

Abstract
STAT transcription factors are regulators of critical cellular processes such as proliferation, survival, and self-renewal. While the activity of these proteins is tightly regulated under physiological conditions, they can become constitutively activated in a broad range of human cancers. This inappropriate STAT activation leads to enhanced transcription of genes that can directly lead to the malignant phenotype. Since STATs are largely dispensable for normal cell function, this has raised the possibility that STATs might be key targets for cancer therapy. Although a number of structure-based strategies have been used to develop STAT inhibitors, an alternate approach is to use cell-based assays that make use of the transcriptional function of STATs. Employing these systems, one can screen large chemical libraries to identify compounds that specifically block the function of a given STAT. This approach can lead to the identification of compounds that inhibit STATs by a variety of mechanisms, and can suggest novel targets for therapy. This type of functional screening strategy has already identified a drug that potently inhibits STAT3, and which is now being evaluated in a clinical trial for patients with chronic lymphocytic leukemia.
Keywords: transcription factors, phosphorylation, drug discovery, cancer therapy, apoptosis
Introduction
Targeted therapy in cancer
After the burst of enthusiasm that followed the clinical development of imatinib mesylate (Gleevec) for the treatment of chronic myelogenous leukemia (CML),1 the clinical impact of targeted therapies for cancer has proceeded at a much more measured pace. This has reflected a number of factors. First, CML is unique, in that its pathogenesis is driven both by the uniform presence of the Bcr-Abl1 fusion tyrosine kinase as well as the critical dependence of the leukemic cells on this one oncogenic event.2 In many other common cancers, tyrosine kinases activated through mutation, fusion or overexpression have been discovered. However, any one oncogenic kinase is much less prevalent. For example, Her2 overexpression in breast cancer, EGF receptor mutation in lung cancer or Flt3 mutation in acute myelogenous leukemia (AML) occur in no more than 20 to 30% of patients. Furthermore, while these oncogenic kinases clearly contribute to the pathogenesis of these cancers, the tumors are not completely dependent on them. Consequently, inhibition of their kinase activity or targeting their extracellular component with antibodies confers only limited therapeutic benefit. Even in cases where kinase inhibition does show efficacy, the rapid emergence of resistance is a recurrent finding and limits the clinical utility. It is becoming increasingly apparent that activation of parallel pathways is a common mechanism by which resistance to kinase inhibitors occurs.
However, the experience gleaned from the clinical development of kinase inhibitors during the past 15 years has provided several key insights. First, targeted kinase inhibitors will only be useful in a small fraction of patients with a given form of cancer. This can still be a powerful approach; for example, although fewer than 5% of patients with non-small cell lung cancer display ALK kinase activation, a high proportion have achieved dramatic results with the ALK inhibitor crizotinib.3 However, it will require that this type of “personalized medicine” be focused on relatively small subsets of patients. Second, kinase inhibitors and other targeted therapies will almost certainly need to be used in combination with other therapies, both to achieve maximal responses (as with monoclonal antibodies such as trastuzumab to Her2 or rituximab to CD20) and to forestall resistance.
These findings have raised the question of whether there is a common convergence point downstream of a variety of kinases and other signaling pathways activated by mutation, such as Ras, Raf or PI3-kinase, which may be targeted therapeutically. Ultimately, these signaling pathways exert most of their effects by regulating the expression or function of transcription factors. In this way, they modulate the expression of genes controlling important cellular processes such as proliferation, survival, invasion and metastasis. Since these oncogenic transcription factors are downstream of a large number of pathways activated through mutations, targeting these proteins holds the promise of extending personalized cancer therapy to a much larger fraction of cancer patients. Furthermore, given the fact that resistance to targeted kinase inhibitors often arises through activation of complementary signaling pathways, targeting transcription factors holds tremendous promise both alone and in conjunction with kinase inhibitors.
STATs as oncogenic transcription factors
Under physiological conditions, transcription factors are generally activated rapidly and transiently in response to cytokines and other stimuli. This allows for tight regulation of the expression of genes whose protein products regulate critical processes such as proliferation, survival, differentiation and invasion. One such group of critical regulators are the STATs, which mediate the effects of a wide variety of cytokines from interferons, to hematologic regulators, to inflammatory mediators.4 Shortly after it became apparent that some family members, like STAT3 and STAT5, were important in signals triggered by hematopoietic growth factors such as erythropoietin and interleukin (IL)-2, it was found that constitutive activation of these proteins is an extremely common finding in nearly all human cancers.5 Consistent with the prediction that oncogenic transcription factors are activated downstream of many activated tyrosine kinases, STATs are activated much more commonly than any single genetic driver mutation.6 For example, in breast cancer, the most commonly activated tyrosine kinase is Her2, whose increased expression and functional activation is driven by genetic amplification. Whereas Her2 is amplified in approximately 25 to 30% of patients, STAT3 is activated constitutively in approximately 70% of patients.7 Thus, while individual analysis is necessary to determine which patients may benefit from STAT3 inhibition, targeting STAT3 or other STAT family members will likely have widespread applicability.
The final consideration in developing an anti-cancer therapy concerns the therapeutic index. While a drug may inhibit a pathway critical for cancer cell proliferation or survival, it is equally important that it not be toxic to normal cells. Evidence from experimental systems to human genetic analyses has provided strong support for the contention that the activity of specific STAT family members can be lost from normal cells without severe consequence, likely due to redundancies in transcriptional regulation under physiological conditions.8,9 Taken together, these findings have suggested that STATs may be particularly worthwhile targets for cancer therapy.
