

Abstract
Numerous disease states are associated with hemolysis or hemorrhage. Because red cells in the extravascular space tend to lyse quickly, hemoglobin (Hb) is released and is prone to autoxidation producing MetHb. Inorganic and organic peroxides may convert Hb and MetHb to higher oxidation states such as ferrylHb. FerrylHb is not a single chemical entity but is a mixture of globin- and porphyrin-centered radicals and covalently cross-linked Hb multimers. Oxidized Hb species are potent prooxidants caused mainly by heme release from oxidized Hb. Moreover, ferrylHb is a strong proinflammatory agonist that targets vascular endothelial cells. This proinflammatory effect of ferrylHb requires actin polymerization, is characterized by the upregulation of proinflammatory adhesion molecules, and is independent of heme release. Deleterious effects of native Hb are controlled by haptoglobin (Hp) that binds cell-free Hb avidly and facilitates its removal from circulation through the CD163 macrophage scavenger receptor-mediated endocytosis. Under circumstances of Hb oxidation, Hp can prevent heme release from MetHb, but unfortunately the Hp-mediated removal of Hb is severely compromised when Hb is structurally altered such as in ferrylHb allowing deleterious downstream reactions to occur even in the presence of Hp.
1. Introduction
The red cell is usually a blessing but sometimes a curse. The normal red cell efficiently binds oxygen from the atmosphere, delivers it to the tissues, and helps remove the product of metabolic combustion, carbon dioxide. Nice. But the same cells can be involved in pathophysiologic mischief upon hemorrhage or intravascular hemolysis. Once outside the vascular system, red cells quickly burst releasing free hemoglobin (Hb). That Hb is prone to spontaneous oxidation (as the scientists trying to develop Hb-based blood substitutes have repeatedly discovered). Even worse, the Hb may be converted to higher oxidation states such as ferrylHb which have potent proinflammatory and prooxidant effects, either directly or via the release of heme which itself is highly prooxidant. Here, we briefly describe what is known regarding the natural mechanisms which have evolved to control this cell-free hemoglobin and what is being learned about the biologic effects of Hb once it is released from the red cell in pathologic states.
2. Antioxidant Network of Erythrocytes
Hemoglobin (Hb) autoxidation is the main source of reactive oxygen species inside erythrocytes. Taking into consideration the high intraerythrocytic concentration of Hb—around 5 mM as tetramer—even a slow rate of autoxidation could generate substantial amounts of ROS. To cope with this challenge, erythrocytes are equipped with highly effective antioxidant defenses [1]. This system includes enzymes such as Cu/Zn superoxide dismutase (SOD1) that convert superoxide anion to hydrogen peroxide (H2O2), catalase (Cat), glutathione peroxidase (Gpx-1), and peroxiredoxins (Prdx1 and Prdx2) which decompose H2O2 to H2O. Nonenzymatic scavengers such as glutathione also contribute to this protection.
Gene knock-out mouse models have been used to estimate the relative importance of these antioxidant defenses. Surprisingly, under steady-state conditions, mice deficient in either Gpx-1 [2] or Cat [3] have a normal phenotype. However, Cat seems to be essential for protection against elevated levels of H2O2 since Cat deficient erythrocytes are very sensitive to exogenous H2O2 [4]. It has been suggested that erythrocyte Cat might function as a “sink” for extra-erythrocytic H2O2 and thus protect somatic cells against exogenous oxidant challenge [5]. In contrast to Gpx1 −/− or Cat −/− mice, Prdx1 −/−, Prdx2 −/−, and SOD1 −/− mice exhibit impaired erythrocyte antioxidant defense. As a result, concentrations of ROS are elevated and accompanied by hemolytic anemia in SOD1 −/− [6], Prdx1 −/− [7], and Prdx2 −/− mice [8].
The idea that Hb itself may have antioxidant properties and thus contribute to the antioxidant network of erythrocytes emerged recently. For example, the Hb β93 cysteine (β93Cys)—highly conserved in vertebrates—has been shown to scavenge superoxide anion produced in the heme pocket of the β-chain of Hb [9]. This reaction may be beneficial as it decreases the rate of Hb autoxidation and reduces heme degradation attributed to the reaction of superoxide with the heme [9]. The contribution of this Hb residue to erythrocyte antioxidant protection might be considerable because of its high concentration. Knock-in mouse models—in which erythrocytes contained wild-type human Hb or human Hb in which the β93 cysteine residue was replaced with Ala—were generated to directly explore the role of the conserved β93Cys [10]. While the authors did not observe hemolysis or hemolytic disorders in the β93Ala mice, they found that β93Ala Hb has higher reactivity toward H2O2 than β93Cys Hb [11]. When β93Ala Hb was reacted with H2O2, elevated formation of high molecular weight, presumably cross-linked Hb species were observed, suggesting that the β93Cys residue plays a protective role in the metabolism of reactive species produced by erythrocytes under stress conditions [11]. This notion was further supported by experiments in which oxidative stress was engendered in vivo in mice by challenging them with lipopolysaccharide (LPS). The authors found that LPS provoked more pronounced lung injury and a greater degree of hypotension in β93Ala versus β93Cys mice. These effects were accompanied by elevated level of erythrocyte reactive oxygen species [11].
Finally, the erythrocyte enzyme, NADH methemoglobin (MetHb) reductase (Aka diaphorase I) also contributes to the antioxidant network of the erythrocyte. Continuous exposure of erythrocyte Hb to oxygen leads to the oxidation of ferrous (Fe2+) heme to ferric (Fe3+) heme. The resulting MetHb is unable to carry oxygen. The function of MetHb reductase is to restore oxygen-binding ability of Hb by reducing ferric ion into ferrous ion (reviewed in [12]).
