

Abstract
Some solid tumors have reduced posttranscriptional RNA editing by adenosine deaminase acting on RNA (ADAR) enzymes, but the functional significance of this alteration has been unclear. Here, we found the primary RNA-editing enzyme ADAR1 is frequently reduced in metastatic melanomas. In situ analysis of melanoma samples using progression tissue microarrays indicated a substantial downregulation of ADAR1 during the metastatic transition. Further, ADAR1 knockdown altered cell morphology, promoted in vitro proliferation, and markedly enhanced the tumorigenicity in vivo. A comparative whole genome expression microarray analysis revealed that ADAR1 controls the expression of more than 100 microRNAs (miRNAs) that regulate many genes associated with the observed phenotypes. Importantly, we discovered that ADAR1 fundamentally regulates miRNA processing in an RNA binding–dependent, yet RNA editing–independent manner by regulating Dicer expression at the translational level via let-7. In addition, ADAR1 formed a complex with DGCR8 that was mutually exclusive with the DGCR8-Drosha complex that processes pri-miRNAs in the nucleus. We found that cancer cells silence ADAR1 by overexpressing miR-17 and miR-432, which both directly target the ADAR1 transcript. We further demonstrated that the genes encoding miR-17 and miR-432 are frequently amplified in melanoma and that aberrant hypomethylation of the imprinted DLK1-DIO3 region in chromosome 14 can also drive miR-432 overexpression.
Introduction
Genetic alterations and dysregulated epigenetic modifications play a role in cancer development and progression (1). Recently, posttranscriptional events, such as perturbation of microRNA (miRNA) expression, are emerging as new players in the development of various human malignancies (2). A-to-I RNA editing is a posttranscriptional process mediated by the adenosine deaminase acting on RNA (ADAR) enzymes, which catalyze nucleotide conversion in RNA transcripts. It was previously reported that many solid tumors generally exhibit lower A-to-I RNA editing (3), but the mechanistic and functional significance has remained obscure.
The deamination of adenosines by ADAR enzymes may occur in coding regions, altering the amino acid sequence, codon reading frames, and splicing pattern (4), or in the noncoding regions, thus affecting the stability of RNA transcripts or their nuclear retention (5). There are 3 members of the ADAR family, ADAR1, ADAR2, and the brain-specific ADAR3 (5). The ubiquitously expressed ADAR1 has 2 isoforms: an interferon-inducible cytoplasmic protein (ADAR1-long, P150) and a constitutive nuclear protein (ADAR1-short, P110), which are synthesized from different translation-initiating methionines (6, 7).
Editing by ADAR1 has been implicated in different physiological processes (6–8), such as embryonic hematopoiesis (9, 10) and development of various non-nervous tissues and in host defense against viral infections by massive hyperediting of viral transcripts (4). In addition, editing of regulatory RNAs (pri- and pre-miRNAs) by ADAR1 has been reported (11). Editing of pri-miRNAs by ADAR1 may interfere with miRNA biogenesis at the precursor stage and thereby alter their homeostasis in the cell (12) or target binding (13). An RNA editing–independent regulation of miR-376a2 by ADAR2 was previously reported (13).
Here, we present what we believe is substantial new evidence that ADAR1 has a fundamental role in the regulation of cancer cell phenotype by controlling nuclear and cytoplasmic processing steps of miRNAs in an isoform-specific, RNA binding–dependent yet RNA editing–independent manner. Moreover, we unravel the underlying genomic and epigenetic events that are responsible for loss of ADAR1 expression in metastatic melanoma cells, which subsequently facilitate the acquisition of an aggressive phenotype. These findings provide insights into the process of cancer development, with potential implications for future translational medicine.
Results
ADAR1 is frequently downregulated in metastatic melanoma cells.
A significant hypoediting effect was reported mainly in brain tumors (3). Since melanocytes are derived from the neural crest, the expression of ADAR1 was investigated in situ in melanoma by using progression tissue microarray (TMA) obtained from National Cancer Institute Cancer Diagnosis Program (NCI CDP). ADAR1 was heterogeneously expressed in the epidermis of normal skin (control), nevi, primary melanomas, and metastasis (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI62980DS1). Staining was mainly observed in the nucleus, which is consistent with the constitutive short form of ADAR1 (ADAR1-P110) (5). There were no significant differences in the nuclear staining among normal epidermis, benign nevi, and primary melanoma specimens (Figure 1A). In contrast, a significantly weaker nuclear staining was observed in the metastatic specimens (Figure 1A). There was almost no cytoplasmic staining in the normal epidermis, as the long form of ADAR1 (ADAR1-P150) is inducible (5). Interestingly, nevi and primary melanoma specimens exhibited a moderate cytoplasmic staining (Figure 1A). While weak cytoplasmic staining could be observed in some metastatic specimens, it was not different in a statistically significant manner from the normal epidermis (Figure 1A). A significant trend toward downregulation of nuclear and cytoplasmic ADAR1 immunoreactivity in metastatic specimens was independently observed in another progression TMA obtained from Massachusetts General Hospital (Supplemental Figure 1, see Methods). There was no significant association between ADAR1 expression level and the BRAF (v-raf murine sarcoma viral oncogene homolog B1: ID 673) or NRAS (neuroblastoma RAS viral [v-ras] oncogene homolog: ID 4893) mutation status (data not shown).
Figure 1. Reduced ADAR1 expression is a common event in melanoma.

(A) Analysis of the immunohistochemical expression levels of ADAR1. Lines in the boxes denote the mean. (B) ADAR1 expression in low-passage metastatic melanoma cultures, as determined by qPCR. Results are expressed as fold above the average values in all normal melanocyte specimens. Cutoffs for overexpression and downregulation were determined as greater than 2 and less than 0.5, respectively. Data represent the mean ± SEM of 2 experiments on independent RNA purifications, each performed in triplicate. (C) ADAR1 expression at the protein level of selected low-passage melanoma cultures (samples A–G in part B), as determined by Western blot. A representative blot is shown. (D) ADAR1 expression at the protein level as determined by Western blot and (E) A-to-I editing rate of BLCAP by ADAR1 as determined by Sequenom MassArray in ADAR1-manipulated 624mel cell system: stable KD of ADAR1 (ADAR1-KD), rescue of ADAR1-P150 or ADAR1-P110 (rescue-P150, rescue-P110, respectively), and transfection with scrambled sequence and empty pcDNA3 (control). Data represent the mean ± SEM of 3 independent experiments, each performed in triplicate. **P < 0.01 (2-tailed t test, ANOVA); ***P < 0.0001 (ANOVA).
In order to quantify the downregulation of ADAR1 in metastatic cells, 34 low-passage patient-derived metastatic melanoma cultures and 4 cultures of normal melanocytes were tested with real-time quantitative PCR (qPCR). ADAR1 was downregulated by at least 2-fold in 62.5% of the metastatic melanoma samples (Figure 1B). ADAR1 levels were almost identical among all cultures of normal melanocytes (Figure 1B). The ratio between ADAR1-P110 and ADAR1-P150 was between 4–12 times in favor of ADAR1-P110 in all melanoma cultures (data not shown). ADAR1 expression levels in selected melanoma cultures as tested by Western blot were in agreement with the qPCR results (Figure 1C).
To study the effect of ADAR1 silencing, 624mel melanoma cells were stably transduced with shRNA that targets both the short and long forms of ADAR1. The transductants were then stably transfected with ADAR1-P110 or ADAR1-P150 bearing silent mutations conferring resistance to the ADAR1-selective shRNA to restore specifically their expression or with an empty pcDNA3 vector (rescue-P110, rescue-P150, and ADAR1-knockdown [ADAR1-KD] and control, respectively) (Figure 1D). Accordingly, reduced editing rate of bladder cancer–associated protein (BLCAP; ID:10904) transcripts, a verified editing target of ADAR1 (14), was observed in the ADAR1-KD cells (9%) as compared with the rescue-P150, rescue-P110, and control cells (18%, 18%, and 19%, respectively) (Figure 1E). The expression level of ADAR2 was similar among the various cells, while the brain-specific ADAR3 was undetected in all melanoma samples tested (data not shown).
ADAR1 controls the aggressiveness of melanoma cells in vitro and in vivo.
Light microscopy revealed distinct morphology of the KD cells (elongated cells with dendrites) when compared with the rescue-P110, rescue-P150, and control cells (short and wide cells) (Figure 2A). Digital image analysis confirmed that ADAR1-KD cells exhibit significantly longer and narrower cell bodies as compared with the rescue and control cells (Supplemental Figure 2).
Figure 2. Reduced expression of ADAR1 enhances cancerous features both in vivo and in vitro.