Strategies for Targeting STATs
It has often been argued that transcription factors are not optimal targets for pharmacological inhibition, because their function is not dependent on small surfaces or pockets to which drug-like organic molecules can bind. However, STATs clearly have discrete domains necessary for their function, including the SH2, DNA binding and N-terminal oligomerization domains.10 These sites can certainly be blocked using a number of strategies, and hold promise for therapeutic development.
Screening approaches to identify STAT inhibitors
An alternate approach to structure-based design of STAT inhibitors is to develop an assay that makes use of the transcriptional activity of a specific STAT. One can then screen compounds for their ability to inhibit that transcriptional function while leaving unaffected transcription factors analyzed in a counter-screen.11 Such an assay would allow the analysis of large chemical libraries for STAT inhibitory activity. This is particularly appealing as many natural products and their derivatives have been found to block STAT3 activation, including the green tea component epigallocatechin-3-gallate (EGCG),12 curcumin, which is an extract of the Indian spice turmeric,13 and triterpenoids.14 Although all of these compounds have other effects on cellular signaling as well, their anti-cancer effects appear to be mediated, at least in part, through their suppression of STAT3-dependent gene expression. Thus, compounds such as these might well be identified through STAT3-dependent screening systems.
One such screening approach to identify inhibitors of STAT3, which is activated widely in human cancers, utilizes a luciferase reporter gene under the control of a high-affinity STAT3-responsive promoter.15 When cells stably transfected with this construct are stimulated with a cytokine that activates STAT3 such as IL-6, luciferase is generated which can be detected by luminometry. This type of strategy is suitable for use in multi-well plates, which allows for high throughput screening of chemical libraries. Many cytokines that activate STAT3, particularly those like IL-6 that signal through the gp130 receptor chain, lead to the activation of STAT1 as well. Since these STATs can bind to the same regulatory elements, this can make it difficult to discern effects related to one STAT vs. the other. To circumvent this problem, human fibrosarcoma cells that lack STAT1 have been used in this system, thereby assuring that all of the transcriptional effects were mediated by STAT3.
Finally, a key component of a screening system such as this is to include a counter-screen to exclude compounds that decrease luciferase activity through a trivial mechanism, such as non-specific cytotoxicity, or global effects on transcription or translation. One such approach is to screen in parallel a cell line in which luciferase activity is under the control of another transcription factor, such as NFκB (Fig. 1).
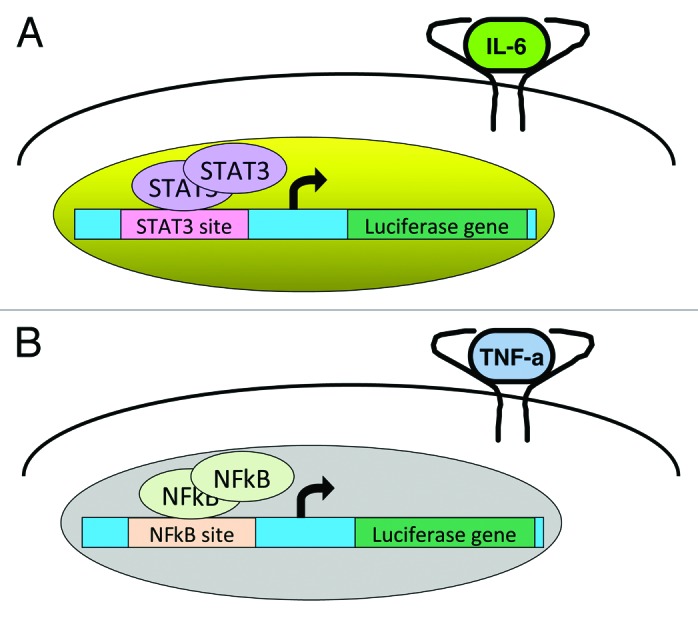
Figure 1. A cell-based screening assay can be designed to test for STAT3 inhibitors in chemical libraries. In one such system, a luciferase reporter gene is ligated to a STAT3-dependent regulatory region, and stably transfected into a cell line (A). When cells are treated with a cytokine that can induce the activation of STAT3, such as IL-6, luciferase is expressed, and its activity can be quantitated by luminometry. In the presence of a STAT3 inhibitor, the induction of luciferase does not occur. To exclude the identification of compounds that are inhibiting STAT3 through a non-specific mechanism, a “counter-screen” is performed on a parallel cell line in which luciferase expression is under the control of an NFκB-responsive promoter that can be induced by tumor necrosis factor (TNF)-α (B). Only compounds that inhibit STAT3-dependent transcription, but not NFκB-dependent transcription, proceed to further validation. This system can also identify compounds that are specific NFκB inhibitors.
When a chemical library, such as the Prestwick collection of approximately 1,200 compounds, is screened with the STAT3 reporter system, the vast majority of compounds have no significant effect on STAT3-dependent luciferase expression (Fig. 2).16 Fewer than 10% decrease STAT3 activity by greater than 25%. However, to exclude non-specific effects, a parallel counter-screen is performed on a similar NFκB-dependent system (Fig. 3). In general, there is little correlation between the effects of compounds in the two assays. However, among the compounds that have the greatest inhibitory effect in the STAT3 assay, many have inhibitory effects in the NFκB assay as well. These compounds most likely are exerting their effects through non-specific mechanisms, and in fact, many are DNA damaging agents and protein synthesis inhibitors. However, a small fraction has significant inhibitory effects in the STAT3 system, and little to no effects in the NFκB system, and these are candidates for further validation.
