

Abstract
We characterized phosphoinositide binding of the S. cerevisiae PROPPIN Hsv2 qualitatively with density flotation assays and quantitatively through isothermal titration calorimetry (ITC) measurements using liposomes. We discuss the design of these experiments and show with liposome flotation assays that Hsv2 binds with high specificity to both PtdIns3P and PtdIns(3,5)P2. We propose liposome flotation assays as a more accurate alternative to the commonly used PIP strips for the characterization of phosphoinositide-binding specificities of proteins. We further quantitatively characterized PtdIns3P binding of Hsv2 with ITC measurements and determined a dissociation constant of 0.67 µM and a stoichiometry of 2:1 for PtdIns3P binding to Hsv2. PtdIns3P is crucial for the biogenesis of autophagosomes and their precursors. Besides the PROPPINs there are other PtdIns3P binding proteins with a link to autophagy, which includes the FYVE-domain containing proteins ZFYVE1/DFCP1 and WDFY3/ALFY and the PX-domain containing proteins Atg20 and Snx4/Atg24. The methods described could be useful tools for the characterization of these and other phosphoinositide-binding proteins.
Keywords: isothermal titration calorimetry, liposome flotation assays, multi-angle laser light scattering, PROPPIN, small unilamellar vesicle
1. Introduction
Phosphoinositides (PtdIns-phosphates) constitute less than 1% of cellular lipids, but are key players in membrane trafficking processes. Their various isoforms are enriched in different cellular membranes, and phosphoinositides are markers to define the identities of membranes by interacting with phosphoinositide-recognizing effector proteins.1 One phosphoinositide effector class is the PROPPPIN protein family.2,3 PROPPINs (β-propellers that bind polyphosphoinositides) interact with PtdIns3P and PtdIns(3,5)P2 dependent on a conserved FRRG motif.2,4,5 We and others recently determined the structure of the PROPPIN Hsv2 (homologous with swollen vacuole phenotype 2).6-8 The FRRG motif is localized on the circumference of the β-propeller, and each arginine points to one of the two phosphoinositide-binding sites. Our isothermal titration calorimetry (ITC) measurements with PtdIns3P-containing liposomes confirmed the 2:1 binding stoichiometry for PtdIns3P binding to PROPPINs.6
PtdIns3P binding of PROPPINs is a requirement for their functions in autophagy. The three yeast PROPPINs, Atg18, Atg21 and Hsv2, are highly homologous, but differ in their autophagic subtype specificities. Atg18 is a core autophagy protein and thus required for macroautophagy, the cytoplasm-to-vacuole targeting (Cvt) pathway and piecemeal microautophagy of the nucleus (PMN).4,9,10 Atg21 is needed for the Cvt pathway and PMN, whereas for Hsv2 only a requirement for efficient PMN has been reported, and its precise function remains unclear.5,11,12 PtdIns(3,5)P2 binding of Atg18 is linked to a nonautophagic function at the vacuole, where it is involved in vesicular transport from the vacuole to the Golgi, and in regulation of the Fab1-containing PtdIns3P 5-kinase complex.2,13,14 Both phosphoinositide binding sites of Atg18 are also required for normal vacuolar morphology.6
Probing the lipid-binding specificity of a phosphoinositide effector protein is an initial step in its characterization. The simplest procedure is a protein lipid overlay assay, where pure phosphoinositides and other lipids are spotted on nitrocellulose membranes. After incubation with isolated proteins, the membranes are treated similar to immunoblots. Commercially available PIP strips (Echelon Biosciences Inc.) contain all seven naturally occurring phosphoinositides. However, a major disadvantage of protein lipid overlay assays is the frequent detection of interactions, which cannot be reproduced with other methods.15 For example, in a genome-wide analysis of pleckstrin homology (PH) domains in S. cerevisiae, 27 of 33 PH domain-containing proteins bound phosphoinositides in dot blot assays. However, binding could be verified for only 10 out of these 27 proteins by alternative methods.16
Other techniques to characterize protein-phosphoinositide binding include surface plasmon resonance measurements, where liposomes are immobilized on a chip, Förster resonance energy transfer-based liposome binding assays, reflectometric interference spectroscopy and ITC measurements as later shown here.6,7,15 The equipment needed for these methods is not available in every laboratory. Here, we describe how liposome flotation assays are a viable alternative to probe phosphoinositide-binding specificities of proteins, requiring only an ultracentrifuge. An additional advantage of using liposomes is that protein binding can be probed at low phosphoinositide concentrations, which more resembles the in vivo situation, unlike the PIP strips, where pure phosphoinositides are spotted on a nitrocellulose membrane resulting in a high local concentration of lipids. We used S. cerevisiae Hsv2 as an example to probe its phosphoinositide binding specificity.
