

Abstract
Perinatal nicotine exposure caused a gender-dependent heightened vascular response to angiotensin II (Ang II) and increased blood pressure in adult male but not female rat offspring. The present study tested the hypothesis that estrogen normalizes perinatal nicotine-induced hypertensive response to Ang II in female offspring. Nicotine was administered to pregnant rats via subcutaneous osmotic minipumps from day 4 of gestation to day 10 after birth. Ovariectomy (OVX) and 17β-estradiol (E2) replacement were performed at 8 weeks old female offspring. At 5 months old, Ang II-induced blood pressure (BP) responses were not changed by nicotine treatment in the sham groups. In contrast, nicotine significantly enhanced Ang II-induced BP responses as compared with saline control in the OVX groups, which was associated with increased Ang II-induced vascular contractions. These heightened responses were abrogated by E2 replacement. In addition, nicotine enhanced Ang II receptor type I (AT1R), NADPH oxidase type 2 (Nox2) protein expressions, and reactive oxygen species (ROS) production of aortas as compared with saline control in the OVX groups. Anti-oxidative agents, both apocynin and tempol, inhibited Ang II-induced vascular contraction and eliminated the differences of contractions between nicotine-treated and control OVX rats. These findings support a key role of estrogen in the sex difference of perinatal nicotine-induced programming of vascular dysfunction, and suggest that estrogen may counteract heightened reactive oxygen species production, leading to protection of females from development programming of hypertensive phenotype in adulthood.
Keywords: nicotine, estrogen, hypertension, programming, angiotensin II, reactive oxygen species
Introduction
An increasing body of evidence suggests that an adverse intrauterine environment plays an important role in the development of common chronic diseases, including hypertension and other cardiovascular and metabolic diseases. Epidemiological studies have shown that fetal exposure to maternal smoking is associated with elevated blood pressure (BP) and increased risk of cardiovascular disease later in life.1,2 As one of the major components in cigarette smoking, nicotine is likely to contribute to developmental programming of cardiovascular disorders.3 Indeed, prenatal exposure to nicotine results in cardiovascular dysfunction and increased blood pressure during adulthood in different animal models.4,5 However, the impact of maternal smoking/nicotine exposure during pregnancy on fetal programming is complex. Pausova et al have demonstrated that prenatal exposure to nicotine does not increase BP in the Brown Norway rats but significantly enhances BP in the spontaneously hypertensive rats.5 Recently, we have also demonstrated that perinatal nicotine exposure reprograms vascular reactivity and causes a gender-specific increase in hypertensive reactivity in adult male but not female offspring.6,7,8 However, the mechanisms underlying the perinatal nicotine-induced, gender-dependent hypertensive reactivity in offspring remain elusive.
Gender differences in development programming of cardiovascular disease and hypertension in humans have been well documented. Epidemiological evidence shows a sex-dependent pathophysiology of hypertension.9,10 Gender differences are also reported in different animal models of fetal programming of adult hypertension. Most of the studies indicate that male offspring develop hypertension whereas female offspring seem to be protected.11–15 Previous studies13,14 in a rat model of fetal programming induced by placental insufficiency during pregnancy have demonstrated marked increases in BP in both pre-pubertal male and female offspring. However, after puberty, only male offspring remain hypertensive whereas female offspring normalize their BP. Furthermore, ovariectomy induces hypertension in adult female offspring which is normalized by estrogen replacement.13 These findings suggest that estrogen may contribute to gender differences in blood pressure response to an adverse intrauterine environment and play an important role in modulating cardiovascular response in adulthood. However, the molecular mechanisms underlying estrogen-induced protective effect on cardiovascular dysfunction in the response to prenatal insults are not clear.
The role of estrogen in regulation of vascular function may not only be dependent on its direct effect on the cardiovascular system, but also dependent on its indirect effect on other systems. Renin-angiotensin system (RAS) is one of the potential targets modulated by estrogen. Numerous studies have demonstrated the role of the RAS in the etiology of hypertension in model of fetal programming.13,14,16–18 Ojeda et al.13 have demonstrated that modulation of the RAS by estrogen may be one mechanism critical to the normalization of blood pressure in adult female growth restricted offspring. In addition, other factors, such as nitric oxide and oxidative stress-mediated signaling, may be modulated by estrogen.
Given the fact that female rats are resistant to developing hypertensive phenotype seen in males after perinatal nicotine treatment in our animal model,8 in the present study we hypothesize that estrogen may normalize perinatal nicotine-induced hypertensive response to Ang II in adult female offspring. To this end, we investigated whether ovariectomy (OVX) uncovers a perinatal nicotine-induced increase in Ang II-induced blood pressure response in female offspring, and whether 17β-estradiol (E2) replacement abrogates nicotine-mediated responses. Furthermore, we determined that OVX-uncovered the BP response in nicotine-treated female offspring which is mediated by increased vascular contractility via heightened reactive oxygen species (ROS) signaling.
