

Abstract
Parkinson's disease is characterized by α‐synuclein pathology in the form of Lewy bodies and Lewy neurites. Braak et al described the spatial and temporal spread of α‐synuclein pathology in Parkinson's disease. Recent experimental studies have demonstrated that α‐synuclein can transfer from cell to cell. In this review, we highlight the involvement of α‐synuclein in Parkinson's disease and in Braak's staging of Parkinson's disease pathology. We discuss whether a prion‐like mechanism of α‐synuclein spread might contribute to Parkinson's disease pathology. We describe recent studies investigating cell‐to‐cell transfer of α‐synuclein and focus our review on the long‐distance axonal transport of α‐synuclein along neurons.
Keywords: α‐Synuclein, Lewy pathology, Parkinson's disease, prion disease, prion‐like aggregation, templated misfolding
Introduction
Pathological accumulation of misfolded α‐synuclein (α‐syn), leading to cell dysfunction and cell death, plays a central role in the pathogenesis of Parkinson's disease (PD). PD is not infectious like prion diseases; however, recent studies suggest the existence of a common mechanism underlying the propagation of α‐syn pathology throughout the brain 2, 24. In experimental paradigms, α‐syn transferring from cell‐to‐cell and initiating the prion‐like spread of pathology in synucleinopathies has been hypothesized. In this review, we highlight the involvement of α‐syn in PD and in the progression of the neuropathology and symptoms of PD. We describe the idea of a prion‐like mechanism contributing to PD pathology, discussing recent studies investigating cell‐to‐cell transfer of α‐syn and focus on the long‐distance axonal transport of α‐syn along neurons. Finally, we address the evaluation of Braak's hypothesis and discuss the prion‐like hypothesis for PD research.
The Involvement of α‐Synuclein in Parkinson's Disease
PD is the second most common neurodegenerative disorder. A clinically important feature of PD neuropathology is the loss of dopaminergic neurons in the substantia nigra pars compacta. It is believed that the resulting depletion of dopamine in the striatum plays a key role for the motor symptoms (eg, bradykinesia, resting tremor, rigidity and postural instability) because “dopamine‐replacement” pharmacotherapy is relatively effective at reducing the motor disturbances. Recent years have, however, shown that several non‐motor symptoms (eg, depression, dementia, anosmia and sleep disturbances) 19 are also a source of major morbidity, and they respond relatively poorly to dopamine replacement therapy 11. Consequently, greater attention is being paid to neurodegenerative changes outside the nigrostriatal pathway in PD. In this context, the presence of intracellular protein inclusion bodies, so‐called Lewy bodies (LB) and Lewy neurites (LN), is now believed to be important. Friedrich H. Lewy (1912) 48 first described these inclusions over a century ago. It was not until 1997 when it was discovered that aggregated α‐syn is the major constituent of LBs and LNs 2, 24, 30, 72 that the modern era of PD neuropathology research was born. Not only is α‐syn the main component of Lewy inclusions in sporadic PD, but missense mutations (A53T, A30P, E46K) in the α‐syn gene are also associated with autosomal dominant PD 19, 41, 67, 78. Furthermore, duplications and triplications in the α‐syn gene lead to a severe neurological syndrome with parkinsonian features 10, 11, 70 and certain single‐nucleotide polymorphisms in the α‐syn gene are associated with increased PD risk 48, 56.
α‐Syn protein is abundantly expressed in the brain as well as in multiple other central and peripheral tissues 4. Maroteaux et al 57 first described the localization of α‐syn to the nucleus and the presynaptic terminal. Although the full function of α‐syn is yet to be defined, it is certainly involved in vesicular trafficking and release, related to its associations with the SNARE complex proteins 9, 60. α‐Syn consists of three domains: an amino‐terminal lipid binding α‐helix, a non‐amyloidogenic core (NAC) domain and an unstructured carboxy‐terminus. These three regions are necessary for the misfolding of the protein 37. α‐Syn is considered natively unfolded, but its amino‐terminus forms α‐helical structures when bound to phospholipids. Recently, investigations of endogenous α‐syn analyzed under non‐denaturing conditions in cell lines and mouse brain tissue revealed that α‐syn might exist as a folded, stable tetramer with a molecular weight of about 58 kDa 5, 76. These results have proven controversial 20. What is more certain is when α‐syn is misfolded, the random coil of the NAC domain forms β‐sheets, which participate in protofibril and fibril formation 68, 74. Importantly, two mutations in α‐syn (E46K, A53T) increase the oligomeric and fibrillar forms of α‐syn 47, further highlighting the importance of the aggregation of α‐syn in its toxicity.
The Braak Hypothesis
What is the connection between α‐syn aggregates and the development of PD? Braak et al 6 suggested that in idiopathic PD post‐mortem brain tissue, LB pathology appears in a stereotypic pattern depending on how advanced the disease is. In stage 1, Lewy pathology (primarily LNs) is found in the olfactory bulb (and anterior olfactory nucleus) and the dorsal motor nucleus of the glossopharyngeal and vagal nerve. In stage 2, the Lewy pathology continues to ascend toward the brainstem, reaching the medulla oblongata and pontine tegmentum. People in stages 1 and 2 do not have clinical PD but might exhibit signs of anosmia and constipation. It is unknown whether these individuals would develop PD later on. In stage 3, the pathology appears in the amygdala and substantia nigra. It is at this stage that marked nigral neurodegeneration is expected to occur and the individual will start to develop motor symptoms. Thus, it is not until Braak stage 3 that people will be clinically diagnosed with PD. The LBs, and to a larger extent LNs, are also found in the forebrain and cerebral cortex in stage 4. In stages 5 and 6, the pathology also appears initially in the anterior association and prefrontal areas of the prefrontal cortex and continues to spread toward the posterior association areas. In summary, Braak et al suggested that α‐syn pathology is initiated in the peripheral nervous system and olfactory bulb, ascends toward the brainstem and into the midbrain, and then eventually spreads to the forebrain. Thus, if α‐syn pathology starts in, for example, the gut, it spreads over very long distances in the nervous system during several years.