3. Oxidation of Hb
Under normal conditions, a very small amount of the oxygen attached to the Hb heme iron dissociates as superoxide anion leaving ferric heme. This explains why, in normal people, 1-2% of Hb is present as MetHb (1). Dismutation of superoxide anion by SODs generates H2O2. Numerous in vitro studies have been performed to describe the peroxide-mediated oxidation of Hb using purified human Hb.
Peroxides like H2O2 may trigger a two-electron oxidation of Hb producing ferryl (Fe4+=O2-) Hb (2), whereas the reaction of MetHb with H2O2 yields ferrylHb radical (Hb∙+(Fe4+ = O2−)) in which the unpaired electron is associated with the globin or the porphyrin ring (3) [13–16]
| (1) |
| (2) |
| (3) |
The high-valence iron compounds are reactive intermediates and decay by several routes [17]. FerrylHb can initiate further production of globin radicals via an intramolecular electron transfer between the ferryl iron and specific amino acid residues of the globin chains resulting in the formation of MetHb globin radical (3). Radical/radical termination of globin- and porphyrin-centered radicals leads to the production of globin-globin (4) or porphyrin-globin cross-links
| (4) |
| (5) |
Considerable effort has been made to explore the mechanism via which the globin radicals are formed and where they are located within the protein. Several amino acids on both α- and β-globin chains were identified as targets of H2O2-triggered oxidation. When Hb was reacted with H2O2 in the presence of the spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO), several DMPO adducts were formed [18, 19]. Identification of the precise amino acid residues trapped by the DMPO by mass spectrometry revealed that radical formation occurs on βCys93, αTyr24, αTyr42, and αHis-20 residues of the globin chains [18]. As part of this latter work, the authors showed that in the absence of a spin trap, the globin-centered radicals can decay by formation of cross-links between the globin chains [18]. Later investigations identified key amino acids that appear to be highly susceptible to H2O2-mediated oxidation [15]. During H2O2-triggered Hb oxidation, βCys93 and βCys112 residues are irreversibly oxidized to cysteic acids, while oxidative modification of βTrp-15 and βMet-55 also occurs [15]. These processes might contribute to the loss of α-helical structure of the β chain surrounding the heme pocket and eventually lead to the deformation and collapse of the β chains [15].
A number of studies have examined whether H2O2-mediated Hb oxidation occurs in intact erythrocytes despite the above-mentioned highly active antioxidant defense systems. Giulivi and Davies provided the first evidence for the formation of ferrylHb in intact red blood cells [20]. Later, the same group detected dityrosine in intact red blood cells exposed to H2O2 and provided a mechanism for its formation. They concluded that dityrosine is a hallmark of globin-centered tyrosyl radical formation and subsequent intermolecular cross-linking [21, 22].
Besides H2O2, oxidized lipids are very important triggers of Hb oxidation. For example, we showed that lipid hydroperoxides of atheroma lipids convert Hb to MetHb [23]. Furthermore, interactions between lipid hydroperoxides of oxLDL or atheroma lipids result in the formation of ferrylHb and covalently cross-linked Hb species [23].
3.1. Hb Oxidation In Vivo
Whether oxidation of Hb leading to the formation of high-valence heme/iron compounds and globin radicals occurs in vivo was a subject of debate until recently. These products are transiently formed and can initiate a range of oxidative reactions with similar reactivity to the hydroxyl radical. The first piece of evidence that the above-mentioned reactions occur in vivo came from the study of Svistunenko et al., in which the authors could detect ferrylHb globin-centered radicals in normal human blood by electron paramagnetic resonance spectroscopy [24]. Furthermore, covalently modified heme in Hb was detected by HPLC in normal human blood [25]. In the latter study, the authors showed that the concentration of covalently modified heme in Hb is increased by exercise, a surrogate model of acute oxidative stress [25].
Oxidation of Hb has been shown in certain pathological conditions. For example, heme-protein cross-linked Hb was detected in cerebrospinal fluid following subarachnoid hemorrhage by reverse phase HPLC [26]. Moreover, recently we found that the content of dityrosine is elevated and globin-globin cross-linked Hb multimers are present in complicated atherosclerotic lesions in humans [23].
4. Control of Free Hb and Hb-Derived Heme
Several pathological conditions are associated with hemolysis when Hb is released from erythrocytes into the extracellular milieu [27]. Extracellular Hb exerts vasoactive effects via scavenging nitric oxide, an important vasodilator and signaling molecule (reviewed in [27]). Moreover, oxidation of extracellular Hb triggers prooxidant (reviewed in [28]) and proinflammatory effects on vascular endothelium [29]. Efficient mechanisms have evolved for the removal of Hb from the circulation to control the deleterious effects of extracellular Hb. Haptoglobin (Hp), an acute-phase protein, is present in plasma in high amounts (0.41–1.65 mg/mL) with the exclusive recognized function of capturing cell-free Hb and chaperoning Hb to macrophages for degradation (reviewed in [30]). Hp binding facilitates the removal of Hb from circulation through the CD163 macrophage scavenger receptor-mediated endocytosis [31] (Figure 1).
Figure 1.
