

Abstract
Acute lymphoblastic leukemia in infants represents an aggressive malignancy associated with a high incidence (approx. 80%) of translocations involving the Mixed Lineage Leukemia (MLL) gene. Attempts to mimic Mixed Lineage Leukemia fusion driven leukemogenesis in mice raised the question whether these fusion proteins require secondary hits. RAS mutations are suggested as candidates. Earlier results on the incidence of RAS mutations in Mixed Lineage Leukemia-rearranged acute lymphoblastic leukemia are inconclusive. Therefore, we studied frequencies and relation with clinical parameters of RAS mutations in a large cohort of infant acute lymphoblastic leukemia patients. Using conventional sequencing analysis, we screened neuroblastoma RAS viral (v-ras) oncogene homolog gene (NRAS), v-Ki-ras Kirsten rat sarcoma viral oncogene homolog gene (KRAS), and v-raf murine sarcoma viral oncogene homolog B1 gene (BRAF) for mutations in a large cohort (n=109) of infant acute lymphoblastic leukemia patients and studied the mutations in relation to several clinical parameters, and in relation to Homeobox gene A9 expression and the presence of ALL1 fused gene 4-Mixed Lineage Leukemia (AF4-MLL). Mutations were detected in approximately 14% of all cases, with a higher frequency of approximately 24% in t(4;11)-positive patients (P=0.04). Furthermore, we identified RAS mutations as an independent predictor (P=0.019) for poor outcome in Mixed Lineage Leukemia-rearranged infant acute lymphoblastic leukemia, with a hazard ratio of 3.194 (95% confidence interval (CI):1.211–8.429). Also, RAS-mutated infants have higher white blood cell counts at diagnosis (P=0.013), and are more resistant to glucocorticoids in vitro (P<0.05). Finally, we demonstrate that RAS mutations, and not the lack of Homeobox gene A9 expression nor the expression of AF4-MLL are associated with poor outcome in t(4;11)-rearranged infants. We conclude that the presence of RAS mutations in Mixed Lineage Leukemia-rearranged infant acute lymphoblastic leukemia is an independent predictor for a poor outcome. Therefore, future risk-stratification based on abnormal RAS-pathway activation and RAS-pathway inhibition could be beneficial in RAS-mutated infant acute lymphoblastic leukemia patients.
Introduction
Acute lymphoblastic leukemia (ALL) in infants (<1 year of age) represents an aggressive, early onset type of leukemia characterized by high relapse rates during treatment, and an unfavorable clinical outcome.1 This poor prognosis is associated with a high incidence of balanced chromosomal translocations involving the Mixed Lineage Leukemia (MLL) gene, which occur in approximately 80% of the infant ALL cases.1 The most common MLL translocation in infant ALL is t(4;11), in which the N-terminus of MLL (chromosome 11q23) fuses to the C-terminus of AF4 (chromosome 4q23). As the joining of MLL and AF4 occurs in-frame, the t(4;11) translocation generates a unique fusion gene encoding the chimeric, and supposedly oncogenic MLL-AF4 fusion protein. Other recurrent in-frame MLL-rearrangements found among infant ALL patients are t(11;19) and t(9;11), giving rise to the fusion proteins MLL-ENL and MLL-AF9, respectively. The presence of an MLL translocation is the strongest independent predictor of an adverse outcome in infant ALL patients.2
Over the past decades, numerous studies have provided important insights into the biology and pathogenesis of MLL-rearranged ALL, but so far in vivo validation of these achievements is hampered by the lack of genuine animal models accurately recapitulating this severe malignancy. Although various attempts have been made to develop mouse models mimicking leukemogenesis of human t(4;11)-positive ALL, these mice displayed propensities towards developing lymphomas or leukemia with phenotypes that differ significantly from those found in humans.3–5 Another discrepancy between murine MLL-AF4 models and t(4;11)-positive ALL in infants is disease latency. In human infants, MLL translocations arise in utero and rapidly lead to the development of overt leukemia, often at or shortly after birth.6 In contrast, most MLL-AF4 mouse models show mean latency periods of approximately 12–14 months.3,5 Moreover, in MLL-rearranged infant ALL, short disease latency is strongly associated with a poor clinical outcome.2,7
Collectively, these inconsistencies form the basis of the question whether MLL fusion proteins (like MLL-AF4) alone are sufficient to induce ALL, or whether these chimeric proteins require co-operative genetic lesions. Bursen et al. recently found that not MLL-AF4 but its reciprocal fusion protein AF4-MLL (independent of the presence of MLL-AF4) was capable of inducing pro-B ALL in mice, suggesting that in t(4;11)-positive ALL both fusions may function as co-operative oncoproteins.8 Tamai et al. showed that in a transgenic mouse model the latency period of MLL-AF4-induced B-cell leukemia/lymphoma can be significantly shortened by the addition of a KRAS mutation.9 Moreover, recent observations demonstrated that the MLL-AF4 fusion protein can activate Elk-1 through the RAS-pathway, which supports the involvement of RAS signaling in the pathogenesis of MLL-rearranged leukemia.10 Based on these findings, it may be hypothesized that RAS mutations represent important secondary ‘hits’. Recent studies on the incidence of RAS mutations in MLL-rearranged ALL demonstrate inconsistent results in limited patient groups. For instance, Liang et al. reported RAS mutations in 10 of 20 (50%) of the cases, while Mahgoub et al. could not identify RAS mutations among 13 MLL-rearranged ALL samples.11,12 Besides, Tamai et al. speculate that the short latency in their KRAS mutation-positive mouse model is likely due to an acceleration of leukemolymphomagenesis by a collaborative upregulation of HOXA9.9HOXA overexpression is often believed to be a hallmark of MLL-rearranged leukemia and has recently been proposed to be required for leukemia survival of MLL-rearranged acute myeloid leukemia (AML) cells.13 Our recent gene expression profiling study revealed the presence of two distinctive subgroups of MLL-AF4 positive ALL cases; those with and those without HOXA expression, with patients lacking HOXA expression being at high risk of disease relapse.14 Based on this finding, as well as on the report demonstrating a prominent oncogenic role for AF4-MLL,8 and the results demonstrating accelerated MLL-AF4-driven leukemogenesis in the presence of a KRAS mutation, Tamai et al. proposed the following subdivision of t(4;11)-positive ALL: one group representing AF4-MLL-driven and HOXA-independent leukemogenesis, and another group displaying MLL-AF4 and HOXA dependence requiring additional genetic hits, such as RAS mutations, to accelerate leukemogenesis.9
However, the precise frequencies and the potential role (in terms of disease aggressiveness) of RAS mutations in MLL-rearranged infant ALL, and their relation with HOXA expression and/or the presence of AF4-MLL remain unclear. Therefore, we screened a large cohort (n>100) of primary infant ALL samples for NRAS, KRAS and BRAF mutations. To further determine the clinical relevance, these mutations were studied in relation to several clinical parameters, as well as to HOXA expression and the presence of AF4-MLL.
