to 2.9% WE. In contrast, the thick layer on leaves of the carnauba palm (Copernicia cerifera) consists of up to 85% of WE (13). Furthermore, WE can also have a structural function as a compound of beeswax (14, 15). In noteworthy amounts, WE appear solely in seeds of the jojoba plant Simmondsia chinensis—which is very special, because plants normally accumulate TAG in their seeds—and in the spermaceti organ in the heads of sperm whales, where it helps to regulate buoyancy (8, 16). The valuable WE were one reason for extensive (and eventually banned) whale hunting; therefore, to date, jojoba and carnauba are major natural sources for WE (13, 17).
TAG and WE in Prokaryotes
Similar to the situation in eukaryotes, TAG are a more common storage lipid than WE in several groups of bacteria, but nevertheless, the majority of all bacteria store PHA rather than TAG or WE. Particularly, species belonging to the Gram-positive actinomycetes share the ability to synthesize large amounts of TAG, e.g., Rhodococcus opacus, with up to more than 80% of the cellular dry weight (CDW) (18). TAG are also the main storage lipid in other genera of the Actinomycetales, such as Mycobacterium, Nocardia, Actinomyces, Arthrobacter, Gordonia, or Dietzia, and in some streptomycetes, such as Streptomyces coelicolor, S. lividans, or Micromonospora echinospora (19–24). Species of the Gram-negative genus Acinetobacter are also able to synthesize TAG, but they accumulate only minor amounts. In contrast to others, these organisms accumulate mainly WE as storage lipids (25–27).
Bacterial WE formation was first discovered over 40 years ago in species of the Gram-negative genus Acinetobacter. Meanwhile, WE were also found in some genera of marine hydrocarbonoclastic bacteria, e.g., Alcanivorax, Marinobacter, or Thalassolituus, as well as in Psychrobacter, Micrococcus, or Moraxella (26, 28–33), and in some Gram-positive actinomycetes, e.g., in species of Corynebacterium or Nocardia (34, 35).
Biotechnological Relevance of TAG and WE
Besides the most obvious use of TAG as edible oils and fats, they have attracted a great and increasing interest for use in the production of fuels such as biodiesel (fatty acid alkyl esters [FAAE]) consisting of TAG-derived fatty acids esterified with short-chain alcohols (mainly methanol) as substitutes for diesel fuel (36). Furthermore, TAG are used as additives for a variety of therapeutic or pharmaceutical purposes. At present, commercially used TAG are obtained almost exclusively from vegetable oils (e.g., from palm, soybean, or rapeseed oil), but microbial lipid producers are considered as an alternative oil source that would not compete with human food supply (21, 37).
WE may also find diverse technical applications, for example, in the commercial production of cosmetics, candles, printing inks, lubricants, and coatings in a range of about 3 million tons per year (38). Currently, the dominant natural source for high-quality WE is jojoba oil, though its high price limits most of its possible applications to cosmetic and medical products. WE may also be produced chemically or with immobilized lipases (39). Thus, there is at present and will be in the future a demand for an economically feasible biotechnological production of inexpensive jojoba oil-like WE from inexpensive substrates (40).
Bacterial WE production not only could be achieved from renewable resources, e.g., sugars or plant-derived fatty acids, but also would enable the synthesis of custom-made WE due to the exceptionally broad substrate range of the responsible acyltransferase AtfA from Acinetobacter baylyi (41). The first studies of WE synthesis in a recombinant strain of Escherichia coli have already been performed, though still with low yields (40). Instead of production of long-chain WE, another promising area of application of the acyltransferase AtfA is the synthesis of FAAE from long-chain acyl-CoAs and ethanol to obtain fatty acid ethyl esters (FAEE), also referred to as “microdiesel” (42). FAEE resemble the established biodiesel fuel. Since FAEE exhibit similar and to some extent even improved properties, they could thus serve as an ecologically friendly, sustainable replacement for biodiesel (43).
Acyltransferase Enzyme Families Involved in TAG and/or WE Synthesis
TAG and membrane glycerophospholipids are both synthesized from the same precursor, phosphatidate (PA), and the two pathways are thus competitive regarding the involved enzymes and carbon flow. As will be outlined in more detail in the next section, glycerol-3-phosphate is successively acylated to 1,2-diacylglycerol (DAG) via the Kennedy pathway (44). The final and committed step from DAG and fatty acyl-coenzyme A (CoA) to TAG is accomplished by acyl-CoA:DAG acyltransferase (DGAT) (EC 2.3.1.20), the only enzyme that is unique to TAG synthesis (45, 46). In the majority of eukaryotes, this reaction is catalyzed by transmembraneous DGAT enzymes belonging to either the DGAT1 or DGAT2 enzyme family, which have evolved separately since the emergence of eukaryotes. Therefore, these families do not show any sequence similarity (47). DGAT1 enzymes are larger, with 6 to 9 transmembrane domains, and share high sequence similarities with eukaryotic sterol:acyl-CoA acyltransferases (ACAT) (EC 2.3.1.26). Enzymes belonging to the DGAT2 family, in contrast, are smaller, with only 1 or 2 transmembrane domains, and include acyl-CoA:monoacylglycerol (MAG) acyltransferases (MGAT) (EC 2.3.1.22) and acyl-CoA wax-alcohol acyltransferases (AWAT) (EC 2.3.1.75) (312).
Wax esters are synthesized by the esterification of long-chain fatty alcohols and CoA-activated fatty acids (acyl-CoA) catalyzed by wax synthases (WS) (EC 2.3.1.75). According to the current level of sequence information, WS fall into three separate groups, as mammalian WS (AWAT1 and AWAT2) are nonhomologous to WS from plants (jojoba type), which are in turn completely unrelated to bacterial WS/DGAT enzymes (7, 9, 17, 48).
As a rough simplification, at least six phylogenetically different families of acyltransferases, which synthesize TAG and/or WE, can be distinguished: (i) the DGAT1 family, (ii) the jojoba WS family, (iii) the DGAT2 family, (iv) an avian WS type, (v) a protozoan DGAT2-related type from Tetrahymena, and (vi) the WS/DGAT (AtfA-type) family of acyltransferases. Figure 2 schematically displays this diversity of enzymes capable of catalyzing DGAT and WS reactions. The jojoba-type WS has no obvious sequence similarities with currently known DGAT1 enzymes, but phylogenetic analyses indicate that the jojoba- and DGAT1-type enzymes share an origin, while recently identified avian and Tetrahymena WS are probably more closely related to DGAT2 than to DGAT1 (49, 50). Furthermore, these phylogenetic analyses clearly demonstrate that the WS/DGAT type of acyltransferase, which was first identified in the prokaryote A. baylyi, is of different origin than jojoba or mammalian WS (49–52). A phylogenetic tree showing the clustering of different WS and DGAT enzymes is provided in Fig. S1 in the supplemental material.
Fig 2.

Different families of acyltransferases involved in TAG and/or WE synthesis in eukaryotes and prokaryotes.
In prokaryotes, only enzymes belonging to the WS/DGAT category, which is shown on the right side in Fig. 2, have been identified so far. Thus, this appears to be the principal and common enzyme type for bacterial WE and TAG synthesis. Recently, two WS from plants belonging to this class have also been identified: PhWS1 from Petunia hybrida (53) and WSD1 from Arabidopsis thaliana (13) exhibit about 20% amino acid similarity to AtfA from A. baylyi. Furthermore, many putative proteins from eukaryotes that are similar to AtfA but yet uncharacterized exist in the database; e.g., A. thaliana possesses both 11 putative WS/DGAT-like enzymes and 12 putative enzymes related to the jojoba WS (13). Moreover, among terrestrial plants, WS/DGAT-homologous protein sequences have been found in, for example, monocotyledons (such as Triticum aestivum) and gymnosperms (such as Pinus taeda) (53).
Further BLAST searches indicate a much wider distribution of AtfA-like WS/DGAT enzymes not only in plants (e.g., medick, grape vine, lycophytes, poplar, grasses, and important crops such as wheat, barley, rice, or soybean) but also in protists and several animals belonging to cnidaria, arthropoda, or hemichordata (see Tables S1 to S4 in the supplemental material). Thus, it seems as though WS and TAG synthesis routes in prokaryotes and eukaryotes have not evolved strictly separated from each other.
AtfA from A. baylyi ADP1: a Bacterial Model Enzyme for WE and TAG Synthesis
AtfA (formerly referred to as WS/DGAT) is the key enzyme for neutral lipid accumulation in A. baylyi strain ADP1. It catalyzes the synthesis of TAG or WE from acyl-CoAs and DAG or fatty alcohols, respectively, as shown in Fig. 3. In 2003, it represented the first characterized member of a new class of acyl-CoA acyltransferases and is regarded as the model enzyme of this class [WS/DGAT (AtfA-type) on the right in Fig. 2] (51). Until now, enzymes similar to AtfA have been identified to be clearly responsible for the synthesis of neutral lipids in several different types of bacteria (Mycobacterium species, marine species of Marinobacter or Alcanivorax, oleaginous species of Rhodococcus, Streptomyces, and others), thereby underlying that the AtfA type is the typical type of acyltransferases essential for bacterial lipid storage.
Fig 3.

Synthesis of wax esters or triacylglycerols from acyl-CoA and fatty alcohol or diacylglycerol, respectively, catalyzed by AtfA.
Biochemical characteristics and substrate range of AtfA.
AtfA from A. baylyi is composed of 458 amino acids and is a 94-kDa homodimer in its native form. The enzyme has already been purified with and without an N-terminal His6 tag to apparent homogeneity in order to accomplish a detailed characterization of the enzyme. The WS reaction follows classical Michaelis-Menten kinetics, and the Km value was determined to be 29 μM for palmitoyl-CoA, with a Vmax of 2.0 μmol mg−1 min−1, whereas the DGAT reaction appeared to follow neither Michaelis-Menten nor cooperative enzyme kinetics. In the enzyme assay, the highest WS and DGAT activities could be measured at 45°C. Furthermore, it was observed that AtfA is inhibited by free CoA (54).
An outstanding property of AtfA is its exceptionally broad substrate range. This is already indicated under natural conditions by its intrinsic ability to synthesize not only WE from long- and straight-chain fatty alcohols but also TAG from more bulky DAG. Using 1-hexadecanol or 1,2-dipalmitoyl-glycerol and 1-palmitoyl-CoA (C16-CoA) as substrates, it was observed that AtfA exhibits approximately 10-fold-higher WS activity than DGAT activity. This value roughly corresponds to the proportions of WE and TAG in A. baylyi actually accumulated under storage conditions, which reach 6.9% (WE) and 1.4% (TAG) of the cellular dry weight, respectively, when the cells are cultivated with an unrelated carbon source (51). Highest enzymatic activities were measured with C16-CoA and linear C14 to C18 fatty alcohols (54). Under natural conditions, WE of 32 to 36 carbon atoms represent the main proportion of WE synthesized in A. baylyi (25, 55).
Additionally, a detailed substrate specificity analysis revealed that there is a great range of accepted (artificial) substrates regarding both the acyl donor and acyl acceptor. As an acyl donor, AtfA accepts saturated or unsaturated acyl-CoA thioesters ranging from C2 to C20. As an acyl acceptor, linear alcohols from C2 to C30 and branched alcohols (such as isoamyl alcohol), as well as cyclic or aromatic alcohols (e.g., cyclohexanol, 2-cyclohexylenethanol, cyclododecanol, and sterols) and mono- and diacylglycerides are accepted as substrates. The DGAT reaction shows a preference for the acylation of the sn-3 position. When the corresponding substrates are supplied, AtfA can also synthesize wax diesters or thio- and dithio-WE from palmitoyl-CoA and long-chain alkanediols or (di)thiols (1-hexadecanethiol, 1,8-octanedithiol, or 1-S-monopalmitoyl-octanedithiol) (56, 57). The enzyme's promiscuity leads to the assumption that the provision of hydrophobic substrates by the host cell is the only limiting factor for a large number of potential products (41). Furthermore, glycidol, a highly reactive and valuable pharmaceutical substrate, can be transformed into the less toxic derivative glycidyl acyl ester (by the esterification with palmitoyl-CoA) by AtfA (58).
While AtfA can acylate the sn-3 or (with lower specificity) the sn-2 position of MAG, it is not able to acylate glycerol or the sn-2 position of lysophosphatidic acid, and thus it does not exhibit acyl-CoA:lysophosphatidic acid acyltransferase (LPAT) activity (54, 56). Furthermore, AtfA does not accept polar substrates such as sugars, organic acids, amino acids, naphthol, amines, or carotenoids. Its catalytic center might be located in a hydrophobic pocket or channel, restricting the accessibility for hydrophilic molecules (59).
Active site and catalytic mechanism of AtfA.
Sequence comparisons with AtfA-homologous proteins of various origins revealed that the short heptapeptide motif HHxxxDG is highly conserved in all members of this class of acyltransferases (60). An HxxxxD-like pattern represents the catalytic active-site motif as part of a conserved condensation domain (Pfam 00668) in several different nonhomologous enzyme classes. In general, these enzymes share the ability to transfer thioester-activated acyl substrates to a hydroxyl or amine acceptor to form an ester or amide bond, such as acyltransferases that synthesize glycerolipids, nonribosomal peptide synthetases, acyltransferases involved in lipid A biosynthesis, polyketide-associated acyltransferases, or chloramphenicol acetyltransferase (CAT). The catalytically active histidine in this motif initiates the deprotonation of a hydroxyl group to enable the nucleophilic attack on the acyl donor (61–67).
Consequently, the assumed mechanism of AtfA starts with the catalytically active histidine residues (His132/133) acting as bases to deprotonate the hydroxyl group of the fatty alcohol or DAG (Fig. 4). Subsequently, initiated by a nucleophilic attack of the generated oxyanion, the oxoester bond of the WE or TAG is formed, while a proton is transferred from histidine to the CoA-S− residue. This results in a release of CoA-SH and a regeneration of the catalytic histidine residues (59).
Fig 4.

Mechanism of wax ester synthesis from acyl-CoA thioester and fatty alcohol catalyzed by AtfA from A. baylyi strain ADP1. (Based on data from reference 59.)
Actually, it was confirmed that this motif is part of the active site of AtfA from A. baylyi strain ADP1 and that, in particular, the second histidine (His133) is crucial for its activity: a replacement of His132 by leucine led to a significant decrease of the enzyme activity, but a complete loss of activity was observed only when both histidines were replaced. Although aspartate and glycine are conserved amino acids in this motif, they seem not to be of major importance for the enzyme activity, because a replacement of either by alanine resulted in no significant decrease of enzyme activity. Their possible structural function still has to be elucidated (59).
Subcellular localization of AtfA.
Due to its isoelectric point of 9.05, the native form of AtfA is positively charged in an environment with a neutral pH. Together with its hydrophobic regions, which are important for the interaction with hydrophobic substrates, this causes an amphiphilic character of the enzyme. This explains why it is partly distributed in the cytoplasm, whereas the main proportion is associated with the membrane or lipid inclusions. It can be speculated that its activity and/or substrate specificity might be influenced depending on whether AtfA is exposed to a hydrophilic (cytoplasm) or hydrophobic (membrane-associated) environment (54, 67). This amphiphilic trait is in sharp contrast to the highly hydrophobic eukaryotic WS, like the enzymes from jojoba or the phytoflagellate Euglena gracilis, harboring several transmembrane domains (17, 52).
Heterologous expression of the atfA gene from A. baylyi ADP1 in prokaryotes and eukaryotes.
The atfA gene from A. baylyi has already been expressed in a functionally active form in a Pseudomonas sp. and E. coli, as well as in the eukaryotic yeast Saccharomyces cerevisiae. In the alkane-degrading bacterium Pseudomonas citronellolis, the heterologous expression of atfA enabled the cells to synthesize WE (but no TAG) when 1-hexadecanol was provided in the medium (51).
In order to reconstruct the WE synthesis pathway from unrelated carbon sources in bacteria that do not naturally accumulate storage lipids, atfA has been introduced in an engineered E. coli strain expressing a bifunctional jojoba acyl-CoA reductase. This enzyme accomplishes the reduction of fatty acyl-CoAs to fatty alcohols in coupled NADPH-dependent reactions, whereas usually the two-step reduction via fatty aldehyde is catalyzed by two independent reductases (68). When cells were cultivated with oleate, the synthesized WE amounted to only 1% of the CDW, although the cells exhibited high WS and DGAT enzyme activities. Additionally, the detection of fatty acid butyl esters indicated that trace amounts of 1-butanol from medium components were also acylated by AtfA. Thus, this study demonstrated that it is in principle feasible to heterologously introduce the WE synthesis pathway in a non-lipid-storing organism, but simultaneously, these experiments also showed that the yield might be strongly restricted due to a weak provision of precursors by the host cell (40).
In 2006, atfA attracted attention as part of the constructed plasmid “pMicrodiesel,” which enabled E. coli to synthesize biodiesel-like FAEE, in particular ethyl oleate. The heterologous coexpression of atfA with pdc and adhB (encoding pyruvate decarboxylase and alcohol dehydrogenase from Zymomonas mobilis, respectively) combined ethanol formation and subsequent acylation. When the engineered strain was cultivated in the presence of glucose and oleic acid, it produced FAEE in amounts of up to 1.28 g/liter or 26% of the CDW (42). Later, this process was scaled up by cultivating the E. coli strain in 20 liters mineral salts medium using glucose or low-price glycerol and sodium oleate. In this pilot-scale production, a cellular FAEE content of approximately 25% (of CDW) was obtained (69).
In contrast to the establishment of WE synthesis in bacteria, the heterologous expression of atfA in S. cerevisiae did not result in WE formation, indicating that the cell could not provide sufficient amounts of fatty alcohols. However, the atfA expression enabled the synthesis of TAG, FAEE, and fatty acid isoamyl esters in a quadruple DGA1 LRO1 ARE1 ARE2 disruption mutant of S. cerevisiae. Steryl ester biosynthesis was not complemented, although crude extracts of the atfA-expressing cells exhibited high acyl-CoA:sterol acyltransferase (ASAT) activity in vitro. Furthermore, it was noticed that AtfA has different substrate specificities depending on whether the expression host is E. coli or S. cerevisiae. On one hand, yeast cells showed a significantly higher DGAT activity (nearly as high as the WS activity) than cells of E. coli. On the other hand, the ASAT activity was considerably higher in E. coli than in S. cerevisiae (70).
S. cerevisiae strains were also investigated for their suitability to produce FAEE biodiesel. For example, the expression of atfA in an engineered S. cerevisiae strain that utilizes endogenously synthesized ethanol (from glycerol) and exogenous fatty acids enabled the formation of up to 0.52 g/liter FAEE (71).
In order to find the optimal WS for an eukaryotic biodiesel production, another study compared recombinant S. cerevisiae strains harboring five different enzymes coming from A. baylyi, Marinobacter hydrocarbonoclasticus, Rhodococcus opacus, Mus musculus, and Psychrobacter arcticus (72). The highest biodiesel yields were obtained with a strain harboring the WS enzyme from M. hydrocarbonoclasticus. This finding was further underlined by in vitro activity measurements in crude cell extracts, as WS from M. hydrocarbonoclasticus showed the highest WS activity with nearly all of the tested substrates (C2, C4, C6, C8, C10, C12, C14, C16, and C18 alcohols) of all studied enzymes. However, AtfA from A. baylyi also accepted all provided alcohols as acyl acceptors, and its substrate range was comparable to those of other tested enzymes with a clear preference for alcohols of longer carbon chain length. This study demonstrated that not only AtfA but also AtfA-like enzymes from diverse origins share an extraordinarily broad substrate range and are therefore interesting candidates for various biotechnological purposes. Therefore, key features of AtfA-like acyltransferases will be discussed in the following sections. Table 1 gives an overview of some properties of AtfA-like acyltransferases that have been characterized so far, which reveals that these homologs vary substantially in size, sequence, or specific activities in natural or artificial hosts. The amino acid identities compared to AtfA range from 22 to 52%, and a multiple-sequence alignment (MSA) discloses the presence of long stretches with a very low degree of conservation (Fig. 5).
TABLE 1.
Characterized AtfA-like acyltransferases from bacteria and plantsa
| Organism | Protein | Accession no. | Size (amino acids) | Maximum % identity (similarity)b | Storagec | Activity in E. colid |
Reference | |
|---|---|---|---|---|---|---|---|---|
| WS | DGAT | |||||||
| A. baylyi ADP1 | AtfA | AAO17391 | 458 | 100 (100) | WE, TAG | ✓ | ✓ | 51 |
| M. tuberculosis H37Rv | Tgs1 | NP_217646 | 463 | 27 (44) | TAG | ✓ | 73 | |
| Tgs2 | NP_218251 | 454 | 38 (58) | (✓) | ✓ | |||
| Tgs3 | NP_217751 | 271 | 29 (47) | ✓ | ||||
| Tgs4 | NP_217604 | 474 | 29 (47) | ✓ | ||||
| M. hydrocarbonoclasticus | WS1 | ABO21020 | 455 | 46 (66) | (ip) WE | ✓ | ✓ | 32 |
| WS2 | ABO21021 | 473 | 39 (60) | ✓ | ||||
| A. borkumensis SK2 | AtfA1 | YP_694462 | 457 | 50 (69) | TAG (WE) | ✓ | ✓ | 26 |
| AtfA2 | YP_693524 | 451 | 41 (60) | ✓ | ||||
| R. opacus PD630 | Atf1 | ACX81314 | 462 | 25 (43) | TAG (WE) | ✓ | 74 | |
| Atf2 | EHI41112 | 453 | 39 (61) | ✓ | ✓ | |||
| S. coelicolor A3(2) | Sco0958 | NP_625255 | 446 | 25 (42) | TAG | ✓ | 75 | |
| Sco1280 | NP_625567 | 413 | 22 (34) | (✓ ?) | ||||
| S. avermitilis MA-4680 | SAV7256 | NP_828432 | 447 | 25 (41) | TAG | ✓ | (✓) | 76 |
| P. arcticus 273-4 | DGAT | YP_263530 | 475 | 52 (72) | ? | ✓ | ? | 72 |
| P. hybrida | PhWS1 | AAZ08051 | 521 | 22 (40) | WE | ✓ | 53 | |
| A. thaliana | WSD1 | AED94163 | 481 | 22 (41) | WE | ✓ | (✓) | 13 |
Abbreviations: WE, wax esters; TAG, triacylglycerols; (ip) WE, isoprenoid wax esters; WS, wax synthase; DGAT, acyl-CoA:diacylglycerol acyltransferase activity.
Maximal percentage of identical or similar amino acids compared to AtfA from A. baylyi strain ADP1.
Natural function of the respective enzyme in its native host; parentheses indicate that WE were synthesized only when precursors (such as hexadecanol) were supplied.
Enzymatic activity of the respective enzyme (catalysis of WS and/or DGAT reactions) upon heterologous expression in E. coli.
Fig 5.

Multiple-sequence alignment of AtfA-like proteins from 17 different organisms (for details, see Table 1). Predicted secondary structural motifs of AtfA are schematically displayed above the AtfA sequence, boxes represent putative α helices, and arrows represent β strands. The active-site motif is marked with a red box.
Other AtfA-Like Acyltransferases in Different Bacteria
Mycobacterium spp.
The important human pathogen Mycobacterium tuberculosis attacks alveolar macrophages and often remains in a nonreplicative, drug-resistant dormancy state. It starts replicating again when the host's immune system is weakened, leading to active tuberculosis (77, 78). To outlast long starvation phases during the dormancy state, the cells seem to rely on lipids as energy reserves. Intracellular TAG inclusions could be detected in M. tuberculosis cells obtained from organ lesions (79, 80). It is assumed that TAG synthesis is induced at the transition to the dormancy state when the cells encounter stressful, hypoxic conditions (73). In the nonpathogenic strain M. smegmatis mc2155, the synthesis of intracellular lipid inclusions, composed mainly of TAG, occurred during the stationary phase, especially in nitrogen-limited medium (79).
The characterization of atfA from A. baylyi in 2003 also paved the way for the identification of 15 atfA-like genes in M. tuberculosis strain H37Rv (with up to 39% amino acid identity to AtfA from A. baylyi) and 8 genes in M. smegmatis mc2155 (with up to 41% amino acid identity to AtfA). This high number of orthologous genes in mycobacteria suggests a great importance for their survival (51, 73). When M. smegmatis is cultivated under storage conditions with glucose, the cells accumulate TAG, although they exhibit both high DGAT and WS activity. However, WE are solely formed in vivo if hexadecanol is provided in the medium. The gene wdh3269, which exhibits the highest similarity to atfA, conferred only very weak WS/DGAT activities when heterologously expressed in E. coli or R. opacus. Therefore, the protein encoded by wdh3269 probably does not have a major contribution to TAG accumulation in M. smegmatis (51).
Reverse transcription-PCR (RT-PCR) analysis revealed that all 15 atfA-like genes of M. tuberculosis H37Rv (designated tgs for “TAG synthases”) are transcribed and that especially tgs1 and, to a lesser extent, tgs2 to -4 are induced under dormancy-inducing conditions. The 15 Tgs proteins significantly differ in their predicted properties, such as the theoretical molecular mass (from 21 to 54 kDa) or the theoretical pI (from 4.7 to 10.4). Furthermore, the predicted subcellular localization differs from membrane bound (8 homologs) to cytoplasmic (6 homologs). The amino acid identities to AtfA ranged from 15 to 39%, and the HHxxxDG motif is modified in 3 homologs. Interestingly, Tgs3 (Rv3234c) exhibited comparatively high DGAT and WS activities, although the second histidine of the active-site motif is replaced by glutamine. In an enzymatic assay of each of the 15 Tgs proteins (with N-terminal His6 tags), significant DGAT activities were detected in only four recombinant E. coli strains, expressing Tgs1 (Rv3130c), Tgs2 (Rv3734c), Tgs3 (Rv3234c), or Tgs4 (Rv3088). However, the authors speculated that the substrate range of Tgs enzymes is likely to differ from the standard substrates applied in the enzyme assay (73). Later, it was shown that Tgs1 prefers C26 acyl-CoA as a substrate. Furthermore, the disruption of tgs1 drastically reduced the ability to accumulate TAG. Thus, Tgs1 is the major contributor to TAG synthesis, and, consequently, C26 is the major constituent of TAG in M. tuberculosis H37Rv (81).
M. ratisbonense SD4 is naturally adapted to degrade alkanes or isoprenoids such as phytane or squalane, and it accumulates TAG or a mixture of isoprenoid WE derived from oxidized intermediates of incomplete isoprenoid catabolism (82, 83). Isoprenoid WE biosynthesis from bulky substrates again demonstrates the broad substrate spectrum of AtfA-like acyltransferases and emphasizes that the type of accumulated lipids depends mainly on the physiological background and on available metabolites rather than on a restricted substrate range of the involved enzymes (83).
Marinobacter hydrocarbonoclasticus.
The Gram-negative, marine bacterium M. hydrocarbonoclasticus is able to utilize various hydrocarbons and isoprenoids as sole carbon and energy sources (84). Furthermore, like some other marine bacteria, M. hydrocarbonoclasticus can accumulate (polyunsaturated) isoprenoid WE upon degradation of phytol, farnesol squalene, or similar chlorophyll-derived compounds, which are abundant in the marine sediment (32, 85–87).
In 2007, four isoprenoid WE biosynthetic genes (together with isoprenoid-CoA synthetases) were identified in the marine bacterium M. hydrocarbonoclasticus by identifying genes similar to atfA in the genome sequence of Marinobacter aquaeolei strain VT8 (32). Meanwhile, it has been proposed to designate M. aquaeolei as M. hydrocarbonoclasticus, too (88). The respective sequences in the genome of M. hydrocarbonoclasticus encode enzymes (termed WS1 to -4) with amino acid identities of 27 to 45% compared to AtfA from A. baylyi (32). WS4 is the product of a truncated pseudogene in M. hydrocarbonoclasticus (but not in M. aquaeolei). N-terminally His6-tagged WS1, WS2, and WS3 were purified in order to analyze their substrate ranges and compare them with that of AtfA (32). It turned out that WS1 and WS2 prefer long-chain-length acyl-CoAs (>C14) to acylate a wide spectrum of fatty alcohols (C10 to C16) or isoprenoid alcohols (phytol or farnesol). Interestingly, if the acyl donor was phytanoyl-CoA instead of (unbranched) acyl-CoA, only equally bulky molecules, phytol and farnesol, were accepted as reaction partners. WS1 and WS2 are able to synthesize not only isoprenoid WE but also hybrid acyl-isoprenoid WE and “normal” WE (Fig. 6). On one hand, regarding WE synthesis, WS2 seems to exhibit a broader substrate range, with a higher preference for longer-chain alcohols as well as a higher activity for isoprenoid WE formation than WS1. On the other hand, only WS1 exhibited DGAT activity. WS3 did not show activity with any of the tested substrates, and thus either it might be an inactive variant or its substrate range is significantly different from those of WS1 and WS2. AtfA from A. baylyi was also included in this test but was unable to catalyze isoprenoid WE formation from phytanoyl-CoA and phytol (32).
Fig 6.