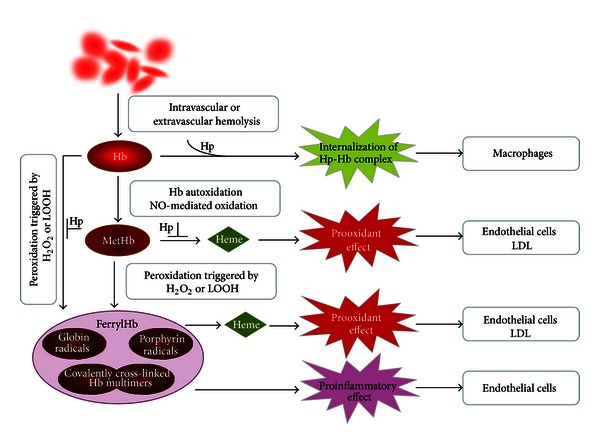
Schematic representation of hemoglobin oxidation and the different biological effects of oxidized Hb species. Hb is released from red blood cells following intra- or extravascular hemolysis. Hb can undergo spontaneous oxidation, or, alternatively, nitric oxide (NO) can trigger Hb oxidation to MetHb. Peroxidation of Hb and MetHb by H2O2 or lipid hydroperoxides (LOOH) leads to the formation of ferrylHb, that is, a mixture of globin radicals, porphyrin radicals, and covalently cross-linked Hb multimers. Haptoglobin (Hp) binds extracellular Hb and facilitates its internalization by macrophages. MetHb and ferrylHb can release heme and induce oxidative modification of lipids such as low-density lipoprotein (LDL), sensitizing cells to oxidant-mediated killing. FerrylHb is a proinflammatory agonist that targets endothelial cells. Heme release from oxidized Hb species and peroxidation of Hb are partially inhibited by Hp.
The formation of the Hp:Hb complex is virtually irreversible, and Hp binding besides facilitating the removal of intravascular cell-free Hb has many additional beneficial effects. Several studies showed that Hb bound to Hp is less prone to H2O2-mediated oxidation than free Hb [32–34]. In fact, the Hb:Hp complex acts as a fairly efficient peroxidase [35]. Further studies proved that Hp prevents H2O2-induced oxidation of amino acids in critical regions of Hb chains—that is, αTyr42, βTyr145, and βCys93—and polymerization of Hb [36]. The recent determination of the crystal structure of the porcine Hp:Hb complex revealed that Hb residues known to be prone to oxidative modifications are buried in the Hp:Hb interface, thereby explaining this direct protective role of Hp against H2O2-induced oxidation [37].
Following internalization by the macrophage, heme is cleaved by heme oxygenase-1 (HO-1) into biliverdin, carbon monoxide, and iron. This mechanism not only provides effective elimination of Hb, but it also assures iron recycling for de novo erythropoiesis under normal circumstances. However, in cases of massive intravascular hemolysis, Hp can be depleted from the circulation in which the case of free Hb is cleared (rather inefficiently) via a low-affinity pathway through CD163 [38] and/or by renal excretion [39, 40]. This latter is accompanied by generation of free iron and organ damage.
The prooxidant effects of Hb are mainly attributed to heme release from oxidized Hb. For example, MetHb but not Hb readily releases its heme prosthetic group which can be taken up by endothelial cells [41]. As a consequence of endothelial heme uptake, cells become extremely sensitive to oxidants like H2O2 or activated inflammatory cells [41]. Endothelial cells exposed to MetHb upregulate HO-1 and ferritin, key molecules responsible for heme degradation and safe storage of iron [41]. Heme release and subsequent endothelial reactions are efficiently blocked by Hp [41]. Heme transfer from oxidized Hb toward low-density lipoprotein (LDL) and consequent lipid peroxidation is also prevented by Hp [42] (Figure 1).
However, the protective effects of Hp are not entirely straightforward. In humans, the Hp gene is polymorphic resulting in the two functional alleles, that is, 1 and 2, which can form three different genotypes: Hp1-1, Hp2-1, and Hp2-2. Because Hp1 is monovalent and Hp2 is bivalent, the structures of protein products in the three genotypes show molecular heterogeneity. Hp1-1 is a small dimeric molecule (86 kDa) whereas Hp2-1 is characterized by linear polymers (86–300 kDa) and Hp2-2 forms large cyclic polymers (170–900 kDa) (reviewed in [43]).
The Hp polymorphism was investigated as a possible genetic determinant in cardiovascular disease. These epidemiologic studies revealed that the Hp2-2 genotype is a risk factor for cardiovascular complications in both type I and type II diabetic patients (reviewed in [44]). Recently, it has been shown that Hp2-2 genotype is associated with elevated amounts of iron in atherosclerotic carotid plaques in diabetic patients [45] which may imply poor stabilization of free Hb. The Hp2-2 genotype was found to be accompanied by increased macrophage infiltration and decreased smooth muscle cell content of the atherosclerotic plaque, two common indicators of plaque instability, in patients with diabetes [46]. In healthy men, Hp2-2 genotype is found to be associated with increased circulating oxLDL levels when compared to Hp1-1 or Hp2-1 genotypes [47]. Recently, the Hp2-2 genotype has been linked to greater risk of vasospasm and of clinical deterioration by delayed cerebral ischemia following subarachnoid hemorrhage [48]. It has been shown that in preeclampsia, pathologies associated with elevated level of cell-free Hb, Hp2-1, and Hp2-2 were associated with increased plasma heme levels and decreased plasma nitrite concentration [49].
Concomitant with these clinical observations, mechanical studies were performed in order to explain the molecular basis of these genetically determined differences. It has been demonstrated that Hp1-1 more efficiently inhibits heme transfer from MetHb to LDL compared to Hp2-2 [50, 51]. It has been reported that Hp2-2:Hb complex exhibits higher functional affinity for the macrophage scavenger receptor CD163 than the Hp1-1:Hb complex [31]. In contrast, Asleh et al. found that clearance rate of Hp1-1:Hb through CD163 is much higher than that of Hp2-2:Hb [52]. There are, as well, conflicting data concerning the different antioxidant properties and different Hb scavenging capacities of Hp1-1 and Hp2-2. In a recent publication, Lipiski et al. [53] did not find differences between Hp1-1 and Hp2-2 in Hb binding and intravascular compartmentalization in vivo. Furthermore, heme transfer from Hb to endothelial cells and LDL were equally blocked by Hp1-1 and Hp2-2 [53]. The apparent association of the Hp2-2 genotype with more severe symptoms in different pathologies might be explained by differences in size and penetration efficiency of Hp1-1 and Hp2-2. Because Hp2-2 is a large molecule, its diffusion into deeper compartments might be limited. Clearly, further investigations are warranted to improve our understanding of the molecular basis of the epidemiological and clinical findings related to the Hp polymorphism.