(A) Morphology of the cells was assessed using a phase-contrast microscope. Original magnification, ×20; scale bar: 100 μm. (B) Net proliferation was monitored by standardized XTT assay in 24-hour intervals for 72 hours. (C) Cells were plated and after 48 hours DNA was extracted, fixed, stained with PI and subsequently analyzed for cell cycle by flow cytometry. (D) Monitoring of tumor growth in SCID-NOD mice (each group comprised 7 mice). (E) Tumors were documented (scale bars: 2 cm) and their paraffin-embedded tissue sections were immunostained with Ki-67 proliferation marker (scale bars: 100 μm). The extent of necrotic area (scale bars: 200 μm) was estimated. Representative tumors are shown. (F) Expression of ADAR1 was confirmed upon termination of the experiment in all tumors with qPCR. Data for A–C represent the mean ± SEM of 3 independent experiments, each performed in triplicate. Results of D–F are of a representative experiment out of 3 performed. *P < 0.05; **P < 0.01; ***P < 0.001 (2-tailed t test).
A remarkably enhanced net proliferation rate following ADAR1-KD was observed in the 624mel cells (Figure 2B) and in 6 other melanoma cell lines tested (Supplemental Figure 3). This effect was corroborated by a significant increase in the population of proliferating cells (G2-M) at the expense of resting cells (GO-G1) in the ADAR1-KD cells as compared with rescue and control cells (Figure 2C). On the other hand, loss of ADAR1 expression had no effect on spontaneous apoptosis (Figure 2C, Sub-G1). Further, there was no effect on the rate of apoptosis induced by different mechanisms, such as interference to microtubule breakdown (Taxol) or DNA crosslinking (Cisplatin) (Supplemental Figure 4). Similar effects on morphology, RNA-editing rate, and proliferation were observed with another system, independently established in another melanoma cell line, 526mel (Supplemental Figure 5). The combined effects suggest that ADAR1 has a fundamental role in the regulation of cancerous features and phenotype.
The various transfectants were injected subcutaneously into SCID-NOD mice. All developed into tumor masses, but ADAR1-KD tumors exhibited a dramatically enhanced growth rate in vivo (Figure 2D). After 45 days, tumors were excised and analyzed macroscopically, histologically, and at the molecular level. Macroscopically, the ADAR1-KD tumors were substantially larger (Figure 2E) and heavier (data not shown), supporting the volume measurements (Figure 2D). Immunostaining for the proliferation marker Ki-67 confirmed a considerable increase in cell proliferation in the ADAR1-KD tumors (Figure 2E). Most proliferating cells were located at the periphery, with extensive central necrosis that was considerably larger in the ADAR1-KD tumors (Figure 2E). Explanted tumors maintained their original ADAR1 expression level (Figure 2F). There were no statistically significant differences between the rescue-P110 and rescue-P150 cells. The in vitro and in vivo observations (Figure 2) concur and strongly suggest that the downregulation of ADAR1 observed in metastatic melanoma promotes tumor growth in vivo by facilitation of cell division and proliferation and not by enhancing resistance to apoptosis.
ADAR1 exerts broad cell-regulation function independently of RNA-editing activity.
A comparative whole-genome expression microarray analysis of ADAR1-KD and control cells revealed 702 differentially expressed genes (DEGs) (Supplemental Table 1). Ten DEGs with key roles in cancer development were validated at the RNA level, and 2 were further verified at the protein level (Supplemental Table 2 and Supplemental Figure 6A). Analysis of the gene clusters showed that “cell growth and proliferation”, “morphology”, and “cell cycle” were among the top 5 most significantly altered clusters (Figure 3A). This concurs with the phenotype demonstrated above (Figure 2). However, RNA-editing site analysis (15) in the DEGs revealed that only 5% (35/702) include putative A-to-I editing sites (Supplemental Table 1), similar to the predictions for the total genome (16). We therefore hypothesized that ADAR1-mediated regulation of the cancerous phenotype is mostly independent of RNA editing.
Figure 3. ADAR1-dependent regulation of proliferation is RNA-editing independent.

(A) Ten functional clusters, determined by IPA, that were significantly affected by ADAR1 downregulation are shown. The significance was calculated by Fisher’s exact test, and is expressed as –log (P value). The number of DEGs that map to a specific pathway is indicated at the top of each column. (B) ADAR1 constructs used for functional assays. Shown are ADAR1-P110, ADAR1-P150, and His-tagged dsRBD, Z-DBD-P110, Z-DBD-P150, ΔCAT-P110, ΔCAT-P150, CAT-MUT-P110, and CAT-MUT-P150. ADAR1 fragments amplified and cloned into pCDNA3 (“truncations”). (C) The His-tagged ADAR1 domains were transfected into 624mel cells, and their expression, relative to mock-control, was detected by intracellular staining in flow cytometry; the impact of “truncation” and overexpression constructs on (D) ADAR1 expression in transductants was determined by qPCR. (E) BLCAP editing rate by ADAR1 was examined by sequenom massarray. (F) Net proliferation was monitored by standardized XTT assay. The number of cells was determined 48 hours after seeding. (G) Cells were plated and after 48 hours DNA was extracted, stained with PI, and subsequently analyzed for cell cycle by flow cytometry. Data represent the mean ± SEM of 3 independent experiments, each performed in triplicate. *P < 0.05; **P < 0.01; ***P < 0.001 (2-tailed t test).
Several His-tagged ADAR1-modified constructs were generated: ADAR1-P150 and ADAR1-P110 bearing the previously described point mutations in the catalytic site H910Y-E912A (17) (CAT-MUT-P150 and CAT-MUT-P110) or devoid of the deamination domain (ΔCAT-P150 and ΔCAT-P110) and isolated catalytic domain (CAT), dsRNA binding domains (dsRBD), or Z-DNA binding domains (Z-DBD-P150 and Z-DBD-P110) (Figure 3B). 624mel cells were stably transfected with all the various constructs or with an empty vector (mock). All His-tagged constructs were expressed at similar levels, as confirmed by intracellular staining with anti-His antibodies (Figure 3C) or with Western blot (data not shown). In addition, all transfectants were tested for endogenous ADAR1 expression level, RNA-editing capacity, and proliferation rate. Mock cells served as negative control and the ADAR1-P150 and ADAR1-110 cells served as positive controls. Importantly, endogenous ADAR1 expression and RNA-editing rate of BLCAP remained similar in all transfectants, except for positive controls (Figure 3, D and E). Concurring with the KD experiments, overexpression of ADAR1-P150 and ADAR1-P110 inhibited melanoma cell proliferation (Figure 3F). Supporting our hypothesis, a similar inhibitory effect was indeed observed in the CAT-MUT-P150, CAT-MUT-P110, ΔCAT-P150, and ΔCAT-P110 cells (Figure 3F). Inhibition of proliferation was corroborated by accumulation of cells with G1 DNA content in cell-cycle analysis, indicative of reduced proliferation (Figure 3G). In addition, ADAR1-P150, ADAR1-P110, CAT-MUT-P150, CAT-MUT-P110, ΔCAT-P150, and ΔCAT-P110 transfectants exhibited similar expression of selected genes (Supplemental Figure 6B). In conclusion, these experiments show that ADAR1 regulates gene expression and proliferation of melanoma cells independently of RNA editing.
ADAR1 controls cancer features by regulation of miRNA expression profile.
The DEGs could not be ascribed to single specific pathways, thus implying that ADAR1 may operate via broad cellular regulators, such as miRNAs. Comparative miRNA expression profiles of ADAR1-KD and control cells demonstrated that the expression of 131 miRNAs (Supplemental Table 3) was significantly changed (defined as >2- or <0.5-fold change), of which 53% (70/131) were already known as oncogenic or tumor-suppressive miRNAs (Supplemental Table 3). Remarkably, KD of ADAR1 increased the expression of 89% (25/28) of the known oncogenic miRNAs and reduced 86% (36/42) of the tumor suppressor miRNAs (Figure 4A). Bioinformatics did not reveal statistically significant differences in the predicted structure of these groups of miRNAs (18, 19). Targets for the 131 miRNAs were predicted with TargetScan 5.1 (20) and crossed with the 702 DEGs. Since miRNAs are expected to suppress gene expression, we disregarded all predicted target genes that were not expressed inversely to the corresponding miRNA. This analysis suggested that the 131 miRNAs target at least 38% of the DEGs (264/702) (Supplemental Table 1).
Figure 4. Reduced ADAR1 expression facilitates malignant activity via miRNAs.

(A) Classification of 131 altered miRNAs into 3 functional groups: oncogenic, tumor suppressors, and unknown, as indicated. The number of miRNAs (n) that map to each group is indicated. The significance was calculated by Fisher’s exact test. (B) Comparison between the expression level (fold change) of miR-21 and miR-34a in ADAR1-KD and control cells using qPCR; ADAR1-KD cell line was (C) transiently transfected with anti-miR-21 oligo (gray squares) or with control–anti-miRNA oligo (black circles) and (D) with miR-34a overexpression plasmid (gray squares) or mock plasmid (black circles). Net proliferation was monitored 24 and 48 hours after seeding by standardized XTT assay. (E) Verification of miR-21 and ADAR1 expression levels in anti–miR-21–transfected ADAR1-KD cells (gray bars) as compared with control oligo (black bars) by qPCR. (F) Verification of miR-34a and ADAR1 expression in miR-34a overexpression plasmid–transduced ADAR1-KD cells (gray bars) as compared with mock (black bars) by qPCR. *P < 0.05; **P < 0.01 (2-tailed t test). Data represent the mean ± SEM of 3 independent experiments, each performed in triplicate.