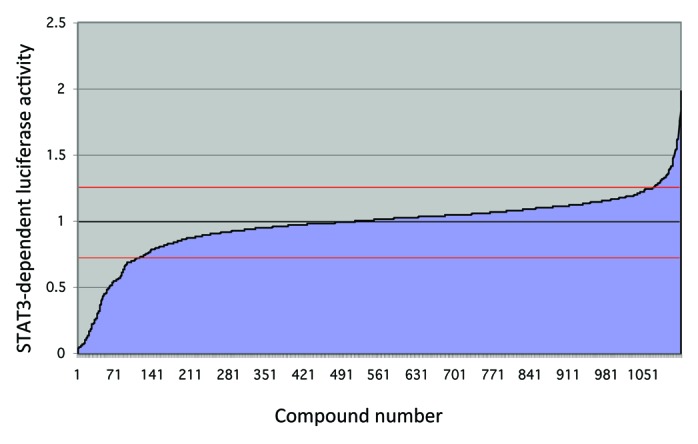
Figure 2. High throughput chemical library screen using a STAT3-dependent luciferase reporter cell line. A value of 1 (black line) represents no change from vehicle-treated cells. Only a small fraction of compounds change IL-6 induced STAT3-dependent luciferase activity by more than 25% (red lines). Of those that do, most are inhibiting luciferase activity, although a small number enhance IL-6 induced luciferase activity.
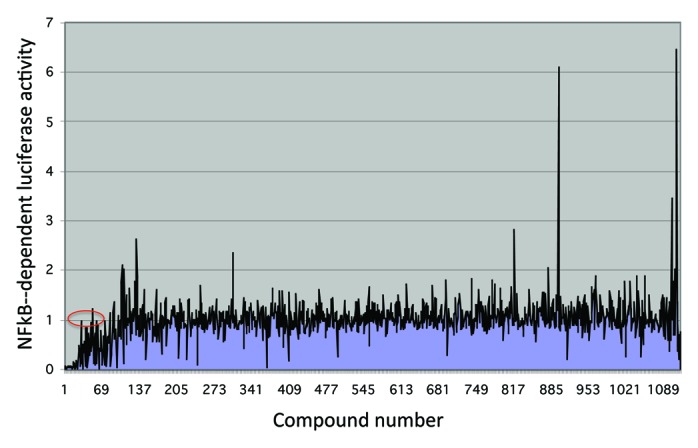
Figure 3. High throughput chemical library screen using an NFκB-dependent luciferase reporter cell line. Compounds are numbered as in Figure 2. Overall, there is a low correlation between activity in the NFκB-dependent assay and the STAT3-dependent assay (Fig. 2). Of the compounds that are inhibitory in the STAT3 assay (compounds 1 through 70), many are inhibitory in the NFκB assay as well, and are likely non-specific. However, a small fraction have no significant effect in the NFκB assay (red oval), and these compounds are appropriate for further validation.
One major strength of this approach is that it views STAT3-dependent transcription from a systems level. This allows effective and specific inhibitors of STAT3 to be discovered that may not directly interact with STAT3 or classical components of the JAK-STAT pathway. For example, if a small molecule specifically inhibited a component of a nuclear import channel that only affected STAT3 dimers, then it would score as a hit. This method has the added advantage of uncovering new targets for therapeutic modulation. Another strength of this approach is that it can also uncover molecules that directly activate or enhance the activation of STAT3 (Fig. 2). While that might not be the direct interest for cancer therapy, it can reveal several key features of STAT signaling. First, it might reveal negative regulators of STAT3 signaling that are important for the biology of this pathway. In addition, activation of STAT3 could be desirable in some situations. For example, increased STAT3 function may protect cardiomyocytes during cardiac ischemia,17 and thus could be a protectant during a myocardial infarction. STAT3 activation may have similar effects in neuronal protection during ischemic stroke.18 Finally, maintaining the pluripotency of embryonic stem (ES) cells can require STAT3 activation,19 and thus a STAT3 activator holds the potential to be important in maintaining ES cells in an undifferentiated state without the need for additional components.
This type of screening strategy also has several shortcomings. First, even if a molecule is uncovered that is a potent and specific inhibitor of STAT3, it can be quite laborious to deconvolute its direct mechanism of action. Although initial biochemical and cellular assays focus on questions such as whether the molecule affects STAT3 tyrosine (or serine) phosphorylation, nuclear localization, DNA binding or transcriptional co-activator recruitment, it may still be difficult to identify the proximal molecular target of an active compound. A second potential shortcoming relates to the mechanism for activating STAT3 in the assay. The use of a cytokine such as IL-6 allows for tightly controlled induction of STAT3 activation during the assay, both in terms of kinetics and amplitude. This is not irrelevant to the constitutive activation of STAT3 occurring in cancer cells, which may be driven by autocrine or paracrine loops involving IL-6 in both epithelial cancers, like breast cancer, and hematological malignancies such as multiple myeloma.20 However, it is possible that active compounds identified in this assay may be specific to IL-6 rather than STAT3.