Methods
Experimental animals
All procedures and protocols were approved by the Institutional Animal Care and Use Committee of Loma Linda University, and followed the guidelines by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Time-dated pregnant Sprague-Dawley rats were randomly divided into two groups: 1) saline control; and 2) nicotine administration through an osmotic minipump at 4 μg/kg/min from day 4 of pregnancy to day 10 after birth, as previously described.6–8 The dose of nicotine resulted in blood levels closely resembling that occurring in moderate human smokers.3 Control rats received saline as the vehicle control. As previously reported,6 the nicotine treatment did not affect the length of gestation, and all of pregnancies reached full term. Newborn pups were kept with their mothers until weaning. At weaning, male and female pups were separated. Since female rats are in a pre-puberty stage before 8 weeks of age, ovariectomy (OVX) was performed at 8 week old female offspring as previously described.13 Briefly, the skin was opened by a ventral midline incision and the ovarian vessels were tied off, and the ovaries were removed. The sham operation consisted of a ventral midline incision, and the ovaries were visualized but not removed. 17β-estradiol (E2) valerate minipellets (7.5 mg for 90-day release) (Innovative Research of American) were used for continuous release of the hormone at a dose to maintain E2 concentrations in the physiologic range.13 All pellets were implanted 7 days after ovariectomy.
Measurement of arterial BP
At 5 months of age, the rats were implanted with a catheter in femoral arteries for recording arterial BP, as described previously.8 The catheter was secured on the back of rat. Two days after recovery from surgery, BP was measured continuously in conscious animal. After the baseline recording for 60 min, animals were received a bolus injection of angiotensin II (Ang II, 10 μg/kg) with subcutaneous injection via an implanted catheter, and BP was recorded for 60 min, as described previously.8,19 Arterial BP responses at this dose of Ang II reached a submaximal level, as determined in our previous studies.8 Arterial systolic BP (SBP) and diastolic BP (DBP), and mean BP (MAP) data were recorded continuously throughout each study with data acquisition software (Powerlab 16/SP and Chart version 4, ADInstruments).
Western immunoblotting
Although aortas and peripheral resistance arteries have different physiologic role in regulation of blood flow, our previous studies in the same animal model have demonstrated that nicotine has similar effect on the vascular reactivity in both vessels.8 Therefore, aortic rings were used for Western blot analysis. Briefly, intact aortic rings were isolated from the different treated offspring. The rings were then homogenized in a lysis buffer. Homogenates were ultrasonicated for 15 sec, and then centrifuged at 4 °C for 10 min at 10,000 g. Supernatants were collected and protein determined. Samples with equal protein were loaded and separated on 10% PAGE-SDS. The membranes were incubated with primary antibody, followed by a secondary horseradish peroxidase-conjugated goat anti-rabbit antibody. The rabbit polyclonal antibodies against Ang II receptor type I (AT1R) (Santa Cruz, CA) and mouse anti-pg91 [phox] (Nox2) (BD Biosciences) were used, respectively. Proteins were visualized with enhanced chemiluminescence reagents, and blots were exposed to Hyperfilm. Results were quantified with the Kodak electrophoresis documentation and analysis system and Kodak ID image analysis software.
Contraction studies
Aortas were isolated from the adult offspring, cut into 4-mm rings and mounted in 10-ml tissue baths containing modified Krebs’ solution equilibrated with a mixture of 95%O2 and 5%CO2. Isometric tensions were measured at 37°C, as described previously.6 Ang II-induced concentration-dependent contractions were obtained by cumulative additions of the agonist in approximate one-half log increments. In certain experiments, tissues were pretreated for 20 min with the inhibitor of NADPH oxidase (apocynin), or the SOD mimetic (tempol), followed by the stimulation with increased concentrations of Ang II. For relaxation studies, the tissues were pre-contracted with submaximal concentration (1 μM) of norepinephrine, followed by acetylcholine (Ach) stimulation, added in a cumulative manner.
Measurement of plasma E2 levels
Plasma estrogen levels were determined with a commercially available Estradiol ELISA kit (Bio-Quant, Inc. San Diego, CA, Biokit, BQ180S).
Measurement of vascular ROS
Total ROS in aortic segments were measured with the Oxiselect™ in vitro ROS/RNS assay kit (Cell Biolabs, Inc. San Diego, CA), following the manufacture’s instruction.
Data analysis
Concentration-response curves were analyzed by computer-assisted nonlinear regression to fit the data using GraphPad Prism (GraphPad Software, San Diego, CA) to obtain pD2 (−log EC50) and the maximum response (Emax). Results were expressed as means ± SEM, and the differences were evaluated for statistical significance (P < 0.05) by ANOVA or by t-test, where appropriate.
Results
Effect of nicotine on basal blood pressure and body weight
As shown in Table 1, the basal arterial blood pressure (BP) and body weight were not significantly different between saline control and nicotine-treated animals in the sham, OVX, and OVX + E2 groups, respectively.
Table 1.
Effect of nicotine on basal arterial BP and body weight.
| Animal group | BW(g) | n | SBP(mmHg) | n | DBP(mmHg) | n | MAP(mmHg) | n | HR(bpm) | n |
|---|---|---|---|---|---|---|---|---|---|---|
| Sham | ||||||||||
| Saline | 228.2±8.1 | 10 | 147.2±4.0 | 6 | 113.0±5.3 | 6 | 124.0±3.5 | 6 | 379.3±16.4 | 6 |
| Nicotine | 292.5±6.2 | 12 | 144.2±4.8 | 6 | 111.0±3.3 | 6 | 122.3±2.5 | 6 | 392.0±16.0 | 6 |
| OVX | ||||||||||
| Saline | 367.0±16.0 | 10 | 146.7±4.7 | 6 | 111.3±5.3 | 6 | 124.4±4.7 | 5 | 367.2±12.9 | 6 |
| Nicotine | 352.7±12.5 | 11 | 159.2±4.9 | 6 | 111.3±1.9 | 6 | 128.0±2.3 | 5 | 365.5±8.7 | 6 |
| OVX + E2 | ||||||||||
| Saline | 301.8±8.7 | 10 | 129.8±8.2 | 6 | 90.3±3.2 | 6 | 102.5±3.9 | 6 | 348.8±6.9 | 6 |
| Nicotine | 314.6±4.9 | 10 | 142.5±5.7 | 6 | 96.7±3.3 | 6 | 112.8±3.9 | 6 | 345.0±7.7 | 6 |
BW, body weight; SBP, systolic blood pressure; DBP, diastolic BP; MAP, mean arterial BP; HR, heart rate; n, animal numbers.
