

Abstract
A speciation process is ongoing in the primary vector of malaria in Africa, Anopheles gambiae. Assortatively mating incipient species known as the M and S forms differentially exploit larval breeding sites associated with different ecological settings. However, some ongoing gene flow between M and S limits significant genomic differentiation mainly to small centromere-proximal regions on chromosomes X and 2L, termed “speciation islands” with the expectation that they contain the genes responsible for reproductive isolation. As the speciation islands exhibit reduced recombination and low polymorphism, more detailed genetic analysis using fine-scale mapping is impractical. We measured global gene expression differences between M and S using oligonucleotide microarrays, with the goal of identifying candidate genes that could be involved in this ongoing speciation process. Gene expression profiles were examined in two independent colonies of both forms at each of three developmental periods of interest: fourth instar larvae, virgin females, and gravid females. Patterns were validated on a subset of genes using quantitative real-time reverse transcription PCR of RNA samples from laboratory colonies and wild mosquitoes collected from Cameroon and Burkina Faso. Considered across all three developmental periods, differentially expressed genes represented ~1-2% of all expressed genes. Although disproportionately represented in the X speciation island, the vast majority of genes were located outside any speciation island. Compared to samples from the other developmental periods, virgin females were characterized by more than twice as many differentially expressed genes, most notably those implicated in olfaction and potentially, mate recognition.
Introduction
Identification of genes that contribute to ecological adaptation and speciation is one of the foremost goals of ecological genomics. Although the challenges are daunting, progress has been fostered by the proliferation of dense genome maps (including whole genome sequences) and powerful genomic tools such as oligonucleotide microarrays. In the absence of a priori candidate genes, three complementary approaches capitalizing on these genomic resources can help to dissect the genic basis of adaptive and species differences. The first, mapping of quantitative trait loci (QTL), relies upon recombination in controlled crosses involving contrasting phenotypes to reveal genomic regions that are tightly associated with those phenotypes (Mackay, 2001). Such regions are likely to contain loci that contribute to the observed phenotypic differences. A second approach employs genome-wide scans to identify the targets of recent selective sweeps. These targets, inferred based on patterns of molecular variation between populations or species (hitchhiking mapping), can be mapped using DNA sequence data (e.g., Akey et al., 2004; Williamson et al., 2007), multilocus microsatellite or AFLP screens (Campbell, Bernatchez, 2004; Kane, Rieseberg, 2007; Schlotterer, 2002), or by the hybridization of genomic DNA to oligonucleotide microarrays (Turner et al., 2005; White et al., 2007). A third approach measures differences in the level and pattern of gene expression, under the hypothesis that at least some expression differences represent phenotypic traits contributing to adaptation or speciation (Ranz, Machado, 2006). The latter two approaches require no prior information about phenotypic differences; as such, their application toward identifying genes that contribute to ecological adaptation in the absence of a known phenotype can be termed “reverse ecology”. All three complementary approaches may reveal a part of the puzzle, though the pieces may not initially overlap.
A case of ongoing speciation has been uncovered in the primary vector for human malaria in sub-Saharan Africa, the mosquito Anopheles gambiae sensu stricto (A. gambiae) (della Torre et al., 2002). Based on fixed nucleotide differences in X-linked ribosomal DNA genes, A. gambiae comprises two molecular forms: M and S (reviewed in Della Torre et al., 2005). The S form is found throughout tropical Africa and is presumed ancestral. Consistent with the classical descriptions of A. gambiae biology (e.g., Gillies, De Meillon 1968), the S form is reproductively active only during the rainy season and breeds in “typical” bare-edged and rain-dependent pools and puddles fully exposed to sunlight. By contrast, the M form occurs only in West and Central Africa (but see Masendu et al. 2004), and is associated with anthropogenic and long-lived breeding sites constructed in conjunction with agricultural activities, such as rice fields and reservoirs impounded for livestock and irrigation. Its association with these permanent or semi-permanent breeding sites suggests a less restrictive seasonal distribution, and indeed, the M form can occupy surprisingly arid climatic zones and unlike S is reproductively active during the dry season. Overall, these observations imply an ongoing process of adaptation to environmental heterogeneities by A. gambiae, with the indirect consequence of increased malaria transmission both spatially and temporally.
In West and Central Africa where their distributions overlap, simultaneously breeding populations of M and S can occur in the same villages. In such areas of sympatry, no discrete differences in breeding habitat or adult resting site have been discovered to date (Edillo et al., 2002; Edillo et al., 2006), though differences in rate of larval development and predator avoidance behavior have been described (Diabate et al., 2005, 2007a). Importantly, although no postmating reproductive isolation exists between M and S forms (Diabate et al., 2007b), there are high levels of assortative mating. Only ~1% of sperm transfer monitored in natural populations shows evidence of matings between forms (Tripet et al., 2001) and mating swarms are generally exclusive to M or S (Diabate et al., 2003, 2006; A. Diabate, T. Lehmann, pers. comm.). Nearly complete premating behavioral isolation also likely explains persistent differences in the frequencies of shared polymorphic chromosomal inversions segregating within sympatric M and S populations (Della Torre et al., 2005; Toure et al., 1998).
On the strength of correlated genetic, cytogenetic, physiological and behavioral evidence, M and S have been considered as nascent species. By definition, fixed rDNA differences mark the reproductive boundaries between them. Nevertheless, the genetic underpinnings of their ecological, behavioral and physiological differences remain entirely unknown. Where in the genome do the differences lie? How many differences exist? The QTL mapping approach mentioned above is powerless to answer these questions for the M and S forms of A. gambiae, as no measurable phenotypic or behavioral differences are known. On the other hand, genome scans delivered a major breakthrough in 2005, with an innovative application of microarray technology. Hybridization of genomic DNA from individual M and S mosquitoes to an A. gambiae oligonucleotide microarray yielded a genome-wide map of significantly diverged regions between the two molecular forms (Turner et al., 2005). Only three small genomic regions of heightened differentiation (referred to as “speciation islands”) emerged from this experiment, two of which were located adjacent to centromeres on chromosomes X and 2L—regions of sharply reduced recombination (Pombi et al., 2006). As predicted from the microarray hybridization results, targeted sequencing within these speciation islands revealed fixed differences between forms and no shared polymorphisms, in contrast to control loci outside of the islands (Stump et al., 2005; Turner, Hahn, 2007; Turner et al., 2005). Because this and previous surveys (reviewed in Krzywinski, Besansky, 2003) generally failed to find genome-wide differentiation between populations of M and S forms of A. gambiae (but see Wondji et al., 2002), these data seem to support a “divergence with gene flow” model of adaptation and speciation in which low-recombination regions resist introgression and preserve sets of alleles adaptive in specific genetic and environmental backgrounds (Hey, 2006). The speciation islands were so-named because these-- as the only regions of divergence detected by this technology—logically should contain many of the genes responsible for differential adaptation and speciation of M and S in an otherwise homogeneous background of shared polymorphism.
Amidst the optimism surrounding the discovery of speciation islands in A. gambiae (e.g., Butlin, Roper, 2005) lays an irony. Though small relative to the entire 260 Mb genome, the speciation islands are not small in absolute terms. They comprise at least 2.8 Mb, and evidence suggests that the X chromosome island alone was grossly underestimated due to the low quality of initial genome assemblies; instead, it likely extends for at least 4 Mb and includes several dozen genes (Stump et al., 2005; White et al, unpublished data). The very pattern that made the genomic islands easy to detect using genomic microarray technology-- namely, an extended footprint of fixed sequence differences with no shared polymorphism-- now makes it virtually impossible to dissect more finely using hitchhiking mapping.
With the goal of identifying candidate genes that could be involved in ecological or behavioral differences associated with ongoing speciation, we adopted the third and complementary approach referred to earlier: screening for global gene expression differences. Using the same platform as Turner et al (2005) (the Affymetrix Anopheles/Plasmodium GeneChip), we examined patterns of gene expression in two independent colonies of both forms at each of three developmental periods of interest: fourth instar larvae, virgin females, and gravid females. Patterns of expression were measured across five intra-colony replicates and were validated on a subset of genes based on quantitative real-time reverse transcription PCR (qRT-PCR) using independent RNA samples from laboratory colonies. Further verification was obtained by performing qRT-PCR on RNA samples extracted from wild M and S mosquitoes collected from Cameroon and Burkina Faso. Considered across all three developmental periods, differentially expressed genes were disproportionately represented in the X speciation island, but the vast majority was located outside any speciation island. Among the three developmental periods compared between M and S, virgin females contained the largest number of differentially expressed genes, most notably those implicated in olfaction and—potentially—mate recognition.