Biosynthesis of (hybrid) isoprenoid wax esters (WE) from phytol and phytanoyl-CoA or acyl-CoA, as catalyzed by WS2 from M. hydrocarbonoclasticus.
When specific activities of the enzymes were compared by means of a spectrophotometric assay, WS2 turned out to be significantly more active than both WS1 and AtfA. For the formation of “normal” WE from hexadecanol and palmitoyl-CoA, its specific activity was about 61.3 mmol/min/mg, in comparison to only 1.3 or 0.4 mmol/min/mg for WS1 or AtfA, respectively. Furthermore, a specific activity of 28.9 mmol/min/mg with phytol and palmitoyl-CoA was detected for WS2, whereas WS1 or AtfA exhibited only about 0.2 or 0.1 mmol/min/mg. For the conversion of isoprenoid substrates (phytol and phytanoyl-CoA), there was no activity detectable with WS1 or AtfA using this assay method, but WS2 still exhibited an activity of approximately 0.4 mmol/min/mg. This value for isoprenoid substrates is comparable to the specific activity of AtfA with its natural acyl substrates. WS1 appears to be quite similar to AtfA; it not only shares the highest amino acid identity (45%) of all M. hydrocarbonoclasticus enzymes but also has similar activity levels and substrate preferences (e.g., it accepts DAG, in contrast to WS2) (32).
WS2 from M. hydrocarbonoclasticus was also included in the already-mentioned comparison of five different acyltransferases for their suitability for biodiesel synthesis in S. cerevisiae, and the strain expressing ws2 achieved the highest biodiesel production. Furthermore, WS2 exhibited highest in vitro activities for nearly all of the tested alcohol substrates, including ethanol. For example, its activity toward ethanol and palmitoyl-CoA was nearly twice as high as the activity of AtfA (72).
In a very recent study, Barney and coworkers compared AtfA and four other bacterial WS/DGATs from Psychrobacter cryohalolentis K5 and Rhodococcus jostii RHA1, as well as two proteins from M. aquaeolei VT8 (which, in principle, equate with WS1 and WS2) (28). The respective genes were expressed in E. coli as dual fusion proteins, harboring an N-terminal maltose binding protein tag and an additional C-terminal His8 tag for increased solubility and easier purification. In in vitro activity assays, the enzyme Ma1 from M. aquaeolei showed by far the highest WS activity with palmitoyl-CoA and dodecanol or hexadecanol. In general, all enzymes exhibited significantly higher WS activities with dodecanol than with hexadecanol, although AtfA showed a higher specificity for hexadecanol in earlier measurements (54). However, as different assay methods were applied, the results might not be comparable. In general, Ma1 from M. aquaeolei turned out to be the most active enzyme. It is also most suitable for a rapid purification process, and hence it is seen as the candidate with greatest potential for future structural and mechanistic studies (28). As mentioned above, it has been proposed that M. aquaeolei and M. hydrocarbonoclasticus belong to the same species (88); thus, Ma1 corresponds to WS1, and Ma2 corresponds to WS2. The deposited protein sequences of WS1 (accession number ABO21020.1) and Ma1 (YP_957462.1) are identical except for position 194, which is aspartate in WS1 and glycine in Ma1, and position 321, where aspartate is conservatively exchanged by glutamate. While the first difference at position 194 lies in a nonconserved area (Fig. 5), the aspartate at position 321 is conserved in many other AtfA-homologous proteins. Between the WS2 (ABO21021.1) and Ma2 (YP_960328.1) sequences, there is only one difference, at position 395, which is glycine in WS2 but aspartate in Ma2. This residue is not conserved in other AtfA homologs (Fig. 5). However, despite there being nearly 100% identity between WS1 and Ma1 and between WS2 and Ma2, their determined activities were greatly divergent. When WS1 and WS2 were compared, the WS activity of WS2 turned out to be approximately 60-fold higher than that of WS1 (32). In contrast, the comparative assay of the WS activities of Ma1 and Ma2 showed that Ma1 is significantly more active than Ma2, i.e., approximately 7.5-fold with dodecanol as the substrate or 3-fold with hexadecanol as the substrate. So far, it is uncertain how these oppositional results for WS1/Ma1and WS2/Ma2 came about and which enzyme, the first or second AtfA homolog from M. hydrocarbonoclasticus, is indeed the more active enzyme.
Another very interesting finding concerning Ma1 from M. aquaeolei was the identification of the small alanine residue at position 360 to be relevant for the fatty alcohol chain length selectivity of Ma1 (89). The replacement of alanine by isoleucine resulted in an increased activity with the shorter-chain-length fatty alcohols nonanol and decanol compared to that of the wild-type enzyme, which is most active with dodecanol. This effect could also be shown for AtfA when replacing the corresponding glycine at position 355 by isoleucine. Apart from that, the ratio of enzyme activities with hexadecanol versus dodecanol remained uninfluenced by the mutations. Therefore, the authors assumed that the incorporation of a larger amino acid residue does not block the access for longer-chain-length alcohols but improves the binding of shorter alcohols to the active site instead (89). The identification of residues affecting the substrate specificities of AtfA-like WS/DGATs is of great relevance for potential biotechnological utilization; hence, this recent study marks a first and important step in this direction.
Alcanivorax borkumensis.
The Gram-negative marine bacterium A. borkumensis is specialized for the utilization of hydrocarbons as sole carbon and energy sources (90). Due to an otherwise very restricted substrate utilization range, A. borkumensis is often exposed to long starvation phases until a sudden occurrence of utilizable hydrocarbon substrates, e.g., oil-polluted water, enables a rapid propagation. A strategy to survive such fluctuating nutrition supply seems to be the accumulation of TAG (to more than 20% of the CDW) and minor amounts of WE. Because large amounts of intracellularly stored TAG are mostly found in Gram-positive bacteria (belonging to the actinobacteria), A. borkumensis is so far outstanding for being the only described Gram-negative bacterium that is able to accumulate TAG in large amounts (26).
In the A. borkumensis genome, two atfA-homologous gene sequences have been identified, designated atfA1 and atfA2, which are both transcribed. The respective enzymes, AtfA1 and AtfA2, share significant amino acid identities (49% and 40%) to AtfA from A. baylyi and a similar degree of identity (46%) to each other. A detailed biochemical characterization of the two enzymes revealed that both are active acyltransferases with broad substrate ranges indeed, but they also differ substantially in their substrate specificities. AtfA1 exhibits high DGAT activity and a nearly 3-fold-higher WS activity and shows a clear preference for 1-MAG than for other MAGs. AtfA2 acts as active WS and accepts all kinds of tested MAGs, but it has solely a residual DGAT activity. Whereas most characterized bacterial WS/DGATs showed a preference for longer-chain-length alcohols, in particular hexadecanol, AtfA1 and AtfA2 prefer medium-chain-length, linear alcohols, such as butanol or decanol, and even showed a higher activity toward cyclic or phenolic alcohols (e.g., cyclohexylethanol or 2-phenylethanol) than toward hexadecanol (26).
By inactivation of both genes, their functional contribution to the storage of neutral lipids in A. borkumensis could be revealed. Although the WS activity is about four times the DGAT activity in wild-type cells, they primarily accumulate TAG when cultivated with pyruvate. WE, besides a greater proportion of TAG, are synthesized only when hexadecane is provided (26, 91). This inability is due to the lack of a fatty acyl-CoA reductase, so that during growth with unrelated carbon sources, there are no fatty alcohols present that could serve for WE synthesis. Inactivation of atfA1 drastically reduced both WS and DGAT activities, but the disruption of atfA2 affected both activities to only a minor degree, although the respective enzyme exhibited significant WS activity in vitro. A double knockout of both genes completely abolished WS activity, but a residual low DGAT activity remained. More detailed analyses confirmed that AtfA1 is the main contributor to TAG and WE accumulation, whereas AtfA2 is dispensable for the storage of lipids. It was speculated that AtfA2 might be involved in synthesis of a yet-unknown fatty acid ester under natural, but not laboratory, conditions. Beyond that, a substantial residual TAG content (between 5 and 10% of the CDW) in the double knockout mutant indicates the existence of an alternative, WS/DGAT-independent TAG biosynthesis pathway (26).
Besides M. hydrocarbonoclasticus and A. borkumensis, other marine hydrocarbonoclastic bacteria are able to accumulate lipids. For example, Alcanivorax jadensis (formerly Fundibacter jadensis) and Thalassolituus oleivorans are known to form intracellular lipid inclusions (TAG, WE, or wax diesters), and they were even reported to export WE when cultivated with n-alkanes (26, 29, 91, 92). However, the responsible acyltransferase has not yet been identified. It can only be speculated that these bacteria also possess one or more AtfA-like acyltransferases.
Rhodococcus opacus.
The Gram-positive, hydrocarbon-degrading soil bacterium R. opacus strain PD630 represents a prime example of bacterial TAG accumulation. Due to its ability to store up to 76 or 87% lipids (of the CDW) when cultivated in nitrogen-limited medium with gluconate or olive oil, respectively, R. opacus PD630 is of great biotechnological interest and serves as a model organism to study bacterial TAG synthesis and accumulation (18, 74).
In order to identify the enzymes involved in TAG biosynthesis in this extraordinary strain, it was searched for atfA-similar sequences in its genome, which was at that time not sequenced. Therefore, nondegenerate primers were designed according to the genome sequence of the closely related R. jostii strain RHA1. In a first survey, 10 atfA-homologous sequences were identified in R. opacus PD630, designated atf1 to atf10 (74). In the genome of R. jostii strain RHA1, there exist 14 atf-homologous genes with amino acid identities of at least 22% to Atf1 from R. opacus.
The heterologous expression of atf1 to atf10 in E. coli revealed that solely Atf1 and Atf2 exhibit relatively low WS and, in the case of Atf2, also DGAT activity. Atf1 was predicted to possess two putative transmembrane domains, whereas Atf2 seems to be a cytoplasmic enzyme. Surprisingly, an atf1 disruption mutant of R. opacus was seriously affected in TAG synthesis (having approximately 50% less TAG accumulation), and its DGAT activity was decreased to approximately one-third of the wild-type activity, whereas the WS activity was not strongly influenced (74). This finding demonstrated again that acyltransferases can exhibit different substrate specificities depending on the expression host, as Atf1 seems to be an important contributor to DGAT activity in cells of R. opacus but not in recombinant E. coli. The finding of very low activities of the majority of Atf proteins in E. coli could be attributed to the expression host as well as to a strongly divergent substrate range that is not covered in standard enzyme assays. However, the high redundancy of atfA-like sequences in Rhodococcus spp. might be a hint for their importance as well as for a presumably complex relationship between the encoded gene products (74). The detection of phenyldecanoic acid as a constituent of WE (e.g., phenyldecyl-phenyldecanoate) and TAG in R. opacus PD630 grown with phenyldecane again demonstrated the ability of AtfA-like enzymes to utilize even bulky substrates (93).
Streptomyces coelicolor and Streptomyces avermitilis.
The genus Streptomyces includes filamentous, Gram-positive soil bacteria, many strains of which produce important antibiotics for clinical use. Furthermore, many Streptomyces strains are also able to synthesize TAG during the postexponential growth phase (94).
In the genome of S. coelicolor, there are genes for three proteins with remote similarities to AtfA from A. baylyi: Sco0958, Sco0123, and Sco1280 (75). Only Sco0958, exhibiting 25% identity to AtfA, contributes to TAG biosynthesis in S. coelicolor strain A3(2): its deletion seriously reduced TAG accumulation to approximately 30% of the S. coelicolor wild-type TAG level, whereas a deletion of the other two candidates had no effect. Furthermore, the TAG leaky phenotype could be complemented by reintroduction of sco0958, and its overexpression resulted in significantly higher TAG accumulation. This protein is the only one harboring an intact active-site motif (HHxxxDG), and it exhibits DGAT but not WS activity in vitro and in vivo when heterologously expressed in E. coli. Although Sco1280 does not seem to play a functional role in TAG storage in S. coelicolor and was not active in E. coli, the expression of two copies of sco1280 in S. coelicolor led to a 20% increased TAG content. This indicates that Sco1280 catalyzes a DGAT reaction but plays only a minor role in S. coelicolor, so that its loss might be easily compensated for by other DGAT enzymes. The authors further speculated that Sco1280 might have a very limited substrate range or that it might need an interaction with an oily surface or another protein for full activity. Sco0123, in contrast, seems to have a completely different physiological role (75).
As was already speculated for other WE- or TAG-synthesizing bacteria, there seems to be an alternative pathway with variable, but generally lower, impact on the overall TAG storage (26, 51). The finding that there was a residual DGAT activity and TAG synthesis left in the triple mutant (sco0958 sco1280 sco0123) of S. coelicolor again supported this hypothesis (75). In S. coelicolor, bacterial phospholipid:DGAT (PDAT) activity could be proven for the first time. This PDAT activity existed at comparable levels in the S. coelicolor A3(2) wild type and in the triple mutant and was thus not affected by the deletion of either of these proteins which are similar to AtfA. Thus, glycerophospholipids can act as acyl donors for TAG biosynthesis via the PDAT pathway in addition to (or as substitute for) the DGAT pathway catalyzed by AtfA-like acyltransferases. So far, the bacterial PDAT-mediating enzyme has not been identified, and the genome of S. coelicolor does not feature any obvious sequence similarity to characterized eukaryotic PDATs (e.g., Lro1 from S. cerevisiae) (75).
In S. avermitilis, more than 60% of the total cellular fatty acids are iso- and anteiso-methyl-branched fatty acids, and, accordingly, the accumulated TAG differ from the common constitution due to their high proportion of such branched fatty acids (e.g., anteiso-pentadecanoic acid) (76). S. avermitilis also is able to synthesize small amounts of WE (less than 5% of the total lipid extract) when cultivated with glucose and hexadecanol. The responsible atfA-homologous gene encodes a protein, termed SAV7256, which strongly resembles the active DGAT enzyme Sco0958 from S. coelicolor (SAV7256 and Sco0958 share 82% identical amino acids). SAV7256 also exhibits the HH(A)xxDG motif and shares 25% identical amino acids with AtfA from A. baylyi. DGAT, but lower WS, activities were detected in crude cell extracts of S. coelicolor strain MA-4680, which is consistent with both high TAG and low or absent WE storage, as well as with the substrate specificity of Sco0958. This DGAT activity increases during cultivation under storage conditions, indicating an induction of sav7256 expression. Remarkably, SAV7256 exhibited an inverse substrate specificity in E. coli. Here, WS activity predominated over DGAT activity, corresponding to 46 and 31% of the activities of AtfA from A. baylyi in E. coli, which was used as positive control. In E. coli crude extracts, SAV7256 preferred 1-decanol rather than C4 or C24 linear alcohols, cyclic or aromatic alcohols, MAG, or DAG. Generally, the substrate range of SAV7256 seemed to be more restricted in comparison to that of AtfA (76).
Alternatives to the AtfA-Catalyzed Pathway for Lipid Synthesis in Bacteria
Studies regarding bacterial enzymes involved in substantial WE and/or TAG accumulation clearly indicate that this function is fulfilled exclusively by acyltransferases belonging to the WS/DGAT (AtfA-like) family, because no other acyltransferases essentially linked to a bacterial lipid synthesis could be detected so far.
However, in some cases there were still residual amounts of TAG detectable after inactivation of the genes similar to atfA. For instance, mutant strains of A. borkumensis still exhibited between 5 to 10% TAG of the CDW or, in the case of S. coelicolor, up to 30% of the wild-type TAG content (26, 75). The genome of A. borkumensis was reported to comprise several putative acyltransferase genes, nonhomologous to atfA, of hitherto-unknown function (95).
Apart from the situations discussed above, it is likely that there are alternative acyl-CoA-independent pathways for TAG synthesis with a rather low contribution to the overall TAG content. This could potentially be a DAG:DAG transacylase which utilizes DAG as both the acyl donor and acceptor. Such a mechanism has been described in plants and animals, although a responsible gene product has not been identified yet (96, 97). An enzyme assay for DAG:DAG transacylase was performed with S. coelicolor crude extracts, but there was no detectable activity (75). Furthermore, the eukaryotic sterol:acyl-CoA acyltransferase from S. cerevisiae also exhibits a certain DGAT side activity (98). Thus, in both cases, it is currently not known whether such enzymatic activities also exist in bacteria. Indeed, as mentioned above, the PDAT pathway as another, acyl-CoA-independent, TAG synthesis route has been detected in S. coelicolor, which had only been described for yeast and plants before (75, 99).
In contrast to alternative TAG synthesis pathways, there seem to be no alternative enzymes for bacterial WE synthesis apart from AtfA-related enzymes. The deletion of atfA-like genes in A. baylyi (51) as well as in A. borkumensis (26) completely abolished their ability to synthesize WE.
To sum up, bacterial accumulation of WE and noteworthy amounts of TAG is essentially linked to WS/DGAT (AtfA-like) enzymes. So far, no alternative acyltransferase type able to catalyze WS or DGAT reactions and to synthesize bulk amounts of these lipids has been identified in bacteria.
AtfA-Homologous Proteins in Eukaryotes
In contrast to the situation in bacteria, there are several different classes of acyltransferases known in eukaryotes which mediate DGAT or WS activity (Fig. 2). However, as mentioned above, database searches suggest that AtfA-homologous proteins are additionally widespread in eukaryotes. However, most of these putative eukaryotic WS/DGAT enzymes, with the exception of the ones from Petunia and Arabidopsis, have not been characterized to date.
Petunia hybrida.
The AtfA-homologous WS from Petunia (designated PhWS1) is composed of 522 amino acids and exhibits 19% identity to AtfA (53). It is predicted to contain two transmembrane domains, with the N terminus, including the active-site motif HHxxxDG, being at the cytoplasmic site. Moreover, PhWS1 activity could be detected only in the membrane fraction of S. cerevisiae cells heterologously expressing PhWS1. In P. hybrida, PhWS1 is mostly expressed within the petals, which are covered by a special mixture of low-molecular-weight WE from very-long-chain fatty acids (VLCFA) and methanol, isoamyl alcohol, and C4 to C12 alcohols. When PhWS1 was heterologously expressed in a TAG-deficient strain of S. cerevisiae, this led to the synthesis of C16-24 fatty acid isoamyl esters but could not restore TAG biosynthesis, indicating that PhWS1 has WS, but not DGAT, activity. The latter could be verified by an in vitro enzyme assay employing PhWS1 with palmitoyl-CoA and MAG or DAG. Furthermore, the activity toward various straight, branched, aromatic, and terpenoid alcohols was determined: in vitro, PhWS1 exhibited the highest specific activity with medium-chain-length dodecanol (C12) and approximately 50% of this activity toward octanol (C8) and decanol (C10). Besides these three substrates, the activity toward other alcohols was significantly lower than that toward dodecanol (e.g., activity toward isoamyl alcohol was ca. 8-fold lower and that toward tetradecanol was 26-fold lower). PhWS1 could not utilize methanol as a substrate. As acyl donors, PhWS1 prefers saturated very-long-chain-length (VLC) acyl-CoAs such as arachidoyl-CoA (C20:0) and behenoyl-CoA (C22:0), with about 6- and 3-fold-increased activities compared to that with palmitoyl-CoA. In comparison to saturated stearoyl-CoA (C18:0), the specific activity was seriously diminished by C-C double bonds, like in oleoyl-CoA (C18:1; 40% less) or linoleoyl-CoA (C18:2; 90% less). The preference for saturated VLC acyl-CoAs is reflected in the composition of waxes composed of saturated VLCFA on petunia petals. However, there must be other WS enzymes that synthesize high-molecular-weight WE-containing VLC alcohols, which could not be synthesized by PhWS1 in vitro (53).
Arabidopsis thaliana.
As mentioned above, database searches revealed that there are 11 putative WS/DGAT enzymes present in Arabidopsis (WSD1 to WSD11), with all of them sharing the conserved HHxxxDG motif in the N-terminal region. WSD1 shares 22% amino acid identity to AtfA from A. baylyi and was first characterized in 2008 (13). The enzyme is located in the membranes forming the endoplasmic reticulum, representing the natural sites for the synthesis of cuticular wax components which are then exported to the cuticle (9, 13). WSD1 disruption mutants of Arabidopsis showed a severely reduced WE content in their total stem wax mixture, which usually contains even-numbered C38 to C54 (mainly C44) WE. In crude protein extracts of E. coli cells heterologously expressing WSD1, the enzyme showed an about 10-fold-higher WS than DGAT activity, which is consistent with the preferences of AtfA from A. baylyi (13, 51). WSD1 enabled recombinant S. cerevisiae cells to synthesize WE when cultivated with palmitic acid (C16) and octadecanol (C18), and even more WE were accumulated from palmitic acid and either tetracosanol (C24) or octacosanol (C28). Furthermore, shorter-chain-length alcohols (ethanol and isoamyl alcohol) present in yeast were not esterified by WSD1, as was reported for AtfA (70) or PhWS1 (53). These findings indicate a preference of WSD1 for VLC alcohols (13). However, WSD1 could not restore the TAG-deficient phenotype of the mutant S. cerevisiae strain, as was the case for recombinant atfA expression. Thus, WSD1 might exhibit DGAT activity only in vitro and not in vivo. Furthermore, Arabidopsis WSD1 mutants showed no diminished TAG contents (for example, in seeds), which also implies that the natural physiological role of WSD1 is stem WE synthesis rather than TAG storage. Others of the 11 putative WS/DGAT homologs of Arabidopsis might take over this role, e.g., WSD9, which is highly expressed in Arabidopsis seeds (13, 100).
GLYCEROL-PHOSPHATE ACYLTRANSFERASES
As mentioned above, membrane glycerophospholipids and TAG are both synthesized from the common precursor phosphatidate (PA), consisting of a 2-fold-acylated glycerol-3-phosphate backbone. Thus, glycerophospholipids and TAG arise from the same PA biosynthesis pathway (the “Kennedy pathway”) embedded in the cytoplasmic membrane (101) (Fig. 7). Different types and common features of bacterial glycerolipid acyltransferases have already been excellently reviewed by Zhang and Rock (66); therefore, this review will present only central aspects necessary to provide a general overview of this important family of acyltransferases in bacteria.
Fig 7.

First two acylation steps in membrane lipid and triacylglycerol synthesis from glycerol-3-phosphate to the central intermediate phosphatidate (Kennedy pathway).
Conserved Motifs of Glycerolipid Acyltransferases
Glycerol-3-phosphate is successively acylated at the sn-1 and sn-2 positions, catalyzed by glycerol-3-phosphate acyltransferase (GPAT) (EC 2.3.1.15) and 1-acylglycerol-3-phosphate acyltransferase (or lysophosphatidic acid acyltransferase [LPAAT] [EC 2.3.1.51]), respectively, yielding PA (102, 103). Glycerolipid acyltransferases (including GPAT and LPAAT enzymes) from bacteria, plants, yeasts, and animals all share the highly conserved active-site motif HxxxxD, consisting of invariant histidine and aspartate residues separated by four less-conserved amino acids. Apart from that, these universally occurring acyltransferases exhibit high sequence variability, and the HxxxxD pattern can be located in different areas (104). The highly conserved region containing the HxxxxD sequence is referred to as block I. Furthermore, there are three short regions (blocks II to IV) exhibiting high conservation in glycerolipid acyltransferases from bacteria, yeasts, and animals (enzymes from plants lack blocks III and IV [105]). Table 2 displays the sequences present in E. coli GPAT and LPAAT (PlsB and PlsC, respectively) as well as consensus sequences for conserved blocks I to IV derived from bacterial, yeast, and animal acyltransferases (105).
TABLE 2.
Blocks with high degree of conservation in acyltransferasesa
| Enzyme | Sequenceb |
|||
|---|---|---|---|---|
| Block I | Block II | Block III | Block IV | |
| E. coli GPAT (PlsB) | VPCHRSHMDYL | GAFFIRR | YFVEGGRSRTGR | ITLIPIYI |
| GPAT consensus | φxxHRSxφDφφ | GxφFIRR | φFφEGTRSRxGK | φφφφPφφφ |
| E. coli LPAAT (PlsC) | IANHQNNYDMV | GNLLLDR | MFPEGTRSR-GR | VPIIPVCV |
| LPAAT consensus | φxNHQSxφDφφ | GxφFIDR | φFPEGTRxxxGx | φPφφPφφφ |
Adapted from reference 105 with permission (copyright 1999 American Chemical Society).
φ, hydrophobic amino acids (V, I, L, F, W, Y, or M). Highly conserved residues are in bold, the catalytically essential histidine and aspartate in block I in italic, and residues for glycerol-3-phosphate binding are underlined. (Accession numbers of E. coli GPAT and LPAAT are P0A7A7 and AAC76054, respectively.)
Catalytic Mechanism of GPAT
In order to investigate the importance of the conserved amino acids of blocks I to IV, E. coli PlsB was analyzed as a representative GPAT acyltransferase by means of site-directed mutagenesis and chemical modifications (104, 105).
An exchange of histidine in block I with alanine completely diminished PlsB activity, indicating its essential role for functionality (104). However, though an exchange with glycine seriously diminished PlsB activity, residual in vitro catalysis of glycerol-3-phosphate acylation was still detectable. This led to the speculation that the flexible nature of the small glycine residue established a great conformational flexibility in this area, thus permitting another amino acid to resume the role of histidine (105). When the negative charge of aspartate in block I was replaced by alanine, the enzyme was completely inactive, but the retention of the negative charge at this position (glutamate) only reduced its activity. In addition, the barely influenced Km value of the Asp311Gly variant indicated that substrate binding was not affected (104, 105). Consequently, it is assumed that histidine and aspartate in HxxxxD form a catalytic dyad in which the negative residue aspartate forms a charge relay system with histidine that abstracted a proton from the hydroxyl group of the substrate. Subsequently, the nucleophilic oxyanion attacks the carbon atom of the acyl-thioester substrate (104).
This assumption was further substantiated when the crystal structure of a distantly related, soluble GPAT variant from plant plastids (squash) became available. In this structure, the HxxxxD motif was folded in a helical turn so that the carboxyl group of aspartate lies in the proper orientation to form an ionic linkage with histidine in such a way that the nonbonding electron pair of the imidazole ring faces the active site to abstract a proton from glycerol-3-phosphate. Furthermore, this GPAT structure exhibits a deep cleft lined with hydrophobic residues and a positively charged pocket near the one end, which most likely represent the binding sites for acyl-CoA and the negatively charged glycerol-phosphate. Both putative substrate binding sites are immediately adjacent to the catalytic HxxxxD pattern (106).
Some of the conserved amino acids in the remaining blocks II to IV were found to be necessary for proper glycerol-3-phosphate binding (underlined in Table 2), due to significantly increased Km values of PlsB variants that have been mutated accordingly. The positively charged arginine in block II seems to play the most critical role in the interaction with the negatively charged phosphate, whereas the phenylalanine in block II, as well as glutamate and serine in block III, are of slightly less importance. The highly hydrophobic block IV is likely to be involved in binding the hydrophobic chain of acyl-CoA. When proline was replaced by polar serine, the Km value significantly increased. Furthermore, the conserved first glycine in block III seems to play a crucial steric role (105).
Figure 8 schematically sums up the different pathways for the synthesis of glycerophospholipids and TAG in bacteria and again illustrates that GPAT and LPAT provide PA as the precursor for both membrane phospholipids and TAG synthesis, if the organism is able to accumulate TAG.
Fig 8.

