

Introduction
The dynamic interaction between the heart and the systemic circulation allows the cardiovascular system to be efficient in providing adequate cardiac output and arterial pressures necessary for sufficient organ perfusion [1]. The cardiovascular system provides adequate pressure and flow to the peripheral organs in different physiological (rest and exercise) and pathological conditions because of the continuous modulation of the arterial system compliance, stiffness and resistance with respect to left ventricular (LV) systolic performance [2]. Cardiac output is the final result of this dynamic modulation. Because LV stroke volume depends on myocardial contractility and loading conditions (preload and afterload), both cardiac and arterial dysfunction can lead to acute hemodynamic decompensation and shock. According to the underlying pathophysiological mechanisms, altered hemodynamic profiles can be classified as primarily reflecting cardiogenic, hypovolemic, obstructive or distributive shock.
Low cardiac output resulting in systemic hypoperfusion requires prompt and adequate treatment to restore cardio vascular function and prevent organ hypoperfusion. Current clinical guidelines recommend resuscitation with intravascular fluid infusions supplemented with selective use of inotropes and vasopressors to reverse shock in critically ill patients [3,4]. However, the treatment of acute he modynamic impairment should be tailored based on the etiological mechanism of the cardiovascular dysfunction. Herein lies a major problem with present guidelines: They cannot distinguish the underlying causes of impaired LV stroke volume leading to impaired cardiac output.
Ventriculo-arterial coupling
The concept that the cardiovascular system works better when the heart and the arterial system are coupled has been well demonstrated [5,6]. When the heart pumps blood into the vascular tree at a rate and volume that matches the capability of the arterial system to receive it, both cardiovascular performance and its associated cardiac energetics are optimal [7,8]. A contractility or arterial tone that is too high or too low decouples these processes and can lead to cardiac failure independent of myocardial ischemia or the toxic effects of sepsis and related systemic disease processes. This optimization means that the LV workload and the arterial system optimally match when the left ventricle ejects the blood into the arterial system and is quantified by ventriculo-arterial (V-A) coupling analysis. This process is optimized without excessive changes in LV pressure, and the mechanical energy of LV ejection is completely transferred from the ventricle to the arterial system [9,10].The role of V-A coupling in the management of critically ill patients with severe hemodynamic instability and shock is becoming increasingly clear.
V-A coupling can be defined as the ratio of the arterial elastance (Ea) to the ventricular elastance (Ees). This ratio was first proposed by Suga [11] as a method to evaluate the mechanical efficiency of the cardiovascular system and the interaction between cardiac performance and vascular function. The Ea/Ees ratio has been consistently demonstrated to be a reliable and effective measure of cardiovascular performance [9,11,12]. Burkhoff and Sagawa, and successively many other investigators, have demonstrated that when the Ea/Es is near unity, the efficiency of the system is optimal. In this case, the left ventricle provides an adequate stroke volume with the lowest possible energetic consumption [13,14]. Thus, V-A coupling is an effective index of the mechanical performance of the left ventricle and of the dynamic modulation of the cardiovascular system. In addition, the Ea/Ees reflects cardiac energetics. The balance between myocardial oxygen consumption and the mechanical energy required to perform this work appears to be optimal when the heart and the peripheral vascular system are coupled.
The area of the LV pressure-volume (P-V) loop (Figure 1) during a single cardiac cycle represents the total mechanical energy of the heart during that beat and has a linear correlation with myocardial oxygen consumption [15]. The energetic efficiency of the cardiovascular system is optimal when all the pulsating energy of the heart is transmitted to the arterial system [12]. Effective understanding of V-A coupling requires an appropriate understanding of the concepts of the determinants, Ea and Ees. And the effective use of these concepts requires an understanding as to how they may be derived at the bedside in critically ill patients.
Figure 1.

The left ventricular (LV) P-V loop. The P-V loop area is highlighted in blue. The slopes of LV elastance (Ees) and arterial elastance (Ea) are shown. ESV: end-systolic volume; ESP: end-systolic pressure; EDV: end-diastolic volume; EDP: end-systolic pressure.