Because the vagus nerve connects the brainstem and the enteric nervous system, Braak et al concluded that PD could in fact begin in the gut and then travel retrogradely toward the brain 27. The appearance of pathology initiating in two separate locations gave rise to the “dual‐hit” hypothesis 28. It was speculated that an unknown pathogen may enter the nervous system through both the olfactory bulb and the enteric plexus of the stomach 27. We, and others, now suspect that a misfolded form of α‐syn might play the role of the unknown pathogen. Misfolded α‐syn could spread from one cell to another and trigger aggregation of α‐syn in the recipient cell. One requirement for this mechanism is the ability of α‐syn to travel from cell‐to‐cell as well as to move from one brain region to another distant structure, acting like a “long distance runner.”
Cell‐to‐Cell Transfer: Evidence from Human Grafting Studies
Mounting evidence points toward α‐syn acting as a prion‐like protein. By suggesting that α‐syn is a prion‐like protein, we mean that misfolded α‐syn could be responsible for the intercellular transmission of PD pathology. We are not implying that the disease can be transmitted between individuals 31. Initial evidence supporting the process in humans became apparent from patients who received embryonic neural tissue grafted into the striatum to replace lost nigral dopaminergic neurons. Autopsy revealed that these young transplanted neurons, introduced stereotaxically into the host brain over a decade prior to death of the patient, contained α‐syn pathology 38, 40, 42, 49, 50. One possible explanation for the presence of LB in young transplanted neurons is that α‐syn can transfer directly from the host brain to grafted cells 7. The question is then, did α‐syn transfer from the host to seed the aggregation of endogenous protein in the grafted cells? Subsequent to the finding of LB pathology in grafted embryonic tissue, model systems have been developed to examine both the transfer and the seeding of α‐syn in vitro and in vivo.
Cell‐to‐Cell Release, Uptake, Transfer and Seeding of α‐Synuclein: Evidence from in vitro and in vivo Studies
The hypothesis of prion‐like transmission of α‐syn pathology relies on the premise that a sick neuron could release its α‐syn or that α‐syn gains access to the extracellular space when the neuron dies. Once in the extracellular space, the misfolded α‐syn could then be free to enter an adjacent neuron and act as a template, seeding the aggregation of numerous α‐syn monomers and initiating the formation of a LB or LN 25 (Figure 1).
Figure 1.
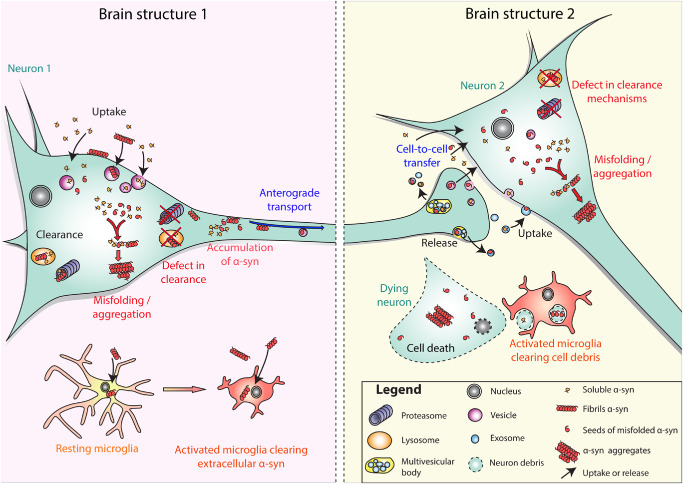
Schematic presentation of the possible mechanisms underlying the spread of synuclein pathology and α‐syn aggregation in Parkinson's disease (PD). In the brain structure on the left, conditions of cellular stress cause α‐syn to misfold within the neuron (neuron 1), or misfolded α‐syn is taken up from the extracellular space. Internalized misfolded α‐syn might be degraded by clearance mechanisms such as the ubiquitin proteasome system, lysosomes and autophagy. Under particular conditions of stress and/or clearance failure, misfolded α‐syn might not be effectively degraded. Thus, the remaining misfolded α‐syn might recruit soluble α‐syn in a seeding mechanism, thereby converting it into misfolded protein, initiating aggregation within neuron 1. The remaining misfolded α‐syn may also undergo intracellular axonal transport, via fast axonal transport or via slow component b axonal transport within the axon of neuron 1. At the terminal of neuron 1, which is located in brain structure 2, transported (misfolded) α‐syn might be released by exocytosis, or in exosomes. The α‐syn released by exocytosis or in exosomes can then be taken up by the surrounding neurons such as neuron 2 (cell‐to‐cell transfer). The same cascade of events including recruitment of endogenous α‐syn, seeding and aggregation, clearance and then failure of clearance is proposed to lead first to the formation of α‐syn aggregates (neurons 1 and 2), and in the end, to the death of the host neuron (dying neuron). Misfolded α‐syn released into the extracellular space from living neurons or dying cells can activate microglia that take up and degrade misfolded α‐syn.