In order to test the effects of ADAR1-regulated miRNAs on cell features, we focused on 2 representative miRNAs in ADAR1-KD cells: miR-21, an oncogenic miRNA (21) that was upregulated (Figure 4B), and miR-34a, a tumor-suppressive miRNA (22) that was downregulated (Figure 4B). ADAR1-KD cells were transiently transfected with anti–miR-21 oligonucleotide or transfected with miR-34a–encoding vector. The appropriate control oligonucleotides or empty vector, respectively, served as negative controls. Reduction in miR-21 expression or enhanced miR-34a expression caused a remarkable decrease in proliferation rate of ADAR1-KD cells (Figure 4, C and D). The expression of ADAR1 remained unchanged following these manipulations (Figure 4, E and F). In conclusion, ADAR1-controlled expression of miRNAs regulates melanoma cell proliferation.
ADAR1 regulates processing of miRNAs in an RNA-editing independent manner.
ADAR1 alters the expression of many miRNAs (Supplemental Table 3), which might shape the metastatic phenotype. It was previously shown that RNA editing of specific pri-miRNA transcripts by ADAR1 affects their biogenesis (11). Here, we hypothesized that ADAR1 regulates miRNA biogenesis independently of RNA editing. So far, such a mechanism was known specifically for miR-376a2 by ADAR2 (13).
We evaluated the expression of pri-miRNA, pre-miRNA, and mature miRNA of miR-21 and miR-34a. The expression of the mature miR-21 was significantly increased in ADAR1-KD cells, while the expression of the pre- and pri-miRNA transcripts was extremely low, as compared with control cells (Figure 5A). The rescue-P110 and rescue-P150 cells displayed an accumulation of either the pri-miRNA or the pre-miRNA transcripts, respectively, with subsequent decrease in the mature miR-21 expression (Figure 5A). miR-34a presented the exact opposite response, as the expression of the mature miRNA decreased in response to KD of ADAR1 expression (Figure 5B). These results strongly suggest that ADAR1 isoforms affect the processing of miRNAs at different steps that coincide with their previously reported subcellular localization. Indeed, ADAR1-110 and Drosha are expressed in the nucleus, while ADAR1-P150 and Dicer are expressed in the cytoplasm (23).
Figure 5. ADAR1 regulates the biogenesis process of miRNAs independently of RNA-editing.

Expression of all miR-21 (A) and miR-34a (B) formats in ADAR1-KD and rescue constructs, as indicated in the figure, was determined by qPCR. Expression of all miR-21 (C) and miR-34a (D) formats in ADAR1 “truncations” system was determined by qPCR. Expression of pre- and mature miR-21 (E) and pre- and mature miR-34a (F) formats in ADAR1 in selected “truncation” constructs was determined by Northern blot. *P < 0.05; #P < 0.01; §P < 0.001 (2-tailed t test). Data represent the mean ± SEM of 3 independent experiments, each performed in triplicate in 3 independent cell systems.
The effects of ADAR1 on miRNA biogenesis were evaluated in the ADAR1 “truncations” system (Figure 3). As expected, overexpression of full-length ADAR1-P110 or ADAR1-P150 similarly decreased the expression of mature miR-21 and increased the expression of mature miR-34a (Figure 5, C and D). However, in line with the results described above, overexpression of ADAR1-P110 resulted in markedly increased pri-miR-21 and decreased pri-miR-34a, while overexpression of ADAR1-P150 affected the corresponding pre-miRNA forms, accordingly (Figure 5, C and D). Importantly, the ΔCAT-P110, ΔCAT-150, CAT-MUT-P110, and CAT-MUT-P150 cells exerted exactly the same effects as their wild-type counterparts (Figure 5, C and D), thus attesting that the regulation of miRNA biogenesis by ADAR1 is mediated mostly via an RNA editing–independent mechanism. The isolated dsRNA-binding domain or Z-DNA–binding domain did not have any significant effect (Figure 5, C and D). These observations were confirmed in Northern blot analyses for mature miRNAs 21 and 34a, as well as for the pre-miR-21 (Figure 5, E and F). The pre-miR-34a was only moderately altered in Northern blot analyses, probably owing to the lesser sensitivity of this method.
These observations suggest that ADAR1 controls the miRNA processing machinery itself. The effect of ADAR1 on 3 key proteins involved in miRNA processing, DGCR8 (DiGeorge syndrome critical region gene 8; ID 54487), Drosha and Dicer, was tested. KD of ADAR1 had no effect on these components at the mRNA level (Figure 6A). However, while Drosha and DGCR8 were unaffected also at the protein level, Dicer protein expression was increased in the ADAR1-KD cells compared with the control and the rescue cells (Figure 6B). This implies on regulation of Dicer at the translation level. Indeed, let-7a, -b, -d, and -e (Figure 6C), which were significantly downregulated following ADAR1-KD (Supplemental Table 3), are known to cause translational repression of Dicer (24–28).
Figure 6. ADAR1 regulates miRNAs processing by affecting the biogenesis proteins.

Expression of Dicer, Drosha, and DGCR8 at the (A) mRNA and (B) protein level as determined by qPCR and Western blot, respectively, in the transfectants indicated. Densitometry quantification of the immunoblots was performed using ImageJ software. Band intensities for Dicer, Drosha, and DGCR8 were determined relative to those of the endogenous control β-actin. All values were normalized to control cells. Data represent the mean ± SEM of 3 independent cell systems. *P < 0.05 (2-tailed t test). (C) Comparison between the expression level (fold change) of let-7 miRNA family members in ADAR1-KD and control cells. (D) Western blot of Dicer, Drosha and DGCR8, ADAR1, and CEACAM1 following pulldown of ADAR1 (right) or CEACAM1 (left). S1–S3 represents lysate samples from 3 independent transfected cell systems extracted both before (total cell lysate) and after IP procedure. (E) Western blotting for ADAR1 (left) or CEACAM1 (right) from total cell lysate or after the respective protein pulldown. Fold values in the total lysate samples represent the fold overexpression of each pri-miRNA over endogenous expression, after normalization to GAPDH. The presence (+) or absence (–) of pri-miRNAs in the pulldown is marked.
It was previously reported that ADAR1 can bind to other RNA-binding proteins (29–31). Thus, HEK 293T cells were transfected with ADAR-P110 or with an irrelevant protein, CEACAM1. Immunoprecipitation of ADAR1 or CEACAM1 was performed, with both proteins efficiently pulled down (Figure 6D). Remarkably, only DGCR8 was coimmunoprecipitated with ADAR1, but not Drosha or Dicer (Figure 6D). Since DGCR8 and Drosha form the active pri-miRNA processing complex (microprocessor) in the nucleus (32), this observation suggests that the complex DGCR8-ADAR1 is mutually exclusive with DGCR8-Drosha, thus unraveling a potentially novel layer of regulation of the Drosha complex. None of these proteins were coimmunoprecipitated with CEACAM1 (Figure 6D).
We further tested the ability of ADAR1 to directly bind pri-miRNAs. HEK 293T cells were transiently transfected with ADAR1 or CEACAM1, along with pri-miR-21 or pri-miR-34a. 293T cells were used in these experiments due to the relatively low transfection efficiency into melanoma cells. Western blot confirmed strong expression of ADAR1 or CEACAM1 in the total cell lysates as well as in the immunoprecipitates following specific protein pull down (Figure 6E). Fold expression of pri-miR-34a and pri-miR-21 was quantified with qPCR, normalized to GAPDH, and determined relative to the parental cells. pri-miR-34a was strongly overexpressed with the cotransfected ADAR1 and similarly with the cotransfected CEACAM1 (Figure 6E). pri-mir-21 was enhanced in a mild manner with both proteins (Figure 6E). The presence of pri-miR-21 and pri-miR-34a in RNA extracted from the immunoprecipitate of ADAR1 or CEACAM1 was tested using qPCR. A specific signal was obtained for pri-miR-34a in the ADAR1 but not CEACAM1 immunoprecipitate, strongly implying on direct binding (Figure 6E). A similar observation for pri-miR-21 could not be made, suggesting that there is no direct binding in this case (Figure 6E). Alternatively, this might be due to significantly weaker expression of pri-miR-21 than pri-miR-34a (Figure 6E). When these experiments were performed on untransfected cells, no signal could be observed (data not shown), probably owing to the minute amounts of RNA extracted from the immunoprecipitates and the overall low sensitivity of the assay. Quantitative analysis is impossible due to the lack of a validated normalizer.
Taken together, our data suggest that ADAR1 affects the biogenesis of miRNAs at various points along the process directly by potentially affecting Drosha complex and indirectly by regulating Dicer via let-7 or by binding to various pri-miRNAs.