Left ventricular elastance (Ees)
When the left ventricle ejects, arterial pressure increases and the LV stroke volume is transferred into the aorta. As LV volume decreases to its end-systolic minimum, the actual end-systolic volume (ESV) is a function of not only intrinsic cardiac contractility, but also arterial pressure. For the same LV end-diastolic volume (EDV) and intrinsic cardiac contractility, if arterial pressure at end-systole were less, then LVESV would also be lower and LV stroke volume greater. Similarly, increasing arterial pressure has the opposite effect to increasing LVESV and decreasing stroke volume. Importantly, the resultant potential endsystolic pressure-volume points possible as arterial pressure is independently varied describe a tight linear relationship called the end-systolic pressure-volume relationship (ESPVR). The slope of the ESPVR is the LV elastance (Ees) (Figure 1) and is determined for the intrinsic contractility of the heart. The steeper the slope, the greater the contractility [16]. Ees (mm Hg/ml) is a useful, load independent index of myocardial contractility and LV inotropic efficiency (end-systolic LV stiffness) [17]. Although myocardial contractility is the main determinant of LV systolic function, biochemical properties of the LV myocytes (stiffness, compliance, fibrosis, etc.), cardiac myocyte contraction synchrony and geometric remodeling of the LV chamber can also significantly influence LV performance [18]. Therefore, Ees should be considered as an integrated measure of LV systolic performance derived from the complex association between the inotropic efficiency and the functional, structural and geometric characteristics of the left ventricle [19].
Arterial elastance (Ea)
Ea (mm Hg/ml) is the expression of the total afterload imposed on the left ventricle and represents the complex association of different arterial properties including wall stiffness, compliance and outflow resistance. Thus, the total arterial compliance, impedance and the systolic and diastolic time interval are significant components of the arterial load [11]. Graphically, Ea can be described as the slope of the line running from the LVEDV to the LV end-systolic pressure (ESP) on the P-V loop (Figure 1). Operationally, Ea can be defined as the capability of the arterial vessels to increase pressure when LV stroke volume increases.
A-V coupling measurement
According to the seminal studies of Frank and Starling, Grossman and colleagues, and subsequently other investigators, LV contractile function can be evaluated by the relationship between the ESP and the ESV [20,21]. After the experimental investigations of Suga and Sugawa on isolated canine hearts, the assessment of V-A coupling in humans has been less successful because of the invasive techniques needed to acquire these measures. Because of the recognized importance of V-A coupling in the management of critically ill patients, much effort has been put into achieving non-invasive measurement approaches [22]. Despite several proposed non-invasive approaches to evaluate Ea/Ees, only the modified single beat method developed by Chen et al. has been validated against the invasive measurement of Ees [23]. The single beat method is based on the assumption that because the time-variation of LV elastance is not influenced by loading conditions or heart rate (HR), the evaluation of the P-V loop can be obtained by a single heart beat [24]. The non-invasive method developed by Chen et al. utilizes echocardiographic measures of LV end-diastolic and end-systolic areas. Chen et al. demonstrated that this method could be used for obtaining the single-beat measure of Ees (Ees[SB]), through the measure of LV ejection fraction (LVEF), stroke volume, pre-ejection time and systolic time interval (Figure 2) when coupled with systolic and diastolic arterial pressure measurements [23].
Figure 2.
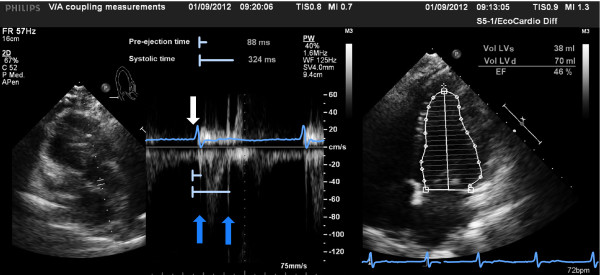
Echocardiographic measurements of left ventricular (LV) time intervals (left) and LV ejection fraction (EF, right). The white arrow indicates the beginning of the QRS complex, the dark blue arrows indicate the aortic valve opening and closure. The light blue lines show the time interval measurements.
Ea is calculated as ESP/stroke volume. From the rearrangement of this equation, Ea is proportional to the sum of HR, arterial resistance and [(ESP - mean arterial pressure[MAP])/stroke volume]. Since end-systolic arterial pressure occurs slightly after peak systolic pressure is achieved, Ea can be calculated as 0.9 × systolic arterial pressure/stroke volume, where 0.9 × systolic arterial pressure equals LVESP [23,25].