Materials and methods
Mosquito colonies
Experiments were conducted on four non-isogenic laboratory colonies of A. gambiae: two of M-form (M-GA-CAM and Mali-NIH) and two of S-form (KIST and Pimperena) (Supplementary Table S1). M-GA-CAM and KIST were derived from parent colonies designated YAOUNDÉ and KISUMU1, respectively, by selection (in 2005) for standard homokaryotypes (i.e., 2L+; 2R+) with respect to all known polymorphic inversions on chromosome 2. All laboratory colonies were maintained in the University of Notre Dame (UND) insectary under controlled conditions of 27±1°C, 85% RH with a 12 h:12 h light:dark cycle that included 1 h dawn and dusk transitions. Colonies were maintained in separate bays to avoid contamination.
Eggs were placed in plastic trays (27 cm × 16 cm × 6.5 cm) containing 1L of R0 (reverse-osmosis purified) water. Larvae were reared at a density of ~100 per pan and fed daily with a 2:1 mixture of finely ground tropical fish pellet:bakers yeast. Pupae were transferred to 0.2 m3 emergence cages. After emergence, adult mosquitoes were supplied absorbent cotton saturated with 20% sucrose solution.
Differential gene expression between M and S colonies was examined at three developmental periods: unsexed late larvae, adult virgin females and gravid females. Late larvae were 4th instar larvae harvested 2-8 h prior to pupation (those harvested 2-4 h prior to the molt to pupa are technically considered pharate pupae but for simplicity we will refer to this sample as late larvae). Virgin females were isolated by sorting newly emerged adults (≤6 h after eclosion) into adjacent sex-specific cages; they were harvested 30 min following the onset of the dusk cycle transition on day 3 post-emergence. To collect gravid females, males and females were maintained in the same cage post-emergence to allow females to become inseminated. On mid-afternoon of day 3, females were offered a bloodmeal on a volunteer’s arm; females that did not successfully engorge were removed. An oviposition cup was placed into each cage 72 h post-bloodfeeding and females were allowed to oviposit overnight. Egg cups were removed the following morning, and females were subject to a second bloodfeeding on a human host mid-afternoon. Gravid females were harvested 82 h following the 2nd bloodmeal, at 4 h post-dusk. This timing, determined through independent experimental trials, represented the window during which the highest density of eggs was laid.
Each of the three developmental periods was represented by five intra-colony replicates. For each colony, replicates were derived from independent RNA samples extracted from different cohorts to ensure that trends were reproducible. In addition, each replicate was derived from larvae/adults drawn from three pans/cages to minimize the contribution of any individual pan/cage to variation between samples. Mosquitoes harvested at each developmental period were immediately snap-frozen in liquid nitrogen and stored at -80°C until RNA extraction.
Field-collected mosquitoes
Sequential collections were made in 2006 from Burkina Faso (26 Sep-13 Oct) and Cameroon (15 Oct-2 Nov) from localities where A. gambiae M and S forms were expected to co-occur (Supplementary Table S2). Within countries, samples came from multiple villages and breeding sites. Larvae (3rd-4th instar) were collected by dipper and transported to local field facilities where they were transferred to basins containing water from the breeding sites freed of mosquito predators, fed with cat or fish pellets, and maintained under ambient temperature, humidity, and photoperiod. Pupae were transferred to netted square cages for adult emergence; adult cages were protected from rain and direct sun by a tarpaulin. Within 12 h after adult eclosion (before mating could occur), sexes were separated into adjacent cages and given access to cotton wool soaked in 20% sucrose. Virgin females were harvested only on days when the sky was clear, 3d post-emergence and mid-way through astronomical twilight (~1.5 creps, as determined by the local Muslim prayer times for sunset and evening). Females were immediately frozen at -20°C, and transferred to individual 1.5 ml tubes of 70% ethanol at -20°C for 10 min to soften the exoskeleton. The legs were dissected and retained in this tube until transport to UND. The remaining carcass was removed, penetrated three times with a needle to break the integrity of the exoskeleton, and transferred to a correspondingly numbered tube containing 500μl of RNAlater RNA Stabilization Reagent (Qiagen, Valencia CA) and stored at -20°C until transport to UND.
At UND, A. gambiae s.s. and its molecular forms were identified using the rDNA-based PCR/RFLP assay of Santolamazza et al. (2004) without prior DNA extraction; a single leg was added directly to the PCR mixture.
RNA isolation
Total RNA was isolated and purified from pools of 15-20 individuals using the RNeasy Mini Kit (Qiagen). RNA quality and quantity were assessed using the NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE) at wavelengths of 230, 260, and 280 nm. The integrity of total RNA was further verified by running 200 ng samples on 1.5% agarose gels. Total RNA (5 μg per sample) was treated with DNase I (Invitrogen, Carlsbad, CA) to remove any contaminating DNA.
GeneChip microarray processing and analysis
RNA samples (5 μg each) were delivered to the Center for Medical Genomics at Indiana University for further processing and hybridization. Single cycle labeling was conducted by synthesizing cDNA using a T7 promoter-dT24 oligonucleotide primer with the SuperScript Choice System (Invitrogen, Carlsbad CA). Products were purified with a GeneChip Sample Cleanup Module (Affymetrix, Santa Clara, CA), and used to prepare biotinylated cRNA with the GeneChip IVT Labeling kit (Affymetrix). Following purification (RNeasy columns; Qiagen), quantification and fragmentation, 15 μg of biotinylated cRNA was hybridized to GeneChip Plasmodium/Anopheles Arrays (Affymetrix) following protocols recommended by Affymetrix. After hybridization, arrays were washed, stained, and scanned using the Affymetrix Model 450 Fluidics Station and Affymetrix Model 3000 scanner controlled by GCOS software. Raw fluorescence intensity values for each probe were obtained using Microarray Suite v5.0 (MAS5) software (Affymetrix). A total of 60 arrays (=3 timepoints × 2 forms × 2 colonies × 5 intra-colony replicates) were run in total. All arrays for a given developmental period (n=20) were processed under identical experimental conditions on the same day.
Files containing all raw (CEL) and normalized data (see below) have been deposited with Array Express in compliance with MIAME and MGED recommendations and are available under accession number E-TABM-344.
CEL files containing the raw intensity values were imported into Bioconductor (www.bioconductor.org), an open-source software project based on the R programming language (www.r-project.org). Data quality was assessed using functions in the affy and affyPLM packages (Bolstad et al., 2005). Background subtraction, normalization, and summarization of probe set data into one expression value were accomplished using the GCRMA function. The subset of expressed genes was obtained in two steps, similar to Hahn and Lanzaro (2005). First, probe sets corresponding to expressed genes were defined as those whose MAS5 normalized hybridization signals exceeded 100 for at least two of the five intra-colony replicates of at least one of the four colony samples. Second, because the Anopheles GeneChip was designed from an early genebuild (Build 2, 2003) such probe sets were re-mapped onto genebuild AgamP3.3. Any probe sets interrogating the same gene were collapsed into a single observation, and those which did not interrogate an annotated gene were omitted. Statistical tests to identify the genes differentially expressed between M and S were conducted on this subset of expressed genes for each developmental period.
Differentially expressed genes between M and S (P<0.05) were identified by applying a linear mixed model analysis of variance (ANOVA) conducted in the nlme package in R. For each gene in this model, the fixed variable was ‘molecular form’, while both ‘colony’ and ‘intra-colony replicate’ were random. Note that all four colonies originated from different parts of Africa and have been inbred to various degrees. Moreover, because intra-colony replicates are not genetically identical, they are not strictly biological replicates (or ‘random’ effects). Thus, gene expression profiles could be influenced by variation in the genetic background of nonisogenic cohorts and colonies, resulting in differences due to genetic background alone (c.f., Sandberg et al., 2000). Because the ANOVA approach for identifying form-specific differences required similar trends in gene expression for both colonies within a form, under the hypothesis that characteristics inherent to M and S (e.g., mate recognition) should be conserved regardless of geographic origin, this is a stringent test that compensates for the problem of nonisogenic colonies. A less conservative statistical approach is to treat each of the samples as biologically independent, though varying amounts of inbreeding within colonies poses similar difficulties for biological and statistical accuracy. Under this alternative approach, gene expression values were subject to additional statistical testing for differences between M and S forms by means of the empirical Bayes-moderated t-test implemented with the limma package in R.
