

Abstract
Sympathetic nerves can regenerate after injury to reinnervate target tissues. Sympathetic regeneration is well documented after chronic cardiac ischemia, so we were surprised that the cardiac infarct remained denervated following ischemia-reperfusion (I-R). We used mice to ask if the lack of sympathetic regeneration into the scar was due to blockade by inhibitory extracellular matrix within the infarct. We found that chondroitin sulfate proteoglycans (CSPGs) were present in the infarct after I-R, but not after chronic ischemia, and that CSPGs caused inhibition of sympathetic axon outgrowth in vitro. Ventricle explants after I-R and chronic ischemia stimulated sympathetic axon outgrowth that was blocked by nerve growth factor antibodies. However, growth in I-R cocultures was asymmetrical, with axons growing toward the heart tissue consistently shorter than axons growing in other directions. Growth toward I-R explants was rescued by adding chondroitinase ABC to the cocultures, suggesting that I-R infarct-derived CSPGs prevented axon extension. Sympathetic ganglia lacking protein tyrosine phosphatase sigma (PTPRS) were not inhibited by CSPGs or I-R explants in vitro, suggesting PTPRS is the major CSPG receptor in sympathetic neurons. To test directly if infarct-derived CSPGs prevented cardiac reinnervation, we performed I-R in ptprs−/− and ptprs+/− mice. Cardiac infarcts in ptprs−/− mice were hyperinnervated, while infarcts in ptprs+/− littermates were denervated, confirming that CSPGs prevent sympathetic reinnervation of the cardiac scar after I-R. This is the first example of CSPGs preventing sympathetic reinnervation of an autonomic target following injury, and may have important consequences for cardiac function and arrhythmia susceptibility after myocardial infarction.
Introduction
Nerve regeneration is often minimal in the CNS, but peripheral nerves can regenerate back to their targets. For example, postganglionic sympathetic nerves reinnervate denervated tissues including the iris (Olson and Malmfors, 1970; Lorez et al., 1975), mesenteric arteries (Aguayo et al., 1973; Hill et al., 1985), pineal gland (Bowers et al., 1984), and skin (Gloster and Diamond, 1992). In addition to regeneration following injury, sympathetic nerves innervating the uterus undergo degeneration and regeneration during each estrous cycle (Zoubina and Smith, 2000). The presence of nerve growth factor (NGF) in the target tissue is important for promoting innervation of tissue during development (Glebova and Ginty, 2004) and reinnervation of a tissue after denervation (Aloe et al., 1985; Gloster and Diamond, 1995; Vo and Tomlinson, 1999).
One of the best characterized targets of the sympathetic nervous system, and sympathetic axon regeneration, is the heart. Release of norepinephrine (NE) from sympathetic nerves stimulates heart rate, conduction velocity, and ventricular contractility. Regeneration of sympathetic nerves in the heart has been well characterized in animal models of chronic cardiac ischemia, where high levels of NGF in the infarcted myocardium lead to nerve sprouting and hyperinnervation (Vracko et al., 1990; Zhou et al., 2004; Hasan et al., 2006; El-Helou et al., 2008). Postmortem analysis of human hearts following heart failure or cardiomyopathy also revealed sympathetic hyperinnervation (Cao et al., 2000). The cardiac sympathetic innervation has been examined in vivo by monitoring uptake of the labeled NE transporter substrates C-11 hydroxyephedrine or iodine-123 meta-iodobenzylguanidine (123I-MIBG). These studies reveal denervation after ischemia-reperfusion (I-R; Stanton et al., 1989) that is followed by reinnervation of peri-infarct myocardium (Hartikainen et al., 1996), and document significant reinnervation in transplanted hearts (Bengel et al., 1999; 2001; Estorch et al., 1999). Imaging studies showing reinnervation of transplanted hearts is complemented by functional responses to exercise (Wilson et al., 2000) and functional responses to drugs that cause NE release or block NE receptors (Bengel et al., 2004). Finally, sympathetic reinnervation of transplants was confirmed postmortem by tyrosine hydroxylase (TH) staining (Kim et al., 2004).
Since sympathetic nerve regeneration is well documented in the heart, we were surprised to discover that the cardiac infarct was not reinnervated following I-R injury (Li et al., 2004). This was particularly unexpected given infarct reinnervation observed after chronic cardiac ischemia (Vracko et al., 1990; Hasan et al., 2006; El-Helou et al., 2008), and evidence of elevated NGF in the scar after I-R (Hiltunen et al., 2001; Zhou et al., 2004). Cardiac I-R triggers an inflammatory response that initiates fibroblast migration and proliferation (Porter and Turner, 2009). Activation of fibroblasts results in production of a collagen-based infarct, or scar, which contains hyaluronic acid (HA) and other extracellular matrix components (Dobaczewski et al., 2006) that are present in glial scars after CNS injury (Sherman and Back, 2008). Here we investigate the possibility that the lack of sympathetic regeneration into the infarct after cardiac I-R is due to blockade of axon growth by inhibitory components of extracellular matrix within the cardiac scar.
Materials and Methods
Animals.
C57BL/6J mice were obtained from The Jackson Laboratory West, and were used for all experiments except those using ptprs transgenic mice. ptprs+/− transgenic mice (BALBC background) were supplied by Michel Tremblay at McGill University (Elchebly et al., 1999), and were bred as heterozygotes. ptprs+/+ and ptprs+/− littermates were used as “wild-type” controls for ptprs−/− studies. All mice were kept on a 12 h light/dark cycle with ad libitum access to food and water. Age and gender-matched male and female mice 12–18 weeks old were used for surgeries, while ganglia from male and female neonatal mice were used for explants and dissociated cultures. All procedures were approved by the Oregon Health and Science University (OHSU) Institutional Animal Care and Use Committee and comply with the Guide for the Care and Use of Laboratory Animals published by the National Academies (8th edition).
Surgery, myocardial I-R.
Anesthesia was induced with 4% isoflurane and maintained with 2% isoflurane. The left anterior descending coronary artery was reversibly ligated for 30 min and then reperfused by release of the ligature. Occlusion was confirmed by sustained S-T wave elevation and regional cyanosis. Reperfusion was confirmed by the return of color to the ventricle distal to the ligation and reperfusion arrhythmia. Core body temperature was monitored by a rectal probe and maintained at 37°C, and a two-lead electrocardiogram was monitored.
Myocardial ischemia.
Chronic ischemia was done in exactly the same manner as described above, but with permanent occlusion of the LAD using 8–0 gauge suture.
Sham surgery.
Sham animals underwent the procedure described above, except for the LAD ligature.
Dissociated primary cell culture with chondroitin sulfate proteoglycan and HA treatment.
Cultures of sympathetic neurons were prepared from superior cervical ganglia (SCG) of newborn mice as described previously (Dziennis and Habecker, 2003). Neurons were plated onto poly-l-lysine (PLL; 0.01%, Sigma-Aldrich) and collagen (10 μg/ml; BD Biosciences)-coated plates, and grown in serum free C2 medium (Lein et al., 1995; Pellegrino et al., 2011) supplemented with 50 ng/ml NGF (BD Biosciences), 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate (Invitrogen). Cells were incubated at 37°C in a humidified 5% CO2 incubator. Cells were maintained for 48 h in the presence of the anti-mitotic agent cytosine arabinoside (1 μm) to reduce the number of non-neuronal cells. Chondroitin sulfate proteoglycan (CSPG) treatments were performed using soluble or fixed CSPGs (Millipore #CC117; mixture includes neurocan, phosphacan, versican, and aggrecan). HA treatments were performed similarly, using mixed molecular weight HA (MP Biomedicals) for soluble treatments. For fixed treatments, high molecular weight (HMW) HA (Lifecore Biomedical) was degraded using bovine testes hyaluronidase (Sigma) to produce low molecular weight (LMW) HA (Generously provided by Dr. Stephen Back, OHSU) (1) Soluble: neurons were grown in 48-well plates coated with PLL and collagen. Vehicle (media), CSPGs (10 ng/ml–20 μg/ml), or HA (10 ng/ml–100 μg/ml) were added to the cultures 24 h after plating, and 24 h after addition of CSPGs, HA, or vehicle, images were acquired for Sholl analysis. (2) Fixed: plates were coated with PLL/collagen, PLL/collagen/CSPGs (100 ng/ml–1 μg/ml), or PLL/collagen/HA (100 ng/ml–1 μg/ml; LMW and HMW) before addition of neurons. Images were acquired for Sholl analysis 24 h after plating. For CSPG degradation experiments, chondroitinase ABC (ChABC, 4 μU/ml; Seikagaku Biobusiness) was added to culture media at the time of plating.