The final potential shortcoming that should be noted relates to the fact that STAT3 may have important biological effects independent of its ability to regulate gene expression. For example, STATs can associate with the cytoskeleton, and may play an important role in cell motility.21,22 In addition, there is evidence that, at least in certain systems, mitochondrial functioning of STAT3 is critical to tumorigenesis, independent of STAT3 tyrosine phosphorylation, nuclear localization or transcriptional modulation. This function of STAT3 may be particularly important in neoplastic transformation mediated by activated Ras.23 Compounds identified in a transcription-based screen may not modulate the cytoskeletal or mitochondrial function of STAT3, although a transcription-based assay may still be able to detect compounds that affect these other aspects of STAT function. For example, the mitochondrial effects of STAT3 appear to require the phosphorylation of STAT3 on serine 727. Although there is conflicting evidence, phosphorylation of this serine residue may also contribute to the transcriptional function of STAT3,24,25 and thus it is possible that some compounds identified in this type of screen would affect both mitochondrial and transcriptional functions of STAT3. Similarly, agents that affect the interaction of STAT3 and the cytoskeleton may also inhibit the transcriptional function of STAT3.26 In any case, it is clear that this type of cell-based functional screen has been very productive in terms of identifying both inhibitors and activators of STATs that may be therapeutically important.
Identification of STAT3 Inhibitors
STAT3 phosphorylation inhibitors: targeting kinases
One of the first published screens using this approach, a diverse chemical library largely comprised of known bioactive molecules was assessed for its ability to inhibit STAT3-dependent luciferase activity.16 Libraries of bioactive compounds provide both structural diversity as well as an opportunity to evaluate compounds that have often been administered to humans, and for which extensive pharmacokinetic data may be available. Using this approach, nifuroxazide, a drug used in many countries to treat diarrheal illnesses, was identified as an inhibitor of STAT3 transcriptional function with an EC50 of approximately 3 μM. At this concentration, nifuroxazide showed no inhibition of NFκB-dependent transcriptional activity in a counter-screen. In analyzing the mechanism of action of nifuroxazide, it was found that this compound inhibited the activating tyrosine phosphorylation of STAT3, while showing no effect on phosphorylation of STAT3 at serine-727. Furthermore, nifuroxazide inhibited the tyrosine phosphorylation of both Tyk2 and JAK2 (though not JAK1), suggesting that it might be acting as a JAK family tyrosine kinase inhibitor. In multiple myeloma cells, a disease driven commonly by STAT3 activation, nifuroxazide decreased expression of the pro-survival gene Mcl-1, and decreased the survival of both myeloma cell lines with activated STAT3 and primary samples from patients. The survival of myeloma cells is enhanced by co-culturing with bone marrow stromal cells.27 Through both cell-cell contact and the release of soluble factors, stromal cells promote the viability and drug resistance of myeloma cells, often through STAT3-dependent pathways.28 Notably, nifuroxazide showed equal effects in inhibiting myeloma cell survival regardless of the presence of stromal cells.
Although inhibition of STAT3 can directly lead to a loss of viability of myeloma cells, it may also decrease the threshold for these cells to undergo apoptosis when simultaneously treated with agents with distinct mechanisms of action. Reflecting this property, co-treatment of myeloma cells with nifuroxazide and either the histone deacetylase inhibitor depsipeptide (romidepsin) or the MEK inhibitor U0126 showed enhanced cytotoxicity. These findings provided proof-of-concept that a transcription based assay for identifying STAT3 inhibition could identify active compounds with clinical relevance.
STAT phosphorylation inhibitors: non-kinase targets
A second inhibitor of STAT tyrosine phosphorylation that was identified through a cell-based transcriptional screen is pimozide.29 Pimozide has been used clinically as a neuroleptic, and is approved by the FDA in the United States for the treatment of symptoms of Tourette syndrome. Pimozide was found to decrease STAT5 phosphorylation in CML cell lines, in which the fusion oncoprotein Bcr-Abl1 leads to constitutive phosphorylation of STAT5 and increased expression of STAT5 target genes. Pimozide also inhibits STAT5 activation driven by mutated JAK2 in myeloproliferative neoplasms.30 However, pimozide is not a kinase inhibitor. It does not inhibit JAKs, Abl1 or Src family members in in vitro kinase assays, nor does it inhibit all signaling pathways downstream of activated kinases. For example, in CML models, kinase inhibitors such as imatinib decrease the phosphorylation and activation not only of STAT5, but of MAP kinase also. By contrast, pimozide does not decrease MAP kinase phosphorylation, and actually leads to an increase in this activating phosphorylation. Although the reason for this effect has not yet been elucidated, it may relate to the fact that many STAT target genes are negative regulators of signaling events,7 and the loss of these regulators leads to increased activity of other pathways. A correlate of this observation is that the combination of a STAT inhibitor such as pimozide and an inhibitor of the MAP kinase pathway, like the MEK inhibitor U0126, would be expected to be particularly effective in killing CML cells. In fact, this combination leads to synergistic killing in cell line models.
