

Abstract
Genomic structural alterations that vary within species, known as large copy number variants, represent an unanticipated and abundant source of genetic diversity that associates with variation in gene expression and susceptibility to disease. Even short insertions and deletions (indels) can exert important effects on genomes by locally increasing the mutation rate, with multiple mechanisms proposed to account for this pattern. To better understand how indels promote genome evolution, we demonstrate that the single nucleotide mutation rate is elevated in the vicinity of indels, with a resolution of tens of base pairs, for the two closely related nematode species Caenorhabditis remanei and C. sp. 23. In addition to indels being clustered with single nucleotide polymorphisms and fixed differences, we also show that transversion mutations are enriched in sequences that flank indels and that many indels associate with sequence repeats. These observations are compatible with a model that reconciles previously proposed mechanisms of indel-associated mutagenesis, implicating repeat sequences as a common driver of indel errors, which then recruit error-prone polymerases during DNA repair, resulting in a locally elevated single nucleotide mutation rate. The striking influence of indel variants on the molecular evolution of flanking sequences strengthens the emerging general view that mutations can induce further mutations.
Keywords: Caenorhabditis, indel, error-prone polymerase, mutation rate, nucleotide diversity
Introduction
Duplication of genetic material has long been recognized as an important driver of phenotypic diversification (Ohno 1970), including during primate evolution (Stankiewicz et al. 2004; Jiang et al. 2007), but only recently have we begun to appreciate the extent to which structural variation occurs in genomes. The discovery of abundant structural variants dramatically changed our view of the sources of genome variation and emphasizes the highly dynamic nature of genome evolution (Iafrate et al. 2004; Sebat et al. 2004; McCarroll et al. 2006; Redon et al. 2006). Such structural variants are traditionally termed copy number variants (CNVs) when larger than 1 kb, and include deletions, insertions, duplications, and inversions that can extend several megabases (Mb) in length. CNVs provide a major class of genomic variants found in the genomes of many organisms (Cutler et al. 2007; Dopman and Hartl 2007; Emerson et al. 2008; Guryev et al. 2008; Fadista et al. 2010; Maydan et al. 2010; Gazave et al. 2011). Approximately 12% of the human genome resides in CNVs (Redon et al. 2006) and as much as 5% of protein-coding genes show structural polymorphism in the nematode Caenorhabditis elegans (Maydan et al. 2010). Consequently, CNVs represent an important cause of phenotypic diversity, in particular affecting gene expression variation (Dopman and Hartl 2007; Stranger et al. 2007; Guryev et al. 2008) and disease susceptibility (Carvalho et al. 2010; Stankiewicz and Lupski 2010).
In addition to large CNVs, genomes harbor a plethora of small insertions and deletions (indels) that contribute substantially to genome sequence divergence and that represent a major constituent of the known heritable human disease burden (Stenson et al. 2003). For instance, perceptions about sequence identity between human and chimpanzee changed drastically upon the discovery that indels contribute more to divergence than do single nucleotide substitutions (Britten 2002). Indeed, indels are far more responsible than nucleotide changes for unmatched sites between closely related genomes (Britten et al. 2003). Indels are also frequent in protein-coding sequences (Wetterbom et al. 2006; Chen et al. 2007) and can alter protein structure (Zhang et al. 2010). One mechanism for the origin of insertion and deletion errors is strand slippage during replication of repetitive sequences. Primer and template strands of DNA can transiently dissociate and form intermediate misalignments that are stabilized by the pairing of the repeat sequences when they re-associate (Kunkel and Bebenek 2000). The length of the repeats also affects the rate of indel formation. DNA polymerases have a proofreading activity resulting from the balance between the rates of primer extension and excision at the primer terminus. When the mismatch is located far from the polymerase active site, because of longer repeat sequences, it does not compromise the rate of polymerization, which results in less efficient proofreading activity (Kunkel 2009).
A more subtle effect of the presence of indels, but with important consequences for genome evolution, is a local elevation of the single nucleotide mutation rate in the regions surrounding indels (Tian et al. 2008). Tian et al. demonstrated that the number of nucleotide substitutions decreases as a function of the distance from indels in the genomes of primate, rodent, rice, fruit fly, and yeast. A recent analysis of nucleotide polymorphism in Drosophila melanogaster revealed that nucleotide changes within populations are more abundant near indels and other variants, although the authors’ interpretation of the data differs (Massouras et al. 2012). Indels have also been found to affect the rate of nucleotide substitutions in bacteria (Zhu et al. 2009; McDonald et al. 2011) and the amount of within-species polymorphism in plants (Hollister et al. 2010). Remarkably, indels influence nucleotide substitution patterns across different time-scales, as evident by their effect on human and chimpanzee divergence, diversity within human populations, and polymorphism between cancer and normal somatic cells of the same individual (De and Babu 2010). Three distinct, but nonmutually exclusive, mechanisms have been proposed to explain the increase of the mutation rate in the vicinity of indels, including a mutagenic effect of heterozygous indels (Tian et al. 2008), the recruitment of low-fidelity polymerases during DNA repair (De and Babu 2010), and low-fidelity polymerase recruitment following polymerase-stalling induced by repeat motifs (McDonald et al. 2011).