Sholl analysis.
To quantify neurite outgrowth in dissociated neurons, the Sholl method was used (Sholl, 1953). A series of concentric circles were superimposed over the cell body using ImageJ, and neurite intersections with the circles were counted. The number of intersections provides an estimate of neurite length and/or increased branching.
Compartmentalized cultures.
To generate microfluidic chambers providing separation of two media compartments (450 μm apart), SYLGARD 184 silicone elastomer (Dow Corning) was poured into a precast mold, and heated at 50−60°C for 2 h. Cleaned chambers were placed in 10 cm culture dishes (Corning) precoated with 0.01% PLL. The axonal compartment was then coated with 10 μg/ml collagen or collagen + 1 μg/ml CSPGs. SCGs were placed in reduced growth factor Matrigel (BD Bioscience) within the cell body compartment. C2 media supplemented with 10 ng/ml NGF was added to both compartments, and cultures were maintained at 37°C in a humidified 5% CO2 incubator. After 24 h, or when axons were first visible in the axonal compartment, images were acquired (t = 0). Additional images were obtained 3 h later (t = 3), and a growth rate was calculated based on the distance extended during that 3 h period.
Heart/ganglia cocultures.
Coculture experiments were performed in 24-well plates by plating pieces of infarcted left ventricle (LV) or corresponding sham tissue, collected 10 d after surgery, with neonatal SCGs. Tissues were plated in 30 μl of reduced growth factor Matrigel, separated by ∼1 mm, and placed at 37°C to solidify the Matrigel before addition of C2 media supplemented with 2 ng/ml NGF. Cocultures were incubated at 37°C in a humidified 5% CO2 incubator for 48 h. Following 48 h in culture, images of the cultures were acquired using phase contrast microscopy (10×) and axon length was analyzed using Nikon Elements. For ChABC experiments, tissue from a single heart was split between the vehicle and ChABC treatment groups to promote consistency. Cocultures were treated from the time of plating with ChABC (4 μU/ml) diluted in culture media, or media alone. For cocultures examining the role of the CSPG receptor protein tyrosine phosphatase sigma (PTPRS), SCGs were dissected from an entire litter of neonatal mice containing the range of PTPRS genotypes (ptprs+/+, ptprs+/−, ptprs−/−). The two ganglia from an individual mouse were divided so that one SCG was cultured with a sham explant and one with an infarct explant. Following 48 h in culture, images were acquired using phase contrast microscopy (10×) and axon length was analyzed using Nikon Elements. Genotyping was completed after image acquisition and analysis so that the experiment was blinded.
Coculture axon length analysis.
Axon length was measured on three cardinal sides of each ganglion (0, 90, and 180°). Growth toward the myocardium was designated 0°, growth perpendicular was 90°, and growth away from the myocardium was 180°. To normalize for interwell variability of growth, a ratio of these measures was used (0/90 or 0/180). Values for the 0/90 or 0/180 ratio near 1 indicated growth was similar in all directions. In contrast, ratios significantly <1 reflected significantly shorter axons on the side of the ganglion projecting toward the heart explant.
Immunohistochemistry.
Hearts were fixed for 1 h in 4% paraformaldehyde (PFA), rinsed in PBS, cryoprotected in 30% sucrose overnight, and frozen in mounting medium for sectioning. Transverse 10 μm sections were cut on a cryostat and thaw mounted onto charged slides. To reduce fixative-induced autofluorescence, sections were rinsed in 10 mg/ml sodium borohydride 3 × 10 min and then rinsed in PBS 3 × 10 min. Sections were then blocked in 3% bovine serum albumin (BSA)/0.3% Triton X-100 in PBS for 1 h. Slides were then incubated with rabbit anti-TH (1:1000; Millipore Bioscience Research Reagents) and mouse anti-chondroitin sulfate (1:300; Sigma CS-56) or sheep anti-fibrinogen (1:500, AbD; Serotec) antibodies overnight at 4°C, rinsed 3 × 10 min in PBS, and incubated 1.5 h with the Alexa Fluor 488-conjugated rabbit IgG-specific antibody (1:1000; Invitrogen) and Alexa Fluor 568-conjugated mouse IgG-specific antibody (1:500) or Alexa Fluor 568-conjugated sheep IgG-specific antibody (1:1000). Due to nonspecific binding of Alexa Fluor 568-conjugated mouse IgG, anti-mouse IgG was added to the blocking solution to prevent nonspecific secondary binding. Sections were rinsed 3 × 10 min in PBS, incubated for 30 min in CuSO4 in 50 mm ammonium acetate to reduce background signal further, rinsed 3 × 10 min in PBS, coverslipped in a 1:1 PBS:glycerol solution, and visualized by fluorescence microscopy.
Cocultures were fixed for 15 min in 4% PFA and rinsed in PBS 3 × 10 min. The tissue was blocked in 3% BSA/0.3% Triton X-100 in PBS for 1 h, then incubated with rabbit anti-TH (1:1000) overnight at 4°C. Tissue was rinsed with PBS and incubated for 1.5 h with the Alexa Fluor 488-conjugated rabbit IgG-specific antibody (1:1000), rinsed again in PBS, and imaged using fluorescence microscopy.
Imaging and threshold analysis.
TH staining was quantified to assess sympathetic innervation density. Images were taken of the infarct and peri-infarct zone of each section, and three sections obtained from a similar level of the base to apex axis were quantified in each heart using threshold discrimination analysis (ImageJ) (Lorentz et al., 2010).
Statistics.
Student's t test was used for comparisons of just two samples. Data with more than two groups were analyzed by one-way ANOVA using the Tukey post hoc test to compare all conditions. For experiments comparing different surgical groups and a second variable (±NGF antibody, PTPRS genotype) two-way ANOVA was performed using the Bonferroni post hoc test. All statistical analyses were performed using Prism 5.0.
Results
Sympathetic denervation following I-R
We observed in previous studies that the infarct (scar) and area adjacent to the infarct remained denervated 7 d after I-R in the rat heart, while hyperinnervation was visible farther away from the developing scar (Li et al., 2004). Subsequent studies have shown hyperinnervation within the infarct after chronic ischemia (Hasan et al., 2006). To investigate further neural remodeling after I-R, we moved to the mouse heart where the cardiac infarct is fully formed and stable 10 d after I-R (Dobaczewski et al., 2006). We examined sympathetic nerve density in the LV 10 d after I-R or sham surgery using TH immunohistochemistry. Sympathetic nerve fibers were detected throughout the LV of sham-operated animals, but no sympathetic fibers were observed within the infarct 10 d after I-R (Fig. 1A,B). A follow-up experiment revealed ongoing denervation of the infarct 20 d after I-R (Fig. 1D). Sympathetic fiber density outside of the infarct was similar to the innervation density in sham animals 10 and 20 d after surgery (Fig. 1C,D). Following 10 d of chronic ischemia (Fig. 1E), however, sympathetic innervation density within the infarct was similar to the surrounding peri-infarct (Fig. 1F).