1.1 General guidelines for the preparation and characterization of liposomes
Lipid stock solutions were prepared with chloroform (see section 3.2 for details). Phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), the phosphoinositide (PtdInsP) isoforms and Texas-Red-phosphatidylethanolamine (Texas-Red-PE) were used for the preparation of small unilamellar vesicles (SUVs). Individual lipids were added in the desired w/w ratio yielding a total of 1 mg lipids and air-dried. We routinely add 2% (w/w) Texas-Red-phosphatidylethanolamine (Texas-Red-PE) to our preparations to visualize the liposomes. Dried lipids were resuspended in 150 µl HP150 buffer (150 mM KCl, 20 mM HEPES pH 7.4) supplemented with 3% (w/v) sodium cholate, loaded onto a self-packed Sephadex G-50 gel filtration column and eluted with HP150 buffer.17 Colored fractions containing the liposomes were pooled and kept at 4°C.
Large unilamellar vesicles (LUVs) were prepared by mixing of the respective lipids with a total weight of 1 mg lipids per preparation. After air drying, the lipid mixture was resuspended in 1 ml HP150 buffer. The lipid suspension was then applied to an extruder using polycarbonate membranes with pore sizes of first 0.4 µm and then 0.1 µm.
We characterized the size distributions of both SUVs and LUVs composed of 2% PtdIns3P, 73% PC, 23% PE and 2% Texas-Red-PE with field-flow fractionation coupled to multi-angle laser light scattering (FFF-MALLS) measurements. With this technique, liposomes of different sizes are first separated according to their diffusion properties (which is related to size, see reference 18 for an in-depth description) followed by conventional static light scattering analysis on monodisperse fractions or bins for absolute size determination. These bins are then compiled to determine the entire size distribution of the liposome sample.
2% PtdIns3P-containing SUVs were homogeneous in size and the average geometric radius was 18 nm (Fig. 1A). This value is in agreement with an average radius of 18 nm determined by electron microscopy measurements of SUVs, prepared with the same protocol.19 In comparison, the 2% PtdIns3P-containing LUVs have radii in the range of 60 nm (Fig. 1B). A diameter of around 100 nm was reported earlier for LUVs prepared with this protocol also using FFF-MALLS measurements for size determination.20
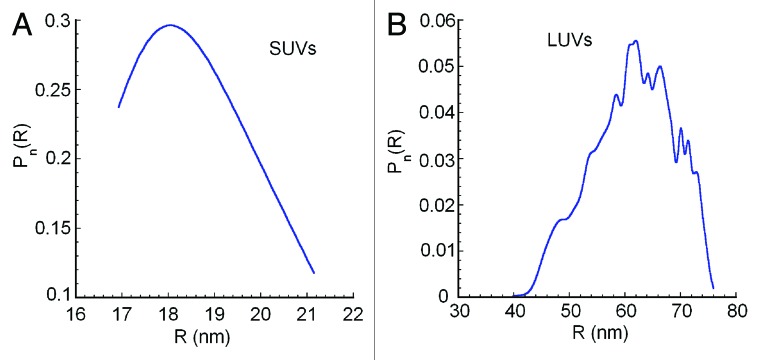
Figure 1. Liposome size distribution obtained by FFF-MALLS. The number size distributions (Pn) portrayed as geometric radii (R) of (A) 2% PtdIns3P-containing SUVs and (B) 2% PtdIns3P-containing LUVs are displayed, confirming the size homogeneity of liposome preparations. Large errors in the determination of the size toward the low end of the elution profile are characteristic at these size ranges and are not computed by the software.
These liposomes can be used without any phospholipid quantification for flotation assays. However, for the ITC analysis the precise phosphoinositide concentrations must be known. We calculate the phosphoinositide concentration from the total phosphate concentration of the liposome-containing solution. We assume that all lipids used for liposome preparation are homogeneously incorporated. For phosphate concentration determination, samples are first treated with perchloric acid and heated to 300°C for 2 h in order to convert the organic bound phosphate to free phosphate before quantification with the phosphomolybdate method as described in detail in section 3.3.21 Since we know the weight ratio of the individual lipids, their molecular weights and the number of phosphorus atoms per phospholipid, the total phosphoinositide concentration present can be calculated. Assuming that the lipids are symmetrically distributed in the lipid bilayer, then 60% of the phosphoinositides are localized in the outer leaflet of small unilamellar vesicles with an average radius of 20 nm, and are accessible for protein binding. In LUVs 50% of phosophoinositides are present on the surface.
1.2 Liposome flotation assays
SUVs and Hsv2 were mixed and overlaid with a noncontinuous Nycodenz gradient and a buffer layer on top. The sample was then spun at 55,000 rpm (275,000 × g) for 90 min. During centrifugation the liposomes migrate to the top buffer layer. If the protein binds to the liposomes, the protein will comigrate in the top buffer layer, but will remain in the lower fractions of the gradient if no binding occurs. After centrifugation, samples were taken from the gradient and analyzed with immunoblotting.