Plasma E2 levels
In the sham groups, there were no significant differences of plasma E2 levels in the comparison of adult female saline control (25.68 ± 5.51 pg/ml, n=7) and adult female nicotine-treated offspring (25.09 ± 5.51 pg/ml, n=7) at 5 months of age. In the OVX groups, E2 levels were significantly decreased in both control (2.13 ± 0.25 pg/ml, n=7) and nicotine-treated offspring (1.87 ± 0.27 pg/ml, n=7) in comparison with their sham counterparts. In the OVX + E2 groups, E2 levels were significantly increased in both control (32.7 ± 5.78 pg/ml, n=7) and nicotine-treated offspring (34.7 ± 3.66 pg/ml, n=7) in comparison with their OVX counterparts. However, E2 levels in the E2 replacement groups were not significant differences in comparison with their sham counterparts.
Effect of nicotine on Ang II-induced blood pressure response
Figure 1 shows Ang II-induced increases in arterial blood pressure response in female offspring. In the sham groups, the SBP, DBP and MAP responses to Ang II were not significantly different between saline control and nicotine-treated animals. However, in the OVX groups, the SBP and MAP, but not DBP, responses to Ang II were significantly increased in the nicotine-treated group as compared with the saline control group (P < 0.05). In the OVX + E2 groups, the Ang II-induced BP responses were not significantly different between the saline control and nicotine-treated animals.
Figure 1. Effect of nicotine on Ang II-induced BP response in sham, OVX, and OVX+E2 offspring.

Diastolic BP (DBP), systolic BP (SBP), and mean arterial BP (MAP) responses to Ang II (10 μg/kg) were measured in sham, ovariectomy (OVX), and OVX + E2 groups of female offspring that had been exposed in utero to saline control or nicotine. All of the data are expressed as means ± SEM from 6 animals in each group. †P<0.05 vs. saline group; *P<0.05 vs. data points in saline group.
Effect of nicotine on vascular AT1R and Nox2 protein expression in adult female offspring
As shown in Figure 2A, there was no significant difference of AT1R protein expression in the aortic arteries of female adult offspring between nicotine-treated and saline control animals. However, in the OVX group, nicotine significantly enhanced vascular AT1R protein expression as compared with the saline control. In the OVX + E2 group, the vascular AT1R protein levels were not significant difference between nicotine-treated and saline control animals (Figure 2A). Since our previous study has shown that AT1R mainly localizes in the vascular smooth muscle cells,8 the measure of AT1R protein expression in whole vessels by Western blot was essentially a measure of VSM expression.
Figure 2. Effect of nicotine on arterial Ang II receptor type 1 (AT1R) and NADPH oxidase type 2 (Nox2) protein expressions in offspring.

AT1R (A) and Nox2 (B) protein levels were determined by Western blot, respectively, in aortas isolated from sham, ovariectomy (OVX), and OVX + E2 groups of female offspring that had been exposed in utero to saline control or nicotine. The protein density was normalized to β-actin, and then expressed as to the relative value of the saline control. All of the data are expressed as means ± SEM of tissues from 4 animals in each group. *P<0.05, nicotine vs. saline control.
Similar to the AT1R protein expression, Nox2 protein levels (Figure 2B) in aortic rings were not significant difference between nicotine-treated and saline control of female intact offspring. However, in the OVX group, nicotine significantly enhanced vascular Nox2 protein levels as compared with the saline control. In the OVX + E2 group, the vascular Nox2 protein levels were not significant difference between nicotine-treated and saline control animals.
Effect of nicotine on Ang II-induced vascular contractions
Figure 3 shows that Ang II-induced vascular contractions of aortas in female offspring were not different between saline control and nicotine-treated rats in sham groups. However, in the OVX groups, the maximal responses of Ang II-induced contractions were significantly increased in aortas of nicotine-treated animals, as compared with that of the control (26.36 ± 1.86 vs. 14.2 ± 1.96 % KCl response, P < 0.05). In the OVX + E2 groups, there was no significant difference in Ang II-induced contractions between saline control and nicotine-treated animals (Figure 3).
Figure 3. Effect of nicotine on Ang II-induced contractions.

Ang II-induced contractions of aortic rings were determined in sham, OVX, and OVX + E2 groups of female offspring that had been exposed in utero to saline control or nicotine. All of the data are expressed as mean ± SEM of tissues from 5–7 animals. The values of the maximal contractile response were presented in the text.
Effect of nicotine on Ach-induced relaxations
As shown in Figure 4, Ach produced dose-dependent relaxations of aortic rings in female offspring. In the sham groups, Ach-induced maximal relaxation in nicotine-treated rats was significantly higher than that in saline control animals (73.37 ± 1.89% vs. 61.1 ± 1.85%, P < 0.05). In the OVX groups, there was no significant difference in Ach-induced relaxations between the saline control and nicotine-treated rats (55.30 ± 2.55% vs. 59.03 ± 2.45%, P > 0.05). In the OVX + E2 animals, there was no significant difference in Ach-induced relaxations between the saline control and nicotine-treated rats (76.7 ± 4.25% vs. 76.31 ± 4.31%, P > 0.05).