Figure 1.
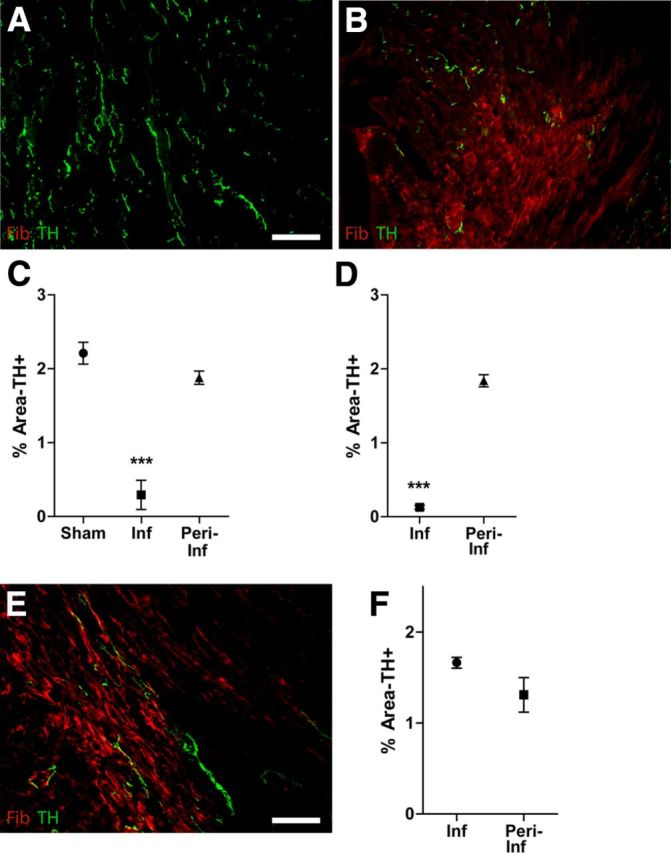
Sympathetic denervation after I-R. Heart sections from Sham (A), I-R (B), and chronic ischemia (E) operated mice were stained for TH (green) to identify sympathetic nerve fibers and fibrinogen (red) to identify the infarct. Scale bars: 100 μm. TH+ fibers were quantified in the infarct and peri-infarct ventricle (or the corresponding area of sham ventricle) 10 d (C) or 20 d (D) after reperfusion, or after 10 d of chronic ischemia (F). Data are the mean ± SEM, n = 4/surgical group; ***p < 0.001 versus sham and versus peri-infarct (C); ***p < 0.001 versus peri-infarct (D); and no significant differences (F).
Cardiac ischemia increases cardiac NGF and enhances axon outgrowth
The lack of scar reinnervation 10 and 20 d after I-R in the mouse heart, while the rest of the heart had normal innervation, suggested that axon regeneration was blocked at the edge of the infarct. This presented a striking contrast to the reinnervation observed in the rat heart 10 d after chronic ischemia (Hasan et al., 2006) (Fig. 1E,F). Coculture of ischemic heart tissue with a sympathetic ganglion leads to enhanced axon outgrowth that is blocked by an anti-NGF antibody (Hasan et al., 2006). To determine whether this apparent discrepancy was due to differential effects of chronic ischemia versus I-R, we performed explant experiments with tissue from both types of injury. We cocultured mouse heart tissue taken 10 d after sham, chronic ischemia, or I-R surgery with neonatal sympathetic ganglia, with or without antibodies to NGF, and measured axon outgrowth after 48 h. Axon length was quantified in three cardinal directions around the ganglion (Fig. 2A) to determine whether explanted myocardium released factors into the media that stimulated axon growth. Heart explants from both types of myocardial infarction, ischemia and I-R, enhanced axon outgrowth compared with sham explants (Fig. 2B). The increase in axon growth was blocked by anti-NGF antibodies, but anti-NGF had no effect on axon length in sham cocultures (Fig. 2B). Thus, NGF is released from the cardiac scar after both ischemia and I-R.
Figure 2.

Infarcted heart explants stimulate axon outgrowth. A, Left, An illustration of the coculture method showing three cardinal directions around the ganglion where axon length was quantified (0, 90, and 180°). Right, Photo of a ganglion cocultured with I-R heart explant. Scale bar, 200 μm. B, SCGs were cocultured with heart tissue from sham, I-R infarcts, or chronic ischemia infarcts, treated with vehicle or a function blocking anti-NGF antibody, and axon length was quantified. Triplicate cocultures were assayed for each condition. Data are mean ± SEM; without anti-NGF, *p < 0.05 and **p < 0.01 versus sham; with anti-NGF, *p < 0.05, and **p < 0.01 versus vehicle, not significant (n.s.) versus sham. C, Directional axon length for the vehicle-treated cocultures quantified in B. Axon length at 0, 90, and 180° is graphed separately rather than pooled together (triplicate cocultures, 6 measurements/direction in each coculture, mean ± SEM, **p < 0.01 vs 0°). For illustration purposes, the length measurements at 0, 90, and 180° were used to generate a diagram of axon growth in all directions. D, Ratio of growth toward the heart/growth away from the heart (0° length/90° length) averaged across three independent coculture experiments performed without NGF antibodies (mean ± SEM; ***p < 0.001). A ratio of 1 is similar growth at 0 and 90°, a ratio <1 indicates shorter axons at 0°.
Although axon growth was enhanced overall in I-R explant cocultures, we noted that growth was more variable in those cocultures compared with sham and chronic ischemia explants. This was due to a consistent asymmetry in axon outgrowth in the I-R-ganglion cocultures, where axons growing directly toward the heart were consistently shorter than axons growing in other cardinal directions (Fig. 2C). Figure 2C shows axon lengths from representative cocultures. In three independent experiments, symmetrical growth was observed in sham and chronic ischemia cocultures, while asymmetrical growth was observed in I-R cocultures, with significantly shorter axons growing directly toward heart explants (Fig. 2D). Our anti-NGF data suggested that NGF was elevated in I-R cocultures, so the consistently shorter axons projecting toward the heart led us to ask whether I-R infarcts might be producing an inhibitory extracellular matrix that formed a barrier to axon extension.
CSPGs are present in the heart following I-R
Nerve regeneration in the CNS is commonly prevented by inhibitory extracellular matrix that includes HA (Tona and Bignami, 1993) and CSPGs (McKeon et al., 1999; Asher et al., 2001; Jones et al., 2002). HA is present in the developing mouse infarct after I-R and is highest at the border zone of the mature scar (Dobaczewski et al., 2006), but it is not known if CSPGs are present in the heart after I-R. We ask whether CSPGs were present within the I-R infarct by carrying out immunohistochemistry with the pan-CSPG antibody CS-56 (Avnur and Geiger, 1984). This antibody binds to chondroitin sulfate (CS) rather than a specific core protein. CS-56 staining was absent in sham hearts (Fig. 3A) and localized to the infarct after I-R (Fig. 3B). Interestingly, CSPGs were undetectable in the chronic ischemia infarct (Fig. 3C). This suggests that CSPGs and HA are present after I-R and are candidates for inhibiting axon growth into the infarct.
Figure 3.

I-R stimulates CSPG production in the heart. Heart sections from Sham (A), I-R (B), and chronic ischemia (C) operated mice were stained 10 d after surgery for TH (green) to identify sympathetic nerve fibers, and for CSPGs using the CS-56 antibody (red). Scale bar, 100 μm. Similar results were obtained in sections from four hearts in each surgical group.