Quantitative real-time RT-PCR
Total RNA (3μg per sample) was extracted from a pool of 15 individuals using the methods already described, and was independent of RNA samples used in the microarray experiment. Each sample was digested with DNase I (Invitrogen) and converted to cDNA using TaqMan Reverse Transcription Reagents (Applied Biosystems, Foster City, CA) as directed. Random hexamers were used as primers in lieu of Oligo d(T)16. Primers targeting exons (Supplementary Table S3) were designed with Primer Express 2.0 software (Applied Biosystems). Forward and reverse primer concentrations of 50, 300, and 900 nM were used to determine optimal conditions for each gene. Quantitative RT-PCR was performed with the AB7500 Real Time PCR system and 96-well optical reaction plates (Applied Biosystems). Ribosomal protein S7 was employed as a control gene using primers given in Dong et al. (2006). All reactions were performed in triplicate in a total volume of 25ul containing 12.5ul of SYBR Green PCR Master Mix and 300 nmol of each primer under the following conditions: 50°C for 2 min, 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 15 s, annealing and extension at 60°C for 1 min, followed by 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s.
Expression levels were measured separately for control and target genes, using four intra-colony replicates for each of four colonies (two M, two S). In addition, each measurement for both target and control genes was based on three technical replicates. Threshold cycle (CT) values reported by the AB7500 Real Time PCR system were normalized and converted to relative log2 fold differences between M and S samples. One-tailed t-tests (P < 0.05) were carried out under the hypothesis that the qRT-PCR expression differences are in the same direction as the array, using the log2 values of candidate genes for statistical validation of differential expression.
Results
Whole-genome high density oligonucleotide microarrays were used to examine gene expression differences between laboratory isolates of the Anopheles gambiae M and S molecular forms, with the goal of identifying candidate genes underlying physiological and behavioral differences. Although no discrete phenotypic differences have been described between forms apart from mating (swarming) behavior, there are quantitative differences in larval development times and behaviors, and in breeding site colonization which could relate to larval competition, predation and/or oviposition preference by gravid females (Diabate et al., 2005, 2007a; Edillo et al., 2002, 2006). Based on these observations, we collected RNA samples at three developmental periods: late (4th instar) larvae, virgin females during the evening twilight when they are reproductively active, and gravid females at night when they seek oviposition substrates. To reduce the likelihood of identifying colony-specific gene expression differences related to either local conditions of the source population or random genetic drift, we sampled from two independent colonies of M and two of S, all originating from different geographic locales. All colonies were maintained under identical controlled insectary conditions. In total, 60 arrays were hybridized, though data from one late larval array (hybridized with a sample from the M-GA-CAM colony), one virgin female array (hybridized with a sample from the Pimperena colony), and one gravid female array (hybridized with a sample from the M-GA-CAM colony) were omitted prior to analysis due to poor quality hybridization.
Small fraction of the genome is differentially expressed
The Anopheles/Plasmodium GeneChip was designed from an early genebuild (Build 2, 2003). After re-mapping to the AgamP3.3 genebuild, the 16,941 A. gambiae probe sets on the Affymetrix GeneChip were found to interrogate only 10,812 predicted genes. Of these, ~81% were detected as expressed in either M or S forms at any of three developmental periods. This proportion was essentially constant across developmental periods: for late larvae, 82% (8903) were expressed; for virgin females, 81% (8754); for gravid females, 79% (8557).
Given the very small fraction (~1%) of the M and S genomes involved in the “speciation islands” (Turner et al., 2005), a correspondingly small fraction of genes might be differentially expressed. To identify the genes whose expression was significantly different between all samples of M versus all samples of S at each developmental period, we applied a linear mixed model ANOVA. The combined total number of genes differentially expressed across all three developmental stages using this approach was 281 at a nominal P<0.05; 23 were differentially expressed in multiple stages. The mixed model testing procedure appears to be highly conservative—only ~2% of expressed genes are significant P<0.05. Applying a less stringent approach (see Methods), the total number of differentially expressed genes at a nominal P<0.05 was 5785. Based on the false discovery rates (FDR; Benjamin, Hochberg, 1995) calculated for each of the three developmental periods, the differentially expressed genes identified with the ANOVA model had FDR values below 0.013 in this second set, suggesting that these genes were truly altered in their expression between M and S. These 281 genes constitute our high confidence gene set, and are verified with qRT-PCR below.
Gene expression differences predominate in virgin females of M and S
The only aspect of M and S biology where discrete differences have been found is premating reproductive isolation; other correlates overlap and seem to be quantitative in nature. It was therefore of interest to examine the distribution of expression differences across the three developmental periods assayed by microarray. Of the transcripts detected, 1.9% (n=164) differed significantly at the virgin female stage between M and S (range 1.17-25 fold), while the percentage that differed between forms at the other stages was smaller by half: 0.7% (n=64) in late larvae (range 1.19-6.08), 0.9% (n=78) in gravid females (range 1.16-21.7) (Figure 1). The excess of differences at the virgin female stage was significant (χ2, P<0.001). For the subset of genes that were significantly differentially expressed at more than twofold (and more than fourfold) between M and S, a similar overrepresentation of virgin female differences was observed: 0.41% (n=36) and 0.09% (n=8) at two- and fourfold expression levels for virgin females versus 0.17% (n=15) and 0.02% (n=2) for late larvae; 0.22% (n=19) and 0.07% (n=6) for gravid females.
Figure 1.
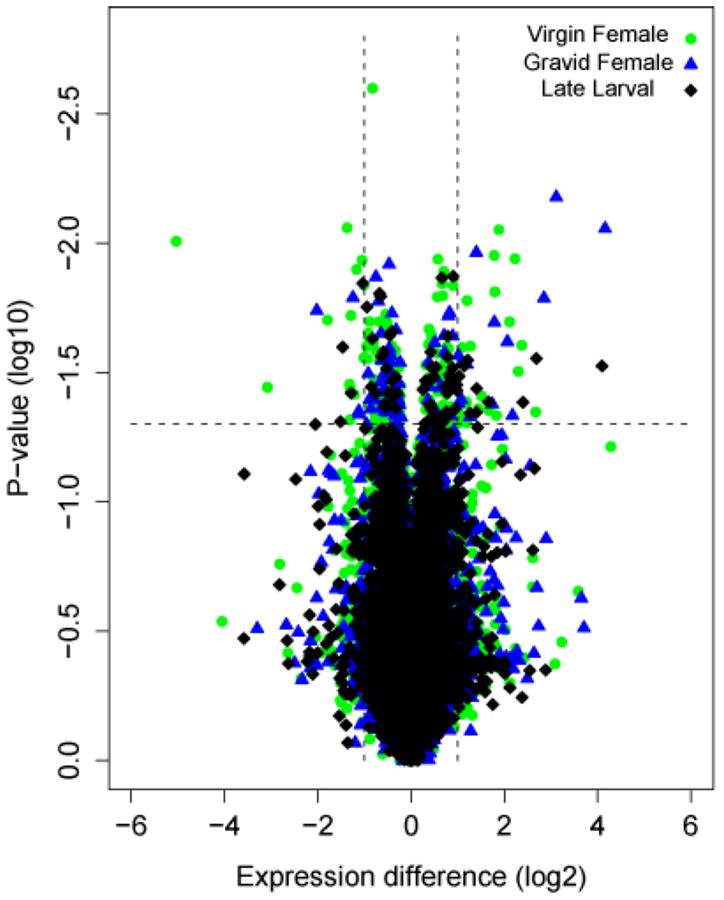
Volcano plot of significance against relative expression differences from microarray comparisons between A. gambiae M and S molecular forms at three developmental periods. Each symbol represents one gene that had detectable expression in either form in late larvae (black diamond), virgin females (green dot), or gravid females (blue triangle). The x-axis displays log2-transformed signal intensity differences between M and S; positive values represent overexpression in S while negative values represent overexpression in M. The Y-axis displays log10-transformed P-values associated with ANOVA tests of differential gene expression. The horizontal dashed line indicates the threshold for significance; the vertical dashed lines indicate thresholds for differential gene expression in excess of twofold.