However, care must be taken with the lipid composition of liposomes to avoid nonspecific interactions of the protein with negatively charged phospholipids.15 Therefore, we first identified conditions where binding of Hsv2 to liposomes does not occur in the absence of phosphoinositides. For that purpose, liposomes containing different PS concentrations were prepared and probed both with wild-type Hsv2 and Hsv2FTTG, where the conserved FRRG motif is mutated and which served as a negative control. For SUVs containing either 10% (w/w) or 20% PS binding of both Hsv2 and Hsv2FTTG was observed due to nonspecific electrostatic interactions. In contrast, neither Hsv2 nor Hsv2FTTG bound to neutral liposomes composed of 75% PC, 23% PE, 2% Texas-Red-PE (Fig. 2A).

Figure 2. Optimization of liposome lipid composition. (A) Hsv2 and Hsv2FTTG bound nonspecifically to SUVs containing either 10% or 20% PS. Both proteins did not interact with neutral liposomes consisting of 0% PS, 75% PC, 23% PE, 2% Texas-Red-PE. (B) PtdIns3P was added stepwise to neutral SUVs consisting of PC, PE and Texas-Red-PE (x% PtdIns3P, (75-x)% PC, 2% Texas-Red-PE, 23% PE). At 1% and 2% PtdIns3P only Hsv2 bound to the liposomes, whereas at a PtdIns3P concentration of 5% and above both wild-type and mutant Hsv2 bound nonspecifically. (C) PtdIns3P concentration was gradually increased in LUVs (x% PtdIns3P, (75-x)% PC, 2% Texas-Red-PE, 23% PE). Wild-type Hsv2 bound completely to 5% and 10% PtdIns3P-containing LUVs. Hsv2FTTG did not bind to LUVs. (D) 1% PtdIns3P-containing SUVs and LUVs with the same PtdIns3P accessible surface concentration in solution were used for flotation assays. (E) Binding of wild type and Hsv2FTTG to 1% PtdIns3P-containing SUVs was analyzed for samples treated with 2 mM β-mercaptoethanol.
We then added increasing amounts of PtdIns3P and tested binding of Hsv2 and Hsv2FTTG. At 1% and 2% PtdIns3P only Hsv2 specifically bound to liposomes, whereas at concentrations of 5% PtdIns3P and 10% PtdIns3P Hsv2FTTG also bound, indicating binding due to nonspecific electrostatic interactions (Fig. 2B).
In order to analyze whether differences in membrane curvature might have an effect on PtdIns3P binding of Hsv2 we also performed liposome flotation assays with LUVs. We tested the same lipid compositions with increasing PtdIns3P concentrations as for the SUVs described above. For the LUVs 5% PtdIns3P was required for complete binding of wild-type Hsv2 and no nonspecific binding of the Hsv2FTTG mutant was detected even at 10% PtdIns3P (Fig. 2C). To exclude concentration-dependent effects due to different liposome concentrations in solution, the experiments were repeated with 1% PtdIns3P-containing LUVs and SUVs preparations having the same surface PtdIns3P concentrations in solution (Fig. 2D). The accessible PtdIns3P concentrations were derived from total phosphate concentration determinations done for both SUVs and LUVs preparations. SUVs and LUVs were diluted so that protein liposome mixtures used for flotation assays contained 0.2 µM wild-type Hsv2 and 4.7 µM surface accessible PtdIns3P, respectively. Complete binding for Hsv2 to 1% PtdIns3P-containing SUVs was observed, whereas most of the protein did not bind to the 1% PtdIns3P-containing LUVs, which is consistent with the previous measurements. The differences in PtdIns3P concentration-dependent binding of Hsv2 to SUVs and LUVs might be due to the fact that head groups are more readily accessible in the more strongly curved SUVs so that complete binding is achieved at lower PtdIns3P concentrations in comparison to LUVs.
Hsv2 was purified without reducing agents. The protein contains eight cysteines, which might become oxidized; therefore, we also performed flotation assays in the presence of β-mercaptoethanol as a control. The protein liposome mixtures were incubated with 2 mM β-mercaptoethanol for 30 min at room temperature before addition of the Nycodenz gradient. Both wild-type and Hsv2FTTG proteins behaved the same as in the absence of reducing agents (Fig. 2E).
Once conditions have been established where nonspecific binding does not occur, one can readily test mutants for phosphoinositide binding with liposome flotation assays. In an earlier study we used 2% PtdIns3P-containing SUVs to probe Hsv2 mutants where residues in the phosphoinositide binding sites 1 or 2 were mutated, and identified conserved residues essential for PtdIns3P binding in both sites.6 Here, we show that liposome flotation assays can also be used to probe the phosphoinositide-binding specificity of Hsv2. For this purpose SUVs were prepared containing 1% (w/w) of any of the seven naturally occurring phosphoinositides, 74% PC, 23% PE, 2% Texas-Red-PE and tested for binding of Hsv2. Hsv2 interacts with PtdIns3P and PtdIns(3,5)P2 (Fig. 3).
