

Abstract
The ability of cytochrome P450 enzymes to catalyze highly regio- and stereospecific hydroxylations makes them attractive alternatives to approaches based on chemical synthesis but they require expensive cofactors, e.g. NAD(P)H, which limits their commercial potential. Ferredoxin (Fdx) is a multifunctional electron carrier that in plants accepts electrons from photosystem I (PSI) and facilitates photoreduction of NADP+ to NADPH mediated by ferredoxin-NAD(P)H oxidoreductase (FdR). In bacteria, the electron flow is reversed and Fdx accepts electrons from NADPH via FdR and serves as the direct electron donor to bacterial P450s. By combining the two systems, we demonstrate that irradiation of PSI can drive the activity of a bacterial P450, CYP124 from Mycobacterium tuberculosis. The substitution of the costly cofactor NADPH with sunlight illustrates the potential of the light-driven hydroxylation system for biotechnology applications.
Keywords: Cytochrome P450, ω-hydroxylation, Light-driven biosynthesis, Lipid hydroxyllase, Photosystem I
Introduction
The introduction of a hydroxyl group at an activated carbon atom in an organic molecule may be achieved using a variety of chemical methods. In contrast, hydroxylation at non-activated positions can only be carried out using harsh reaction conditions that typically lack the desired regio- and stereospecificity. The ability of mono-oxygenases to catalyze regio- and stereospe-cific hydroxylations of complex organic molecules at non-activated positions therefore makes such enzymes particularly useful biocatalysts. Cytochromes P450 (P450) are heme-containing monooxygenases present in mammals, plants, fungi and bacteria. P450 s typically catalyze C-hydroxylations but they may also function as N- or S-hydroxylases or facilitate epoxidation, dealkylation, oxidation, deamination, dehalogenation and isomerization reactions (Sono et al. 1996). Besides their regio- and stereospecificity, other advantages of using P450 s as biocatalysts are their efficiency at low molar concentrations and optimum temperatures in the 20–40°C range, which minimize undesired side reactions (Loughlin 2000).
P450 systems can be classified based on the redox partners required for catalytic activity (Hannemann et al. 2007). Most bacterial P450s are dependent on the action of an NAD(P)H-dependent ferredoxin reductase (FdR) and ferredoxin (Fdx), but the classification system is complicated as some bacterial P450s can function with a variety of electron donor systems (Goni et al. 2009). A shared feature for all P450 systems is their dependence on external electron donors, e.g. NAD(P)H, which is also one of the main obstacles for using P450s as biocatalysts. The stoichiometric amount of NAD(P)H needed for hydroxylation of substrate molecules has a major impact on production costs, and different approaches have been suggested to either circumvent the use of, or to regenerate, the reduced cofactors. The most frequent approach is enzymatic regeneration of NADPH using glucose-6-phosphate dehydrogenase or formate dehydrogenase (Tishkov et al. 1999; van der Donk and Zhao 2003). Alternative methods with the aim to provide cost-effective reducing equivalents to P450 are the use of peroxides (Cirino and Arnold 2003), chemical regeneration of NAD(P)H using inorganic catalysts (Hollmann et al. 2002) and direct or indirect cathodic reduction of NAD(P)+ (Reipa et al. 1997; Udit et al. 2006). An interesting approach to circumvent NADPH consumption is electrochemical reduction of P450 redox partners and subsequent donation of electrons to P450 s but these methods are not easily applicable given a complex experimental setup (Krishnan et al. 2011; Wirtz et al. 2000).