Oxidation of Hb leads to the formation of structurally altered (e.g., covalently cross-linked) Hb species. It was hypothesized that these structural changes might be associated with the impairment of the endogenous scavenging pathways. Recent studies have revealed that the elimination of oxidized Hb species via both high-affinity and low-affinity pathways can be severely compromised [38, 54].
Heme can be released from Hb upon oxidation. Hemopexin (Hx) is an acute-phase plasma protein that binds heme with the highest affinity of any known protein [55]. Hx inhibits the catalytic activity of heme in oxidative reactions [56–58]. Hx-heme complexes are taken up via the scavenger receptor LDL receptor-related protein 1/CD91 [59]. Although CD91 is expressed by many different cell types, among them hepatocytes and macrophages in liver and spleen are the most important cells in scavenging the circulating Hx-heme complexes [60]. Following endocytosis, heme is degraded by HO-1 and iron is stored by ferritin [61].
Hemoglobin and heme uptake pathways as well as heme degradation and iron storage mechanisms must function efficiently to control cell-free Hb- and heme-triggered cellular and organ damage and to limit renal iron exposure/loss during hemolysis or hemorrhage. Hp knock-out and Hx knock-out mice suffer from oxidative renal injury and elevated iron loading in the kidney after a strong hemolytic stress [62, 63]. Surprisingly, Hp/Hx double-null mice show reduced Hb accumulation in the kidney and improved survival after a lethal hemolytic stimulus [64]. This suggests that other, presently uncharacterized, protective mechanisms may be present in Hp/Hx double-null mice which provide tolerance in hemolytic stress in a similar way as sickle hemoglobin confers tolerance to plasmodium infection [65].
5. Biological Effects of Oxidized Hb
5.1. Oxidized Hb as a Prooxidant
Iron compounds can facilitate the production of hydroxyl radicals from reactive oxygen species via the Fenton reaction. Sadrzadeh et al. hypothesized that Hb might provide catalytically active iron for the Fenton reaction and thus mediate hydroxyl radical generation [66]. They showed that Hb readily promotes hydroxyl radical formation in the presence of a superoxide anion-generating system [66]. They also examined the prooxidant effect of Hb on the central nervous system in vivo. They found that Hb injected into the spinal cord of cats mediates peroxidation of lipids in the central nervous system. The iron chelator desferrioxamine prevented the Hb-induced damage, suggesting that free iron derived from Hb/heme is the proximate toxic species [67].
Similarly, we later showed that MetHb but not Hb induces oxidative modification of LDL [42]. This effect of MetHb was inhibited by the heme-scavenging protein Hx and by Hp or cyanide, agents that either bind free heme or strengthen the heme-globin bond, highlighting the role of heme release in this process [42, 68]. Recently, we showed that interactions between Hb and lipids derived from atheromatous lesions lead to a feed-forward process of Hb oxidation: conversion of oxyHb to MetHb, spontaneous heme release, oxidative heme lysis, iron release, and further lipid peroxidation [23]. We tested whether highly oxidized forms of Hb such as ferrylHb might induce lipid peroxidation and found that ferrylHb like MetHb readily initiates oxidative modification of LDL and lipids from atheromatous lesions (Potor et al. unpublished observation) (Figure 1).
Transition metals, particularly iron, markedly potentiate oxidant damage to cells. Balla and coworkers showed that MetHb but not Hb synergizes damage from reactive oxygen species or activated inflammatory cell-mediated damage on vascular endothelial cells [41] (Figure 1). As might be expected, this effect of MetHb could be abrogated by the heme scavenger hemopexin or by strengthening the interaction between the heme and globin moieties, once again emphasizing the critical impact of heme release [41].
5.2. Oxidized Hb as an Inflammatory Mediator
Hemolytic or hemorrhagic episodes are often associated with inflammation even when infectious agents are absent [69]. Considerable effort has been made to define the mediators that trigger such inflammatory response. Endothelium, the interface between blood and tissue, has a pivotal role in the inflammatory response mainly by inducing the leukocyte adhesion cascade to facilitate the transmigration of inflammatory cells to the inflamed tissue. It has been shown that endothelial cells exposed to heme (100 μmol/L) upregulate the expression of adhesion molecules: intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin [70]. While searching for other mediators of hemolysis-associated inflammation, we found that ferrylHb but not native Hb or MetHb triggers the upregulation of the same proinflammatory adhesion molecules. [29] (Figure 1). In fact, ferrylHb seems to be a strong agonist of this inflammatory response, because it induces adhesion molecules at concentrations as low as 10 μmol/L, a concentration at which heme has no effect. Endothelial cells exposed to ferrylHb show rearrangement of the actin cytoskeleton leading to disruption of the endothelial cell monolayer, intercellular gap formation, and increased permeability of the monolayer [29]. Actin polymerization is required for ferrylHb-induced inflammatory response and involves the activation of the c-Jun N-terminal kinase and the p38 mitogen-activated protein kinase signal transduction pathways [29]. Induction of inflammation is a unique property of the heavily oxidized ferrylHb because neither Hb nor MetHb triggers these effects [29] (Figure 1).
Heme itself has been shown to induce inflammation in mice [71]. In that work, mice were injected intravenously with heme to obtain 750–1000 μmol/L intravascular heme concentration (≈55–75 μmol/kg). Heme enhanced vascular permeability and increased leukocytes migration into inflammatory areas which was accompanied by the upregulation of the adhesion molecules in liver and pancreas [71]. Heme has been shown to be chemotactic for neutrophils when is injected into the peritoneal cavity of mice at a dose of only 2–20 μmol/kg [72]. We found that ferrylHb injected intraperitoneally to mice at a dose of 2 μmol/kg induced a robust inflammatory response leading to the recruitment of PMN cells in the peritoneum [29]. This effect was not observed when mice were challenged with Hb or MetHb [29].