The disproportionate contribution of the virgin female stage to gene expression differences between M and S is illustrated diagrammatically in Figure 2. This Venn diagram also emphasizes the relatively small overlap of gene expression differences across multiple developmental stages. Only 12-19% of genes differentially expressed at one stage were also different at other stages, and only two genes (<1%) were differentially expressed across all three developmental periods. With one exception, differences shared between developmental periods showed the same direction of differential expression across periods, and roughly half involved genes with the largest differential expression (more than twofold differentially expressed).
Figure 2.
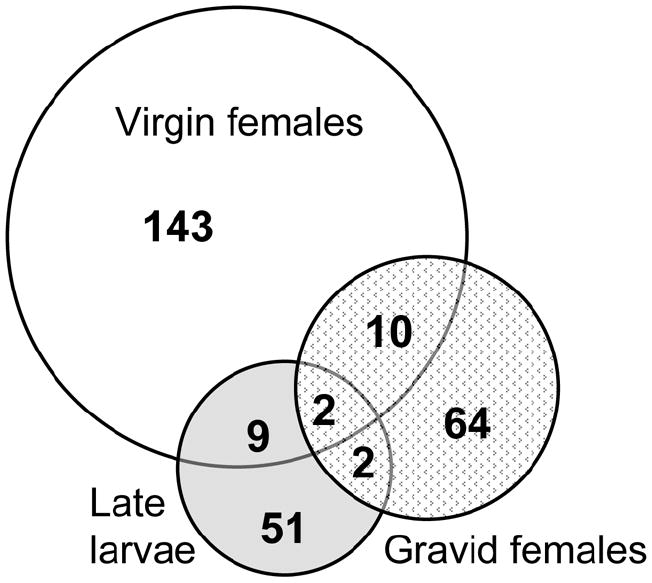
Venn diagram of genes differentially expressed between A. gambiae M and S based on microarray comparisons at three developmental periods. Shown are the numbers of nonoverlapping and overlapping genes between M and S virgin females (white circle), gravid females (stippled circle), and late larvae (gray circle).
Gene expression differences primarily involve overexpression in S
The relative contribution of M and S to differential gene expression was examined in the high confidence gene set at each developmental period. To be clear about terminology, differentially expressed genes whose expression was greater in S relative to M will be referred to as “overexpressed” in S (and vice versa); this term was chosen as the most neutral, given that we do not know whether the true biological basis for this difference is upregulation in S or downregulation in M. Among the set of all differentially expressed genes, no clear trend emerged. In virgin and gravid females, only 48% and 42% of genes were overexpressed in S; in late larvae, 61% were overexpressed in S. However, when consideration was limited to the subset of genes differentially expressed by at least twofold between M and S, three times as many genes were overexpressed in S than in M at all three developmental periods. In this subset, overexpression in S accounts for 72%, 79% and 73% of the differences at virgin, gravid and late larval periods, respectively.
The trend toward overexpression in S relative to M raises an important question: to what extent do differences in signal intensity reflect sequence differences rather than gene expression levels? A potential problem with using a microarray designed from one species (or molecular form) to measure gene expression in other closely related species (or forms) is that gene expression differences can be confounded by sequence mismatches to probes on the array (Gilad et al., 2005). This issue is especially pertinent for regions of the genome exhibiting the greatest level of nucleotide divergence between species or emerging species. Thus, high differentiation between M and S at genes in the speciation islands (average nucleotide divergence is ~1-2% in the islands; Stump et al., 2005; Turner, Hahn, 2007) could be mistakenly interpreted as high levels of differential expression between forms.
Three lines of evidence allay this concern as it applies to our results. First, if the trend toward overexpression in S was due exclusively to nucleotide divergence rather than differential gene expression between forms, we would expect that the same genes would be implicated at all three developmental periods (given that their expression was detected in both forms throughout-- which it was), yet only 2 of 281 differentially expressed genes were shared between the three developmental periods. Second, the Anopheles/Plasmodium GeneChip was designed based on reference sequence determined from the PEST colony (Holt et al., 2002). Although this colony descended from initial crosses between M and S forms, it is M-like in the X chromosome island (see Stump et al., 2005). Thus if nucleotide divergence were the sole factor explaining overexpression in S (i.e., if the S-bias was due to fewer mismatches between probes on the chip and genomic targets in one form), the bias should have been in the opposite direction to that observed: M would be expected to be overexpressed. The third and most definitive evidence comes from independent microarray experiments in which genomic DNA from individual M or S mosquitoes was hybridized to the same platform to assess sequence divergence (White et al, unpublished data). Probe-level sequence divergence data for all differentially expressed genes located in the speciation islands was examined. The vast majority of genes (>95%) were not significantly differentiated at targets corresponding to the oligonucleotide probes, implying that differential gene expression is not an artifact driven by DNA sequence divergence. While we can not completely exclude this effect, its role must be relatively minor and cannot by itself explain the trend toward overexpression in S at any developmental period.
Candidate genes include those involved in olfaction
Functional annotation of the A. gambiae genome is incomplete and uneven in quality. For this reason, we attempted no formal quantitative analysis of differentially expressed genes by function; our approach was exploratory. Using putative functions already assigned to genes, or assigning possible functions based on orthologs predicted in the Ensembl gene report and gene ontology (GO) terms mapped to the genes, we placed genes from each developmental period into various functional categories. This was not possible for ~25% of genes from each period, as they could not be assigned any function. The proportion of differentially expressed genes at each stage that were found in eight categories (other categories not shown) is given in Figure 3. Among the most populous categories, particularly in the virgin female samples, were “transcription” (a category that contains nucleic acid binding proteins potentially acting as transcription and splicing factors) and “sensory perception & response.” In virgin females, the latter category included a striking number of genes potentially involved in olfaction, a process that is likely to play a role in mate recognition. Among these genes were four odorant binding proteins (OBP49, OBP52, OBPjj9, OBP25) and an antennal carrier protein (AP-1). Also included was a cuticular protein (CPF3), a member of a small cuticle family (Togawa et al., 2007). Previous studies of CPF3 indicated that mRNA from this gene was abundant in pharate adults of the A. gambiae G3 strain, and the protein did not have the chitin-binding capacity found with the more numerous CPR family of proteins. These properties led to the supposition that CPF3 was located in the epicuticle (Togawa et al., 2007). A structural model suggests that it could bind an unbranched lipoidal compound similar to the cuticular hydrocarbons that serve as sex pheromones in Drosophila (S. Hamodrakas, pers comm). In addition, several other genes in this category (two G-protein coupled receptors, three GTPases, syntaxin and three glutathione-S-transferases) may have roles as odorant/taste receptors, signal transduction components, mediators of synaptic vesicle docking and odor degrading enzymes (Rutzler, Zwiebel, 2005). Three other genes—two included in the “sensory perception” category that are similar to the Drosophila genes lingerer and doublesex, and the third in “fatty acid metabolism” (a delta-9-desaturase)—have roles in courtship behavior (lingerer; Kuniyoshi et al., 2002) and the control or production of sex pheromones (doublesex and desaturase 1 and 2; Dallerac et al., 2000; Jallon et al., 1988) in Drosophila.
Figure 3.
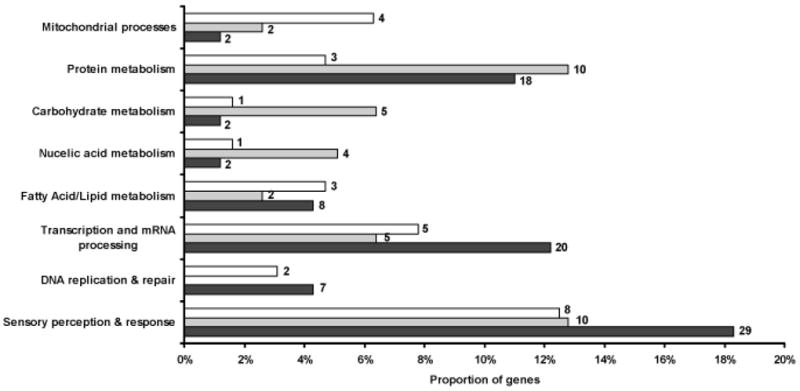
Distribution of differentially expressed genes among functional categories at each developmental period. Bars indicate the proportion of genes in each category: black, virgin females; gray, gravid females; white, late larvae. Number of genes in each category is given beside each bar. Percentages do not total to 100 as not all categories are shown.