Based on the concept of synthetic biology that every biological part may be considered as an independent bio-brick, we have recently generated a system able to utilize light to drive the P450 catalytic cycle (Jensen et al. 2011). This system was based on the use of photosystem I (PSI) isolated from barley (Hordeum vulgare L.) and the plant P450 CYP79A1 isolated from sorghum (Sorghum bicolor Moench), thereby combining the catalytic properties of two membrane complexes, which in nature are localized in the thylakoid membrane of the chloroplast and in the endoplasmic reticulum, respectively. The two main electron acceptors of PSI, Fdx and the FMN-containing flavodoxin (Fld), were both able to mediate direct electron transfer from PSI (Naver et al. 1996; Scheller et al. 1989) to CYP79A1, avoiding the use of NADPH. The PSI-Fdx-CYP79A1 system supported a twofold higher turnover rate of CYP79A1 as observed following reconstitution with its native electron donor, the membrane-bound NADPH-dependent cytochrome P450 reductase (CPR) (Jensen and Møller 2010).
To study the versatility of plant PSI as electron donor for biotechnologically useful P450s, the soluble bacterial CYP124 from Mycobacterium tuberculosis was chosen. CYP124 is a methyl-branched lipid ω-hydroxylase that uses spinach FdR and the [2Fe-2S] iron-sulfur protein, Fdx, as an electron transport system (Johnston et al. 2009). The ω-hydroxylation of hydrocarbon chains is potentially a highly desirable biotechnological reaction with the product formed being hard to obtain by traditional chemical synthesis. Thus, CYP124 is likely to become an interesting industrial enzyme. The use of spinach derived Fdx and FdR for the initial biochemical characterization of CYP124 enables a direct comparison to the efficiency of the light driven PSI-Fdx electron transport system in which spinach Fdx also is the electron carrier of choice (Fig. 1). This experimental setup excludes potential ambiguities arising from differences in electron carrier affinity towards CYP124.
Fig. 1.
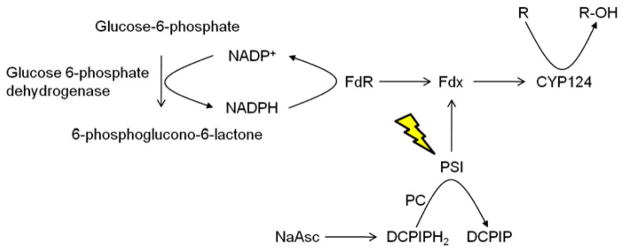
Two different regenerative electron transport systems for CYP124. Following electron donation to P450, the photooxidized PSI reaction center is re-reduced by electron transfer from plastocyanin (PC) with sodium ascorbate and DCPIP serving as the ultimate electron donors to PC
Materials and methods
Materials
Hordeum vulgare Photosystem I (PSI) was isolated as described previously (Jensen et al. 2011). Spinach Fdx and FdR were purchased from Sigma Aldrich. Mycobacterium tuberculosis CYP124 was isolated as reported elsewhere (Johnston et al. 2009). N, O-bis(Trimethylsilyl)trifluoroacetamide (BSTFA) was purchased from Pierce.
Light-driven CYP124 enzymatic assays
PSI-Fdx-CYP124 catalytic experiments were carried out at 25°C in 50 mM potassium phosphate buffer (pH 7.5) supplemented with 0.11 mM 2,6-dichlorophenolindo-phenol (DCPIP) and 3.6 mM sodium ascorbate in a total volume of 200 μl. CYP124 (31 pmol), substrate and the PSI electron transport system consisting of 0.42 μM PSI, 5.5 μM spinach plastocyanin, 21 μg ml−1 Fdx and 25 μg ml−1 catalase (Sigma-Aldrich, C9332) were added to the buffer and allowed to equilibrate to 25°C in a foil-covered tube. Reactions were initiated by irradiation with a Schott KL 1500 light source fitted with two red filters (Schott RG660 and Corning 2–58) and one gray filter (Schott NG4), resulting in a light intensity of 151 μmol quanta m−2 s−1. After 10 min, reactions were stopped and analyzed as previously described (Johnston et al. 2009; Johnston et al. 2010). In short, reactions were quenched by adding an equal volume of 1 M HCl. After quenching, approx. 8 vol. tert-butyl methyl ether (TBME) were added and the phases were separated by centrifugation. The organic layer was reserved and the hydrophilic layer was extracted twice more with 5 vol. TBME. Samples were dried under N2 and resuspended in 50 μl of the derivatization reagent BSTFA. The derivatization reaction was allowed to proceed for at least 30 min at room temperature to ensure complete derivatization. The samples were subsequently analyzed by GC–MS. All assays were repeated at least three times.