Design and Methods
Patient samples and cell lines
Bone marrow or peripheral blood samples from untreated infants (< 1 year of age) diagnosed with ALL were collected at the institutes participating in the international collaborative INTERFANT protocol.2 Informed consent was obtained according to the Declaration of Helsinki, and approved by the Institutional Review Board of the Erasmus University Medical Center. All samples were processed as described before.15
The t(4;11)-positive cell lines SEM, RS4;11, and MV4-11 were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany), BEL-1 was a kind gift from Dr. Tang (University Paris, France). The t(11;19)-positive cell line KOPN-8 was purchased from The Global Biosource Center (ATCC, Middlesex, UK). All cell lines were maintained as suspension cultures in RPMI 1640 with L-Alanyl-L-Glutamine (Invitrogen Life Technologies, Breda, The Netherlands) supplemented with 10% FCS (Integro, Zaandam, The Netherlands).
DNA and RNA extraction
Genomic DNA and RNA were extracted from approximately 5×106 leukemic cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions, and quantified on a Nanodrop ND-1000 spectrophotometer (Isogen). The integrity of DNA and RNA was assessed on standard 0.8% or 1.5% agarose gels, respectively.
Detection of NRAS, KRAS and BRAF mutations
Using PCR and sequence analysis, mutation hotspots were screened in NRAS and KRAS exon one and two, and in BRAF exon 15.11,16 Amplicons were generated on a 2720 Thermal cycler (Applied Biosystems, Foster City, CA, USA), polymerase chain reaction (PCR) mixture and cycling conditions are described in the Online Supplementary Table S1. Primer sequences were adapted from previous publications11,16 and modified by additional M13 tags (Online Supplementary Table S1). Sequence analysis of both sense and antisense strands was carried out using M13 primers, and the BigDye terminator v1.1 Cycle Sequencing kit (Applied Biosystems) according to the manufacturers’ recommendations, and analyzed on an Applied Biosystems 3130x/Genetic Analyzer. The CLC Workbench software (CLCbio, Aarhus, Denmark) was used to analyze the sequences, references are listed in Online Supplementary Table S2. All mutations were confirmed in replicate sequences.
In vivo prednisone and in vitro prednisolone responses
In vivo prednisone responses, assessed during a prophase of one week of daily systemic prednisone (60 mg/m2) administration before preceding combination chemotherapy, were available for a subset of patients. Responses are defined as good when blast counts in the peripheral blood dropped below 1000 cells/μL, and poor when more than 1000 cells/μL remained detectable.2,17
In vitro drug cytotoxicity of prednisolone (the active metabolite of prednisone) and dexamethasone was available for a subset of patients. The in vitro drug cytotoxcity was determined using 4-day MTT assays as described elsewhere.18
Gene expression profiles
Due to our recently published gene expression profiling (GEP) study,14 microarray data (Affymetrix HU133plus2.0) was available for a part of the patient samples used in this study. Generation of these gene expression profiles has been described before.14 Data was deposited in the GEO database19 under accession number GSE19475. Because of our interest in the relation of HOXA expression and RAS mutations, we extracted and studied the expression of HOXA9 from the existing dataset (probe sets: 209905_at, and 214651_s_at). GEP data was available for 27 of the 38 t(4;11)-positive infant ALL cases.
Statistical analysis
Fisher’s exact test was used to compare mutation frequencies in different patient groups and Mann-Whitney U-Test to compare the median age at diagnosis.
Event-free survival (EFS) and overall survival (OS) curves were estimated using the Kaplan-Meier method and analyzed by log rank (Mantel-Cox’s) tests. EFS is defined as time from diagnosis to death in induction, disease relapse, the emergence of secondary malignancies, or death in complete remission. OS is defined as time from diagnosis to death from any cause. Cumulative incidence of relapse (CIR) is defined as time from complete remission to disease relapse, adjusted for death as competing risk. Patients who did not achieve complete remission were allocated an event at time-point zero in the EFS and CIR analyses. Multivariate analysis of prognostic factors was performed by Cox’s regression model based on EFS and the Wald Backward Test (entry probability P=0.05 and removal probability P=0.10) was used for the joint analysis of age at diagnosis, white blood cell (WBC) counts, in vitro prednisolone response (LC50: lethal concentration to 50% of the leukemic cells), in vivo prednisone response, and RAS mutations. RAS mutations and in vivo prednisone response analyzed as dichotomous variables, the other variables as continuous.