Here, we investigate the role of indels on nucleotide variation in nematodes, using dense polymorphism and divergence data from two closely related species of Caenorhabditis nematodes that each harbor very high levels of polymorphism (Cutter et al. 2006; Jovelin et al. 2009; Dey et al. 2012). We demonstrate that indel and single nucleotide variants cluster together and that single nucleotide variation is increased with closer proximity to indels. A large proportion of indels have associated nearby short sequence repeats. Moreover, nucleotide substitutions close to indels are biased toward transversions, a signature of error-prone polymerases. However, we do not find evidence that repeats alone increase the single nucleotide mutation rate. These results are compatible with the combined effects of previously proposed mechanisms of indel-associated mutation (De and Babu 2010; McDonald et al. 2011), suggesting that repeat sequences encourage indel formation, with subsequent recruitment of error-prone polymerases that incidentally create single nucleotide mutations during DNA repair.
Materials and Methods
We investigated the effect of indels on single base-pair mutations using a polymorphism data set that targeted nucleotide variation around all known miRNA genes in the C. remanei genome (Jovelin R, Cutter AD, unpublished data). Polymorphism data were collected using Sanger sequencing of both DNA strands. We controlled for data quality and the potential for sequencing errors in the following two ways: 1) Primers were designed such that forward and reverse sequences strongly overlap, resulting in all loci being sequenced on both strands, and 2) all single nucleotide polymorphisms (SNPs) were verified by visual inspection of the sequence chromatograms, increasing confidence for the discovered polymorphisms. Sequences were deposited in GenBank under accession numbers KC867968-KC869320. For this study, we masked out the miRNA sequences themselves, including the entire pre-miRNA fold because of possible biases introduced by the strong purifying selection operating directly on these regulatory RNA genes. The final data set includes 217 sequence fragments, approximately 180-bp long on average, sequenced in 8 to 20 strains of C. remanei (n < 10 for six fragments, median n = 11), as well as 103 orthologous fragments sequenced in two strains of the closely related species, C. sp. 23. A total of 130 orthologous fragments were used to investigate nucleotide divergence between species, using C. remanei strain PB4641 (reference genome) and C. sp. 23 strain VX0082 (or VX0087 if amplification failed for strain VX0082). For each fragment, alleles were manually aligned using BioEdit (Hall 1999), and nucleotide diversity (Nei 1987) was quantified using DnaSp v5.10 (Librado and Rozas 2009). Interspecies divergence was measured with a Jukes–Cantor distance in MEGA 5 (Tamura et al. 2011), or with DnaSP for the sliding window analysis. We also generated automated multiple sequence alignments using CLUSTAL W with default parameters (Thompson et al. 1994) to test for consistency with results from our manually curated alignments.
To investigate the effect of indels on nucleotide variation, we analyzed nonoverlapping windows of 10-bp width starting at position −1 or +1 relative to the indel (supplementary fig. S1, Supplementary Material online). When multiple indels were present, half of the indel-bound region was ascribed to each flanking indel to avoid double-counting of polymorphisms. Because in most cases the length of the DNA fragment was not a multiple of 10, we retained the last window only if it contained ≥7 nucleotides. Windows less than 10 bp immediately adjacent to an indel were also excluded (e.g., an indel-bound region shorter than 20 bp). Single nucleotide differences were then averaged across all windows of a given distance from an indel to assess nucleotide polymorphism and divergence as a function of distance from indels. Window distances with a sample size less than 20 loci were discarded.
To control for differential selective constraints on DNA fragments, we used C. sp. 23 as an outgroup to determine the derived and ancestral state of indel mutations in C. remanei. We then computed Di, the amount of single nucleotide divergence between the outgroup and a C. remanei strain carrying the derived indel mutation, and Dni, the amount of single nucleotide divergence between the outgroup and a strain without the indel. If indels locally increase the mutation rate, then more substitutions are expected to accumulate on the lineage harboring the derived indel mutation (Tian et al. 2008; Zhu et al. 2009; McDonald et al. 2011). We then plotted Di and Dni as a function of the distance from the indel using nonoverlapping windows, as described earlier. For simplicity, we restricted this analysis to fragments containing only a single indel in C. remanei. The background level of divergence, Db, is the average level of divergence across all windows.
We also investigated selective constraints by comparing the excess of derived low-frequency variants between indel-containing fragments and nonindel fragments relative to a neutral reference. The excess of derived low-frequency variants is E (%) = [100 × (fr – fn)]/fn where fr is the fraction of variants in the region of interest (with-indel or no-indel fragments) that have a derived allele frequency (DAF) below a given cut-off, and fn is the fraction of variants in the neutral reference with the same DAF cut-off (Mu et al. 2011). As a neutral reference we used derived polymorphisms at synonymous sites from 20 protein-coding genes (Dey et al. 2012). We applied five DAF cut-off values from 0.1 to 0.3 in increments of 0.05.
We followed the scheme of McDonald et al. (2011) to investigate the sequence context around indels. In particular, we identified homopolymeric repeats of four nucleotides or longer (maximum observed 8 bp long within C. remanei) and designated indels as “contiguous” to a repeat if the indel was immediately adjacent to it, or if it occurred inside the repeat. Multi-nucleotide repeat motifs were too rare in this data set to consider in addition to the mononucleotide repeats. Indels were designated as ‘proximal’ if they occurred within 5 bp of a repeat sequence. Repeats interrupted by a mutation in one allele that would result in conserved homopolymers shorter than four nucleotides were not counted in the repeat analysis.