Figure 4. Effect of nicotine on Ach-induced relaxations.
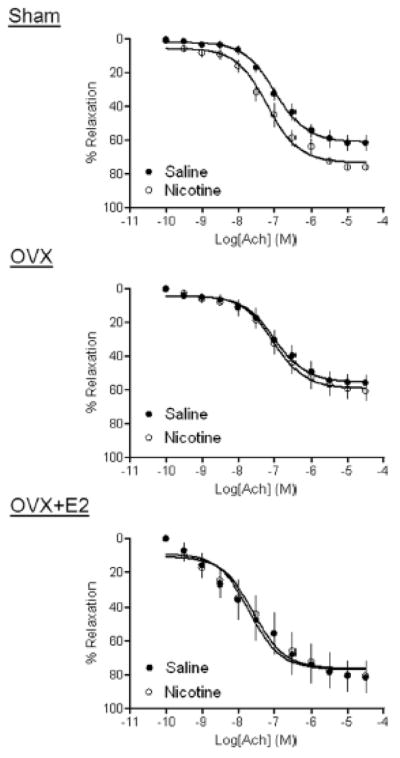
Ach-induced relaxations of aortic rings were determined in sham, OVX, and OVX + E2 groups of female offspring that had been exposed in utero to saline control or nicotine. All of the data are expressed as mean ± SEM of tissues from 5–7 animals. The values of the maximal relaxation response were presented in the text.
Effect of apocynin and tempol on Ang II-induced contractions
Given the present finding that perinatal nicotine treatment significantly enhanced Ang II-induced contractions in OVX females (Figure 4), we determined whether ROS play a role in the heightened vascular reactivity to Ang II in OVX females. As shown in Figure 5, NADPH oxidase inhibitor apocynin significantly inhibited Ang II-induced contractions of aortic rings isolated from both saline control (14.17 ± 1.9% vs. 6.35 ± 0.84%, P < 0.05) and nicotine treated (26.36 ± 1.86% vs. 8.94 ± 0.81%, P < 0.05) OVX females. While in the absence of apocynin, Ang II-induced contractions were significantly greater in nicotine-treated as compared with control animals (26.36 ± 1.86% vs. 14.17 ± 1.9%, P < 0.05), in the presence of apocynin, there was no significant difference in Ang II-induced contractions between nicotine-treated and control animals (8.94 ± 0.81% vs. 6.35 ± 0.84%, P > 0.05) (Figure 5). E2 replacement completely blocked the effect of apocynin in both saline control and nicotine-treated animals (Figure 5). Similar findings were obtained with SOD mimetic tempol (Figure 6)
Figure 5. Effect of apocynin on Ang II-induced contractions.

Pregnant rats were treated with saline (control) or nicotine, and Ang II-induced contractions in the absence or presence of apocynin were determined in aortic rings isolated from sham, OVX, and OVX + E2 groups of female offspring. Data are means ± SEM of tissues from 5–7 animals.
Figure 6. Effect of tempol on Ang II-induced contractions.

Pregnant rats were treated with saline (control) or nicotine, and Ang II-induced contractions in the absence or presence of tempol were determined in aortic rings isolated from sham, OVX, and OVX + E2 groups of female offspring. Data are means ± SEM of tissues from 5–7 animals.
Effect of nicotine on ROS production
To determine whether nicotine-induced changes in vascular contractile function are associated with the oxidative stress levels, we measured the total ROS production in aortic rings isolated from the different groups of female offspring. As shown in figure 7, OVX and OVX plus E2 replacement had no significant effect on ROS production in comparison with the sham group in the saline control offspring. However, in the nicotine-treated offspring, OVX significantly enhanced ROS production as compared with the sham group which was reversed by E2 replacement. In addition, the ROS production was significantly higher in the nicotine-treated OVX offspring than in the saline control OVX offspring.
Figure 7. Effect of nicotine on ROS production in aortic segments.
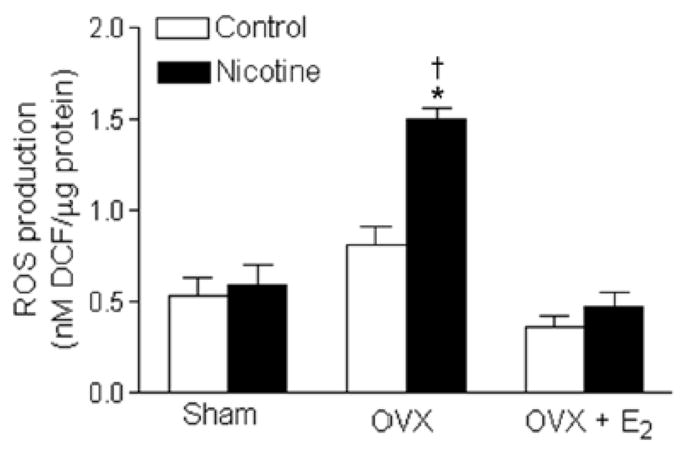
Total ROS productions were measured in aortic segments isolated from sham, ovariectomy (OVX), and OVX + E2 groups of female offspring that had been exposed in utero to saline control or nicotine. All of the data are expressed as means ± SEM from 5 animals per group. *P < 0.05 vs. sham counterparts. †P < 0.05 vs. OVX control.