A disproportionate number of differentially expressed genes are found in the X island
We searched for nonrandom patterns in the genomic distribution of genes differentially expressed between M and S (Table 1). As a first step, we asked whether differentially expressed genes were disproportionately X-linked or otherwise overrepresented on a given chromosome arm relative to the number expected given arm length and the number of expressed genes per arm. Considering each developmental period individually, there was no overrepresentation of differentially expressed genes on any arm in the virgin or gravid female samples. However, in late larval samples there was a significant excess of differentially-expressed genes on the X chromosome (10) compared to elsewhere in the genome (54) (χ2, P = 0.048). In these larval samples, autosomal genes that were differentially expressed between M and S were uniformly distributed across the four autosome arms.
Table 1.
Chromosomal distribution of 281 genes differentially expressed between M and S at three developmental periods1.
| Developmental Period
|
|||
|---|---|---|---|
| Chromosome
|
Late Larval | Virgin Female | Gravid Female |
| 2L | 10 (1) | 32 (2) | 22 (1) |
| 2R | 23 | 56 | 22 |
| 3L | 11 | 32 | 13 |
| 3R | 10 | 31 | 14 |
| X | 10 (2) | 13 (3) | 7 (0) |
Genes in parentheses are located inside speciation islands on 2L and X.
The next step was to assess whether the differentially expressed genes were disproportionately represented in the speciation islands. The size of the speciation islands, roughly estimated from hybridization of genomic DNA from individual M and S to oligonucleotide microarrays (after Turner et al., 2005) was ~4.2 Mb and ~3.0 Mb for X and 2L, respectively [White et al, unpublished; note that sizes of the islands differ from Turner et al. (2005) due to improved AgamP3 assembly in centromere-proximal regions]. Using a χ2 test, the observed numbers of differentially expressed genes found within and outside the boundaries of one or both islands was compared to the numbers expected in the two partitions, given the total number of expressed genes and the length of each partition. Across all three developmental periods and considering both islands together, there was a significant overrepresentation of differentially expressed genes in the islands (8 genes; P = 0.0002). Considering the islands separately, only the X island carried a significant excess of differentially expressed genes (5 genes; P = 0.00003). With the exception of the gravid female samples in which no excess was found in either or both islands, the other developmental periods when analyzed separately also showed a significant excess of differentially expressed genes only in the X island. The results were unchanged when the analyses were repeated using speciation islands whose length was increased by 1 Mb each.
Quantitative real-time RT-PCR validates microarray results
Correct interpretation of our microarray results rests on the assumption that the GCRMA-normalized expression values are correlated with actual RNA levels in the samples under consideration. To validate this assumption, we measured gene expression levels using an independent technique: quantitative real-time RT-PCR (qRT-PCR). A total of 14 genes were targeted for validation primarily because of the evident importance of the virgin female period in differential gene expression. Other criteria were their map location within (or near) speciation islands on X or 2L, and/or functional annotation suggesting possible roles in prezygotic isolation (Table 2).
Table 2.
Comparison of average gene expression levels as measured by oligonucleotide microarray and qRT-PCR.
| Fold-change ratio of S/M1
|
|||||
|---|---|---|---|---|---|
| Affymetrix ID | Vectorbase ID | Band | Description (biological process) | Microarray | qRT-PCR |
| 2L.387.0_CDS_at | AGAP004690 | 20A | CPF3 cuticular protein | -24.96 | -27.2*** |
| 3R.26.0_CDS_at | AGAP009194 | 33C | GST-E2 | -7.81 | -6.37*** |
| UNKN.168.0_CDS_at | AGAP001091 | 6 | protein kinase | 4.7 | 1.05NS |
| X.212.0_CDS_at | AGAP001030 | 5D | signal transduction | 3.47 | 2.5*** |
| 2L.25.0_CDS_at | AGAP004799 | 20C | AP-1 antennal carrier protein | 2.83 | 3.4** |
| 3L.14.0_CDS_at | AGAP012320 | 46B | OBP25 odorant binding protein | 2.09 | 2.8* |
| 2R.1354.0_CDS_a_at | AGAP004572 | 19C | fatty acid desaturase | 2.03 | 1.92** |
| X.213.0_CDS_a_at | AGAP001036 | 5D | aminopeptidase | 1.86 | 2.2*** |
| 2R.3535.0_CDS_at | AGAP004050 | 17C | doublesex | 1.77 | 0.93NS |
| X.1091.0_CDS_at | AGAP000959 | 5D | unknown | 1.47 | 2.42*** |
| UNKN.50.2_CDS_a_at | AGAP001082 | 6 | saposin (lipid metabolism) | -1.42 | -1.51* |
| 2L.543.1_a_at | AGAP004817 | 20C | lingerer (copulation) | -1.41 | -2.17** |
| UNKN.137.0_UTR_a_at | AGAP001090 | 6 | unknown | 1.30 | 1.31NS |
| Ag.2L.94.0_CDS_at | AGAP006076 | 23C | OBP50 | 1.08NS | 1.90** |
Positive and negative values are S-biased and M-biased, respectively.
NS, not significant;
= P<0.05,
= P<0.01,
= P<0.001.
The direction of differential gene expression was consistent between the microarray and qRT-PCR methods, as expected. In addition, a strong correlation was found for the magnitude of the fold change ratios derived from the two methods (Table 2; Figure 4). The correlation was highly significant not only for the complete data set of 14 genes (Spearman’s coefficient rs = 0.98; P<0.000001), but also after removing the gene (CPF3) with the most biased expression (Spearman’s coefficient rs = 0.74; P=0.004).
Figure 4.
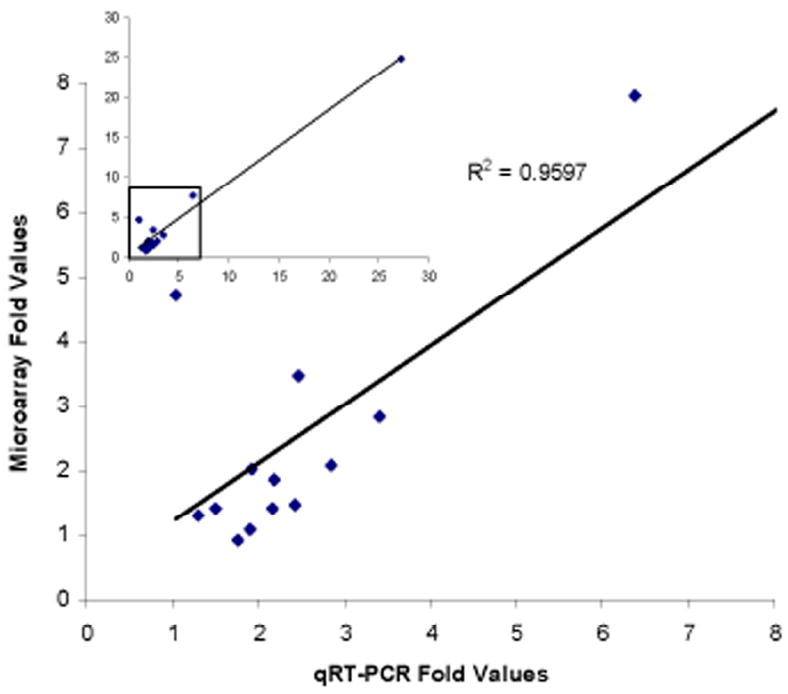
Correlation between microarray and qRT-PCR estimates of gene expression differences between A. gambiae M and S virgin females. Inset at upper left shows the plot for all 14 genes; the box enclosing data from 13 of the genes is magnified in the plot below.
Although correlation between the fold change values estimated by the two methods was high, not every gene identified as significantly differentially expressed using microarray data was confirmed by qRT-PCR. Of the 14 genes we screened, 13 were significantly differentially expressed from our “high confidence” microarray set. We found that ten of these (77%) were also significantly differentially expressed by qRT-PCR. The three disagreements (23%) are comparable to the level of discrepancy (13-16%) seen in a study devoted to this issue (Dallas et al., 2005), and have several possible explanations. Assuming that they were not false positives from the microarray analysis, discrepancies can nevertheless arise due to different hybridization kinetics of the probe sets/primers, or to the qRT-PCR primers interrogating a different transcript(s) than the one interrogated by the microarray probe. The last possibility could apply in the case of AGAP001090, as different exons are targeted by the probes and primers. Although this explanation is not consistent with current AgamP3.4 annotation for the other two genes, it cannot be discounted. The presence of annotation errors is suggested by the fact that the qRT-PCR primers targeting AGAP001030 (Supplementary Table S3)-- a strongly biased gene by both microarray and qRT-PCR methods-- anneal to a region annotated as intronic in AgamP3.4.