Infant ALL patients without MLL-rearrangements were excluded from these analyses as the prognosis of these patients is significantly more favorable.2 CIR was computed with the statistical environment R version 2.14.0 using Bioconductor packages (R Development Core Team, 2011). The other analyses were performed with SPSS Statistics version 17.0 (SPSS Inc. Chicago, IL, USA). All tests were two-tailed and P<0.05 was considered significant.
Results
RAS and BRAF mutations in infant ALL
RAS and BRAF mutation screening was performed in 109 primary infant ALL samples, as well as in an additional 4 matched relapsed samples. Patients’ characteristics are listed in Online Supplementary Table S3. Overall, in 15 of 109 (13.8%) of the patients a RAS mutation was detected, comprising 7 of 109 (6.4%) patients carrying an NRAS mutation, and 8 of 109 (7.2%) patients bearing a KRAS mutation (Table 1, Figure 1). No BRAF mutations were found. Among patients carrying NRAS mutations, 2 harbored an exon one mutation at codon 12, and 5 an exon two mutation at codon 61. All observed KRAS mutations were located in exon one, of which four at codon 12 and four in codon 13. (Table 1, Figure 1). One mutation was found among the 4 matched relapse samples and displayed an NRAS Gln61Lys mutation that was not present in the corresponding primary diagnosis sample.
Table 1.
RAS mutations.
Figure 1.
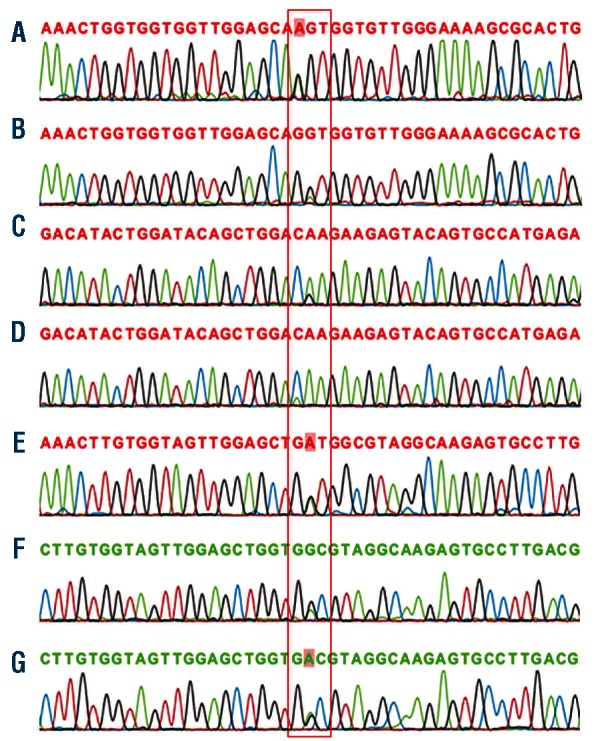
RAS mutations. (A) NRAS exon1 condon12 (Gly>Ser) mutation, corresponding with Patient 2, (B) NRAS exon1 condon12 (Gly>Asp) mutation, corresponding with Patient 14, (C) NRAS exon2 condon61 (Gln>Arg) mutation, corresponding with Patient 4, (D) NRAS exon2 condon61 (Gln>Lys) mutation, corresponding with Patient 13, (E) KRAS exon1 codon12 (Gly>Asp) mutation, corresponding with KOPN-8 cell line, (F) KRAS exon1 condon13 (Gly>Asp) mutation, corresponding with Patient 6, (G) KRAS exon1 condon13 (Gly> Asp) mutation, corresponding with Patient 7.
For the MLL-rearranged ALL cell lines, only KOPN-8 carried a KRAS mutation at exon one, at codon 12 (Gly12Asp) (Figure 1E).
Frequency of RAS mutations among different infant ALL subtypes
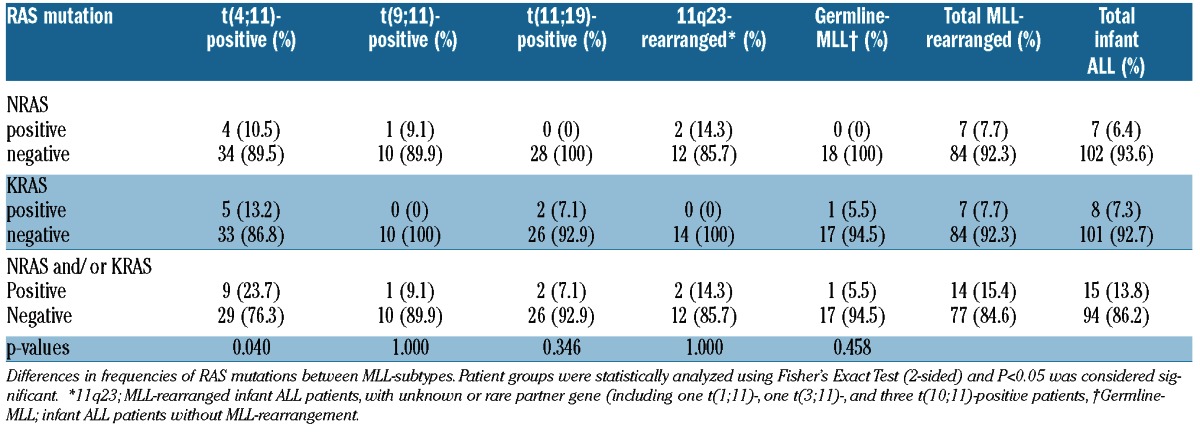
Next we compared the frequencies of RAS mutations among different infant ALL subtypes including patients with t(4;11), t(11;19), t(9:11), and infant ALL patients without MLL translocations. Interestingly, we found a significantly higher frequency of 9 of 38 (23.7%) RAS mutations in t(4;11)-positive infants (P=0.04) compared to the remaining infant ALL cases, with a frequency of 6 of 71 (7.8%). In the other infant ALL subtypes, the frequencies did not differ significantly from the total patient cohort (Table 2).