Overview of pathways for membrane phospholipid and triacylglycerol biosynthesis in bacteria. G3P, glycerol-3-phosphate; FAex, exogenous fatty acids; LPA, lysophosphatidic acid; PA, phosphatidic acid; DAG, diacylglycerol; TAG, triacylglycerol.
LPA Biosynthesis in E. coli and Other Gammaproteobacteria (via PlsB)
In the first and most detailed analyzed model system of E. coli and other gammaproteobacteria, glycerol-3-phosphate is acylated to lysophosphatidate (LPA) by PlsB and further acylated to PA by PlsC (left side of Fig. 8). Both membrane-bound enzymes of E. coli utilize either acyl-ACP or acyl-CoA (107, 108), of which the latter enables the cell to utilize exogenous fatty acids (after CoA activation by FadD) as an energy-saving alternative (66). PlsB is quite selective for C16:0 and C18:1 fatty acyl chains but usually does not accept substrates with a C16:1 carbon chain (109, 110). PlsB-homologous GPATs have also been identified in mammals and plants (111, 112). However, during recent years it has turned out that there exists an alternative for the first acylation step in the vast majority of bacteria, as shown on the right side of Fig. 8 (113). A comprehensive analysis of bacterial genomes revealed that this alternative PlsX/PlsY pathway is most widely distributed, whereas the PlsB pathway seems to be restricted primarily to gammaproteobacteria. While many of the latter organisms (e.g., Pseudomonas aeruginosa) possess both alternatives, the Xanthomonadales have solely PlsB (66).
LPA Biosynthesis in Other Bacteria (via the PlsX/PlsY System)
PlsY represents a unique class of GPAT, as it not only has a completely different sequence and size but also uses acyl-phosphate as a novel and unique acyl donor. This precursor is provided by the soluble phosphotransacetylase PlsX, which catalyzes the formation of acyl-phosphate from acyl-ACP (114) (on the right in Fig. 8). Although the crystal structure of PlsX has been determined, the residues specifically involved in substrate binding and catalysis still remain to be elucidated (115).
PlsY has been characterized as a remarkably small (23-kDa) membrane-bound enzyme in comparison to PlsB (93 kDa). It is predicted to possess five membrane-spanning segments and three cytoplasmic domains. An alignment with over 400 bacterial PlsY sequences showed that this enzyme family includes highly conserved sequences but has no eukaryotic homologs. PlsY enzymes lack the characteristic conserved blocks of GPAT and LPAAT enzymes and exhibit three different, highly conserved sequence regions instead (motifs 1 to 3) located in each cytoplasmic loop (Table 3) (66).
TABLE 3.
Consensus sequences of highly conserved motifs 1 to 3 in the PlsY family of glycerolipid acyltransferasesa
| Motif | Sequence | Proposed function |
|---|---|---|
| 1 | GSGNxGxTNxxR | Acyl-P binding |
| 2 | FxGGKxVA | G3P binding |
| 3 | HxxNxxxxxxxxE | Catalysis |
Based on data from reference 66.
The glycine-rich motif 1 is located in the first cytoplasmic loop, and serine was found to be crucial for PlsY activity. Therefore, it can be speculated that PlsY functions through an acyl-enzyme intermediate like serine esterases. Asparagine and arginine are putatively involved in acyl-phosphate binding. The second cytoplasmic loop contains the conserved motif 2 and represents the glycerol-3-phosphate binding site with an essential, positively charged lysine. The conservative exchange of the glycines with alanine strongly affected substrate binding due to steric hindrance. Motif 3 is located in the C-terminal cytoplasmic domain and comprises the catalytically essential histidine and asparagine residues. As has been described for GPATs, the assumed role of His185 of PlsY is to initiate the catalytic mechanism by abstracting a proton from the hydroxyl group of glycerol-3-phosphate to facilitate the nucleophilic attack on the phosphoanhydride bond of acyl-phosphate. A replacement of glutamate in motif 3 prevented proper folding and/or membrane assembly of the PlsY mutant, which was therefore also inactive. Nevertheless, it can also be speculated that glutamate is essentially involved in establishing a charge relay system with histidine. The fact, that PlsY is inhibited by acyl-CoA additionally underlines that it functions in a different way than PlsB-like acyltransferases (113).
PA Biosynthesis (via PlsC)
While the initial step in glycerophospholipid and/or TAG synthesis is realized in different ways via PlsB or PlsY, the second acylation step is always catalyzed by PlsCs (LPAATs), which are ubiquitously distributed in all bacteria. Nevertheless, there are differences between PlsCs from Gram-positive bacteria, such as Bacillus subtilis, which are dependent on acyl-ACP as the essential acyl donor (115), and PlsC from E. coli, which readily utilizes both acyl-ACP and acyl-CoA (103, 114). This is in accordance with the fact that most Gram-negative bacteria that belong to the gammaproteobacteria have the opportunity to incorporate exogenous fatty acids into their lipids, whereas the remaining bacteria depend on de novo fatty acid synthesis as a source for membrane synthesis. As mentioned above, LPAAT enzymes share the common highly conserved blocks I to IV with GPATs; therefore, it is assumed that they use a similar catalytic mechanism. However, no mutagenesis experiments or membrane topology analyses have been conducted to verify these predictions for PlsC, which are solely based on sequence resemblance to PlsB (66).
Lpx ACYLTRANSFERASES FOR SYNTHESIS AND MODIFICATION OF LIPID A
The outer membrane of Gram-negative bacteria represents an asymmetric bilayer, with an inner monolayer formed by glycerophospholipids and an outer monolayer composed mainly of lipopolysaccharides (LPS). LPS are complex glycolipids which not only provide protection to the cell as a physical barrier but also act as endotoxins in case of Gram-negative pathogens (116, 117). LPS molecules are composed of three domains: lipid A, core, and O antigen. Lipid A is the bioactive component of LPS, which is specifically recognized by the innate immune system and responsible for the endotoxin property of LPS causing inflammatory response-induced endotoxicity leading to host tissue damage or septic shock. Thus, knowledge about its biosynthesis and its possible repression represents an important basis for the development of antimicrobial substances or vaccines to combat important Gram-negative pathogens (118–120).
Lipid A functions as the hydrophobic anchor of LPS and is essential for outer membrane integrity; thus, this portion of LPS is indispensable for the viability of most Gram-negative bacteria (120). This essential anchor is composed of a glucosamine-derived disaccharide backbone substituted with phosphate at positions 1 and 4′ and is integrated in the outer membrane via several attached (3-hydroxy-)fatty acyl chains (116, 117). Although the overall structure of lipid A is conserved among Gram-negative bacteria, different species modify this complex molecule upon environmental stimuli by removing or modifying the phosphate groups or acyl chains, respectively. As a consequence, the toxicities caused by lipid A molecules from different species show widely varying extents (116, 120).
Transfer of Primary Acyl Chains by “Early Acyltransferases” LpxA and LpxD
UDP-N-acetylglucosamine-O-acyltransferases (LpxA and LpxD), which catalyze the first two acylation steps in the lipid A biosynthesis pathway, are soluble cytosolic enzymes and usually require 3-hydroxyacyl chains attached to the essential acyl donor ACP (120–124). The first step of the overall lipid A biosynthesis route, displayed in Fig. 9, is the reversible O-acylation of the 3-hydroxyl group of the sugar nucleotide UDP-N-acetylglucosamine (GlcNAc) catalyzed by LpxA (125, 126).
Fig 9.

Lipid A biosynthesis pathway of Escherichia coli (details are described in the text).
Substrate specificities of LpxA acyltransferases.
LpxA acyltransferases are highly selective for the acyl chain length of their ACP substrate. This specificity is species dependent and is consistent with the composition of lipid A isolated from the respective bacterium. While LpxA from E. coli (LpxAEc) has about a 1,000-fold-higher preference for 3-hydroxy-C14-ACP rather than for 3-hydroxy-C10-ACP, the P. aeruginosa homolog (LpxAPa) has an opposite specificity (121). Although the specificity for 3-hydroxy-C14-ACP of LpxAEc is very high, it can additionally incorporate odd-chain acyl residues differing by one carbon atom unit when the respective fatty acid substrate is available (123, 127). The respective enzymes from Neisseria meningitidis and Helicobacter pylori prefer 3-hydroxy-C12-ACP and 3-hydroxy-C16-ACP as substrates, respectively (128, 129). In contrast to that, LpxAs from Bordetella species are less restrictive regarding the acyl chain length and likewise accept 3-hydroxyacyl residues with a length of C10, C12, or C14 (130). Similarly, the LpxA acyltransferase from Porphyromonas gingivalis utilizes 3-hydroxy-C16- and 3-hydroxy-C14-ACPs (127). Lipid A from Chlamydia trachomatis exhibits an unusual structure, as the 3- and 3′-hydroxyl groups of glucosamine are attached to a myristoyl residue. LpxA from this bacterium is at present the only known variant that does not require 3-hydroxyacyl chains, but it shows a clear preference for myristoyl-ACP (131). Upon heterologous expression of different lpxA genes in E. coli, it became obvious that the observed acyl chain specificities are restricted by the respective LpxA enzyme variant, possessing precise, internal hydrocarbon rulers. The substrate specificities of LpxAEc and LpxAPa can be swapped by single amino acid substitutions (Gly173Met or Met169Gly, respectively) (124).
Three-dimensional (3D) structure of LpxA.
Native LpxAEc is a trimer composed of three identical (28-kDa) LpxA subunits, whereof each subunit exhibits a C-terminal domain with four α helices and an N-terminal domain forming a left-handed helix composed of short parallel sheets. This unusual fold captures approximately two-thirds of the enzyme and forms a left-handed parallel β helix (LβH). The N-terminal portion of the LpxA sequence comprises 30 tandem repeats of a hexapeptide sequence motif, and three contiguous hexapeptide repeats specify one turn of the helix (122, 132). This repeating motif was also detected in other transferases and is characterized by an aliphatic residue (Ile, Val, or Leu, often directly followed by Gly) and a small residue (Ala, Ser, Cys, Val, Thr, or Asn) preceding the hydrophobic residue (132, 133). The side chains of the repeating aliphatic residues are directed inwards, with the triangular helix forming a hydrophobic, solvent-free core (132).
Catalytic mechanism of LpxA.
LpxAs from diverse bacterial species share many conserved basic amino acid residues which are positioned around the cleft in between adjacent subunits and participate in substrate binding and catalysis. His125 is essential for activity of LpxAEc and is thus believed to be the catalytic base that attacks the 3-hydroxyl group of GlcNAc. His122, His144, His160, and Arg204 are also of high importance for activity and are presumably involved in substrate binding (134). The analysis of the crystal structure of LpxAEc in complex with its product revealed that each of the three active-site regions in the clefts between the three LβHs accommodates one product molecule. In addition, His125 of each LpxAEc subunit lies within hydrogen-bonding distance to the respective 3-O atom of the glucosamine ring. The position of the product is stabilized by multiple hydrogen bonds to conserved amino acid residues; e.g., His122 and Gln73 form a hydrogen bond to the 3-hydroxyl group of the acyl chain. Furthermore, parts of the UDP-GlcNAc molecule are hydrogen bonded by His144, Lys76 (6-hydroxyl group of glucosamine ring), Leu75(acetyl group), Gln161 (phosphate residues), Asn198, and Arg205 (uridine moiety). The acyl chain lies within the cleft in a position parallel to a β helix. When comparing LpxA-bound products with a carbon chain length of C10 or C14, it became obvious that the end of the 3-hydroxy-C14 residue extends far more into the cleft, with its end reaching beyond Gly173. In contrast, the chain of a 3-hydroxy-C10 residue ends before reaching Gly173. Thus, when Gly173 is replaced by a more bulky amino acid (such as methionine, as mentioned above), there would not be enough space for the longer C14 chain anymore. Methionine at this position might in turn increase the affinity to the terminal methyl group of the shorter C10 chain through additional hydrophobic interactions. Analogously, a replacement of His191, which could restrict the ability of LpxAEc to utilize acyl chains longer than C14, by arginine might be the reason why LpxA from Helicobacter can use 3-hydroxy-C16-ACP as the substrate. Thus, steric hindrance in the active-site clefts, representing the fatty acid binding grooves, determines the generally very restricted acyl chain selectivity of LpxA acyltransferases. Shorter-chain-length acyl-ACP substrates, which should not encounter steric hindrances in the more distal part of the cleft, are probably utilized at significantly lower rates, because their shorter hydrophobic tail contributes less binding energy (122).
LpxA transfers acyl chains by using a general acid-base catalysis mechanism. Similar to the mechanism described for acyltransferase AtfA from A. baylyi, (Fig. 4), the free electron pair of nitrogen of His125 (in this case NE2) activates the glucosamine 3-hydroxyl group by abstracting a proton and generating an oxoanion, which then acts as nucleophile to attack the thioester linkage of (hydroxy)acyl-ACP. A hydrogen bond between the side chain of conserved Asp126 and the proton probably helps to position and/or stabilize His125. Furthermore, the backbone nitrogen of Gly143 is assumed to function as an oxyanion hole during the acid-base catalysis to stabilize a tetrahedral enzyme-substrate intermediate (122, 134).
The LpxA variant of Leptospira interrogans has also been structurally studied, because it is restricted not only to 3-hydroxy-C12-ACP but also to an UDP-GlcNAc analog which harbors an amino instead of a hydroxyl group at the 3′ position of the glucosamine ring. This represents an unusual characteristic, as LpxAEc can equally utilize both variants. This selectivity of the L. interrogans enzyme might be due to a differing orientation of the Gln68 backbone carbonyl group in comparison to the respective Gln73 in LpxAEc, providing an additional hydrogen bond acceptor for the 3-amino group. The chain length specificity is probably determined by the Lys171 side chain in the fatty acid binding cleft, which again substantiates the hypothesis that this cleft in between two subunits of LpxA functions as a hydrocarbon ruler and restricts the carbon chain length of the substrate (135).
LpxA homologs not only are very widespread among (nearly all) Gram-negative bacteria but also occur in the majority of higher plants, although their function remains unclear (136). A. thaliana has an LpxA homolog with 38% identical (and 53% similar) amino acid residues compared to LpxA from E. coli. This enzyme also seems to have a preference for 3-hydroxy-C14-ACP, and its three-dimensional structure is generally very similar to that of LpxAEc. Not only the conserved catalytic but also the chain length-determining positions align well with the respective residues of LpxAEc (137).
LpxD acyltransferases catalyzing the second and committed N-acylation step.
The LpxA product is deacetylated by LpxC to UDP-3-acylglucosamine, and subsequently a second acyl chain is attached via an amide linkage at position 2 of the glucosamine ring by the N-acyltransferase LpxD, yielding UDP-2,3-diacylglucosamine (Fig. 9). LpxD and LpxA are encoded in the same operon and share significant sequence homology (45% similarity) to each other. Therefore, it is supposed that both enzymes result from gene duplications (125, 138, 139). Like LpxAEc, LpxDs from E. coli and from other Enterobacteriaceae also prefer 3-hydroxy-C14-ACP, whereas LpxD from P. aeruginosa prefers 3-hydroxy-C12-ACP (like LpxAPa) (123). Native LpxD is also a trimer composed of three identical (36-kDa) LpxD subunits, and its structure is comparable to that of LpxA, including the characteristic LβH motif (140, 141). An additional N-terminal uridine binding domain containing highly conserved amino acids could be identified, placing the glucosamine ring near the catalytic center which lies in the cleft between two adjacent subunits (140). Due to a high binding affinity of 3-hydroxy-C14-ACP for free LpxD, it is proposed that the acyl-ACP substrate binds prior to the UDP-3-acylglucosamine substrate and that the product UDP-2,3-diacylglucosamine dissociates earlier than ACP, which in turn acts as a competitive inhibitor against acyl-ACP (142). A conserved histidine residue (His239 in LpxDEc) is seen as the catalytic base facilitating the generation of a nucleophile (amine group at position 2 of the glucosamine ring) which attacks the carbonyl carbon of the acyl chain. Furthermore, an oxyanion hole at Gly257 probably stabilizes the tetrahedral intermediate (140, 142). The length of the hydrophobic cleft, which represents the fatty acyl chain binding groove, is restricted by the orientation of a certain amino acid side chain (Met290 in LpxDEc) at its distal end, thus preventing the incorporation of acyl chains with more than 14 carbon atoms. The observation that an exchange of Met290 with alanine enabled LpxDEc to utilize longer-chain 3-hydroxy-C16-ACP rather than 3-hydroxy-C14-ACP strongly supports the length-restricting function of Met290. A comparison of 19 LpxD sequences further indicated that enzymes utilizing C12 or C14 acyl substrates exhibit a bulky residue (e.g., methionine or leucine, but never glycine) at the position corresponding to Met290 of LpxDEc, whereas enzymes that are presumed to bind longer-chain-length substrates tend to possess a small residue such as glycine at this position (141).
Addition of Secondary Acyl Chains by “Late Acyltransferases” LpxL and LpxM
After the β-1′,6-glycosidic linkage between two lipid X monomers is formed (by LpxB), the lipid A precursor is further modified by phosphorylation (LpxK) and attachment of two 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) moieties to the 6′ position (WaaA) (Fig. 9) (120). Lipid A synthesis is completed by secondary O-acylations at hydroxyl groups of attached 3-hydroxy fatty acids, catalyzed by LpxL-like acyltransferases. This group of acyltransferases comprises membrane-bound enzymes which do not exhibit any similarity to soluble LpxA and LpxD. While in E. coli and related bacteria the two successive acylations (first laurate at position 2′ then myristate at position 3′) are carried out by homologous LpxL (HtrB) and LpxM (MsbB) (Fig. 9), other Gram-negative bacteria possess solely one or more lpxL-orthologous sequences but no lpxM gene (139, 143, 144). P. aeruginosa, for example, possesses two LpxL versions, each of which is presumed to transfer one of the secondary lauroyl groups of to lipid A of P. aeruginosa (116). The lpxM gene is a product of lpxL gene duplication, and thus LpxM shares approximately 47% similarity to LpxL and represents a more recently emerged, specialized version characterized as myristoyl transferase (139, 144).
In addition to lpxM, lpxP represents a third lpxL ortholog in E. coli and closely related bacteria (139). It encodes a palmitoleoyl transferase with a high similarity to LpxL (74% similar amino acids). As a response to low growth temperatures (12°C), the function of LpxL can be replaced by LpxP. LpxP from E. coli attaches an unsaturated palmitoleoyl (C16:1) residue instead of laurate at position 2′ to lipid A, which probably helps to adapt the outer membrane fluidity to low temperatures (145, 146).
LpxL and LpxM from E. coli both require the lipid A-Kdo2 disaccharide, to which they add a secondary lauroyl and then a secondary myristoyl group, respectively (143, 147). However, in Neisseria meningitidis and P. aeruginosa, the respective enzymes do not show this specificity; they fully acylate lipid A, even if Kdo is not attached. The lipid A molecules isolated from the latter microorganisms show a more symmetrical structure than lipid A from E. coli, because a lauryol moiety is attached to each glucosamine unit at the 2 and the 2′ positions (148–150).
The structure of LpxL-like acyltransferases has not been elucidated yet. However, LpxL from E. coli has been purified and studied in detail (64). Purified LpxL not only was able to perform the first lauroylation step but could also replace LpxM by performing a slow second lauroylation in vitro. Furthermore, although lauroyl-ACP is obviously the naturally preferred substrate of LpxL, it can alternatively utilize several acyl-CoAs instead of acyl-ACPs at a lower rate in vitro. For example, C10-CoA, to a minor extent C14-CoA, and to a very low extent also C16-CoA are used. However, the utilization of acyl-CoAs strongly depends on their concentration, as they can also act as inhibitors (64). This indicates that LpxL does not exhibit a structure comparable to that of LpxA or LpxD, which are characterized by their strict hydrocarbon rulers.
The sequence of the 36-kDa LpxL enzyme shows a predicted N-terminal transmembrane domain. Although LpxL-like acyltransferases have no detectable homology to any other known protein, they are distantly related to the GPAT family, as approximately 150 amino acids of their sequences exhibit a weak homology to LPAAT, including the catalytic motif in conserved block I (see above) (64, 105). An alignment of 47 different LpxL-like acyltransferases revealed that one histidine (His132 in LpxLEc) is strictly conserved and that all sequences possess either Asp or Glu at the position corresponding to Glu137 of LpxLEc. Thus, all LpxL-like acyltransferases exhibit a highly conserved HxxxxD/E motif consisting of an essential histidine and an acidic residue. Apart from that, the aligned sequences shared only one additional, highly conserved arginine (Arg169 in LpxLEc). Based on a comparison with three further well-conserved blocks of GPATs (II to IV), putative catalytically important residues of LpxL could be selected and exchanged with alanine by site-directed mutagenesis. The activities of LpxA variants with mutated His132 or Glu137 were most strongly impaired (over 1,000- and 3,000-fold-reduced activity compared to that of the wild-type LpxA, respectively). Consequently, the HxxxxD/E motif was identified as the catalytic dyad of LpxL acyltransferases, with histidine acting as the catalytic base by activating the hydroxyl group for its nucleophilic attack on the carbonyl carbon atom of the acyl donor (64).
Site-directed mutagenesis of Arg169 or Asp200 also significantly reduced the LpxL activity. These two residues are therefore assumed to represent the counterparts of two conserved arginine and glutamine residues in GPATs which are known to be involved in substrate binding and which are conserved in GPAT blocks II and III, respectively (64, 105).
Modification of Lipid A by Additional Attachment of an Acyl Chain Catalyzed by PagP
After its transport across the membrane, the lipid A structure can be further modified by the cell as an adaptation to changes of the environmental conditions. A mechanism involving additional acylation is exerted by the outer membrane palmitoyl transferase PagP. This enzyme catalyzes the esterification of palmitoyl residues derived from glycerophospholipids to hydroxyl groups of the 3-hydroxymyristoyl chains at position 2. The resulting hepta-acylated lipid A can provide a more tightly packed LPS layer and higher integrity of the outer membrane (151, 152). PagP was identified in a rather narrow group of mainly pathogenic Gram-negative bacteria (153). It forms an eight-stranded antiparallel β-barrel with the putative active site located at the extracellular site of the outer membrane (154). The interior of the PagP region, which is exposed to the inner leaflet of the membrane, is mostly polar, whereas the interior of the other, LPS-exposed, region is strictly hydrophobic. Punctuations of the β-strands by proline residues enable the lateral access of lipid substrates from the inner leaflet of the membrane (155–157). The enzyme possesses something similar to a hydrocarbon ruler, which became obvious when Gly88 was replaced by alanine, methionine, or cysteine, thereby restricting the selectivity to shorter acyl chains (155). However, although the structure and dynamics of PagP have been studied in detail, its exact catalytic mechanism remains uncertain (158). The putative catalytically active residues His33, Asp76, and Ser77 were initially thought to exert a mechanism similar to that of classical serine esterases as a catalytic triad. However, detailed structural analysis revealed that they are not organized correctly to form such a triad (155). Moreover, it was observed that typical inhibitors acting against serine esterases were not effective against PagP (158). It is assumed that PagP can exist in different conformational states and performs an induced-fit mechanism. Thus, the catalytic mechanism will probably become clear only when there is detailed (structural) information about the catalytically competent state (153, 158).
POLYKETIDE-ASSOCIATED PROTEINS
Polyketides (PK) form a family of complex natural products with high functional and structural diversity. They include pharmaceutically highly interesting compounds with antibiotic, anticancer, or immunosuppressive activities (e.g., erythromycin, rifamycin, or rapamycin). PK biosynthesis catalyzed by multimodular PK synthases (PKS) is closely related to de novo fatty acid synthesis by fatty acid synthases (FAS) and also includes stepwise chain extensions from a common pool of simple precursors. During synthesis, the growing PK chain remains bound to PKS (159). The synthesis of methyl-branched fatty acids is achieved by elongation of linear fatty acids using methylmalonyl-CoA (160).
Pathogenic mycobacteria possess a unique lipid-rich cell wall as protection against harmful environmental influences, such as therapeutic agents. Components thereof act as offensive, defensive, or adaptation effectors which additionally contribute to mycobacterial virulence. This thick wall contains various complex PK-containing (glyco)lipids embedded into a mycolic acid layer which is covalently linked to the arabinogalactan-peptidoglycan structure (161, 162).
Examples of mycobacterial PK-containing lipids are esters of two multimethyl-branched fatty acid residues such as mycocerosate linked to a (phenol)phthiocerol (POL) or phthiodiolone (PONE) backbone containing two free hydroxyl groups to form phthiocerol dimycocerosates (PDIMs) (Fig. 10A). The phenolphthiocerol backbone may be further linked to a complex glycosyl moiety, forming a phenolic glycolipid (PGL) (Fig. 10B). Another class of mycobacterial glycolipids share a trehalose core that is acylated and further modified, e.g., by sulfate groups or complex glycosidic attachments. Three representatives of this class are shown in Fig. 11. Sulfolipid 1 (SL-1) (Fig. 11A) and polyacyltrehalose (PAT), containing five long acyl chains (Fig. 11B), are synthesized by M. tuberculosis. Lipooligosaccharides (LOS), composed of a highly complex glycan backbone and the acyltrehalose part (Fig. 11C), are synthesized by the close relative Mycobacterium marinum and contribute to its virulence (62, 163–167).
Fig 10.

Chemical structures of the polyketide-containing complex lipids PDIM (A) and PGL (B) synthesized by PapA5 via esterification of two mycocerosyl residues (green) with both hydroxyl groups of the (phenol)phthiocerol backbone (black).
Fig 11.

Chemical structures of trehalose-based polyketide-containing glycolipids synthesized by mycobacteria. R, complex and variable glycan moiety.
Commonly, each set of enzymes required for synthesis of any of these PK-containing structures is encoded in one biosynthetic cluster. These clusters contain, for example, the respective pks genes for the synthesis of the branched long-chain fatty acid building blocks or membrane spanning proteins for the translocation to the outer surface of the lipid cell wall. Each cluster usually encodes another type of enzyme. These putative gene products were designated “polyketide-associated proteins” (Pap) because their function remained unclear over a long time.
In 2004, Onwueme and coworkers characterized such a Pap (designated PapA5) from M. tuberculosis as a first member of a new subfamily of acyltransferases which are essential for the acylation of complex mycobacterial PK-derived compounds (62). The M. tuberculosis genome comprises five such genes, papA1 to papA5, and homologous genes could be identified in other mycobacterial species, as well as in Amycolatopsis mediterranei or S. coelicolor, and all of these sequences share the highly conserved motif HxxxD(x14Y). PapA5 from M. tuberculosis exhibits the extended motif HHxxxDG, which is also highly conserved in AtfA-like acyltransferases (62). Table 4 shows the relevant sequence region containing the highly conserved motif in PapA proteins that have already been characterized.
TABLE 4.
Characterized Pap acyltransferases from Mycobacterium or Amycolatopsis and sequence region containing the proposed active-site motif HxxxDx14Y
| Protein | Species | HxxxDx14Y motif region | Reference |
|---|---|---|---|
| PapA1 | Mycobacterium tuberculosis | SIDHLHADGQFVGVGLMEFQSMYTAL | 165 |
| PapA2 | M. tuberculosis | SIAHLCVDPMIVGVLFIEIHMMYSAL | |
| PapA3 | M. tuberculosis | SVDHVHVDAMIVGVTLMEFHLMYAAL | 164 |
| PapA4 | M. marinum | SVDHLNVDAMFISAVFWEIEAMYNTL | 167 |
| PapA5 | M. tuberculosis | YLHHCMADGHHGAVLVDELFSRYTDA | 62 |
| PapA5 | M. marinum | YVHHSLADAHHLGSLLDELLSRYTDV | 163 |
| Rif-Orf20 | Amycolatopsis mediterranei | AIQHSVADAHHATAILSALWSCYTDV | 168 |
Strictly conserved residues are in bold; residues resembling the AtfA active-site motif (HHxxxDG) are underlined.
PapA5, a suspected membrane-associated enzyme, has been analyzed in most detail, and its 3D structure is known. As revealed by papA5 deletion mutants of M. tuberculosis (a PDIM producer) and M. marinum (a PDIM and PGL producer), this acyltransferase is crucial for the diesterification of phthiocerol with two residues of mycocerosate, as shown in green in Fig. 10 (62, 163). The highly conserved second histidine (His124) and aspartate (Asp128) residues are essential for enzyme activity and are therefore considered the catalytically active-site residues, with histidine as a catalytic base and aspartate as an active-site stabilizer, as described above. In contrast, His123 and Tyr143 (the latter also being highly conserved) are obviously not of crucial importance for PapA5 (62). This conclusion could be further strengthened by analysis of the crystal structure of PapA5 (169). The enzyme folds into a two-domain structure, with both domains being structurally related to each other but potentially movable independently from each other. Although the respective sequence identities are rather low, each PapA5 domain exhibits a structural similarity to the chloramphenicol acetyltransferase (CAT) monomer. Furthermore, PapA5 displays a certain structural similarity to dihydrolipoyl transacetylase, carnitine acetyltransferase, and VibH, a nonribosomal peptide synthetase, as all of them share the same active site (the condensation domain) and fold into two tandem domains similar to CAT (169–171). The active-site motif is located in the N-terminal domain (domain 1) at the interface between the two domains. One hydrophobic channel, which leads to the active site, is likely to represent the binding site for the artificial substrate 1-octanal or the respective part of phthiocerol. The modeling of putative substrates further strongly suggests that His124 acts as a catalytic base, because it would be in close proximity to the hydroxyl group of the substrate. However, the PapA5 structure exhibits an unusual α helix, which is not present in the structure of CAT or carnitine acetyltransferase, thereby blocking the access to this putative substrate binding channel. This indicates that conformational changes, e.g., upon phosphorylation, might be necessary in order to activate the enzyme (169). Indeed, more recent studies indicated that PapA5 may be reversibly phosphorylated at a threonine residue(s) by serine/threonine protein kinase PknB, although the exact phosphorylation sites are still unknown. This modification might induce the necessary conformational change to make the active site accessible for substrates, or it might be relevant for an interaction with other components of PDIM or PGL biosynthesis (172).
In contrast to AtfA-like acyltransferases, harboring an HHxxxDG active-site region and exhibiting broad substrate spectra, PapA5 showed a rather narrow substrate range when tested with various nucleophiles (short-, medium-, and long-chain-length alcohols, diols, hydroxy esters, acids, amines, or thiols) and acyl-CoAs (with chain lengths ranging from C2 to C26 and benzoyl-CoA). Of these artificial substrates, PapA5 showed a clear preference for medium-chain-length alcohols (C8 > C10 > C9) and exhibited O-specificity, as it did not accept amines or thiols. Of the selection of acyl-CoAs, ester formation was highest with long-chain acyl-CoAs (C16:0-CoA, followed by C18:1- and C17:0-CoA), whereas C14:0-CoA seemed to inhibit the enzyme (62). When PapA5 (from M. marinum) was incubated with C16:0-CoA and glycosyl-phenolphthiocerol (GPPOL), one of its supposed physiological substrates, GPPOL-monopalmitate but not GPPOL-dipalmitate was built in vitro. However, as mycocerosyl residues and not C16:0-CoA are supposed to be the physiological acyl substrate and relevant factors such as protein interaction partners or association with a membrane are missing in vitro, it is assumed that PapA5 is responsible for the acylation of both hydroxyl groups of (phenol)phthiocerol in vivo (163). Furthermore, the physiological acyl donors for Pap acyltransferases remain unknown. Although they utilize CoA thioesters in vitro, they might transfer polyketide-fatty acyl chains directly from a PKS component in vivo. Alternatively, the residues could be transferred to ACP in between (165). Thus, the in vitro measured catalytic behavior and activity of Pap acyltransferases are not necessarily an exact image of their functionality in vivo, where different (and much more complex) substrates that are potentially linked to alternative acyl donors are present.
As mentioned above, PAT, SL-1, and LOS are characterized by an acylated trehalose core and constitute another mycobacterial virulence-enhancing class of PK-containing glycolipids (Fig. 11). The respective biosynthesis gene clusters also contain one or more papA genes encoding the acyltransferases responsible for acylation of the trehalose core.
PapA1 and PapA2, for example, are essential for SL-1 biosynthesis in M. tuberculosis (165, 173) (Fig. 11A). In an enzyme assay with trehalose-2-sulfate (T2S) and C16-CoA, it could be shown that PapA2 performs the first acylation of T2S at the 2′ position, while it did not accept unsulfated trehalose as the substrate. Subsequently, T2S-2′-palmitate is acylated by PapA1. Remarkably, although PapA1 and PapA2 share significant sequence similarity, they are both very specific for their respective acyl acceptor. However, it still needs to be clarified whether the remaining two acylations for the completion of SL-1 are catalyzed by other, yet-unidentified, enzymes, or whether PapA1 and/or PapA2 can also catalyze these reactions in vivo (165).
Biosynthesis of PAT, a penta-acylated trehalose-based glycolipid of M. tuberculosis, depends on PapA3. This enzyme, in contrast to PapA2, is highly specific for trehalose and does not acylate other, structurally related, disaccharides, including sulfated trehalose. Hence, it can be excluded that there is some kind of cross talk between the biosynthesis pathways of SL-1 and PAT in M. tuberculosis. Moreover, a papA3 knockout mutant of M. tuberculosis does not synthesize PAT anymore, whereas its SL-1 biosynthesis is not negatively affected. In vitro enzyme assays employing purified PapA3 from recombinant E. coli cells with C16-CoA and trehalose as substrates resulted in the formation of trehalose-2-palmitate and trehalose-2,3-dipalmitate with both palmitoyl residues attached to a single pyranose ring of trehalose. It could be clearly demonstrated that the 2 position is acylated first, because trehalose-3-palmitate is not accepted as a substrate. Thus, PapA3 is assumed to install at least the palmitoyl and mycolipenoyl groups at positions 2 and 3 in vivo (marked in blue in Fig. 11B). However, under physiological conditions, the enzyme might additionally be capable of transferring the remaining mycolipenoyl residues found in PAT (164).
LOS comprises a third type of mycobacterial glycolipids with an acylated trehalose section which is further glycosylated in a complex manner (174, 175). In M. marinum, there are four LOS subtypes (I to IV) that differ in the glycan moieties but share a similar acylation pattern, with two 2,4-dimethylhexadecanoate residues linked to positions C4 and C2 of the terminal and internal glucose residue, respectively. Furthermore, an unsaturated 2,4-dimethyl-2-pentadecenoate chain is attached to the C6 position of the terminal glucose ring (Fig. 11C). The LOS biosynthesis cluster strongly resembles the SL-1 and PAT synthesis gene clusters, and the encoded PapA4 was recently identified as crucial for the assembly of the complete, acylated LOS structure (167).
In the genome of A. mediterranei, a producer of the antibiotic rifamycin B, a distant gene homologous to the PapA5 gene was identified, whose gene product was designated Rif-Orf20 (168). Rifamycin B has antibiotic activity primarily against Gram-positive bacteria due to its inhibition of the prokaryotic DNA-dependent RNA polymerase (176). The molecule is composed of a PK framework with several side chains, for example, an acetyl group at C25 (marked in red in Fig. 12). The in vitro formation of an intermediate of rifamycin biosynthesis during an enzyme assay employing Rif-Orf20 and acetyl-CoA revealed that this enzyme is responsible for rifamycin acetylation at position C25 (Fig. 12). Besides acetyl-CoA, the enzyme also accepted propionyl-CoA with about 80% less efficiency, which enables the synthesis of a rifamycin derivative. The ability to use acetyl-CoA is in sharp contrast to the case for PapA5, which was completely inactive with such short-chain-length acyl chains. However, the substrate range of Rif-Orf20 seems to be shifted, but comparably narrow, as it did not show activity with butyryl-, isobutyryl-, palmitoyl-, or benzoyl-CoA (168).
Fig 12.