The release of α‐synuclein
The first step in this hypothesis requires that there is cellular release of α‐syn. Under physiological conditions, small amounts of α‐syn are released from cells in the absence of membrane damage, despite the lack of a secretory peptide sequence on α‐syn 44. Endogenous monomeric and aggregated forms of α‐syn can be secreted into the culture medium of differentiated human neuroblastoma cells and rat primary cortical neurons 14, 24, 44 and are detected in human cerebrospinal fluid and plasma 17. The secretion of α‐syn maybe due to its association with vesicle trafficking as α‐syn can induce the aggregation of yeast Rab GTPase proteins and block endoplasmic reticulum‐Golgi trafficking 12, 23, 71. Release of α‐syn from neurons is partially mediated by exocytosis and is increased under conditions of misfolded proteins 24, 32, 44, 45, 46, 63, 75. Also, α‐syn can be released from enteric neurons in a neuronal activity‐regulated manner 63. α‐Syn is secreted by non‐classical exocytic or endocytic pathways 44. α‐Syn can be directly integrated into secretory vesicles and subsequently released by exocytosis or be translocated to early endosomes 1, 18. From early endosomes in neuronal cultures, α‐syn protein can either be released to the extracellular space through the recycling endosome or incorporated to intraluminal vesicles of multivesicular bodies 26. Multivesicular body cargo, including α‐syn, can be directed for degradation by fusion with lysosomes or secretion by fusion with the plasma membrane, upon release of exosomal vesicles 26.
Uptake of α‐synuclein
The ability of cells in culture to take up α‐syn is dependent on the cell type and the species of α‐syn 13, 18, 44. In a cell culture study investigating α‐syn uptake via endocytosis, higher order oligomers were the species of α‐syn that entered cells more readily than monomers, highlighting the importance of α‐syn oligomerization in cell‐to‐cell propagation 13. In cultured neuronal cells, both oligomeric and fibrillar forms of α‐syn are internalized through endocytosis and targeted to the lysosome for degradation 24, 46, 54, 61, 75, 77 (Figure 1). Further support for endocytosis as a mechanism of α‐syn uptake is demonstrated in studies employing endocytosis inhibitors 24, 45. For a thorough review on the uptake of α‐syn into cells via endocytosis, please see 16.
The transfer and seeding of α‐synuclein
The in vivo data supporting cell‐to‐cell transfer and seeding of α‐syn pathology are striking. Several groups have succeeded in producing models that display transfer of α‐syn from the host brain to grafted cells similar to that hypothesized to occur in PD patients 39, 49. Desplats et al transplanted mouse neuronal progenitor cells into the hippocampus of transgenic animals overexpressing human α‐syn 15 and discovered that 1‐week post‐injection, grafted cells had human α‐syn immunoreactivity, and by 4 weeks, some of the grafted cells contained aggregates, which expressed human α‐syn. In another study, wild‐type mouse nigral tissue was grafted into the striatum of transgenic mice overexpressing human α‐syn, and small amounts of host‐derived human α‐syn were observed in around 1% of the grafted dopamine neurons 24. Evidence for transfer and seeding of endogenous α‐syn was presented 24. In a next step, rats were engineered to markedly overexpress human α‐syn in the nigrostriatal pathway and then transplanted with embryonic rat nigral tissue. In this case, over 20% of the grafted dopamine neurons displayed host‐derived human α‐syn. Labeling with species‐specific α‐syn antibodies revealed evidence that the imported human α‐syn seeded aggregation of endogenous rat α‐syn in these naïve neurons 2. Thus, sometimes, a small area immunoreactive for human α‐syn was enveloped by a larger area immunoreactive for rat α‐syn derived from the host neuron 2.
To further investigate the phenomena of α‐syn propagation, Kordower et al 39 grafted fetal rat brain tissue into 6‐hydroxydopamine lesioned adult rats. After the transplant, viruses containing human α‐syn were injected distal to the grafted cells. Close examination revealed a number of grafted neurons expressing human α‐syn. Transferred α‐syn can therefore not only propagate from neuron to neuron but can also be modified, aggregate and form pathogenic species. For a more in‐depth description of the concept of cell‐to‐cell transfer in PD and the subsequent seeding of α‐syn aggregates, please see the recent review by Dunning et al 62.
Transferred α‐syn can play a pathogenic role. In young transgenic (A53T) α‐syn mice inoculated intracerebrally with brain tissue from old transgenic (A53T) α‐syn mice, early signs of motor impairment were detected. This sign of disease was associated with insoluble phosphorylated α‐syn and dystrophic neurites in the raphe nucleus and the lateral vestibular nucleus in these animals 58. Importantly, inoculation with old transgenic brain tissue decreased lifespan with death occurring significantly earlier than in transgenic mice inoculated with brain homogenate from young healthy transgenic mice. In contrast, wild‐type mice lacking the α‐syn locus, inoculated with brain homogenate from old transgenic (A53T) mice, survived the longest 58.