Since all these measures can be readily made at the bedside, it is possible to assess V-A coupling in critically ill patients.
Ventriculo-arterial decoupling in altered hemodynamic states
Although the occurrence of V-A decoupling is generally associated with pathological states, in some physiological conditions, such as during exercise or with increased age, a mismatch between LV performance and arterial system function can occur.
According to the physiological changes occurring during exercise, the cardiovascular system modulates its performance to provide adequate pressure and flow to peripheral organs in the presence of increased metabolic demand. Because the Ea/Ees is normally coupled in healthy humans at rest, during exercise Ees increases more than Ea; consequently, the ratio of Ea to Ees decreases [26]. Because LV performance and, therefore, Ees normally increases during exercise, Ea can increase, decrease or be unchanged. The Ea modifications impact Ea/Ees in conditions of increased LV work. Ea generally increases in elderly people as a consequence of the structural changes in the arterial properties (compliance, stiffness, pulse wave). The mismatch between Ea and Ees during exercise can be blunted in elderly people because of a reduction in the cardiovascular reserve. Furthermore, cardiovascular diseases (hypertension, coronary artery disease, diabetes mellitus), which have an impact on V-A coupling, frequently occur in elderly patients.
Cardiovascular impairment leading to acute systemic hypotension and shock can occur in many different clinical scenarios. Cardiovascular dysfunction is generally characterized by an altered Ea/Ees due to either the decrease in Ees or to the significant increase in Ea. Operationally, the system becomes uncoupled when the ratio of arterial to ventricular elastance is > 1 (Ea/Ees > 1), while an Ea increase is the consequence of an increase of the arterial load.
Ventriculo-arterial decoupling due to decreased Ees
According to the hemodynamic profile of the underlying pathology, an Ees decrease indicates an impairment in myocardial contractility, resulting in reduction of LV performance. Although myocardial ischemia and acute myocardial infarction are the most common causes of LV contractile dysfunction, other pathologies, such as myocarditis and severe cardiomyopathy (dilative and hypertrophic), can be characterized by myocardial depression and diminished Ees [27,28]. The acute decompensation of LV function can lead to severe hemodynamic impairment and shock.
Acute heart failure
Acute failure of LV systolic function is characterized by the inability of the heart to provide an adequate stroke volume to match the metabolic demands of the body. Acute systolic cardiac dysfunction results in both a low ejection fraction and increased filling pressures in the attempt to maintain a sufficient stroke volume. For this reason, the LV ESPVR is shifted downward and rightward, and Ees decreases. At the same time, Ea significantly increases because of increased sympathetic tone as the normal response of the autonomic nervous system to compensate for the reduced LV stroke volume. Thus, one sees tachycardia and increased arterial tone [2]. Hence, the cardiovascular system is generally uncoupled in acute heart failure, and Ea/Ees increases up to three or four fold [29]. Therefore, the treatment of acute heart failure is usually to decrease Ea by calcium channel blockers, beta-blockers and other vasodilating agents, whereas Ees is increased by inotrope infusions. Traditionally, the management of acute heart failure is based on inotropic agents aimed to improve myocardial contractility and to restore organ perfusion [30]. The inotropic agents commonly employed in the treatment of acute heart failure (catecholamines and phosphodiesterase III inhibitors) induce well-described adverse effects, such as increased myocardial oxygen consumption, arrhythmias and increased mortality [12]. In recent years, the therapeutic use of the novel calcium sensitizer, levosimendan, has been progressively emphasized because of its inotropic and vasodilator properties [31,32]. Because of its intrinsic performance, levosimendan impacts both Ea and Ees and improves V-A coupling [12]. Moderate systemic arterial hypotension can be induced by levosimendan, which then requires the administration of low doses of norepinephrine. The association between the inotropic and vasopressor action seems to improve cardiovascular performance by matching Ea and Ees.
Cardiac surgery
Cardiogenic shock is the most severe clinical manifestation of perioperative acute heart failure after cardiac surgery and leads to systemic hypoperfusion and organ dysfunction that generally requires either pharmacological or mechanical support. The acute reduction in Ees, from many different causes that range from inadequate myocardial protection to myocardial stunning, is usually the main reason for the uncoupled Ea/Ees ratio.