Infectious diseases that cause hemolysis are among the most threatening human diseases. Recently, new mechanisms have been proposed which might help explain why the combination of hemolysis and infection is so dangerous. It has been shown that heme, released from oxidized Hb upon hemolysis, amplifies the innate immune response to microbial molecules such as LPS [73]. This effect of heme is dependent on the generation of reactive oxygen species [73]. Moreover, heme activates toll-like receptor 4, a receptor of the innate immune system that recognizes pathogen-associated molecular patterns such as LPS [74]. In the last couple of years, heme has been implicated in different disease models (e.g., malaria and sepsis) as a molecule that can negatively modify the tolerance of the host to invading pathogens [65, 75–79].
Overall, oxidized Hb appears to be a two-edged sword, with both edges unfortunately pointed in the wrong direction. Not only does free Hb promote oxidative damage triggered by exogenous and endogenous oxidants, but highly oxidized forms such as ferryl Hb also cause the upregulation of proinflammatory adhesion molecules. The net effect of such upregulation is the recruitment of inflammatory cells such as neutrophils and macrophages with resultant promotion of local inflammatory reactions. Teleologically, such responses to oxidized Hb may make sense; the presence of a hemorrhagic focus implies that the integument has been breached. Such an incident may permit the entry of adventitious bacteria, most of which can use Hb iron as fertilizer and with which they can grow rapidly [80]. In this light, it may be no surprise that Hp can suppress the Hb-driven growth of pathogenic bacteria [44].
6. Conclusions
Our growing understanding of the pathophysiologic effects of extracellular Hb is leading to an improved understanding of normal and pathologic responses to various forms of Hb after release from red cells. For most of human history, the bruise was viewed as simply an interesting lesion, the color of which evolved over a period of days from red (red cells and some free oxyHb) to brown (lysed red cells, MetHb) to green/yellow (biliverdin/bilirubin). Now, it seems there may be more going on than meets the eye, especially the formation of different forms of oxidized Hb with varying biological activities. The complex system which has evolved to control and dispose of cell-free Hb generally works well unless overwhelmed by excessive hemorrhage or hemolysis. It is now becoming clear that cell-free, variously oxidized Hb is capable of promoting oxidation and inflammatory responses. This occurs through (1) the release of free heme (a toxic prooxidant molecule), (2) increasing expression of vascular adhesion molecules, and (3) the subsequent promotion of inflammatory processes. Fuller understanding of this chain of events may lead to the development of improved diagnostics and therapeutics meant to interrupt the pathologic effects of cell-free Hb.
Conflict of Interests
The authors have no conflict of interests.
Acknowledgments
The research group is supported by the Hungarian Academy of Sciences (11003). This work was supported by Hungarian Government Grants OTKA-K75883 (György Balla), OTKA-K83478 (József Balla), OTKA- PD83435 (Viktória Jeney), European Reintegration Grant FP7-PEOPLE-2010-268332 (Viktória Jeney), and by the TÁMOP-4.2.2.A-11/1/KONV-2012-0045 and TÁMOP-4.2.2/B-10/1-2010-0024 Projects. The project is cofinanced by the European Union and the European Social Fund. It is also supported by the National Institutes of Health (RO1AA018016) and the Commonwealth of Kentucky Research Challenge Trust Fund (John W. Eaton).
References
- 1.Siems WG, Sommerburg O, Grune T. Erythrocyte free radical and energy metabolism. Clinical Nephrology. 2000;53(1):S9–S17. [PubMed] [Google Scholar]
- 2.Ho YS, Magnenat JL, Bronson RT, et al. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. The Journal of Biological Chemistry. 1997;272(26):16644–16651. doi: 10.1074/jbc.272.26.16644. [DOI] [PubMed] [Google Scholar]
- 3.Ho YS, Xiong Y, Ma W, Spector A, Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. The Journal of Biological Chemistry. 2004;279(31):32804–32812. doi: 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- 4.Jacob HS, Ingbar SH, Jandl JH. Oxidative hemolysis and erythrocyte metabolism in hereditary acatalasia. The Journal of Clinical Investigation. 1965;44:1187–1199. doi: 10.1172/JCI105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agar NS, Sadrzadeh SMH, Hallaway PE, Eaton JW. Erythrocyte catalase. A somatic oxidant defense? The Journal of Clinical Investigation. 1986;77(1):319–321. doi: 10.1172/JCI112294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iuchi Y, Okada F, Onuma K, et al. Elevated oxidative stress in erythrocytes due to a SOD1 deficiency causes anaemia and triggers autoantibody production. Biochemical Journal. 2007;402(2):219–227. doi: 10.1042/BJ20061386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neumann CA, Krause DS, Carman CV, et al. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424(6948):561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 8.Lee TH, Kim SU, Yu SL, et al. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003;101(12):5033–5038. doi: 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- 9.Balagopalakrishna C, Abugo OO, Horsky J, Manoharan PT, Nagababu E, Rifkind JM. Superoxide produced in the heme pocket of the β-chain of hemoglobin reacts with the β-93 cysteine to produce a thiyl radical. Biochemistry. 1998;37(38):13194–13202. doi: 10.1021/bi980941c. [DOI] [PubMed] [Google Scholar]