Table 2.
Frequencies of RAS mutations in MLL-subtypes of infant ALL.
Time of disease onset and RAS mutations
Early onset in MLL-rearranged infant ALL is associated with a poor clinical outcome.2,7 There was no difference in median age at diagnosis between primary RAS mutation-negative MLL-rearranged infant ALL patients (3.8 months; range 0.0–11.5 months) and the RAS-mutated group (5.3 months; range 0.8–11.8 months) (P=0.89). Likewise, RAS mutations did not seem to affect disease latency when we analyzed t(4;11)-positive infant ALL patients alone. Also, dividing patients by their age at diagnosis in the following ordinal categories: <3 months, 3–6 months, 6–9 months, 9–12 months, demonstrated no increased frequencies in any of the age groups for neither the total MLL-rearranged cohort (P=0.51), nor for t(4;11)-positive patients (P=0.31).
WBC count at diagnosis and RAS mutations
High WBC counts at diagnosis has previously been identified as a poor prognostic factor in infant ALL.2 Interestingly, RAS-mutated MLL-rearranged infants appeared to have significantly higher WBCs at diagnosis (P=0.013). Approximately 82% (9 of 11) of the RAS mutation-positive cases showed WBCs higher than 300×109 cells/L, compared to approximately 45% (33 of 73) of the RAS mutation-negative infants. Similarly, among t(4;11)-positive cases, WBCs higher than 300 × 109 cells/L were found in 87.5% (7 of 8) of the mutated cases, and in 41.4% (12 of 29) of the mutation-negative cases (P=0.018).
Drug resistance of RAS-mutated infant ALL patients
A poor in vivo response to prednisone represents an adverse prognostic factor in MLL-rearranged infant ALL,17 and MLL-rearranged infant ALL patients cells are highly resistant to prednisolone and dexamethasone in vitro.20MLL-rearranged infant ALL cells bearing an RAS mutation at diagnosis appeared significantly (P<0.05) more resistant to both glucocortocoids (Figure 2A and B). A comparable trend was only observed for t(4;11)-positive samples, although the differences did not reach statistical significance (Figure 2C and D). No differences were found between the in vivo prednisone response of RAS-mutated and non-mutated MLL-rearranged infant ALL patients (P=0.451), nor by comparing RAS-mutated and non-mutated t(4;11)-positive cases alone (P=0.635). Besides, studying the control cells (without glucocorticoid treatment) in our in vitro cytotoxicity assays, we found RAS-mutated MLL-rearranged infant ALL cells to display significantly (P=0.022) higher endogeneous viability (Online Supplementary Figure S1). Furthermore, we asked whether exposure to glucocorticoids would invoke a positive selection for RAS-mutated cells in samples that ostensibly carry subclonal mutations. Therefore, we performed a time lapse prednisolone exposure experiment and sequenced the RAS mutations in order determine whether the sequence graphs revealed a positive selection of the mutated clone. However, we did not find any signs of positive selection in both patients: the intensity of the peak corresponding to the mutated nucleotide remained equal throughout the experiment (Online Supplementary Figure S2). Suggesting that either the subclone was stable during the experiment or that these mutations may not have been sub-clonal.
Figure 2.

Drug cytotoxicity of RAS-mutated and non-mutated infant ALL patients. (A) In vitro prednisolone cytotoxicity in MLL-rearranged infant ALL patients, (B) In vitro dexamethasone cytotoxicity in MLL-rearranged infant ALL patients, (C) In vitro prednisolone cytotoxicity in t(4;11)-rearranged infant ALL patients, (D) In vitro dexamethasone cytotoxicity in t(4;11)-rearranged infant ALL patients. Mean in vitro cytotoxicity responses in RAS-mutated and non-mutated infant ALL patients were statistically analyzed using Mann-Whitney U-test. Error bars represent standard error of the mean. Cytotoxicity data for prednisolone and dexamethasone was available for 63 and 44 MLL-rearranged infants ALL patients and 26 and 18 t(4;11)-rearranged infants, respectively.
Clinical outcome of RAS-mutated infant ALL patients
Clinical outcome data was available for 79 MLL-rearranged infant ALL cases, with 33 of them being t(4;11)-positive. The presence of a RAS mutation at diagnosis was associated with poor outcome in both the MLL-rearranged infant ALL patients, as well as in t(4;11)-positive cases alone. Among all MLL-rearranged infant ALL patients, the 5-year EFS rates for the RAS mutation-positive and negative cases was 0.0±0.0% versus 32.7±6.0% (P=0.042), and the 5-year OS was 11.1±10.5% versus 45.3±6.0% (P=0.08), respectively (Figure 3A and B). CIR analysis showed a slight tendency towards a higher relapse risk for RAS-mutated cases, with a 3-year CIR of 66.7±15.7% versus 48.1±6.1% in RAS wild-type patients (P=0.119) (Figure 3C). Among the t(4;11)-positive cases comparable, but more distinctive, results were found for the 5-year EFS (P=0.019), 5-year OS (P=0.020), and 3-year CIR (P=0.012) (Figure 3D-F).