Results and Discussion
Indels Contribute Significantly to Sequence Divergence
Caenorhabditis nematodes provide an increasingly valuable model in evolutionary genetics but, until recently, analyses of molecular evolution have been limited by the extreme sequence divergence between known species. Here, we investigate the effects of indels on genome evolution by taking advantage of both the high nucleotide polymorphism within species and the modest divergence of C. remanei to the recently discovered close relative C. sp. 23 (Graustein et al. 2002; Cutter et al. 2006; Jovelin et al. 2009; Dey et al. 2012). Using a population genetic data set of 217 sequence fragments from C. remanei and 130 orthologous fragments from C. sp. 23, we identified 2,033 SNPs and 292 indels across the 39.5 kb of sequence in C. remanei. We also identified 268 SNPs and 35 indels in the 18.7 kb of sequence from C. sp. 23 (table 1). The ratio of indels to SNPs (I/S) is much less than 1 in both C. remanei and C. sp. 23, similar to the ratio previously reported on a genome-wide scale between two strains of C. elegans (Wicks et al. 2001) (table 2). In stark contrast, direct detection of new mutations in C. elegans from sequencing of mutation accumulation lines show that new indel mutations occur more frequently than single nucleotide changes (Denver et al. 2004), indicating that selection disproportionately removes indels from populations (Chen et al. 2009). However, comparisons of the number of unmatched nucleotides due to indels with those due to nucleotide changes showed that indels dominate sequence divergence among closely related organisms (Britten et al. 2003). Although our estimates of the ratio of unpaired nucleotides from indels to nucleotide changes are less than 1 and are smaller than estimates reported for other species, they nevertheless implicate a significant contribution of indels to sequence divergence in Caenorhabditis and particularly within C. remanei (table 2).
Table 1.
Summary of Indel and Nucleotide Diversity Identified in Our Data Set
| Scale of Divergence | DNA Fragments | N | L (bp) | Average L (bp) | I (per bp) | R | S | D (%) |
|---|---|---|---|---|---|---|---|---|
| Within C. remanei | All | 217 | 39,502 | 182.04 | 292 (0.007) | 768 | 2,033 | 1.931 |
| No indel | 92 | 13,688 | 148.78 | 0 (0) | 265 | 482 | 1.418 | |
| With indel | 125 | 25,814 | 206.51 | 292 (0.011) | 503 | 1,551 | 2.309 | |
| Within C. sp. 23 | All | 103 | 18,674 | 181.30 | 35 (0.002) | 427 | 268 | 1.427 |
| No indel | 80 | 14,241 | 178.01 | 0 (0) | 305 | 123 | 0.812 | |
| With indel | 23 | 4,433 | 192.74 | 35 (0.008) | 122 | 145 | 3.604 | |
| C. remanei vs. C. sp. 23 | All | 130 | 24,008 | 184.68 | 252 (0.010) | 432 | 1,703 | 8.341 |
| No indel | 32 | 5,120 | 160 | 0 (0) | 78 | 208 | 4.265 | |
| With indel | 98 | 18,888 | 192.73 | 252 (0.013) | 354 | 1,495 | 9.672 |
Note.—N, sample size; L, length; I, number of indels; R, number of homopolymeric repeats of 4 bp or longer; S, number of polymorphic sites for within species variation and number of substitutions for between species divergence; D, nucleotide divergence, measured by the index of nucleotide diversity π for within species variation and by the number of substitutions per site K for between species variation.
Table 2.
Summary of the Ratios of Indel and SNP (or Substitution) Counts and the Ratios of Unmatched Nucleotides (Ru) Attributable to Indels and to Those Attributable to SNPs (or Substitutions)
| Scale of Divergence | I/S | Ru |
|---|---|---|
| Within Caenorhabditis remanei | 0.144 | 0.971 |
| Within C. sp. 23 | 0.131 | 0.317 |
| Within C. elegansa | 0.332 | 0.548 |
| C. remanei vs. C. sp. 23 | 0.148 | 0.654 |
aFrom Britten et al. (2003).
Structural Alterations and Single Nucleotide Variants Cluster Together
For this sample of the C. remanei genome, indels and SNPs co-occur nonrandomly at the scales of both variation within species and divergence between species. The presence of at least one indel significantly increases the amount of SNP by 1.6-fold (C. remanei) to 4.4-fold (C. sp. 23), and increases the observed sequence divergence between the two species by a factor of 2.3 (fig. 1A). Remarkably, single nucleotide differences also correlate positively with the number of indels per DNA fragment, again regardless of whether the scale of comparison is between individuals of the same species or between different species (C. remanei: Spearman’s ρ = 0.398, P < 0.0001; C. sp. 23: Spearman’s ρ = 0.465, P < 0.0001; between species: Spearman’s ρ = 0.431, P < 0.0001) (fig. 1B).
Fig. 1.—

(A) Higher nucleotide diversity is associated with the presence of indels at different time scales, within and between species. NI, no indel; I, indel. Means are represented ± 1 SEM (standard error of the mean). ***Wilcoxon two-sample test P < 0.0001. (B) Nucleotide variation increases with the number of indels per DNA fragment. Comparison of mean nucleotide polymorphism and divergence in DNA fragments having different number of indels. Error bars represent ± 1 SEM.