Treatment with inodilator drugs in post-cardiotomy acute heart failure has been demonstrated to be effective in restoring V-A coupling and improving LV performance [33,34].
Tachycardia
Tachycardia induced by stress conditions and during exercise is generally well tolerated by healthy people. Indeed, the increase in HR is a compensatory mechanism that enhances myocardial contractility and LV performance [2]. This force-frequency relationship has beneficial effects on cardiac performance, which contribute to maintain the efficiency of the cardiovascular system and provide optimal V-A coupling. The intrinsic mechanism underlying the force-frequency relationship is most likely the dynamic balance of the intracellular Ca2+ concentration [35]. In acute heart failure patients, baseline LV V-A uncoupling can be significantly worsened by tachycardia because the normal force-frequency relationship is impaired.
Acute systemic hypotension
Several different etiological mechanisms can cause hypotension, but systemic arterial hypotension is generally the first and often life-threatening symptom of acute cardiovascular dysfunction. Either heart failure or circulatory impairment can lead to systemic arterial hypotension and hemodynamic decompensation. Because peripheral organ perfusion strictly depends on the MAP, systemic arterial hypotension should be promptly reversed. According to recommended treatments, fluid resuscitation is generally the first therapeutic approach [3,4]. Indeed, the hemodynamic response to fluid administration depends on either the increase in stroke volume consequent to fluid resuscitation or on other factors, including arterial tone [1,36]. MAP is the product of the interaction between LV stroke volume and arterial system function expressed by the Ea. Recently, we proposed a bedside method to evaluate the dynamic arterial elastance (Eadyn) to determine which patients are responders to fluid resuscitation [37]. In mechanically ventilated patients, Eadyn corresponds to the ratio of pulse pressure variation (PPV) to stroke volume variation (SVV) during a single positive pressure breath. The bedside evaluation of the Eadyn should allow either functional assessment of arterial tone or of fluid responsiveness in severely hypotensive patients. Further investigations are required because of both the limitations of the published studies and the small populations enrolled, but the predictive value of Eadyn suggests a useful application in clinical practice.
Although MAP is the main determinant of organ perfusion, it can also be in fluenced by flow distribution and tissue oxygenation. Despite the restoration of a normal MAP, patients with cardiovascular dysfunction can suffer from decreased organ oxygenation and perfusion [38]. In this context, a sufficient MAP should be maintained in hemodynamically instable patients to prevent any further tissue in jury [3].
Ventriculo-arterial decoupling due to increased Ea
Acute systemic hypertension
Increased arterial pressure represents the most common cause of pressure overload of the left ventricle and plays a key role in the evolution of ventricular hypertrophy, dysfunction and failure. Hypertensive crisis is characterized by a sudden increase in blood pressure associated with or followed by signs of impairment in target organs, such as heart, kidney and brain. Most patients arriving at the emergency department with elevated blood pressure and acute pulmonary edema have a normal LVEF, suggesting that acute heart failure in these situations is due to diastolic dysfunction [39]. However, LV systolic dysfunction has also been reported in association with uncontrolled systemic hypertension, which may be responsible for the development of acute heart failure and pulmonary edema [40]. In these situations, the acute LV decompensation dramatically improves with rapid blood pressure normalization.
The mechanism by which a hypertensive crisis may cause acute and severe LV systolic dysfunction, even causing acute pulmonary edema, consists of acute V-A uncoupling because of an abrupt and exaggerated increase in Ea. Such afterload mismatch leads to an exhaustion of the preload reserve of the left ventricle [40]. The rapid reduction in elevated blood pressure by aggressive treatment usually induces a regression of myocardial dysfunction by restoring Ea and, therefore, a coupling of the Ea/Ees ratio. Thus, consideration of the pathophysiological role of V-A decoupling allows the true pathogenic mechanism of acute heart failure in a patient with normal LV systolic function to be determined.
Ventriculo-arterial decoupling in septic shock
The hemodynamic profile of septic shock is primarily characterized by generalized vasodilatation resulting in severe hypotension with systemic hypoperfusion [41]. The occurrence of myocardial depression and eventual cardiac dysfunction has been largely recognized in patients with septic shock, although the pathogenetic mechanisms of the myocardial injury are not yet completely understood [42,43].