Figure 3.

Survival of RAS-mutated and non-mutated infant ALL patients. (A) 5-year event-free survival (EFS), (B) 5-year overall survival (OS), (C) 3-year cumulative incidence of relapse (CIR) for RAS-mutated MLL-rearranged infant ALL patients. Survival data were available for 79 of the 91 MLL-rearranged infant ALL cases. (D) 5-year EFS, (E) 5-year OS, (F) 3-year CIR for RAS-mutated t(4;11)-positive infant ALL patients. Survival data were available for 33 of the 38 t(4;11)-positive infant ALL cases.
RAS mutations in relation to AF4-MLL and HOXA expression in t(4;11)-rearranged infants
We studied the relation between the presence of AF4-MLL and HOXA9 expression in t(4;11)-positive infant ALL samples and the incidence of RAS mutations. The occurrence of RAS mutations did not differ significantly between cases with AF4-MLL (3 of 15) or without AF4-MLL (6 of 23). Re-analyzing our previously published gene expression profiling data we found that all RAS mutation-positive cases lacked HOXA9 expression (Online Supplementary Figure S3). Our earlier observations suggested that t(4;11)-positive infants lacking HOXA expression have a worse prognosis than patients expressing high HOXA levels.14 However, when excluding the RAS mutation-positive cases from this analysis, the association of HOXA expression and clinical outcome was lost (P=0.857). Also, no association between AF4-MLL expression and clinical outcome was detected (P=0.354), even after excluding the RAS-mutated t(4;11)-positive infants (P=0.177). Thus, neither the level of HOXA nor the presence of AF4-MLL expression, but the presence of RAS mutations seems to dictate the poor prognosis in these patients. Next, we asked whether RAS mutations influenced the previously reported prognostic value of high-level FLT3 expression as well.21 Therefore, we studied the overlap between high FLT3 expression and the presence of RAS mutations in our patient cohort, but we could not find any correlation between FLT3 expression and RAS mutations at all. The RAS-mutated infants are equally divided between the patients with either FLT3 high or low expression. Because of this equal distribution, we had no rationale for re-analyzing the previously published prognosis data for FLT3 expression in the same manner as we did with the HOXA expression, where all RAS-mutated patients had low HOXA expression.
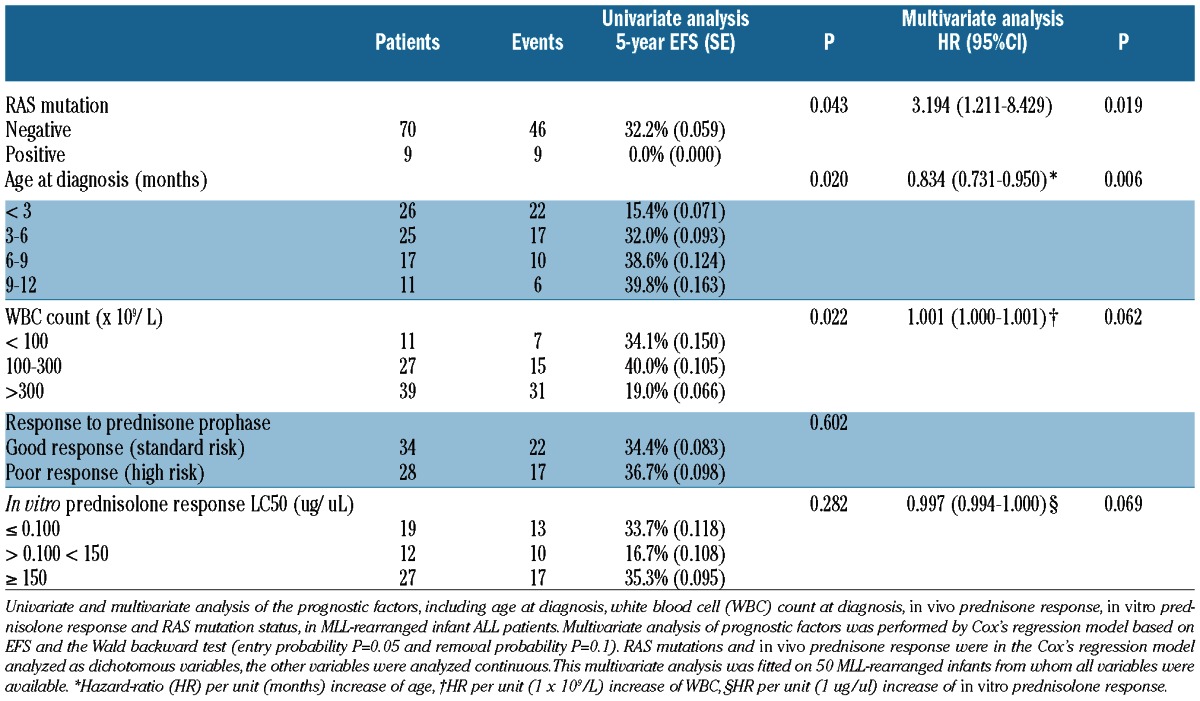
Multivariate analysis of RAS mutations and clinical parameters
Because the previously described clinical parameters in this study are interdependent, we performed a Cox’s regression multivariate analysis, to evaluate the independent prognostic value of RAS mutations. This multivariate analysis was fitted on MLL-rearranged infants (n=50) from whom all prognostic variables were available. We identified the presence of an RAS mutation at diagnosis as an independent predictor (P=0.019) for poor outcome in MLL-rearranged infant ALL, with a hazard-ratio (HR) of 3.194 (95% confidence interval (CI): 1.211–8.429) (Table 3). Besides RAS mutations, low age at diagnosis was identified as an independent predictor (P=0.006, HR: 0.834, 95%CI: 0.731–0.950) for poor outcome in our MLL-rearranged infant ALL cohort. Other variables in the final model were WBC counts at diagnosis (P=0.062, HR: 1.001, 95%CI: 1.000–1.001) and in vitro prednisolone response (P=0.069, HR: 0.997, 95%CI: 0.997–1.000) (Table 3).