We tested the robustness of the results from our curated alignment procedure by comparing them to an analysis of automated multiple sequence alignments, and found the same association between indels and nucleotide variation (supplementary fig. S2, Supplementary Material online). We also found little quantitative difference between estimates of nucleotide variation for indel-containing DNA fragments that were either manually or automatically aligned (C. remanei: Spearman’s ρ = 0.980; C. sp. 23: ρ = 0.999; between species: ρ = 0.922) and for all DNA fragments (C. remanei: Spearman’s ρ = 0.990; C. sp. 23: ρ = 0.999; between species: ρ = 0.945), indicating that the two procedures generate very similar multiple sequence alignments. Because our results are not sensitive to alternative alignment procedures, we used our manually curated alignments for further analysis. Moreover, these results for Caenorhabditis are entirely consistent with the clustering of indel and SNP mutations observed in prokaryotes and other eukaryotes (Hardison et al. 2003; Longman-Jacobsen et al. 2003; Wetterbom et al. 2006; Tian et al. 2008; Zhu et al. 2009; De and Babu 2010; Hollister et al. 2010; McDonald et al. 2011).
Single Nucleotide Variation Increases near Indels
Recently, Tian et al. determined that the single nucleotide mutation rate is higher in the vicinity of indels in the genomes of primate, rodent, rice, fruit fly, and yeast (Tian et al. 2008). To test for such an effect in Caenorhabditis, we quantified single nucleotide differences in nonoverlapping 10-bp windows for each indel-containing DNA fragment and analyzed the change in nucleotide polymorphism and divergence as a function of the distance from the nearest indel (supplementary fig. S1, Supplementary Material online). We restricted analysis to nucleotide variation within C. remanei and to divergence between species, owing to the few indels detected within the smaller C. sp. 23 population sample (table 1). We found that the 10-bp window immediately adjacent to the indel has the greatest nucleotide polymorphism in C. remanei, and that nucleotide polymorphism declines as a function of the distance from the indel (fig. 2A). This proximity to an indel exerts its influence over the long term, as well, with nucleotide divergence between C. remanei and C. sp. 23 also declining with distance from the indel (fig. 2B).
Fig. 2.—
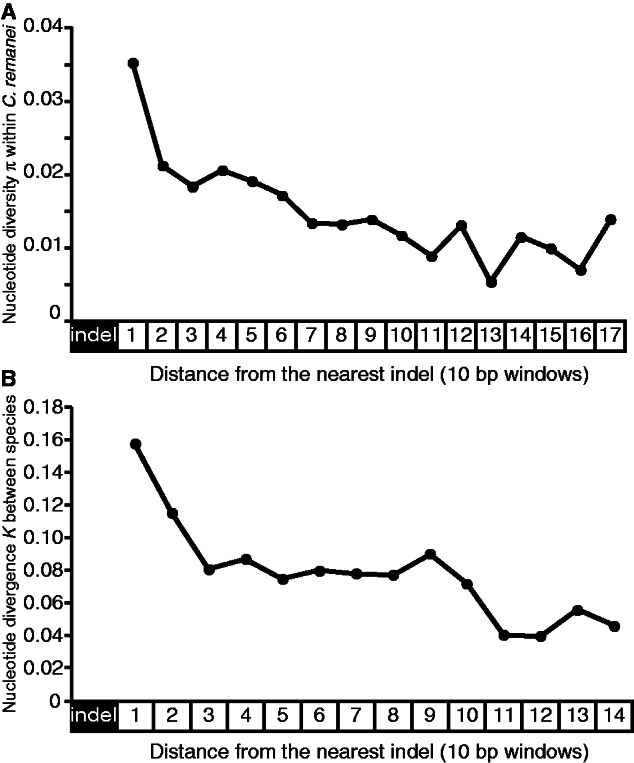
(A) Nucleotide diversity within Caenorhabditis remanei decreases as a function of the distance from the nearest indel. (B) Nucleotide divergence between C. remanei and C. sp. 23 decreases as the distance from the nearest indel increases. Each point in (A) and (B) represents the average nucleotide diversity in nonoverlapping windows of 10 bp equally distant from their nearest indel.
However, these patterns of greater SNP and divergence near indels could result either from a direct effect of indels on the mutation rate or from lower selective constraints permitting the accumulation of both indels and SNPs. The data analyzed for this study were originally collected for a survey of sequence variation at miRNA loci. Although here we only analyzed regions flanking the miRNAs, owing to strong purifying selection on the miRNA sequences themselves, regions downstream of the miRNAs tend to have higher nucleotide variation than upstream flanking regions (Jovelin R, Cutter AD, unpublished data). Nevertheless, this cannot explain the association between indels and SNPs because downstream fragments are not enriched for indels (χ2 test: C. remanei: P = 0.23; C. sp. 23: P = 0.51; between species: P = 0.54) and because the nonrandom distribution of indels and SNPs is observed both in upstream and downstream sequence fragments (supplementary fig. S3, Supplementary Material online). We therefore further investigated differential selective constraints among our collection of DNA fragments irrespective of their position relative to the miRNAs.
Purifying selection is expected to skew the site frequency spectrum toward rare variants (Fay et al. 2001). However, we do not find a significant difference in the Tajima’s D summary of the site frequency spectrum (Tajima 1989) between those fragments containing at least one indel and those fragments lacking indels (mean Tajima’s D: D(nonindel) = −0.0586, D(indel) = −0.1601; Wilcoxon two-sample P = 0.63). To further explore the intensity of purifying selection using allele frequencies, we compared the excess of derived low-frequency variants between indel-containing fragments and nonindel fragments relative to a neutral reference (Mu et al. 2011), using polymorphisms within C. remanei polarized with C. sp. 23 as the outgroup. However, nonindel fragments do not harbor significantly more low-frequency variants, regardless of which DAF cut-off value we chose, suggesting that nonindel fragments and indel-containing fragments experience similar levels of purifying selection (fig. 3A).