Like kinase inhibitors, pimozide decreases expression of STAT5 target genes, and decreases viability of CML cell lines and primary CML cells through induction of apoptosis. Since STAT5 is involved in the response to cytokines in normal hematopoiesis, the question arises as to whether inhibition of STAT5 would be expected to have toxicity to normal cells. The survival of normal mature hematopoietic cells, such as peripheral blood mononuclear cells, is unaffected by pimozide. Furthermore, in vitro colony formation by CD34+ cells from healthy donors is only minimally affected by pimozide. These findings also reflect the fact that in clinical use pimozide has not been found to have hematopoietic toxicity.
The effects of pimozide are not limited to CML, and this drug has shown efficacy in other myeloid malignancies. For example, in AML, another disease in which aberrant STAT activation (STAT5 and/or STAT3) is common, pimozide can also decrease STAT5 phosphorylation and STAT5-dependent gene expression.31 Similar to CML, in AML cell lines driven by an activating mutation in the cell surface tyrosine kinase Flt3, pimozide shows synergistic effects on loss of viability and induction of apoptosis when combined with kinase inhibitors targeting Flt3, including midostaurin (PKC412) and the multi-kinase inhibitor sunitinib. Notably, pimozide was non-toxic and highly active in a murine model of mutant Flt3-driven leukemia.
Among the advantages of targeting a STAT is the ability to overcome the resistance to kinase inhibitors that commonly develops in clinical practice.32 In CML, a range of point mutations arising within Bcr-Abl1 can mediate resistance to imatinib. One mutation in particular, T315I, confers resistance to all of the presently available approved kinase inhibitors. If STAT5 is a key downstream mediator of the effects of Bcr-Abl1, then it would be predicted that inhibition of STAT5 would be equally effective in the presence of such a mutation. In fact, the sensitivity to pimozide of hematopoietic cells rendered growth factor independent by transfection of Bcr-Abl1 is identical regardless of whether the cells express the wild-type fusion protein or the mutant form carrying T315I.29 This ability to overcome the effects of even these so-called “gatekeeper” mutations is a principal advantage of a STAT inhibitor like pimozide. In contrast to kinases, naturally occurring mutations in STATs are extremely rare, perhaps reflecting the limited tolerance of transcription factors to structural modification. This suggests that resistance to drugs targeting STATs may be much less likely to occur through mutations in the STATs themselves.
It is sometimes suggested that blocking sequential steps in the same biological pathway is unlikely to provide any therapeutic benefit over complete inhibition of a single step in that pathway. However, one might predict that the combination of a STAT inhibitor and an inhibitor of an upstream kinase might have several benefits. First, if there is incomplete inhibition of either target, perhaps related to pharmacokinetic considerations, then the combination may be more likely to suppress the entire pathway more completely. In fact, the combination of imatinib and pimozide leads to a synergistic induction of apoptosis and decrease in viable cell number.33 This is similar to what is seen when pimozide is combined with Flt3 kinase inhibitors in models of AML, or with JAK2 inhibitors in models of myeloproliferative neoplasms.30,31 Second, co-administration of a kinase inhibitor and a STAT5 inhibitor would also be expected to remove the selective pressure for the emergence of resistance-causing kinase mutations or mutational activation of another kinase. Thus, dual inhibition of STATs and upstream kinases may be a particularly effective therapeutic combination.
One final consideration concerns the fact that patients with CML generally need to continue treatment with kinase inhibitors indefinitely to suppress the leukemia. The concern is that a small population of self-renewing cells, sometimes referred to as leukemic stem cells, is able to persist in the presence of kinase inhibitors. These cells may be relatively quiescent or they may be able to maintain activation of STAT5-dependent survival pathways through alternative signaling pathways. If this latter hypothesis is correct, then it would be predicted that an inhibitor directed at STAT5 specifically might have efficacy against this critical population. In fact, pimozide completely inhibits the development of in vitro colonies from CD34+ cells from patients with CML,33 raising the possibility that STAT-directed therapy might be able to eradicate these putative tumor stem cells.
One other aspect of pharmacological STAT inhibition has important implications for understanding both biology and therapeutic strategies. In developing drugs, a key pharmacological issue is what drug levels must be achieved to functionally inhibit the target, and for what period of time is it necessary to maintain these levels to achieve a therapeutic effect. Since STATs regulate pro-survival genes whose continued expression is necessary to maintain the viability of a cancer cell, it may be that even intermittent inhibition of STAT-dependent transcriptional activity is sufficient to have therapeutic benefit.30 This can be important for drugs that have short half-lives in vivo or that may have an unfavorable side effect profile. There is evidence that even transient exposure to pimozide may be sufficient to trigger cell death in myeloid neoplasms, and further understanding of this therapeutic interval is likely to be important for clinical translation of this approach.30
STAT inhibitors that do not affect phosphorylation
Although much effort in the development of STAT inhibitors has focused on compounds that block the activating tyrosine phosphorylation, a particular advantage of a transcription-based screening approach is that it can identify molecules that specifically inhibit the function of STATs without altering tyrosine phosphorylation. This has a number of advantages. First, it can help identify indirect modulators of STAT transcriptional function, which can reveal new insights about other aspects of this signaling pathway, and may reveal unappreciated targets for therapeutic development. One such inhibitor that emerged from a screen of known bioactive compounds is pyrimethamine, an anti-microbial drug.11,34 Pyrimethamine, which is FDA-approved for the treatment of malaria and toxoplasmosis, inhibits STAT3 transcriptional activity without affecting STAT3 tyrosine phosphorylation at low micromolar concentrations. Pyrimethamine also shows no effects on the highly homologous transcription factor STAT5. Reflecting its mechanism-specific effects, pyrimethamine causes a loss of viability of multiple myeloma cells characterized by constitutive STAT3 activation, but not those that lack this molecular hallmark. Furthermore, as predicted by its good safety record in humans, pyrimethamine has no effect on the viability of peripheral blood mononuclear cells (PBMCs) from healthy donors. Finally, high throughput profiling of human cancer cell lines has suggested that pyrimethamine may be effective against a broad spectrum of cancers.11
Given that pyrimethamine can inhibit STAT3 transcriptional function independent of STAT3 tyrosine phosphorylation, it would be expected to have activity in cancers characterized by STAT3 that is transcriptionally active through an alternate mechanism like serine phosphorylation, as is seen in chronic lymphocytic leukemia (CLL).35,36 Given that pyrimethamine is already known to be safe in humans, and that concentrations sufficient to block STAT3 function can be achieved with standard dosing regimens, a clinical trial of pyrimethamine in CLL has been initiated (ClinicalTrials.gov identifier: NCT01066663). Since most patients with CLL have large numbers of circulating leukemia cells, a clinical trial in this disease also affords the opportunity to recover tumor cells from patients on therapy to determine whether STAT3-dependent transcriptional function is being inhibited in the patients. This information will be critical for designing subsequent studies employing STAT inhibition.