Discussion
Our previous studies have demonstrated that perinatal nicotine exposure causes a gender-dependent increase in blood pressure response in adult male but not female rat offspring.8 The present study provides new evidence that female sex hormone estrogen plays a key role in protecting females against perinatal nicotine-mediated developmental programming of hypertensive phenotype in adult offspring. The major findings of present study are: 1) OVX uncovered nicotine-induced programming of heightened vascular reactivity to Ang II in female offspring, 2) estrogen replacement abrogated the nicotine-induced response, 3) OVX uncovered nicotine-heightened AT1R gene expression associated with increase in NADPH oxidase (Nox2) expression and ROS production, which was reversed by E2 replacement, 4) inhibition of ROS production by apocynin and tempol abolished the heightened vascular reactivity resulted from perinatal nicotine exposure in OVX females.
In the present study, we found that Ang II-induced blood pressure responses in female adult offspring were not significantly different between saline control and nicotine-treated groups, which is consistent with our previous findings that perinatal nicotine exposure enhances blood pressure response in male but not female offspring in the same animal model.8 The gender differences in fetal programming of adult cardiovascular diseases have been reported in different animal models. Most of the studies indicate that male offspring develop hypertension, whereas female offspring seem to be protected.11–15 In a model of intrauterine growth-restriction (IUGR) induced by placental insufficiency in rats, both male and female offspring developed hypertension at pre-pubertal ages. However, after puberty, only male but not female offspring remained hypertensive, suggesting that female sex hormones might protect females from the development of hypertension.13 The present study further addressed the contribution of female sex hormone estrogen to the observed sex differences in prenatal nicotine-mediated hypertension. We found that removal of ovaries (OVX), the main source of circulating estrogen in females, significantly facilitated the development of higher blood pressure response in nicotine-treated rats compared with the saline control ones and this effect was abolished by estrogen replacement. Our findings further suggest that estrogen plays an important role in protecting female offspring from development of hypertensive phenotype in response to maternal nicotine exposure. Similar findings have been reported in the animal model of placental insufficiency that blood pressure is significantly increased following ovariectomy in the adult female offspring with no effect on blood pressure in adult female control offspring.13 Furthermore, estrogen replacement normalizes blood pressure in ovariectomized adult growth restricted females.13 However, the mechanisms underlying estrogen induced counteraction of nicotine-induced hypertensive response are far more complex. In contrast to previous findings that direct nicotine treatment alters the circulating estrogen levels,20 in present study we found that there were no differences of plasma E2 levels between saline control and nicotine-treated offspring. Consistent with our finding, previous studies also indicated that in utero adverse environmental exposure did not affect the circulating estrogen levels in adult female offspring.13 These finding suggest that the protective role of estrogen in the regulation of cardiovascular function may be not only dependent on estrogen levels, but also dependent on its effect on downstream signaling or other regulatory systems. One potential target for modulation by estrogen is the renin-angiotensin system (RAS).21–23 Estrogen may activate the RAS by augmenting levels of renin and angiotensinogen or their downstream signaling pathways.13,24 Estrogen can shift the vasoconstrictor-vasodilator balance of the renin-angiotensin system, resulting in protection against hypertension.24 In addition, previous studies in OVX rats have demonstrated estrogen replacement directly decreases hypothalamic AT1 receptor expression and binding affinity in female rats.23 Our previous studies have demonstrated that prenatal nicotine exposure enhances vascular AT1R expression in male adult offspring.8 However, in present study, the vascular AT1R expression was not changed by nicotine exposure in adult female offspring, suggesting that the effect of nicotine on vascular AT1R gene expression may be abrogated by sex hormones. Indeed, the current findings that the vascular AT1R protein levels in the OVX group but not in the OVX + E2 group were significantly higher in nicotine-treated rats than in control animals, further support a key role of estrogen in prevention of nicotine-induced exaggerated AT1R gene expression in female offspring. Therefore, estrogen-induced counteraction of nicotine-mediated heightened vascular AT1R gene expression may be one of the key mechanisms for the sex difference in development hypertensive phenotype.
It is likely that multiple mechanisms are involved in OVX-mediated increased susceptibility of BP responses to Ang II in nicotine-treated rats. In the present study, we found that nicotine significantly enhanced Ang II-induced contractions as compared with saline control in the OVX groups but not in the OVX + E2 group. This suggests that estrogen might directly regulate vascular contractility and counteract nicotine-induced exaggerated vascular contractility and elevated blood pressure response. The molecular and cellular mechanisms underlying the protective effect of estrogen on nicotine-mediated Ang II-induced hypertensive response are unknown. Previous studies with estrogen have suggested that many of the cardiovascular effects of sex hormones may be related to their role in the modulation of oxidative stress.25–27 Our previous studies have shown that perinatal nicotine exposure-induced hypertensive response to Ang II in adult male offspring is associated with an increase in vascular NADPH oxidative-mediated ROS production,28 suggesting that nicotine-mediated heightened vascular ROS may play a key role in programming of hypertensive phenotype in adult male offspring. In current study, we found that the vascular ROS levels had no significant differences between the saline control and nicotine-treated offspring, but were significantly enhanced in nicotine-treated offspring as compared with the control offspring in the OVX groups, which was abolished by E2 replacement. These findings suggest that nicotine-mediated vascular ROS production may be modulated by estrogen and the enhanced ROS may be one of the key mechanisms mediating nicotine-induced hypertensive responses in the OVX groups. An increasing body of evidence suggests that Ang II/AT1R directly regulates the NADPH oxidase/ROS signaling pathway, leading to enhanced vasoconstriction in pathophysiological conditions.16,29–31 Our previous studies have also suggested nicotine-mediated up-regulation of Ang II/AT1R system may play an important role in vascular NADPH oxidase-derived ROS production in male adult offspring.8,28 Apocynin is commonly used as an inhibitor of NADPH oxidase. Previous studies have demonstrated that apocynin can selectively inhibit NADPH oxidase and ROS production in smooth muscle cells and non-smooth muscle cells.32,33 The present finding that apocynin significantly inhibited Ang II-induced contractions of aortic rings in both saline control and nicotine-treated OVX rats suggests that NADPH oxidase-derived ROS plays a role in Ang II-mediated contractions. However, our current finding that the membrane permeable SOD mimetic tempol inhibited Ang II-induced contractions only in nicotine-treated OVX rats but not in saline control OVX rats suggests that nicotine may alter the sensitivity of SOD, i.e. oxidase defense system to the effect of ovariectomy. Furthermore, in the present study we found that both apocynin and tempol eliminated Ang II-induced contractions between the saline control and nicotine-treated OVX rats. These findings suggest that ovariectomy enhanced nicotine-mediated contractions may be due to heightened vascular oxidative stress.