Fig. 3.—
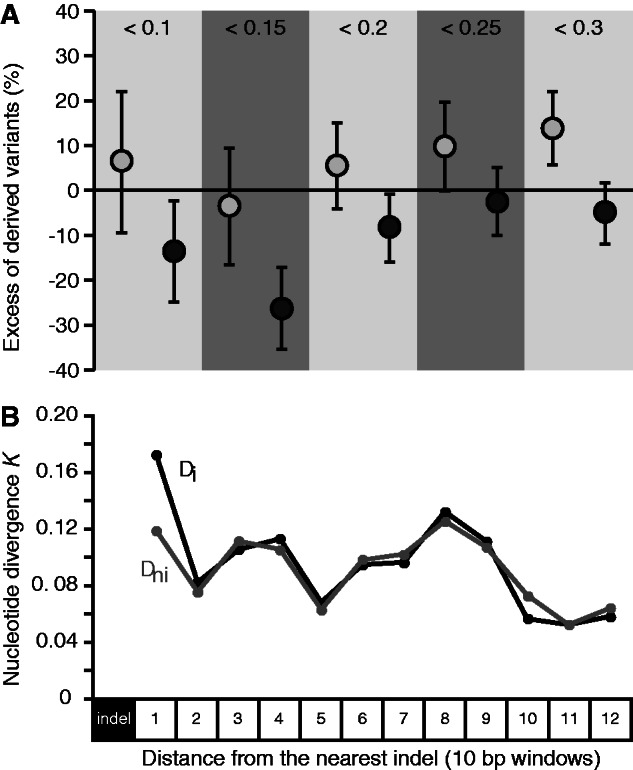
(A) Comparison of the mean excess of derived variants relative to a neutral reference at different frequency cut-offs between indel-containing fragments (dark gray) and nonindel fragments (light gray). DNA fragments without indels do not have significantly more derived variants than fragments with indels, suggesting that selective constraints operating on the two types of fragments are similar and cannot solely explain the increased nucleotide variation in indel-containing fragments. Error bars represent the standard error of the mean. (B) Comparison of nucleotide divergence between indel-containing alleles (black) and nonindel alleles (gray). Nucleotide divergence between Caenorhabditis sp. 23 and the C. remanei strain carrying the indels is higher immediately next to the indel suggesting that indels increase the local mutation rate.
Another way to control for the potential effect of selective constraints is to compare the number of substitutions that are specific to the lineages with and without indels. A higher number of substitutions is expected in the lineage carrying the derived indel allele if indels result in increased nucleotide mutations, and rate differences between the lineages with and without indels cannot be attributed to differences in selective constraints because the regions compared are strictly orthologous (Tian et al. 2008; Zhu et al. 2009; McDonald et al. 2011). We again used C. sp. 23 as the outgroup to determine the derived and ancestral state of indel mutations in C. remanei. We then computed nucleotide divergence between C. sp. 23 and a C. remanei strain containing an indel mutation (Di), and between C. sp. 23 and a C. remanei strain lacking the indel mutation (Dni). Plotting the level of divergence as a function of the distance from the indel, we found that, as expected if indels increase the mutation rate, Di is nominally higher than Dni in the window immediately adjacent to the indel (fig. 3B). Although the difference between Di and Dni is not significant in window 1 (Wilcoxon two-sample P = 0.557), Di in this window is significantly higher than the background divergence (Db) whereas Dni is not (Di vs. Db: Wilcoxon two-sample P = 0.041; Dni vs. Db: P = Wilcoxon two-sample P = 0.199).
A mutagenic effect of indels predicts that the frequencies of nucleotide polymorphisms linked to derived indels would reflect the age of the indel allele. Derived indel variants at high frequency are likely to be older than low-frequency indels. Consequently, high frequency derived indels should occur on haplotypes with nucleotide polymorphisms at a broad range of frequencies, reflecting mutation over its history, and low frequency derived indels should occur on haplotypes with only low frequency SNPs, thus producing a positive correlation between indel and linked SNP frequencies (in contrast to the hypothesis of Massouras et al. 2012). Supporting this prediction, we found a significant positive correlation between the frequencies of derived indels and derived SNPs in C. remanei (Spearman’s ρ = 0.23, P = 5.5 × 10−9). When examined as a function of the distance from indels, we find this positive correlation in the two windows immediately flanking an indel, but the correlation becomes weak and not significant at the most distant windows from the indel (not shown). Altogether, these results are inconsistent with differential selective constraints among DNA regions having caused the clustering between indels and SNPs in Caenorhabditis. Instead, they implicate a higher mutation rate associated with the presence of the indel.