The most recent developments in the field of α‐syn transfer and seeding are from Virginia Lee, John Trojanowski, and colleagues. In their first paper of 2012, the authors injected recombinant human α‐syn preformed fibrils or brain lysate from symptomatic transgenic mice overexpressing A53T α‐syn [M83 mice, 22] into the cortex and striatum of asymptomatic transgenic mice. Induction of α‐syn pathology in recipient mice as early as 30 days post‐injection was observed and the pathology progressively spread to interconnecting brain regions 55. The site of injection produced differential patterns of α‐syn pathology in the recipient mice. The pattern was consistent with the neuronal connections to and from the site of injection. Taking their findings a step further, Luk et al demonstrated transfer and seeding of α‐syn using synthetic preformed fibrillar mouse α‐syn injected into wild‐type mice 53. In this study, preformed fibrils assembled from recombinant mouse α‐syn were injected into the dorsal striatum. Phosphorylated α‐syn (indicating recruitment of endogenous α‐syn that had undergone post‐translational modification) was observed in neurons at the injection site 30 days post‐injection. The authors also described LB‐like structures containing α‐syn in some brain regions interconnected with the striatum, such as the neocortex. However, there was no phosphorylated α‐syn in brain regions that do not project directly to the striatum, suggesting that, in fact, there was no trans‐synaptic transmission of α‐syn aggregation in this paradigm. Notably, dopaminergic neurons in the substantia nigra, which are one population that project to the injection site, frequently exhibited α‐syn aggregates. The consequences of these changes were striking. The accumulating α‐syn led to a gradual loss of dopaminergic cells and depletion of striatal dopamine, accompanied with motor deficits on the rotarod and wire hang test 53. In agreement with the study by Mougenot et al 58, injections of preformed α‐syn fibrils did not give rise to α‐syn aggregates when injected into α‐syn null mice, confirming that recruitment of endogenous α‐syn is crucial for the pathogenic process.
Crucial Factors Affecting the Capacity of α‐Synuclein to Act in a Prion‐Like Fashion
Based on the in vitro and in vivo studies described earlier, what factors affect the capacity of α‐syn to act in a prion‐like fashion? Clearly, several steps are crucial in the process. For example, for α‐syn to be an effective prion‐like protein, it has to be released by cells; not be cleared and degraded by microglia; but taken up by neighboring neurons; evade intracellular protein degradation and promote seeding of aggregates in the cytosol, and importantly, undergo long‐distance transport from one brain structure to another region so that the neuropathology can spread over long distances.
Recent immunization studies in α‐syn transgenic mice indicate that antibodies targeting α‐syn will promote its uptake and degradation by microglia and thereby reduce the likelihood of intercellular α‐syn transfer 3. As expected, studies on intracerebral injections of α‐syn indicate that both the molecular species of α‐syn and the α‐syn protein homeostasis in the mice receiving the injections will influence the outcome. Luk et al 53 used recombinant mouse α‐syn in their studies on intracerebral injections of α‐syn. Not only was the protein synthetic, the fibrils were also sonicated to create a mixture of very small seeds of fibrillar α‐syn (please see the electron micrographs in supplementary Figure 1B in reference (53)).
The efficacy at which monomeric, oligomeric or fibrillar forms of α‐syn are taken up by cells vary in cell culture 45 and the same most probably applies in vivo, too. This should significantly impact the likelihood of pathological aggregates forming inside the neurons in the injected brains. Although monomeric, oligomeric and fibrillar forms of α‐syn can all be taken up by cells from the extracellular space, and the uptake of monomers is efficient 24, in the study by Luk et al, monomers did not induce pathology 53. It was not determined whether the injected monomeric α‐syn was still present in the brains after 30 days. It is likely that the monomeric α‐syn was taken up, transported away from the injection site and potentially cleared by the first (30 days) time point investigated. Alternatively, the injected monomeric α‐syn was impossible to distinguish from the host protein because mouse recombinant α‐syn was injected into mice 53. In the studied paradigm, only aggregated α‐syn would be possible to detect and as an increased load of monomeric α‐syn apparently was not sufficient to promote aggregation, this readout was negative.
The studies on injections of synthetic preformed fibrillar mouse α‐syn in wild‐type mice demonstrate that the presence of endogenous α‐syn is crucial in the seeding of aggregates 53, 58. Thus, α‐syn knock‐out mice did not develop pathology when preformed α‐syn fibrils were injected. Furthermore, the degree of homology between the “α‐syn seed” and the α‐syn in the recipient neuron can be important. Human α‐syn shares 95.3% homology with mouse α‐syn, which carries the native rodent sequence 43. Perhaps the rodent sequence of α‐syn is more permissive to seeding and aggregation 51, 59. It would be interesting to investigate how well mouse and human fibrillar α‐syn species interact, directly comparing the time delay in cellular uptake and the lag time to produce α‐syn aggregate pathology as well as the transport of these species to interconnected brain regions in vivo.
Axonal Transport of α‐Synuclein: is α‐Synuclein the Long Distance Runner?
α‐Synuclein is transported via fast and slow component axonal transport
α‐Syn is actively transported in both directions in axons. Proteins are transported along axons via either the fast transport component (FC, 100–400 mm/day), slow transport component a (SCa, 0.1–2 mm/day) or slow transport component b (SCb, 2–10 mm/day) 29, 69. Similar to other proteins, α‐syn can be transported along axons 35, 36, 52, 73. A study utilized rat visual pathways as a model system 36 and found that approximately 25% of wild‐type α‐syn travels using FC axonal transport, presumably after binding the membranes of vesicles. A majority (approximately 75%) of rat α‐syn is transported in the SC (approximately 15% in SC a and approximately 60% in SC b) 36. According to studies in mouse hippocampal neurons, this α‐syn is transported slowly along axons to synaptic terminals 73.