In most of the patients with septic shock, cardiovascular efficiency is impaired, and the Ea/Ees becomes uncoupled (Ea/Ees > 1). The hemodynamic profile is characterized by both the significant increase in Ea and the decrease in Ees. Because the increase in Ea is generally induced by pharmacological vasoconstriction (norepinephrine) and the consequent increase in arterial tone, a decrease in Ees generally depends on the reduction in myocardial contractility. Whatever the underlying mechanism, when A-V uncoupling occurs in septic shock, the cardiac energetics are unfavorable and are often sacrificed to maintain tissue perfusion. Despite the severe hemodynamic dysfunction, Ea/Ees can be normally coupled in patients with septic shock (Ea/Ees = 1). In these cases, the proper interaction between LV performance and arterial system function can depend on both a normal cardiac function and on a proper therapeutic approach with fluid administration, inotropes and vasoconstrictors.
According to international guidelines, management of septic shock requires either fluid resuscitation or the employment of vasopressors (norepinephrine) to maintain an adequate cardiac output and tissue perfusion [4]. The use of inotropes is recommended when cardiac dysfunction and an increase in filling pressures occur. Recently, the use of levosimendan has been suggested for the treatment of myocardial dysfunction associated with septic shock [44,45].
Right ventricular dysfunction and V-A coupling
The cardiovascular system can be acutely decompensated by right ventricular (RV) failure with consequent severe hemodynamic instability and systemic hypoperfusion [46]. The RV dysfunction can be due to intrinsic mechanisms that occur in myocardial infarction or can be secondary to an increase in the RV afterload caused by pulmonary hypertension. Despite the underlying etiological mechanisms, RV uncoupling can occur [47].
Because the right ventricle contributes to the efficiency of the cardiovascular system through right V-A coupling, the assessment of the Ea/Ees of the right side of the circulation can play a role in the management of critically ill patients [48]. RV V-A coupling expresses the optimal interaction between RV performance and function of the pulmonary vascular system. When Ees decreases (RV acute myocardial infarction, septic shock) or Ea increases (pulmonary hypertension, acute respiratory distress syndrome [ARDS]), the right side of the cardiovascular apparatus becomes uncoupled.
Although evaluation of RV elastance requires invasive techniques with significant limitations for clinical practice, non-invasive models to assess the RV Ea/Ees have recently been proposed. Pulmonary artery catheterization and cardiac magnetic resonance may be useful in determining RV V-A coupling [48,49].
Conclusion
Although the management of shock patients has traditionally been based on advanced hemodynamic monitoring, the role of echocardiography in the evaluation of patients with acute hemodynamic decompensation has been progressively expanding in the last decades.
The management of critically ill patients with acutely altered hemodynamic states can benefit from the assessment of V-A coupling. Since the introduction of echocardiographic methods to assess cardiovascular function, the bedside evaluation of the Ea/Ees has become available and reliable.
The diagnostic function of echocardiographic assessment is enhanced by its capability to contribute to an evaluation of the Ea/Ees ratio and, therefore, V-A coupling in patients with acute hemodynamic impairment. The assessment of cardiovascular function by evaluation of the Ea/Ees ratio can offer an adjunctive perspective for understanding the pathophysiology of altered hemodynamic profiles, and for guiding therapeutic strategies and testing the effectiveness of treatments. However, this concept still needs to be tested in large trials.
Competing interests
The authors declare that they have no competing interests.
List of abbreviations used
ARDS: acute respiratory distress syndrome; Ea: arterial elastance; Eadyn: dynamic arterial elastance; Ees: ventricular elastance; ESP: end-systolic pressure; ESPVR: end-systolic pressure-volume relationship; ESV: end-systolic volume; HR: heart rate; LV: left ventricular; LVEF: left ventricular ejection fraction; MAP: mean arterial pressure; PPV: pulse pressure variation; P-V: pressure-volume; RV: right ventricular; SVV: stroke volume variation; V-A: ventriculo-arterial.
This article is one of ten reviews selected from the Annual Update in Intensive Care and Emergency Medicine 2013 and co-published as a series in Critical Care. Other articles in the series can be found online at http://ccforum.com/series/annualupdate2013. Further information about the Annual Update in Intensive Care and Emergency Medicine is available from http://www.springer.com/series/8901.
Contributor Information
Fabio Guarracino, Email: fabiodoc64@hotmail.com.
Rubia Baldassarri, Email: rubiabaldassarri@gmail.com.