ADAR1 expression is regulated by an additive cofunction of miR-17-5p and miR-432.
miR-17-5p is part of the miR-17-92 cluster, which plays an oncogenic role in other malignancies (33), but has not been studied in melanoma. miR-432 has never been investigated before. A putative binding site for miR-17-5p and 2 sites for miR-432 were identified in the 3′ UTR of ADAR1 (Supplemental Figure 7A). Preliminary analysis of 2 melanoma cell lines, C8161 highly aggressive (HAG) and the C81-61 poorly aggressive (PAG) cell lines (22), showed that ADAR1 was expressed 8-fold higher in the PAG cells, while miR-17-5p and miR-432 were expressed 5.5-fold and 20-fold higher in HAG cells (Supplemental Figure 7B).
A portion of ADAR1 3′ UTR containing the putative binding site for miR-17-5p (UTR) was cloned downstream to Renilla luciferase in a dual luciferase reporting system. The putative binding site was altered with 3 point mutations (UTR-MUT). miR-17-5p was cloned into the pQCXIP expression vector. Empty psiCheck2 (NO-UTR) and pQCXIP (mock) served as negative controls. The various constructs were cotransfected into C81-61 cells (PAG), which express moderate endogenous levels of miR-17-5p (Supplemental Figure 7). The luciferase signal of cells cotransfected with both empty vectors served as point of reference. Forced expression of miR-17-5p with the UTR construct significantly inhibited the luciferase signal, while the inhibitory effect was abolished when the UTR-MUT construct was tested (Figure 7A). This suggests that miR-17-5p binds directly to the 3′ UTR of ADAR1. In order to study the role of endogenous miR-17-5p, C8161 (HAG) cells, which express high levels of miR-17-5p (Supplemental Figure 7), were transiently transfected with anti–miR-17-5p oligonucleotides or with control oligonucleotides. miR-17-5p was efficiently silenced (Figure 7B), while the expression of ADAR1 was significantly enhanced both at RNA (Figure 7C) and protein (Figure 7D) levels. Both ADAR1-P150 and ADAR1-P110 were enhanced (Figure 7D). Functionally, cell proliferation rate (Figure 7E) as well as RNA-editing rate (data not shown) were inhibited significantly.
Figure 7. ADAR1 expression is directly controlled by miR-17-5p and miR-432 in an additive manner.

(A) UTR and MUT-UTR denote ADAR1 3′ UTR segments containing the reference sequence or mutated sequence in the miR-17-5p–binding site, respectively. Those were cotransfected with miR-17-5p (miR-17-5p OX) or with an empty vector (mock) to PAG cells. Relative luciferase results were normalized to the values of MUT-UTR transductants. Expression of (B) miR-17-5p and (C) ADAR1 in HAG cells following indicated manipulation was determined by qPCR and (D) Western blot for ADAR1. (E) Net proliferation 48 hours after seeding was determined by standardized XTT assay. (F) UTR, UTR-mutA, -mutB, or -mutAB denote ADAR1 3′ UTR segments containing the reference sequence or mutated sequence in miR-432–binding sites. Those were cotransfected with miR-432 (miR-432 OX) or with an empty vector (mock) to 293T cells. Relative luciferase results were normalized to the values of MUT-UTR transductants. Expression of (G) miR-432 and (H) ADAR1 in HAG cells following indicated manipulation was determined by qPCR and (I) Western blot. (J) Net proliferation 48 hours after seeding was determined by standardized XTT assay. Relative expression of (K) miR-17-5p, miR-432, and (L) ADAR1 in PAG cells transfected with mock plasmid, miR-17-5p, or miR-432 overexpression plasmids or both as determined by qPCR and (M) Western blot. (N) Net proliferation 48 hours after seeding was determined by standardized XTT assay. *P < 0.05; #P < 0.01; §P < 0.001 (2-tailed t test). Data represent the mean ± SEM of 3 independent experiments, each performed in triplicate.
A similar approach was implemented for miR-432. An ADAR1 3′ UTR fragment that includes both putative sites for miR-432 was cloned. Constructs containing separately mutated sites (UTR-mutA and UTR-mutB) or both (UTR-mutAB) were tested as described above in PAG cells, which exhibit moderate miR-432 expression (Supplemental Figure 7). Transfection of miR-432 with UTR demonstrated a clear decrease in luciferase signal (Figure 7F), attesting for the direct binding of miR-432 to 3′ UTR of ADAR1. The presence of both intact binding sites is required for effective inhibition (Figure 7F). miR-432 was expressed in similar levels with all reporting constructs (data not shown). Effective silencing of miR-432 in HAG cells (Figure 7G) enhanced ADAR1 expression both at RNA (Figure 7H) and protein levels (Figure 7I) and accordingly inhibited cell proliferation (Figure 7J).
The cofunction between miR-17-5p and miR-432 was studied by transfecting PAG cells with each miRNA separately or simultaneously, with half the amount of each miRNA (Figure 7K). The simultaneously transfected cells indeed overexpressed the miRNAs at lower levels than the cells transfected with each individual miRNA (Figure 7K). Nevertheless, ADAR1 expression was similarly downregulated at the RNA (Figure 7L) and protein (Figure 7M) levels in all transfectants. Accordingly, a similar enhancement in net proliferation was observed (Figure 7N). Taken together, it is shown that miR-17-5p and miR-432 are direct endogenous regulators of ADAR1, which cofunction in an additive manner.
Cancer cells employ genomic and epigenetic mechanisms to downregulate ADAR1 via miR-17-5p and miR-432.
Expression analysis of both miRNAs in 26 patient-derived low-passage metastatic melanoma cultures and 4 normal melanocyte cultures showed that about 30% of melanoma cases significantly upregulated miR-17-5p expression, while more than 95% strongly upregulated miR-432 (Figure 8A). Both miRNAs exhibited a significant inverse correlation with ADAR1 expression (miR-17-5p: P = 0.0013, R = –0.58; miR-432: P = 0.0014, R = –0.55) (Figure 8B). There was no correlation between ADAR1 and 6 other miRNAs (miR-20a, -133a, -184, -185, -31, and -204) (Supplemental Figure 8). These observations hint of the physiological relevance of miR-17 and miR-432 in cancer development. Indeed, chromosomal copy number analysis in 12 metastatic melanoma cell lines and 3 normal cell samples (melanocytes, HUVEC, and keratinocytes), showed a very common and significant increase both in miR-17-92 cluster (60%, 3–6 copies) and in miR-432 (92%, 3–7 copies) (Figure 8C). Thus, genomic copy number amplification of ADAR1-repressive miRNAs may account for downregulation of ADAR1 in cancer cells. However, although genomic copy numbers correlated with miRNA expression (miR-17-5p: P = 0.015, R = 0.68; miR-432: P = 0.0008, R = 0.81), the significant differences between miR-17-5p and miR-432 (Figure 8A) both in percentage and intensity suggest the existence of another, differential mechanism.
Figure 8. Genomic and epigenetic regulation of miRNAs that target ADAR1.

(A) Fold expression of miR-17-5p vs. miR-432 in 26 melanoma cultures (black diamonds) and 4 normal melanocyte cultures (white squares). Threshold is set to the value of 2. (B) Normalized miR-17-5p, miR-432, and ADAR1 expression level in low-passage primary cultures of metastatic melanoma and normal melanocytes presented as 1/ΔCt. Correlation was calculated using Pearson’s test. (C) Genomic amplification of miR-17-5p and miR-432 expression in 12 melanoma cell lines (black triangles) and 3 normal (control) cell samples (black circles). y axis denotes number of genomic copies. Values higher than 2 are defined as amplification, while values lower than 2 are considered as deletion of genomic copies. Data represent the mean ± SEM are indicated. Two melanoma cell lines were treated with 5′-aza-2-deoxycitidine and PBA (treatment) or with medium (control) for 2–6 days. Expression of (D) miR-17-5p and (E) miR-432 was tested by qPCR, and (F) ADAR1 expression level of day 2–6 was determined by (F) qPCR and (G) Western blot (results of a representative day are presented) (T, treatment; C, control). *P < 0.05; #P < 0.01; §P < 0.0001 (2-tailed t test). Data represent the mean ± SEM of 2 independent experiments, each performed in triplicate.