CSPGs inhibit sympathetic axon outgrowth in vitro, but HA does not
CSPGs (McKeon et al., 1991; Davies et al., 1997) and HA (Tona and Bignami, 1993) inhibit axon regeneration in the CNS, but they have not been examined in sympathetic nerve regeneration. To examine sympathetic axon responses to CSPGs, dissociated sympathetic neurons were grown in serum free medium to limit formation of dendrites (Lein et al., 1995), and treated for 24 h with increasing concentrations of CSPGs diluted in media. Neurites were analyzed using the Sholl method. Soluble CSPGs inhibited process outgrowth in a dose-dependent manner (Fig. 4B). Similar results were obtained when neurons were grown on plates precoated with increasing concentrations of CSPGs (Fig. 4C). CSPG-induced inhibition of axon outgrowth was prevented by treatment of CSPGs with ChABC (data not shown). While these experiments indicated that CSPGs inhibited axon outgrowth when in contact with the entire neuron, they do not reveal whether local contact of the growing axon with CSPGs would block extension. To determine whether CSPG interaction with axons blunted outgrowth, compartmentalized microfluidic chambers were used, and only the axonal compartment was coated with CSPGs. When axons encountered CSPGs, growth slowed significantly, and axon bundles formed that were absent in the control cultures (Fig. 4E). Axon growth was restored by degradation of CSPGs by ChABC, indicating that sympathetic axon outgrowth was inhibited by CSPGs in the axonal compartment (Fig. 4F). In contrast to the strong inhibition of axon outgrowth by CSPGs, soluble HA had no effect on sympathetic axon outgrowth (Fig. 5A). This was also true when both HMW and LMW forms of HA were precoated onto plates (Fig. 5B).
Figure 4.

CSPGs inhibit sympathetic axon outgrowth in vitro. A, A representative image of a sympathetic neuron, overlaid with a series of concentric circles for Sholl analysis. Scale bar, 20 μm. Dissociated sympathetic neurons were treated with soluble CSPGs (B), or plated onto dishes precoated with CSPGs (C), and neurite crosses were quantified by Sholl analysis. Data are the mean ± SEM of at least 10 neurons/condition, **p < 0.01; ***p < 0.001. Similar results were obtained in three independent experiments. D, Illustration of a microfluidic chamber with a ganglion explant on one side and axons growing through grooves into a second chamber that was coated with collagen or CSPGs. E, Representative micrographs of axons growing at t = 0 and t = 3 h on collagen or CSPGs (1 μg/ml). White arrowheads indicate the leading edge of the axon in the t = 0 image, while black arrowheads indicate the final point of the axon at t = 3 h. Scale bar, 20 μm. Note bundling of the axons growing on CSPGs. F, Quantification of axon growth in microfluidic chambers where the distal compartment was coated with collagen, collagen/CSPGs, or collagen/CSPGs + ChABC. Data are mean ± SEM for at least eight axons per condition, **p < 0.01, and similar results were obtained in three independent experiments.
Figure 5.

HA does not inhibit sympathetic axon outgrowth in vitro. Dissociated sympathetic neurons were treated with soluble HA (A), or plated onto dishes precoated with LMW or HMW HA (B), and neurite crosses were quantified by Sholl analysis. Data are the mean ± SEM least 10 neurons/condition. Similar results were obtained in three independent experiments.
Enzymatic degradation of infarct-derived CSPGs restores axon outgrowth
We observed CSPGs in infarcted myocardium after I-R, and found that CSPGs inhibited sympathetic axon outgrowth. To test if infarct-derived CSPGs prevented sympathetic axon extension, we performed coculture experiments with or without ChABC to degrade CSPGs. Sham or I-R myocardium was split into two pieces and cocultured with a sympathetic ganglion in the presence of vehicle or ChABC. In vehicle-treated I-R cocultures, axons growing directly toward the heart were shorter than axons growing in other directions (Fig. 6A). However, in the ChABC-treated I-R cocultures, axons growing toward the heart were not significantly shorter than axons growing in other directions (Fig. 6B). This suggests that I-R explants secreted inhibitory CSPGs into the surrounding Matrigel and these CSPGs were degraded by ChABC. Treatment with ChABC had no effect on axon growth in sham cocultures (data not shown). These data can be expressed as a ratio of growth toward the heart/growth away from the heart (0°/90°) to control for different amounts of total growth in each individual coculture, and data combined across multiple cocultures (Fig. 6C). ChABC consistently increased axon growth toward I-R explants, providing strong evidence that CSPGs produced by infarcted myocardium after I-R inhibit sympathetic regeneration into the infarct.
Figure 6.

Enzymatic degradation of infarct-derived CSPGs restores axon outgrowth. SCGs were cocultured with heart tissue after sham or I-R surgery, and treated with either vehicle (A) or ChABC (B). Tissue from a single heart was split between the vehicle and ChABC groups. Axon length was quantified at 0, 90, and 180° (6 measurements/direction) and triplicate cocultures were averaged (mean ± SEM, *p < 0.05 vs 90 and 180°). C, Axon length normalized as the ratio of growth toward the heart/growth away from the heart (0° length/90° length) averaged from three independent experiments, each assayed in triplicate (mean ± SEM; ***p < 0.001).
Absence of PTPRS restores axon outgrowth
Enzymatic degradation of CSPGs proved sufficient to overcome infarct-induced inhibition of sympathetic axon outgrowth in vitro. To further test the role of infarct-derived CSPGs, we used mice that lack the CSPG receptor PTPRS (Elchebly et al., 1999; Shen et al., 2009). First, we confirmed that CSPGs did not inhibit sympathetic outgrowth in neurons lacking PTPRS. Sympathetic ganglia from ptprs+/− and ptprs−/− mice were treated with or without (1 μg/ml) CSPGs, and the growth rate was quantified. ptprs+/− axon growth was inhibited by CSPGs, but ptprs−/− axons were not affected (Fig. 7A). Sympathetic ganglia from ptprs+/− and ptprs −/− mice were then cultured with sham or I-R explants to determine whether the lack of ptprs would allow axon growth through the CSPGs release by I-R explants. Figure 7B shows an example of tissue from a single heart cocultured with ptprs−/− and ptprs+/− ganglia. Infarcted myocardium inhibited the growth of ptprs+/+ and ptprs+/− axons but did not inhibit growth of ptprs−/− axons (Fig. 7C). Similar results were obtained in three independent experiments, averaged in Figure 7D. Together, these in vitro experiments strongly support the idea that CSPGs are the primary agents responsible for inhibiting sympathetic axon regeneration in the heart after I-R, and that PTPRS is the major receptor for CSPGs in these neurons.
Figure 7.