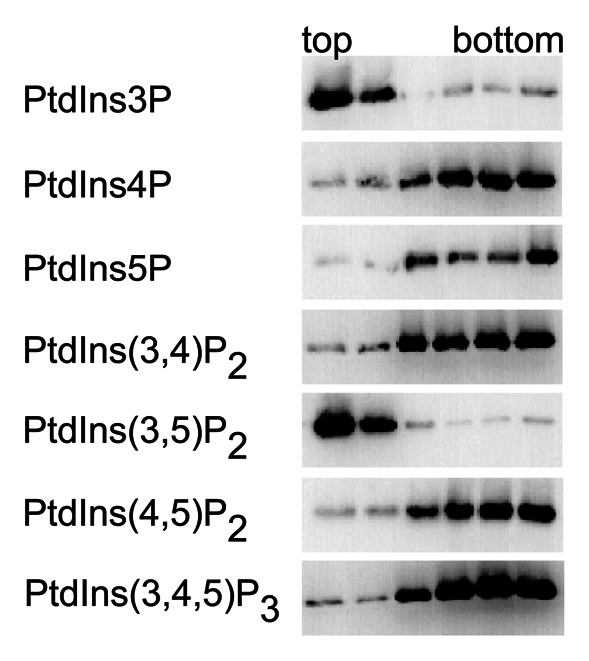
Figure 3. PI binding specificity of Hsv2. Liposome flotation assays were done with SUVs containing one of the seven phosphoinositide isoforms (1% phosphoinositide, 74% PC, 23% PE, 2% Texas-Red-PE).
Phosphoinositide binding of Hsv2 has been characterized previously with PIP strips (Echelon Biosciences Inc.), and two groups reported different observations. For maltose binding protein-tagged Hsv2 strong binding to PtdIns3P, weaker binding for PtdIns4P and PtdIns5P, and a very weak signal for PtdIns(3,5)P2 were reported.5 We previously analyzed binding of GST-Hsv2 to PIP strips and observed strong signals for both PtdIns3P and PtdIns(3,5)P2 and weaker signals for PtdIns4P, PtdIns5P and PtdIns(3,4)P2.6 In contrast, in the liposome flotation assays described here we observed specific binding of Hsv2 to PtdIns3P and PtdIns(3,5)P2 but not to the other isoforms.
1.3 Isothermal titration calorimetry
We quantitatively analyzed PtdIns3P binding of Hsv2 with ITC measurements. Here the ligand is added stepwise to the interaction partner and heat released or absorbed as a result of binding is recorded. The dissociation constant, stoichiometry, enthalpy and entropy of binding can be determined with this method. We first performed ITC measurements with the water soluble short fatty acid chain analog dibutanoyl-PtdIns3P. During these titrations no significant heat changes were detected and a very similar curve was observed when buffer was titrated into buffer in a control experiment (Fig. 4A). We concluded that Hsv2 does not bind to dibutanoyl-PtdIns3P freely in solution.

Figure 4. ITC measurements with Hsv2. (A) Titration of dibutyl-PtdIns3P into Hsv2 (black line). Red trace shows the control experiment where buffer is titrated into buffer. (B) Titration of Hsv2 into 2% PtdIns3P-containing SUVs (black line). Red line shows titration of buffer. (C) Titration of Hsv2 into 2% PtdIns3P-containing LUVs (black trace). Hsv2 binds to the liposomes, but LUVs were prone to precipitation. As a control, buffer was titrated into buffer (red line).
We then performed ITC measurements with Hsv2 and SUVs as used in the flotation assays. The liposomes were composed of 2% PtdIns3P, 73% PC, 23% PE, 2% Texas-Red-PE in HP150 buffer. Hsv2 (concentration range of 50–90 µM) was titrated into liposomes with an accessible PtdIns3P concentration range of 5 to 10 µM. During these measurements large enthalpy changes were observed during the first titration steps, which decreased until saturation was reached (Fig. 4B). We determined a dissociation constant KD of 0.67 ± 0.02 µM and a stoichiometry of 0.50 ± 0.03 for Hsv2 binding to PtdIns3P, showing that Hsv2 has two PtdIns3P binding sites.6 The enthalpy for binding is -18.4 ± 0.8 kcal/mol. Values are the average from four measurements and SEM are given.
We also tried ITC measurements with LUVs composed of 2% PtdIns3P, 73% PC, 23% PE, 2% Texas-Red-PE. During measurements we observed enthalpy changes, but samples precipitated during the titrations (Fig. 4C). We assume that stirring of the sample during ITC measurements lead to precipitation of the LUVs. In contrast no precipitation occurred during ITC measurements with the SUVs.