NADPH-driven CYP124 enzymatic assays
NADPH-FdR-Fdx-CYP124 catalytic experiments were carried out in glass testtubes at 25°C in 50 mM potassium phosphate buffer (pH 7.5) in 0.5 ml. CYP124 (94 pmol) was preincubated with substrate (see individual experiments for concentration) at RT for 30 min. After incubation, MgCl2 (10 mM), catalase (25 μg ml−1), Fdx (21 μg ml−1), FdR (0.08 units), glucose-6-phosphate dehydrogenase (1 unit) and glucose-6-phosphate (1.2 mM) were added to the reaction. The reaction was started by addition of NADP+ (0.08 mM). The reaction was allowed to proceed for 30 min at room temperature with gentle agitation. Reaction was quenched and analyzed as described for the light-driven CYP124 enzymatic assay.
NADP+ photoreduction
NADP+ photoreduction activity was determined from the absorbance change at 340 nm in a reaction mixture (total vol 500 μl) containing TNM buffer [20 mM Tricine (pH 7.5), 40 mM NaCl, 8 mM MgCl2, 0.1% (v/v) n-dodecyl-β-D-maltopyranoside (DDM), 2 mM sodium ascorbate, 0.06 mM DCPIP], 0.5 mM NADP+, 1.1 nmol spinach plastocyanin, 0.38 μM spinach FNR (Sigma), and 84 pmol PSI. Spinach Fd (Sigma) (76 pmol) was added to the reaction. The light-induced production of NADPH was measured using a Cary 1E dual-beam spectrophotometer at 340 nm. The sample was kept at 25°C and irradiated from the top with a Schott KL 1,500 light source fitted with two red filters (Schott RG660 and Corning 2–58). NADPH production was quantified using the molar extinction coefficient of 6620 M−1 cm−1. Catalase and/or superoxide dismutase was added to 20 μg ml−1.
Results and discussion
Methyl-branched lipids are substrates of CYP124 with substrate binding affinity being affected by the extent of methyl branching, lipid chain length and presence of a polar functional group (Johnston et al. 2009). Furthermore, CYP124 is able to hydroxylate the cholesterol metabolite, cholest-4-en-3-one (Johnston et al. 2010). To test the capability of PSI and Fdx to serve as an electron generator and donor system, CYP124 was incubated with various lipids, including isoprenoid alcohols, fatty acids and sterols that díffer in chain length and in the degree of methyl branching, e.g. palmitic acid, 15-methyl-palmitic acid, farnesol, geranylgeraniol and cholest-4-en-3-one. PSI and spinach Fdx were added and product formation was analyzed after irradiation (151 μmol quanta m−2 s−1 for 10 min). Hydroxylation of cholest-4-en-3-one and of the isoprenoids farnesol and geranylgeraniol was readily detected but at a lower efficiency than previously reported for the NADPH-FdR-Fdx-CYP124 system (Johnston et al. 2009). As expected, only very low activity towards palmitic acid was observed. In contrast to previously published data, the oxidation rate of 15-methyl-palmitic acid was also severely diminished (Johnston et al. 2009).