Inappropriate STAT3 activation is also found in a hyperproliferative though non-neoplastic disease, polycystic kidney disease (PCKD). Pyrimethamine shows therapeutic effects in in vitro and animal models of this disease, though at concentrations in the mid-micromolar range.34 At these concentrations, pyrimethamine does cause some decrease in STAT3 tyrosine phosphorylation and a corresponding increase in STAT1 phosphorylation, so this drug may have overlapping concentration-dependent mechanisms for inhibiting STAT3 function.
Activation of STAT1 as a Therapeutic Target
STAT1, which mediates the effects of interferons, can exert cytostatic, pro-apoptotic and anti-angiogenic effects.37,38 Consequently, STAT1 can oppose some of the pro-tumorigenic effects of STAT3, and may exert a therapeutic effect in cancer. The nature of transcription-based screens makes it equally easy to identify compounds that activate or enhance transcriptional function as those that inhibit it. This approach was used to identify activators of STAT1-dependent transcription from a chemical library.39 One compound identified through this approach, 2-(1,8-naphthyridin-2-yl)phenol (2-NP), increases expression of endogenous STAT1 target genes. 2-NP exerts this effect by prolonging the time that STAT1 remains phosphorylated after being activated. Related to this effect, 2-NP enhances the ability of interferon-γ to inhibit the proliferation of tumor cells. As predicted by its mechanism, it only exerts this effect in cancer cells expressing STAT1, but not in cancer cells that lack expression of this transcription factor.
Alternative Approaches to Screens for STAT3 Inhibitors
Cell-based assays for drug discovery have generally focused on mammalian cells, for their obvious relevance to human cellular physiology and disease. However, cells obtained from other model organisms may have certain advantages, such as reduced genomic redundancy, that may be beneficial. A rudimentary STAT signaling system is present in Drosophila, in which ligands of the Unpaired family interact with a single receptor to activate one JAK, which can phosphorylate and activate a single STAT. Using Drosophila cells with a stably introduced STAT-responsive luciferase reporter, a compound library of polysubstituted imidopiperidines was screened.40 To exclude non-specific toxicity, a Renilla luciferase reporter, whose activity can be distinguished from the standard firefly luciferase, was also introduced into these cells. Using this approach, a compound termed AUH-6–96 was identified that decreased STAT transcriptional activity through inhibition of phosphorylation. This compound was then found to decrease IL-6 induced STAT3 phosphorylation as well as the constitutive STAT3 phosphorylation found in the Hodgkin lymphoma cell line L540, the breast cancer cell line MDA-MB-468 and the prostate cancer cell line DU145. AUH-6–96 decreased expression of STAT3-dependent pro-survival genes such as Bcl-xL and Bcl-2 and induced apoptosis in L540 cells. Suggesting that its effects are mechanism-specific, this compound decreased viability of cancer cell lines with activated STAT3, but not those without. These findings suggest that a broad array of cellular systems may be useful in identifying signaling inhibitors.
Although a transcription-based assay has been very productive and has the capability of uncovering diverse mechanisms to modulate STAT function, other cell-based approaches have also been successfully employed to identify STAT3 inhibitors. One strategy is to use antibodies specific for the tyrosine phosphorylated form of STAT3 to perform a well-based “cytoblot” to detect compounds that inhibit this activating phosphorylation event. Using this approach to screen the National Cancer Institute Diversity Set of 1992 compounds, JSI-124 (curcurbitacin I), was identified and validated as a STAT3 phosphorylation inhibitor, which showed therapeutic activity in cell culture-based and animal models of STAT3-driven tumors.41
A second approach that has been used employs microscopic imaging to identify compounds that block cytokine-induced nuclear translocation of a STAT3 construct that expresses a fluorescent protein. This approach cannot only identify inhibitors of STAT3 tyrosine phosphorylation, but it can also potentially identify inhibitors targeting proteins necessary for the nuclear translocation of STAT3. Using this approach to screen the National Cancer Institute Diversity Set, SD-1029 was identified, and found to be an inhibitor of STAT3 tyrosine phosphorylation.42 SD-1029 appears to act by inhibiting the kinase JAK2, which mediates STAT3 tyrosine phosphorylation in a range of tumor types. In addition, this compound decreases expression of STAT3 target genes and induces apoptosis in breast and ovarian cancer cell lines. Reflecting the fact that many STAT3 target genes promote survival and resistance to chemotherapy, SD-1029 enhances apoptosis when combined with the cytotoxic drug paclitaxel. These types of combinations may be an important strategy for the optimal clinical translation of STAT inhibitors, which may not be sufficient to induce maximal cell killing on their own.