The results of the current work showed that E2 administration to nicotine-treated OVX rats significantly decreased Ang II-induced contractions which were not further altered by the treatment of apocynin and tempol. These findings suggest that estrogen and apocynin and tempol may regulate nicotine-mediated vascular contractions through the common mechanism, i.e. ROS signaling pathway. Indeed, a growing body of studies has demonstrated that estrogen can act as an antioxidant to regulate cardiovascular function.34–37 In a spontaneous hypertensive rat model, ovariectomy increased ROS excretion in females, suggesting that estrogen suppresses levels of ROS.38 In contrast to our previous findings that perinatal nicotine enhances vascular NADPH oxidase (Nox2) protein expression in male adult offspring,28 current findings that nicotine had no effect on Nox2 expression in female offspring, suggest a sex difference in nicotine-induced programming of ROS-related gene expression. Furthermore, our present findings that nicotine enhanced vascular Nox2 protein expression compared with the saline control in the OVX groups but not in the OVX + E2 groups, suggest that estrogen abolishes nicotine-induced programming of the heightened NADPH oxidase expression. In agreement with our findings, previous studies have demonstrated that estrogen directly inhibits NADPH oxidase activity via the estrogen receptor to attenuate ischemic oxidative damage.39 From these multiple lines of evidence, it is reasonable to hypothesize that estrogen is serving as an antioxidant to inhibit ROS sensitivity to the effect of nicotine and may contribute to the sex differences in the effects of prenatal nicotine exposure on vascular function and blood pressure in adult offspring.
In addition to modulation of Ang II/ATR/ROS signaling, estrogen may modulate nicotine-mediated NO-dependent signaling. Our previous studies6 in the same animal model have demonstrated that prenatal nicotine treatment attenuates endothelium-dependent relaxations in male offspring but enhances them in females without alteration of vascular eNOS gene expression, which suggests that prenatal nicotine selectively decrease eNOS activity which may be reversed and further enhanced by estrogen, resulting in altered NO-mediated vasodilatation in a gender-dependent manner. This is further supported by the present finding that Ach-induced relaxations of aortas were significantly increased in female offspring of nicotine-treated animals. However, the present findings that Ach-induced relaxations were not significant differences between OVX saline control and OVX nicotine-treated offspring suggest that the exaggerated vasoconstrictions but not relaxations may contribute to the enhanced BP response.
Perspectives
Sex differences are demonstrated in human hypertension and also are apparent in several animal models of fetal programming of hypertension. Our previous studies have shown that perinatal nicotine exposure causes a gender dependent increase in the risk of development of hypertensive phenotype in adult male but not female rat offspring. The present study provides novel evidence that estrogen plays a key role in the sex difference of fetal programming of adult hypertension. Findings from this study suggest that estrogen may function as an antioxidant to counteract nicotine-induced heightened ROS, resulting in protection of females from development of hypertensive phenotype in adulthood. However, the cellular and molecular mechanisms that how estrogen modifies nicotine-induced heightened vascular ROS, resulting in counteraction of nicotine-mediated hypertensive responses are still unknown, and whether the modifications of ROS by estrogen are due to direct regulation of Ang II/receptor-mediated signaling is unknown. Therefore, future studies are needed to clarify the mechanisms involved in initiating programming of Ang II/receptor-mediated ROS signaling in utero, as well as the signaling interactions with sex hormones secondary to the development of hypertension. Understanding these mechanisms will allow for development of gender-specific strategies for prevention and treatment of fetal originated cardiovascular diseases.
Novelty and Significance.
What Is New
Perinatal nicotine exposure causes a gender-dependent development of hypertensive phenotype in adult male but not female offspring.
OVX uncovers nicotine-induced programming of hypertensive reactivity in female offspring which is abrogated by estrogen replacement.
OVX uncovered nicotine-heightened vascular reactivity associated with increase in AT1R and Nox2-derived ROS production, and inhibition of ROS attenuates the heightened vascular reactivity.
What Is Relevant?
In utero nicotine exposure is one of the important risk factors of hypertension and other cardiovascular disease.
Our findings show a sex differences in fetal programming of hypertension.
Estrogen may function as an antioxidant to counteract nicotine-induced heightened Ang II/ATR-mediated ROS, resulting in protecting females from development of hypertensive phenotype in adulthood.
Summary
Our studies provide new insights and contribute to the novel concept that fetal programming of adult hypertension may be modulated by sex hormones via Ang II/ATR-mediated ROS signaling. Identifying and understanding the gender specific modification of Ang II/ATR/ROS signaling may provide new leads in the development of preventive diagnosis and therapeutic strategies of fetal programming of hypertension and other cardiovascular dysfunction.