Accumulation of reactive oxygen species (ROS) can occur during the catalytic cycles of PSI and P450s, if the electron transfer from electron donor to terminal acceptor is not tightly coupled. PSI is susceptible to damage by ROS (Asada 1999; Powles 1984), resulting in a reduction in electron donation to CYP124 and reduced overall activity if PSI is inactivated by ROS. During light induction of PSI, ROS could arise either directly by PSI electron transfer to oxygen (Tjus et al. 2001) or from the high reactivity of excess reduced Fdx. Photoreduction of O2 by PSI is only a matter of concern in absence of oxidized Fdx, as PSI-Fdx interactions are kinetically favored compared to direct electron transfer from PSI to O2 (Asada 1999). The 89% amino acid sequence identity between spinach Fdx and H. vulgare (barley) Fdx indicates that the interaction between spinach Fdx and barley PSI is highly specific, preventing photoreduction of O2. Fdx delivers electrons to FdR, which catalyzes the reduction of NADP+ to NADPH. In the presence of FdR, O2 reduction by Fdx is greatly suppressed (Furbank and Badger 1983). NADP+ photoreduction was not affected by the presence or absence of superoxide dismutase and/or catalase (Fig. 2), indicating that the PSI-Fdx reaction is tightly coupled and PSI is fully functional.
Fig. 2.

The rate of NADP+ photoreduction by PSI in presence of catalase and superoxide dismutase was determined. NADP+ photoreduction was measured as described in the “Materials and methods”
In contrast to the detrimental effect of ROS accumulation on PSI activity, P450 activity can be increased as ROS can serve as alternative electron sources for P450 hydroxylations (Cirino and Arnold 2003; Denisov et al. 2005; Joo et al. 1999). The native electron donor for CYP124 has not been identified, but several Fdxs’ have been identified in the M. tubercolosis genome, any of which could be a potential electron donor for CYP124 (Ouellet et al. 2010). Instead, CYP124 was characterized with spinach Fdx, which potentially results in suboptimal interactions with CYP124 and increased potential for uncoupling and ROS generation. Addition of catalase to the PSI-Fdx-CYP124 setup increased CYP124 activity threefold (Fig. 3). The presence of catalase had no effect on the rate of NADP+ photoreduction by PSI, indicating that accumulation of ROS is caused by uncoupling in the catalytic cycle of CYP124, leading to degradation of PSI and subsequent reduced electron flow to CYP124. Addition of superoxide dismutase did not further improve CYP124 activity.
Fig. 3.

Reactive oxygen species are generated in the PSI-Fdx-CYP124 system in absence of catalase, thereby damaging PSI. Assays were done as described in the “Materials and methods” in the presence of either 15 μM farnesol or 20 μM cholest-4-en-3-one
Determination of the kinetic constants for farnesol and cholest-4-en-3-one (Fig. 4) oxidation in the PSI-Fdx-CYP124 system was performed in presence of catalase (Table 1). The low KM value in the light-driven system suggests that CYP124 is saturated with substrate at relatively low concentrations compared to the FdR-Fdx-CYP124 system, and will act at a more or less constant rate, even at low substrate concentrations. The lower kcat values observed using the PSI-Fdx-CYP124 system compared to the FdR-Fdx-CYP124 system suggest that uncoupling between spinach Fdx and CYP124 is not the main contributor to the reduced activity of the PSI-Fdx-CYP124 system, but rather the presence of interfering factors in the PSI-driven system.
Fig. 4.

Chemical structures of a Farnesol, b Cholest-4-en-3-one and c n-dodecyl-β-D-maltopyranoside (DDM)
Table 1.