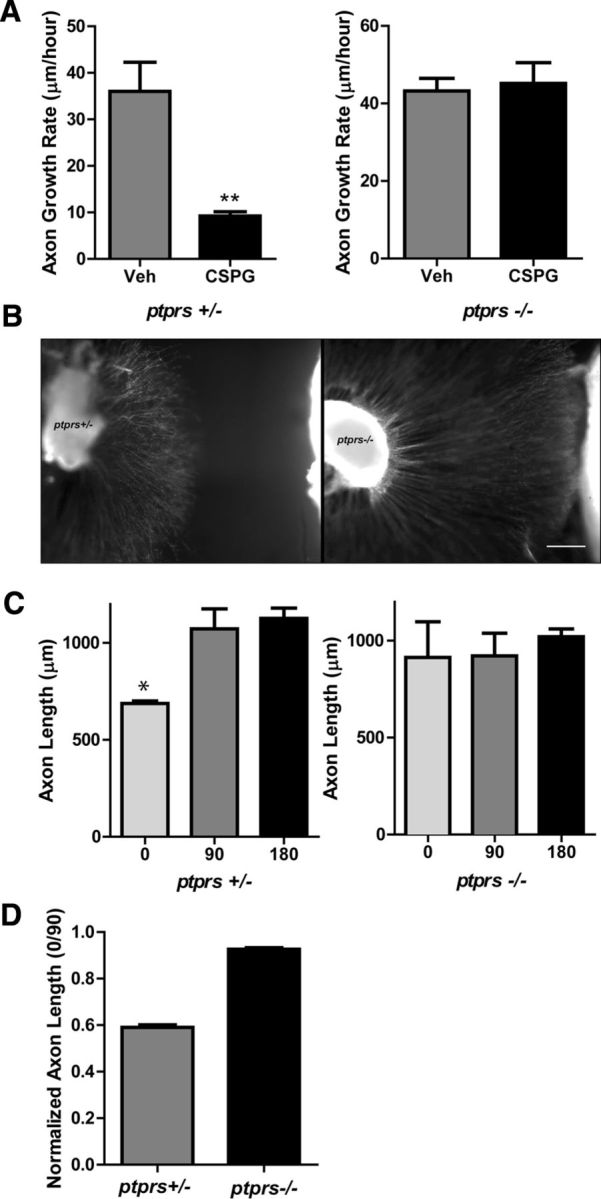
Loss of PTPRS restores axon outgrowth in vitro. A, Axon growth rate in ptprs +/− and ptprs −/− SCG explants treated with 1 μg/ml CSPGs. Data are mean ± SEM; n = 3, **p < 0.01, and are representative of three independent experiments. B, Micrographs of a ptprs+/− and ptprs−/− SCG cocultured with I-R infarct tissue from the same heart. The culture was fixed and stained for TH to highlight axons. Scale bar, 200 μm. C, ptprs+/− and ptprs−/− SCG explants were cocultured with sham or I-R infarcted heart tissue. Axon length was quantified at 0, 90, and 180° (6 measurements/direction) and triplicate cocultures were averaged (mean ± SEM, *p < 0.05 vs 90 and 180°). D, Axon length normalized as the ratio of growth toward the heart/growth away from the heart (0° length/90° length) averaged from three independent experiments, each assayed in triplicate.
To test the role of CSPGs in vivo, ptprs−/− and ptprs+/− mice underwent I-R or sham procedures. Ten days after surgery, heart sections were double labeled for TH to identify sympathetic neurons and fibrinogen to identify the infarct. Sham animals showed no significant fibrinogen staining, and TH-positive sympathetic fibers were evenly distributed in the sham LVs of both genotypes (Fig. 8A,C). Likewise, TH-positive fiber densities outside the infarct were similar in both genotypes 10 d after I-R (Fig. 8E). However, the infarct was devoid of sympathetic innervation in ptprs+/− mice, while ptprs−/− hearts exhibited significant hyperinnervation within the infarct (Fig. 8B,D). The hyperinnervation is consistent with coculture experiments indicating high levels of NGF in the scar, and confirms that CSPGs prevent reinnervation of the infarct after cardiac I-R.
Figure 8.
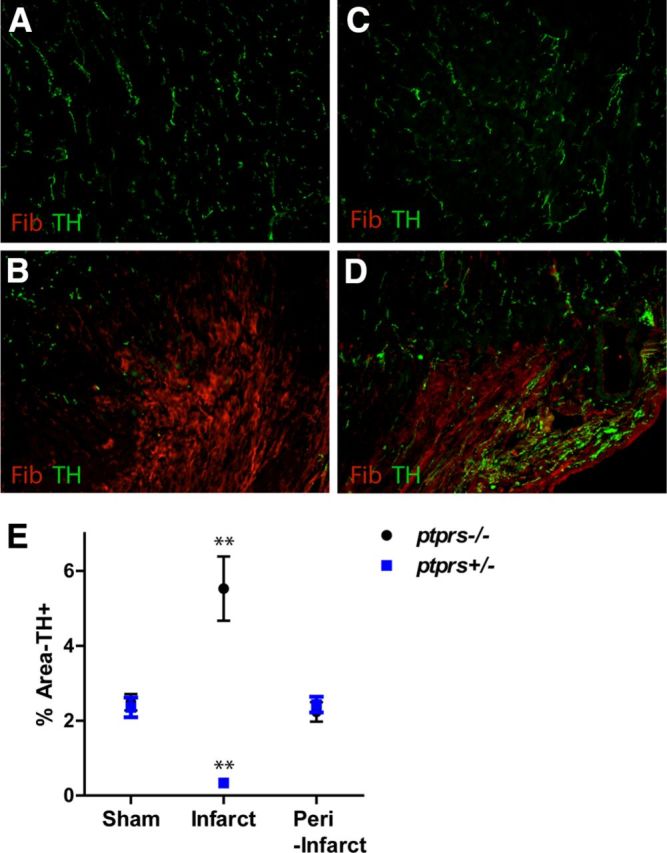
Loss of CSPG receptor PTPRS restores axon outgrowth in vivo. Heart sections from Sham (A, C) or I-R (B, D) operated ptprs +/− (A, B) and ptprs −/− (C, D) mice were stained for TH (green) to identify sympathetic nerve fibers and fibrinogen (red) to identify the infarct. Scale bar, 100 μm. Sham hearts of both genotypes (A, C) exhibit normal innervation. In contrast, the infarct is denervated in ptprs+/− hearts (B), but hyperinnervated in ptprs−/− hearts (D). E, Quantification of TH-positive fiber density within the LV of sham and I-R operated (infarct and peri-infarct) animals 10 d post surgery (mean ±SEM, n = 5 mice/group; two-way ANOVA, **p < 0.01, ***p < 0.001).
Discussion
Sympathetic neurons typically regenerate after injury, in contrast to central neurons whose regeneration is often limited. Reinnervation of the heart after chronic ischemia (Vracko et al., 1990; Zhou et al., 2004; Hasan et al., 2006; El-Helou et al., 2008) or cardiac transplantation (Bengel et al., 1999, 2001, 2002; Estorch et al., 1999) fit this model of robust sympathetic regeneration. However, we found that a common injury affecting over a million people each year in the United States (Roger et al., 2012), cardiac I-R, generates a scar that is refractory to reinnervation. In this study, we found that regeneration into the cardiac scar is prevented by CSPGs, which also inhibit axon regeneration in the CNS (McKeon et al., 1991; Davies et al., 1997). We further showed that CSPGs prevent scar reinnervation by acting through PTPRS on sympathetic neurons, and that the absence of PTPRS led to hyperinnervation of the scar in vivo. This is the first demonstration of injury-induced CSPGs preventing sympathetic reinnervation of a target innervated by the autonomic nervous system.
We investigated a role for CSPGs in cardiac remodeling after I-R, because the lack of scar reinnervation was reminiscent of the lack of axon regeneration in the CNS after injury. Neurons in the CNS are prevented from regenerating by inhibitory molecules of the extracellular matrix including CSPGs, Tenascin C (TnC), and HA (for review, see Properzi et al., 2003; Silver and Miller, 2004; Fitch and Silver, 2008; Sherman and Back, 2008). These inhibitory matrix molecules are generated by reactive astrocytes and are key components of the “glial scar.” CSPGs are particularly important for preventing axon regeneration in the CNS, and removal of CS chains from core proteins with the enzyme ChABC can restore axon regeneration in culture and in vivo (Lin et al., 2008; Massey et al., 2008; Nakamae et al., 2009; Tom et al., 2009; Lee et al., 2010). Formation of a cardiac scar shares some similarities with formation of a glial scar. Cardiac I-R triggers a significant inflammatory response including production of cytokines and infiltration of neutrophils and monocytes, a process that initiates fibroblast proliferation and migration (Porter and Turner, 2009). Similar to astrocytes in CNS injury, activation of fibroblasts results in the production of extracellular matrix and maturation of a collagen-based scar that contains TnC (Tamaoki et al., 2005) and HA (Dobaczewski et al., 2006). HA inhibits myelination of central axons (Back et al., 2005), but at least some of its effects on axon outgrowth are due to interactions with CSPGs (Sherman and Back, 2008). Our data indicate that LMW and HMW HA had no effect on sympathetic axon outgrowth, and that CSPGs are the major source of axon inhibition. It is not yet clear if fibroblasts are the source of CSPGs in the I-R infarct, or why CSPGs are not present in the chronic ischemia infarct, which is also characterized by fibrosis (Pfeffer and Braunwald, 1990), but future studies can address these issues.