One gene not considered differentially expressed between M and S based on the microarray analysis (OBP50) proved significantly overexpressed in S by qRT-PCR. This result may reflect the greater sensitivity of qRT-PCR. It also adds a fifth odorant binding protein to the set of candidate genes differentially expressed between M and S virgin females.
Gene expression differences are consistent between lab and field
Our study design included two independent colonies of each A. gambiae molecular form from different parts of Africa, an approach adopted in an effort to minimize the likelihood of finding gene expression differences related to local adaptation or genetic drift within either colonies or geographic regions. Nevertheless, the reliance on laboratory colonies raises doubts about whether these gene expression differences reflect patterns in natural populations. To address this issue, we also sampled virgin females from sympatric populations of M and S from two locations in West and Central Africa: Burkina Faso and Cameroon. Based on RNA extracted from these individuals in both locales, we performed qRT-PCR on a subset of five genes that were previously found to be differentially expressed based on both microarray and qRT-PCR analyses of laboratory colonies. As shown in Figure 5, qRT-PCR results from natural populations were consistent between both parts of Africa, and also with our laboratory results. Although the size of fold-changes in expression between M and S differed, gene expression differences were in the same direction and statistically significant (all at P<0.05).
Figure 5.
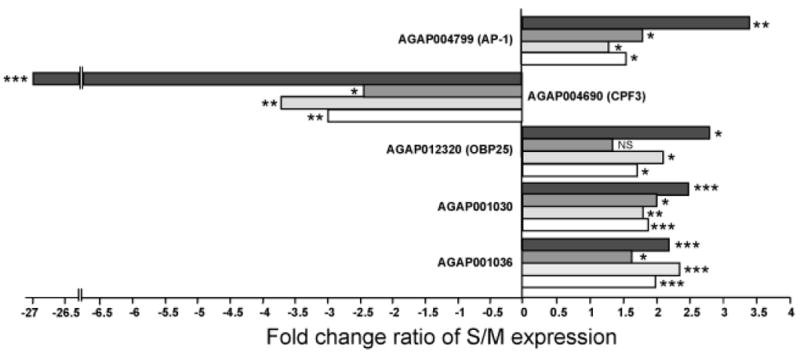
Differential gene expression assessed for five genes by qRT-PCR in A. gambiae M and S samples from laboratory colonies and natural populations. Horizontal bars represent average fold change ratio between S and M samples from laboratory colonies (black bars) or field-collected samples from Burkina Faso (dark gray bars), Cameroon (light gray bars), and Burkina Faso + Cameroon (white bars). Positive values represent overexpression in S; negative values represent overexpression in M. P-values are indicated as NS, not significant; *, P<0.05; **, P<0.01; ***, P<0.001.
Discussion
Although the M and S forms of A. gambiae are widely considered to be nascent species, no specific phenotypic differences-- morphological, behavioral or ecological-- have been identified (in the case of morphology) or characterized in detail (in the case of behavior and ecology). The absence of detailed phenotypic information discourages any a priori candidate gene approach and precludes a QTL mapping approach to understanding this ongoing process of speciation. Instead, we adopted a “reverse ecology” approach to identify candidate genes. By generating whole-genome profiles of transcription at key developmental periods, we have explored gene expression itself as a phenotype to identify genes with potential roles in species isolation. Our approach rests on the premise that differences in gene regulation are likely to contribute to species differences, an idea that is not new (King, Wilson, 1975) and which has received increasing empirical support (Borneman et al., 2007; Wray, 2007). Our experiments provide the first genome-wide description of gene expression differences between M and S in late larvae, virgin females, and gravid females, developmental periods that we hypothesized would be most likely to show differences based on what is known of M and S biology. The candidate gene list appears robust, based on successful validation using an alternative method of RNA quantification. Real-time qRT-PCR successfully verified most of the tested candidate genes from independent RNA samples collected not only from the same four laboratory colonies used for the microarray experiments, but also from two natural populations. Yet, the fact that fully one-fourth of the candidate genes have no similarity to others in the public databases and therefore no known function is sobering and demonstrates the extent of difficult work that lies ahead.
Previous application of oligonucleotide microarrays to map nucleotide divergence between nascent M and S species revealed that only ~1% of the genome was significantly differentiated (Turner et al., 2005). Moreover, detectable differentiation was largely confined to two centromere-proximal regions of low recombination on chromosomes 2L and X, named “speciation islands” based on the expectation that their combined content of 67 predicted genes would include the “speciation genes” responsible for ecological and behavioral (reproductive) isolation between M and S. Our findings based on differences in transcript abundance between M and S at different developmental periods partially support this result. Relative to differentiated genomic sequences, a comparably small fraction of the transcriptome was differentially expressed between forms at any of the developmental periods examined (1-2%), and considering all periods collectively, we do find as many as 8 candidate genes in the X chromosome speciation island—a disproportionately high number. However, candidate genes were not disproportionately represented in the 2L island, despite the four interesting candidates located there (CPF3) or adjacent (antennal carrier protein AP-1, lingerer, and a putative transcription factor). No genes were found to be differentially expressed in the 2R island defined by Turner et al. (2005), consistent with the fact that this island does not show nucleotide differences between M and S outside of Cameroon (Turner, Hahn 2007). The vast majority (>93%) of the combined total of 281 significantly differentially expressed genes did not map within or even adjacent to either the 2L or X chromosome speciation islands, and were dispersed across all chromosome arms. It is possible that these genes are controlled by a trans-acting factor located within one of the islands, whether it is differentially expressed or not. Unfortunately, the functional annotation of the A. gambiae genome is too incomplete—particularly in the repetitive DNA-rich and difficult-to-assemble centric regions containing the speciation islands-- to provide insight on this point. Further investigation is required to determine the functional roles and possible interactions of these gene products, as well as their contribution to ecological or behavioral isolation between M and S forms.
The timing of many mosquito behaviors such as larval-pupal ecdysis, adult emergence, host seeking, swarming and oviposition is governed by endogenous circadian rhythms entrained to the natural cycle of light and dark (reviewed by Clements, 1999). A. gambiae is a crepuscular and nocturnal species. Adults are mostly inactive during daylight, but attain peak flight activity at dusk when swarming and mating occur, and remain active at night when host seeking and oviposition occur. As the experimental light regime with gradual dawn and dusk transitions mimicked that found in nature, we assumed that at least one of the cues (reduction in light intensity) that stimulate and condition the sequence of behaviors entailed in mate seeking and oviposition was received by the virgin and gravid adult females, respectively, despite the absence of males or oviposition-site attractants at the time that RNA was collected. Our results from the virgin female samples in particular appear to support that hypothesis. Twice as many genes were differentially expressed between M and S forms at the virgin female developmental period than either of the other developmental periods. This was not only a statistically significant result based on our laboratory colonies, but one which may have some biological significance in field populations as well, because the virgin females were assayed at the chronological age and the diel time when mating normally occurs. If some of the genes differentially expressed in M and S virgin females contribute to courtship behavior, our results would agree with previous studies suggesting that courtship phenotypes are among the first traits to evolve among incipient species (Gleason, Ritchie, 1998; Mackay et al., 2005; Mullen et al., 2007). Evidence consistent with this interpretation is discussed below.