Kinetic constants of the light-driven system compared to the compared to the NADPH-dependent system
| Substrate | PSI-Fdx-CYP124
|
NADPH-FdR-Fdx-CYP124
|
||
|---|---|---|---|---|
| KM (μM) | kcat (min−1) | KM (μM) | kcat (min−1) | |
| Farnesol | 3.4 ± 1.1 | 3.1 ± 0.2 | 36 ± 3a | 15.5 ± 2.8b |
| Cholest-4-en-3-one | 8.5 ± 2.2 | 2.8 ± 0.2 | 21 ± 2.1a | 11.7 ± 1.3b |
The farnesol and cholest-4-en-3-one concentrations used were 0–50 and 0–75 μM, respectively. Otherwise, the experimental setup for determining the kinetic constants was identical to the procedure described in the materials and methods section
Values from Johnston et al. (2010)
Values from Johnston et al. (2009)
The PSI multi-subunit protein complex was solubilization from the thylakoid membranes by DDM (Fig. 4) and isolated by sucrose density gradient centrifugation. The isolated PSI complex was stored in 0.085% (w/v) DDM which was further diluted to 0.015% (w/v) (~300 μM DDM) in the assay mixture. DDM is a non-ionic detergent with a long non-branched alkyl chain, reminiscent of the linear fatty acids substrates for CYP124, e.g., lauric acid and palmitic acid (Johnston et al. 2009). Inclusion of 0.015% (w/v) DDM in the assay mixtures containing CYP124, farnesol, Fdx, FdR and NADPH reduced farnesol conversion to 2.9 ± 0.5% compared to the 16.1 ± 0.8% conversion of farnesol in the absence of DDM. Apparently DDM inhibits CYP124 activity which explains the lower kcat values observed for the light driven PSI-Fdx-CYP124 system (Table 1). Inhibition was most pronounced towards palmitic acid and 15-methyl palmitic acid. The already low affinity of CYP124 for palmitic acid (KD >100 μM) may explain the strong inhibitory effect of DDM. CYP124 binds 15-methyl-palmitic acid with higher affinity (KD of ~ 1 μM) but its structural similarity to DDM may explain why DDM inhibits hydroxylation of 15-methyl-palmitic acid more than hydroxylation of farnesol or geranylgeraniol, which have similar dissociation constants.
Given the promiscuous substrate specificity of CYP124, the choice of alternative detergents for solubilizing PSI which do not structurally resemble known CYP124 substrates was limited. Common detergents include SDS, Triton X-100 and Tween 20 which unfortunately all resemble known substrates for CYP124. Even a steroid group on the otherwise structurally very different detergent, digitonin at 0.1% (w/v), was sufficient to inhibit CYP124 oxidation of cholest-4-en-3-one (3.2 ± 0.07% conversion in the presence and 16.3 ± 0.8% in the absence of digitonin). A future approach to avoid inhibition of CYP124 and other lipid-preferring P450 s by detergents would be to reconstitute PSI using for example nanodiscs (Ham et al. 2010; Nath et al. 2007) although nanodiscs able to solubilize proteins the size of PSI have so far not been successfully assembled.
Despite the slightly reduced activity of the light-driven PSI-Fdx-CYP124 system, the system does offer several advantages: (I) No requirement for the expensive cofactor NAD(P)H(II) Spinach Fdx is a promiscuous redox partner and can in many instances substitute for the native electron donor for bacterial P450s, thereby high-lighting the potential of using a PSI-Fdx system as a general electron donor system for P450s. The target range of the PSI based electron transport system can be further expanded by the possibility of using Fld as an electron carrier. Cyanobacterial Fld can interact with plant PSI (Medina et al. 1992) and has previously been shown to facilitate electron transfer to P450s (Goni et al. 2009; Jensen et al. 2011). (III) The light-driven system is extremely versatile illustrated by its ability to function using natural sunlight or intense filtered light. For example, exposure of the PSI-Fdx-CYP124 system (200 μl in a 7 ml glass test tube) to sunlight for 30 min at noon (November 4th, 2010, San Francisco) resulted in 20.5% catalytic conversion of 60 μM farnesol.
In summary, we have established a light-driven electron transport system in which PSI can function as an electron donor for CYP124 via Fdx as electron carrier. The diversity of the light-driven PSI electron transport system and the avoidance of the costly cofactor NADPH highlight its potential for carrying our chemically complex hydroxylations in vitro, illustrated here by the ω-hydroxylation of branched hydrocarbon chains, simply using light to provide reducing equivalents.