The striking hyperinnervation observed in ptprs−/− hearts after I-R confirms the central role for CSPGs inhibiting regeneration, as well as the presence of high NGF in the scar. This hyperinnervation is interesting for several reasons. First, multiple receptors have now been identified for CSPGs (Shen et al., 2009; Fisher et al., 2011; Dickendesher et al., 2012), and our data suggest that PTPRS is the major receptor in sympathetic neurons. Second, removal of CSPGs in the spinal cord is not sufficient to restore growth without also adding neurotrophins (Jones et al., 2003; García-Alías et al., 2011). Our data show that in the heart, simply removing inhibitory CSPG signaling results in hyperinnervation of the scar, making this an interesting model for testing CSPG-targeted therapeutics. Third, proNGF was recently identified as elevated in the heart after I-R, where it contributes to expansion of the infarct after reperfusion (Siao et al., 2012). ProNGF, which is elevated in the mouse heart 1 and 3 d after I-R (Siao et al., 2012), stimulates axon degeneration rather than outgrowth in adult sympathetic neurons in vivo (Al-Shawi et al., 2008). Given that we performed the mouse I-R procedure identically in the proNGF study (Siao et al., 2012) and the current study, our data suggest that sometime between days 3 and 10 after injury, the balance shifts between proNGF and NGF so that NGF effects predominate and hyperinnervation occurs. Finally, high levels of NGF are sufficient to support sensory neuron growth over CSPGs in vitro (Zhou et al., 2006), but our data indicate that NGF levels in vivo are not sufficiently high to stimulate sympathetic regeneration through CSPGs and into the cardiac scar.
The functional consequences of reinnervating the infarct are not yet understood. While reinnervation of the infarct and presumptive restoration of NE release following I-R will not enhance contractile function in the scar, it may stimulate contractility of myocytes surrounding the infarct, and thus increase cardiac output in ptprs−/− mice. It is likely, however, that reinnervation of the scar will have a greater impact on arrhythmia susceptibility than on contractile function or cardiac output. Heterogeneity of sympathetic transmission after MI, and subsequent electrical remodeling of cardiac myocytes, is a major contributor to the development of arrhythmias and sudden cardiac death in humans (Rubart and Zipes, 2005). Early studies mapping sympathetic innervation in human hearts after I-R with 123I-MIBG imaging identified denervation (Stanton et al., 1989) followed by some reinnervation of peri-infarct myocardium (Hartikainen et al., 1996). Two newer studies of 123I-MIBG imaging in postinfarct patients implanted with cardioverter defibrillators concluded that a larger area of sympathetic denervation soon after I-R is associated with a greater probability of ventricular arrhythmias (Boogers et al., 2010; Nishisato et al., 2010). Recent detailed mapping studies in intact human hearts revealed sympathetic denervation of the normal myocardium directly adjacent to the scar, which is thought to contribute to arrhythmia generation (Vaseghi et al., 2012). The denervation led to electrical remodeling, so that the activation recovery interval at the border zone differed from that of the scar and surrounding viable myocardium. Dispersion of activation recovery interval is indicative of electrical instability in the heart, and significantly increases the probability of arrhythmia (Kuo et al., 1983). Given the secretion of CSPGs from infarct explants, the denervation at the border zone in human hearts may be due to the presence of inhibitory CSPGs. Thus, degrading CSPGs in vivo with ChABC might restore innervation to the border zone and decrease arrhythmia susceptibility.
In summary, this study provides the first example of CSPGs preventing sympathetic reinnervation of an autonomic target following injury, despite high levels of NGF in the infarct. We show that CSPGs act through PTPRS to block sympathetic axon outgrowth, and that the absence of PTPRS results in hyperinnervation of the infarct. This may have important consequences for cardiac function and arrhythmia susceptibility for people who suffer a myocardial infarction.
Footnotes
This work was supported by National Institutes of Health (NIH) HL093056 (B.A.H.) and an Oregon Brain Institute Neurobiology of Disease Fellowship (R.T.G.). We thank Dr. Stephen Back (OHSU, Portland OR) for high and low molecular weight hyaluronan, Dr. Michael Cohen (OHSU, Portland OR) for assistance with microfluidic chambers, and Dr. Michel Tremblay for PTPRS mice. We also thank Dr. Aviva Symes (Uniformed Services University of Health Sciences, Bethesda MD) for the suggestion to examine CSPGs in the cardiac scar, Dr. Herb Geller (National Institute of Neurological Disorders and Stroke, Bethesda MD) for helpful comments on this manuscript, and Alexandra Brown for technical assistance.
The authors declare to competing financial interests.
References
- Aguayo AJ, Peyronnard JM, Bray GM. A quantitative ultrastructural study of regeneration from isolated proximal stumps of transected unmyelinated nerves. J Neuropathol Exp Neurol. 1973;32:256–270. doi: 10.1097/00005072-197304000-00006. [DOI] [PubMed] [Google Scholar]
- Aloe L, Cozzari C, Levi-Montalcini R. Cyclocytidine-induced release of nerve growth factor from mouse submandibular glands enhances regeneration of sympathetic fibers in adult mice. Brain Res. 1985;332:259–265. doi: 10.1016/0006-8993(85)90595-5. [DOI] [PubMed] [Google Scholar]
- Al-Shawi R, Hafner A, Olsen J, Olson J, Chun S, Raza S, Thrasivoulou C, Lovestone S, Killick R, Simons P, Cowen T. Neurotoxic and neurotrophic roles of proNGF and the receptor sortilin in the adult and ageing nervous system. Eur J Neurosci. 2008;27:2103–2114. doi: 10.1111/j.1460-9568.2008.06152.x. [DOI] [PubMed] [Google Scholar]
- Asher RA, Morgenstern DA, Moon LD, Fawcett JW. Chondroitin sulphate proteoglycans: inhibitory components of the glial scar. Prog Brain Res. 2001;132:611–619. doi: 10.1016/S0079-6123(01)32106-4. [DOI] [PubMed] [Google Scholar]
- Avnur Z, Geiger B. Immunocytochemical localization of native chondroitin-sulfate in tissues and cultured cells using specific monoclonal antibody. Cell. 1984;38:811–822. doi: 10.1016/0092-8674(84)90276-9. [DOI] [PubMed] [Google Scholar]
- Back SA, Tuohy TM, Chen H, Wallingford N, Craig A, Struve J, Luo NL, Banine F, Liu Y, Chang A, Trapp BD, Bebo BF, Jr, Rao MS, Sherman LS. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;11:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- Bengel FM, Ueberfuhr P, Ziegler SI, Nekolla S, Reichart B, Schwaiger M. Serial assessment of sympathetic reinnervation after orthotopic heart transplantation. A longitudinal study using PET and C-11 hydroxyephedrine. Circulation. 1999;99:1866–1871. doi: 10.1161/01.CIR.99.14.1866. [DOI] [PubMed] [Google Scholar]
- Bengel FM, Ueberfuhr P, Schiepel N, Nekolla SG, Reichart B, Schwaiger M. Effect of sympathetic reinnervation on cardiac performance after heart transplantation. N Engl J Med. 2001;345:731–738. doi: 10.1056/NEJMoa010519. [DOI] [PubMed] [Google Scholar]