Table 3.
Univariate and multivariate analysis of prognostic factors of MLL-rearranged infant ALL patients.
Discussion
Activating RAS mutations, resulting in a proliferative advantage, have been observed in several hematopoietic malignancies including ALL, AML, chronic myelomonocytic leukemia, and juvenile chronic myelogenous leukemia.22–28 Here we report a RAS mutation frequency of approximately 14% in a large (n>100) cohort of infant ALL cases, and a frequency of approximately 24% in infant ALL patients carrying MLL translocation t(4;11). These results are not consistent with previously published studies that reported either high RAS mutation frequency of 50%, or a total absence of RAS mutations in MLL-rearranged ALL.11,12 The observed frequencies in these studies may have been compromised by the small patient numbers. However, these frequencies are in agreement with the previously reported frequencies of 6–20.8% RAS mutations found in childhood ALL.11,29–31
To determine the role of RAS mutations in respect of aggressiveness in MLL-rearranged infant ALL, we compared several clinical parameters in RAS mutation-positive and negative patients. Early onset of a KRAS mutation in an MLL-AF4-positive transgenic mouse model was associated with an early disease onset, and, therefore, seemed to represent a more aggressive leukemia.9 We could not confirm an association between the presence of RAS mutations and an early onset of MLL-rearranged infant ALL. However, our data showed that RAS mutations independently contribute to a poor outcome in MLL-rearranged infant ALL patients. Moreover, MLL-rearranged infant ALL patients carrying RAS mutations also display significantly higher WBC counts at diagnosis, and appeared significantly more resistant to glucocorticoids in vitro.
Although conventional Sanger sequencing certainly is not quantitative, 4 of 7 (57%) of the NRAS mutations and 5 of 8 (62%) of the KRAS mutations appeared to be subclonal in our sequencing graphs. Repeated sequence runs on these samples persistently showed that the peak corresponding to the mutated nucleotide remained markedly smaller than the wild-type nucleotide (e.g. Figure 1D). If indeed a relatively high number of RAS mutations is subclonal, suggesting that not all leukemic cells carry the genetic abnormality, it seems plausible that RAS mutations are acquired as secondary hits after the MLL-fusions arise (e.g. during an MLL-fusion-positive pre-leukemic state, or even during overt leukemia). An alternative explanation could be that RAS mutations are necessary for leukemogenesis and that patients harboring the wild-type RAS gene carry mutations in other genes supporting MLL fusion driven leukemogenesis. As we only use highly pure leukemic samples (>90% leukemic blasts), this supposed subclonality may not only indicate that a certain portion of the leukemic cells remained unaffected, but also it shows that these mutations are leukemia-specific and are unlikely to be present in germline. Unfortunately, we had no opportunity to validate this, as no germ-line samples were available. Nonetheless, although several of the identified RAS mutations may suggest subclonality, we did not find any differences in clinical parameters or outcome between patients harboring ‘subclonal’ or ‘clonal’ RAS mutation (data not shown). In order to confirm subclonality of the RAS mutations as implied by our Sanger sequencing results, we used TOPO® TA Cloning (Invitrogen Life Technologies, Breda, The Netherlands) to sequence single PCR-amplified DNA fragments in three patient samples (Online Supplementary Table S4). We found that in all patients the number of mutated fragments was lower than the expected percentage of approximately 50% in case the mutation would have been clonal. Hence, these results demonstrate that RAS mutations in infant ALL patients can indeed be of a subclonal nature.
The observed presence of a RAS mutation in one of the relapse samples, which was not present in the patient-matched primary diagnostic sample, supports the hypothesis of RAS mutations secondary hits. In line with this, Case et al. recently demonstrated that in matched presentation/relapse samples of childhood ALL patients, KRAS mutations are predominantly found at relapse, and were observed at very low levels in the matched diagnostic samples.32 In combination, these data could suggest that RAS mutations represent secondary hits and that RAS-mutated clones may very well contribute to disease aggressiveness, progression, and relapse.
Finally, our data indicate that RAS-pathway inhibition could be beneficial for infants. Therapy with specific RAS-inhibitors would eradicate the RAS-mutated leukemic clones, but possibly leave the non-mutated MLL-rearranged leukemic cells unaffected, especially in the infants that seem to harbor subclonal RAS mutations. Although specific RAS-pathway inhibitors may not eradicate all leukemic clones, we strongly believe, based on our data, that targeting the RAS-mutated clones could lead to a less aggressive disease period and increased survival-rates. Therefore, we would not suggest RAS-pathway inhibition as a monotherapy, but alongside the current infant ALL therapy. Interestingly, several RAS-pathway inhibitors, like tipifarib and sorafenib, are already available and currently studied in hematologic malignancies in phase I/II trials. Both compounds are well tolerated; however, tipifarib activity did not seem to correlate with RAS mutations or RAS pathway-dependent activation.33 On the other hand, phase I/II studies using sorafenib in AML and myelodysplastic syndrome patients, showed promising results and targeted inhibition of both ERK phosphorylation, as well as FLT3 signaling.34–36 A combined inhibitory effect on both RAS and FLT3 signaling may well be highly effective in the treatment of MLL-rearranged infant ALL as the majority of these patients are also characterized by constitutive FLT3 activation.15
In conclusion, we demonstrate that RAS mutations frequently occur in MLL-rearranged infant ALL cases and especially in t(4;11)-positive infant ALL patients, and their presence represents an independent poor prognostic factor. Therefore, the RAS-signaling pathway could be a potential target for therapeutic intervention, but also provides a rationale for future risk-stratification strategies. However, although RAS mutation-positive patients are at high risk of relapse, the prognosis for RAS mutation-negative patients remains far from favorable. Thus, a continued search for additional mutations, for example in other components of the RAS pathway, that typify an unfavorable outcome, may be beneficial.