Aerial swarming by A. gambiae males is crepuscular, beginning about 10 minutes after dusk and continuing for about 20 minutes (Clements, 1999). Sexually receptive virgin females (typically those older than 60 h; Charlwood, Jones, 1979) fly individually to the swarms. Females entering the swarm are grasped by males, and mating lasting a few seconds is initiated in flight. The existence of a premating barrier between M and S forms is beyond doubt, given the rarity of inter-form sperm transfer (~1%) even where both forms are currently breeding in the same location (Tripet et al., 2001). However, the mechanistic basis of premating behavioral isolation is completely unknown. Swarms are usually exclusive to one form, suggesting a spatial and/or temporal component to isolation (Diabate et al., 2003; Diabate et al., 2006). Nevertheless, mixed swarms are not sufficiently rare (4 of 26 swarms were mixed in one survey; Diabate et al., 2006) for swarm segregation to serve as the sole isolating factor. Specific mate recognition systems operating within swarms may be more important for sexual isolation (Diabate et al., 2006; Tripet et al., 2004). Until recently, male choice has been emphasized by researchers, given that males are known to be stimulated by, fly toward and attempt to seize and clasp females (or any object) with the appropriate flight tone (Charlwood, Jones, 1979; Clements, 1999). However, the extent of auditory-based male choosiness has been questioned, as they respond to frequencies from 350-600 Hz (Charlwood, Jones, 1979). Moreover, the hypothesis that flight tone is the basis for specific mate recognition by males has not stood up to precise measurements which revealed extensive overlap in the distribution of amplitudes between molecular forms of A. gambiae (Tripet et al., 2004). Instead, it has been suggested that contact pheromones may serve as recognition cues, and that females may be the more selective of the sexes in mate discrimination (Tripet et al., 2004). Females are capable of rejecting the copulation attempts of males by violent kicking (Charlwood, Jones, 1979). Female A. gambiae also may have more at stake in choosing mates correctly given that they mate only once (Goma, 1963; Jones, Gubbins, 1978) in contrast to males which swarm every day of post teneral life (Nielsen, Haeger, 1960). Our data are suggestive in this regard. A surprising number of candidate genes at the virgin female stage are plausibly involved in scent detection, possibly related to mate recognition. Among these are five odorant binding proteins and an antennal carrier protein whose roles may be the discrimination of male odor. Unfortunately, corresponding gene expression profiles of males were not assessed in this study. Future studies should fill this gap to gain a more complete picture of gene expression differences at the time of peak activity in both males and females of A. gambiae M and S forms.
In contrast to the virgin females, gene expression differences between late larvae and gravid females of M and S were not immediately suggestive of behavioral or ecological processes that might distinguish the forms, though we expect that ecological and adaptive divergence is fundamental to the speciation process in A. gambiae (Coluzzi, 1982; Manoukis et al., in press). More comprehensive sampling of different developmental periods and more detailed investigation of specific candidate genes already identified is clearly necessary to give more insight into the biology of differences between molecular forms. This study is an important first step in that process, as it has identified candidate genes that could not have been anticipated based on current levels of understanding in this system. The gene expression microarray approach is a powerful one, but two limitations should be acknowledged. First, it can only probe genes that are present on the microarray, and not all are present in the case of the A. gambiae genome. Indeed, representation on the microarray is especially poor in the very regions of the genome of greatest interest—the centromere proximal ones—and ribosomal RNA is absent altogether. Second, its success at identifying genes that contribute to ecological and reproductive isolation between these incipient species is premised on the notion that the basis for isolation involves gene expression differences. This need not be the case; important differences may arise through other means, including changes in coding sequence and post-transcriptional processes. Nevertheless, the candidate genes identified here, particularly those which might play a role in mate recognition, provide important leads. Implicating these genes in the process of premating behavioral isolation—a long-term goal—will be a multidisciplinary process that ultimately depends upon detailed understanding of mating behavior and the development of behavioral assays, a much neglected yet fundamentally important area of vector biology.
Supplementary Material
Acknowledgments
We thank R. Bruggner and E. Stinson for computer assistance. S. Christley and E. Paradis provided assistance with R. We are grateful to J.-B. Ouedraogo and R. Dabire of IRSS, Burkina Faso and the entomology teams at IRSS and OCEAC, Cameroon who assisted with field collections. Special thanks to IRD, Cameroon for providing “semi-field” insectary facilities. M. Rockman first coined the term “reverse ecology” in conversation with MWH. This work was funded by National Institutes of Health grant number AI63508 to NJB. The microarray studies were carried out at the Center for Medical Genomics (CMG) at Indiana University School of Medicine under the direction of H. Edenberg. The CMG is supported in part by the Indiana Genomics Initiative at Indiana University, which is supported in part by the Lilly Endowment, Inc.
References
- Akey JM, Eberle MA, Rieder MJ, et al. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biology. 2004;2:e286. doi: 10.1371/journal.pbio.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate – A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society B. 1995;57:289–300. [Google Scholar]
- Bolstad BM, Irizarry RA, Gautier L, Wu Z. Preprocessing high-density oligonucleotide arrays. In: Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S, editors. Bioinformatics and computational biology solutions using R and Bioconductor. Springer-Verlag; New York: 2005. pp. 13–32. [Google Scholar]
- Borneman AR, Gianoulis TA, Zhang ZD, et al. Divergence of transcription factor binding sites across related yeast species. Science. 2007;317:815–819. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- Butlin R, Roper C. Evolutionary genetics: microarrays and species origins. Nature. 2005;437:199–201. doi: 10.1038/437199a. [DOI] [PubMed] [Google Scholar]
- Campbell D, Bernatchez L. Generic scan using AFLP markers as a means to assess the role of directional selection in the divergence of sympatric whitefish ecotypes. Molecular Biology and Evolution. 2004;21:945–956. doi: 10.1093/molbev/msh101. [DOI] [PubMed] [Google Scholar]
- Charlwood JD, Jones MDR. Mating behaviour in the mosquito, Anopheles gambiae s.l. I. Close range and contact behaviour. Physiological Entomology. 1979;4:111–120. [Google Scholar]
- Clements AN. The Biology of Mosquitoes. CABI Publishing; New York: 1999. [Google Scholar]
- Coluzzi M. Spatial distribution of chromosomal inversions and speciation in anopheline mosquitoes. In: Barigozzi C, editor. Mechanisms of Speciation. Alan R. Liss, Inc.; New York: 1982. pp. 143–153. [PubMed] [Google Scholar]
- Dallas PB, Gottardo NG, Firth MJ, et al. Gene expression levels assessed by oligonucleotide microarray analysis and quantitative real-time RT-PCR -- how well do they correlate? BMC Genomics. 2005;6:59. doi: 10.1186/1471-2164-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallerac R, Labeur C, Jallon JM, et al. A delta 9 desaturase gene with a different substrate specificity is responsible for the cuticular diene hydrocarbon polymorphism in Drosophila melanogaster. Proceedings of the National Academy of Sciences U S A. 2000;97:9449–9454. doi: 10.1073/pnas.150243997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- della Torre A, Costantini C, Besansky NJ, et al. Speciation within Anopheles gambiae--the glass is half full. Science. 2002;298:115–117. doi: 10.1126/science.1078170. [DOI] [PubMed] [Google Scholar]
- Della Torre A, Tu Z, Petrarca V. On the distribution and genetic differentiation of Anopheles gambiae s.s molecular forms. Insect Biochemistry and Molecular Biology. 2005;35:755–769. doi: 10.1016/j.ibmb.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Diabate A, Baldet T, Brengues C, et al. Natural swarming behaviour of the molecular M form of Anopheles gambiae. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2003;97:713–716. doi: 10.1016/s0035-9203(03)80110-4. [DOI] [PubMed] [Google Scholar]
- Diabate A, Dabire KR, Heidenberger K, et al. Evidence for divergent selection between the molecular forms of Anopheles gambiae: role of predation. BMC Evolutionary Biology. 2007a;7 doi: 10.1186/1471-2148-8-5. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diabate A, Dabire RK, Kengne P, et al. Mixed swarms of the molecular M and S forms of Anopheles gambiae (Diptera: Culicidae) in sympatric area from Burkina Faso. Journal of Medical Entomology. 2006;43:480–483. doi: 10.1603/0022-2585(2006)43[480:msotmm]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Diabate A, Dabire RK, Kim EH, et al. Larval development of the molecular forms of Anopheles gambiae (Diptera: Culicidae) in different habitats: a transplantation experiment. Journal of Medical Entomology. 2005;42:548–553. doi: 10.1093/jmedent/42.4.548. [DOI] [PubMed] [Google Scholar]