A likely explanation as to why Hsv2 only interacts with membrane-incorporated PtdIns3P is the requirement of loop 6CD for membrane association. Deletion and replacement of loop 6CD with a GS-linker in K. lactis Hsv2 (KlHsv2259–274GSGSGS) abolishes membrane binding.7 In line with these data, loop 6CD inserts into the membrane in our model for membrane binding of PROPPINs based on docking studies with PtdIns3P and PtdIns(3,5)P2.6
Hsv2 localizes to perivacuolar puncta and endosomes in yeast, and is required for efficient PMN, but no other functions are yet known for the protein.22 Here we showed that Hsv2 binds with high specificity to both PtdIns3P and PtdIns(3,5)P2 with liposome flotation assays, and our ITC measurements proved the 2:1 binding stoichiometry for PtdIns3P binding of PROPPINs. We conclude that the ITC measurements using SUVs and liposome flotation assays described here are useful tools to characterize phosphoinositide binding of PROPPINs that can also be applied to other phosphoinositide-binding proteins.
2. Materials
The following lipids were purchased from Avanti Polar Lipids: PC: L-α-phosphatidylcholine from egg, chicken (840051P); PE: L-α-phosphatidylethanolamine from brain, porcine (840022P); PS: L-α-phosphatidylserine from brain, porcine (840032P); 18:1 PtdIns3P: 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′-phosphate) (850150P); PtdIns4P: L-α-phosphatidylinositol-4-phosphate ammonium salt from brain, porcine (840045P); 18:1 PtdIns5P: 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-5′-phosphate) ammonium salt (850152P); 18:1 PtdIns(3,5)P2: 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,5′-bisphosphate) ammonium salt (850154P); 18:1 PtdIns(4,5)P2: 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-4′,5′-bisphosphate) ammonium salt (850155P); 18:1 PtdIns(3,5)P2: 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,5′-bisphosphate) ammonium salt (850154P); 18:1 PtdIns(3,4,5)P3; 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,4′,5′-trisphosphate) ammonium salt (850156P). Texas-Red-PE: 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (T-1395MP) was bought from Life Technologies. Dibutanoyl phosphatidylinositol-3-phosphate was purchased from MoBiTec (P3004-EC).
Preparations of liposomes were done with the following chemicals: chloroform (Merck, 1.02445.1000), sodium cholate (Sigma Aldrich, C1254), Sephadex G-50 (Sigma Aldrich, G5050) and Nycodenz (Progen, 1002424).
NaH2PO4 (Sigma Aldrich, S-0751), H2SO4 (Merck, 1.00731), HClO4 (AppliChem, A0539), ammonium molybdate (Sigma Aldrich, 09880) and 0.12% (w/v) Triton X-100 (Sigma Aldrich, X100RS) were used for measuring phosphate concentrations.
An Äkta Purifier 10 (GE Healthcare, 28-4062-64), a 5 ml GSTrap FF column (GE Healthcare, 17-5131-01) and a Superdex 75 16/60 column (GE Healthcare, 28-9893-33) were used for protein purification. For cleavage of GST-tag thrombin (MP Biomedicals, 154163) was used. Lysis of E. coli for protein purification was done with a microfluidizer M-110L (Microfluidics Corporation).
Large unilamellar vesicles were prepared with a Mini-extruder (Avanti Polar Lipids, 610000) using polycarbonate membranes with pore sizes of 0.4 µm (Whatman, 800282) and 0.1 µm (Whatman, 800309). For the liposome flotation assays a Sorvall Discovery M150 SE analytical ultracentrifuge (Thermo Scientific) was used with a S55-S swinging bucket rotor (Thermo Scientific, 45594) and 230 µl polycarbonate tubes (Beckman Coulter, 343775). ITC measurements were done with a VP ITC MicroCalorimeter (GE Healthcare). FFF-MALLS measurements were performed with an Eclipse 2 system connected to a DAWN EOS multi-angle scattering setup, both from Wyatt Technology Europe and an Agilent 1100 series HPLC pump with an autosampler (Agilent Technologies).
3. Detailed Protocols
3.1 Purification of Hsv2
Full-length S. cerevisiae GST-Hsv2 was expressed from pGEX-4T-3 in E. coli BL21 (DE3). Bacteria were grown in 4.5 L ZYM-5052 auto-inducing medium for 3 h at 37°C and then 22°C overnight.23 After harvesting, cells were resuspended in 60 ml standard buffer (300 mM NaCl, 30 mM HEPES pH 7.0). Standard buffer was used for all purification steps. Bacteria were lysed in a microfluidizer M-110L (Microfluidics Corporation) and spun at 14,000 rpm for 1 h at 4°C to remove cell debris. Supernatant was loaded onto a 5 ml GSTrap FF column previously equilibrated with at least 5 column volumes (CV) of standard buffer. After loading the supernatant fraction the column was washed with 20 CV standard buffer. GST-Hsv2 was eluted with 5 CV standard buffer supplemented with 20 mM glutathione. To cleave off the GST tag, 200 µl of 1 U/µl thrombin was added to the protein and dialyzed against 1 L standard buffer overnight at 4°C. Dialyzed sample was reapplied onto the GSTrap FF column to remove free GST and uncleaved protein. Hsv2 was then concentrated to a volume of approx. 5 ml and loaded onto a Superdex 75 16/60 column. Purified Hsv2 was concentrated, aliquoted and flash frozen in liquid nitrogen, and stored at −80°C.