Acknowledgments
The authors would like to thank the members and participating institutes of the INTERFANT-99 study for generously providing leukemic samples.
Footnotes
The online version of this paper has a Supplementary Appendix.
Members of the INTERFANT-99 study are as follows: M. Campbell (Programa Infantil Nacional de Drogas Atineoplasicas (PINDA)), M. Felice (Argentina), A. Ferster (Children’s Leukemia Group (CLCG)), I. Hann and A. Vora (UK Children’s Cancer Study Group (UKCCSG)), L. Hovi (Nordic Society of Paediatric Haematology and Oncology (NOPHO)), G. Janka-Schaub (Cooperative Study Group for Treatment of ALL (COALL)), C.K. Li (Hong Kong), G. Mann (Berlin-Frankfurt-Munster Group-Austria (BFM-A)), T. LeBlanc (French ALL Group (FRALLE)), R. Pieters (Dutch Childhood Oncology Group (DCOG)), G. de Rossi and A. Biondi (Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP)), J. Rubnitz (St Jude Children’s Research Hospital (SJCRH)), M. Schrappe (Berlin-Frankfurt-Munster Group-Germany (BFM-G)), L. Silverman (Dana-Farber Cancer Institute (DFCI)), J. Stary (Czech Paediatric Haematology (CPH)), R. Suppiah (Australian and New Zealand Children’s Haematology/Oncology Group (ANZCHOG)), T. Szczepanski (Polish Paediatric Leukemia and Lymphoma Study Group (PPLLSG)), and M. Valsecchi and P. de Lorenzo (Trial Operating Center (CORS)).
Funding
This study was financially supported by research funding from KIKA (Kinderen Kankervrij). Furthermore, RWS was financially supported by the Dutch Cancer Society (KWF Kankerbestrijding).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Greaves MF. Infant leukaemia biology, aetiology and treatment. Leukemia. 1996;10(2): 372–7 [PubMed] [Google Scholar]
- 2.Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007; 370(9583):240–50 [DOI] [PubMed] [Google Scholar]
- 3.Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood. 2006;108(2):669–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14(5):355–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metzler M, Forster A, Pannell R, Arends MJ, Daser A, Lobato MN, et al. A conditional model of MLL-AF4 B-cell tumourigenesis using invertor technology. Oncogene. 2006; 25(22):3093–103 [DOI] [PubMed] [Google Scholar]
- 6.Greaves M. In utero origins of childhood leukaemia. Early Hum Dev. 2005;81(1):123–9 [DOI] [PubMed] [Google Scholar]
- 7.van der Linden MH, Valsecchi MG, De Lorenzo P, Moricke A, Janka G, Leblanc TM, et al. Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-99 protocol. Blood. 2009;114(18):3764–8 [DOI] [PubMed] [Google Scholar]
- 8.Bursen A, Schwabe K, Ruster B, Henschler R, Ruthardt M, Dingermann T, et al. The AF4. MLL fusion protein is capable of inducing ALL in mice without requirement of MLL. AF4. Blood. 2010;115(17):3570–9 [DOI] [PubMed] [Google Scholar]
- 9.Tamai H, Miyake K, Takatori M, Miyake N, Yamaguchi H, Dan K, et al. Activated K-Ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia. 2011;25:888–91 [DOI] [PubMed] [Google Scholar]
- 10.Ng MH, Ng RK, Kong CT, Jin DY, Chan LC. Activation of Ras-dependent Elk-1 activity by MLL-AF4 family fusion oncoproteins. Exp Hematol. 2010;38(6):481–8 [DOI] [PubMed] [Google Scholar]
- 11.Liang DC, Shih LY, Fu JF, Li HY, Wang HI, Hung IJ, et al. K-Ras mutations and N-Ras mutations in childhood acute leukemias with or without mixed-lineage leukemia gene rearrangements. Cancer. 2006;106(4): 950–6 [DOI] [PubMed] [Google Scholar]
- 12.Mahgoub N, Parker RI, Hosler MR, Close P, Winick NJ, Masterson M, et al. RAS mutations in pediatric leukemias with MLL gene rearrangements. Genes Chromosomes Cancer. 1998;21(3):270–5 [PubMed] [Google Scholar]
- 13.Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M, et al. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood. 2009;113(11):2375–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stam RW, Schneider P, Hagelstein JA, van der Linden MH, Stumpel DJ, de Menezes RX, et al. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood. 2010;115(14):2835–44 [DOI] [PubMed] [Google Scholar]
- 15.Stam RW, den Boer ML, Schneider P, Nollau P, Horstmann M, Beverloo HB, et al. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood. 2005;106(7):2484–90 [DOI] [PubMed] [Google Scholar]
- 16.Xu X, Quiros RM, Gattuso P, Ain KB, Prinz RA. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res. 2003;63(15): 4561–7 [PubMed] [Google Scholar]
- 17.Dordelmann M, Reiter A, Borkhardt A, Ludwig WD, Gotz N, Viehmann S, et al. Prednisone response is the strongest predictor of treatment outcome in infant acute lymphoblastic leukemia. Blood. 1999;94(4): 1209–17 [PubMed] [Google Scholar]