Biosynthesis of rifamycin B, a polyketide antibiotic produced by Amycolatopsis mediterranei. The acetyl moiety (marked in red) is attached by the PapA5 homolog Rif-Orf20.
CHLORAMPHENICOL ACETYLTRANSFERASE
Chloramphenicol acetyltransferases (CATs) (acetyl-CoA:chloramphenicol O3-acetyltransferase; EC 2.3.1.28) catalyze the transfer of an acetyl residue from acetyl-CoA to one of the two hydroxyl groups of chloramphenicol (Fig. 13). CATs are responsible for bacterial resistance to the antibiotic chloramphenicol, which inhibits the ribosomal peptidyl-transferase activity. The respective CAT-encoding gene is widespread among Gram-positive and Gram-negative bacteria and is often plasmid borne (177, 178).
Fig 13.

Acetylation of chloramphenicol catalyzed by chloramphenicol acetyltransferase (CAT) by transfer of the acetyl group from acetyl-CoA to the primary hydroxyl group of chloramphenicol, forming 3-acetylchloramphenicol.
At least 16 distinct subgroups of CAT genes have been identified so far, which encode proteins sharing more than 80% identical amino acids (179). The structures of native CATs are formed by three identical subunits, each of which is 207 to 238 amino acids in size and 24 to 26 kDa in mass (178, 179). To date, CAT type III (CATIII) from E. coli represents the most extensively studied CAT variant, as its 3D structure was solved almost 25 years ago (177).
As mentioned before, CATs possess the highly conserved HHxxxDG sequence, in which histidine and aspartate have been identified as crucial active-site residues (61, 180, 181). His195 (of CATIII) acts as a general base and deprotonates the C3 hydroxyl of chloramphenicol, which in turn attacks the carbonyl carbon atom of the thioester bond in acetyl-CoA (as described for AtfA [Fig. 4]). Thus, an oxyanion tetrahedral intermediate is formed, and the reaction does not follow the alternative, so-called ping-pong mechanism (182).
The monomer structure of CATIII resembles the open-face sandwich which is also found in other, nonhomologous, proteins. Three monomers constitute a compact disc-shaped trimer which exhibits substrate binding pockets at the interface between two subunits, which are predominantly lined with hydrophobic residues. As most of the pocket is formed by one subunit but the respective catalytically essential histidine residue is provided by the second subunit, CAT monomers cannot be catalytically active. A chloramphenicol molecule binds in the deep pocket in a manner that its primary C3 hydroxyl group can form a hydrogen bond to the N3 nitrogen of the imidazole ring of the active-site histidine. Furthermore, the crystal structure revealed a possible role for Ser128 as a transition state stabilizer for the oxyanion. The highly conserved Asp199 forms a salt bridge to an arginine but is not assumed to stabilize the catalytic imidazole ring of histidine. His195 is stabilized by a hydrogen bond of the protonated ring to its own backbone carbonyl carbon atom. The analysis of a CATIII Asp199Asn mutant structure revealed that the replacement of aspartate by asparagine results in severe structural changes. The main chain is dislocated, and the network of stabilizing hydrogen bonds as well as the salt bridge between aspartate and arginine is interrupted. The replacement of Asp199 by alanine did not influence the activity dramatically but significantly increased the thermolability of the enzyme, which also underlines the proposed structure-stabilizing effect of the buried charge of Asp199 (177, 183, 184).
The second substrate, acetyl-CoA, must approach the catalytic site through an alternative tunnel from a different direction than chloramphenicol, because the latter blocks the access to the active site in the pocket. This tunnel from the protein's surface to the catalytic side is lined with hydrophobic residues that contribute to the overall binding energy (177, 184–186).
As the nitro group and one of the two chlorines are exposed to the outside of the pocket, chloramphenicol analogs with substitutions at these positions are also acetylated by CATIII. However, CAT shows a rather strict substrate specificity, and it catalyzes the acyl transfer from CoA thioesters only to structures similar to chloramphenicol (184). Thus, although CAT and AtfA-like acyltransferases employ the same catalytic mechanism, they most likely have different conformations and substrate binding areas because they differ so much regarding their substrate range. While in CAT the active-site motif is deeply buried in a pocket, it may be assumed that in AtfA-like acyltransferases this catalytic site must be located in a more accessible area or at the interface of two more flexible subunits, which allows the esterification of even bulky substrates.
RtxC ACYLTRANSFERASES THAT ACTIVATE RTX LEUKOTOXINS
Members of the large and diverse bacterial protein family of RTX (repeats-in-toxin) exoproteins are characterized by the presence of repeating glycine- and aspartate-rich short motifs typically of nine amino acid residues, designated RTX repeats, forming a unique calcium binding structure (parallel β helix) (187, 188). Another common trait of RTX proteins is their special single-step translocation mechanism across both the inner and outer membranes via a type I secretion system, initiated by a C-terminal secretion signal (189, 190).
The family of RTX proteins, comprehensively reviewed by Linhartová et al. (191), encompasses pore-forming leukotoxins, multifunctional enzymatic toxins, bacteriocins, nodulation proteins, surface layer proteins, and secreted hydrolytic enzymes. Although the functions of many proteins belonging to this family still remain unknown, most of them are believed to exert host cell damage or somehow support virulence of pathogens.
Numerous important Gram-negative animal pathogens or commensals secrete proteinaceous pore-forming leukotoxins with molecular masses typically between 100 and 120 kDa, possessing similar structural and functional domains, encoded in rtx operons (Table 5) (191). They constitute a unique class of bacterial RTX proteins and, apart from the common RTX protein features mentioned above, exhibit a hydrophobic domain to infiltrate the target cell membrane and to form cation-selective pores (209–214). Furthermore, these toxins are synthesized as inactive protoxins, requiring one or two posttranslational acylations in order to exert their full cytotoxic activity once they are translocated and folded correctly in the extracellular space. The essential acylation of ε-amino groups of internal lysine residues of the protoxin is catalyzed by coexpressed RtxC acyltransferases (192, 215, 216). Three different RtxC lysine acyltransferases, HlyC from E. coli, CyaC from Bordetella pertussis, and LtxC from Aggregatibacter (Formerly Actinobacillus) actinomycetemcomitans have been characterized.
TABLE 5.
Pathogenic bacteria synthesizing HlyA and HlyA-homologous RTX toxinsa
| Bacterium | RTX toxin | RtxC acyltransferase (accession no.)b | Reference(s)c |
|---|---|---|---|
| Uropathogenic strain of E. coli | HlyA | HlyC (P06736) | 192, 193 |
| Enterohemorrhagic strain of E. coli | EhxA | 194 | |
| Bordetella pertussis | CyaA | CyaC (AAA22971) | 195, 196 |
| Mannheimia/Pasteurella haemolytica | LktA | LktC | 197 |
| Serotype A1 | LktA | LktC (P16533) | |
| Serotype T3 | LktA | LktC (P55120) | |
| Serotype A11 | LktA | LktC (P55121) | |
| Pasteurella aerogenes | PaxA | PaxC (AAF15369) | 198 |
| Proteus vulgaris | PvxA | 199 | |
| Morganella morganii | MmxA | 200 | |
| Aggregatibacter actinomycetemcomitans | LtxA | LtxC (P16461) | 201, 202 |
| Actinobacillus pleuropneumoniae | ApxIA | ApxIC (P55132) | 203, 204 |
| ApxIIA | ApxIIC (P15376) | 205, 204 | |
| ApxIIIA | ApXIIIC (Q04474) | 206 | |
| Actinobacillus equuli | AqxA | AqxC (AAM45565) | 207 |
| Moraxella bovis | MbxA | MbxC (AAP74652) | 208 |
Partially adapted from reference 191 with permission of the publisher (copyright 2010 Federation of European Microbiological Societies.)
If possible, the respective RtxC acyltransferases (and NCBI accession numbers) are supplied. Characterized acyltransferases are marked in bold.
First references correspond to the identification of RTX toxin; secondly listed references correspond to the respective acyltransferase.
HlyC from E. coli
The first characterized pore-forming RTX leukotoxin was HlyA from a pathogenic hemolytic E. coli strain, and the respective acyltransferase, HlyC, has been studied in the most detail as a model RtxC acyltransferase to elucidate their unique mechanism of acylation (192, 193, 217–220). HlyC appears to be structurally and functionally distinct from other characterized eukaryotic or prokaryotic acyltransferase types, to which it does not show significant sequence similarities (221). In vitro, HlyC utilizes several acyl-ACPs (C12, C14, C16, C16:1, C18, and C18:1) as acyl donors, with C14-ACP being the most effective substrate (193, 222). A similar preference is observed in vivo, as myristic acid accounts for the majority (68%) of acyl chains covalently attached to native HlyA. The remaining acyl chains were found to be odd C15 and C17 saturated fatty acids (223). The purpose of these usually extremely rare fatty acids and a mechanism determining the specificity of HlyC are still unknown (191). HlyC is clearly acyl-ACP dependent, as C16-CoA is not accepted as an acyl donor. Free ACP, but not free fatty acids or acyl-CoAs, inhibits the reaction (222). A survey of homologous RtxC acyltransferases revealed that 21% of 170 amino acid residues were highly conserved, whereas the N- and C-terminal regions do not exhibit obvious conservation. Conserved amino acid residues which might transitionally carry an acyl moiety during acyl transfer, such as amino acids with hydroxyl, sulfhydryl, imidazole, or carboxyl groups, were chemically modified or mutated in order to identify the acylation site in HlyC essential for the formation of an acyl-HlyC intermediate during catalysis. These studies showed that His23 is absolutely essential for activity and that Ser20 seems to exert a crucial function during catalysis. When His23 was replaced by alanine, cysteine, serine, aspartate, or lysine, HlyC was completely inactive, while a replacement of His23 by tyrosine resulted in a minimally active HlyC variant (220, 224). The fact that His23 was the only conserved residue capable of bearing an acyl group whose mutation completely abolished HlyC activity, as well as the finding that a His23Lys variant forms the acyl-HlyC complex but is unable to transfer the acyl group to pro-HlyA, substantiated the assumed catalytic role of histidine. Consequently, it was postulated that the acylation followed a so-called “ping-pong” mechanism consisting of two independent, partial reaction steps (Fig. 14). First, the acyl chain from acyl-ACP is transferred to HlyC, initiated by a nucleophilic attack of His23, and free ACP is released. Second, from this covalent acyl-HlyC heterocomplex, the acyl chain is transferred to ε-amino nucleophiles of lysine residues Lys564 and Lys690 in pro-HlyA (Fig. 14). There is no complex formed between all three components, pro-HlyA, HlyC, and acyl-ACP (220, 225). This postulated mechanism is analogous to the internal acyl group transfer via an acyl-imidazole intermediate during catalysis of the covalent binding of human complement component C4. Solely the replacement of the crucial histidine residue of C4 by tyrosine could sustain a reduced activity, as was also found for HlyC (226, 227). It is assumed that the phenolic group of tyrosine may function as a less effective nucleophile than the imidazole group of histidine. When His23 of HlyC is replaced by lysine, as mentioned above, the ε-amino group of Lys23 can serve as an internal acylation site, but in contrast to the wild type, it is probably not reactive enough to allow a subsequent transfer of the acyl moiety to the protoxin's amino group (220).
Fig 14.

Catalytic mechanism of HlyC acyltransferase. Details are described in the text. HD, hydrophobic region; RTX, repeats-in-toxin.
CyaC from Bordetella pertussis
B. pertussis produces a bifunctional toxin, CyaA, which harbors a cell-invasive adenylate cyclase domain at the N terminus that catalyze an uncontrolled conversion of ATP to cyclic AMP (cAMP) in the host cell (195). CyaA is fully activated by a single palmitoylation at Lys983, although a second lysine residue, Lys860, may additionally be acylated. However, the acylation of Lys860 alone does not confer a cell-invasive activity on CyaA (196, 228–231). The respective acyltransferase CyaC, which acylates pro-CyaA, functions analogously to HlyC. In CyaC, Ser30 and His33 are essential for the catalysis of the acyl transfer to CyaA. Furthermore, a replacement of Ala140 influenced the selectivity of CyaC for the two alternative acylation sites. When alanine was replaced by glycine, the enzyme generated a mixture of mono- and (10 to 15%) biacylated CyaAs, and either only Lys860 or both Lys860 and Lys983 were modified. When Ala140 was replaced by valine, a nearly 50:50 mixture of non- and monoacylated CyaAs was generated, and Lys983 was used as the principal acylation site. Thus, even conservative substitutions of Ala140 drastically reduced the ability to synthesize a biacylated form of CyaA (196). In its native host, CyaC transfers palmitoyl residues almost exclusively onto Lys983, whereas the selectivity in recombinant E. coli cells was different. Recombinant CyaA was acylated by palmitoleoyl, palmitoyl, and myristoyl residues at Lys983 and partly also at Lys860 (228, 230).
LtxC from Aggregatibacter (Formerly Actinobacillus) actinomycetemcomitans
So far, LtxC from A. actinomycetemcomitans has not been characterized biochemically. However, it is expected that this acyltransferase exhibits a reaction mechanism similar to that of HlyC. It can acylate not only pro-LtxA but also pro-LktA toxins (from Mannheimia haemolytica) (232). Furthermore, pro-LktA can also be acylated by HlyC from E. coli or CyaC from B. pertussis (216). This indicates a certain redundancy in the characteristic acylation domains of homologous RTX leukotoxins. Although most of the RtxC acyltransferases listed in Table 5 have not been studied yet, they are likely to share the same catalytic mechanism with HlyC, as outlined above (Fig. 14).
LIPASES
Lipases are ubiquitous, mostly extracellular enzymes belonging to the enzyme class of hydrolases (EC 3.1.1.3). Their natural function is to hydrolyze the carboxyl ester bonds in acylglycerols, with long-chain TAG being their predominant natural substrate. Thus, lipases do not represent acyltransferases in the classical sense. However, they exhibit a high promiscuity and can also catalyze reverse reactions of hydrolysis such as (trans)esterifications at low water concentrations, e.g., in organic solvents (233–235). Figure 15 gives an overview of the different types of reactions catalyzed by lipases. Lipases represent the most widely used biocatalysts in various application fields. Besides their already mentioned high versatility, they are easily produced in large amounts and thus at a relatively low price and are, furthermore, comparatively stable. About 1,000 tons of lipases are added to detergents each year, together with proteases, amylases, and cellulases (236). In addition, their chemo-, regio-, and/or stereoselectivity has promoted their use for the production of biodiesel (43), (chiral) fine chemicals, agrochemicals, cosmetics, or flavorings (237). A high number of lipases have already been studied in detail, and as this research field, especially concerning the utilization of lipases for biodiesel production, has already been reviewed extensively (235, 238), this review will focus on the catalytic mechanism of hydrolysis and transesterification catalyzed by lipases, which differs completely from that for the acyltransferases discussed above.
Fig 15.

Hydrolysis and (trans)esterification reactions catalyzed by lipases.
Although they have a rather low sequence similarity, all lipases share the common, highly conserved α/β hydrolase fold. In the majority of cases, this widely distributed fold consists of eight β strands forming a left-handed, superhelical twisted β sheet partly connected and surrounded by α helices. Enzymes belonging to the α/β hydrolases exhibit a catalytic triad composed of a nucleophilic residue, such as serine, cysteine, or aspartate, an acid residue, and a histidine residue. The nucleophilic residue is always positioned in the middle of the pentapeptide motif Gly-X-Ser-X-Gly, called the lipase box, which forms a highly conserved structure with a sharp, γ-like turn, the so-called nucleophile elbow. Thereby, the key nucleophilic residue is easily accessible during catalysis. The position of the acid residue, which participates in the catalytic triad by forming a hydrogen bond with the active-site histidine, seems to be more variable. Similarly, the catalytic histidine is positioned in a structurally more or less variable loop after the eighth β strand (233, 239–241). Since in lipases the nucleophilic residue is usually serine, their catalytic mechanism is comparable to that of serine proteases. However, although chemically similar, the seryl hydroxyl group is structurally positioned differently in the two enzyme types (242). In general, the reaction can be divided into two steps, each of which involves the formation of a tetrahedral intermediate. First, a covalent acyl-enzyme ester is formed and, second, this ester bond is broken by the attack of a second nucleophile (239). This second nucleophile can either be a water molecule, leading to hydrolysis, or an alcoholic compound, which results in transesterification of the fatty acid (235) (Fig. 16).
Fig 16.

Catalytic mechanism of lipases. A detailed description is provided in the text. (Based on data from reference 239.)
In order to initiate the reaction, the residues that form the catalytic triad have to interact to increase the nucleophilic strength of serine. Histidine, on one hand, abstracts a proton from the hydroxyl group of serine. The acid residue, on the other hand, interacts with histidine and thereby properly adjusts its imidazole ring and furthermore balances the emerging positive charge. The alkoxide ion Ser-O− is now able to perform a nucleophilic attack on the substrate's carbonyl carbon atom of the lipid ester bond (Fig. 16, step 1). A tetrahedral intermediate is formed, and its negatively charged oxyanion is stabilized by hydrogen bonds to the peptide backbone in the oxyanion hole (Fig. 16, step 2). The proton which was deposited at the histidine residue is subsequently used to release the alcohol component as the first product, leaving the acid component covalently attached to the nucleophilic serine residue. The following deacylation step of this acyl-enzyme complex is facilitated by a second nucleophilic attack. Therefore, histidine abstracts a proton from either water or an alcohol molecule. The resulting strong nucleophile again attacks the carbonyl carbon atom, which is covalently linked to the enzyme's serine residue (Fig. 16, step 3). During a second, transient tetrahedral intermediate step, histidine transfers this proton to the oxygen atom of serine to release the second product, which is a free fatty acid in the case of water as the second nucleophile or a FAAE in the case of an alcohol as the second nucleophile. This step also regenerates the active site of the enzyme (Fig. 16, step 4) (239).
Recently, two lipases, Tgl3p and Tgl5p, that contain not only the highly conserved lipase box motif (Gly-X-Ser-X-Gly) but also the HxxxxD acyltransferase motif, which is usually absent from lipases, have been identified in yeast (243). Besides acting as lipases, both enzymes were found to catalyze the acyl-CoA-dependent acylation of lysophospholipids. Tgl3p can specifically acylate lysophosphatidylethanolamine (LPE) and, to lesser extents, lysophosphatidic acid (LPA), lysophosphatidylcholine (LPC), and lysophosphatidylserine. Tgl5p, in turn, is more specific for LPA. Thus, these extraordinary enzymes are involved in both the degradation of lipids and the biosynthesis of glycerophospholipids.
Site-directed mutagenesis of the HxxxxD motif could clearly prove that histidine is crucial for acyltransferase activity, whereas a replacement of aspartate led to only a moderate decrease of this activity. It could additionally be shown that the lipase and acyltransferase activities act independently from each other: on one hand, a mutation of the catalytic serine belonging to the lipase box did not influence acyltransferase activity, and on the other hand, a mutated HxxxxD motif did not affect lipase activity. However, neither mutation of the histidine in HxxxxD nor mutation of the serine in the lipase box could completely abolish acyltransferase or lipase activity, respectively, as the mutant activities were still approximately 30% or 40% of the corresponding wild-type activities (243).
PHA SYNTHASES
The term PHA comprises a diverse group of naturally occurring polyesters composed of 3-hydroxyalkanoic acids. Over 150 different polyester constituents have been described so far, and the carbon chain length of the alkyl groups at the C3 (or β) position can vary from methyl to tridecyl (see Fig. 1). The molecular masses of PHA polymers can range from 50 to 1,000 kDa or even above, and they may exhibit variable physical properties according to their monomer composition (244, 245). The lack of specificity of PHA synthases is further underlined by their ability to synthesize polythioesters (PTE) by generating thioester bonds between 3-mercaptopropionic acid or other thiolic compounds (246, 247).
In the past decades, PHA have attracted public interest as a promising solution for the continuously increasing plastic waste problem. As PHA polyesters are water insoluble, nontoxic, and thermoplastically deformable, they can serve as adequate substitutes for conventional plastics (e.g., for packaging materials) with the advantage of being biodegradable and thus in principle also compostable (248). In contrast, artificial and nonbiodegradable PTE could find applications in the medical or automotive industry (249). In contrast to the omnipresent conventional plastics, which are produced from mineral oil, PHA or PTE can be produced in a sustainable way using renewable resources such as carbohydrates derived from surplus plant material. Thus, it is expected that microbiological plastic production will be capable of competing with conventional plastics in the future, as oil prices will continue to increase (250).
PHA are usually classified by their monomer chain length: short-chain-length PHA (PHASCL, with C3 to C5 monomers) are synthesized by a wide range of eubacteria and even some archaea (248, 251, 252). The most common type of PHASCL is poly(3-hydroxybutyrate) (PHB), which is particularly prominent as it is accumulated in very large amounts of up to 90% (of the CDW) by the Gram-negative (“Knallgas”) bacterium Ralstonia eutropha (also designated as Cupriavidus necator) (253). Medium-chain-length PHA (PHAMCL, with C6 to C14 monomers) are synthesized mainly by pseudomonads using pathways which are closely linked to the fatty acid metabolism (245). So-called structurally related substrates such as alkanes or fatty acids are CoA activated, are optionally shortened by β-oxidation, and can then directly be incorporated into PHA (254). Additionally, the majority of pseudomonads of the rRNA homology group I, such as Pseudomonas putida or P. aeruginosa, can synthesize PHAMCL from unrelated substrates. They are catabolized to the central metabolite acetyl-CoA first, followed by a metabolic route which involves de novo fatty acid synthesis to provide 3-hydroxyacyl-CoAs as activated precursors for PHA synthesis, which will be outlined below (255, 256).
Unspecific PHA synthases (PhaC) polymerize a wide range of 3-hydroxyalkanoic acids and belong to the serine hydrolase superfamily. Thus, they cannot be considered as classical acyltransferases, although they handle hydroxy fatty acids. Studies concerning PHA synthases have been reviewed elsewhere (257), and this review will hence focus only on a description of their proposed catalytic mechanism in order to compare it with the functionality of other acyl-transferring enzymes.
The 3D structure of PHA synthases has not been elucidated yet, but it has been observed that the enzyme is a homodimer in its active form and that its dimerization is induced by the presence of substrate (258, 259). PHA synthases catalyze the polymerization at the surface of the growing PHA granule (260, 261). Their significantly increased activity at a hydrophobic-hydrophilic interface strongly resembles the behavior of lipases, which are characterized by a phenomenon called interfacial activation at the lipid-water interface. Furthermore, in silico analyses revealed sequence identities to lipases, and all PHA synthases contain the slightly modified lipase box motif Gly-X-Cys-X-Gly with cysteine instead of serine. Structure models of type I, II, and III PHA synthases indicate the formation of a catalytic triad by the highly conserved residues cysteine (located at the nucleophile elbow), histidine, and aspartate (257, 262–265).
From these clues, a model for the catalytic mechanism which is similar to the lipase mechanism has been developed. Two thiol groups, or one hydroxyl group and one thiol group, might function as a loading site for 3-hydroxyalkanoic acids and as a priming/elongation site, respectively (257, 266). Figure 17 shows the proposed catalytic mechanism of initiation and elongation of a PHA chain. The one or two PhaC subunits are schematically shown in blue with the involved catalytic amino acids histidine, cysteine, and aspartate. Steps 1 to 3 illustrate the loading of a 3-hydroxyalkanoic acid monomer to one subunit, while steps 4 to 6 show the first elongation step, consisting of the connection of two monomers. In order to initiate the loading of the substrate to one of the two subunits, histidine is believed to abstract a proton from the thiol group of cysteine, which enables the nucleophilic attack of the carbonyl carbon atom of a 3-hydroxyalkanoic acid (Fig. 17, step 1). The resulting tetrahedral intermediate is broken by the release of free CoA (Fig. 17, step 2). The substrate is then covalently linked to the PhaC subunit (Fig. 17, step 3). The second subunit probably loads a primer molecule consisting of a short chain of several 3-hydroxyalkanoate moieties, following the same mechanism (Fig. 17, step 4). The 3-hydroxyl group of the substrate is then activated by aspartate. Thus, aspartate is proposed to act as second general base catalyst next to histidine, which is different from the classical lipase mechanism. The activated 3-hydroxyl group can subsequently attack the covalent acyl-enzyme intermediate at the other subunit (Fig. 17, step 4), which involves the formation of a second tetrahedral intermediate (Fig. 17, step 5) and results in the elongation of the PHA polymer by one monomer. The second thiol group and the negative charge at aspartate are regenerated, and another substrate molecule can be loaded (Fig. 17, step 6) (257, 266). The unstable oxyanions of the tetrahedral intermediates might be stabilized by an oxyanion hole provided by serine, e.g., Ser297 in PhaC1 from Pseudomonas sp. USM 4-55, which is a conserved residue in type II PHA synthases (266).
Fig 17.

Proposed catalytic mechanism of PHA synthases. A detailed description is provided in the text. (Based on data from references 257 and 266.)
TRANSACYLASE PhaG INVOLVED IN PHA SYNTHESIS
As mentioned above, many pseudomonads are able to synthesize PHAMCL from unrelated carbon sources by employing de novo fatty acid synthesis. The connection between the pathways is accomplished by a transacylase which transfers the 3-hydroxyacyl moieties from ACP to CoA, thereby preparing their polymerization by PhaC. This enzyme is termed 3-hydroxyacyl-ACP:CoA transacylase (PhaG). As PhaC can polymerize only 3-hydroxyacyl-CoA and not 3-hydroxyacyl-ACP precursors, the mediator PhaG is essential for PHA accumulation from unrelated carbon sources using de novo fatty acid synthesis (256). Figure 18 displays this biosynthesis pathway starting from the central metabolite acetyl-CoA.
Fig 18.