Michael R Pinsky, Email: pinskymr@upmc.edu.
References
- Pinsky MR. Both perfusion pressure and flow are essential for adequate resuscitation. Sepsis. 2000;4:143–146. [Google Scholar]
- Chantler PD, Lakatta EG. Arterial-ventricular coupling with aging and disease. Front Physio. 2012;3:90. doi: 10.3389/fphys.2012.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli M, Levy M, Andrews PJ. Hemodynamic monitoring in shock and implications for management. International Consensus Conference. Intensive Care Med. 2007;33:575–590. doi: 10.1007/s00134-007-0531-4. [DOI] [PubMed] [Google Scholar]
- Dellinger RP, Levy MM, Carlet JM. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock. Intensive Care Med. 2008;34:17–60. doi: 10.1007/s00134-007-0934-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkley PF, Van Fossen DB, Nunziala E, Unverferth DV, Leier CV. Influence of positive inotropic therapy on pulsatile hydraulic load and ventricular-vascular coupling in congestive heart failure. J Am Coll Cardiol. 1990;15:1127–1135. doi: 10.1016/0735-1097(90)90253-L. [DOI] [PubMed] [Google Scholar]
- Elzinga G, Westerhof N. Matching between ventricle and arterial load. Circ Res. 1991;68:1495–1500. doi: 10.1161/01.RES.68.6.1495. [DOI] [PubMed] [Google Scholar]
- Kass DA, Kelly RP. Ventriculo-arterial coupling: concepts, assumptions, and applications. Ann Biomed Eng. 1992;20:41–62. doi: 10.1007/BF02368505. [DOI] [PubMed] [Google Scholar]
- Starling MR. Left ventricular-arterial coupling relations in the normal human heart. Am Heart J. 1993;125:1659–1666. doi: 10.1016/0002-8703(93)90756-Y. [DOI] [PubMed] [Google Scholar]
- Sunagawa K, Maughan WL, Burkhoff D, Sagawa K. Left ventricular interaction with arterial load studied in isolated canine ventricle. Am J Physiol. 1983;245:H773–H780. doi: 10.1152/ajpheart.1983.245.5.H773. [DOI] [PubMed] [Google Scholar]
- Kelly RP, Ting CT, Yang TM, Liu CP, Maughan WL, Chang MS, Kass DA. Effective arterial elastance as index of arterial vascular load in humans. Circulation. 1992;86:513–521. doi: 10.1161/01.CIR.86.2.513. [DOI] [PubMed] [Google Scholar]
- Suga H. Time course of left ventricular pressure-volume relationship under various end-diastolic volume. Jap Heart J. 1969;10:509–515. doi: 10.1536/ihj.10.509. [DOI] [PubMed] [Google Scholar]
- Guarracino F, Cariello C, Danella A, Doroni L, Lapolla F, Stefani M, Baldassarri R, Vullo C. Effect of levosimendan on ventriculo-arterial coupling in patients with ischemic cardiomyopathy. Acta Anaesthesiol Scand. 2007;51:1217–1224. doi: 10.1111/j.1399-6576.2007.01428.x. [DOI] [PubMed] [Google Scholar]
- Sunagawa K, Maughan WL, Sagawa K. Optimal arterial resistance for the maximal stroke work studied in isolated canine left ventricle. Circ Res. 1985;56:586–595. doi: 10.1161/01.RES.56.4.586. [DOI] [PubMed] [Google Scholar]
- Burkhoff D, Sagawa K. Ventricular efficiency predicted by an analytical model. Am J Physiol. 1986;250:1021–1027. doi: 10.1152/ajpregu.1986.250.6.R1021. [DOI] [PubMed] [Google Scholar]
- Hayash K, Shigemi K, Shishido T, Sugimachr M, Sunagawa K. Single-beat Estimation of Ventricular End-systolic Elastance-effective Arterial Elastance as an Index of Ventricular Mechanoenergetic Performance. Anesthesiology. 2000;92:1769–1776. doi: 10.1097/00000542-200006000-00037. [DOI] [PubMed] [Google Scholar]
- Blaudszun G, Morel DR. Relevance of the volume-axis intercept, V0, compared with the slope of end-systolic pressure-volume relationship in response to large variations in inotropy and afterload in rats. Exp Physiol. 2011;96:1179–1195. doi: 10.1113/expphysiol.2011.059881. [DOI] [PubMed] [Google Scholar]