3.2 Liposome preparation
Stock solutions of the lipids were prepared with chloroform: 10 mg/ml PC, 25 mg/ml PS, 25 mg/ml PE, 1 mg/ml Texas-Red PE, 1 mg/ml PtdIns3P, 1 mg/ml PtdIns4P, 1 mg/ml PtdIns5P, 1 mg/ml PtdIns(3,5)P2, 1 mg/ml PtdIns(3,4)P2, 1 mg/ml PtdIns(4,5)P2, 1 mg/ml PtdIns(3,4,5)P3 and stored at -20°C.
Small unilamellar vesicles were prepared by adding the individual lipids in the desired w/w ratio yielding a total of 1 mg lipids in a 2 ml microcentrifuge tube. We routinely add 2% (w/w) Texas-Red-PE to stain the liposomes. The lipids are air-dried to evaporate the chloroform. Dried lipids are then resuspended in 150 µl HP150 buffer (150 mM KCl, 20 mM HEPES pH 7.4) supplemented with 3% (w/v) sodium cholate. To remove the cholate dissolved lipids are loaded onto a self-packed Sephadex G-50 gel filtration column (Sigma Aldrich, G5050) with a column volume of approx. 5 ml and eluted with approx. 4 ml HP150 buffer. Pink fractions containing the liposomes (approx. 500 µl) are pooled and kept at 4°C.
Large unilamellar vesicles were prepared with a Mini-extruder (Avanti Polar Lipids, 610000) using polycarbonate membranes with pore sizes of 0.4 µm and 0.1 µm. Lipids were mixed and dried in the same way as for the preparation of SUVs. Then lipids were directly resuspended in 1 ml HP150 buffer and subjected to the Mini-extruder with a 0.4 µm membrane for approx. 25 strokes. In a second step the membrane was exchanged with a 0.1 µm pore size membrane and another 25 strokes were performed.
3.3 Phosphate concentration determination
For phosphate concentration determination, 10 µl of liposomes were diluted with 90 µl water to yield an approx. phospholipid concentration of 0.2 mM, then 20 µl of 70% (w/v) HClO4 and 100 µl 7.2 M H2SO4 are added. The mixture is heated in an open glass test tube at 300°C for 2 h to release the organic bound phosphate. After cooling to room temperature, 1 ml water is added to the sample to dissolve the formed phosphate and 100 µl of a solution containing 3% (w/v) ammonium molybdate and 0.12% (w/v) Triton X-100 is added and mixed. Absorbance is measured at 660 nm precisely 20 min after addition of the ammonium molybdate solution.
For the preparation of a calibration curve we use a 0.2 mM NaH2PO4 stock solution. The measurement points of the curve range from 0- to 30-nmol phosphate. For example, for the 5 nmol data point, 25 µl of 0.2 mM NaH2PO4 are mixed with 20 µl of 70% HClO4, and 100 µl 7.2 M H2SO4 are added and treated in the same manner as described for the liposomes.
3.4 Liposome size determination by field-flow fractionation coupled to multi-angle laser light scattering
The size distribution of liposomes can be conveniently measured by FFF-MALLS. Liposomes of different sizes are first separated according to their diffusion properties, which are related to size, followed by conventional static light scattering analysis on monodisperse fractions or bins for absolute size determination.18 These bins are then compiled to determine the entire size distribution of the liposome sample.
Liposome size distributions are measured essentially as described in references 24, and 25 using an Eclipse 2 system from Wyatt Technology with exactly the same FFF chamber specifications and using 0.1 µm filtered buffer as the eluent [20 mM HEPES, 150 mM KCl, pH 7.4, 0.02% (w/v) NaN3]. The Eclipse system is connected to a DAWN EOS multi-angle light scattering setup and an Agilent 1100 series HPLC pump and autosampler. Briefly, liposomes are diluted 1:50 with HP150 buffer and injected into the FFF using the Agilent autosampler (50 µl). The channel flow is set to 1.0 ml/min and liposomes are injected with a flow rate of 0.20 ml/min and concentrated with a focus flow of 3.0 ml/min for 3 min. Immediately after focus, liposomes are fractionated with a cross-flow of 0.75 ml/min which is reduced linearly to 0 during the course of 35 min.