- 10.Isbell TS, Sun CW, Wu LC, et al. SNO-hemoglobin is not essential for red blood cell-dependent hypoxic vasodilation. Nature Medicine. 2008;14(7):773–777. doi: 10.1038/nm1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vitturi DA, Sun CW, Harper VM, et al. Antioxidant functions for the hemoglobin beta93 cysteine residue in erythrocytes and in the vascular compartment in vivo. Free Radical Biology & Medicine. 2013;55:119–129. doi: 10.1016/j.freeradbiomed.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Percy MJ, McFerran NV, Lappin TRJ. Disorders of oxidised haemoglobin. Blood Reviews. 2005;19(2):61–68. doi: 10.1016/j.blre.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Harel S, Kanner J. The generation of ferryl or hydroxyl radicals during interaction of haemproteins with hydrogen peroxide. Free Radical Research Communications. 1988;5(1):21–33. doi: 10.3109/10715768809068555. [DOI] [PubMed] [Google Scholar]
- 14.Patel RP, Svistunenko DA, Darley-Usmar VM, Symons MCR, Wilson MT. Redox cycling of human methaemoglobin by H2O2 yields persistent ferryl iron and protein based radicals. Free Radical Research. 1996;25(2):117–123. doi: 10.3109/10715769609149916. [DOI] [PubMed] [Google Scholar]
- 15.Jia Y, Buehler PW, Boykins RA, Venable RM, Alayash AI. Structural basis of peroxide-mediated changes in human hemoglobin: a novel oxidative pathway. The Journal of Biological Chemistry. 2007;282(7):4894–4907. doi: 10.1074/jbc.M609955200. [DOI] [PubMed] [Google Scholar]
- 16.Alayash AI, Patel RP, Cashon RE. Redox reactions of hemoglobin and myoglobin: biological and toxicological implications. Antioxidants and Redox Signaling. 2001;3(2):313–327. doi: 10.1089/152308601300185250. [DOI] [PubMed] [Google Scholar]
- 17.Reeder BJ, Cutruzzola F, Bigotti MG, Hider RC, Wilson MT. Tyrosine as a redox-active center in electron transfer to ferryl heme in globins. Free Radical Biology and Medicine. 2008;44(3):274–283. doi: 10.1016/j.freeradbiomed.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 18.Deterding LJ, Ramirez DC, Dubin JR, Mason RP, Tomer KB. Identification of free radicals on hemoglobin from its self-peroxidation using mass spectrometry and immuno-spin trapping: observation of a histidinyl radical. The Journal of Biological Chemistry. 2004;279(12):11600–11607. doi: 10.1074/jbc.M310704200. [DOI] [PubMed] [Google Scholar]
- 19.Ramirez DC, Chen YR, Mason RP. Immunochemical detection of hemoglobin-derived radicals formed by reaction with hydrogen peroxide: involvement of a protein-tyrosyl radical. Free Radical Biology and Medicine. 2003;34(7):830–839. doi: 10.1016/s0891-5849(02)01437-5. [DOI] [PubMed] [Google Scholar]
- 20.Giulivi C, Davies KJA. A novel antioxidant role for hemoglobin. The comproportionation of ferrylhemoglobin with oxyhemoglobin. The Journal of Biological Chemistry. 1990;265(32):19453–19460. [PubMed] [Google Scholar]
- 21.Giulivi C, Davies KJA. Mechanism of the formation and proteolytic release of H2O2-induced dityrosine and tyrosine oxidation products in Hemoglobin and red blood cells. The Journal of Biological Chemistry. 2001;276(26):24129–24136. doi: 10.1074/jbc.M010697200. [DOI] [PubMed] [Google Scholar]
- 22.Giulivi C, Davies KJA. Dityrosine and tyrosine oxidation products are endogenous markers for the selective proteolysis of oxidatively modified red blood cell hemoglobin by (the 19 S) proteasome. The Journal of Biological Chemistry. 1993;268(12):8752–8759. [PubMed] [Google Scholar]
- 23.Nagy E, Eaton JW, Jeney V, et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(7):1347–1353. doi: 10.1161/ATVBAHA.110.206433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Svistunenko DA, Patel RP, Voloshchenko SV, Wilson MT. The globin-based free radical of ferryl hemoglobin is detected in normal human blood. The Journal of Biological Chemistry. 1997;272(11):7114–7121. doi: 10.1074/jbc.272.11.7114. [DOI] [PubMed] [Google Scholar]
- 25.Vollaard NBJ, Reeder BJ, Shearman JP, Menu P, Wilson MT, Cooper CE. A new sensitive assay reveals that hemoglobin is oxidatively modified in vivo. Free Radical Biology and Medicine. 2005;39(9):1216–1228. doi: 10.1016/j.freeradbiomed.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 26.Reeder BJ, Sharpe MA, Kay AD, Kerr M, Moore K, Wilson MT. Toxicity of myoglobin and haemoglobin: oxidative stress in patients with rhabdomyolysis and subarachnoid haemorrhage. Biochemical Society Transactions. 2002;30(4):745–748. doi: 10.1042/bst0300745. [DOI] [PubMed] [Google Scholar]
- 27.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. Journal of the American Medical Association. 2005;293(13):1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 28.Balla J, Vercellotti GM, Jeney V, et al. Heme, heme oxygenase and ferritin in vascular endothelial cell injury. Molecular Nutrition and Food Research. 2005;49(11):1030–1043. doi: 10.1002/mnfr.200500076. [DOI] [PubMed] [Google Scholar]
- 29.Silva G, Jeney V, Chora A, Larsen R, Balla J, Soares MP. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. The Journal of Biological Chemistry. 2009;284(43):29582–29595. doi: 10.1074/jbc.M109.045344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alayash AI. Haptoglobin: old protein with new functions. Clinica Chimica Acta. 2011;412(7-8):493–498. doi: 10.1016/j.cca.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 31.Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409(6817):198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 32.Buehler PW, Abraham B, Vallelian F, et al. Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood. 2009;113(11):2578–2586. doi: 10.1182/blood-2008-08-174466. [DOI] [PubMed] [Google Scholar]