Elucidating the Mechanism of Action of Screen-Derived Compounds
As noted, chemical library screens have been very productive in identifying molecules that modulate transcription factor function, and have even allowed the identification of a STAT3 inhibitor that is currently in a clinical trial. Given that the assay is agnostic as to the mechanism of action of a compound, it allows the identification of molecular probes that modulate a variety of mechanisms by which STATs are regulated. A major shortcoming of this approach, however, is that the direct target of the compounds identified may not be immediately apparent. The target of compounds that act through kinase inhibition, like nifuroxazide, can be deconvoluted relatively rapidly. However, the proximal target of a compound like pimozide, which decreases STAT tyrosine phosphorylation without obvious kinase inhibitory activity, may be more obscure. Such an agent may inhibit a co-activator of a kinase or it may enhance the activity of a negative regulator of STAT signaling, such as a phosphatase. It can be even more difficult to elucidate the target of a compound like pyrimethamine, which decreases STAT3 transcriptional function without necessarily affecting STAT tyrosine phosphorylation or DNA binding. Presumably compounds in this class are inhibiting a co-activator of transcription that partners with STAT3.
A number of strategies can be employed to identify the direct target of these compounds including testing of individual candidates, using derivatized drugs for affinity isolation of targets, performing RNA interference screens to identify gene products necessary for the function of a compound, or analyzing cell lines selected for resistance to the effect of an agent. However, these approaches can be time-consuming, and do not guarantee success. As is the case with pyrimethamine, one can argue that it is not always necessary to identify a direct target to proceed to a clinical trial with a functionally active STAT inhibitor. However, both to understand the biological significance of a chemical probe and to fully exploit its therapeutic potential, direct identification of a target will almost always be a necessary, though challenging, component of evaluation of compounds identified through this approach.
Conclusion
An abundance of evidence supports the roles of STATs, particularly STAT3 and STAT5, as key mediators of oncogenic signaling pathways. Since STATs can be inhibited in normal cells without significant consequence, STATs represent very appealing targets for the rational molecular therapy of cancer. Cell-based functional assays of STAT transcriptional activity can allow the identification of specific and effective inhibitors of individual STATs. Furthermore, this approach can allow the identification of unappreciated regulators of STAT signaling, novel targets for therapeutic intervention and lead compounds for clinical trials evaluating the efficacy of this approach.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/22662
References
- 1.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 3.Kwak EL, Bang Y-J, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darnell JE., Jr. STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 5.Frank DA. STAT signaling in the pathogenesis and treatment of cancer. Mol Med. 1999;5:432–56. [PMC free article] [PubMed] [Google Scholar]
- 6.Levy DE, Inghirami G. STAT3: a multifaceted oncogene. Proc Natl Acad Sci U S A. 2006;103:10151–2. doi: 10.1073/pnas.0604042103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65:5054–62. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- 8.Garcia R, Yu C-L, Hudnall A, Catlett R, Nelson KL, Smithgall T, et al. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997;8:1267–76. [PubMed] [Google Scholar]
- 9.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 10.Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–7. doi: 10.1016/S0955-0674(00)00199-X. [DOI] [PubMed] [Google Scholar]
- 11.Nelson EA, Sharma SV, Settleman J, Frank DA. A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Oncotarget. 2011;2:518–24. doi: 10.18632/oncotarget.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masuda M, Suzui M, Weinstein IB. Effects of epigallocatechin-3-gallate on growth, epidermal growth factor receptor signaling pathways, gene expression, and chemosensitivity in human head and neck squamous cell carcinoma cell lines. Clin Cancer Res. 2001;7:4220–9. [PubMed] [Google Scholar]
- 13.Kim HY, Park EJ, Joe EH, Jou I. Curcumin suppresses Janus kinase-STAT inflammatory signaling through activation of Src homology 2 domain-containing tyrosine phosphatase 2 in brain microglia. J Immunol. 2003;171:6072–9. doi: 10.4049/jimmunol.171.11.6072. [DOI] [PubMed] [Google Scholar]
- 14.Liby K, Voong N, Williams CR, Risingsong R, Royce DB, Honda T, et al. The synthetic triterpenoid CDDO-Imidazolide suppresses STAT phosphorylation and induces apoptosis in myeloma and lung cancer cells. Clin Cancer Res. 2006;12:4288–93. doi: 10.1158/1078-0432.CCR-06-0215. [DOI] [PubMed] [Google Scholar]