Elution of the liposomes is detected by the DAWN EOS, and the scattering data are recorded using the manufacturer’s ASTRA software. After baseline correction for detectors 3 through 18, the angle-dependent light scattering intensities of the liposome elution fractions are fitted with the coated-sphere model (real refractive index = 1.33, coating refractive index = 1.45, and coating thickness = 4 nm). The geometric radius obtained from this analysis corresponds to the outer diameter of a spherical liposome. The algorithms in the ASTRA software are then used to reconstruct the number size distribution, that is, the relative number of liposomes with a given radius R.
3.5 Liposome flotation assays
30% and 80% Nycodenz stock solutions are prepared with HP150 buffer. First, 5 µl 2 µM Hsv2 were mixed with 45 µl of the liposomes and incubated for 10 min at room temperature. Then 50 µl 80% (w/w) Nycodenz (Progen, 1002424) in HP150 buffer were added and thoroughly mixed with the protein-liposome sample in 230-µl thick-walled polycarbonate tubes (Beckman Coulter, 343775). Next 50 µl of 30% (w/w) Nycodenz in HP150 were overlaid and finally 30 µl HP150 buffer was added as top layer. Samples are spun at 55,000 rpm (275000 × g) for 90 min at 4°C in a S55-S swinging bucket rotor (Thermo Scientific) in a Sorvall Discovery M150 SE analytical ultracentrifuge (Thermo Scientific). After centrifugation, 30 µl aliquots are taken starting from the top of the gradient and analyzed with SDS-PAGE and western blotting. We used a polyclonal rabbit antibody raised against a C-terminal peptide from S. cerevisiae Hsv2 (CGEPTRWELVRESWREL). We used both the primary anti-Hsv2 antibody and the secondary goat anti-rabbit IgG HRP-labeled antibody (BioRad, 170-6515) in dilutions of 1:5000. Each condition tested was measured three times.
3.6 Isothermal titration calorimetry measurements
Mesurements were done with a VP ITC MicroCalorimeter, which has a cell volume of 1.8 ml and the syringe volume is 500 µl. As a rule of thumb, the concentration of the ligand in the syringe needs to be approx. 7–10 times higher than that of the interaction partner in the cell. Hsv2 was dialyzed against HP150 buffer. For titrations with the soluble PtdIns3P analog dibutanoyl phosphatidylinositol-3-phosphate (MoBiTec, P3004-EC) was dissolved in HP150 buffer at a concentration of 50 µM. The concentration of Hsv2 in the cell was 2 µM. The ligand dibutanoyl-PtdIns3P was added stepwise by 20 injections with 15 µl ligand with a spacing time of 250 sec between injections. Measurements were done at 25°C.
For titrations with the liposomes, SUVs were prepared as described above. Concentrated Hsv2 (concentration range 50–90 µM) was titrated into the liposomes (accessible PtdIns3P concentration range 5–10 µM) with a first injection of 3 µl and followed by 19 injections of 15 µl each with a waiting period of 500 sec between injections to reach baseline level. Data were fitted with a single-site binding model using the MicroCal Origin 7.0 software.
Acknowledgments
We thank Ursel Ries for measuring the phosphate concentrations, Angel Pérez-Lara for help with the preparation of the ITC figures and Reinhard Jahn for his support. This work was funded by a grant of the SFB860 to K.K. and M.T.
Glossary
Abbreviations:
- Cvt
cytoplasm-to-vacuole targeting
- Hsv2
homologous with swollen vacuole phenotype 2
- ITC
isothermal titration calorimetry
- LUVs
large unilamellar vesicles
- FFF-MALLS
field-flow fractionation multi-angle laser light scattering
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PtdIns-phosphates
phosphoinositides
- PtdIns3P
phosphatidylinositol-3-phosphate
- PMN
piecemeal microautophagy of the nucleus
- PROPPINs
β-propellers that bind polyphosphoinositides
- PS
phosphatidylserine
- RifS
reflectometric interference spectroscopy
- SUVs
small unilamellar vesicles
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/23978
References
- 1.Kutateladze TG. Translation of the phosphoinositide code by PI effectors. Nat Chem Biol. 2010;6:507–13. doi: 10.1038/nchembio.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dove SK, Piper RC, McEwen RK, Yu JW, King MC, Hughes DC, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. EMBO J. 2004;23:1922–33. doi: 10.1038/sj.emboj.7600203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dove SK, Dong K, Kobayashi T, Williams FK, Michell RH. Phosphatidylinositol 3,5-bisphosphate and Fab1p/PIKfyve underPPIn endo-lysosome function. Biochem J. 2009;419:1–13. doi: 10.1042/BJ20081950. [DOI] [PubMed] [Google Scholar]
- 4.Krick R, Tolstrup J, Appelles A, Henke S, Thumm M. The relevance of the phosphatidylinositolphosphat-binding motif FRRGT of Atg18 and Atg21 for the Cvt pathway and autophagy. FEBS Lett. 2006;580:4632–8. doi: 10.1016/j.febslet.2006.07.041. [DOI] [PubMed] [Google Scholar]