Acknowledgments
Sources of Funding
This work was supported by the California Tobacco-related Disease Research Program Award #18KT-0024 (DX), and by National Institutes of Health Grants HL83966 (LZ), HL110125 (LZ), HL89012 (LZ), and DA032510 (DX).
Footnotes
Disclosures
None
References
- 1.Beratis NG, Panagoulias D, Varvarigou A. Increased blood pressure in neonates and infants whose mothers smoked during pregnancy. J Pediatr. 1996;128:806–812. doi: 10.1016/s0022-3476(96)70333-5. [DOI] [PubMed] [Google Scholar]
- 2.Blake KV, Gurrin LC, Evans SF, Beilin LJ, Landau LI, Stanley FJ, Newnham JP. Maternal cigarette smoking during pregnancy, low birth weight and subsequent blood pressure in early childhood. Early Hum Dev. 2000;57:137–147. doi: 10.1016/s0378-3782(99)00064-x. [DOI] [PubMed] [Google Scholar]
- 3.Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse? J Pharmacol Exp Ther. 1998;285:931–945. [PubMed] [Google Scholar]
- 4.Gao YJ, Holloway AC, Su LY, Takemori K, Lu C, Lee RM. Effects of fetal and neonatal exposure to nicotine on blood pressure and perivascular adipose tissue function in adult life. Eur J Pharmacol. 2008;590:264–268. doi: 10.1016/j.ejphar.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 5.Pausova Z, Paus T, Sedova L, Berube J. Prenatal exposure to nicotine modifies kidney weight and blood pressure in genetically susceptible rats: a case of gene-environment interaction. Kidney Int. 2003;64:829–835. doi: 10.1046/j.1523-1755.2003.00172.x. [DOI] [PubMed] [Google Scholar]
- 6.Xiao D, Huang X, Lawrence J, Yang S, Zhang L. Fetal and neonatal nicotine exposure differentially regulates vascular contractility in adult male and female offspring. J Pharmacol Exp Ther. 2007;320:654–661. doi: 10.1124/jpet.106.113332. [DOI] [PubMed] [Google Scholar]
- 7.Lawrence J, Xaio D, Xue Q, Rejali M, Yang S, Zhang L. Prenatal Nicotine Exposure Increases Heart Susceptibility to Ischemia/Reperfusion Injury in Adult Offspring. J Pharmacol Exp Ther. 2008;324:331–341. doi: 10.1124/jpet.107.132175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao D, Xu Z, Huang X, Longo LD, Yang S, Zhang L. Prenatal gender-related nicotine exposure increases blood pressure response to angiotensin II in adult offspring. Hypertension. 2008;51:1239–1247. doi: 10.1161/HYPERTENSIONAHA.107.106203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubey PK, Oparil S, Imthum B, Jackson EK. Sex hormones and hypertension. Cardiovasc Res. 2002;53:688–708. doi: 10.1016/s0008-6363(01)00527-2. [DOI] [PubMed] [Google Scholar]
- 10.Kotchen JM, Mckean HE, Kotchen TA. Blood pressure trends with aging. Hypertension. 1982;4:128–134. doi: 10.1161/01.hyp.4.5_pt_2.iii128. [DOI] [PubMed] [Google Scholar]
- 11.Brawley L, Itoh S, Torrens C, Bark A, Bertram C, Poston C, Hanson M. Dietary Protein restriction in pregnancy induces hypertension and vascular defects in rat male offspring. Pediatr Res. 2003;54:83–90. doi: 10.1203/01.PDR.0000065731.00639.02. [DOI] [PubMed] [Google Scholar]
- 12.Hemmings DG, Williams SJ, Davidge ST. Increased myogenic tone in 7-month-old adult male but not female offspring from rat dams exposed to hypoxia during pregnancy. Am J Physiol Heart Circ Physiol. 2005;289:H674–H682. doi: 10.1152/ajpheart.00191.2005. [DOI] [PubMed] [Google Scholar]
- 13.Ojeda NB, Grigore D, Robertson EB, Alexander BT. Estrogen protects against increased blood pressure in postpubertal female growth restricted offspring. Hypertension. 2007;50:679–685. doi: 10.1161/HYPERTENSIONAHA.107.091785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ojeda NB, Grigore D, Yanes LL, Iliescu R, Robertson EB, Zhang H, Alexander BT. Testosterone contributes to marked elevations in mean arterial pressure in adult male intrauterine growth restricted offspring. Am J Physiol Regul Integr Comp Physiol. 2007;292:R758–R763. doi: 10.1152/ajpregu.00311.2006. [DOI] [PubMed] [Google Scholar]
- 15.Grigore D, Ojeda NB, Alexander BT. Sex differences in the fetal programming of hypertension. Gend Med. 2008;5:S121–S132. doi: 10.1016/j.genm.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yzydorczyk C, Gobeil F, Jr, Cambonie G, Lahaie I, Le NL, Samarani S, Ahmad A, Lavoie JC, Oligny LL, Pladys P, Hardy P, Nuyt AM. Exaggerated vasomotor response to Ang II in rats with fetal programming of hypertension associated with exposure to a low-protein diet during gestation. Am J Physiolo Regul Integr Comp Physiol. 2006;291:R1060–R1068. doi: 10.1152/ajpregu.00798.2005. [DOI] [PubMed] [Google Scholar]
- 17.Manning J, Vehaskai VM. Low birth weight associated adult hypertension in the rat. Pediatr Nephrol. 2001;16:471–422. doi: 10.1007/s004670000560. [DOI] [PubMed] [Google Scholar]