- 33.Banerjee S, Jia Y, Siburt CJ, et al. Haptoglobin alters oxygenation and oxidation of hemoglobin and decreases propagation of peroxide-induced oxidative reactions. Free Radical Biology & Medicine. 2012;53:1317–1326. doi: 10.1016/j.freeradbiomed.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 34.Miller YI, Altamentova SM, Shaklai N. Oxidation of low-density lipoprotein by hemoglobin stems from a heine- initiated globin radical: antioxidant role of haptoglobin. Biochemistry. 1997;36(40):12189–12198. doi: 10.1021/bi970258a. [DOI] [PubMed] [Google Scholar]
- 35.Kapralov A, Vlasova II, Feng W, et al. Peroxidase activity of hemoglobin·haptoglobin complexes. Covalent aggreation and oxidative stress in plasma and macrophages. The Journal of Biological Chemistry. 2009;284(44):30395–30407. doi: 10.1074/jbc.M109.045567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pimenova T, Pereira CP, Gehrig P, Buehler PW, Schaer DJ, Zenobi R. Quantitative mass spectrometry defines an oxidative hotspot in hemoglobin that is specifically protected by haptoglobin. Journal of Proteome Research. 2010;9(8):4061–4070. doi: 10.1021/pr100252e. [DOI] [PubMed] [Google Scholar]
- 37.Andersen CB, Torvund-Jensen M, Nielsen MJ, et al. Structure of the haptoglobin-haemoglobin complex. Nature. 2012;489:456–459. doi: 10.1038/nature11369. [DOI] [PubMed] [Google Scholar]
- 38.Schaer DJ, Schaer CA, Buehler PW, et al. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood. 2006;107(1):373–380. doi: 10.1182/blood-2005-03-1014. [DOI] [PubMed] [Google Scholar]
- 39.Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti, Hemolysis a GM. nd free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121:1276–1284. doi: 10.1182/blood-2012-11-451229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray RK, Connell GE, Pert JH. The role of haptoglobin in the clearance and distribution of extracorpuscular hemoglobin. Blood. 1961;17:45–53. [PubMed] [Google Scholar]
- 41.Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(20):9285–9289. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeney V, Balla J, Yachie A, et al. Pro-oxidant and cytotoxic effects of circulating heme. Blood. 2002;100(3):879–887. doi: 10.1182/blood.v100.3.879. [DOI] [PubMed] [Google Scholar]
- 43.Goldenstein H, Levy NS, Levy AP. Haptoglobin genotype and its role in determining heme-iron mediated vascular disease. Pharmacological Research. 2012;66:1–6. doi: 10.1016/j.phrs.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costacou T, Levy AP. Haptoglobin genotype and its role in diabetic cardiovascular disease. Journal of Cardiovascular Translational Research. 2012;5:423–435. doi: 10.1007/s12265-012-9361-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lioupis C, Barbatis C, Drougou A, et al. Association of haptoglobin genotype and common cardiovascular risk factors with the amount of iron in atherosclerotic carotid plaques. Atherosclerosis. 2011;216(1):131–138. doi: 10.1016/j.atherosclerosis.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 46.Lioupis C, Barbatis C, Lazari P, et al. Macrophage infiltration and smooth muscle cells content associated with haptoglobin genotype in human atherosclerotic carotid plaques. Angiology. 2012;63:178–183. doi: 10.1177/0003319711410051. [DOI] [PubMed] [Google Scholar]
- 47.Brouwers A, Langlois M, Delanghe J, et al. Oxidized low-density lipoprotein, iron stores, and haptoglobin polymorphism. Atherosclerosis. 2004;176(1):189–195. doi: 10.1016/j.atherosclerosis.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 48.Ohnishi H, Iihara K, Kaku Y, et al. Haptoglobin phenotype predicts cerebral vasospasm and clinical deterioration after aneurysmal subarachnoid hemorrhage. Journal of Stroke and Cerebrovascular Diseases. 2013 doi: 10.1016/j.jstrokecerebrovasdis.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 49.Sandrim VC, Palei ACT, Sertorio JT, Cavalli RC, Duarte G, Tanus-Santos JE. Effects of eNOS polymorphisms on nitric oxide formation in healthy pregnancy and in pre-eclampsia. Molecular Human Reproduction. 2010;16(7):506–510. doi: 10.1093/molehr/gaq030.gaq030 [DOI] [PubMed] [Google Scholar]
- 50.Bamm VV, Tsemakhovich VA, Shaklai M, Shaklai N. Haptoglobin phenotypes differ in their ability to inhibit heme transfer from hemoglobin to LDL. Biochemistry. 2004;43(13):3899–3906. doi: 10.1021/bi0362626. [DOI] [PubMed] [Google Scholar]
- 51.Melamed-Frank M, Lache O, Enav BI, et al. Structure-function analysis of the antioxidant properties of haptoglobin. Blood. 2001;98(13):3693–3698. doi: 10.1182/blood.v98.13.3693. [DOI] [PubMed] [Google Scholar]
- 52.Asleh R, Marsh S, Shilkrut M, et al. Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circulation Research. 2003;92(11):1193–1200. doi: 10.1161/01.RES.0000076889.23082.F1. [DOI] [PubMed] [Google Scholar]
- 53.Lipiski M, Deuel JW, Baek JH, Engelsberger WR, Buehler PW, Schaer DJ. Human Hp1-1 and Hp2-2 phenotype-specific haptoglobin therapeutics are both effective in vitro and in guinea pigs to attenuate hemoglobin toxicity. Antioxidants & Redox Signaling. 2013 doi: 10.1089/ars.2012.5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vallelian F, Pimenova T, Pereira CP, et al. The reaction of hydrogen peroxide with hemoglobin induces extensive α-globin crosslinking and impairs the interaction of hemoglobin with endogenous scavenger pathways. Free Radical Biology and Medicine. 2008;45(8):1150–1158. doi: 10.1016/j.freeradbiomed.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 55.Hrkal Z, Vodrazka Z, Kalousek I. Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. European Journal of Biochemistry. 1974;43(1):73–78. doi: 10.1111/j.1432-1033.1974.tb03386.x. [DOI] [PubMed] [Google Scholar]