- 15.Nelson EA, Walker SR, Kepich A, Gashin LB, Hideshima T, Ikeda H, et al. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood. 2008;112:5095–102. doi: 10.1182/blood-2007-12-129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson E, Hideshima T, Gashin L, Walker SR, Lynch RA, Chauhan D, et al. Nifuroxazide Inhibits STAT3 Function and Shows Potent Anti-Tumor Activity Against Multiple Myeloma. Blood. 2006;108:3450. [Google Scholar]
- 17.Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, et al. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation. 2001;104:979–81. doi: 10.1161/hc3401.095947. [DOI] [PubMed] [Google Scholar]
- 18.Yamashita T, Sawamoto K, Suzuki S, Suzuki N, Adachi K, Kawase T, et al. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J Neurochem. 2005;94:459–68. doi: 10.1111/j.1471-4159.2005.03227.x. [DOI] [PubMed] [Google Scholar]
- 19.Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–60. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9. doi: 10.1016/j.ccr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Perez M, Salazar EP. A role for the cytoskeleton in STAT5 activation in MCF7 human breast cancer cells stimulated with EGF. Int J Biochem Cell Biol. 2006;38:1716–28. doi: 10.1016/j.biocel.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Ma X, Sayeski PP. Identification of tubulin as a substrate of JAK2 tyrosine kinase and its role in JAK2-dependent signaling. Biochemistry. 2007;46:7153–62. doi: 10.1021/bi700101n. [DOI] [PubMed] [Google Scholar]
- 23.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuringa JJ, Schepers H, Vellenga E, Kruijer W. Ser727-dependent transcriptional activation by association of p300 with STAT3 upon IL-6 stimulation. FEBS Lett. 2001;495:71–6. doi: 10.1016/S0014-5793(01)02354-7. [DOI] [PubMed] [Google Scholar]
- 25.Wen Z, Darnell JE., Jr. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997;25:2062–7. doi: 10.1093/nar/25.11.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker SR, Chaudhury M, Nelson EA, Frank DA. Microtubule-targeted chemotherapeutic agents inhibit signal transducer and activator of transcription 3 (STAT3) signaling. Mol Pharmacol. 2010;78:903–8. doi: 10.1124/mol.110.066316. [DOI] [PubMed] [Google Scholar]
- 27.Mitsiades CS, Mitsiades NS, Munshi NC, Richardson PG, Anderson KC. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: interplay of growth factors, their receptors and stromal interactions. Eur J Cancer. 2006;42:1564–73. doi: 10.1016/j.ejca.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 28.Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NFkappa B. Blood. 1996;87:1104–12. [PubMed] [Google Scholar]
- 29.Nelson EA, Walker SR, Kepich A, Terrell S, Gashin L, Frank DA. Pimozide Inhibits STAT5 Signaling in Chronic Myelogenous Leukemia and Reduces the Viability of Both Imatinib Sensitive and Imatinib Resistant Cells. Blood. 2007;110:2953. [Google Scholar]
- 30.Bar-Natan M, Nelson EA, Walker SR, Kuang Y, Distel RJ, Frank DA. Dual inhibition of JAK2 and STAT5 enhances killing of myeloproliferative neoplasia cells. Leukemia. 2012;26:1407–10. doi: 10.1038/leu.2011.338. [DOI] [PubMed] [Google Scholar]
- 31.Nelson EA, Walker SR, Xiang M, Weisberg E, Bar-Natan M, Barrett R, et al. The STAT5 inhibitor pimozide displays efficacy in models of AML driven by FLT3 mutations. Genes Cancer. 2012 doi: 10.1177/1947601912466555. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–5. doi: 10.1182/blood.V99.9.3472. [DOI] [PubMed] [Google Scholar]
- 33.Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117:3421–9. doi: 10.1182/blood-2009-11-255232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takakura A, Nelson EA, Haque N, Humphreys BD, Zandi-Nejad K, Frank DA, et al. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum Mol Genet. 2011;20:4143–54. doi: 10.1093/hmg/ddr338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank DA, Mahajan S, Ritz J. B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J Clin Invest. 1997;100:3140–8. doi: 10.1172/JCI119869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, et al. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010;115:2852–63. doi: 10.1182/blood-2009-10-230060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–92. doi: 10.2174/1566524023362456. [DOI] [PubMed] [Google Scholar]
- 38.Battle TE, Lynch RA, Frank DA. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Res. 2006;66:3649–57. doi: 10.1158/0008-5472.CAN-05-3612. [DOI] [PubMed] [Google Scholar]
- 39.Lynch RA, Etchin J, Battle TE, Frank DA. A small-molecule enhancer of signal transducer and activator of transcription 1 transcriptional activity accentuates the antiproliferative effects of IFN-gamma in human cancer cells. Cancer Res. 2007;67:1254–61. doi: 10.1158/0008-5472.CAN-06-2439. [DOI] [PubMed] [Google Scholar]
- 40.Kim BH, Yin C-H, Guo Q, Bach EA, Lee H, Sandoval C, et al. A small-molecule compound identified through a cell-based screening inhibits JAK/STAT pathway signaling in human cancer cells. Mol Cancer Ther. 2008;7:2672–80. doi: 10.1158/1535-7163.MCT-08-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–9. [PubMed] [Google Scholar]
- 42.Duan Z, Bradner JE, Greenberg E, Levine R, Foster R, Mahoney J, et al. SD-1029 inhibits signal transducer and activator of transcription 3 nuclear translocation. Clin Cancer Res. 2006;12:6844–52. doi: 10.1158/1078-0432.CCR-06-1330. [DOI] [PubMed] [Google Scholar]