Reports on the effects of point mutations in α‐syn on its propensity to undergo axonal transport are conflicting. Mutant A30P α‐syn does not undergo the same FC axonal transport as the wild‐type form in the rat visual system, possibly due to its reduced membrane binding 36. Another study investigating the axonal transport of α‐syn in peripheral nerve demonstrated that the transport of human and mouse α‐syn is not affected by the A30P and A53T α‐syn mutations 52. With increasing age, however, the rate of α‐syn axonal transport decreases 52.
α‐Synuclein is transported along axons in both directions in the central nervous system
Cell culture studies using microfluidic chambers to isolate and separate neuronal cell bodies from their terminals have demonstrated that the α‐syn that is taken up can also be transported along the axons of neurons. In mouse primary cortical and hippocampal neurons, α‐syn can travel in both retrograde 14, 75 and anterograde directions 21, 75 (Figure 1). Recently, we studied the fate of various molecular species of human α‐syn (soluble α‐syn or fibrils) after injection into the olfactory bulb of wild‐type mice. We asked whether α‐syn is transferred to interconnected structures within a few hours after injection. As a control protein, we injected bovine serum albumin (BSA). We detected the proteins we injected using antibodies that recognize specifically human α‐syn (syn211) or BSA (Figure 2). Within 20 minutes, human α‐syn was taken up by mitral cells, which are the relay cells of the olfactory bulb that project to other olfactory structures. Less than an hour after injection, we found injected soluble α‐syn was present in multiple structures directly connected to the olfactory bulb, for example, piriform cortex and amygdala. Consistent with the in vitro studies mentioned earlier, our detailed anatomical analyses suggest that the α‐syn we had injected was transported both in anterograde and in retrograde directions. By contrast, our control protein, BSA, was rarely taken up by olfactory bulb cells and did not transfer to other brain structures, suggesting that soluble forms of α‐syn are both exceptionally good and fast long distance runners.
Figure 2.
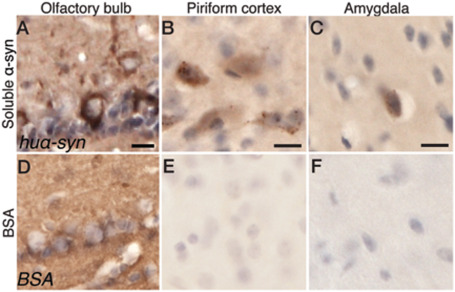
Uptake of α‐syn in the olfactory bulb and transfer of soluble α‐syn to interconnected regions in the mouse brain. Human α‐syn staining in the injected olfactory bulb (A), the ipsilateral piriform cortex (B) and the ipsilateral amygdala (C) after injection of soluble α‐syn. Bovine serum albumin (BSA) staining in the injected olfactory bulb (D), the ipsilateral piriform cortex (E) and the ipsilateral amygdala (F) after injection of BSA. The scale bar represents 10 μm. (Data from Rey et al. 2013, submitted.)
Can α‐synuclein be transported along peripheral nerves from the gut to the brain?
Is there any evidence that misfolded α‐syn can undergo long‐distance transfer from the gut to the brain, in keeping with Braak's proposed dual‐hit hypothesis? A recent study explored in some anatomical details how misfolded α‐syn might spread from the enteric nervous system to the central nervous system 65. Pan‐Montojo et al followed up their earlier report suggesting that following mitochondrial inhibition, by giving rotenone locally in the gut, α‐syn levels are increased in enteric neurons and aggregated protein is released 64. Eventually, this misfolded α‐syn reaches the central nervous system where it was suggested to cause degeneration of dopamine neurons accompanied by motor deficits 64. They suggested that it is the sympathetic and parasympathetic nerves that take up the extracellular α‐syn and retrogradely transport it to the soma of the autonomic nervous system neurons. In this most recent study, the investigators severed the sympathetic and parasympathetic nerves in the same animal model of PD 65. Resection of sympathetic and parasympathetic nerves halted the progression of PD‐like α‐syn pathology to the previously connected neurons within the intermediolateral column of the spinal cord, the vagal dorsal motor nuclei and the substantia nigra. The claim that α‐syn can be transported along long autonomic nervous system structures supports the notion that α‐syn is a long distance runner with the ability to spread pathology from one structure to another.
An Evaluation of the Braak Staging
The Braak hypothesis that Lewy pathology spreads throughout the brain in regions that are connected to the peripheral nerves or the olfactory system is not uncontroversial. One reason why the hypothesis remains contentious is partly due to the fact that Braak staging and severity of PD symptoms do not always correlate 8. Certainly, some individuals with relatively severe α‐syn pathology discovered post‐mortem were never diagnosed clinically with PD prior to death 66. Moreover, in some PD cases, the distribution of Lewy pathology does not match any of the Braak stages 33, 34. In Braak's stages 1 and 2, lower brainstem synucleinopathy is designated to represent “early” PD. However, criticism arose as to whether these individuals would develop PD later on 8. When considering these cases, it is important to note that Braak's staging uses the presence of LBs and LNs as the identifiable hallmark. Possibly, smaller aggregates of α‐syn that do not qualify as LBs or LNs contribute to the variation in Braak's staging in some cases.