- 5.Strømhaug PE, Reggiori F, Guan J, Wang CW, Klionsky DJ. Atg21 is a phosphoinositide binding protein required for efficient lipidation and localization of Atg8 during uptake of aminopeptidase I by selective autophagy. Mol Biol Cell. 2004;15:3553–66. doi: 10.1091/mbc.E04-02-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krick R, Busse RA, Scacioc A, Stephan M, Janshoff A, Thumm M, et al. Structural and functional characterization of the two phosphoinositide binding sites of PROPPINs, a β-propeller protein family. Proc Natl Acad Sci U S A. 2012;109:E2042–9. doi: 10.1073/pnas.1205128109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baskaran S, Ragusa MJ, Boura E, Hurley JH. Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy. Mol Cell. 2012;47:339–48. doi: 10.1016/j.molcel.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe Y, Kobayashi T, Yamamoto H, Hoshida H, Akada R, Inagaki F, et al. Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J Biol Chem. 2012;287:31681–90. doi: 10.1074/jbc.M112.397570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan J, Stromhaug PE, George MD, Habibzadegah-Tari P, Bevan A, Dunn WA, Jr., et al. Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol Biol Cell. 2001;12:3821–38. doi: 10.1091/mbc.12.12.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barth H, Meiling-Wesse K, Epple UD, Thumm M. Autophagy and the cytoplasm to vacuole targeting pathway both require Aut10p. FEBS Lett. 2001;508:23–8. doi: 10.1016/S0014-5793(01)03016-2. [DOI] [PubMed] [Google Scholar]
- 11.Krick R, Muehe Y, Prick T, Bremer S, Schlotterhose P, Eskelinen E-L, et al. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell. 2008;19:4492–505. doi: 10.1091/mbc.E08-04-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meiling-Wesse K, Barth H, Voss C, Eskelinen E-L, Epple UD, Thumm M. Atg21 is required for effective recruitment of Atg8 to the preautophagosomal structure during the Cvt pathway. J Biol Chem. 2004;279:37741–50. doi: 10.1074/jbc.M401066200. [DOI] [PubMed] [Google Scholar]
- 13.Jin N, Chow CY, Liu L, Zolov SN, Bronson R, Davisson M, et al. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J. 2008;27:3221–34. doi: 10.1038/emboj.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Efe JA, Botelho RJ, Emr SD. Atg18 regulates organelle morphology and Fab1 kinase activity independent of its membrane recruitment by phosphatidylinositol 3,5-bisphosphate. Mol Biol Cell. 2007;18:4232–44. doi: 10.1091/mbc.E07-04-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narayan K, Lemmon MA. Determining selectivity of phosphoinositide-binding domains. Methods. 2006;39:122–33. doi: 10.1016/j.ymeth.2006.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu JW, Mendrola JM, Audhya A, Singh S, Keleti D, DeWald DB, et al. Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol Cell. 2004;13:677–88. doi: 10.1016/S1097-2765(04)00083-8. [DOI] [PubMed] [Google Scholar]
- 17.Schuette CG, Hatsuzawa K, Margittai M, Stein A, Riedel D, Küster P, et al. Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc Natl Acad Sci U S A. 2004;101:2858–63. doi: 10.1073/pnas.0400044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korgel BA, van Zanten JH, Monbouquette HG. Vesicle size distributions measured by flow field-flow fractionation coupled with multiangle light scattering. Biophys J. 1998;74:3264–72. doi: 10.1016/S0006-3495(98)78033-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, Jahn R. One SNARE complex is sufficient for membrane fusion. Nat Struct Mol Biol. 2010;17:358–64. doi: 10.1038/nsmb.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park Y, Hernandez JM, van den Bogaart G, Ahmed S, Holt M, Riedel D, et al. Controlling synaptotagmin activity by electrostatic screening. Nat Struct Mol Biol. 2012;19:991–7. doi: 10.1038/nsmb.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eibl H, Lands WE. A new, sensitive determination of phosphate. Anal Biochem. 1969;30:51–7. doi: 10.1016/0003-2697(69)90372-8. [DOI] [PubMed] [Google Scholar]
- 22.Krick R, Henke S, Tolstrup J, Thumm M. Dissecting the localization and function of Atg18, Atg21 and Ygr223c. Autophagy. 2008;4:896–910. doi: 10.4161/auto.6801. [DOI] [PubMed] [Google Scholar]
- 23.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Castorph S, Schwarz Henriques S, Holt M, Riedel D, Jahn R, Salditt T. Synaptic vesicles studied by dynamic light scattering. Eur Phys J E Soft Matter. 2011;34:1–11. doi: 10.1140/epje/i2011-11063-2. [DOI] [PubMed] [Google Scholar]
- 25.Hernandez JM, Stein A, Behrmann E, Riedel D, Cypionka A, Farsi Z, et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science. 2012;336:1581–4. doi: 10.1126/science.1221976. [DOI] [PMC free article] [PubMed] [Google Scholar]