Metabolic link between de novo fatty acid synthesis and PHA synthesis in pseudomonads. (Based on data from reference 300.)
As a first step, the fatty acid synthase multienzyme system (FASII) converts acetyl-CoA to malonyl-ACP units, which are utilized for subsequent condensation reactions. Each reaction cycle consists of four steps: condensation, reduction, dehydration, and a final reduction. During the condensation reaction between malonyl-ACP and acyl-ACP, one carbon atom is released as carbon dioxide so that the growing acyl chain is elongated by two carbon atoms during each cycle. Furthermore, 3-hydroxyacyl-ACPs with increasing chain lengths occur as intermediates in each cycle (267). PhaG transfers the 3-hydroxyacyl moiety from these ACP-bound intermediates to CoA and thus channels them into PHAMCL synthesis (256).
In vitro enzyme assays with PhaG and the common PHAMCL precursor 3-hydroxydecanoyl-ACP or -CoA demonstrated that the conversion is reversible and that MgCl2 seems to be an important cofactor for the reaction (Fig. 19). However, it could also be shown that PhaG does not accept straight-chain, nonhydroxylated substrates such as decanoyl-CoA (256).
Fig 19.
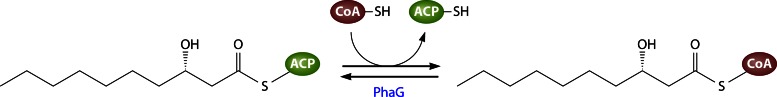
Conversion of 3-hydroxydecanoyl-ACP to 3-hydroxydecanoyl-CoA catalyzed by PhaG.
The first identification and description of PhaG were achieved by the analysis of P. putida mutants that were significantly impaired in PHA accumulation when gluconate was the sole carbon source but not when octanoate was provided as the substrate. However, residual PHA amounts (of up to 3% of the CDW) synthesized from unrelated carbon sources in those mutants indicate the presence of alternative routes to bypass the loss of PhaG activity to a limited extent. This could be a release of free fatty acids by a thioesterase and a subsequent, unspecific esterification with CoA. Alternatively, small amounts of 3-hydroxyacyl-CoA intermediates might emerge from β-oxidation (256). Especially in P. aeruginosa, such alternative pathways seem to play a more important role than in P. putida, as the PHA accumulation in a P. aeruginosa phaG mutant is reduced to only about 40% of the wild-type PHA content (268).
PhaG from Pseudomonas putida as a Model Enzyme
The member of the PhaG enzyme family characterized first and in most detail is PhaG from P. putida (PhaGPp). The 34-kDa enzyme consists of 295 amino acids and is localized in the soluble cytosolic cell fraction (269). Although its amino acid sequence contains the short pattern HxxxxD, representing the catalytically active-site motif for various ester- or amide-forming enzymes as described above, PhaG does not exhibit a significant overall sequence homology to other known transacylases or acyltransferases.
Biochemical properties of PhaGPp.
In order to purify PhaGPp and to characterize its biochemical properties, Hoffmann et al. overexpressed a PhaG-His6 fusion protein in E. coli and refolded active enzymes from inclusion bodies (269). The enzyme's geometry seemed to accept only a C-terminal His tag, whereas an N-terminal His tag resulted in a strong inactivation. This interference between an N-terminal His tag and the correct folding or catalysis of PhaG indicates a more important functional role of the N terminus than of the C terminus. PhaG might form a homodimer when high enzyme concentrations are present. Furthermore, PhaG can be specifically inhibited by the addition of 2-bromooctanoic acid to P. fluorescens cells (270, 271).
Two- and three-dimensional structures of PhaGPp.
The secondary structure of PhaGPp was predicted by means of two different approaches: an analysis of circular dichroism (CD) spectra of the purified enzyme and a software-based prediction of the protein structure. The resulting predicted proportions of α helix (28 to 29%), β sheet (22 to 24%), β turn (18 to 18.5%), and random coil (30 to 31%) from both methods were in very good agreement. Furthermore, the peptide sequence of PhaGPp was searched for domains with known structure. As a part of the sequence exhibits 22% identity to the conserved α/β hydrolase fold domain, it was speculated that PhaG is a member of the α/β hydrolase superfamily. A 3D structure was predicted based on the known structures of the conserved α/β hydrolase fold and the mouse epoxide hydrolase (269).
The applied method, called “protein threading” (or “fold recognition”), aligns the amino acids of a protein of interest with the amino acid backbone coordinates of a known structural fold. This method is applied when there are no homologous structures available and it is based solely on similarities to known and common protein folds (272, 273). It thus represents an alternative to “homology (or comparative) modeling,” which relies on template structures exhibiting an evident homology to the studied protein (274).
The generated PhaGPp threading model suggests that an α/β hydrolase fold builds the core, with Ser102, His251, and Asp223 acting together as a catalytic triad. These residues are underlined in the PhaGPp sequence in the MSA shown in Fig. 20. The putative active-site Ser102 sits at the nucleophile elbow, as described for lipases (269). Another predicted PhaGPp structure, which was generated with a bacterial epoxide hydrolase as the template, confirmed the model generated by Hoffmann and coworkers (275). The essential roles of Ser102, His251, and Asp223 were confirmed by the observation that a replacement of each of these amino acids with alanine diminished PhaG activity (269). This type of catalytic triad with serine is found in enzymes belonging to the serine hydrolase superfamily, which act, for example, as lipases, PHA depolymerases, serine hydrolases, (thio-)esterases, or fatty acid or PK synthases (233, 276–281). As mentioned above, the carbonyl carbon atom of the acyl group is covalently attached to the serine hydroxyl group of the enzyme and is subsequently released or, in case of PhaG, transesterified to ACP or CoA. The catalytic mechanism of PhaG is thought to be quite similar to the mechanism exerted by the FASII component malonyl-CoA:ACP transacylases (MCAT) (269). MCAT transfers the malonyl moiety to ACP, providing the malonyl-ACP intermediates for the initiation of type II fatty acid synthesis in bacteria. Its 3D structure reveals a core similar to that of α/β hydrolases (281). However, although the catalytic core seems to be organized in a similar manner, the very low sequence similarities between PhaG and other kinds of acyltransferases or serine hydrolases are remarkable (269).
Fig 20.

Multiple-sequence alignment of different PhaG proteins (generated with ClustalW). Amino acid residues identical to those of the PhaGPp reference protein from Pseudomonas putida are displayed as dots. PhaG protein sequences are derived from the following bacteria: Pp, P. putida; Pn, P. nitroreducens; Pm, P. mendocina; Po, P. oleovorans; Bc, Burkholderia caryophylli; Ps, Pseudomonas sp. 61-3; Pf, P. fluorescens; Pa, P. aeruginosa (for details, see Table 6).
Assuming that PhaG has a catalytic mechanism similar to that of serine hydrolases, the catalytic function of the identified HxxxxD/A motif remains obscure. PhaGPp variants with a mutated histidine in this pattern were inactive, whereas a replacement of the aspartate residue only reduced the activity and slightly shifted the substrate specificity. This residue might therefore be involved in substrate binding at the protein's surface (269).
Distribution of PhaGs in Bacteria
So far, PhaG-homologous proteins have been identified in eight different Pseudomonas species with amino acid similarities to PhaGPp ranging from 77 to 100% (Table 6). In addition to the eight already-described PhaG proteins, a BLAST search reveals the presence of PhaG homologs encoded in the genomes of additional Pseudomonas species (Table 6). Applying a PCR-based strategy, phaG genes were even identified in nonpseudomonads, such as Burkholderia caryophylli or Aeromonas hydrophila (275, 282), although the detection of a gene similar to phaG in A. hydrophila is questionable, because its genome sequence, which was published later, does not have a sequence similar to phaG. There remains uncertainty regarding a possible occurrence of phaG genes in the genus Burkholderia, as the ability to link de novo fatty acid synthesis with PHAMCL synthesis via PhaG was originally considered to be unique to Pseudomonas species. A BLAST search of the available Burkholderia genome sequences with the PhaGPp sequence as a query identifies solely the identified PhaG homolog of B. caryophylli (PhaGBc) (Table 6).
TABLE 6.
PhaG homologs in various bacteria sorted according to their amino acid identity to PhaG from Pseudomonas putida KT2440a
| Organism | Protein size (amino acids) | % Identity (similarity)b | Special features | Reference |
|---|---|---|---|---|
| P. putida strain KT2440 | 295 | 100 | 256 | |
| P. stutzeri strain 1317 | 295 | 100 | 282 | |
| P. nitroreducens strain 0802 | 295 | 99 (99) | Cryptic but active | 282 |
| P. mendocina strain LZ | 293 | 99 (99) | Inactive | 275 |
| P. oleovorans | 295 | 95 (98) | Cryptic but active | 283 |
| Burkholderia caryophylli | 294 | 78 (87) | 282 | |
| Pseudomonas sp. 61-3 | 294 | 70 (82) | HxxxxAE motif | 284 |
| P. fluorescens strain BM07 | 294 | 69 (82) | HxxxxAE motif | 270 |
| P. aeruginosa | 300 | 59 (77) | 268 | |
| P. pseudoalcaligenes | 295 | 99 (99) | Uncharacterized | |
| Pseudomonas sp. TJI-51 | 295 | 92 (95) | Uncharacterized | |
| P. entomophila | 294 | 89 (95) | Uncharacterized | |
| Pseudomonas sp. M47T1 | 294 | 72 (84) | Uncharacterized | |
| P. chlororaphis | 294 | 71 (83) | Uncharacterized | |
| P. synxantha | 294 | 68 (83) | Uncharacterized | |
| Pseudomonas sp. Ag1 | 294 | 67 (81) | Uncharacterized | |
| Pseudomonas sp. PAMC 25886 | 294 | 66 (81) | Uncharacterized | |
| P. brassicacearum subsp. brassicacearum NFM421 | 294 | 68 (80) | Uncharacterized | |
| P. extremaustralis 14-3 | 294 | 68 (82) | Uncharacterized | |
| Pseudomonas sp. USM 4-55 | 295 | 57 (74) | Uncharacterized | |
| P. fulva | 292 | 54 (72) | Uncharacterized | |
| P. savastanoi | 287 | 52 (74) | Uncharacterized | |
| Various P. syringae pathovars | 293 | 66 (80) | Uncharacterized |
The first nine proteins have been described in the literature, whereas the remaining, uncharacterized ones have been found via BLAST searches. In case of multiple hits for proteins similar to PhaG in different strains of one species, the maximal amino acid identity to PhaGPp in this species is given.
Maximal percentage of identical or similar amino acids in comparison to the reference sequence of PhaGPp.
Further hits in the genomes of, for example, B. pseudomallei, B. oklahomensis, B. thailandensis, B. mallei, B. gladioli, or B. glumae, showing maximal amino acid identities of about 40% to PhaGPp as well as to PhaGBc, and these proteins have been annotated as subunit A of rhamnosyltransferase I in the database. This enzyme is homologous to PhaG, as will be outlined below. Thus, as the complete genome sequence of B. caryophylli is not available yet, it remains to be elucidated whether the single PCR-identified phaG sequence represents a unique trait of B. caryophylli and whether it is related to PHAMCL biosynthesis in this bacterium at all. Most studied Burkholderia species exhibit a PHB synthesis pathway resembling that of R. eutropha, where PhaG is not necessary because the substrates are not derived from de novo fatty acid synthesis (285). However, the detection of an additional PHAMCL biosynthesis pathway in B. caryophylli resembling that of pseudomonads (with phaC1 and phaC2) indicates that this bacterium may be distinguished from other Burkholderia species in this regard (286). It can thus be speculated that B. caryophylli is more related to Pseudomonas spp. regarding its PHA metabolism than other Burkholderia species, which would explain why it also possess a PhaG homolog.
The sequences of nine characterized PhaG proteins (the first nine proteins in Table 6) were aligned in order to visualize the conservation of certain domains. As the overall similarity is quite high, the interpretability was advanced by displaying amino acid residues identical to the PhaGPp reference as dots (Fig. 20). The MSA shows a higher degree of conservation of the N termini of PhaG proteins than of the C termini, which is in accordance with previous PhaG alignments and the assumption that the N terminus of PhaG is functionally more important, as noted above (269).
PCR-identified phaG genes from P. stutzeri and P. nitroreducens, which are nearly identical to the PhaGPp gene, could be functionally expressed in a P. putida strain (282). Apart from that, two other PhaG versions (from P. aeruginosa and Pseudomonas sp. 61-3) have been analyzed in more detail. In the following, characterized PhaG enzymes from species other than P. putida will be described. Furthermore, an inactive PhaG variant and the occurrence of cryptic phaG genes or the untypical absence of phaG genes in Pseudomonas species are outlined.
P. aeruginosa.
PhaGPa from P. aeruginosa shares about 59% identical amino acids with PhaGPp and is approximately half as active as PhaGPp when heterologously expressed in a phaG mutant of P. putida (268). Another observation strengthening the assumption that PhaGPa is less active than PhaGPp is a significantly (40%) increased PHA accumulation in P. aeruginosa upon phaGPp expression (256). However, a phaG insertion mutant of P. aeruginosa is still able to accumulate a significant proportion of about 40% of the wild-type PHA content, implying that alternative pathways to provide 3-hydroxyacyl-CoA precursors have a higher contribution to the overall PHA synthesis in comparison to P. putida (268). Consequently, bearing a less active PhaG version might not be a burden for P. aeruginosa because it harbors compensatory pathways.
Pseudomonas sp. 61-3.
The PhaG version of Pseudomonas sp. 61-3, PhaGPs, shares 70% identical amino acids with PhaGPp and is also involved in channeling 3-hydroxyacyl-CoAs derived from unrelated carbon sources in PHA synthesis. However, this strain is additionally able to synthesize PHB from acetyl-CoA via the PhaA/PhaB-mediated pathway involving the β-ketothiolase-catalyzed condensation of two acetyl-CoA molecules and its subsequent reduction by acetoacetyl-CoA reductase to 3-hydroxybutyryl-CoA. Furthermore, there seem to be alternative metabolic routes to provide 3-hydroxyacyl-CoAs that can bypass PhaG, as described for P. aeruginosa (284). As can be seen in Table 6, in PhaGPs the HxxxxD-like motif is modified to HxxxxAE. Via site-directed mutagenesis, Matsumoto and coworkers (284) investigated to what extent the HxxxxD-like motif is relevant for PhaG activity. As verified by Hoffmann and coworkers (269), the conserved histidine, His177, seems to be crucial for PhaG functionality, but a replacement of neither Ala182 nor Glu183 influenced its activity. On one hand, a reconstruction of the conserved motif HxxxxD harboring the acidic residue aspartate instead of neutral alanine did not increase the activity. On the other hand, the acidic glutamate obviously did not inherit the function of aspartate in the modified motif. Thus, the authors came to the conclusion that an acidic residue as part of the conserved motif HxxxxD is not essential and that the PhaG mechanism must therefore differ from that of glycerolipid acyltransferases (284).
P. mendocina.
A catalytically inactive PhaG protein was identified in P. mendocina (275). The protein is nearly identical to PhaGPp but exhibits two crucial modifications: an exchange of Ser78 with cysteine and a short deletion of the two successive amino acids Ala111 and Leu112 (Fig. 20). Although phaG is expressed during growth with gluconate as the sole carbon source by P. mendocina, the cells can synthesize only residual amounts of PHA (1%). To elucidate the harmful impact of each of the two mutations, the authors introduced them separately into PhaGPp and measured the resulting activity of the enzyme. The Ser78Cys variant of PhaGPp showed an approximately 50% reduced activity in comparison to wild-type PhaGPp, indicating that the hydroxyl group of serine might play a supporting role in structure maintenance or catalysis. However, site-directed mutagenesis of PhaGPm to exchange the mutated cysteine with serine or threonine did not result in an active enzyme. Deleting either Leu110, Ala111, Leu112, or both Ala111 and Leu112 in PhaGPp diminished its activity completely. Correspondingly, an insertion of Ala111 together with Leu112 in PhaGPm recovered its activity to about 50% (comparable to that of the PhaGPp Ser78Cys mutant). Comparative modeling of a 3D structure of the inactive PhaGPm variant suggested that the mutation at position 78 does not alter the overall protein structure compared to that of PhaGPp but that the short deletion (ΔAla111ΔLeu112) dramatically disordered the structure. The predicted structural impacts would result in surface exposure of the aspartate belonging to the catalytic triad and thus in a loss of the essential steric adjustment of the catalytically active residues to each other. Additionally, the deletion and a consequent distortion eliminate the hydrophobic environment surrounding the catalytic triad (275).
P. oleovorans and P. nitroreducens.
Although the genomes of P. oleovorans and P. nitroreducens encode PhaGs with high similarities to PhaGPp (95% and 99% identical amino acids, respectively), these organisms are unable to utilize sugars for PHA accumulation. The respective genes were found to be cryptic, as they are not transcribed properly under natural conditions (282, 283). PHA accumulation could be restored in P. oleovorans by reintroducing the phaGPo gene under the control of the lacZ promoter (283).
Pseudomonas species lacking PhaG.
The PhaG-mediated biosynthesis of PHAMCL from unrelated carbon sources appears to be a widespread strategy among various pseudomonads, but several species are not able to exploit this route. For example, P. fragi and P. jessenii are known to synthesize only very small amounts of PHA from sugars (287, 288). A BLAST search reveals that their genomes do not encode a protein similar to PhaG. This again underlines that PhaG is the essential connecting link between the central metabolism and PHA synthesis and thus is important for a noteworthy accumulation of PHAMCL from nonrelated substrates.
SYNTHESIS OF RHAMNOLIPIDS
Rhamnolipids (RL) are extracellular glycolipids, composed of 3-hydroxy fatty acids which are glycosidically linked to one or two l-rhamnose units (displayed at the bottom of Fig. 21), that naturally appear in a great diversity of structures, with about 60 congeners and homologs, and may fulfill various physiological functions (comprehensively reviewed in references 289 and 290).
Fig 21.

Synthesis of rhamnolipids from 3-hydroxydecanoyl-ACP derived from de novo fatty acid synthesis.
Functions of Rhamnolipids
The amphiphilic, surface-active RL act as emulsifiers and thereby, for example, facilitate the uptake and assimilation of poorly soluble hydrocarbon substrates (291–293). As RL reduce the surface tension and act as wetting agent, they promote swarming or sliding of bacterial colonies (294, 295). Additionally, RL synthesis is involved in bacterial biofilm development by altering the cell surface hydrophobicity and facilitates cell adhesion (294, 296). The ability of RL to intercalate into cell membranes and permeabilize them is assumed to cause their antimicrobial properties (297). RL of pathogenic microbes are furthermore considered to play an important virulence-enhancing or even hemolytic role and induce various immune responses (289). Indeed, their importance for invasion of respiratory epithelia and establishment of infection could be proven for the opportunistic pathogen P. aeruginosa (298, 299).
Rhamnolipid-Producing Bacteria
To date, most studied RL producers belong to the genus Pseudomonas, of which especially P. aeruginosa has extensively been studied as a model system for RL synthesis. During the last years, the ability to synthesize and secrete RL or RL-like compounds has been detected in an increasing number of natural isolates covering a broader taxonomic range. For example, RL synthesis seems to be a more widely distributed trait in the Proteobacteria, as RL-producing strains belonging to the genera Acinetobacter, Pseudoxanthomonas, Enterobacter, Pantoea (gammaproteobacteria), Burkholderia (betaproteobacteria), or Myxococcus (deltaproteobacteria) have been described. Furthermore, RL-synthesizing isolates were even reported for other phyla, such as the Actinobacteria (Renibacterium, Cellulomonas, or Nocardioides) or the Firmicutes (Tetragenococcus). However, some of these findings need to be confirmed by more detailed measurements and taxonomic characterizations, and in most cases, a physiological function of RL in these bacteria is still unknown (289).
Rhamnolipid Biosynthesis
Similarly to PHAMCL, RL are also synthesized from 3-hydroxy fatty acyl-ACPs provided by de novo fatty acid synthesis. Thus, in those pseudomonads which are able to accumulate PHAMCL and secrete RL, both biosynthesis pathways compete for the same precursors. This becomes obvious in mutants defective in RL synthesis, as they show elevated PHAMCL accumulation (300, 301). Figure 21 illustrates the biosynthesis of mono- or dirhamnolipids from 3-hydroxyacyl-ACPs derived from de novo fatty acid synthesis. In P. aeruginosa, the lipid part of RL is composed predominantly of 3-hydroxydecanoyl moieties (302).
First, two molecules of 3-hydroxydecanoyl-ACP are linked by RhlA (3-hydroxyacyl-ACP:3-hydroxyacyl-ACP O-3-hydroxyacyltransferase) to a dimer. In general, dimerization products from two 3-hydroxyacyl residues are termed 3-(3-hydroxyalkanoyloxy)alkanoates (HAAs) (294, 303). The second step is the condensation of HAA with dTDP-l-rhamnose catalyzed by rhamnosyltransferase I, RhlB, leading to mono-RL (l-rhamnosyl-3-hydroxydecanoyl-3-hydroxydecanoate in Fig. 21) (304). Finally, a second rhamnose moiety can optionally be attached to the first by rhamosyltransferase II, RhlC, resulting in di-RL (l-rhamnosyl-l-rhamnosyl-3-hydroxydecanoyl-3-hydroxydecanoate in Fig. 21) (305). The first two enzymes of this pathway, RhlA and RhlB, are encoded in one operon, rhlAB (304).
For several years, some aspects regarding a direct connection between de novo fatty acid synthesis and RL synthesis remained uncertain. It had been shown that PHAMCL and RL syntheses both compete for the same source of FASII-derived substrates, and it was speculated that a putative β-ketoacyl reductase, RhlG, provided substrates for RL synthesis by reduction of β-ketodecanoyl-ACP to β-hydroxydecanoyl-ACP (300, 306). However, later it turned out that RhlG does not have the catalytic capability to carry out the proposed reaction, and its indispensability for RL synthesis could not be clearly proven (303, 307). RhlA from P. aeruginosa and PhaGs from various Pseudomonas species exhibit significant sequence homology, i.e., 41 to 48% identical and 60 to 65% similar amino acids, which first led to the assumption that RhlA, like PhaG, might also catalyze a transacylation of 3-hydroxydecanoyl moieties from ACP to CoA (294). The speculation that PhaC might provide HAA dimers was also disproven by the finding that PhaC-negative mutants still synthesize RL (306, 308). In vitro enzyme assays could finally clarify the physiological role of RhlA as an acyltransferase that forms HAA by directly utilizing 3-hydroxydecanoyl-ACPs. Thus, RhlA links de novo fatty acid synthesis with RL synthesis and therefore competes directly with its distant homolog PhaG for substrates. Purified RhlA appeared to be a 34-kDa monomer in solution (303). The enzyme is assumed to be attached or bound to the cytoplasmic membrane (305). In vitro, purified RhlA is highly selective for the 3-hydroxydecanoyl-ACP intermediate and exhibited only approximately 10 or 5% of this activity when 3-hydroxy-C8- or 3-hydroxy-C12-ACP was supplied as the substrate, respectively. This high specificity of RhlA seems to control the acyl chain composition of RL, as reflected in the aforementioned predominance of C10 residues in RL synthesized by P. aeruginosa (303).
However, although the role of RhlA appears to be clarified, it still remains to be elucidated how RhlA catalyzes the esterification between the 3-hydroxyl group of one and the ACP-thioester bond of the other 3-hydroxydecanoyl-ACP molecule. Until now, no studies have been performed concerning conserved and catalytically important residues or a putative structure of RhlA. Furthermore, publications concerning RhlA do not clearly specify whether the second ACP moiety is detached from HAA during its formation (as shown in Fig. 21) or remains attached during RL synthesis. On the basis of the moderate amino acid sequence similarity between RhlA and PhaG, it might be speculated that the catalytic mechanisms are similar. In RhlA protein sequences from strains of P. aeruginosa, B. pseudomallei, and Pantoea ananatis, the amino acids which form a catalytic triad in PhaG proteins are also conserved.
Rhamnose, the hydrophilic moiety of amphiphilic RL, is attached to HAA by rhamnosyltransferases I and II. The membrane-bound, 47-kDa rhamnosyltransferase I, RhlB, transfers a dTDP-activated l-rhamnose moiety to the free 3-hydroxyl group of HAA via an O-glycosidic linkage (Fig. 21) (304).
RhlC transfers a second rhamnose moiety to a fraction of mono-RL, also referred to as rhamnolipid 1, via an α-1,2-glycosidic linkage, yielding di-RL (Fig. 21). As RhlB and RhlC catalyze the formation of glycosidic linkages, they cannot be classified as acyltransferases. There is no sequence similarity between RhlB or RhlC and PhaG, as it is the case for RhlA. The amino acid sequence of the 36-kDa RhlC exhibits significant homology to other rhamnosyltransferases that attach l-rhamnose to terminal l-rhamnosyl or other sugar moieties (e.g., in the LPS). By deletion of the rhlC gene, it was demonstrated that RhlC is essential for di-RL synthesis. It is assumed that mono- and di-RL are synthesized at the cytoplasmic site of the membrane and subsequently transported out of the cell (305). However, the exact mechanism for secretion of RL is not known yet (309). Likewise, a putative catalytic mechanism of both rhamnosyltransferases has not been proposed yet. RL synthesis is tightly regulated at the transcriptional level and influenced by numerous factors. In general, it depends on cell density, which is quorum sensing regulated via the RhlR regulator, and limitation of nutrients, such as phosphate and nitrogen (310, 311). However, this complex transcriptional regulatory network lies beyond the scope of this review.
While the majority of P. aeruginosa-secreted RL are composed of 3-hydroxydecanoyl-3-hydroxydecanoate linked to one or two rhamnose moieties, there exist diverse structures in other RL-producing bacteria. The lipid proportion of RL is most often composed of one or two (in a few cases three) 3-hydroxy fatty acyl chains that can be saturated, monosaturated, or polyunsaturated and vary from C8 to C16 in length. The distal carboxyl group most often remains free, but in some cases it is esterified with a short alkyl residue. The free 2-hydroxyl group of the second rhamnose moiety can also be rarely further acylated with long-chain alkenoic acid (289).
Industrial Relevance of Rhamnolipids
There is a diverse range of possible industrial applications for RL due to their physicochemical properties, e.g., as detergents or emulsifiers in the cosmetic, pharmaceutical, food, and detergent industries. Furthermore, they might be applied for bioremediation, enhanced oil recovery, and biodegradation. Key advantages compared to conventional petrochemical-based surfactants is that RL can be synthesized biotechnologically from renewable resources and that they are biodegradable (309). Due to their great range of potential applications and variety in structure, it can be expected that RL biosynthesis will be the subject of extensive research and that more detailed biochemical knowledge of the corresponding enzymes will become available soon. Finding nonpathogenic production strains as alternatives to the RL model producer P. aeruginosa or the establishment of high-yield recombinant RL synthesis also represents an important aspect to facilitate large-scale biotechnological RL production.
CONCLUSIONS AND COMPARISON OF ACYL TRANSFER REACTIONS IN BACTERIA
The aim of this review was to provide a representative survey of the wide spectrum of acyl transfer-catalyzing enzymes in prokaryotes. From a variety of acyl residue-transferring enzymes, a part of which is still poorly analyzed, several enzyme types have been selected due to their relevance and/or current state of research. Some key features of each type are summarized in Table 7.
TABLE 7.
Different types of bacterial acyltransferases presented in this reviewa
| Enzyme type | Model enzyme(s) | Physiological function(s) | Reaction catalyzed | Acyl donor(s) | Acyl acceptor(s) | Catalytic site |
|---|---|---|---|---|---|---|
| WS/DGAT | AtfA from A. baylyi | WE and/or TAG storage | O-Acylation of (fatty) alcohol or DAG with acyl-CoA | Saturated or unsaturated acyl-CoAs (C2-C20; preferred, C16) | Linear alcohols (C2-C30; preferred, C14-C18); branched, cyclic, or aromatic alcohols; MAG or DAG, long-chain alkanediols, thiols, and dithiols; glycidol | HHxxxDG motif with catalytically active His |
| GPAT | PlsB from E. coli (but most bacteria have PlsY instead) | 1-Acylglycerol-3-phosphate (lysophosphatidate) synthesis | O-Acylation of sn-1 position of glycerol-3-phosphate with acyl-CoA or -ACP | C16 or C18:1 acyl-CoA or -ACP (PlsY, acylphosphate) | Glycerol-3-phosphate | HxxxxD motif catalytic dyad with catalytically active His (PlsY, catalytic His) |
| LPAAT | PlsC from E. coli | Phosphatidate synthesis | O-Acylation of sn-2 position of lysophosphatidate | Acyl-CoA or -ACP | 1-Acylglycerol-3-phosphate (lysophosphatidate) | HxxxxD motif (like GPATs) |
| Lpx acyltransferases | LpxA and LpxD from E. coli | First two acylation steps in lipid A biosynthesis | O- and N-acylation of UDP-N-acetylglucosamine and UDP-3-acylglucosamine | 3-Hydroxyacyl-ACP (C14) | UDP-N-acetylglucosamine and UDP-3-O-(3-hydroxymyristoyl)-glucosamine | Catalytically active His (putatively stabilized by adjacent Asp) |
| LpxL from E. coli (homologs: LpxM, LpxP) | Secondary acylation steps in lipid A biosynthesis | O-Acylation of 3-hydroxy fatty acids attached to lipid A precursor (Kdo2-IVA) | Acyl-ACP (C12) (LpxL, acyl-ACP or -CoA; LpxP, C16:1 acyl-ACP) | Kdo2-IVA (tetra-acylated lipid A intermediate) | HxxxxD/E motif (like GPATs) | |
| Polyketide-associated acyltransferases | PapA5 from M. tuberculosis | Synthesis of polyketide-containing complex lipids (e.g., PGL or PDIM) | O-Acylations of phthiocerol with two mycocerosate residues | Unknown donor for mycocerosyl residues; in vitro, long-chain acyl-CoAs (preferred, C16) | (Phenol)phthiocerol, glycosylphenolphthiocerol, linear alcohols (C8–C10; preferred, C8) | (H)HxxxD(G)x13-14Y motif with catalytically active His |
| PapA1 and PapA2 from M. tuberculosis | Synthesis of sulfolipid 1 (SL-1) | O-Acylation of trehalose-2-sulfate | Unknown donor for hydroxyphthioceranoyl residue, C16 acyl-CoA | Trehalose-2-sulfate and trehalose-2-sulfate-2′-palmitate | HxxxD(G)x13-14Y motif | |
| PapA3 from M. tuberculosis | Synthesis of polyketide-containing polyacyltrehalose (PAT) | Successive (?) O-acylations of a trehalose core | Unknown donor for mycolipenoyl residue, C16 acyl-CoA | (Acylated) trehalose | HxxxDx14Y motif | |
| PapA4 from M. marinum | Synthesis of polyketide-containing lipooligosaccharides (LOS) | Successive (?) O-acylations of the trehalose section of LOS | Unknown donor for 2,4-dimethylhexadecanoate and 2,4-dimethyl-2-pentadecenoate residues | (Acylated) trehalose | HxxxDx14Y motif | |
| Rif-Orf20 from A. mediterranei | Rifamycin B biosynthesis | O-Acetylation of a rifamycin B intermediate | Acetyl-CoA (propionyl-CoA) | Rifamycin intermediate | HxxxDx14Y motif | |
| Chloramphenicol acetyltransferase | Type III CAT from E. coli | Inactivation of chloramphenicol | O-Acetylation of chloramphenicol | Acetyl-CoA | Chloramphenicol | HHxxxDG motif catalytically active His |
| RtxC acyltransferases | HlyA from E. coli | Synthesis of (proteinaceous) leukotoxins | N-Acylation of lysine residues of the protoxin | Acyl-ACPs (C12-C18, C16:1, C18:1; preferred, C14) | ε-Amino groups of lysine | Catalytically active His and crucial Ser residue (covalent acyl-HlyC heterocomplex) |
| Lipase | ? | Hydrolysis of acylglycerols | ? | ? | ? | Catalytic triad composed of histidine, serine (in lipase box), and aspartate |
| PHA synthase | ? | Polymerization of 3-hydroxyalkanoic acids to PHA | ? | ? | ? | Catalytic triad composed of histidine, cysteine (in modified lipase box), and aspartate |
| PhaG transacylase | PhaG from P. putida | Metabolic link between de novo fatty acid synthesis and PHAMCL synthesis | (Reversible) transfer of 3-hydroxyacyl-chain from ACP to CoA | 3-Hydroxyacyl-ACP (or -CoA); preferred, 3-hydroxy-C10 | CoA (or ACP) | HxxxxD/A motif, but different mechanism via catalytic triad (composed of Ser, His, and Asp residues) |
| RhlA | RhlA from P. aeruginosa | Metabolic link between de novo fatty acid synthesis and rhamnolipid synthesis (synthesis of HAA) | O-Acylation of the 3-hydroxy group of 3-hydroxyacyl-ACP | 3-Hydroxyacyl-ACPs (preferred, 3-hydroxy-C10) | (3-Hydroxy group of a) 3-hydroxyacyl-residue | Potentially similar mechanism as for PhaG with a catalytic triad |
The given catalyzed reactions and (preferred) acyl donors or acceptors refer to the respective model enzyme of each type.
Regarding their catalytic mechanism, the enzyme types can roughly be divided into two groups: the majority of enzymes discussed in this review are characterized by a conserved Hxxx(x)D/E-like active-site motif with a catalytically active histidine residue and a noncovalent transition state, whereas RtxC acyltransferases, lipases, PHA synthases, or PhaG transacylases form a covalent intermediate during catalysis. Apart from these enzymes, all of the “real” acyltransferases, which have been discussed in this review, seem not to function via a catalytic triad but have a crucial catalytically active histidine residue in common. In some of the Hxxx(x)D/E-like motifs, histidine and the negatively charged residue aspartate or glutamate seem to form an essential catalytic dyad. In GPAT-, LpxA-, LpxL-, PapA5-, and CATIII-like acyltransferases, aspartate is thought to stabilize the protonated histidine residue during catalysis and/or to have an essential structural function for the catalytic core. In contrast, the highly conserved aspartate residue does not seem to be of major importance for catalysis or structure in AtfA-like WS/DGATs, as an exchange of this residue with a nonpolar amino acid did not influence the enzyme's activity dramatically. Thus, it can be speculated that the actual spatial arrangement of residues belonging to this kind of active-site motif might be different in these enzyme types.
Although PhaG transacylases also possess a conserved HxxxxD/A sequence motif, it could be shown that they use a completely different catalytic mechanism than, for example, AtfA-like WS/DGATs. Due to sequence homology and structural resemblance, they have been assigned to the α/β hydrolase superfamily and possess an essential catalytic triad like that described for lipases and PHA synthases. In contrast to AtfA-like WS/DGATs, which acylate free hydroxyl groups, PhaG transfers the acyl residue by separating it from an activated thioester linkage and subsequently forming another thioester linkage. This explains why it resembles a (thio)esterase, and thus hydrolase, rather than an acyltransferase. The phenomenon of a simultaneous presence of a catalytic triad and the HxxxxD motif has also been described for the lipases Tgl3p and Tgl5p from yeast, which were shown to harbor two independent activities and function as both lipase and acyltransferase.
It should be noted that HlyC-like acyltransferases exert a completely different kind of “ping-pong” mechanism which distinguishes them from AtfA-like acyltransferases as well as from lipases, PhaG transacylases, or PHA synthases. In HlyC, the catalytic histidine residue directly attacks the thioester carbonyl carbon atom and forms a covalent acyl-enzyme heterocomplex, whereas histidine in the HxxxxD-like motif or catalytic triad first attacks the hydroxyl group of the alcohol or serine, respectively, to create a strong nucleophile which then can, in turn, attack the thioester carbonyl carbon atom.
To sum up, the transfer of hydrophobic acyl chains is essential for a multitude of biological functions and can be realized by enzymes with completely different structures and/or catalytic mechanisms. Despite this variety, it can be noted that all involved mechanisms known so far directly or indirectly rely on a catalytically active histidine residue. Furthermore, in “real” acyltransferases, this histidine residue is most often part of an Hxxx(x)D-like sequence motif together with acidic aspartate or glutamate.
Supplementary Material
ACKNOWLEDGMENT
We are very grateful to the Rahn-Quade-Stiftung for financial support from a fellowship to Annika Röttig (project T381 of the Deutsches Stiftungszentrum in Essen).
Biographies