What else could explain the differences between the findings of Braak and some other investigators? Notably, the methodology varies between different studies. Braak et al 6 used 100‐μm‐thick sections sampling greater volumes than in commonly used 10‐μm‐thick sections. Differences in post‐mortem time before tissue fixation and variations in fixation and immunostaining protocols are other confounders. The clinical data on neurological and psychiatric symptoms prior to death can also be inconsistent between studies. Until the technology exists to definitively image insoluble α‐syn longitudinally in living people, it is impossible to evaluate fully the Braak hypothesis.
Conclusions
The spreading of α‐syn pathology in PD, as suggested by Braak et al, would require that α‐syn can travel along long unmyelinated axons and seed aggregation in new neurons at the destination during a slow process that takes years or even decades. To date, no study has unequivocally demonstrated α‐syn transport from one brain structure to another in a living human or animal. The development of in vivo imaging techniques to visualize the movement of different molecular forms of α‐syn would help us address the Braak hypothesis and understand if α‐syn can act like a prion‐like protein in PD. Assuming that this is the case, identifying the mechanisms of cell‐to‐cell spread and transport of α‐syn might allow for the development of agents to block these processes. Such agents could represent a new generation of therapeutics in the fight against PD and provide the first truly disease‐modifying agents that can slow the progression of PD.
Conflict of interest
The authors declare that they have no conflict of interest related to this article.
Acknowledgments
Our work was supported by the PRISTINE‐PD European Research Council Advanced Award; Michael J. Fox Foundation for Parkinson's Research; Swedish Brain Foundation; Swedish Parkinson Foundation; Swedish Research Council, including the Linnaeus grant Bagadilico; European Research Area Network of European Funding for Neuroscience Research Program MIPROTRAN; Human Frontier Science Program; and Swedish Brain Power. All authors are active in the Strong Research Environment Multipark (Multidisciplinary research in Parkinson's disease at Lund University).
References
- 1. Alvarez‐Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM (2011) Lysosomal dysfunction increases exosome‐mediated alpha‐synuclein release and transmission. Neurobiol Dis 42:360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Angot E, Steiner JA, Lema Tomé CM, Ekström P, Mattsson B, Björklund A, Brundin P (2012) Alpha‐synuclein cell‐to‐cell transfer and seeding in grafted dopaminergic neurons in vivo . PLoS ONE 7:e39465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY et al (2012) Antibody‐aided clearance of extracellular‐synuclein prevents cell‐to‐cell aggregate transmission. J Neurosci 32:13454–13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baltic S, Perovic M, Mladenovic A, Raicevic N, Ruzdijic S, Rakic L, Kanazir S (2004) α‐Synuclein is expressed in different tissues during human fetal development. J Mol Neurosci 22:199–204. [DOI] [PubMed] [Google Scholar]
- 5. Bartels T, Choi JG, Selkoe DJ (2011) α‐Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477:107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braak H, Rüb U, Gai WP, Del Tredici K (2003) Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110:517–536. [DOI] [PubMed] [Google Scholar]
- 7. Brundin P, Li JY, Holton JL, Lindvall O, Revesz T (2008) Research in motion: the enigma of Parkinson's disease pathology spread. Nat Rev Neurosci 9:741–745. [DOI] [PubMed] [Google Scholar]
- 8. Burke RE, Dauer WT, Vonsattel JPG (2008) A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 64:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC (2010) Alpha‐synuclein promotes SNARE‐complex assembly in vivo and in vitro . Science 329:1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chartier‐Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S et al (2004) Alpha‐synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364:1167–1169. [DOI] [PubMed] [Google Scholar]
- 11. Chaudhuri KR, Yates L, Martinez‐Martin P (2005) The non‐motor symptom complex of Parkinson's disease: a comprehensive assessment is essential. Curr Neurol Neurosci Rep 5:275–283. [DOI] [PubMed] [Google Scholar]
- 12. Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B et al (2006) Alpha‐synuclein blocks ER‐Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313:324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A et al (2007) Different species of alpha‐synuclein oligomers induce calcium influx and seeding. J Neurosci 27:9220–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Danzer KM, Ruf WP, Putcha P, Joyner D, Hashimoto T, Glabe C et al (2011) Heat‐shock protein 70 modulates toxic extracellular α‐synuclein oligomers and rescues trans‐synaptic toxicity. FASEB J 25:326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L et al (2009) Inclusion formation and neuronal cell death through neuron‐to‐neuron transmission of alpha‐synuclein. Proc Natl Acad Sci U S A 106:13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dunning CJR, Reyes JF, Steiner JA, Brundin P (2012) Can Parkinson's disease pathology be propagated from one neuron to another? Prog Neurobiol 97:205–219. [DOI] [PubMed] [Google Scholar]
- 17. El‐Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ et al (2003) Alpha‐synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. FASEB J 17:1945–1947. [DOI] [PubMed] [Google Scholar]
- 18. Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH et al (2010) Cell‐produced alpha‐synuclein is secreted in a calcium‐dependent manner by exosomes and impacts neuronal survival. J Neurosci 30:6838–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fahn S (2003) Description of Parkinson's disease as a clinical syndrome. Ann N Y Acad Sci 991:1–14. [DOI] [PubMed] [Google Scholar]