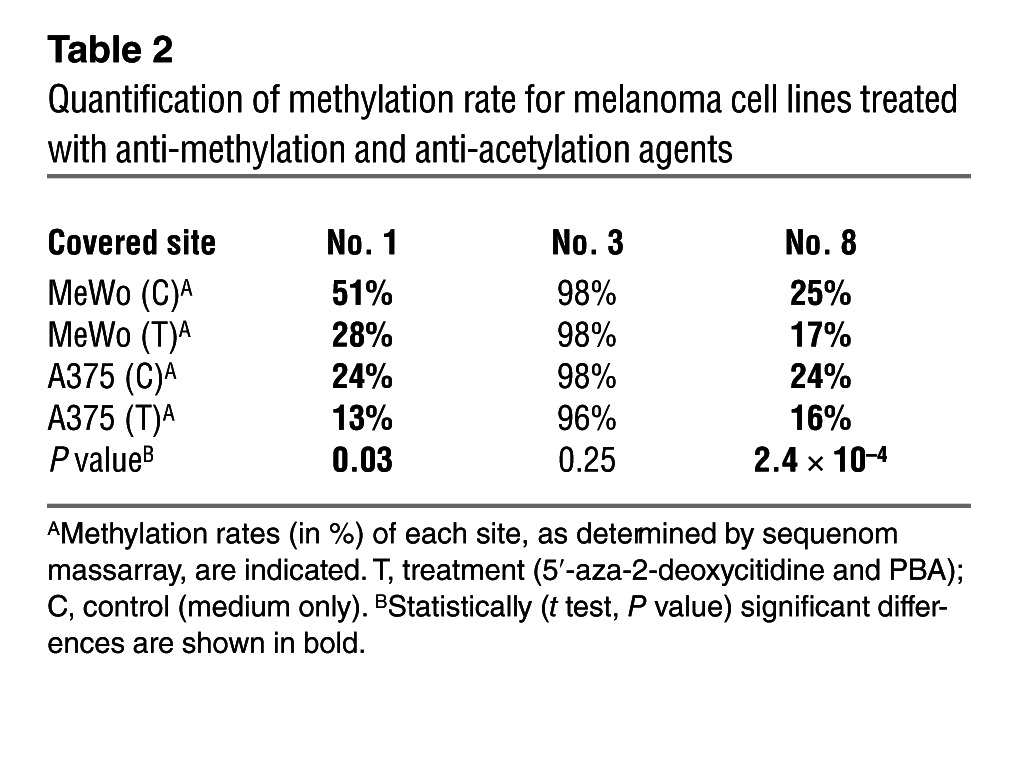
miR-432 is encoded in chromosome 14 in the DLK1-DIO3 region (Supplemental Figure 9), which has been shown to be imprinted (34), and was therefore suspected to be regulated epigenetically. Quantification of genomic methylation in more than 10 sites in RTL1 comparing 12 melanoma cell lines and 3 normal cell samples confirmed a robust hypomethylation in this region in the cancer samples (Table 1). Moreover, a striking hypomethylation was observed within the sequence of miR-432 and upstream of its genomic location (Table 1). The effect is site specific, as some sites in both RTL and miR-432 remained hypermethylated among both normal and cancer samples (Table 1). Methylation indeed accounts for repression of miR-432 expression because it was increased in a time-dependent manner in 2 different melanoma cell lines tested, A375 and MeWo, when exposed to epigenetic modifiers 5′-aza-2-deoxycitidine and 4-phenylbutyric acid (PBA) (Figure 8D). Demethylation was clearly demonstrated in sites no. 1 and no. 8 in the miR-432 region (Table 2). There were no predicted methylation sites in the region of the miR-17-92 cluster, and indeed, the expression of miR-17-5p remained constant (Figure 8E). Finally, concurring with the robust upregulation in miR-432 expression, a drastic downregulation of ADAR1 expression was observed after treatment with demethylating agents (Figures 8, F and G). These combined results highlight the imprinted RTL1 region as a target for hypomethylation in cancer, which in turn controls ADAR1 expression via hypomethylation of miR-432.
Table 1.
Quantification of methylation rate for RTL1 and miR-432
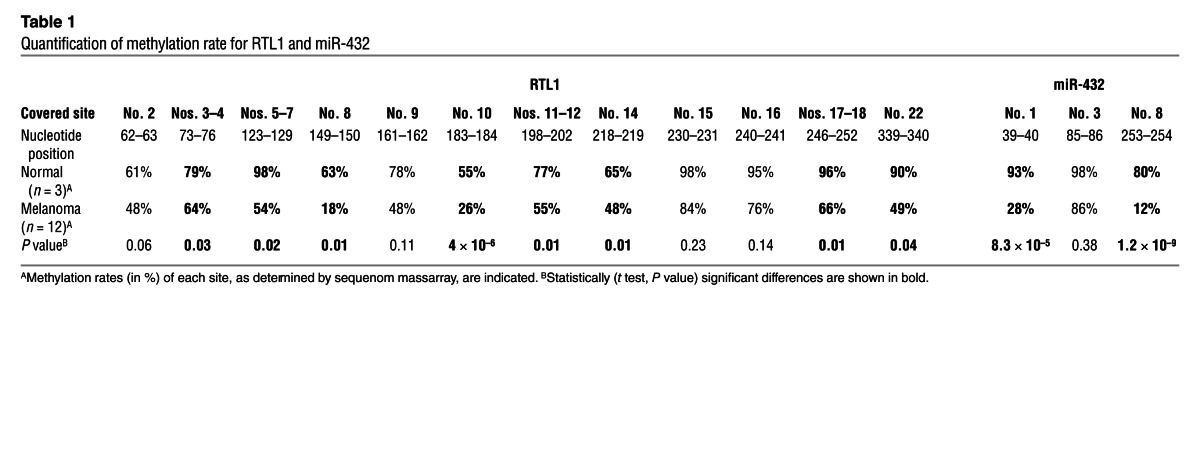
Table 2.
Quantification of methylation rate for melanoma cell lines treated with anti-methylation and anti-acetylation agents
Discussion
The role of ADAR1 as an RNA-editing enzyme is well established (12). Many studies emphasize the significance of A-to-I RNA editing in regulation of gene expression (35), viral host defense (36), embryonic development (37), miRNA processing (38), alternative splicing (1), and the function of several receptors located in the CNS (39). Lower RNA-editing rates were described in various human cancers (3, 40); however, whether reduced editing rate facilitates cancer or whether it is a consequence of another process remained obscure. Here, we present what we believe to be novel mechanistic evidence for the roles of ADAR1 in regulation of cancerous features and for how it is lost in metastatic melanoma cells to facilitate the acquisition of an aggressive phenotype.
Metastatic melanoma cells exhibit a common and significant downregulation of both ADAR1-P110 and ADAR1-P150 expression as compared with normal melanocytes, nevi, and primary melanoma tumors (Figure 1). Therefore, it seems that loss of both isoforms is associated with the metastatic transition and could be explained by the upregulation of miR-17 and miR-432 in metastatic cells (Figure 8), which target them both (Figure 7). Series of experimental ADAR1 manipulations in melanoma cell lines demonstrate that ADAR1 controls the expression of hundreds of genes and fundamentally suppresses their malignant phenotype in vitro and in vivo (Figures 2–4). It should be noted that a weak, selective induction of the inducible ADAR1-P150 was observed only in nevi and primary tumors as compared with normal epidermis (Figure 1). Given the suppressive features of ADAR1-P150 (Figures 2 and 3), this could hint of its potential role as an inherent resistance mechanism to melanocyte transformation. Another melanoma suppressive protein, IGFBP7, which promotes apoptosis and senescence, operates in nevi bearing the BRAFV600E mutation and is then silenced by methylation in metastatic cells (41). It should be noted that an opposite role for ADAR1-P150 was reported in HeLa cells (42) and that these differences might be due to cell-specific miRNA and target gene expression profiles. In addition, an upregulation of ADAR1 was observed in some other malignancies, such as lobular breast cancer (43) and B cell acute lymphoblastic leukemia (44).The robust dominance of ADAR1-P110 probably dictates the final cellular outcome. The corroboration between the robust cell regulatory roles of ADAR1 with its common low expression in melanoma metastasis strongly points to a central involvement of ADAR1 in melanoma progression.
We hypothesized that the diffuse DEG profile observed following experimental KD of ADAR1 suggests that ADAR1 regulates miRNAs, broad-acting cellular mediators. Indeed, KD of ADAR1 altered the expression of 131 miRNAs (Supplemental Table 3), many of which are known cancer regulators (Supplemental Table 3), and target 38% of the DEGs (Supplemental Table 1). The rest of the DEGs, which are not predicted to be targeted by the ADAR1-controlled miRNAs, might be secondarily altered. Correction of exemplar upregulated miR-21 (by silencing) or of downregulated miR-34a (by forced expression) partially reversed the enhanced proliferation of ADAR1-KD cells (Figure 4). This attests for the mechanistic role of ADAR1-controlled miRNAs in determining the phenotype of cancer cells. Interestingly, most altered miRNAs with known oncogenic or tumor-suppressive properties were upregulated or downregulated, respectively, following ADAR1-KD (Figure 4). However, there were no structural differences among the groups, based on bioinformatics predictions (18, 19), which could explain this phenomenon. Further, crossing of TargetScan 5.1 (20) predicted targets for miR-21 or miR-34a with downregulated or upregulated DEGs, respectively, indicated that the increase in miR-21 was potentially responsible for the downregulation of 7 genes, 6 of them by 3.3- to 10-fold, while the decrease in miR-34a was potentially responsible for the upregulation of 10 genes, 4 of them by 4- to 9-fold (Supplemental Table 5). In light of these observations, the differences between the herein-reported DEGs in human melanoma cells and the interferon-related DEG pattern in hematopoietic cells reported in ADAR1-knockout mice (45) are not surprising, as they can be attributed to the different cell types, which endogenously express different sets of miRNAs.