Annika Röttig has been a Ph.D. student in the Institute for Molecular Microbiology and Biotechnology at the University of Münster in Germany since 2012. Her bachelor's degree thesis dealt with the microbial acyltransferase-based production of biodiesel-like compounds. In her master's thesis and continuing during her Ph.D. studies, she has focused on a bacterial model acyltransferase and investigating the biochemistry of this enzyme family and its potential for biotechnological applications.

Alexander Steinbüchel has been a full professor and the executive director of the Institute for Molecular Microbiology and Biotechnology at the University of Münster since 1994. He obtained his doctoral degree under the supervision of Hans-Günther Schlegel at the University of Göttingen in 1983. Afterwards, he studied the metabolism of protozoa at the Rockefeller University in New York as a postdoctoral fellow. Back in Germany, he joined the University of Göttingen in 1991. His major research interests concern the biosynthesis of biopolymers, microbial lipids, the biodegradation of rubber, and the microbial production of industrially relevant compounds. His laboratory identified and characterized several key enzymes and regulatory mechanisms of these metabolic pathways and also established and optimized various production processes.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MMBR.00010-13.
REFERENCES
- 1. White SW, Zheng J, Zhang YM, Rock CO. 2005. The structural biology of the type II fatty acid biosynthesis. Annu. Rev. Biochem. 74:791–831 [DOI] [PubMed] [Google Scholar]
- 2. Black PN, DiRusso CC. 2003. Transmembrane movement of exogenous long-chain fatty acids: proteins, enzymes, and vectorial esterification. Microbiol. Mol. Biol. Rev. 67:454–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan DI, Vogel HJ. 2010. Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem. J. 430:1–19 [DOI] [PubMed] [Google Scholar]
- 4. Murphy DJ. 1993. Structure, function and biogenesis of storage lipid bodies and oleosins in plants. Prog. Lipid Res. 32:247–280 [DOI] [PubMed] [Google Scholar]
- 5. Wältermann M, Steinbüchel A. 2005. Neutral lipid bodies in prokaryotes: recent insights into structure, formation, and relationship to eukaryotic lipid depots. J. Bacteriol. 187:3607–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson AJ, Dawes EA. 1990. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol. Rev. 54:450–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng JB, Russell DW. 2004. Mammalian wax biosynthesis. II. Expression cloning of wax synthase cDNAs encoding a member of the acyltransferase gene family. J. Biol. Chem. 279:37798–37807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murphy DJ. 2001. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog. Lipid Res. 40:325–438 [DOI] [PubMed] [Google Scholar]
- 9. Samuels L, Kunst L, Jetter R. 2008. Sealing plant surfaces: cuticular wax formation by epidermal cells. Annu. Rev. Plant Biol. 59:683–707 [DOI] [PubMed] [Google Scholar]
- 10. Voet D, Voet JG. 1990. Lipids and membranes, p 271–314 In Voet D, Voet JG. (ed), Biochemistry. Wiley, New York, NY [Google Scholar]
- 11. Zweytick D, Athenstaedt K, Daum G. 2000. Intracellular lipid particles in eukaryotic cells. Biochim. Biophys. Acta 1469:101–120 [DOI] [PubMed] [Google Scholar]
- 12. Post-Beittenmiller D. 1996. Biochemistry and molecular biology of wax production in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47:405–430 [DOI] [PubMed] [Google Scholar]
- 13. Li F, Wu X, Lam P, Bird D, Zheng H, Samuels L, Jetter R, Kunst L. 2008. Identification of the wax ester synthase/acyl-coenzyme A:diacylglycerol acyltransferase WSD1 required for stem wax ester biosynthesis in Arabidopsis. Plant Physiol. 148:97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blomquist GJ, Chu AJ, Remaley S. 1980. Biosynthesis of wax in the honeybee, Apis mellifera L. Insect Biochem. 10:313–321 [Google Scholar]
- 15. Tulloch AP. 1971. Beeswax: structure of the esters and their component hydroxy acids and diols. Chem. Phys. Lipids. 6:235–265 [Google Scholar]
- 16. Clarke MR. 1970. Function of the spermaceti organ of the sperm whale. Nature 228:873–874 [DOI] [PubMed] [Google Scholar]
- 17. Lardizabal KD, Metz JG, Sakamoto T, Hutton WC, Pollard MR, Lassner MW. 2000. Purification of a Jojoba embryo wax synthase, cloning of its cDNA, and production of high levels of wax in seeds of transgenic Arabidopsis. Plant Physiol. 122:645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alvarez HM, Mayer F, Fabritius D, Steinbüchel A. 1996. Formation of intracytoplasmic lipid inclusions by Rhodococcus opacus strain PD630. Arch. Microbiol. 165:377–386 [DOI] [PubMed] [Google Scholar]
- 19. Akao T, Kusaka T. 1976. Solubilization of diglyceride acyltransferase from membrane of Mycobacterium smegmatis. J. Biochem. 80:723–728 [DOI] [PubMed] [Google Scholar]
- 20. Alvarez HM, Kalscheuer R, Steinbüchel A. 1997. Accumulation of storage lipids in species of Rhodococcus and Nocardia and effects of inhibitors and polyethylene glycol. Fett/Lipid. 99:239–246 [Google Scholar]
- 21. Alvarez HM, Steinbüchel A. 2002. Triacylglycerols in prokaryotic microorganisms. Appl. Microbiol. Biotechnol. 60:367–376 [DOI] [PubMed] [Google Scholar]
- 22. Barksdale L, Kim KS. 1977. Mycobacterium. Bacteriol. Rev. 41:217–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koval'schuk LP, Donets AP, Razumovskii PN. 1973. Lipid biosynthesis by actinomycetes cultivated on different media. Mikrobiol. Int. 42:567–571 [PubMed] [Google Scholar]
- 24. Wayman M, Jenkins AD, Kormendy AG. 1984. Bacterial production of fats and oils, p 129–143 In Ratledge C, Dawson P, Rattray J. (ed), Biotechnology for the oils and fat industry. American Oil Chemists Society, Champaign, IL [Google Scholar]
- 25. Fixter LM, Nagi MN, McCormack JG, Fewson CA. 1986. Structure, distribution and function of wax esters in Acinetobacter calcoaceticus. J. Gen. Microbiol. 132:3147–3157 [Google Scholar]
- 26. Kalscheuer R, Stöveken T, Malkus U, Reichelt R, Golyshin PN, Sabirova JS, Ferrer M, Timmis KN, Steinbüchel A. 2007. Analysis of storage lipid accumulation in Alcanivorax borkumensis: evidence for alternative triacylglycerols biosynthesis routes in bacteria. J. Bacteriol. 189:918–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Makula RA, Lockwood PJ, Finnerty WR. 1975. Comparative analysis of lipids of Acinetobacter species grown on hexadecane. J. Bacteriol. 121:303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barney BM, Wahlen BD, Garner EL, Wei J, Seefeldt LC. 2012. Differences in substrate specificities of five bacterial wax ester synthases. Appl. Environ. Microbiol. 78:5734–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bredemeier R, Hulsch R, Metzger JO, Berthe-Corti L. 2003. Submersed culture production of extracellular wax esters by the marine bacterium Fundibacter jadensis. Mar. Biotechnol. 5:579–588 [DOI] [PubMed] [Google Scholar]
- 30. Bryn K, Jantzen E, Bovre K. 1977. Occurrence and patterns of waxes in Neisseriaceae. J. Gen. Microbiol. 102:33–43 [DOI] [PubMed] [Google Scholar]
- 31. Gallagher IHC. 1971. Occurrence of waxes in Acinetobacter. J. Gen. Microbiol. 68:245–247 [DOI] [PubMed] [Google Scholar]
- 32. Holtzapple E, Schmidt-Dannert C. 2007. Biosynthesis of isoprenoid wax ester in Marinobacter hydrocarbonoclasticus DSM 8798: identification and characterization of isoprenoid coenzyme A synthetase and wax ester synthases. J. Bacteriol. 189:3804–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russell NJ, Volkman JK. 1980. The effect of growth temperature and wax ester composition in the psychrophilic bacterium Micrococcus cryophilus ATCC 15174. J. Gen. Microbiol. 118:131–141 [Google Scholar]
- 34. Bacchin P, Robertiello A, Viglia A. 1974. Identification of n-decane oxidation products in Corynebacterium cultures by combined gas chromatography mass spectrometry. Appl. Microbiol. 28:737–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raymond RL, Davies JB. 1960. n-Alkane utilization and lipid formation by a Nocardia. Appl. Microbiol. 8:329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adamczak M, Bornscheuer UT, Bednarski W. 2009. The application of biotechnological methods for the synthesis of biodiesel. Eur. J. Lipid Sci. Technol. 111:808–813 [Google Scholar]
- 37. Rude MA, Schirmer A. 2009. New microbial fuels: a biotech perspective. Curr. Opin. Microbiol. 12:274–281 [DOI] [PubMed] [Google Scholar]
- 38. Rontani JF. 2010. Production of wax esters by bacteria, p 460–470 In Timmis KN. (ed), Handbook of hydrocarbon and lipid microbiology. Springer-Verlag, Berlin, Germany [Google Scholar]
- 39. Hills G. 2003. Industrial use of lipases to produce fatty acid esters. Eur. J. Lipid Sci. Technol. 105:601–607 [Google Scholar]
- 40. Kalscheuer R, Stöveken T, Luftmann H, Malkus U, Reichelt R, Steinbüchel A. 2006. Neutral lipid biosynthesis in engineered Escherichia coli: Jojoba oil-like wax esters and fatty acid butyl esters. Appl. Environ. Microbiol. 72:1373–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stöveken T, Steinbüchel A. 2008. Bacterial acyltransferases as an alternative for lipase-catalyzed acylation for the production of oleochemicals and fuels. Angew Chem. Int. Ed. Engl. 47:3688–3694 [DOI] [PubMed] [Google Scholar]
- 42. Kalscheuer R, Stölting T, Steinbüchel A. 2006. Microdiesel: Escherichia coli engineered for fuel production. Microbiology 152:2529–2536 [DOI] [PubMed] [Google Scholar]
- 43. Röttig A, Wenning L, Bröker D, Steinbüchel A. 2010. Fatty acid alkyl esters: perspectives for production of alternative biofuels. Appl. Microbiol. Biotechnol. 85:1713–1733 [DOI] [PubMed] [Google Scholar]
- 44. Ohlrogge J, Browse J. 1995. Lipid biosynthesis. Plant Cell 7:957–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bell RM, Coleman RA. 1980. Enzymes of glycerolipid synthesis in eukaryotes. Annu. Rev. Biochem. 49:459–487 [DOI] [PubMed] [Google Scholar]
- 46. Cases S, Smith SJ, Zhen YW, Myers HM, Lear SR, Sande E, Novak S, Collins C, Welch CB, Lusis AJ, Erickson SK, Farese RV., Jr 1998. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. U. S. A. 95:13018–13023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yen CLE, Stone SJ, Koliwad S, Harris C, Farese RV. 2008. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 49:2283–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Turkish AR, Henneberry AL, Cromley D, Padamsee M, Oelkers P, Bazzi H, Christiano AM, Billheimer JT, Sturley SL. 2005. Identification of two novel human acyl-CoA wax alcohol acyltransferases. J. Biol. Chem. 280:14755–14764 [DOI] [PubMed] [Google Scholar]
- 49. Biester EM, Hellenbrand J, Frentzen M. 2012. Multifunctional acyltransferases from Tetrahymena thermophila. Lipids 47:371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Biester EM, Hellenbrand J, Gruber J, Hamberg M, Frentzen M. 2012. Identification of avian wax synthases. BMC Biochem. 13:4–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kalscheuer R, Steinbüchel A. 2003. A novel bifunctional wax ester synthase/acyl-CoA:diacylglycerol acyltransferase mediates wax ester and triacylglycerol biosynthesis in Acinetobacter calcoaceticus ADP1. J. Biol. Chem. 278:8075–8082 [DOI] [PubMed] [Google Scholar]
- 52. Teerawanichpan P, Qiu X. 2010. Fatty acyl-CoA reductase and wax synthase from Euglena gracilis in the biosynthesis of medium-chain wax esters. Lipids 45:263–273 [DOI] [PubMed] [Google Scholar]
- 53. King A, Nam JW, Han J, Hilliard J, Jaworski JG. 2007. Cuticular wax biosynthesis in petunia petals: cloning and characterization of an alcohol-acyltransferase that synthesizes wax-esters. Planta 226:381–394 [DOI] [PubMed] [Google Scholar]
- 54. Stöveken T, Kalscheuer R, Malkus U, Reichelt R, Steinbüchel A. 2005. The wax ester synthase/acyl-CoA:diacylglycerol acyltransferase from Acinetobacter sp. strain ADP1: characterization of a novel type of acyltransferase. J. Bacteriol. 187:1369–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ervin JL, Geigert J, Neidleman SL, Wadsworth J. 1984. Substrate-dependent and growth temperature dependent changes in the wax ester compositions produced by Acinetobacter sp. HO1-N, p 217–222 In Ratledge C, Dawson P, Rattray L. (ed), Biotechnology of the oil and fats industry. American Oil Chemists Society, Champaign, IL [Google Scholar]
- 56. Kalscheuer R, Uthoff S, Luftmann H, Steinbüchel A. 2003. In vitro and in vivo biosynthesis of wax diesters by an unspecific bifunctional wax ester synthase/acyl-CoA:diacylglycerol acyltransferase from Acinetobacter calcoaceticus ADP1. Eur. J. Lipid Sci. Technol. 105:578–584 [Google Scholar]
- 57. Uthoff S, Stöveken T, Weber N, Vosmann K, Klein E, Kalscheuer R, Steinbüchel A. 2005. Thio wax ester biosynthesis utilizing the unspecific bifunctional wax ester synthase/acyl coenzyme A:diacylglycerol acyltransferase of Acinetobacter sp. strain ADP1. Appl. Environ. Microbiol. 71:790–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim OB, Luftmann H, Steinbüchel A. 2009. Biotransformation of glycidol by the unspecific wax ester synthase/acyl-CoA:diacylglycerol acyltransferase of Acinetobacter baylyi ADP1. Eur. J. Lipid Sci. Technol. 111:972–978 [Google Scholar]
- 59. Stöveken T, Kalscheuer R, Steinbüchel A. 2009. Both histidine residues of the conserved HHXXXDG motif are essential for wax ester synthase/acyl-CoA:diacylglycerol acyl-transferase catalysis. Eur. J. Lipid Sci. Technol. 111:112–119 [Google Scholar]
- 60. Wältermann M, Stöveken T, Steinbüchel A. 2007. Key enzymes for biosynthesis of neutral lipid storage compounds in prokaryotes: properties, function and occurrence of wax ester synthases/acyl-CoA:diacylglycerol acyltransferases. Biochimie 89:230–242 [DOI] [PubMed] [Google Scholar]
- 61. Murray IA, Lewendon A, Kleanthous C, Shaw WV. 1986. Catalytic mechanism of chloramphenicol acetyltransferase investigated by site-directed mutagenesis. Biochem. Soc. Trans. 14:1227–1228 [Google Scholar]
- 62. Onwueme KC, Ferreras JA, Buglino J, Lima CD, Quadri LEN. 2004. Mycobacterial polyketide-associated proteins are acyltransferases: proof of principle with Mycobacterium tuberculosis PapA5. Proc. Natl. Acad. Sci. U. S. A. 101:4608–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen LO. 2007. Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure 15:781–792 [DOI] [PubMed] [Google Scholar]
- 64. Six DA, Carty SM, Guan Z, Raetz CRH. 2008. Purification and mutagenesis of LpxL, the lauroyltransferase of Escherichia coli lipid A biosynthesis. Biochemistry 47:8623–8637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Stachelhaus T, Mootz HD, Bergendahl V, Marahiel MA. 1998. Peptide bond formation in nonribosomal peptide biosynthesis. J. Biol. Chem. 273:22773–22781 [DOI] [PubMed] [Google Scholar]
- 66. Zhang YM, Rock CO. 2008. Acyltransferases in bacterial glycerophospholipid synthesis. J. Lipid Res. 49:1867–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wältermann M, Hinz A, Robenek H, Troyer D, Reichelt R, Malkus U, Galla HJ, Kalscheuer R, Stöveken T, von Landenberg P, Steinbüchel A. 2005. Mechanism of lipid-body formation in prokaryotes: how bacteria fatten up. Mol. Microbiol. 55:750–763 [DOI] [PubMed] [Google Scholar]
- 68. Pollard MR, McKeon T, Gupta LM, Stumpf PK. 1979. Studies on biosynthesis of waxes by developing jojoba seed. II. The demonstration of wax biosynthesis by cell-free homogenates. Lipids 14:651–662 [Google Scholar]
- 69. Elbahloul Y, Steinbüchel A. 2010. Pilot-scale production of fatty acid ethyl esters by an engineered Escherichia coli strain harboring the p(Microdiesel) plasmid. Appl. Environ. Microbiol. 76:4560–4565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kalscheuer R, Luftmann H, Steinbüchel A. 2004. Synthesis of novel lipids in Saccharomyces cerevisiae by heterologous expression of an unspecific bacterial acyltransferase. Appl. Environ. Microbiol. 70:7112–7125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yu KO, Jung J, Kim SW, Park CH, Han SO. 2012. Synthesis of FAEEs from glycerol in engineered Saccharomyces cerevisiae using endogenously produced ethanol by heterologous expression of an unspecific bacterial acyltransferase. Biotechnol. Bioeng. 109:110–115 [DOI] [PubMed] [Google Scholar]
- 72. Shi S, Valle-Rodríguez JO, Khoomrung S, Siewers V, Nielsen J. 2012. Functional expression and characterization of five wax ester synthases in Saccharomyces cerevisiae and their utility for biodiesel production. Biotechnol. Biofuels 5:7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Daniel J, Deb C, Dubey VS, Sirakova TD, Abomoelak B, Morbidoni HR, Kolattukudy PE. 2004. Introduction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J. Bacteriol. 186:5017–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Alvarez AF, Alvarez HM, Kalscheuer R, Wältermann M, Steinbüchel A. 2008. Cloning and characterization of a gene involved in triacylglycerol biosynthesis and identification of additional homologous genes in the oleaginous bacterium Rhodococcus opacus PD630. Microbiology 154:2327–2335 [DOI] [PubMed] [Google Scholar]
- 75. Arabolaza A, Rodriguez E, Altabe S, Alvarez H, Gramajo H. 2008. Multiple pathways for triacylglycerol biosynthesis in Streptomyces coelicolor. Appl. Environ. Microbiol. 74:2573–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kaddor C, Biermann K, Kalscheuer R, Steinbüchel A. 2009. Analysis of neutral lipid biosynthesis in Streptomyces avermitilis MA-4680 and characterization of an acyltransferase involved herein. Appl. Microbiol. Biotechnol. 84:143–155 [DOI] [PubMed] [Google Scholar]
- 77. Smith I. 2003. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 16:463–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wayne LG, Sohaskey CD. 2001. Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55:139–163 [DOI] [PubMed] [Google Scholar]
- 79. Garton NJ, Christensen H, Minnikin DE, Adegbola RA, Barer MR. 2002. Intracellular lipophilic inclusions of mycobacteria in vitro and in sputum. Microbiology 148:2951–2958 [DOI] [PubMed] [Google Scholar]
- 80. Russell DG. 2003. Phagosomes, fatty acids and tuberculosis. Nat. Cell Biol. 5:776–778 [DOI] [PubMed] [Google Scholar]
- 81. Sirakova TD, Dubey VS, Deb C, Daniel J, Korotkova TA, Abomoelak B, Kolattukudy PE. 2006. Identification of a diacylglycerol acyltransferase gene involved in accumulation of triacylglycerol in Mycobacterium tuberculosis under stress. Microbiology 152:2717–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Berekaa MM, Steinbüchel A. 2000. Microbial degradation of the multiply branched alkane 2,6,10,15,19,23-hexamethyltetracosane (squalane) by Mycobacterium fortuitum and Mycobacterium ratisbonense. Appl. Environ. Microbiol. 66:4462–4467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Silva RA, Grossi V, Alvarez HM. 2007. Biodegradation of phytane (2,6,10,14-tetramethylhexadecane) and accumulation of related isoprenoid wax esters by Mycobacterium ratisbonense strain SD4 under nitrogen-starvation conditions. FEMS Microbiol. Lett. 272:220–228 [DOI] [PubMed] [Google Scholar]
- 84. Gauthier MJ, Lafay B, Christen R, Fernandez L, Acquaviva M, Bonin P, Bertrand JC. 1992. Marinobacter hydrocarbonoclasticus gen. nov., sp. nov., a new, extremely halotolerant, hydrocarbon-degrading marine bacterium. Int. J. Syst. Bacteriol. 42:568–576 [DOI] [PubMed] [Google Scholar]
- 85. Klein B, Grossi V, Bouriat P, Goulas P, Grimaud R. 2008. Cytoplasmic wax ester accumulation during biofilm-driven substrate assimilation at the alkane-water interface by Marinobacter hydrocarbonoclasticus SP17. Res. Microbiol. 159:137–144 [DOI] [PubMed] [Google Scholar]
- 86. Rontani JF, Bonin P, Volkman JK. 1999. Production of wax esters during aerobic growth of marine bacteria on isoprenoid compounds. Appl. Environ. Microbiol. 65:221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rontani JF, Mouzdahir A, Michotey V, Caumette P, Bonin P. 2003. Production of a polyunsaturated isoprenoid wax ester during aerobic metabolism of squalene by Marinobacter squalenivorans sp. nov. Appl. Environ. Microbiol. 69:4167–4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Márquez MC, Ventosa A. 2005. Marinobacter hydrocarbonoclasticus Gauthier et al. 1992 and Marinobacter aquaeolei Nguyen et al. 1999 are heterotypic synonyms. Int. J. Syst. Evol. Microbiol. 55:1349–1351 [DOI] [PubMed] [Google Scholar]
- 89. Barney BM, Mann RL, Ohlert JM. 19 October 2012. Identification of a residue affecting fatty alcohol selectivity in wax ester synthase. Appl. Environ. Microbiol. doi: 10.1128/AEM.02523-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yakimov MM, Golyshin PN, Lang S, Moore ERB, Abraham WR, Lünsdorf H, Timmis KN. 1998. Alcanivorax borkumensis gen. nov., sp. nov., a new, hydrocarbon-degrading and surfactant-producing marine bacterium. Int. J. Syst. Bacteriol. 48:339–348 [DOI] [PubMed] [Google Scholar]
- 91. Manilla-Pérez E, Lange AB, Luftmann H, Robenek H, Steinbüchel A. 2011. Neutral lipid production in Alcanivorax borkumensis SK2 and other marine hydrocarbonoclastic bacteria. Eur. J. Lipid Sci. Technol. 113:8–17 [Google Scholar]
- 92. Manilla-Pérez E, Lange AB, Hetzler S, Wältermann M, Kalscheuer R, Steinbüchel A. 2010. Isolation and characterization of a mutant of the marine bacterium Alcanivorax borkumensis SK2 defective in lipid biosynthesis. Appl. Environ. Microbiol. 76:2884–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Alvarez HM, Luftmann H, Silva RA, Cesari AC, Viale A, Wältermann M, Steinbüchel A. 2002. Identification of phenyldecanoic acid as a constituent of triacylglycerols and wax ester produced by Rhodococcus opacus PD630. Microbiology 148:1407–1412 [DOI] [PubMed] [Google Scholar]
- 94. Olukoshi ER, Packter NM. 1994. Importance of stored triacylglycerols in Streptomyces: possible carbon source for antibiotics. Microbiology 140:931–943 [DOI] [PubMed] [Google Scholar]
- 95. Schneiker S, Martins dos Santos VAP, Bartels D, Bekel T, Brecht M, Buhrmester J, Chernikova TN, Denaro R, Ferrer M, Gertler C, Goesmann A, Golyshina OV, Kaminski F, Khachane AN, Lang S, Linke B, McHardy AC, Meyer F, Nechitaylo T, Pühler A, Regenhardt D, Rupp O, Sabirova JS, Selbitschka W, Yakimov MM, Timmis KN, Vorhölter FJ, Weidner S, Kaiser O, Golyshin PN. 2006. Genome sequence of the ubiquitous hydrocarbondegrading marine bacterium Alcanivorax borkumensis. Nat. Biotechnol. 24:997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lehner R, Kuksis A. 1993. Triacylglycerol synthesis by an sn-1,2(2,3)-diacylglycerol transacylase from rat intestinal microsomes. J. Biol. Chem. 268:8781–8786 [PubMed] [Google Scholar]
- 97. Stobart K, Mancha M, Lenman M, Dahlqvist A, Stymne S. 1997. Triacylglycerols are synthesized and utilized by transacylation reactions in microsomal preparations of developing safflower (Carthamus tinctorius L.) seeds. Planta 203:58–66 [Google Scholar]
- 98. Sandager L, Gustavsson MH, Ståhl U, Dahlqvist A, Wiberg E, Banas A, Lenman M, Ronne H, Stymne S. 2002. Storage lipid synthesis is non-essential in yeast. J. Biol. Chem. 277:6478–6482 [DOI] [PubMed] [Google Scholar]
- 99. Dahlqvist A, Ståhl U, Lenman M, Banas A, Lee M, Sandager L, Ronne H, Stymne S. 2000. Phospholipid:diacylglycerol acyltransferase: an enzyme that catalyzes the acyl-CoA-independent formation of triacylglycerol in yeast and plants. Proc. Natl. Acad. Sci. U. S. A. 97:6487–6492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Schölkopf B, Weigel D, Lohmann JU. 2005. A gene expression map of Arabidopsis thaliana development. Nat. Genet. 37:501–506 [DOI] [PubMed] [Google Scholar]
- 101. Lehner R, Kuksis A. 1996. Biosynthesis of triacylglycerols. Prog. Lipid Res. 35:169–201 [DOI] [PubMed] [Google Scholar]
- 102. Wilkison WO, Bell RM. 1997. sn-Glycerol-3-phosphate acyltransferase from Escherichia coli. Biochim. Biophys. Acta 1348:3–9 [DOI] [PubMed] [Google Scholar]
- 103. Coleman J. 1990. Characterization of Escherichia coli cells deficient in 1-acyl-sn-glycerol-3-phosphate acyltransferase activity. J. Biol. Chem. 265:17215–17221 [PubMed] [Google Scholar]
- 104. Heath RJ, Rock CO. 1998. A conserved histidine is essential for glycerolipid acyltransferase catalysis. J. Bacteriol. 180:1425–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lewin TM, Wang P, Coleman RA. 1999. Analysis of amino acid motifs diagnostic for the sn-glycerol-3-phosphate acyltransferase reaction. Biochemistry 38:5764–5771 [DOI] [PubMed] [Google Scholar]