- 20. Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT et al (2012) α‐Synuclein in the central nervous system and from erythrocytes, mammalian cells and E. coli exists predominantly as a disordered monomer. J Biol Chem 287:15345–15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y et al (2012) Neuron‐to‐neuron transmission of α‐synuclein fibrils through axonal transport. Ann Neurol 72:517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002) Neuronal alpha‐synucleinopathy with severe movement disorder in mice expressing A53T human alpha‐synuclein. Neuron 34:521–533. [DOI] [PubMed] [Google Scholar]
- 23. Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ et al (2008) The Parkinson's disease protein alpha‐synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A 105:145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G et al (2011) alpha‐Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hardy J (2005) Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: “permissive templating” as a general mechanism underlying neurodegeneration. Biochem Soc Trans 33:578–581. [DOI] [PubMed] [Google Scholar]
- 26. Hasegawa T, Konno M, Baba T, Sugeno N, Kikuchi A, Kobayashi M et al (2011) The AAA‐ATPase VPS4 regulates extracellular secretion and lysosomal targeting of α‐synuclein. Plos ONE 6:e29460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hawkes CH, Del Tredici K, Braak H (2007) Parkinson's disease: a dual‐hit hypothesis. Neuropathol Appl Neurobiol 33:599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hawkes CH, Del Tredici K, Braak H (2009) Parkinson's disease: the dual hit theory revisited. Ann N Y Acad Sci 1170:615–622. [DOI] [PubMed] [Google Scholar]
- 29. Hirokawa N (1997) The mechanisms of fast and slow transport in neurons: identification and characterization of the new kinesin superfamily motors. Curr Opin Neurobiol 7:605–614. [DOI] [PubMed] [Google Scholar]
- 30. Irizarry MC, Growdon W, Gomez‐Isla T, Newell K, George JM, Clayton DF, Hyman BT (1998) Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson's disease and cortical Lewy body disease contain alpha‐synuclein immunoreactivity. J Neuropathol Exp Neurol 57:334–337. [DOI] [PubMed] [Google Scholar]
- 31. Irwin DJ, Abrams JY, Schonberger LB, Werber Leschek E, Mills JL, Lee VMY, Trojanowski JQ (2013) Potential infectivity of neurodegenerative disease associated proteins. Arch Neurol. doi: 10.1001/jamaneurol.2013.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ (2010) Non‐classical exocytosis of alpha‐synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 113:1263–1274. [DOI] [PubMed] [Google Scholar]
- 33. Jellinger KA (2011) Neuropathology of sporadic Parkinson's disease: evaluation and changes of concepts. Mov Disord 27:8–30. [DOI] [PubMed] [Google Scholar]
- 34. Jellinger KA (2009) Critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 67:550–550. [DOI] [PubMed] [Google Scholar]
- 35. Jensen PH, Li JY, Dahlström A, Dotti CG (2008) Axonal transport of synucleins is mediated by all rate components. Eur J Neurosci 11:3369–3376. [DOI] [PubMed] [Google Scholar]
- 36. Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M (1998) Binding of alpha‐synuclein to brain vesicles is abolished by familial Parkinson's disease mutation. J Biol Chem 273:26292–26294. [DOI] [PubMed] [Google Scholar]
- 37. Jo E (2000) Alpha‐synuclein membrane interactions and lipid specificity. J Biol Chem 275:34328–34334. [DOI] [PubMed] [Google Scholar]
- 38. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body‐like pathology in long‐term embryonic nigral transplants in Parkinson's disease. Nat Med 14:504–506. [DOI] [PubMed] [Google Scholar]
- 39. Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L et al (2011) Transfer of host‐derived α‐synuclein to grafted dopaminergic neurons in rat. Neurobiol Dis 43:552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB (2008) Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord 23:2303–2306. [DOI] [PubMed] [Google Scholar]
- 41. Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S et al (1998) Ala30Pro mutation in the gene encoding alpha‐synuclein in Parkinson's disease. Nat Genet 18:106–108. [DOI] [PubMed] [Google Scholar]
- 42. Kurowska Z, Englund E, Widner H, Lindvall O, Li JY, Brundin P (2011) Signs of degeneration in 12–22‐year old grafts of mesencephalic dopamine neurons in patients with Parkinson's disease. J Park Dis 1:83–92. [DOI] [PubMed] [Google Scholar]
- 43. Lavedan C, Buchholtz S, Auburger G, Albin RL, Athanassiadou A, Blancato J et al (1998) Absence of mutation in the beta‐ and gamma‐synuclein genes in familial autosomal dominant Parkinson's disease. DNA Res 5:401–402. [DOI] [PubMed] [Google Scholar]
- 44. Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha‐synuclein and its aggregates. J Neurosci 25:6016–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee H‐J, Suk J‐E, Bae E‐J, Lee J‐H, Paik SR, Lee S‐J (2008) Assembly‐dependent endocytosis and clearance of extracellular α‐synuclein. Int J Biochem Cell Biol 40:1835–1849. [DOI] [PubMed] [Google Scholar]
- 46. Lee H‐J, Suk J‐E, Bae E‐J, Lee S‐J (2008) Clearance and deposition of extracellular α‐synuclein aggregates in microglia. Biochem Biophys Res Commun 372:423–428. [DOI] [PubMed] [Google Scholar]
- 47. Lewis KA, Yaeger A, DeMartino GN, Thomas PJ (2010) Accelerated formation of alpha‐synuclein oligomers by concerted action of the 20S proteasome and familial Parkinson mutations. J Bioenerg Biomembr 42:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lewy FH (1912) Paralysis agitans. I. Pathologische anatomie. In: Handbuch der Neurologie, Vol. 3. Lewandowsky M, Abelsdorff G (eds), pp. 920–933. Springer‐Verlag: Berlin. [Google Scholar]