A-to-I RNA editing of miRNAs has been shown to either lead to recognition of new targets due to altered seed sequence or to maturation defects due to dysregulated processing (46–48). Recently, 2 comprehensive bioinformatics studies revealed only a limited number of editing events in mature miRNA sequences (49, 50). These reports, combined with the robust alteration of more than 130 miRNAs following ADAR1-KD, further support our argument that ADAR1 regulates both cell proliferation and miRNA processing independently of RNA editing. Indeed, the effects exerted by ADAR1 bearing a mutated or truncated catalytic deamination domain (CAT-MUT or ΔCAT, respectively) were similar to the full ADAR1 enzymes (Figures 3 and 5). While the effects of ADAR1 on miRNA processing, expression, and thereby functions were independent of RNA editing, they did depend on RNA-binding capacity. We show that both the dsRBD, essential for binding to the target RNA molecule and/or other RNA-binding proteins (51), and the Z-DBD, necessary for ADAR1 effects on NF-90 activity (30), nuclear export, and binding to specific sequences based on their conformation (52, 53), are required. ADAR1 truncation mutants lacking the dsRBD or ZDBD failed to exert any impact either on cellular proliferation or miRNA biogenesis (Figures 3 and 5). In addition, we demonstrated that ADAR1 is capable of directly binding pri-miRNAs (Figure 6E). RNA editing–independent roles of ADAR1 were previously reported, for example, by creating specific protein-protein complexes with NF-90 (30), decreasing PKR kinase activity at the translation level (31). In addition, it was reported that ADAR2 regulates the processing of miR-376a2 independently of RNA-editing activity (13). Our findings expand the known scope of ADAR1-mediated regulation of miRNA biogenesis by direct RNA editing of specific pri-miRNAs (23, 48), and postulate a broad regulatory scope for ADAR1, which operates via systematic control of the miRNA-processing machinery.
A systematic regulation of the miRNA processing machinery by ADAR1 is supported by the specific effect of ADAR1-P110 and ADAR1-P150 on the processing of the pri- and pre-miRNA precursors, respectively, which ultimately have the same end result on mature miRNA expression (Figure 5). Several mechanisms might be involved here: (a) ADAR1 creates complexes with DGCR8 as they are coimmunoprecipitated (Figure 6D); this might occur via their dsRNA-binding domains, as previously reported for ADAR1 and Exportin-5 or NF-90 (29, 54); the lack of ADAR1-Drosha complexes (Figure 6D) suggests that the formation of DGCR8-ADAR1 and DGCR8-Drosha complexes are mutually exclusive; thus ADAR1 could affect the availability of DGCR8 for Drosha, thereby altering the efficiency of the microprocessor in general; (b) ADAR1 regulates Dicer expression at the translation level (Figure 6B). This probably occurs via Let-7 miRNAs, which have been recently shown to regulate Dicer at the translation level (24–28) and are strongly controlled by ADAR1 (Supplemental Table 3); (c) ADAR1 can directly bind some miRNA precursors and compete over the dsRNA substrates with the enzymatic processors; (d) the expression of SMAD1 and NF-90, which were recently shown to regulate microprocessor activity (23), is also controlled by ADAR1 (Supplemental Tables 1 and 2) in a yet to be defined mechanism. Collectively, these results imply that the previously described cancer-associated diminished RNA editing (3) probably reflects the mere downregulation of ADAR, although a contribution of RNA editing to the malignant phenotype of cancer cannot be entirely excluded.
Finally, we identified miR-17-5p and miR-432 as the direct, independent, endogenous cellular regulators of ADAR1. Together, these miRNAs regulate ADAR1 expression in an additive manner (Figure 7). Our findings suggest that loss of ADAR1 in cancer cells and the subsequent increase in malignant features is mediated by overexpression of these miRNAs (Figure 8). miR-17-5p, an miRNA with known oncogenic properties, is overexpressed in various cancers, mainly in hematopoietic malignancies (33), due to amplification of miR-17HG, a noncoding genomic segment that contains the miR-17-92 cluster (13q31.3) (55). In contrast, there are only scarce data on miR-432.
Genomic and epigenetic mechanisms account for the overexpression of miR-17-5p and miR-432 in melanoma cells. We show that amplification of the genomic segment encoding miR-17-5p frequently occurs in melanoma, thereby facilitating the malignant phenotype by directly targeting ADAR1 (Figures 7 and 8). The overexpression of miR-432 in melanoma occurs due to frequent genomic amplification and aberrant hypomethylation patterns of the DLK1-DIO3 locus in chromosome 14 (Figure 8 and Table 1). Cancer development and progression involve aberrant methylation in general, with most studies focusing on hypermethylation of tumor-suppressive genes (56). Epigenetic regulation of miRNAs has started gaining the focus of cutting edge research very recently (57). The regulation of miR-432 by hypomethylation is novel for the following reasons: (a) the miR-432 gene is found within the DLK1-DIO3 imprinted domain (58), which has been poorly studied in cancer; (b) one of the methylation sites is found within the sequence of the miR-432, which represents the first such example. The mechanisms by which the imprinting patterns of the DLK1-DIO3 region are destroyed in cancer could include, for example, DNA methyl transferase dysregulation or acquisition of somatic mutations in methylation sites and should be the focus of future investigations.
The ADAR1-dependent and RNA editing–independent roles presented here as well as the miRNA-mediated mechanisms of ADAR1 downregulation are probably applicable to additional types of cancer. These findings could be translated into innovative lines of diagnosis and therapy in a broad range of malignancies.
Methods
Cells and antibodies.
The melanoma lines 624mel 526mel, C8161 (HAG) and C81-61 (PAG), SKmel2, SKmel24, MalMe3M, G361, WN115, MeWo, A375, WM266-4, HEK293T cell lines (ATCC), normal human epidermal melanocytes (PromoCell) and normal melanocyte cultures hmel-p-16, Nohm-4 and Hermes-2B were maintained as previously described (22, 59–61). The 34 primary cultures derived from surgically removed metastatic melanoma specimens were established and cultured as described (59). Stably transfected cell lines were cultured with 1 μg/ml puromycin (Calbiochem) or 2 mg/ml G418 (Alexis Biochemicals)
The following primary antibodies were used: PE-His (Miltenyi Biotec), annexin V–FITC (Bender Medsystems), Ki67 (DAKO), ADAR1 (Sigma-Aldrich), β-actin (MP Biochemicals), Dicer (Abcam), Drosha (Abcam), DGCR8 (Abcam), and HRP-conjugated secondary antibodies (Jackson Immunoresearch).
RNA isolation and real-time qPCR analysis.
Total RNA was isolated with Tri Reagent (Sigma-Aldrich) according to the manufacturer’s instructions. cDNA was synthesized using high-capacity or Taq-Man miRNA reverse transcription kit (Applied Biosystems).
qPCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) with gene-specific primers (listed in Supplemental Table 4) or Taq-Man Universal Master Mix (Applied Biosystems) with custom primer/probe mixtures (Applied Biosystems) according to the manufacturer’s instructions. The real-time PCR (qPCR) reactions were normalized to GAPDH, HPRT, or U6 endogenous control as reported previously (60).
Genomic amplification assay.
Genomic DNA (gDNA) was extracted from melanoma using a Wizard genomic purification kit (Promega) according to the manufacturer’s instructions. Expression analysis was performed using TaqMan Copy Number Variation Assay (Applied Biosystems) along with custom ordered probes (Applied Biosystems). The qPCR results were normalized to 2 TaqMan copy number reference assays: TERT and RNase-P (Applied Biosystems).
Expression constructs and stable transfections.
The expression systems used in this work were pSuper.puro, pCDNA3.neo, pQCXIP.puro, and psiCheck2. Identification of the various primers that were designed for cloning and introduction of mutations is available in Supplemental Table 4. Transfections were performed with Turbofect (Fermentas) according to the manufacturer’s instructions. Retroviral transductions were performed as previously described (22). Site-directed mutagenesis was performed using QuikChange kit (Stratagene) according to the manufacturer’s instructions.
Anti-miRNA oligos and transient transfection.
The LNA anti-miRNA oligos (unmodified) along with the proper control oligos (miRcury LNA, Exiqon) were used for miRNA silencing. The various oligos were transiently transfected with Turbofect (Fermentas) and the cells were tested for miRNA expression 48 hours after transfection.
Western blotting.
Lysates of 5 × 106 cells were analyzed by SDS-PAGE. Western blot for ADAR1 and actin with specific antibodies, which was developed with standard ECL reaction, was performed using standard protocols.
Quantification of RNA editing.
PCR amplifications and analysis of editing levels were carried out as described (61).
Quantification of cell growth.
Cells (5 × 103) were seeded in triplicate in 96-well flat-bottom microplates, and designated wells were harvested daily for 72 hours. Net proliferation was tested via a standardized XTT (Biological Industries) colorimetric assay, as previously described (22). Standard curve was performed individually for each treatment in all experiments.
Flow cytometry.
Staining for extracellular and intracellular antigens was performed according to standard protocols, as reported previously (60). Cell-cycle analysis was performed on 1 × 106 cells with standard overnight 70% ethanol fixation, permeabilization with 0.05% Triton X-100 (Sigma-Aldrich),and PI staining. All experiments were performed using a FACSCalibur instrument (BD Biosciences), and data were analyzed using FlowJo software (Tree Star Inc.).