- Sagawa K, Suga H, Shoukas AA, Bakalar KM. End-systolic pressure/volume ratio: a new index of ventricular contractility. Am J Cardiol. 1997;40:748–753. doi: 10.1016/0002-9149(77)90192-8. [DOI] [PubMed] [Google Scholar]
- Borlaug BA, Kass DA. Ventricular-vascular interaction in heart failure. Heart Fail Clin. 2008;4:23–36. doi: 10.1016/j.hfc.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass DA. Age-related changes in ventricular-arterial coupling: pathophysiologic implications. Heart Fail Rev. 2002;7:51–62. doi: 10.1023/A:1013749806227. [DOI] [PubMed] [Google Scholar]
- Grossman W, Braunwald E, Mann T, McLaurin LP, Green LH. Contractile state of the left ventricle in man as evaluated from end-systolic pressure volume relations. Circulation. 1977;56:845–852. doi: 10.1161/01.CIR.56.5.845. [DOI] [PubMed] [Google Scholar]
- Sagawa K. The end-systolic pressure-volume relation of the ventricle: definition modifications and clinical use. Circulation. 1981;63:1223–1227. doi: 10.1161/01.CIR.63.6.1223. [DOI] [PubMed] [Google Scholar]
- Senzaki H, Chen CH, Kass DA. Single-beat estimation of end-systolic pressure-volume relation in humans: a new method with the potential for noninvasive application. Circulation. 1996;94:2497–2506. doi: 10.1161/01.CIR.94.10.2497. [DOI] [PubMed] [Google Scholar]
- Chen CH, Fetics B, Nevo E, Rochitte CE, Chiou KR, Ding PA, Kawaguchi M, Kass DA. Noninvasive single-beat determination of left ventricular end-systolic elastance in humans. J Am Coll Cardiol. 2001;38:2028–2034. doi: 10.1016/S0735-1097(01)01651-5. [DOI] [PubMed] [Google Scholar]
- Shishido T, Hayashi K, Shigemi K, Sato T, Sugimachi M, Sunagawa K. Singlebeat estimation of end-systolic elastance using bilinearly approximated time-varying elastance curve. Circulation. 2000;102:1983–1989. doi: 10.1161/01.CIR.102.16.1983. [DOI] [PubMed] [Google Scholar]
- Zanon F, Aggio S, Baracca E, Pastore G, Corbucci G, Boaretto G, Braggion G, Piergenili C, Rigatelli G, Roncon L. Ventricular-arterial coupling in patients with heart failure treated with cardiac resynchronization therapy: may we predict the long-term clinical response? Eur J Echocardiogr. 2009;10:106–111. doi: 10.1093/ejechocard/jen184. [DOI] [PubMed] [Google Scholar]
- Najjar SS, Schulman SP, Gerstenblith G, Fleg JL, Kass DA, O'Connor F, Becker LC, Lakatta EG. Age and gender affect ventricular-vascular coupling during aerobic exercise. J Am Coll Cardiol. 2004;44:611–617. doi: 10.1016/j.jacc.2004.04.041. [DOI] [PubMed] [Google Scholar]
- Widyastuti Y, Stenseth R, Berg KS, Pleym H, Wahba A, Videm V. Preoperative and intraoperative prediction of risk of cardiac dysfunction following open heart surgery. Eur J Anaesthesiol. 2012;29:143–151. doi: 10.1097/EJA.0b013e32834de368. [DOI] [PubMed] [Google Scholar]
- Buja LM. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol. 2005;14:170–175. doi: 10.1016/j.carpath.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Fox JM, Maurer MS. Ventriculo-vascular coupling in systolic and diastolic heart failure. Curr Heart Fail Rep. 2005;2:204–211. doi: 10.1007/BF02696651. [DOI] [PubMed] [Google Scholar]
- Majure DT, Teerlink JR. Update on the management of acute decompensated heart failure. Curr Treat Options Cardiovasc Med. 2011;13:570–585. doi: 10.1007/s11936-011-0149-2. [DOI] [PubMed] [Google Scholar]
- Toller WG, Stranz C, Nieminen MS. Levosimendan, a new inotropic and vasodilator agent. Anaesthesiology. 2006;104:556–569. doi: 10.1097/00000542-200603000-00024. [DOI] [PubMed] [Google Scholar]