Supplementary Material
Acknowledgments
KJ was supported by a Ph.D. stipend from the Faculty of Life Sciences, University of Copenhagen and sponsored by a travel stipend from Bilateral network activities, Visiting Scientist Grants 2010: Improving plants for food, fuel and bioactive natural products (09-075501). JBJ was supported by a Heiser Postdoctoral Fellowship for Research in Leprosy and Tuberculosis sponsored by The New York Community Trust. This work was also funded by the Villum Foundation to the research centre “Pro-Active Plants” and from “Center for Synthetic Biology”, funded by the UNIK research initiative of the Danish Ministry of Science, Technology and Innovation (BLM) and NIH grants GM25515 and AI074824 (P.O.M.).
Contributor Information
Kenneth Jensen, Email: keje@life.ku.dk, Plant Biochemistry Laboratory, Department of Plant Biology and Biotechnology, University of Copenhagen, 40 Thorvaldsensvej, 1871 Copenhagen, Frederiksberg C, Denmark.
Jonathan B. Johnston, Department of Pharmaceutical Chemistry, University of California San Francisco, 600 16th Street, Genentech Hall Box 2280, San Francisco, CA 94158-2280, USA
Paul R. Ortiz de Montellano, Department of Pharmaceutical Chemistry, University of California San Francisco, 600 16th Street, Genentech Hall Box 2280, San Francisco, CA 94158-2280, USA
Birger Lindberg Møller, Email: blm@life.ku.dk, Plant Biochemistry Laboratory, Department of Plant Biology and Biotechnology, University of Copenhagen, 40 Thorvaldsensvej, 1871 Copenhagen, Frederiksberg C, Denmark.
References
- Asada K. The water–water cycle in chloroplasts: scavenging of active oxygens and dissipation of excess photons. Annu Rev Plant Physiol Plant Mol Biol. 1999;50:601–639. doi: 10.1146/annurev.arplant.50.1.601. [DOI] [PubMed] [Google Scholar]
- Cirino PC, Arnold FH. A self-sufficient peroxide-driven hydroxylation biocatalyst. Angew Chem Int Edit. 2003;42:3299–3301. doi: 10.1002/anie.200351434. [DOI] [PubMed] [Google Scholar]
- Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- Furbank RT, Badger MR. Oxygen exchange associated with electron transport and photophosphorylation in spinach thylakoids. Biochim Biophys Acta Bioenerg. 1983;723:400–409. [Google Scholar]
- Goni G, Zollner A, Lisurek M, Velazquez-Campoy A, Pinto S, Gomez-Moreno C, Hannemann F, Bernhardt R, Medina M. Cyanobacterial electron carrier proteins as electron donors to CYP106A2 from Bacillus megaterium ATCC 13368. Biochim Biophys Acta. 2009;1794:1635–1642. doi: 10.1016/j.bbapap.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Ham MH, Choi JH, Boghossian AA, Jeng ES, Graff RA, Heller DA, Chang AC, Mattis A, Bayburt TH, Grinkova YV, Zeiger AS, Van Vliet KJ, Hobbie EK, Sligar SG, Wraight CA, Strano MS. Photoelectrochemical complexes for solar energy conversion that chemically and autonomously regenerate. Nat Chem. 2010;2:929–936. doi: 10.1038/nchem.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannemann F, Bichet A, Ewen KM, Bernhardt R. Cytochrome P450 systems–biological variations of electron transport chains. Biochim Biophys Acta. 2007;1770:330–344. doi: 10.1016/j.bbagen.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Hollmann F, Witholt B, Schmid A. [Cp*Rh(bpy)−(H2O)](2 +): a versatile tool for efficient and non-enzymatic regeneration of nicotinamide and flavin coenzymes. J Mol Catal B Enzym. 2002;19:167–176. [Google Scholar]
- Jensen K, Møller BL. Plant NADPH-cytochrome P450 oxidoreductases. Phytochemistry. 2010;71:132–141. doi: 10.1016/j.phytochem.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Jensen K, Jensen PE, Møller BL. Light-driven cytochrome P450 hydroxylations. ACS Chem Biol. 2011;6:533–539. doi: 10.1021/cb100393j. [DOI] [PubMed] [Google Scholar]