Human melanoma xenografts in SCID-NOD mice.
Eight-week-old male SCID-NOD mice were used for xenograft studies. Approximately 5 × 106 viable tumor cells were resuspended in 100 μl PBS solution and injected subcutaneously. Mice were monitored 3 times a week for tumor volume by caliper measurements (longest diameter × shortest diameter2 × 0.52). On termination of each experiment, the tumors were extracted, documented, weighed, and preserved. Immunostaining was performed on the formalin-preserved tumors, and total RNA was extracted from the cryopreserved tumors according to standard procedures.
Immunohistochemistry.
Immunohistochemical staining was performed on 5-μm sections of paraffin-embedded tissues or progression TMA slides (see below) according to standard procedures. Tissues were stained for Ki-67 or ADAR1, followed by hematoxylin (Sigma-Aldrich) counterstaining. Isotype-matched or preimmune rabbit serum served as negative control. The percentage of Ki-67–positive cells was determined by an expert pathologist (I. Barshack), who was blinded to the experimental groups.
Progression TMA.
TMA slides were provided by the NCI CDP and included 66 benign nevi, 90 primary tumors, and 74 metastases. Another TMA set was provided by the NCI SPORE program and included 66 melanocytic nevi, 66 primary melanomas, and 75 metastases. For each sample, intensity of ADAR1 expression (nuclear and cytoplasmic) was scored from 0 (negative) to 3, and percentages of expression were defined as 0 to 3 for 0%–5%, 6%–25%, 26%–75%, and 76%–100%, respectively. Cells were examined over superficial and deep dermal foci in primary melanomas and nevi in order to detect possible “maturation” changes within the lesions.
Luciferase reporter assay.
C81-61 (PAG) cells were cotransfected with 1 μg of psiCheck2-ADAR1 3′ UTR (UTR), psiCheck2-ADAR1 mutated 3′ UTR (UTR-MUT), or psiCheck2-empty vector (No-UTR) and 0.1 μg of the pQCXIP-miR-17 (miR-17-5p) or pQCXIP-empty vector (mock) as control. HEK 293T cells were cotransfected with 1 μg of psiCheck2-ADAR1 3′ UTR plasmids (UTR), different psiCheck2-ADAR1 mutated 3′ UTR (UTR-mutA, UTR-mutB, and UTR-mutAB), or psiCheck2–empty vector (No-UTR) and 0.1 μg of the pQCXIP-miR-432 (miR-432) or pQCXIP–empty vector (mock) as control. Cells were harvested 48 hours after transfection and assayed with Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions.
Microarray expression analysis.
Total RNA was extracted and used as template to generate cDNA and subsequent biotinylated target cRNA that was processed by an Affymetrix GeneChip Instrument System (Affymetrix) according to the manufacturer’s recommendations ( http://affymetrix.com/support/technical/manual.affx). The DEGs were analyzed by Ingenuity Pathway Analysis ( http://www.ingenuity.com), while the miRNAs were analyzed using Partek Genomic Suite (Partek Inc.) and TargetScan 5.1 (Whitehead Institute for Biomedical Research). Full microarray data are deposit in NCBI GEO archives (GSE31963).
Bioinformatic analysis.
MC-Fold (62) was applied for prediction of miRNA secondary structure and ΔG values.
DNA methylation analysis.
gDNA of melanoma cell lines was extracted as described above. Bisulfate treatment was carried out on gDNA using EZ-DNA Methylation Gold Kit (Zymo) according to the manufacturer’s instructions. Next, PCR was performed using bisulfate-treated DNA as a template with specifically designed primers (Supplemental Table 4).
Following PCR amplification, a shrimp alkaline phosphate (SAP) treatment, in vitro transcription, and RNaseA cleavage (MASScleave) were performed. The samples were purified by resin treatment, spotted on 384-well spectroCHIP, and analyzed by a spectral acquisition on a MASSarray Analyser (Sequenom). The average methylation ratio for each CpG position was calculated by averaging the ratios of the melanoma versus normal samples. Non-template control (NTC) sample used as negative control (for details see Supplemental Methods).
Demethylation experiments.
Melanoma cell lines A375 and MeWo were treated for 2, 4, and 6 days with 3 μM anti-methylation reagent 5′-Aza aza-2-deoxycitidine (Sigma-Aldrich) dissolved in 50% acetic acid and 3 mM anti-acetylation agent (PBA) (Sigma-Aldrich) dissolved in 50% EtOH. Control samples were treated with medium containing the same volume of dissolvent. The growth medium containing these reagents or dissolvent only was changed daily. The treated cells and the control samples were harvested at days 2, 4, and 6. RNA, cell lysates, and gDNA were extracted from the cells and analyzed, as previously mentioned.
Immunoprecipitation.
HEK 293T transfected cells were harvested and total cell lysates were extracted and assayed following incubation with either anti-ADAR1 or anti-CEACAM1 with Dynabeads Protein-G beads (Sigma-Aldrich) according to the manufacturer’s instructions. Both total cell lysates and immunoprecipitates were analyzed by Western blot according to standard protocols. Total RNA was isolated and analyzed by qPCR as described above (for details see Supplemental Methods).
Northern blot.
RNA samples were electrophoresed, transferred, and hybridized as previously described (63) using P32-labeled probes. The oligonucleotides used as probes are the complementary sequences of the mature miRNA (miRbase Registry). U6 was used to normalize expression levels (for details see Supplemental Methods).
Determination of apoptosis.
Melanoma ADAR1-KD, ADAR1-rescue-P110, ADAR1-rescue-P150, and control cells (104 cells/well) were seeded in 96-well plates and untreated (control) or treated with either 100 μM cisplatin (Abiplatin; ABIC) or 50 μM taxol (Ebetaxel; Ebewe Pharma). Cells were collected 48 hours after treatment, washed with PBS, and stained with both annexin V–FITC and PI according to the manufacturer’s instructions (eBioscience). Apoptosis rate was further determined by FACSCalibur instrument (BD Biosciences), and data analysis was performed using FlowJo software (Tree Star Inc.).
Statistics.
Data were analyzed using the unpaired 2-tailed Student’s t test, the 1-way ANOVA test, and Fisher’s exact test. Correlations were examined with Pearson’s correlation test. P ≤ 0.05 was considered significant.
Study approval.
Animal studies were approved and performed in accordance with the Institutional Review Board (no. 504/2009), Sheba Medical Center. The primary melanoma specimens were obtained according to Israel Ministry of Health approval no. 3518/2004 (ClinicalTrails.gov Identifier NCT00287131). NCI CDP TMA slides were provided by the NCI CDP. Other investigators may have received slides from these same array blocks. The NCI SPORE TMA used in these studies was generated with the support of M.D. Anderson SPORE in skin cancer (NIH p50CA093459).
Supplementary Material
Acknowledgments
G. Markel is supported by the Israel Science Foundation (grant 1489/10) and the Carol Fingerut Foundation for Cancer Research (grant 2008). D.M. Kallenberg is supported by Wellcome Trust (grant 078327). G. Rechavi is supported by the I-CORE Program of the Planning and Budgeting Committee and the Israel Science Foundation (grant no. 41/11.1), the Flight Attendants Medical Research Institute (FAMRI), the I-CORE Program of the Planning and Budgeting Committee and the Israel Science Foundation (grant no. 41/11.1), and the Israel Ministry for Science and Technology (Scientific Infrastructure Program, grant 2010). G. Rechavi holds the Djerassi Chair in Oncology, Tel Aviv University. E. Galun is supported by an I-CORE ISF grant (grant no. 41/11.1) and an SFB DFG grant (grant no. 841). The authors would like to thank Michael Aronson for his support in this work. This work was performed in partial fulfillment of the requirements for a Ph.D. degree for Yael Nemlich, Sackler Faculty of Medicine, Tel Aviv University.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(6):2703–2718. doi:10.1172/JCI62980.