- Landoni G, Mizzi A, Biondi-Zoccai G, Bignami E, Prati P, Ajello V, Marino G, Guarracino F, Zangrillo A. Levosimendan reduces mortality in critically ill patients. A meta-analysis of randomized controlled studies. Minerva Anestesiol 2. 010;76:276–286. [PubMed] [Google Scholar]
- De Santis V, Vitale D, Tritapepe L. Levosimendan and cardiac surgery. J Cardiothorac Vasc Anesth. 2010;24:210. doi: 10.1053/j.jvca.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Toller W, Algotsson L, Guarracino F. Perioperative use of levosimendan: Best practice in operative settings. J Cardiothorac Vasc Anesth. in press . [DOI] [PubMed]
- Masutani S, Cheng HJ, Tachibana H, Little WC, Cheng CP. Levosimendan restores the positive force-frequency relation in heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H488–H496. doi: 10.1152/ajpheart.01116.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monge García MI, Gil Cano A, Gracia Romero M. Dynamic arterial elastance to predict arterial pressure response to volume loading in preload-dependent patients. Crit Care. 2011;15:R15. doi: 10.1186/cc9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinsky MR. In: Yearbook of Intensive Care and Emergency Medicine. Vincent JL, editor. Springer, Heidelberg; 2002. Functional hemodynamic monitoring: applied physiology at the bedside; pp. 534–551. [Google Scholar]
- Lamia B, Chemla D, Richard C, Teboul JL. Clinical review: interpretation of arterial pressure wave in shock states. Crit Care. 2005;9:601–606. doi: 10.1186/cc3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little WC. Hypertensive pulmonary oedema is due to diastolic dysfunction. Eur Heart J. 2001;22:1961–1964. doi: 10.1053/euhj.2001.2665. [DOI] [PubMed] [Google Scholar]
- Ross J. On variations in the cardiac hypertrophic response to pressure overload. Circulation. 1997;95:1349–1351. doi: 10.1161/01.CIR.95.6.1349. [DOI] [PubMed] [Google Scholar]
- Vieillard-Baron A. Septic cardiomyopathy. Ann Intensive Care. 2011;1:6. doi: 10.1186/2110-5820-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD, Doddi M. Sepsis and the heart. Br J Anaesth. 2010;104:3–11. doi: 10.1093/bja/aep339. [DOI] [PubMed] [Google Scholar]
- Young JD. The heart and circulation in severe sepsis. Br J Anaesth. 2004;93:114–120. doi: 10.1093/bja/aeh171. [DOI] [PubMed] [Google Scholar]
- De BD, Taccone FS, Radermacher P. Levosimendan in septic shock: another piece in the puzzle, but many pieces are still lacking. Intensive Care Med. 2007;33:403–405. doi: 10.1007/s00134-006-0520-z. [DOI] [PubMed] [Google Scholar]
- Pinto BB, Rehberg S, Ertmer C, Westphal M. Role of levosimendan in sepsis and septic shock. Curr Opin Anaesthesiol. 2008;21:168–177. doi: 10.1097/ACO.0b013e3282f43c56. [DOI] [PubMed] [Google Scholar]
- Price LC, Wort SJ, Finney SJ, Marino PS, Brett SJ. Pulmonary vascular and right ventricular dysfunction in adult critical care: current and emerging options for management: a systematic literature review. Crit Care. 2010;14:R169. doi: 10.1186/cc9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevin LG, Barnard M. Right ventricular failure. Contin Educ Anaesth Crit Care Pain. 2007;7:89–94. doi: 10.1093/bjaceaccp/mkm016. [DOI] [Google Scholar]
- Grignola JC, Ginés F, Bia D, Armentano R. Improved right ventricular-vascular coupling during active pulmonary hypertension. Int J Cardiol. 2007;115:171–182. doi: 10.1016/j.ijcard.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Sanz J, García-Alvarez A, Fernández-Friera L, Nair A, Mirelis JG, Sawit ST, Pinney S, Fuster V. Right ventriculo-arterial coupling in pulmonary hypertension: a magnetic resonance study. Heart. 2012;98:238–243. doi: 10.1136/heartjnl-2011-300462. [DOI] [PubMed] [Google Scholar]