- Bengel FM, Ueberfuhr P, Hesse T, Schiepel N, Ziegler SI, Scholz S, Nekolla SG, Reichart B, Schwaiger M. Clinical determinants of ventricular sympathetic reinnervation after orthotopic heart transplantation. Circulation. 2002;106:831–835. doi: 10.1161/01.CIR.0000025631.68522.9D. [DOI] [PubMed] [Google Scholar]
- Bengel FM, Ueberfuhr P, Karja J, Schreiber K, Nekolla SG, Reichart B, Schwaiger M. Sympathetic reinnervation, exercise performance and effects of beta-adrenergic blockade in cardiac transplant recipients. Eur Heart J. 2004;25:1726–1733. doi: 10.1016/j.ehj.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Boogers MJ, Borleffs CJ, Henneman MM, van Bommel RJ, van Ramshorst J, Boersma E, Dibbets-Schneider P, Stokkel MP, van der Wall EE, Schalij MJ, Bax JJ. Cardiac sympathetic denervation assessed with 123-iodine metaiodobenzylguanidine imaging predicts ventricular arrhythmias in implantable cardioverter-defibrillator patients. J Am Coll Cardiol. 2010;55:2769–2777. doi: 10.1016/j.jacc.2009.12.066. [DOI] [PubMed] [Google Scholar]
- Bowers CW, Baldwin C, Zigmond RE. Sympathetic reinnervation of the pineal gland after postganglionic nerve lesion does not restore normal pineal function. J Neurosci. 1984;4:2010–2015. doi: 10.1523/JNEUROSCI.04-08-02010.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS, Chen LS. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000;101:1960–1969. doi: 10.1161/01.CIR.101.16.1960. [DOI] [PubMed] [Google Scholar]
- Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J. Regeneration of adult axons in white matter tracts of the central nervous system. Nature. 1997;390:680–683. doi: 10.1038/37776. [DOI] [PubMed] [Google Scholar]
- Dickendesher TL, Baldwin KT, Mironova YA, Koriyama Y, Raiker SJ, Askew KL, Wood A, Geoffroy CG, Zheng B, Liepmann CD, Katagiri Y, Benowitz LI, Geller HM, Giger RJ. NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat Neurosci. 2012;15:703–712. doi: 10.1038/nn.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324:475–488. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- Dziennis S, Habecker BA. Cytokine suppression of dopamine-beta-hydroxylase by extracellular signal-regulated kinase-dependent and -independent pathways. J Biol Chem. 2003;278:15897–15904. doi: 10.1074/jbc.M212480200. [DOI] [PubMed] [Google Scholar]
- Elchebly M, Wagner J, Kennedy TE, Lanctôt C, Michaliszyn E, Itié A, Drouin J, Tremblay ML. Neuroendocrine dysplasia in mice lacking protein tyrosine phosphatase sigma. Nat Genet. 1999;21:330–333. doi: 10.1038/6859. [DOI] [PubMed] [Google Scholar]
- El-Helou V, Proulx C, Gosselin H, Clement R, Mimee A, Villeneuve L, Calderone A. Dexamethasone treatment of post-MI rats attenuates sympathetic innervation of the infarct region. J Appl Physiol. 2008;104:150–156. doi: 10.1152/japplphysiol.00663.2007. [DOI] [PubMed] [Google Scholar]
- Estorch M, Campreciós M, Flotats A, Marí C, Bernà L, Catafau AM, Ballester M, Narula J, Carri ó I. Sympathetic reinnervation of cardiac allografts evaluated by 123I-MIBG imaging. J Nucl Med. 1999;40:911–916. [PubMed] [Google Scholar]
- Fisher D, Xing B, Dill J, Li H, Hoang HH, Zhao Z, Yang XL, Bachoo R, Cannon S, Longo FM, Sheng M, Silver J, Li S. Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. J Neurosci. 2011;31:14051–14066. doi: 10.1523/JNEUROSCI.1737-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Silver J. CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol. 2008;209:294–301. doi: 10.1016/j.expneurol.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Alías G, Petrosyan HA, Schnell L, Horner PJ, Bowers WJ, Mendell LM, Fawcett JW, Arvanian VL. Chondroitinase ABC combined with neurotrophin NT-3 secretion and NR2D expression promotes axonal plasticity and functional recovery in rats with lateral hemisection of the spinal cord. J Neurosci. 2011;31:17788–17799. doi: 10.1523/JNEUROSCI.4308-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glebova NO, Ginty DD. Heterogeneous requirement of NGF for sympathetic target innervation in vivo. J Neurosci. 2004;24:743–751. doi: 10.1523/JNEUROSCI.4523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster A, Diamond J. Sympathetic nerves in adult rats regenerate normally and restore pilomotor function during an anti-NGF treatment that prevents their collateral sprouting. J Comp Neurol. 1992;326:363–374. doi: 10.1002/cne.903260305. [DOI] [PubMed] [Google Scholar]
- Gloster A, Diamond J. NGF-dependent and NGF-independent recovery of sympathetic function after chemical sympathectomy with 6-hydroxydopamine. J Comp Neurol. 1995;359:586–594. doi: 10.1002/cne.903590406. [DOI] [PubMed] [Google Scholar]
- Hartikainen J, Kuikka J, Mäntysaari M, Länsimies E, Pyörälä K. Sympathetic reinnervation after acute myocardial infarction. Am J Cardiol. 1996;77:5–9. doi: 10.1016/S0002-9149(97)89125-4. [DOI] [PubMed] [Google Scholar]
- Hasan W, Jama A, Donohue T, Wernli G, Onyszchuk G, Al-Hafez B, Bilgen M, Smith PG. Sympathetic hyperinnervation and inflammatory cell NGF synthesis following myocardial infarction in rats. Brain Res. 2006;1124:142–154. doi: 10.1016/j.brainres.2006.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CE, Hirst GD, Ngu MC, van Helden DF. Sympathetic postganglionic reinnervation of mesenteric arteries and enteric neurones of the ileum of the rat. J Auton Nerv Syst. 1985;14:317–334. doi: 10.1016/0165-1838(85)90079-7. [DOI] [PubMed] [Google Scholar]
- Hiltunen JO, Laurikainen A, Väkevä A, Meri S, Saarma M. Nerve growth factor and brain-derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J Pathol. 2001;194:247–253. doi: 10.1002/path.878. [DOI] [PubMed] [Google Scholar]
- Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH. NG2 is a major chondroitin sulfate proteoglycan produced after spinal cord injury and is expressed by macrophages and oligodendrocyte progenitors. J Neurosci. 2002;22:2792–2803. doi: 10.1523/JNEUROSCI.22-07-02792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Sajed D, Tuszynski MH. Axonal regeneration through regions of chondroitin sulfate proteoglycan deposition after spinal cord injury: a balance of permissiveness and inhibition. J Neurosci. 2003;23:9276–9288. doi: 10.1523/JNEUROSCI.23-28-09276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DT, Luthringer DJ, Lai AC, Suh G, Czer L, Chen LS, Chen PS, Fishbein MC. Sympathetic nerve sprouting after orthotopic heart transplantation. J Heart Lung Transplant. 2004;23:1349–1358. doi: 10.1016/j.healun.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Kuo CS, Munakata K, Reddy CP, Surawicz B. Characteristics and possible mechanism of ventricular arrhythmia dependent on the dispersion of action potential durations. Circulation. 1983;67:1356–1367. doi: 10.1161/01.CIR.67.6.1356. [DOI] [PubMed] [Google Scholar]