- 49. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ et al (2008) Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host‐to‐graft disease propagation. Nat Med 14:501–503. [DOI] [PubMed] [Google Scholar]
- 50. Li JY, Englund E, Widner H, Rehncrona S, Bjorklund A, Lindvall O, Brundin P (2010) Characterization of Lewy body pathology in 12‐ and 16‐year‐old intrastriatal mesencephalic grafts surviving in a patient with Parkinson's disease. Mov Disord 25:1091–1096. [DOI] [PubMed] [Google Scholar]
- 51. Li J, Uversky VN, Fink AL (2001) Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha‐synuclein. Biochemistry 40:11604–11613. [DOI] [PubMed] [Google Scholar]
- 52. Li W, Hoffman PN, Stirling W, Price DL, Lee MK (2004) Axonal transport of human alpha‐synuclein slows with aging but is not affected by familial Parkinson's disease‐linked mutations. J Neurochem 88:401–410. [DOI] [PubMed] [Google Scholar]
- 53. Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VMY (2012) Pathological α‐synuclein transmission initiates Parkinson‐like neurodegeneration in nontransgenic mice. Science 338:949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR et al (2009) Exogenous α‐synuclein fibrils seed the formation of Lewy body‐like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 106:20051–20056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VMY (2012) Intracerebral inoculation of pathological α‐synuclein initiates a rapidly progressive neurodegenerative α‐synucleinopathy in mice. J Exp Med 209:975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA et al (2005) High‐resolution whole‐genome association study of Parkinson disease. Am J Hum Genet 77:685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maroteaux L, Campanelli JT, Scheller RH (1988) Synuclein: a neuron‐specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8:2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L et al (2012) Prion‐like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 33:2225–2228. [DOI] [PubMed] [Google Scholar]
- 59. Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D et al (1999) Both familial Parkinson's disease mutations accelerate alpha‐synuclein aggregation. J Biol Chem 274:9843–9846. [DOI] [PubMed] [Google Scholar]
- 60. Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK et al (2010) Increased expression of alpha‐synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M (2010) Seeded aggregation and toxicity of α‐synuclein and tau: cellular models of neurodenegerative diseases. J Biol Chem 285:34885–34898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Olanow CW, Prusiner SB (2009) Is Parkinson's disease a prion disorder? Proc Natl Acad Sci U S A 106:12571–12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Paillusson S, Clairembault T, Biraud M, Neunlist M, Derkinderen P (2012) Activity‐dependent secretion of alpha‐synuclein by enteric neurons. J Neurochem. doi: 10.1111/jnc.12131. [DOI] [PubMed] [Google Scholar]
- 64. Pan‐Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R et al (2010) Progression of Parkinson's disease pathology is reproduced by intragastric administration of rotenone in mice. Plos ONE 5:e8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pan‐Montojo F, Schwarz M, Winkler C, Arnhold M, O'Sullivan GA, Pal A et al (2012) Environmental toxins trigger PD‐like progression via increased alpha‐synuclein release from enteric neurons in mice. Sci Rep 2:898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Parkkinen L, Pirttilä T, Alafuzoff I (2008) Applicability of current staging/categorization of α‐synuclein pathology and their clinical relevance. Acta Neuropathologica 115:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al (1997) Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. [DOI] [PubMed] [Google Scholar]
- 68. Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA (2000) Fiber diffraction of synthetic alpha‐synuclein filaments shows amyloid‐like cross‐beta conformation. Proc Natl Acad Sci U S A 97:4897–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shah JV, Cleveland DW (2002) Slow axonal transport: fast motors in the slow lane. Curr Opin Cell Biol 14:58–62. [DOI] [PubMed] [Google Scholar]
- 70. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J et al (2003) alpha‐Synuclein locus triplication causes Parkinson's disease. Science 302:841. [DOI] [PubMed] [Google Scholar]
- 71. Soper JH, Kehm V, Burd CG, Bankaitis VA, Lee VMY (2011) Aggregation of α‐synuclein in S. cerevisiae is associated with defects in endosomal trafficking and phospholipid biosynthesis. J Mol Neurosci 43:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha‐synuclein in Lewy bodies. Nature 388:839–840. [DOI] [PubMed] [Google Scholar]
- 73. Tang Y, Das U, Scott DA, Roy S (2012) The slow axonal transport of alpha‐synuclein—mechanistic commonalities amongst diverse cytosolic cargoes. Cytoskeleton 69:506–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Uversky VN, Eliezer D (2009) Biophysics of Parkinson's disease: structure and aggregation of alpha‐synuclein. Curr Protein Pept Sci 10:483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Volpicelli‐Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A et al (2011) Exogenous α‐synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J et al (2011) A soluble α‐synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 108:17797–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Waxman EA, Giasson BI (2010) A novel, high‐efficiency cellular model of fibrillar α‐synuclein inclusions and the examination of mutations that inhibit amyloid formation. J Neurochem 113:374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zarranz JJ, Alegre J, Gomez‐Esteban JC, Lezcano E, Ros R, Ampuero I et al (2004) The new mutation, E46K, of alpha‐synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. [DOI] [PubMed] [Google Scholar]