- Diabate A, Dabire RK, Millogo N, Lehmann T. Evaluating the effect of postmating isolation between molecular forms of Anopheles gambiae (Diptera: Culicidae) Journal of Medical Entomology. 2007b;44:60–64. doi: 10.1603/0022-2585(2007)44[60:eteopi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Dong Y, Taylor HE, Dimopoulos G. AgDscam, a hypervariable immunoglobulin domain-containing receptor of the Anopheles gambiae innate immune system. PLoS Biology. 2006;4:e229. doi: 10.1371/journal.pbio.0040229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edillo FE, Toure YT, Lanzaro GC, Dolo G, Taylor CE. Spatial and habitat distribution of Anopheles gambiae and Anopheles arabiensis (Diptera: Culicidae) in Banambani village, Mali. Journal of Medical Entomology. 2002;39:70–77. doi: 10.1603/0022-2585-39.1.70. [DOI] [PubMed] [Google Scholar]
- Edillo FE, Tripet F, Toure YT, et al. Water quality and immatures of the M and S forms of Anopheles gambiae s.s. and An arabiensis in a Malian village. Malaria Journal. 2006;5:35. doi: 10.1186/1475-2875-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Rifkin SA, Bertone P, Gerstein M, White KP. Multi-species microarrays reveal the effect of sequence divergence on gene expression profiles. Genome Research. 2005;15:674–680. doi: 10.1101/gr.3335705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies MT, De Meillon B. The Anophelinae of Africa South of the Sahara. 2. South African Institute for Medical Research; Johannesburg: 1968. [Google Scholar]
- Gleason JM, Ritchie MG. Evolution of courtship song and reproductive isolation in the Drosophila willistoni species complex; do sexual signals diverge the most quickly? Evolution. 1998;52 doi: 10.1111/j.1558-5646.1998.tb02031.x. [DOI] [PubMed] [Google Scholar]
- Goma LKH. Tests for multiple insemination in Anopheles gambiae Giles. Nature. 1963;197:99–100. [Google Scholar]
- Hahn MW, Lanzaro GC. Female-biased gene expression in the malaria mosquito Anopheles gambiae. Current Biology. 2005;15:R192–193. doi: 10.1016/j.cub.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Hey J. Recent advances in assessing gene flow between diverging populations and species. Current Opinion in Genetics and Development. 2006;16:592–596. doi: 10.1016/j.gde.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Holt RA, Subramanian GM, Halpern A, et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002;298:129–149. doi: 10.1126/science.1076181. [DOI] [PubMed] [Google Scholar]
- Jallon JM, Lauge G, Orssaud L, Antony C. Female pheromones in Drosophila melanogaster are controlled by the doublesex locus. Genetical Research. 1988;51:17–22. [Google Scholar]
- Jones MDR, Gubbins SJ. Changes in the circadian flight activity of the mosquito Anopheles gambiae in relation to insemination, feeding and oviposition. Physiological Entomology. 1978;3:213–220. [Google Scholar]
- Kane NC, Rieseberg LH. Selective sweeps reveal candidate genes for adaptation to drought and salt tolerance in common sunflower, Helianthus annuus. Genetics. 2007;175:1823–1834. doi: 10.1534/genetics.106.067728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- Krzywinski J, Besansky NJ. Molecular systematics of Anopheles: from subgenera to subpopulations. Annual Review of Entomology. 2003;48:111–139. doi: 10.1146/annurev.ento.48.091801.112647. [DOI] [PubMed] [Google Scholar]
- Kuniyoshi H, Baba K, Ueda R, et al. lingerer, a Drosophila gene involved in initiation and termination of copulation, encodes a set of novel cytoplasmic proteins. Genetics. 2002;162:1775–1789. doi: 10.1093/genetics/162.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TF. The genetic architecture of quantitative traits. Annual Review of Genetics. 2001;35:303–339. doi: 10.1146/annurev.genet.35.102401.090633. [DOI] [PubMed] [Google Scholar]
- Mackay TF, Heinsohn SL, Lyman RF, et al. Genetics and genomics of Drosophila mating behavior. Proceedings of the National Academy of Sciences U S A. 2005;102:6622–6629. doi: 10.1073/pnas.0501986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoukis NC, Powell JR, Touré MB, et al. A test of the chromosomal theory of ecotypic speciation in Anopheles gambiae. Proceedings of the National Academy of Sciences U S A. 2008;105 doi: 10.1073/pnas.0709806105. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masendu HT, Hunt RH, Govere J, et al. The sympatric occurrence of two molecular forms of the malaria vector Anopheles gambiae Giles sensu stricto in Kanyemba, in the Zambezi Valley, Zimbabwe. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2004;98:393–396. doi: 10.1016/j.trstmh.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Mullen SP, Mendelson TC, Schal C, Shaw KL. Rapid evolution of cuticular hydrocarbons in a species radiation of acoustically diverse Hawaiian crickets (Gryllidae: trigonidiinae: Laupala) Evolution. 2007;61:223–231. doi: 10.1111/j.1558-5646.2007.00019.x. [DOI] [PubMed] [Google Scholar]
- Nielsen ET, Haeger JS. Swarming and mating in mosquitoes. Miscellaneous Publications of the Entomological Society of America. 1960;1:71–95. [Google Scholar]
- Pombi M, Stump AD, Della Torre A, Besansky NJ. Variation in recombination rate across the X chromosome of Anopheles gambiae. American Journal of Tropical Medicine and Hygiene. 2006;75:901–903. [PubMed] [Google Scholar]
- Ranz JM, Machado CA. Uncovering evolutionary patterns of gene expression using microarrays. Trends in Ecology and Evolution. 2006;21:29–37. doi: 10.1016/j.tree.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Rutzler M, Zwiebel LJ. Molecular biology of insect olfaction: recent progress and conceptual models. Journal of Comparative Physiology A. 2005;191:777–790. doi: 10.1007/s00359-005-0044-y. [DOI] [PubMed] [Google Scholar]
- Sandberg R, Yasuda R, Pankratz DG, et al. Regional and strain-specific gene expression mapping in the adult mouse brain. Proc Natl Acad Sci U S A. 2000;97:11038–11043. doi: 10.1073/pnas.97.20.11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santolamazza F, Della Torre A, Caccone A. Short report: A new polymerase chain reaction-restriction fragment length polymorphism method to identify Anopheles arabiensis from An. gambiae and its two molecular forms from degraded DNA templates or museum samples. American Journal of Tropical Medicine and Hygiene. 2004;70:604–606. [PubMed] [Google Scholar]
- Schlotterer C. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics. 2002;160:753–763. doi: 10.1093/genetics/160.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump AD, Fitzpatrick MC, Lobo NF, et al. Centromere-proximal differentiation and speciation in Anopheles gambiae. Proceedings of the National Academy of Sciences U S A. 2005;102:15930–15935. doi: 10.1073/pnas.0508161102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togawa T, Augustine Dunn W, Emmons AC, Willis JH. CPF and CPFL, two related gene families encoding cuticular proteins of Anopheles gambiae and other insects. Insect Biochemistry and Molecular Biology. 2007;37:675–688. doi: 10.1016/j.ibmb.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Toure YT, Petrarca V, Traore SF, et al. The distribution and inversion polymorphism of chromosomally recognized taxa of the Anopheles gambiae complex in Mali, West Africa. Parassitologia. 1998;40:477–511. [PubMed] [Google Scholar]
- Tripet F, Dolo G, Traore S, Lanzaro GC. The “wingbeat hypothesis” of reproductive isolation between members of the Anopheles gambiae complex (Diptera: Culicidae) does not fly. Journal of Medical Entomology. 2004;41:375–384. doi: 10.1603/0022-2585-41.3.375. [DOI] [PubMed] [Google Scholar]
- Tripet F, Toure YT, Taylor CE, et al. DNA analysis of transferred sperm reveals significant levels of gene flow between molecular forms of Anopheles gambiae. Molecular Ecology. 2001;10:1725–1732. doi: 10.1046/j.0962-1083.2001.01301.x. [DOI] [PubMed] [Google Scholar]
- Turner TL, Hahn MW. Locus- and population-specific selection and differentiation between incipient species of Anopheles gambiae. Molecular Biology and Evolution. 2007;24:2132–2138. doi: 10.1093/molbev/msm143. [DOI] [PubMed] [Google Scholar]
- Turner TL, Hahn MW, Nuzhdin SV. Genomic islands of speciation in Anopheles gambiae. PLoS Biology. 2005;3:e285. doi: 10.1371/journal.pbio.0030285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BJ, Hahn MW, Pombi M, et al. Localization of candidate regions maintaining a common polymorphic inversion (2La) in Anopheles gambiae. PLoS Genet. 2007;3:e217. doi: 10.1371/journal.pgen.0030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson SH, Hubisz MJ, Clark AG, et al. Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007;3:e90. doi: 10.1371/journal.pgen.0030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondji C, Simard F, Fontenille D. Evidence for genetic differentiation between the molecular forms M and S within the Forest chromosomal form of Anopheles gambiae in an area of sympatry. Insect Molecular Biology. 2002;11:11–19. doi: 10.1046/j.0962-1075.2001.00306.x. [DOI] [PubMed] [Google Scholar]
- Wray GA. The evolutionary significance of cis-regulatory mutations. Nature Reviews Genetics. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.