- 18.Langley-Evans SC, Jackson AA. Captopril normalizes systolic blood pressure in rats with hypertension induced by fetal exposure to maternal low protein diets. Comp Biochem Physiol A Physiol. 1995;110:223–228. doi: 10.1016/0300-9629(94)00177-u. [DOI] [PubMed] [Google Scholar]
- 19.Matys T, Pawlak R, Kucharewicz I, Chabielska E, Buczko W. Hypotensive effect of angiotensin II after AT1-receptor blockade with losartan. J Physiol Pharmacol. 2000;51:161–166. [PubMed] [Google Scholar]
- 20.EI-Seweidy MM, Mohamed HE, Asker ME, Atteia HH. Nicotine and vascular endothelial dysfunction in female oavriectomized rats: role of estrogen replacement therapy. J Pharm Pharmacol. 2012;64:108–119. doi: 10.1111/j.2042-7158.2011.01377.x. [DOI] [PubMed] [Google Scholar]
- 21.Fischer M, Baessler A, Schunkert H. Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc Res. 2002;53:672–677. doi: 10.1016/s0008-6363(01)00479-5. [DOI] [PubMed] [Google Scholar]
- 22.Nickening G, Baumer AT, Grohe C, Kahlert S, Strehlow K, Rosenkranz S, Stablein A, Beckers F, Smits JF, Daemen MJ, Vetter H, Bohm M. Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation. 1998;97:2197–2201. doi: 10.1161/01.cir.97.22.2197. [DOI] [PubMed] [Google Scholar]
- 23.Kisley LR, Sakai RR, Fluharty SJ. Estrogen decreases hypothalamic angiotensin II AT1 receptor binding and mRNA in the female rat. Brain Res. 1999;844:34–42. doi: 10.1016/s0006-8993(99)01815-6. [DOI] [PubMed] [Google Scholar]
- 24.Brosnihan KB, Li P, Ganten D, Ferrario CM. Estrogen protects transgenic hypertensive rats by shifting the vasoconstrictor-vasodilator balance of RAS. Am J Physiol. 1997;273:R1908–R1915. doi: 10.1152/ajpregu.1997.273.6.R1908. [DOI] [PubMed] [Google Scholar]
- 25.Barp J, Araujo AS, Fernandes TR, Rigatto KV, Llesuy S, Bello-Klein A, Singal P. Myocardial antioxidant and oxidative stress changes due to sex hromones. Braz J Med Nio Res. 2002;35:1075–1081. doi: 10.1590/s0100-879x2002000900008. [DOI] [PubMed] [Google Scholar]
- 26.Hernandez I, Delgado JL, Diaz J, Quesada T, Teruel MJ, Llanos MC, Carbonell LF. 17betal-estradiol prevents oxidative stress and decreases blood pressure in ovariectomized rats. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1599–R1605. doi: 10.1152/ajpregu.2000.279.5.R1599. [DOI] [PubMed] [Google Scholar]
- 27.Morimoto K, Uji M, Ueyama T, Kimura H, Kohno T, Takamata A, Yano S, Yoshida K. Estrogen replacement suppresses pressor response and oxidative stress induced by cage-switch stress in ovariectomized rats. Ann N Y Acad Sci. 2008;1148:213–218. doi: 10.1196/annals.1410.045. [DOI] [PubMed] [Google Scholar]
- 28.Xiao D, Huang X, Yang S, Zhang L. Anternatal nicotine induces heightened oxidative stress and vascular dysfunction in rat offspring. Br J Pharmacol. 2011;164:1400–1409. doi: 10.1111/j.1476-5381.2011.01437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 30.Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- 31.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44:248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 32.Ahmad M, Kelly MR, Zhao X, Kandhi S, Wolin MS. Roles for Nox4 in the contractile response of bovine pulmonary arteries to hypoxia. Am J Physiol Heart Circ Physiol. 2010;298:H1879–H1988. doi: 10.1152/ajpheart.01228.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stefanska J, Pawliczak R. Apocynin: molecular aptitudes. Mediators Inflamm. 2008;2008:106507. doi: 10.1155/2008/106507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res. 2010;106:1681–1691. doi: 10.1161/CIRCRESAHA.109.213645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mooradian AD. Antioxidant properties of steroids. J Steroid Biochem Mol Biol. 1993;45:509–511. doi: 10.1016/0960-0760(93)90166-t. [DOI] [PubMed] [Google Scholar]
- 36.Grundt A, Grundt C, Knoth K, Lemmer B. Rhythmic supplement of 17beta-extradiol according to the physiological estrous cycle: Effect on blood pressure in females ovariectomized rats. Horm Metab Res. 2010;42:130–136. doi: 10.1055/s-0029-1241807. [DOI] [PubMed] [Google Scholar]
- 37.Siqueira R, Campos C, Colombo R, Becker CU, Fernandes TR, Araujo AS, Bello-Klein A. Influence of estrogen on pulmonary arterial hypertension: role of oxidative stress. Cell Biochem Funct. 2011;29:543–548. doi: 10.1002/cbf.1784. [DOI] [PubMed] [Google Scholar]
- 38.Sullivan JC, Sasser JM, Pollock JS. Sexual Dimorphism in oxidant status in spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2007;292:R764–R768. doi: 10.1152/ajpregu.00322.2006. [DOI] [PubMed] [Google Scholar]
- 39.Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, Vadlamudi RK, Brann DW. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J Neurosci. 2009;29:13823–13836. doi: 10.1523/JNEUROSCI.3574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]