- 106. Turnbull AP, Rafferty JB, Sedelnikova SE, Slabas AR, Schierer TP, Kroon JT, Simon JW, Fawcett T, Nishida I, Murata N, Rice DW. 2001. Analysis of the structure, substrate specificity, and mechanism of squash glycerol-3-phosphate (1)-acyltransferase. Structure 9:347–353 [DOI] [PubMed] [Google Scholar]
- 107. Coleman J. 1992. Characterization of the Escherichia coli gene for 1-acyl-sn-glycerol-3-phosphate acyltransferase (plsC). Mol. Gen. Genet. 232:295–303 [DOI] [PubMed] [Google Scholar]
- 108. Green PR, Merrill AH, Bell RM., Jr 1981. Membrane phospholipid synthesis in Escherichia coli: purification, reconstitution, and characterization of sn-glycerol-3-phosphate acyltransferase. J. Biol. Chem. 256:11151–11159 [PubMed] [Google Scholar]
- 109. Rock CO, Goelz SE, Cronan JE., Jr 1981. Phospholipid synthesis in Escherichia coli. Characteristics of fatty acid transfer from acyl-acyl carrier protein to sn-glycerol-3-phosphate. J. Biol. Chem. 256:736–742 [PubMed] [Google Scholar]
- 110. Jackson MB, Cronan JE., Jr 1978. An estimate of the minimum amount of fluid lipid required for the growth of Escherichia coli. Biochim. Biophys. Acta 512:472–479 [DOI] [PubMed] [Google Scholar]
- 111. Ganesh BB, Wang P, Kim JH, Black TM, Lewin TM, Fiedorek FT, Coleman RA. 1999. Rat sn-glycerol-3-phosphate acyltransferase: molecular cloning and characterization of the cDNA and expressed protein. Biochim. Biophys. Acta 1439:415–423 [DOI] [PubMed] [Google Scholar]
- 112. Hanke C, Wolter FP, Coleman J, Peterek G, Frentzen M. 1995. A plant acyltransferase involved in triacylglycerol biosynthesis complements an Escherichia coli sn-1-acylglycerol-3-phosphate acyltransferase mutant. Eur. J. Biochem. 232:806–810 [PubMed] [Google Scholar]
- 113. Lu YJ, Zhang F, Grimes KD, Lee RE, Rock CO. 2007. Topology and active site of PlsY: the bacterial acylphosphate:glycerol-3-phosphate acyltransferase. J. Biol. Chem. 282:11339–11346 [DOI] [PubMed] [Google Scholar]
- 114. Lu YJ, Zhang YM, Grimes KD, Qi J, Lee RE, Rock CO. 2006. Acyl-phosphates initiate membrane phospholipid synthesis in Gram-negative pathogens. Mol. Cell 23:765–772 [DOI] [PubMed] [Google Scholar]
- 115. Paoletti L, Lu YL, Schujman GE, de Mendoza D, Rock CO. 2007. Coupling of fatty acid and phospholipid synthesis in Bacillus subtilis. J. Bacteriol. 189:5816–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. King JD, Kocíncová D, Westman EL, Lam JS. 2009. Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 15:261–312 [DOI] [PubMed] [Google Scholar]
- 117. Raetz CRH. 1986. Molecular genetics of membrane phospholipid synthesis. Annu. Rev. Genet. 20:253–295 [DOI] [PubMed] [Google Scholar]
- 118. Galanos C, Lüderitz O, Rietschel ET, Westphal O, Brade H, Brade L, Freudenberg M, Schade U, Imoto M, Yoshimura H, Kusumoto S, Shiba T. 1985. Synthetic and natural Escherichia coli free lipid A express identical endotoxic activities. Eur. J. Biochem. 148:1–5 [DOI] [PubMed] [Google Scholar]
- 119. Miller SI, Ernst RK, Bader MW. 2005. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 3:36–46 [DOI] [PubMed] [Google Scholar]
- 120. Trent MS. 2004. Biosynthesis, transport, and modification of lipid A. Biochem. Cell Biol. 82:71–86 [DOI] [PubMed] [Google Scholar]
- 121. Dotson GD, Kaltashov IA, Cotter RJ, Raetz CR. 1998. Expression cloning of a Pseudomonas gene encoding a hydroxydecanoyl-acyl carrier protein-dependent UDP-GlcNAc acyltransferase. J. Bacteriol. 180:330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Williams AH, Raetz CRH. 2007. Structural basis for the acyl chain selectivity and mechanism of UDP-N-acetylglucosamine acyltransferase. Proc. Natl. Acad. Sci. U. S. A. 104:13543–13550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Williamson JM, Anderson MS, Raetz CR. 1991. Acyl-acyl carrier protein specificity of UDP-GlcNAc acyltransferases from Gram-negative bacteria: relationship to lipid A structure. J. Bacteriol. 173:3591–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wyckoff TJ, Lin S, Cotter RJ, Dotson GD, Raetz CR. 1998. Hydrocarbon rulers in UDP-N-acetylglucosamine acyltransferases. J. Biol. Chem. 273:32369–32372 [DOI] [PubMed] [Google Scholar]
- 125. Coleman J, Raetz CR. 1988. First committed step of lipid A biosynthesis in Escherichia coli: sequence of the lpxA gene. J. Bacteriol. 170:1268–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Galloway SM, Raetz CR. 1990. A mutant of Escherichia coli defective in the first step of endotoxin biosynthesis. J. Biol. Chem. 265:6394–6402 [PubMed] [Google Scholar]
- 127. Bainbridge BW, Karimi-Naser L, Reife R, Blethen F, Ernst RK, Darveau RP. 2008. Acyl chain specificity of the acyltransferases LpxA and LpxD and substrate availability contribute to lipid A fatty acid heterogeneity in Porphyromonas gingivalis. J. Bacteriol. 190:4549–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lee BI, Suh SW. 2003. Crystal structure of UDP-N-acetylglucosamine acyltransferase from Helicobacter pylori. Proteins 53:772–774 [DOI] [PubMed] [Google Scholar]
- 129. Odegaard TJ, Kaltashov IA, Cotter RJ, Steeghs L, van der Ley P, Khan S, Maskell DJ, Raetz CR. 1997. Shortened hydroxyacyl chains on lipid A of Escherichia coli cells expressing a foreign UDP-N-acetylglucosamine O-acyltransferase. J. Biol. Chem. 272:19688–19696 [DOI] [PubMed] [Google Scholar]
- 130. Sweet CR, Preston A, Toland E, Ramirez SM, Cotter RJ, Maskell DJ, Raetz CR. 2002. Relaxed acyl chain specificity of Bordetella UDP-N-acetylglucosamine acyltransferases. J. Biol. Chem. 277:18281–18290 [DOI] [PubMed] [Google Scholar]
- 131. Sweet CR, Lin S, Cotter RJ, Raetz CR. 2001. A Chlamydia trachomatis UDP-N-acetylglucosamine acyltransferase selective for myristoyl-acyl carrier protein. Expression in Escherichia coli and formation of hybrid lipid A species. J. Biol. Chem. 276:19565–19574 [DOI] [PubMed] [Google Scholar]
- 132. Raetz CRH, Roderick SL. 1995. A left-handed parallel beta helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science 270:997–1000 [DOI] [PubMed] [Google Scholar]
- 133. Vaara M. 1992. Eight bacterial proteins, including UDP-N-acetylglucosamine acyltransferase (LpxA) and three other transferases of Escherichia coli, consist of a six-residue periodicity theme. FEMS Microbiol. Lett. 97:249–254 [DOI] [PubMed] [Google Scholar]
- 134. Wyckoff TJO, Raetz CRH. 1999. The active site of Escherichia coli UDP-N-acetylglucosamine acyltransferase. Chemical modification and site-directed mutagenesis. J. Biol. Chem. 274:27047–27055 [DOI] [PubMed] [Google Scholar]
- 135. Robins LI, Williams AH, Raetz CRH. 2009. Structural basis for the sugar nucleotide and acyl-chain selectivity of Leptospira interrogans LpxA. Biochemistry 48:6191–6201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Li C, Guan Z, Liu D, Raetz CRH. 2011. Pathway for lipid A biosynthesis in Arabidopsis thaliana resembling that of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 108:11387–11392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Joo SH, Chung HS, Raetz CRH, Garrett TA. 2012. Activity and crystal structure of Arabidopsis thaliana UDP-N-acetylglucosamine acyltransferase. Biochemistry 51:4322–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kelly TM, Stachula SA, Raetz CRH, Anderson MS. 1993. The firA gene of Escherichia coli encodes UDP-3-O-(R-3-hydroxymyristoyl)-glucosamine N-acyltransferase. The third step of endotoxin biosynthesis. J. Biol. Chem. 268:19866–19874 [PubMed] [Google Scholar]
- 139. Opiyo SO, Pardy RL, Moriyama H, Moriyama EN. 2010. Evolution of the Kdo2-lipid A biosynthesis in bacteria. BMC Evol. Biol. 10:362–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Buetow L, Smith TK, Dawson A, Fyffe S, Hunter WN. 2007. Structure and reactivity of LpxD, the N-acyltransferase of lipid A biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 104:4321–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bartling CM, Raetz CRH. 2009. Crystal structure and acyl chain selectivity of Escherichia coli LpxD, the N-acyltransferase of lipid A biosynthesis. Biochemistry 15:8672–8683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Bartling CM, Raetz CRH. 2008. Steady-state kinetics and mechanism of LpxD, the N-acyltransferase of lipid A biosynthesis. Biochemistry 47:5290–5302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Clementz T, Bednarski JJ, Raetz CRH. 1996. Function of the htrB high temperature requirement gene of Escherchia coli in the acylation of lipid A: HtrB catalyzed incorporation of laurate. J. Biol. Chem. 271:12095–12102 [DOI] [PubMed] [Google Scholar]
- 144. Clementz T, Zhou Z, Raetz CRH. 1997. Function of the Escherichia coli msbB gene, a multicopy suppressor of htrB knockouts, in the acylation of lipid A. J. Biol. Chem. 272:13353–13360 [DOI] [PubMed] [Google Scholar]
- 145. Carty SM, Sreekumar KR, Raetz CRH. 1999. Effect of cold shock on lipid A biosynthesis in Escherichia coli. Induction at 12 degrees C of an acyltransferase specific for palmitoleoyl-acyl carrier protein. J. Biol. Chem. 274:9677–9685 [DOI] [PubMed] [Google Scholar]
- 146. Vorachek-Warren MK, Ramirez S, Cotter RJ, Raetz CRH. 2002. A triple mutant of Escherichia coli lacking secondary acyl chains on lipid A. J. Biol. Chem. 277:14194–14205 [DOI] [PubMed] [Google Scholar]
- 147. Brozek KA, Raetz CRH. 1990. Biosynthesis of lipid A in Escherichia coli. Acyl carrier protein-dependent incorporation of laurate and myristate. J. Biol. Chem. 265:15410–15417 [PubMed] [Google Scholar]
- 148. Goldman RC, Doran CC, Kadam SK, Capobianco JO. 1988. Lipid A precursor from Pseudomonas aeruginosa is completely acylated prior to addition of 3-deoxy-d-manno-octulosonate. J. Biol. Chem. 263:5217–5223 [PubMed] [Google Scholar]
- 149. Mohan S, Raetz CRH. 1994. Endotoxin biosynthesis in Pseudomonas aeruginosa: enzymatic incorporation of laurate before 3-deoxy-d-manno-octulosonate. J. Bacteriol. 176:6944–6951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Tzeng YL, Datta A, Kolli VK, Carlson RW, Stephens DS. 2002. Endotoxin of Neisseria meningitidis composed only of intact lipid A: inactivation of the meningococcal 3-deoxy-d-manno-octulosonic acid transferase. J. Bacteriol. 184:2379–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. 2000. Transfer of palmitate from phospholipids to lipid A in outer membranes of Gram-negative bacteria. EMBO J. 19:5071–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Brozek KA, Bulawa CE, Raetz CRH. 1987. Biosynthesis of lipid A precursors in Escherichia coli. A membrane-bound enzyme that transfers a palmitoyl residue from a glycerophospholipid to lipid X. J. Biol. Chem. 262:5170–5179 [PubMed] [Google Scholar]
- 153. Raetz CRH, Reynolds CM, Trent MS, Bishop RE. 2007. Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 76:295–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Kanipes MI, Lin S, Cotter RJ, Raetz CR. 2001. Ca2+-induced phosphoethanolamine transfer to the outer 3-deoxy-d-manno-octulosonic acid moiety of Escherichia coli lipopolysaccharide. A novel membrane enzyme dependent upon phosphatidylethanolamine. J. Biol. Chem. 12:1156–1163 [DOI] [PubMed] [Google Scholar]
- 155. Ahn VE, Lo EI, Engel CK, Chen L, Hwang PM, Kay LE, Bishop RE, Privé GG. 2004. A hydrocarbon ruler measures palmitate in the enzymatic acylation of endotoxin. EMBO J. 23:2931–2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Evanics F, Hwang PM, Cheng Y, Kay LE, Prosser RS. 2006. Topology of an outer-membrane enzyme: measuring oxygen and water contacts in solution NMR studies of PagP. J. Am. Chem. Soc. 128:8256–8264 [DOI] [PubMed] [Google Scholar]
- 157. Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CRH, Privé GG, Bishop RE, Kay LE. 2002. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc. Natl. Acad. Sci. U. S. A. 99:13560–13565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Bishop RE. 2005. The lipid A palmitoyltransferase PagP: molecular mechanisms and role in bacterial pathogenesis. Mol. Microbiol. 57:900–912 [DOI] [PubMed] [Google Scholar]
- 159. Staunton J, Weissman KJ. 2001. Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 18:380–416 [DOI] [PubMed] [Google Scholar]
- 160. Chopra T, Gokhale RS. 2009. Polyketide versatility in the biosynthesis of complex mycobacterial cell wall lipids. Methods Enzymol. 459:259–294 [DOI] [PubMed] [Google Scholar]
- 161. Brennan PJ, Nikaido H. 1995. The envelope of mycobacteria. Annu. Rev. Biochem. 64:29–63 [DOI] [PubMed] [Google Scholar]
- 162. Daffé M, Draper P. 1998. The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39:131–203 [DOI] [PubMed] [Google Scholar]
- 163. Chavadi SS, Onwueme KC, Edupuganti UR, Jerome J, Chatterjee D, Soll CE, Quadri LEN. 2012. The mycobacterial acyltransferase PapA5 is required for biosynthesis of cell wall-associated phenolic glycolipids. Microbiology 158:1379–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Hatzios SK, Schelle MW, Holsclaw CM, Behrens CR, Botyanszki Z, Lin FL, Carlson BL, Kumar P, Leary JA, Bertozzi CR. 2009. PapA3 is an acyltransferase required for polyacyltrehalose biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 284:12745–12751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kumar P, Schelle MW, Jain M, Lin FL, Petzold CJ, Leavell MD, Leary JA, Cox JS, Bertozzi CR. 2007. PapA1 and PapA2 are acyltransferases essential for the biosynthesis of the Mycobacterium tuberculosis virulence factor sulfolipid-1. Proc. Natl. Acad. Sci. U. S. A. 104:11221–11226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Onwueme KC, Vos CJ, Zurita J, Ferreras JA, Quadri LEN. 2005. The dimycocerosate ester polyketide virulence factors of mycobacteria. Prog. Lipid Res. 44:259–302 [DOI] [PubMed] [Google Scholar]
- 167. Rombouts Y, Alibaud L, Carrère-Kremer S, Maes E, Tokarski C, Elass E, Kremer L, Guérardel Y. 2011. Fatty acyl chains of Mycobacterium marinum lipooligosaccharides. J. Biol. Chem. 286:33678–33688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Xiong Y, Wu X, Mahmud T. 2005. A homologoue of the Mycobacterium tuberculosis PapA5 protein, Rif-Orf20, is an acetyltransferase involved in the biosynthesis of antitubercular drug rifamycin B by Amycolatopsis mediterranei S699. Chembiochem. 6:834–837 [DOI] [PubMed] [Google Scholar]
- 169. Buglino J, Onwueme KC, Ferreras JA, Quadri LE, Lima CD. 2004. Crystal structure of PapA5, a phthiocerol dimycocerosyl transferase from Mycobacterium tuberculosis. J. Biol. Chem. 279:30634–30642 [DOI] [PubMed] [Google Scholar]
- 170. Jogl G, Tong L. 2003. Crystal structure of carnitine acetyltransferase and implications for the catalytic mechanism and fatty acid transport. Cell 112:113–122 [DOI] [PubMed] [Google Scholar]
- 171. Keating TA, Marshall CG, Walsh CT, Keating AE. 2002. The structure of VibH represents nonribosomal peptide synthetase condensation, cyclization and epimerization domains. Nat. Struct. Biol. 9:522–526 [DOI] [PubMed] [Google Scholar]
- 172. Gupta M, Sajid A, Arora G, Tandon V, Singh Y. 2009. Forkhead-associated domain-containing protein Rv0019c and polyketide-associated protein PapA5, from substrates of serine/threonine protein kinase PknB to interacting proteins of Mycobacterium tuberculosis. J. Biol. Chem. 284:34723–34734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Bhatt K, Gurcha SS, Bhatt A, Besra GS, Jacobs WR., Jr 2007. Two polyketide-synthase-associated acyltransferase are required for sulfolipid biosynthesis in Mycobacterium tuberculosis. Microbiology 153:513–520 [DOI] [PubMed] [Google Scholar]
- 174. Rombouts Y, Burguière A, Maes E, Coddeville B, Elass E, Guérardel Y, Kremer L. 2009. Mycobacterium marinum lipooligosaccharides are unique caryophyllose-containing cell wall glycolipids that inhibit tumor necrosis factor-alpha secretion in macrophages. J. Biol. Chem. 284:20975–20988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Rombouts Y, Elass E, Biot C, Maes E, Coddeville B, Burguière A, Tokarski C, Buisine E, Trivelli X, Kremer L, Guérardel Y. 2010. Structural analysis of an unusual bioactive N-acylated lipo-oligosaccharide LOS-IV in Mycobacterium marinum. J. Am. Chem. Soc. 132:16073–16084 [DOI] [PubMed] [Google Scholar]
- 176. Floss HG, Yu TW. 2005. Rifamycin—mode of action, resistance, and biosynthesis. Chem. Rev. 105:621–632 [DOI] [PubMed] [Google Scholar]
- 177. Leslie AGW, Moody PCE, Shaw WV. 1988. Structure of chloramphenicol acetyltransferase at 1.75-Å resolution. Proc. Natl. Acad. Sci. U. S. A. 85:4133–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Murray IA, Shaw WV. 1997. O-Acetyltransferases for chloramphenicol and other natural products. Antimicrob. Agents Chemother. 41:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Schwarz S, Kehrenberg C, Doublet B, Cloeckaert A. 2004. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 28:519–542 [DOI] [PubMed] [Google Scholar]
- 180. Kleanthous C, Cullis PM, Shaw WV. 1985. 3-(Bromoacetyl)chloramphenicol, an active site-directed inhibitor for chloramphenicol acetyltransferase. Biochemistry 24:5307–5313 [DOI] [PubMed] [Google Scholar]
- 181. Lewendon A, Murray IA, Shaw WV. 1994. Replacement of catalytic histidine-195 of chloramphenicol acetyltransferase: evidence for a general base role for glutamate. Biochemistry 33:1944–1950 [DOI] [PubMed] [Google Scholar]
- 182. Kleanthous C, Shaw WV. 1984. Analysis of the mechanism of chloramphenicol acetyltransferase by steady-state kinetics. Evidence for a ternary-complex mechanism. Biochem. J. 223:211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Lewendon A, Murray IA, Klenthous C, CuIlis PM, Shaw WV. 1988. Substitutions in the active-site of chloramphenicol acetyltransferase—role of a conserved aspartate. Biochemistry 27:7385–7390 [DOI] [PubMed] [Google Scholar]
- 184. Shaw WV, Leslie AGW. 1991. Chloramphenicol acetyltransferase. Annu. Rev. Biophys. Biophys. Chem. 20:363–386 [DOI] [PubMed] [Google Scholar]
- 185. Day PJ, Shaw WV. 1992. Acetyl coenzyme A binding by chloramphenicol acetyltransferase. J. Biol. Chem. 267:5122–5127 [PubMed] [Google Scholar]
- 186. Leslie AGW. 1990. Refined structure of type III chloramphenicol acetyltransferase at 1.75 Å resolution. J. Mol. Biol. 213:167–186 [DOI] [PubMed] [Google Scholar]
- 187. Baumann U, Wu S, Flaherty KM, McKay DB. 1993. Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: a two-domain protein with a calcium binding parallel beta roll motif. EMBO J. 12:3357–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Welch RA. 2001. RTX toxin structure and function: a story of numerous anomalies and few analogies in toxin biology. Curr. Top. Microbiol. Immunol. 257:85–111 [DOI] [PubMed] [Google Scholar]
- 189. Šebo P, Ladant D. 1993. Repeat sequences in the Bordetella pertussis adenylate cyclase toxin can be recognized as alternative carboxy-proximal secretion signals by the Escherichia coli α-haemolysin translocator. Mol. Microbiol. 9:999–1009 [DOI] [PubMed] [Google Scholar]
- 190. Stanley P, Koronakis V, Hughes C. 1991. Mutational analysis supports a role for multiple structural features in the C terminal secretion signal of Escherichia coli haemolysin. Mol. Microbiol. 5:2391–2403 [DOI] [PubMed] [Google Scholar]
- 191. Linhartová I, Bumba L, Masín J, Basler M, Osicka R, Kamanová J, Procházková K, Adkins I, Hejnová-Holubová J, Sadílková L, Morová J, Šebo P. 2010. RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 34:1076–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Goebel W, Hedgepeth J. 1982. Cloning and functional characterization of the plasmid-encoded hemolysin determinant of Escherichia coli. J. Bacteriol. 151:1290–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Issartel JP, Koronakis V, Hughes C. 1991. Activation of Escherichia coli prohaemolysin to the mature toxin by acyl carrier protein-dependent fatty acylation. Nature 351:759–761 [DOI] [PubMed] [Google Scholar]
- 194. Schmidt H, Beutin L, Karch H. 1995. Molecular analysis of the plasmid-encoded hemolysin of Escherichia coli O157:H7 strain EDL 933. Infect. Immun. 63:1055–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Glaser P, Ladant D, Sezer O, Pichot F, Ullmann A, Danchin A. 1988. The calmodulin-sensitive adenylate cyclase of Bordetella pertussis: cloning and expression in Escherichia coli. Mol. Microbiol. 2:19–30 [PubMed] [Google Scholar]
- 196. Basar T, Havlicek V, Bezouskova S, Hackett M, Šebo P. 2001. Acylation of lysine 983 is sufficient for toxin activity of Bordetella pertussis adenylate cyclase. Substitutions of alanine 140 modulate acylation site selectivity of the toxin acyltransferase CyaC. J. Biol. Chem. 276:348–354 [DOI] [PubMed] [Google Scholar]
- 197. Lo RY, Strathdee CA, Shewen PE. 1987. Nucleotide sequence of the leukotoxin genes of Pasteurella haemolytica A1. Infect. Immun. 55:1987–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Kuhnert P, Heyberger-Meyer B, Nicolet J, Frey J. 2000. Characterization of PaxA and its operon: a cohemolytic RTX toxin determinant from pathogenic Pasteurella aerogenes. Infect. Immun. 68:6–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Welch RA. 1987. Identification of two different hemolysin determinants in uropathogenic Proteus isolates. Infect. Immun. 55:2183–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Koronakis V, Cross M, Senior B, Koronakis E, Hughes C. 1987. The secreted hemolysins of Proteus mirabilis, Proteus vulgaris, and Morganella morganii are genetically related to each other and to the α-hemolysin of Escherichia coli. J. Bacteriol. 169:1509–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Lally ET, Golub EE, Kieba IR, Taichman NS, Rosenbloom J, Rosenbloom JC, Gibson CW, Demuth DR. 1989. Analysis of the Actinobacillus actinomycetemcomitans leukotoxin gene. Delineation of unique features and comparison to homologous toxins. J. Biol. Chem. 264:15451–15456 [PubMed] [Google Scholar]
- 202. Balashova NV, Shah C, Patel JK, Megalla S, Kachlany SC. 2009. Aggregatibacter actinomycetemcomitans LtxC is required for leukotoxin activity and initial interaction between toxin and host cells. Gene 443:42–47 [DOI] [PubMed] [Google Scholar]
- 203. Frey J, Meier R, Gygi D, Nicolet J. 1991. Nucleotide sequence of the hemolysin I gene from Actinobacillus pleuropneumoniae. Infect. Immun. 59:3026–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Lin L, Bei W, Sha Y, Liu J, Guo Y, Liu W, Tu S, He Q, Chen H. 2007. Construction and immunogenicity of a ΔapxIC/ΔapxIIC double mutant of Actinobacillus pleuropneumoniae serovar 1. FEMS Microbiol. Lett. 274:55–62 [DOI] [PubMed] [Google Scholar]
- 205. Chang YF, Young R, Struck DK. 1989. Cloning and characterization of a hemolysin gene from Actinobacillus (Haemophilus) pleuropneumoniae. DNA 8:635–647 [DOI] [PubMed] [Google Scholar]
- 206. Jansen R, Briaire J, Kamp EM, Gielkens AL, Smits MA. 1993. Cloning and characterization of the Actinobacillus pleuropneumoniae-RTX-toxin III (ApxIII) gene. Infect. Immun. 61:947–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Berthoud H, Frey J, Kuhnert P. 2002. Characterization of Aqx and its operon: the hemolytic RTX determinant of Actinobacillus equuli. Vet. Microbiol. 87:159–174 [DOI] [PubMed] [Google Scholar]
- 208. Angelos JA, Hess JF, George LW. 2003. An RTX operon in hemolytic Moraxella bovis is absent from nonhemolytic strains. Vet. Microbiol. 92:363–377 [DOI] [PubMed] [Google Scholar]
- 209. Basler M, Knapp O, Masin J, Fiser R, Maier E, Benz R, Šebo P, Osicka R. 2007. Segments crucial for membrane translocation and pore-forming activity of Bordetella adenylate cyclase toxin. J. Biol. Chem. 282:12419–12429 [DOI] [PubMed] [Google Scholar]
- 210. Benz R, Maier E, Ladant D, Ullmann A, Šebo P. 1994. Adenylate cyclase toxin (CyaA) of Bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with HlyA of Escherichia coli. J. Biol. Chem. 269:27231–27239 [PubMed] [Google Scholar]
- 211. Cruz WT, Young R, Chang YF, Struck DK. 1990. Deletion analysis resolves cell-binding and lytic domains of the Pasteurella leukotoxin. Mol. Microbiol. 4:1933–1939 [DOI] [PubMed] [Google Scholar]
- 212. Menestrina G, Pederzolli C, Dalla Serra M, Bregante M, Gambale F. 1996. Permeability increase induced by Escherichia coli hemolysin A in human macrophages is due to the formation of ionic pores: a patch clamp characterization. J. Membr. Biol. 149:113–121 [DOI] [PubMed] [Google Scholar]