- 18.Pieters R, Loonen AH, Huismans DR, Broekema GJ, Dirven MW, Heyenbrok MW, et al. In vitro drug sensitivity of cells from children with leukemia using the MTT assay with improved culture conditions. Blood. 1990;76(11):2327–36 [PubMed] [Google Scholar]
- 19.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramakers-van Woerden NL, Beverloo HB, Veerman AJ, Camitta BM, Loonen AH, van Wering ER, et al. In vitro drug-resistance profile in infant acute lymphoblastic leukemia in relation to age, MLL rearrangements and immunophenotype. Leukemia. 2004;18(3):521–9 [DOI] [PubMed] [Google Scholar]
- 21.Stam RW, Schneider P, de Lorenzo P, Valsecchi MG, den Boer ML, Pieters R. Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood. 2007;110(7): 2774–5 [DOI] [PubMed] [Google Scholar]
- 22.Bar-Eli M, Ahuja H, Foti A, Cline MJ. N-RAS mutations in T-cell acute lymphocytic leukaemia: analysis by direct sequencing detects a novel mutation. Br J Haematol. 1989;72(1):36–9 [DOI] [PubMed] [Google Scholar]
- 23.Bos JL, Verlaan-de Vries M, van der Eb AJ, Janssen JW, Delwel R, Lowenberg B, et al. Mutations in N-ras predominate in acute myeloid leukemia. Blood. 1987;69(4):1237–41 [PubMed] [Google Scholar]
- 24.Cogswell PC, Morgan R, Dunn M, Neubauer A, Nelson P, Poland-Johnston NK, et al. Mutations of the ras protooncogenes in chronic myelogenous leukemia: a high frequency of ras mutations in bcr/abl rearrangement-negative chronic myelogenous leukemia. Blood. 1989;74(8):2629–33 [PubMed] [Google Scholar]
- 25.Janssen JW, Steenvoorden AC, Lyons J, Anger B, Bohlke JU, Bos JL, et al. RAS gene mutations in acute and chronic myelocytic leukemias, chronic myeloproliferative disorders, and myelodysplastic syndromes. Proc Natl Acad Sci USA. 1987;84(24):9228–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyauchi J, Asada M, Sasaki M, Tsunematsu Y, Kojima S, Mizutani S. Mutations of the N-ras gene in juvenile chronic myelogenous leukemia. Blood. 1994;83(8):2248–54 [PubMed] [Google Scholar]
- 27.Needleman SW, Kraus MH, Srivastava SK, Levine PH, Aaronson SA. High frequency of N-ras activation in acute myelogenous leukemia. Blood. 1986;67(3):753–7 [PubMed] [Google Scholar]
- 28.Rodenhuis S, Bos JL, Slater RM, Behrendt H, van 't Veer M, Smets LA. Absence of oncogene amplifications and occasional activation of N-ras in lymphoblastic leukemia of childhood. Blood. 1986;67(6):1698–704 [PubMed] [Google Scholar]
- 29.Lubbert M, Mirro J, Jr, Miller CW, Kahan J, Isaac G, Kitchingman G, et al. N-ras gene point mutations in childhood acute lymphocytic leukemia correlate with a poor prognosis. Blood. 1990;75(5):1163–9 [PubMed] [Google Scholar]
- 30.Wiemels JL, Zhang Y, Chang J, Zheng S, Metayer C, Zhang L, et al. RAS mutation is associated with hyperdiploidy and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia. 2005;19(3): 415–9 [DOI] [PubMed] [Google Scholar]
- 31.Yokota S, Nakao M, Horiike S, Seriu T, Iwai T, Kaneko H, et al. Mutational analysis of the N-ras gene in acute lymphoblastic leukemia: a study of 125 Japanese pediatric cases. Int J Hematol. 1998;67(4):379–87 [DOI] [PubMed] [Google Scholar]
- 32.Case M, Matheson E, Minto L, Hassan R, Harrison CJ, Bown N, et al. Mutation of genes affecting the RAS pathway is common in childhood acute lymphoblastic leukemia. Cancer Res. 2008;68(16):6803–9 [DOI] [PubMed] [Google Scholar]
- 33.Lancet JE, Duong VH, Winton EF, Stuart RK, Burton M, Zhang S, et al. A phase I clinical-pharmacodynamic study of the farnesyltransferase inhibitor tipifarnib in combination with the proteasome inhibitor bortezomib in advanced acute leukemias. Clin Cancer Res. 2011;17(5):1140–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crump M, Hedley D, Kamel-Reid S, Leber B, Wells R, Brandwein J, et al. A randomized phase I clinical and biologic study of two schedules of sorafenib in patients with myelodysplastic syndrome or acute myeloid leukemia: a NCIC (National Cancer Institute of Canada) Clinical Trials Group Study. Leuk Lymphoma. 2010;51(2):252–60 [DOI] [PubMed] [Google Scholar]
- 35.Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, Konopleva MY, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28(11):1856–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borthakur G, Kantarjian H, Ravandi F, Zhang W, Konopleva M, Wright JJ, et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica. 2011;96(1):62–8 [DOI] [PMC free article] [PubMed] [Google Scholar]