Regional Sequence Context and Indel-Associated Mutagenesis
It was first suggested that indels might be mutagenic because indel heterozygotes could affect chromosomal pairing during meiosis, resulting in synthesis errors associated with DNA repair (Tian et al. 2008). Consistent with this hypothesis, the effect of indel-associated mutagenesis varies with the mating system of plant species and is lower in self-fertilizing species that have reduced indel heterozygosity (Hollister et al. 2010). However, the mechanism responsible for the association between indels and nucleotide variants remains elusive. Several lines of evidence suggest that the mutagenic-when-heterozygous effect of indels might be transient and, alone, is insufficient to account for the patterns of elevated single nucleotide variation near indels. First, nucleotide variation is slightly increased relative to background divergence near the location of the indel in the lineage that does not carry the indel (Tian et al. 2008; Zhu et al. 2009; Hodgkinson and Eyre-Walker 2011; McDonald et al. 2011). Second, the association between indels and nucleotide variation is also observed within the same individual between normal and cancer somatic cells (De and Babu 2010). Third, the proportion of nucleotide divergence attributable to the presence of an indel decreases over time, indicating that indels cause a short burst of nucleotide diversity but only transiently (McDonald et al. 2011).
In addition to a direct mutagenic effect of heterozygous indels, recruitment of low-fidelity DNA polymerases to indels could increase the likelihood of synthesis error during DNA repair (De and Babu 2010; McDonald et al. 2011). McDonald et al. also suggested that the presence of repeat sequences leads to stalling during replication, which gets restarted by error-prone polymerases, thereby locally increasing the mutation rate. Because repeat sequences also can induce strand slippage and the creation of indels, and because polymerase-stalling motifs can lead to double-stranded DNA breaks, the elevated mutation rate may ultimately depend on the sequence context and the presence of repeat sequences (McDonald et al. 2011).
Therefore, we searched for homopolymeric repeats in the vicinity of indels and found that 20.2% of indels are contiguous with a repeat motif in C. remanei (34.8% in C. sp. 23), and that 34.3% of C. remanei indels are located within 5 bp of a repeat (57.1% in C. sp. 23). Similarly, 33.7% of indels occur within 5 bp of a repeat sequence that is present in both species; 23.4% of indels are immediately adjacent to a repeat (fig. 4). Thus, many indels are associated with homopolymeric repeats in Caenorhabditis, in agreement with the results reported for other species by McDonald et al. (2011). However, an increasing number of repeats does not yield higher nucleotide diversity in DNA fragments that lack indels, as we found either no correlation or a negative correlation between nucleotide variation and repeat density (C. remanei: Spearman’s ρ = −0.224, P = 0.032; C. sp. 23: ρ = −0.061, P = 0.588; between species: ρ = 0.116, P = 0.528). Similar results were found for all DNA fragments, with or without indels (supplementary table S1, Supplementary Material online). In addition, the significant positive correlation between the number of indels and repeats supports the hypothesis that repeat sequences may promote the creation of indels (supplementary table S1, Supplementary Material online).
Fig. 4.—
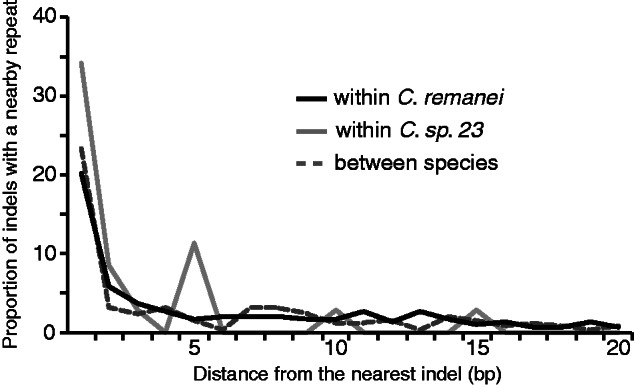
Indels are often associated with homopolymeric repeat sequences. The 20 bp surrounding indels and the proportion of indels associated with a repeat are shown. Only the position of the nearest repeat was scored when multiple repeats were found in the proximity of an indel.
An intriguing feature of the mutations close to indels is that they are enriched for transversions, despite a genomic transition mutation bias (fig. 5). This pattern is reminiscent of the mutation bias toward transversions of some error-prone polymerases (Tian et al. 2008; McDonald et al. 2011). We analyzed the ratio of transitions to transversions as a function of the distance from indels for mutations segregating within C. remanei and for substitutions between C. remanei and C. sp. 23. Polymorphisms in C. remanei are only slightly enriched for transversions immediately next to an indel, but this trend is exacerbated for substitutions between the species (fig. 5). The human genome exhibits a similar contrast, in which polymorphisms do not seem to be enriched for transversions near indels (De and Babu 2010), despite more numerous transversion substitutions between human and chimpanzee near indels (Tian et al. 2008). These findings are consistent with the accumulation over evolutionary time of mutations induced by error-prone polymerases.
Fig. 5.—
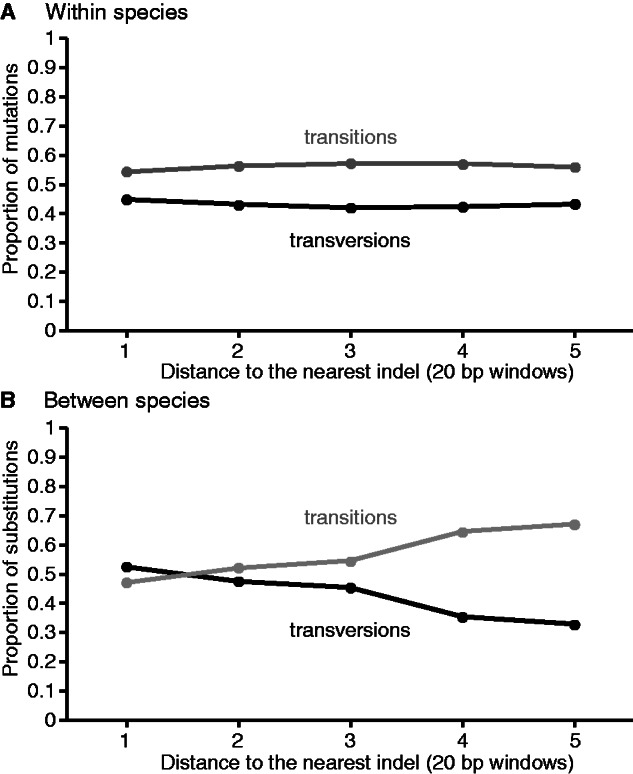
The proportion of transversion mutations (black) segregating in Caenorhabditis remanei is slightly increased near indels (A), whereas this trend is more pronounced for transversion substitutions between C. sp. 23 and C. remanei (B). Windows with less than 50 mutations or substitutions were discarded to avoid variation in the transition/transversion ratio due to low sampling. The trend is smoothened in (A) and (B) by using windows of 20 bp instead of windows of 10 bp.