- 56.Gutteridge JMC, Smith A. Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochemical Journal. 1988;256(3):861–865. doi: 10.1042/bj2560861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vincent SH, Grady RW, Shaklai N, Snider JM, Muller-Eberhard U. The influence of heme-binding proteins in heme-catalyzed oxidations. Archives of Biochemistry and Biophysics. 1988;265(2):539–550. doi: 10.1016/0003-9861(88)90159-2. [DOI] [PubMed] [Google Scholar]
- 58.Balla G, Jacob HS, Eaton JW, Belcher JD, Vercellotti GM. Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arteriosclerosis and Thrombosis. 1991;11(6):1700–1711. doi: 10.1161/01.atv.11.6.1700. [DOI] [PubMed] [Google Scholar]
- 59.Hvidberg V, Maniecki MB, Jacobsen C, Højrup P, Møller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. 2005;106(7):2572–2579. doi: 10.1182/blood-2005-03-1185. [DOI] [PubMed] [Google Scholar]
- 60.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. The Journal of Clinical Investigation. 2001;108(6):779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alam J, Smith A. Receptor-mediated transport of heme by hemopexin regulates gene expression in mammalian cells. The Journal of Biological Chemistry. 1989;264(30):17637–17640. [PubMed] [Google Scholar]
- 62.Fagoonee S, Gburek J, Hirsch E, et al. Plasma protein haptoglobin modulates renal iron loading. American Journal of Pathology. 2005;166(4):973–983. doi: 10.1016/S0002-9440(10)62319-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tolosano E, Hirsch E, Patrucco E, et al. Defective recovery and severe renal damage after acute hemolysis in hemopexin-deficient mice. Blood. 1999;94(11):3906–3914. [PubMed] [Google Scholar]
- 64.Tolosano E, Fagoonee S, Hirsch E, et al. Enhanced splenomegaly and severe liver inflammation in haptoglobin/hemopexin double-null mice after acute hemolysis. Blood. 2002;100(12):4201–4208. doi: 10.1182/blood-2002-04-1270. [DOI] [PubMed] [Google Scholar]
- 65.Ferreira A, Marguti I, Bechmann I, et al. Sickle hemoglobin confers tolerance to plasmodium infection. Cell. 2011;145(3):398–409. doi: 10.1016/j.cell.2011.03.049. [DOI] [PubMed] [Google Scholar]
- 66.Sadrzadeh SMH, Graf E, Panter SS. Hemoglobin. A biologic Fenton reagent. The Journal of Biological Chemistry. 1984;259(23):14354–14356. [PubMed] [Google Scholar]
- 67.Sadrzadeh SMH, Anderson DK, Panter SS. Hemoglobin potentiates central nervous system damage. The Journal of Clinical Investigation. 1987;79(2):662–664. doi: 10.1172/JCI112865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miller YI, Smith A, Morgan WT, Shaklai N. Role of hemopexin in protection of low-density lipoprotein against hemoglobin-induced oxidation. Biochemistry. 1996;35(40):13112–13117. doi: 10.1021/bi960737u. [DOI] [PubMed] [Google Scholar]
- 69.Arruda MA, Graça-Souza AV, Barja-Fidalgo C. Heme and innate immunity: new insights for an old molecule. Memorias do Instituto Oswaldo Cruz. 2005;100(7):799–803. doi: 10.1590/s0074-02762005000700022. [DOI] [PubMed] [Google Scholar]
- 70.Wagener FADTG, Feldman E, De Witte T, Abraham NG. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Experimental Biology and Medicine. 1997;216(3):456–463. doi: 10.3181/00379727-216-44197. [DOI] [PubMed] [Google Scholar]
- 71.Wagener FADTG, Eggert A, Boerman OC, et al. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood. 2001;98(6):1802–1811. doi: 10.1182/blood.v98.6.1802. [DOI] [PubMed] [Google Scholar]
- 72.Porto BN, Alves LS, Fernández PL, et al. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. The Journal of Biological Chemistry. 2007;282(33):24430–24436. doi: 10.1074/jbc.M703570200. [DOI] [PubMed] [Google Scholar]
- 73.Fernandez PL, Dutra FF, Alves L, et al. Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. The Journal of Biological Chemistry. 2010;285(43):32844–32851. doi: 10.1074/jbc.M110.146076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Figueiredo RT, Fernandez PL, Mourao-Sa DS, et al. Characterization of heme as activator of toll-like receptor 4. The Journal of Biological Chemistry. 2007;282(28):20221–20229. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 75.Larsen R, Gozzelino R, Jeney V, et al. A central role for free heme in the pathogenesis of severe sepsis. Science Translational Medicine. 2010;2(51) doi: 10.1126/scitranslmed.3001118.51ra71 [DOI] [PubMed] [Google Scholar]
- 76.Ferreira A, Balla J, Jeney V, Balla G, Soares MP. A central role for free heme in the pathogenesis of severe malaria: the missing link? Journal of Molecular Medicine. 2008;86(10):1097–1111. doi: 10.1007/s00109-008-0368-5. [DOI] [PubMed] [Google Scholar]
- 77.Pamplona A, Ferreira A, Balla J, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nature Medicine. 2007;13(6):703–710. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- 78.Seixas E, Gozzelino R, Chora A, et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(37):15837–15842. doi: 10.1073/pnas.0903419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gozzelino R, Andrade BB, Larsen R, et al. Metabolic adaptation to tissue iron overload confers tolerance to malaria. Cell Host & Microbe. 2012;12:693–704. doi: 10.1016/j.chom.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 80.Telang S, Vimr E, Mahoney JR, et al. Strain-specific iron-dependent virulence in Escherichia coli . Journal of Infectious Diseases. 2001;184(2):159–165. doi: 10.1086/322017. [DOI] [PubMed] [Google Scholar]