- Lee H, McKeon RJ, Bellamkonda RV. Sustained delivery of thermostabilized chABC enhances axonal sprouting and functional recovery after spinal cord injury. Proc Natl Acad Sci U S A. 2010;107:3340–3345. doi: 10.1073/pnas.0905437106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein P, Johnson M, Guo X, Rueger D, Higgins D. Osteogenic protein-1 induces dendritic growth in rat sympathetic neurons. Neuron. 1995;15:597–605. doi: 10.1016/0896-6273(95)90148-5. [DOI] [PubMed] [Google Scholar]
- Li W, Knowlton D, Van Winkle DM, Habecker BA. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Heart Circ Physiol. 2004;286:H2229–H2236. doi: 10.1152/ajpheart.00768.2003. [DOI] [PubMed] [Google Scholar]
- Lin R, Kwok JC, Crespo D, Fawcett JW. Chondroitinase ABC has a long-lasting effect on chondroitin sulphate glycosaminoglycan content in the injured rat brain. J Neurochem. 2008;104:400–408. doi: 10.1111/j.1471-4159.2007.05066.x. [DOI] [PubMed] [Google Scholar]
- Lorentz CU, Alston EN, Belcik T, Lindner JR, Giraud GD, Habecker BA. Heterogeneous ventricular sympathetic innervation, altered beta-adrenergic receptor expression, and rhythm instability in mice lacking the p75 neurotrophin receptor. Am J Physiol Heart Circ Physiol. 2010;298:H1652–H1660. doi: 10.1152/ajpheart.01128.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorez HP, Kuhn H, Bartholini G. Degeneration and regeneration of adrenergic nerves in mesenteric blood vessels, iris and atrium of the rat after 6-hydroxydopamine injection. J Neurocytol. 1975;4:157–176. doi: 10.1007/BF01098780. [DOI] [PubMed] [Google Scholar]
- Massey JM, Amps J, Viapiano MS, Matthews RT, Wagoner MR, Whitaker CM, Alilain W, Yonkof AL, Khalyfa A, Cooper NG, Silver J, Onifer SM. Increased chondroitin sulfate proteoglycan expression in denervated brainstem targets following spinal cord injury creates a barrier to axonal regeneration overcome by chondroitinase ABC and neurotrophin-3. Exp Neurol. 2008;209:426–445. doi: 10.1016/j.expneurol.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Jurynec MJ, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci. 1999;19:10778–10788. doi: 10.1523/JNEUROSCI.19-24-10778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamae T, Tanaka N, Nakanishi K, Kamei N, Sasaki H, Hamasaki T, Yamada K, Yamamoto R, Mochizuki Y, Ochi M. Chondroitinase ABC promotes corticospinal axon growth in organotypic cocultures. Spinal Cord. 2009;47:161–165. doi: 10.1038/sc.2008.74. [DOI] [PubMed] [Google Scholar]
- Nishisato K, Hashimoto A, Nakata T, Doi T, Yamamoto H, Nagahara D, Shimoshige S, Yuda S, Tsuchihashi K, Shimamoto K. Impaired cardiac sympathetic innervation and myocardial perfusion are related to lethal arrhythmia: quantification of cardiac tracers in patients with ICDs. J Nucl Med. 2010;51:1241–1249. doi: 10.2967/jnumed.110.074971. [DOI] [PubMed] [Google Scholar]
- Olson L, Malmfors T. Growth characteristics of adrenergic nerves in the adult rat. Fluorescence histochemical and 3H-noradrenaline uptake studies using tissue transplantations to the anterior chamber of the eye. Acta Physiol Scand. 1970;(Suppl 348):1–112. [PubMed] [Google Scholar]
- Pellegrino MJ, Parrish DC, Zigmond RE, Habecker BA. Cytokines inhibit norepinephrine transporter expression by decreasing Hand2. Mol Cell Neurosci. 2011;46:671–680. doi: 10.1016/j.mcn.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.CIR.81.4.1161. [DOI] [PubMed] [Google Scholar]
- Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Properzi F, Asher RA, Fawcett JW. Chondroitin sulphate proteoglycans in the central nervous system: changes and synthesis after injury. Biochem Soc Trans. 2003;31:335–336. doi: 10.1042/BST0310335. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, et al. Heart disease and stroke statistics–2012 update: a report from the Am Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–2315. doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, He Z, Silver J, Flanagan JG. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 2009;326:592–596. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman LS, Back SA. A ‘GAG’ reflex prevents repair of the damaged CNS. Trends Neurosci. 2008;31:44–52. doi: 10.1016/j.tins.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Siao CJ, Lorentz CU, Kermani P, Marinic T, Carter J, McGrath K, Padow VA, Mark W, Falcone DJ, Cohen-Gould L, Parrish DC, Habecker BA, Nykjaer A, Ellenson LH, Tessarollo L, Hempstead BL. ProNGF, a cytokine induced after myocardial infarction in humans, targets pericytes to promote microvascular damage and activation. J Exp Med. 2012;209:2291–2305. doi: 10.1084/jem.20111749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Stanton MS, Tuli MM, Radtke NL, Heger JJ, Miles WM, Mock BH, Burt RW, Wellman HN, Zipes DP. Regional sympathetic denervation after myocardial infarction in humans detected noninvasively using I-123-metaiodobenzylguanidine. J Am Coll Cardiol. 1989;14:1519–1526. doi: 10.1016/0735-1097(89)90391-4. [DOI] [PubMed] [Google Scholar]
- Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167:71–80. doi: 10.1016/S0002-9440(10)62954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom VJ, Sandrow-Feinberg HR, Miller K, Santi L, Connors T, Lemay MA, Houlé JD. Combining peripheral nerve grafts and chondroitinase promotes functional axonal regeneration in the chronically injured spinal cord. J Neurosci. 2009;29:14881–14890. doi: 10.1523/JNEUROSCI.3641-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tona A, Bignami A. Effect of hyaluronidase on brain extracellular matrix in vivo and optic nerve regeneration. J Neurosci Res. 1993;36:191–199. doi: 10.1002/jnr.490360209. [DOI] [PubMed] [Google Scholar]
- Vaseghi M, Lux RL, Mahajan A, Shivkumar K. Sympathetic stimulation increases dispersion of repolarization in humans with myocardial infarction. Am J Physiol Heart Circ Physiol. 2012;302:H1838–H1846. doi: 10.1152/ajpheart.01106.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo PA, Tomlinson DR. The regeneration of peripheral noradrenergic nerves after chemical sympathectomy in diabetic rats: effects of nerve growth factor. Exp Neurol. 1999;157:127–134. doi: 10.1006/exnr.1999.7018. [DOI] [PubMed] [Google Scholar]
- Vracko R, Thorning D, Frederickson RG. Fate of nerve fibers in necrotic, healing, and healed rat myocardium. Lab Invest. 1990;63:490–501. [PubMed] [Google Scholar]
- Wilson RF, Johnson TH, Haidet GC, Kubo SH, Mianuelli M. Sympathetic reinnervation of the sinus node and exercise hemodynamics after cardiac transplantation. Circulation. 2000;101:2727–2733. doi: 10.1161/01.CIR.101.23.2727. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Walzer M, Wu YH, Zhou J, Dedhar S, Snider WD. Neurotrophins support regenerative axon assembly over CSPGs by an ECM-integrin-independent mechanism. J Cell Sci. 2006;119:2787–2796. doi: 10.1242/jcs.03016. [DOI] [PubMed] [Google Scholar]
- Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, Fishbein MC, Sharifi B, Chen PS. Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res. 2004;95:76–83. doi: 10.1161/01.RES.0000133678.22968.e3. [DOI] [PubMed] [Google Scholar]
- Zoubina EV, Smith PG. Axonal degeneration and regeneration in rat uterus during the estrous cycle. Auton Neurosci. 2000;84:176–185. doi: 10.1016/S1566-0702(00)00209-5. [DOI] [PubMed] [Google Scholar]