Conclusion
Indels in the genomes of Caenorhabditis nematodes associate nonrandomly with nucleotide variants and this association cannot be explained solely by relaxed purifying selection on the afflicted sequence regions. Our results suggest that error-prone DNA repair could explain indel-associated mutation, although the data do not preclude a role for indels having a mutational effect when heterozygous. A formal alternative is that a single complex mutation comprising both indels and single nucleotide changes might create clustering of and linkage disequilibrium between indels and SNPs (Hodgkinson and Eyre-Walker 2011), although it is unclear whether this mechanism could enrich the nearby SNPs in transversions. Instead, our analysis supports a model in which regional sequence context, in particular homopolymeric repeats, increases the likelihood of indel creation and the subsequent recruitment of low-fidelity DNA polymerases for DNA repair, resulting in a locally elevated single nucleotide mutation rate and a local bias toward transversions. Although here we focused on the association between indels and SNPs, our results support the emerging general view that mutations can themselves induce other mutations (Amos 2010; Hodgkinson and Eyre-Walker 2010, 2011; Schrider et al. 2011). The mutagenic properties of mutations may have nontrivial consequences on genome evolution and may therefore represent a significant source of the heterogeneity in nucleotide variation across genomes. Moreover, when sign epistasis—the genetic background-dependent effect of a mutation on fitness—is prevalent (Weinreich et al. 2005; Wagner 2008), this local mutagenic effect of mutations could profoundly affect the rate of adaptation by bringing together tightly linked combinations of mutations that, under some circumstances, could either enhance or hamper adaptation.
Supplementary Material
Supplementary figures S1–S3 and table S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank two anonymous reviewers for comments on the manuscript. The authors thank Alivia Dey for sharing data in advance of publication. This work was supported by a fellowship from the Ontario Ministry of Research and Innovation to R.J. and the Natural Sciences and Engineering Research Council of Canada and the Canada Research Chair program grants to A.D.C.
Literature Cited
- Amos W. Even small SNP clusters are non-randomly distributed: is this evidence of mutational non-independence? Proc Biol Sci. 2010;277:1443–1449. doi: 10.1098/rspb.2009.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ. Divergence between samples of chimpanzee and human DNA sequences is 5%, counting indels. Proc Natl Acad Sci U S A. 2002;99:13633–13635. doi: 10.1073/pnas.172510699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ, Rowen L, Williams J, Cameron RA. Majority of divergence between closely related DNA samples is due to indels. Proc Natl Acad Sci U S A. 2003;100:4661–4665. doi: 10.1073/pnas.0330964100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Zhang F, Lupski JR. Evolution in health and medicine Sackler colloquium: genomic disorders: a window into human gene and genome evolution. Proc Natl Acad Sci U S A. 2010;107(1 Suppl):1765–1771. doi: 10.1073/pnas.0906222107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FC, Chen CJ, Li WH, Chuang TJ. Human-specific insertions and deletions inferred from mammalian genome sequences. Genome Res. 2007;17:16–22. doi: 10.1101/gr.5429606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JQ, et al. Variation in the ratio of nucleotide substitution and indel rates across genomes in mammals and bacteria. Mol Biol Evol. 2009;26:1523–1531. doi: 10.1093/molbev/msp063. [DOI] [PubMed] [Google Scholar]
- Cutler G, Marshall LA, Chin N, Baribault H, Kassner PD. Significant gene content variation characterizes the genomes of inbred mouse strains. Genome Res. 2007;17:1743–1754. doi: 10.1101/gr.6754607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Baird SE, Charlesworth D. High nucleotide polymorphism and rapid decay of linkage disequilibrium in wild populations of Caenorhabditis remanei. Genetics. 2006;174:901–913. doi: 10.1534/genetics.106.061879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Babu MM. A time-invariant principle of genome evolution. Proc Natl Acad Sci U S A. 2010;107:13004–13009. doi: 10.1073/pnas.0914454107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Morris K, Lynch M, Thomas WK. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature. 2004;430:679–682. doi: 10.1038/nature02697. [DOI] [PubMed] [Google Scholar]
- Dey A, Jeon Y, Wang GX, Cutter AD. Global population genetic structure of Caenorhabditis remanei reveals incipient speciation. Genetics. 2012;191:1257–1269. doi: 10.1534/genetics.112.140418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopman EB, Hartl DL. A portrait of copy-number polymorphism in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2007;104:19920–19925. doi: 10.1073/pnas.0709888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JJ, Cardoso-Moreira M, Borevitz JO, Long M. Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science. 2008;320:1629–1631. doi: 10.1126/science.1158078. [DOI] [PubMed] [Google Scholar]