References
- 1.Scholzová E, Malík R, Sevcík J, Kleibl Z. RNA regulation and cancer development. Cancer Lett. 2007;246(1–2):12–23. doi: 10.1016/j.canlet.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 2.Galeano F, Tomaselli S, Locatelli F, Gallo A. A-to-I RNA editing: The “ADAR” side of human cancer. Semin Cell Dev Biol. 2012;23(3):244–250. doi: 10.1016/j.semcdb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Paz N, et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007;17(11):1586–1595. doi: 10.1101/gr.6493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keegan LP, Gallo A, O’Connell MA. The many roles of an RNA editor. Nat Rev Genet. 2001;2(11):869–878. doi: 10.1038/35098584. [DOI] [PubMed] [Google Scholar]
- 6.Keegan LP, Leroy A, Sproul D, O’Connell MA. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol. 2004;5(2):209. doi: 10.1186/gb-2004-5-2-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valente L, Nishikura K. Progress in Nucleic Acid Research and Molecular Biology. 2005. ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. In: Kivie M, ed. pp. 299–338. Waltham, Massachusetts, USA: Academic Press; [DOI] [PubMed] [Google Scholar]
- 8.Dominissini D, Moshitch-Moshkovitz S, Amariglio N, Rechavi G. Adenosine-to-inosine RNA editing meets cancer. Carcinogenesis. 2011;32(11):1569–1577. doi: 10.1093/carcin/bgr124. [DOI] [PubMed] [Google Scholar]
- 9.Iizasa H, Nishikura K. A new function for the RNA-editing enzyme ADAR1. Nat Immunol. 2009;10(1):16–18. doi: 10.1038/ni0109-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.XuFeng R, et al. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc Natl Acad Sci U S A. 2009;106(42):17763–17768. doi: 10.1073/pnas.0903324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wulff BE, Nishikura K. Modulation of microRNA expression and function by ADARs. Curr Top Microbiol Immunol. 2012;353:91–109. doi: 10.1007/82_2011_151. [DOI] [PubMed] [Google Scholar]
- 12.Zinshteyn B, Nishikura K. Adenosine-to-inosine RNA editing. Wiley Interdiscip Rev Syst Biol Med. 2009;1(2):202–209. doi: 10.1002/wsbm.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heale BSE, et al. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009;28(20):3145–3156. doi: 10.1038/emboj.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galeano F, et al. Human BLCAP transcript: new editing events in normal and cancerous tissues. Int J Cancer. 2010;127(1):127–137. doi: 10.1002/ijc.25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levanon EY, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotech. 2004;22(8):1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 16.Levanon EY, et al. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Research. 2005;33(4):1162–1168. doi: 10.1093/nar/gki239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai F, Drakas R, Nishikura K. Mutagenic analysis of double-stranded RNA adenosine deaminase, a candidate enzyme for RNA editing of glutamate-gated ion channel transcripts. J Biol Chem. 1995;270(29):17098–17105. doi: 10.1074/jbc.270.29.17098. [DOI] [PubMed] [Google Scholar]
- 18. Cancer Research Center at Sheba. Down-Regulated. Sheba Web site. http://sheba-cancer.org.il/yael/mirs_down/down.html . Accessed April 2, 2013.
- 19. Cancer Research Center at Sheba. Up-Regulated. Sheba Web site. http://sheba-cancer.org.il/yael/mirs_up/up.html . Accessed April 2, 2013.
- 20.Lewis BP, ShihIhJones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 21.Krichevsky AM, Gabriely G. miR-21: a small multi-faceted RNA. J Cell Mol Med. 2009;13(1):39–53. doi: 10.1111/j.1582-4934.2008.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenberg E, et al. Regulation of cancer aggressive features in melanoma cells by microRNAs. PLoS One. 2011;6(4):e18936. doi: 10.1371/journal.pone.0018936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. doi: 10.1038/nri3049. [DOI] [PubMed] [Google Scholar]
- 24.Jakymiw A, et al. Overexpression of dicer as a result of reduced let-7 microRNA levels contributes to increased cell proliferation of oral cancer cells. Genes Chromosomes Cancer. 2010;49(6):549–559. doi: 10.1002/gcc.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci U S A. 2008;105(39):14879–14884. doi: 10.1073/pnas.0803230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paroo Z, Ye X, Chen S, Liu Q. Phosphorylation of the human microRNA-generating complex mediates MAPK/Erk signaling. Cell. 2009;139(1):112–122. doi: 10.1016/j.cell.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455(7209):58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 28.Tokumaru S, Suzuki M, Yamada H, Nagino M, Takahashi T. let-7 regulates Dicer expression and constitutes a negative feedback loop. Carcinogenesis. 2008;29(11):2073–2077. doi: 10.1093/carcin/bgn187. [DOI] [PubMed] [Google Scholar]
- 29.Fritz J, Strehblow A, Taschner A, Schopoff S, Pasierbek P, Jantsch MF. RNA-regulated interaction of transportin-1 and exportin-5 with the double-stranded RNA-binding domain regulates nucleocytoplasmic shuttling of ADAR1. Mol Cell Biol. 2009;29(6):1487–1497. doi: 10.1128/MCB.01519-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nie Y, Ding L, Kao PN, Braun R, Yang J-H. ADAR1 interacts with NF90 through double-stranded RNA and regulates NF90-mediated gene expression independently of RNA editing. Mol Cell Biol. 2005;25(16):6956–6963. doi: 10.1128/MCB.25.16.6956-6963.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Samuel CE. Adenosine deaminase ADAR1 increases gene expression at the translational level by decreasing protein kinase PKR-dependent eIF-2alpha phosphorylation. J Mol Biol. 2009;393(4):777–787. doi: 10.1016/j.jmb.2009.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6(5):376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 33.Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol. 2010;42(8):1348–1354. doi: 10.1016/j.biocel.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seitz H, et al. Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet. 2003;34(3):261–262. doi: 10.1038/ng1171. [DOI] [PubMed] [Google Scholar]
- 35.Gommans WM, Maas S. Characterization of ADAR1-mediated modulation of gene expression. Biochem Biophys Res Commun. 2008;377(1):170–175. doi: 10.1016/j.bbrc.2008.09.109. [DOI] [PubMed] [Google Scholar]
- 36.Samuel CE. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 2011;411(2):180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Q, et al. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem. 2004;279(6):4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- 38.Nishikura K. Editor meets silencer: crosstalk between RNA editing and RNA interference. Nat Rev Mol Cell Biol. 2006;7(12):919–931. doi: 10.1038/nrm2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amariglio N, Rechavi G. A-to-I RNA editing: A new regulatory mechanism of global gene expression. Blood Cells Mol Dis. 2007;39(2):151–155. doi: 10.1016/j.bcmd.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Cenci C, et al. Down-regulation of RNA editing in pediatric astrocytomas. J Biol Chem. 2008;283(11):7251–7260. doi: 10.1074/jbc.M708316200. [DOI] [PubMed] [Google Scholar]
- 41.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132(3):363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Hou Z, Wu Y, Ma X, Luo X. p150 ADAR1 isoform involved in maintenance of HeLa cell proliferation. BMC Cancer. 2006;6:282. doi: 10.1186/1471-2407-6-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461(7265):809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 44.Ma C-H, et al. Abnormal expression of ADAR1 isoforms in Chinese pediatric acute leukemias. Biochem Biophys Res Commun. 2011;406(2):245–251. doi: 10.1016/j.bbrc.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 45.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10(1):109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8(8):763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315(5815):1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang W, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13(1):13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alon S, et al. Systematic identification of edited microRNAs in the human brain. Genome Res. 2012;22(8):1533–1540. doi: 10.1101/gr.131573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawahara Y, et al. Frequency and fate of microRNA editing in human brain. Nucleic Acids Research. 2008;36(16):5270–5280. doi: 10.1093/nar/gkn479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fierro-Monti I, Mathews MB. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem Sci. 2000;25(5):241–246. doi: 10.1016/S0968-0004(00)01580-2. [DOI] [PubMed] [Google Scholar]
- 52.George CX, Gan Z, Liu Y, Samuel CE. Adenosine deaminases acting on RNA, RNA editing, and interferon action. J Interferon Cytokine Res. 2011;31(1):99–117. doi: 10.1089/jir.2010.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herbert A, et al. The Zα domain from human ADAR1 binds to the Z-DNA conformer of many different sequences. Nucleic Acids Research. 1998;26(15):3486–3493. doi: 10.1093/nar/26.15.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakamoto S, et al. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol Cell Biol. 2009;29(13):3754–3769. doi: 10.1128/MCB.01836-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ernst A, et al. De-repression of CTGF via the miR-17-92 cluster upon differentiation of human glioblastoma spheroid cultures. Oncogene. 2010;29(23):3411–3422. doi: 10.1038/onc.2010.83. [DOI] [PubMed] [Google Scholar]
- 56.Ren J, et al. DNA hypermethylation as a chemotherapy target. Cell Sig. 2011;23(7):1082–1093. doi: 10.1016/j.cellsig.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 57.Agirre X, Martínez-Climent Jρ, Odero MD, Prósper F. Epigenetic regulation of miRNA genes in acute leukemia. Leukemia. 2012;26(3):395–403. doi: 10.1038/leu.2011.344. [DOI] [PubMed] [Google Scholar]
- 58.da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008;24(6):306–316. doi: 10.1016/j.tig.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 59.Besser MJ, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2010;16(9):2646–2655. doi: 10.1158/1078-0432.CCR-10-0041. [DOI] [PubMed] [Google Scholar]
- 60.Markel G, et al. Systemic dysregulation of CEACAM1 in melanoma patients. Cancer Immunol Immunother. 2010;59(2):215–230. doi: 10.1007/s00262-009-0740-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koren-Michowitz M, et al. A new MALDI-TOF-based assay for monitoring JAK2 V617F mutation level in patients undergoing allogeneic stem cell transplantation (allo SCT) for classic myeloproliferative disorders (MPD). Leuk Res. 2008;32(3):421–427. doi: 10.1016/j.leukres.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 62.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452(7183):51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 63.Pall GS, Hamilton AJ. Improved northern blot method for enhanced detection of small RNA. Nat Protoc. 2008;3(6):1077–1084. doi: 10.1038/nprot.2008.67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.