- Johnston JB, Kells PM, Podust LM, Ortiz de Montellano PR. Biochemical and structural characterization of CYP124: a methyl-branched lipid omega-hydroxylase from Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2009;106:20687–20692. doi: 10.1073/pnas.0907398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JB, Ouellet H, Ortiz de Montellano PR. Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J Biol Chem. 2010;285:36352–36360. doi: 10.1074/jbc.M110.161117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo H, Lin Z, Arnold FH. Laboratory evolution of peroxide-mediated cytochrome P450 hydroxylation. Nature. 1999;399:670–673. doi: 10.1038/21395. [DOI] [PubMed] [Google Scholar]
- Krishnan S, Wasalathanthri D, Zhao LL, Schenkman JB, Rusling JF. Efficient bioelectronic actuation of the natural catalytic pathway of human metabolic cytochrome P450 s. J Am Chem Soc. 2011;133:1459–1465. doi: 10.1021/ja108637s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin WA. Biotransformations in organic synthesis. Bioresource Technol. 2000;74:49–62. [Google Scholar]
- Medina M, Hervas M, Navarro JA, De la Rosa MA, Gomez-Moreno C, Tollin G. A laser flash absorption spectroscopy study of Anabaena sp. PCC 7119 flavodoxin photoreduction by photosystem I particles from spinach. FEBS Lett. 1992;313:239–242. doi: 10.1016/0014-5793(92)81200-6. [DOI] [PubMed] [Google Scholar]
- Nath A, Atkins WM, Sligar SG. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- Naver H, Scott MP, Golbeck JH, Møller BL, Scheller HV. Reconstitution of barley photosystem I with modified PSI-C allows identification of domains interacting with PSI-D and PSI-A/B. J Biol Chem. 1996;271:8996–9001. doi: 10.1074/jbc.271.15.8996. [DOI] [PubMed] [Google Scholar]
- Ouellet H, Johnston JB, de Ortiz Montellano PR. The Mycobacterium tuberculosis cytochrome P450 system. Arch Biochem Biophys. 2010;493:82–95. doi: 10.1016/j.abb.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles SB. Photoinhibition of photosynthesis induced by visible-light. Annu Rev Plant Phys. 1984;35:15–44. [Google Scholar]
- Reipa V, Mayhew MP, Vilker VL. A direct electrode-driven P450 cycle for biocatalysis. Proc Natl Acad Sci USA. 1997;94:13554–13558. doi: 10.1073/pnas.94.25.13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller HV, Svendsen I, Møller BL. Subunit composition of photosystem I and identification of center X as a [4Fe-4S] iron-sulfur cluster. J Biol Chem. 1989;264:6929–6934. [PubMed] [Google Scholar]
- Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- Tishkov VI, Galkin AG, Fedorchuk VV, Savitsky PA, Rojkova AM, Gieren H, Kula MR. Pilot scale production and isolation of recombinant NAD +- and NADP +-specific formate dehydrogenases. Biotechnol Bioeng. 1999;64:187–193. [PubMed] [Google Scholar]
- Tjus SE, Scheller HV, Andersson B, Møller BL. Active oxygen produced during selective excitation of photosystem I is damaging not only to photosystem I, but also to photosystem II. Plant Physiol. 2001;125:2007–2015. doi: 10.1104/pp.125.4.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udit AK, Hill MG, Gray HB. Electrochemistry of cytochrome P450BM3 in sodium dodecyl sulfate films. Langmuir. 2006;22:10854–10857. doi: 10.1021/la061162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Donk WA, Zhao H. Recent developments in pyridine nucleotide regeneration. Curr Opin Biotechnol. 2003;14:421–426. doi: 10.1016/s0958-1669(03)00094-6. [DOI] [PubMed] [Google Scholar]
- Wirtz M, Klucik J, Rivera M. Ferredoxin-mediated electrocatalytic dehalogenation of haloalkanes by cytochrome P450(cam) J Am Chem Soc. 2000;122:1047–1056. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.