- Fadista J, Thomsen B, Holm LE, Bendixen C. Copy number variation in the bovine genome. BMC Genomics. 2010;11:284. doi: 10.1186/1471-2164-11-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazave E, et al. Copy number variation analysis in the great apes reveals species-specific patterns of structural variation. Genome Res. 2011;21:1626–1639. doi: 10.1101/gr.117242.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graustein A, Gaspar JM, Walters JR, Palopoli MF. Levels of DNA polymorphism vary with mating system in the nematode genus Caenorhabditis. Genetics. 2002;161:99–107. doi: 10.1093/genetics/161.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guryev V, et al. Distribution and functional impact of DNA copy number variation in the rat. Nat Genet. 2008;40:538–545. doi: 10.1038/ng.141. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hardison RC, et al. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 2003;13:13–26. doi: 10.1101/gr.844103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson A, Eyre-Walker A. Human triallelic sites: evidence for a new mutational mechanism? Genetics. 2010;184:233–241. doi: 10.1534/genetics.109.110510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson A, Eyre-Walker A. Variation in the mutation rate across mammalian genomes. Nat Rev Genet. 2011;12:756–766. doi: 10.1038/nrg3098. [DOI] [PubMed] [Google Scholar]
- Hollister JD, Ross-Ibarra J, Gaut BS. Indel-associated mutation rate varies with mating system in flowering plants. Mol Biol Evol. 2010;27:409–416. doi: 10.1093/molbev/msp249. [DOI] [PubMed] [Google Scholar]
- Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Jiang Z, et al. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet. 2007;39:1361–1368. doi: 10.1038/ng.2007.9. [DOI] [PubMed] [Google Scholar]
- Jovelin R, Dunham JP, Sung FS, Phillips PC. High nucleotide divergence in developmental regulatory genes contrasts with the structural elements of olfactory pathways in Caenorhabditis. Genetics. 2009;181:1387–1397. doi: 10.1534/genetics.107.082651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA. Evolving views of DNA replication (in)fidelity. Cold Spring Harb Symp Quant Biol. 2009;74:91–101. doi: 10.1101/sqb.2009.74.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Longman-Jacobsen N, Williamson JF, Dawkins RL, Gaudieri S. In polymorphic genomic regions indels cluster with nucleotide polymorphism: quantum genomics. Gene. 2003;312:257–261. doi: 10.1016/s0378-1119(03)00621-8. [DOI] [PubMed] [Google Scholar]
- Massouras A, et al. Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet. 2012;8:e1003055. doi: 10.1371/journal.pgen.1003055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maydan JS, Lorch A, Edgley ML, Flibotte S, Moerman DG. Copy number variation in the genomes of twelve natural isolates of Caenorhabditis elegans. BMC Genomics. 2010;11:62. doi: 10.1186/1471-2164-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, et al. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- McDonald MJ, Wang WC, Huang HD, Leu JY. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 2011;9:e1000622. doi: 10.1371/journal.pbio.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu XJ, Lu ZJ, Kong Y, Lam HY, Gerstein MB. Analysis of genomic variation in non-coding elements using population-scale sequencing data from the 1000 Genomes Project. Nucleic Acids Res. 2011;39:7058–7076. doi: 10.1093/nar/gkr342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Ohno S. Evolution by gene duplication. New-York: Springer-Verlag; 1970. [Google Scholar]
- Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Hourmozdi JN, Hahn MW. Pervasive multinucleotide mutational events in eukaryotes. Curr Biol. 2011;21:1051–1054. doi: 10.1016/j.cub.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Shaw CJ, Withers M, Inoue K, Lupski JR. Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res. 2004;14:2209–2220. doi: 10.1101/gr.2746604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- Stranger BE, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, et al. Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature. 2008;455:105–108. doi: 10.1038/nature07175. [DOI] [PubMed] [Google Scholar]
- Wagner A. Neutralism and selectionism: a network-based reconciliation. Nat Rev Genet. 2008;9:965–974. doi: 10.1038/nrg2473. [DOI] [PubMed] [Google Scholar]
- Weinreich DM, Watson RA, Chao L. Perspective: sign epistasis and genetic constraint on evolutionary trajectories. Evolution. 2005;59:1165–1174. [PubMed] [Google Scholar]
- Wetterbom A, Sevov M, Cavelier L, Bergstrom TF. Comparative genomic analysis of human and chimpanzee indicates a key role for indels in primate evolution. J Mol Evol. 2006;63:682–690. doi: 10.1007/s00239-006-0045-7. [DOI] [PubMed] [Google Scholar]
- Wicks SR, Yeh RT, Gish WR, Waterston RH, Plasterk RH. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat Genet. 2001;28:160–164. doi: 10.1038/88878. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Huang J, Wang Z, Wang L, Gao P. Impact of indels on the flanking regions in structural domains. Mol Biol Evol. 2010;28:291–301. doi: 10.1093/molbev/msq196. [DOI] [PubMed] [Google Scholar]
- Zhu L, Wang Q, Tang P, Araki H, Tian D. Genomewide association between insertions/deletions and the nucleotide diversity in bacteria. Mol Biol Evol. 2009;26:2353–2361. doi: 10.1093/molbev/msp144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.