- 213. Osickova A, Osicka R, Maier E, Benz R, Šebo P. 1999. An amphipathic alpha-helix including glutamates 509 and 516 is crucial for membrane translocation of adenylate cyclase toxin and modulates formation and cation selectivity of its membrane channels. J. Biol. Chem. 274:37644–37650 [PubMed] [Google Scholar]
- 214. Schindel C, Zitzer A, Schulte B, Gerhards A, Stanley P, Hughes C, Koronakis V, Bhakdi S, Palmer M. 2001. Interaction of Escherichia coli hemolysin with biological membranes. A study using cysteine scanning mutagenesis. Eur. J. Biochem. 268:800–808 [DOI] [PubMed] [Google Scholar]
- 215. Šebo P, Glaser P, Sakamoto H, Ullmann A. 1991. High-level synthesis of active adenylate cyclase toxin of Bordetella pertussis in a reconstructed Escherichia coli system. Gene 104:19–24 [DOI] [PubMed] [Google Scholar]
- 216. Westrop G, Hormozi K, da Costa N, Parton R, Coote J. 1997. Structure-function studies of the adenylate cyclase toxin of Bordetella pertussis and the leukotoxin of Pasteurella haemolytica by heterologous C protein activation and construction of hybrid proteins. J. Bacteriol. 179:871–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Ludwig A, Garcia F, Bauer S, Jarchau T, Benz R, Hoppe J, Goebel W. 1996. Analysis of the in vivo activation of hemolysin (HlyA) from Escherichia coli. J. Bacteriol. 178:5422–5430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Stanley P, Packman LC, Koronakis V, Hughes C. 1994. Fatty acylation of two internal lysine residues required for the toxic activity of Escherichia coli hemolysin. Science 266:1992–1996 [DOI] [PubMed] [Google Scholar]
- 219. Stanley P, Koronakis V, Hughes C. 1998. Acylation of Escherichia coli hemolysin: a unique protein lipidation mechanism underlying toxin function. Microbiol. Mol. Biol. Rev. 62:309–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Worsham LMS, Trent MS, Earls L, Jolly C, Ernst-Fonberg ML. 2001. Insights into the catalytic mechanism of HlyC, the internal protein acyltransferases that activates Escherichia coli hemolysin toxin. Biochemistry 40:13607–13616 [DOI] [PubMed] [Google Scholar]
- 221. Stanley P, Hyland C, Koronakis V, Hughes C. 1999. An ordered reaction mechanism for bacterial toxin acylation by the specialized acyltransferases HlyC: formation of a ternary complex with acylACP and protoxin substrates. Mol. Microbiol. 34:887–901 [DOI] [PubMed] [Google Scholar]
- 222. Trent MS, Worsham SMS, Ernst-Fonberg ML. 1998. The biochemistry of hemolysin toxin activation: characterization of HlyC, an internal protein acyltransferase. Biochemistry 37:4644–4652 [DOI] [PubMed] [Google Scholar]
- 223. Lim KB, Walker CRB, Guo L, Pellett S, Shabanowitz J, Hunt DF, Hewlett EL, Ludwig A, Goebel W, Welch RA, Hackett M. 2000. Escherichia coli α-hemolysin (HlyA) is heterogeneously acylated in vivo with 14-, 15-, and 17-carbon fatty acids. J. Biol. Chem. 275:36698–36702 [DOI] [PubMed] [Google Scholar]
- 224. Trent MS, Worsham LMS, Ernst-Fonberg ML. 1999. HlyC, the internal protein acyltransferase that activates hemolysin toxin: roles of various conserved residues in enzymatic activity as probed by site-directed mutagenesis. Biochemistry 38:9541–9548 [DOI] [PubMed] [Google Scholar]
- 225. Worsham LMS, Langston KG, Ernst-Fonberg ML. 2005. Thermodynamics of a protein acylation: activation of Escherichia coli hemolysin toxin. Biochemistry 44:1329–1337 [DOI] [PubMed] [Google Scholar]
- 226. Dodds AW, Ren XD, Willis AC, Law SKA. 1996. The reaction mechanism of the internal thioester in the human complement component C4. Nature 379:177–179 [DOI] [PubMed] [Google Scholar]
- 227. Ren XD, Dodds AW, Enghild JJ, Chu CT, Law SKA. 1995. The effect of residue 1106 on the thioester-mediated covalent binding reaction of human complement protein C4 and the monomeric rat α-macroglobulin α1I3. FEBS Lett. 368:87–91 [DOI] [PubMed] [Google Scholar]
- 228. Basar T, Havlicek V, Bezouskova S, Halada P, Hackett M, Šebo P. 1999. The conserved lysine 860 in the additional fatty acylation site of Bordetella pertussis adenylate cyclase is crucial for toxin function independently of its acylation status. J. Biol. Chem. 274:10777–10783 [DOI] [PubMed] [Google Scholar]
- 229. Hackett M, Guo L, Shabanowitz J, Hunt DF, Hewlett EL. 1994. Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science 266:433–435 [DOI] [PubMed] [Google Scholar]
- 230. Hackett M, Walker CB, Guo L, Gray MC, Van Cuyk S, Ullmann A, Shabanowitz J, Hunt DF, Hewlett EL, Šebo P. 1995. Hemolytic, but not cell-invasive activity, of adenylate cyclase toxin is selectively affected by differential fatty-acylation in Escherichia coli. J. Biol. Chem. 270:20250–20253 [DOI] [PubMed] [Google Scholar]
- 231. Havlicek V, Higgins L, Chen W, Halada P, Šebo P, Sakamoto H, Hackett M. 2001. Mass spectrometric analysis of recombinant adenylate cyclase toxin from Bordetella pertussis strain 18323/pHSP9. J. Mass. Spectrom. 36:384–391 [DOI] [PubMed] [Google Scholar]
- 232. Lally ET, Golub EE, Kieba IR. 1994. Identification and immunological characterization of the domain of Actinobacillus actinomycetemcomitans leukotoxin that determines its specificity for human target cells. J. Biol. Chem. 269:31289–31295 [PubMed] [Google Scholar]
- 233. Jaeger KE, Ransac S, Dijkstra BW, Colson C, van Heuvel M, Misset O. 1994. Bacterial lipases. FEMS Microbiol. Rev. 15:29–63 [DOI] [PubMed] [Google Scholar]
- 234. Kapoor M, Gupta MN. 2012. Lipase promiscuity and its biochemical applications. Process Biochem. 47:555–569 [Google Scholar]
- 235. Nielsen PM, Brask J, Fjerbaek L. 2008. Enzymatic biodiesel production: technical and economical considerations. Eur. J. Lipid Sci. Technol. 110:692–700 [Google Scholar]
- 236. Hasan F, Shah AA, Javed S, Hameed A. 2010. Enzymes used in detergents: lipases. Afr. J. Biotechnol. 9:4836–4844 [Google Scholar]
- 237. Jaeger KE, Eggert T. 2002. Lipases for biotechnology. Curr. Opin. Biotechnol. 13:390–397 [DOI] [PubMed] [Google Scholar]
- 238. Bajaj A, Lohan P, Jha PN, Mehrotra R. 2010. Biodiesel production through lipase catalyzed transesterification: an overview. J. Mol. Catal. B Enzym. 62:9–14 [Google Scholar]
- 239. Jaeger KE, Dijkstra BW, Reetz MT. 1999. Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu. Rev. Microbiol. 53:315–351 [DOI] [PubMed] [Google Scholar]
- 240. Ollis DL, Shea E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A. 1992. The α/β hydrolase fold. Protein Eng. 5:197–211 [DOI] [PubMed] [Google Scholar]
- 241. Schrag JD, Cygler M. 1997. Lipases and α/β hydrolase fold. Methods Enzymol. 284:85–107 [DOI] [PubMed] [Google Scholar]
- 242. Dodson GG, Lawson DM, Winkler FK. 1992. Structural and evolutionary relationships in lipase mechanism and activation. Faraday Discuss. 93:95–105 [DOI] [PubMed] [Google Scholar]
- 243. Rajakumari S, Daum G. 2010. Janus-faced enzymes yeast Tgl3p and Tgl5p catalyze lipase and acyltransferase reactions. Mol. Biol. Cell 21:501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Madison LL, Huisman GW. 1999. Metabolic engineering of poly(3-hydroxyalkanoates): from DNA to plastic. Microbiol. Mol. Biol. Rev. 63:21–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Rehm BHA. 2010. Bacterial polymers: biosynthesis, modifications and applications. Nat. Rev. Microbiol. 8:578–592 [DOI] [PubMed] [Google Scholar]
- 246. Lütke-Eversloh T, Bergander K, Luftmann H, Steinbüchel A. 2001. Identification of a new class of biopolymer: bacterial synthesis of a sulfur-containing polymer with thioester linkages. Microbiology 147:11–19 [DOI] [PubMed] [Google Scholar]
- 247. Lütke-Eversloh T, Steinbüchel A. 2004. Microbial polythioesters. Macromol. Biosci. 4:165–174 [DOI] [PubMed] [Google Scholar]
- 248. Steinbüchel A. 1996. Synthesis and production of biodegradable thermoplastics and elastomers: current state and outlook. Kautschuk Gummi Kunststoffe 49:120–124 [Google Scholar]
- 249. Steinbüchel A. 2005. Non-biodegradable biopolymers from renewable resources: perspectives and impacts. Curr. Opin. Biotechnol. 16:607–613 [DOI] [PubMed] [Google Scholar]
- 250. Shah AA, Hasan F, Hameed A, Ahmed S. 2008. Biological degradation of plastics: a comprehensive review. Biotechnol. Adv. 26:246–265 [DOI] [PubMed] [Google Scholar]
- 251. Hezayen FF, Rehm BHA, Eberhardt R, Steinbüchel A. 2000. Polymer production by two newly isolated extremely halophilic archaea: application of a novel corrosion-resistant bioreactor. Appl. Microbiol. Biotechnol. 54:319–325 [DOI] [PubMed] [Google Scholar]
- 252. Lillo JG, Rodriguez-Valera F. 1990. Effects of culture conditions on poly(β-hydroxybutyric acid) production by Haloferax mediterranei. Appl. Environ. Microbiol. 56:2517–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Schlegel HG, Gottschalk G, Von Bartha R. 1961. Formation and utilization of poly-beta-hydroxybutyric acid by knallgas bacteria (Hydrogenomonas). Nature 191:463–465 [DOI] [PubMed] [Google Scholar]
- 254. Huisman GW, de Leeuw O, Eggink G, Witholt B. 1989. Synthesis of poly-3-hydroxyalkanoates is a common feature of fluorescent pseudomonads. Appl. Environ. Microbiol. 55:1949–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Timm A, Steinbüchel A. 1990. Formation of polyesters consisting of medium-chain-length 3-hydroxyalkanoic acids from gluconate by Pseudomonas aeruginosa and other fluorescent pseudomonads. Appl. Environ. Microbiol. 56:3360–3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Rehm BHA, Krüger N, Steinbüchel A. 1998. A new metabolic link between fatty acid de novo synthesis and polyhydroxyalkanoic acid synthesis. J. Biol. Chem. 273:24044–24051 [DOI] [PubMed] [Google Scholar]
- 257. Rehm BHA. 2003. Polyester synthases: natural catalysts for plastics. Biochem. J. 376:15–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Rehm BHA, Qi QS, Beermann BB, Hinz HJ, Steinbüchel A. 2001. Matrix-assisted in vitro folding of Pseudomonas aeruginosa class II polyhydroxyalkanoate synthase from inclusion bodies produced in recombinant Escherichia coli. Biochem. J. 358:263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Wodzinska J, Snell KD, Rhomberg A, Sinskey AJ, Biemann K, Stubbe J. 1996. Polyhydroxybutyrate synthase: evidence for covalent catalysis. J. Am. Chem. Soc. 118:6319–6320 [Google Scholar]
- 260. Liebergesell M, Sonomoto K, Madkour M, Mayer F, Steinbüchel A. 1994. Purification and characterization of the poly(hydroxyalkanoic acid) synthase from Chromatium vinosum and localization of the enzyme at the surface of poly(hydroxyalkanoic acid) granules. Eur. J. Biochem. 226:71–80 [DOI] [PubMed] [Google Scholar]
- 261. Mayer F, Madkour MH, Pieper-Fürst U, Wieczorek R, Gesell ML, Steinbüchel A. 1996. Electron microscopic observations on the macromolecular organization of the boundary layer of bacterial PHA inclusion bodies. J. Gen. Appl. Microbiol. 42:445–455 [Google Scholar]
- 262. Amara AA, Rehm BHA. 2003. Replacement of the catalytic nucleophile Cys-296 by serine in class II polyhydroxyalkanoate synthase from Pseudomonas aeruginosa-mediated synthesis of a new polyester: identification of catalytic residues. Biochem. J. 374:413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Jia Y, Kappock TJ, Frick T, Sinskey AJ, Stubbe J. 2000. Lipases provide a new mechanistic model for polyhydroxybutyrate (PHB) synthases: characterization of the functional residues in Chromatium vinosum PHB synthase. Biochemistry 39:3927–3936 [DOI] [PubMed] [Google Scholar]
- 264. Qi Q, Rehm BH. 2001. Polyhydroxybutyrate biosynthesis in Caulobacter crescentus: molecular characterization of the polyhydroxybutyrate synthase. Microbiology 147:3353–3358 [DOI] [PubMed] [Google Scholar]
- 265. Rehm BHA, Antonio RV, Spiekermann P, Amara AA, Steinbüchel A. 2002. Molecular characterization of the poly(3-hydroxybutyrate) (PHB) synthase from Ralstonia eutropha: in vitro evolution, site-specific mutagenesis and development of a PHB synthase protein model. Biochim. Biophys. Acta 1594:178–190 [DOI] [PubMed] [Google Scholar]
- 266. Wahab HA, Ahmad Khairudin NB, Samian MR, Najimudin N. 2006. Sequence analysis and structure prediction of type II Pseudomonas sp. USM 4-55 PHA synthase and an insight into its catalytic mechanism. BMC Struct. Biol. 6:23–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Magnuson K, Jackowski S, Rock CO, Cronan JE., Jr 1993. Regulation in fatty acid biosynthesis in Escherichia coli. Microbiol. Rev. 57:522–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Hoffmann N, Steinbüchel A, Rehm BHA. 2000. The Pseudomonas aeruginosa phaG gene product is involved in the synthesis of polyhydroxyalkanoic acid consisting of medium-chain-length constituents from non-related carbon sources. FEMS Microbiol. Lett. 184:253–259 [DOI] [PubMed] [Google Scholar]
- 269. Hoffmann N, Amara AA, Beermann BB, Qi A, Hinz HJ, Rehm BHA. 2002. Biochemical characterization of the Pseudomonas putida 3-hydroxyacyl ACP:CoA transacylase, which diverts intermediates of fatty acid de novo biosynthesis. J. Biol. Chem. 277:42926–42936 [DOI] [PubMed] [Google Scholar]
- 270. Lee HJ, Choi MH, Kim TU, Yoon SC. 2001. Accumulation of polyhydroxyalkanoic acid containing large amounts of unsaturated monomers in Pseudomonas fluorescens BM07 utilizing saccharides and its inhibition by 2-bromooctanoic acid. Appl. Environ. Microbiol. 67:4963–4974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Lee HJ, Rho JK, Noghabi KA, Lee SE, Choi MH, Yoon SC. 2004. Channeling of intermediates derived from medium-chain fatty acids and de novo-synthesized fatty acids to polyhydroxyalkanoic acid by 2-bromooctanoic acid in Pseudomonas fluorescens BM07. J. Microbiol. Biotechnol. 14:1256–1266 [Google Scholar]
- 272. Jones DT, Taylor WR, Thornton JM. 1992. A new approach to protein fold recognition. Nature 358:86–89 [DOI] [PubMed] [Google Scholar]
- 273. Ginalski K. 2006. Comparative modeling of protein structure prediction. Curr. Opin. Struct. Biol. 16:172–177 [DOI] [PubMed] [Google Scholar]
- 274. Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modeling. Bioinformatics 22:195–201 [DOI] [PubMed] [Google Scholar]
- 275. Zheng LZ, Li Z, Tian HL, Li M, Chen GQ. 2005. Molecular cloning and functional analysis of (R)-3-hydroxyacyl-acyl carrier protein:coenzyme A transacylase from Pseudomonas mendocina LZ. FEMS Microbiol. Lett. 252:299–307 [DOI] [PubMed] [Google Scholar]
- 276. Braaz R, Handrick R, Jendrossek D. 2003. Identification and characterisation of the catalytic triad of the alkaliphilic thermotolerant PHA depolymerase PhaZ7 of Paucimonas lemoignei. FEMS Microbiol. Lett. 224:107–112 [DOI] [PubMed] [Google Scholar]
- 277. Derewenda ZS, Derewenda U. 1991. Relationships among serine hydrolases—evidence for a common structural motif in triacylglyceride lipases and esterases. Biochem. Cell Biol. 69:842–851 [DOI] [PubMed] [Google Scholar]
- 278. Lee LC, Lee YL, Leu RJ, Shaw JF. 2006. Functional role of catalytic triad and oxyanion hole-forming residues on enzyme activity of Escherichia coli thioesterases I/protease I/phospholipase L1. Biochem. J. 397:69–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Natarajan S, Kim JK, Jung TK, Doan TTN, Ngo HPT, Hong MK, Kim S, Tan VP, Ahn SJ, Lee SH, Han Y, Ahn YJ, Kang LW. 2012. Crystal structure of malonyl CoA-acyl carrier protein transacylase from Xanthomonas oryzae pv. oryzae and its proposed binding with ACP. Mol. Cells 33:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Serre L, Verbree EC, Dauter Z, Stuitje AR, Derewenda ZS. 1995. The Escherichia coli malonyl-CoA:acyl carrier protein transacylase at 1.5-Å resolution. J. Biol. Chem. 270:12961–12964 [DOI] [PubMed] [Google Scholar]
- 281. Zhang L, Liu WL, Xiao J, Hu T, Chen J, Chen K, Jiang H, Shen X. 2007. Malonyl-CoA:acyl carrier protein transacylase from Helicobacter pylori: crystal structure and its interaction with acyl carrier protein. Protein Sci. 16:1184–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Zheng Z, Chen JC, Tian HL, Bei FF, Chen GQ. 2005. Specific identification of (R)-3-hydroxyacyl-ACP:CoA transacylase gene from Pseudomonas and Burkholderia strains by polymerase chain reaction. Chin. J. Biotechnol. 21:19–24 [PubMed] [Google Scholar]
- 283. Hoffmann N, Steinbüchel A, Rehm BHA. 2000. Homologous functional expression of cryptic phaG from Pseudomonas oleovorans established the transacylase-mediated polyhydroxyalkanoate biosynthetic pathway. Appl. Microbiol. Biotechnol. 54:665–670 [DOI] [PubMed] [Google Scholar]
- 284. Matsumoto K, Matsusaki H, Taguchi S, Seki M, Doi Y. 2001. Cloning and characterization of the Pseudomonas sp. 61-3 phaG gene involved in polyhydroxyalkanoate biosynthesis. Biomacromolecules 2:142–147 [DOI] [PubMed] [Google Scholar]
- 285. Rodrigues MF, Valentin HE, Berger PA, Tran M, Asrar J, Gruys KJ, Steinbüchel A. 2000. Polyhydroxyalkanoate accumulation in Burkholderia sp.: a molecular approach to elucidate the genes involved in the formation of two homopolymers consisting of short-chain-length 3-hydroxyalkanoic acids. Appl. Microbiol. Biotechnol. 53:453–460 [DOI] [PubMed] [Google Scholar]
- 286. Hang X, Zhang G, Wang G, Zhao X, Chen GQ. 2002. PCR cloning of polyhydroxyalkanoate biosynthesis genes from Burkholderia caryophylli and their functional expression in recombinant Escherichia coli. FEMS Microbiol. Lett. 210:49–54 [DOI] [PubMed] [Google Scholar]
- 287. Fiedler S, Steinbüchel A, Rehm BHA. 2000. PhaG-mediated synthesis of poly(3-hydroxyalkanoates) consisting of medium-chain-length constituents from nonrelated carbon sources in recombinant Pseudomonas fragi. Appl. Environ. Micbrobiol. 66:2117–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Tobin KM, O'Leary NDO, Dobson ADW, O'Connor KE. 2007. Effect of heterologous expression of phaG [(R)-3-hydroxyacyl-ACP-CoA transferase] on polyhydroxyalkanoate accumulation from the aromatic hydrocarbon phenylacetic acid in Pseudomonas species. FEMS Microbiol. Lett. 268:9–15 [DOI] [PubMed] [Google Scholar]
- 289. Abdel-Mawgoud AM, Lépine F, Déziel E. 2010. Rhamnolipids: diversity of structures, microbial origins and roles. Appl. Microbiol. Biotechnol. 86:1323–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Lang S, Wullbrandt D. 1999. Rhamnose lipids—biosynthesis, microbial production and application potential. Appl. Microbiol. Biotechnol. 51:22–32 [DOI] [PubMed] [Google Scholar]
- 291. Beal R, Betts WB. 2000. Role of rhamnolipid biosurfactants in the uptake and mineralization of hexadecane in Pseudomonas aeruginosa. J. Appl. Microbiol. 89:158–168 [DOI] [PubMed] [Google Scholar]
- 292. Hommel R. 1994. Formation and function of biosurfactants for degradation of water-insoluble substrates, p 63–87 In Ratledge C. (ed), Biochemistry of microbial degradation. Kluwer Academic Publishers, London, United Kingdom [Google Scholar]
- 293. Noordman WH, Janssen DB. 2002. Rhamnolipid stimulates uptake of hydrophobic compounds by Pseudomonas aeruginosa. Appl. Environ. Microbiol. 68:4502–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Déziel E, Lépine F, Milot S, Villemur R. 2003. rhlA is required for the production of a novel biosurfactant promoting swarming motility in Pseudomonas aeruginosa: 3-(3-hydroxyalkanoyloxy)alkanoic acids (HAAs), the precursors of rhamnolipids. Microbiology 149:2005–2013 [DOI] [PubMed] [Google Scholar]
- 295. Murray TS, Kazmierczak BI. 2008. Pseudomonas aeruginosa exhibits sliding motility in the absence of type IV pili and flagella. J. Bacteriol. 190:2700–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Al-Tahhan RA, Sandrin TR, Bodour AA, Maier RM. 2000. Rhamnolipid induced removal of lipopolysaccharide from Pseudomonas aeruginosa: effect on cell surface properties and interaction with hydrophobic substrates. Appl. Environ. Microbiol. 66:3262–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Sotirova AV, Spasova DI, Galabova DN, Karpenko E, Shulga A. 2008. Rhamnolipid-biosurfactant permeabilizing effects on gram-positive and gram-negative bacterial strains. Curr. Microbiol. 56:639–644 [DOI] [PubMed] [Google Scholar]
- 298. Van Gennip M, Christensen LD, Alhede M, Phipps R, Jensen PO, Christophersen L, Pamp SJ, Moser C, Mikkelsen PJ, Koh AY, Tolker-Nielsen T, Pier GB, Hoiby N, Givskov M, Bjarnsholt T. 2009. Inactivation of the rhlA gene in Pseudomonas aeruginosa prevents rhamnolipid production, disabling the protection against polymorphonuclear leukocytes. Apmis 117:537–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Zulianello L, Canard C, Köhler T, Caille D, Lacroix JS, Meda P. 2006. Rhamnolipids are virulence factors that promote early infiltration of primary human airway epithelia by Pseudomonas aeruginosa. Infect. Immun. 74:3134–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Rehm BHA, Mitsky TA, Steinbüchel A. 2001. Role of fatty acid de novo biosynthesis in polyhydroxyalkanoic acid (PHA) and rhamnolipids synthesis by pseudomonads: establishment of the transacylase (PhaG)-mediated pathway for PHA biosynthesis in Eschericia coli. Appl. Environ. Microbiol. 67:3102–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Choi MH, Xu J, Gutierrez M, Yoo T, Cho YH, Yoon SC. 2011. Metabolic relationship between polyhydroxyalkanoic acid and rhamnolipid synthesis in Pseudomonas aeruginosa: comparative 13C NMR analysis of the products in wild-type and mutants. J. Biotechnol. 151:30–42 [DOI] [PubMed] [Google Scholar]
- 302. Déziel E, Lépine F, Dennie D, Boismenu D, Mamer OA, Villemur R. 1999. Liquid chromatography/mass spectrometry analysis of mixtures of rhamnolipids produced by Pseudomonas aeruginosa strain 57RP grown on mannitol or naphthalene. Biochim. Biophys. Acta 1440:244–252 [DOI] [PubMed] [Google Scholar]
- 303. Zhu K, Rock CO. 2008. RhlA converts β-hydroxyacyl-acyl carrier protein intermediates in fatty acid synthesis to the β-hydroxydecanoyl-β-hydroxydecanoate component of rhamnolipids in Pseudomonas aeruginosa. J. Bacteriol. 190:3147–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Ochsner UA, Fiechter A, Reiser J. 1994. Isolation, characterization, and expression in Escherichia coli of the Pseudomonas aeruginosa rhlAB genes encoding a rhamnosyltransferase involved in rhamnolipid biosurfactant synthesis. J. Biol. Chem. 269:19787–19795 [PubMed] [Google Scholar]
- 305. Rahim R, Ochsner UA, Olvera C, Graninger M, Messner P, Lam JS, Soberón-Chávez G. 2001. Cloning and functional characterization of the Pseudomonas aeruginosa rhlC gene that encodes rhamnosyltransferase 2, an enzyme responsible for di-rhamnolipid biosynthesis. Mol. Microbiol. 40:708–718 [DOI] [PubMed] [Google Scholar]
- 306. Campos-Garcia J, Caro AD, Najera R, Miller-Maier RM, Al-Tahhan RA, Soberon-Chavez G. 1998. The Pseudomonas aeruginosa rhlG gene encodes an NADPH-dependent β-ketoacyl reductase which is specifically involved in rhamnolipid synthesis. J. Bacteriol. 180:4442–4451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Miller DJ, Zhang YM, Rock CO, White SW. 2006. Structure of RhlG, an essential β-ketoacyl reductase in the rhamnolipid biosynthetic pathway of Pseudomonas aeruginosa. J. Biol. Chem. 281:18025–18032 [DOI] [PubMed] [Google Scholar]
- 308. Pham TH, Webb JS, Rehm BHA. 2004. The role of polyhydroxyalkanoate biosynthesis by Pseudomonas aeruginosa in rhamnolipid and alginate production as well as stress tolerance and biofilm formation. Microbiology 150:3405–3413 [DOI] [PubMed] [Google Scholar]
- 309. Müller MM, Hausmann R. 2011. Regulatory and metabolic network of rhamnolipid biosynthesis: traditional and advanced engineering towards biotechnological production. Appl. Microbiol. Biotechnol. 91:251–264 [DOI] [PubMed] [Google Scholar]
- 310. Medina G, Juarez K, Valderrama B, Soberón-Chávez G. 2003. Mechanism of Pseudomonas aeruginosa RhlR transcriptional regulation of the rhlAB promoter. J. Bacteriol. 185:5976–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Soberón-Chavez G, Lépine F, Déziel E. 2005. Production of rhamnolipids by Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 68:718–725 [DOI] [PubMed] [Google Scholar]
- 312. Turchetto-Zolet AC, Maraschin F, de Morais GL, Cagliari A, Andrade CMB, Margis-Pinheiro M, Margis R. 2011. Evolutionary view of acyl-CoA diacylglycerol acyltransferase (DGAT), a key enzyme in neutral lipid biosynthesis. BMC Evol. Biol. 11:263 doi: 10.1186/1471-2148-11-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.