

Abstract
Visual refractive errors (REs) are complex genetic traits with a largely unknown etiology. To date, genome-wide association studies (GWASs) of moderate size have identified several novel risk markers for RE, measured here as mean spherical equivalent (MSE). We performed a GWAS using a total of 7280 samples from five cohorts: the Age-Related Eye Disease Study (AREDS); the KORA study (‘Cooperative Health Research in the Region of Augsburg’); the Framingham Eye Study (FES); the Ogliastra Genetic Park-Talana (OGP-Talana) Study and the Multiethnic Study of Atherosclerosis (MESA). Genotyping was performed on Illumina and Affymetrix platforms with additional markers imputed to the HapMap II reference panel. We identified a new genome-wide significant locus on chromosome 16 (rs10500355, P = 3.9 × 10−9) in a combined discovery and replication set (26 953 samples). This single nucleotide polymorphism (SNP) is located within the RBFOX1 gene which is a neuron-specific splicing factor regulating a wide range of alternative splicing events implicated in neuronal development and maturation, including transcription factors, other splicing factors and synaptic proteins.
INTRODUCTION
Refractive error (RE) is the most common human eye disorder and includes three phenotypes: myopia, astigmatism and hyperopia (1). RE, usually assessed as a quantitative measurement, mean spherical equivalent (MSE), refers to the dioptric power of optical lenses necessary to achieve proper distance correction. Myopia, by convention represented by negative values of RE, affects more than one in four individuals over age 40 in the United States and Western Europe, while hyperopia (positive values of RE) is present in about 10% of individuals in the same age group. In Asia, the prevalence of myopia is even higher, exceeding 70% in some Asian countries, which makes it an even stronger public health concern (2,3). Worldwide, more than 150 million people are estimated to be visually impaired because of uncorrected RE, of whom 8 million are functionally blind (4).
Both environmental and genetic factors are known to be involved in the development of RE. In particular, several studies have shown that reading and other near work tasks are risk factors for myopia development. Outdoor activity is thought to be protective against myopia (5–9). Despite wide agreement that REs are heavily influenced by environmental and behaviorial factors, a large fraction of the variance of refraction within populations is accounted for by heritable factors. Heritability estimates for RE are consistently high across a wide spectrum of ethnic groups despite varying inter-ethnic differences in the prevalence of REs and predisposing environmental factors for myopia development. For example, the heritability of RE in an adult Old Order Amish population (a cultural and genetic isolate with a low incidence of myopia) was found to be 70% (10). High heritabilities, greater than 50%, have also been recorded in several other populations of diverse ancestries including Caucasian Americans (11,12), Europeans (13–16) and African Americans (11). Familial aggregation studies have estimated sibling recurrence risk ratios, λs (defined as the ratio of disease manifestation, given that one's sibling is affected, compared with disease prevalence in the general population) to range from 2 to 5.6 for myopia and 1.6 to 4.9 for hyperopia in US Caucasians (10,11,17). In addition, various monogenic connective tissue disorders such as Marfan and Stickler syndromes are associated with myopia (18). Over 16 susceptibility loci have been mapped in familial myopia by linkage analysis (18). This evidence strongly supports a large contribution of genetic factors to the physiopathology of RE.
Identification of common genetic variants that are associated with RE has also come through the use of genome-wide high-density single nucleotide polymorphism (SNP) array genotyping. Three different genome-wide association studies (GWASs) of myopia in Chinese cohorts have identified common variants located on chromosomes 13q12, 4q25 and 5p15 (19–21). Two GWASs of RE in populations of European ancestry have identified common variants located on chromosomes 15q25 and 15q14 (22,23), the latter replicating across various studies representing diverse ethnic groups (24). A GWAS on myopia in a Japanese cohort has identified common variants at 11q24 (25) while a GWAS on axial length in Singapore adults identified a locus at 1q41 (26). Because of unequal linkage disequilibrium structures across different populations and potential interactions between genetic variants and environmental factors, effect sizes of common genetic variants may differ between populations. This difference in effect size is demonstrated by the inconsistent signal for RE across different ethnicities for 15q25. Hence, we conducted an independent GWAS on MSE in Caucasian individuals of European descent and perforned a meta analysis of MSE in five cohorts to increase sample size and the likelihood of identifying additional genetic variants associated with RE. Replication was also performed across a large series of additional cohorts.
RESULTS
Baseline characteristics for subjects from the five studies: Age-Related Eye Disease Study (AREDS), Framingham Eye Study (FES), Ogliastra Genetic Park-Talana (OGP-Talana) Study, Multiethnic Study of Atherosclerosis (MESA), and Cooperative Health Research in the Region of Augsburg (KORA) are shown in Table 1, with further characteristics of each study sample more fully described in supplementary files.
Table 1.
Baseline characteristics of the five discovery samples (mean ± standard deviation or percentage)
| AREDS | KORA | FES | MESA | OGP-Talana | |
|---|---|---|---|---|---|
| N | 1877 | 1869 | 1389 | 1462 | 683 |
| Mean age (SD) | 68 (4.7) | 55.6 (11.8) | 55.6 (8.9) | 61.9 (9.4) | 42.2 (19.1) |
| Average MSE (SD) | 0.6 (2.1) | −0.8 (7.3) | 0.2 (2.4) | −0.3 (2.6) | −0.2 (1.8) |
| Sex (% male) | 41 | 50 | 43 | 49 | 42 |
| Lambda | 1.005 | 1.014 | 1.037 | 1.014 | 1.164 |
In the discovery stage, testing for population stratification using EIGENSOFT and principal components analysis (PCA) found no evidence of population stratification in the KORA data but significant principal components were detected in the AREDS, FES and MESA samples. These were adjusted for in the subsequent genome-wide association analysis by including the three most significant principal components from the PCA analysis as covariates in our regression models. The OGP-Talana data were adjusted jointly for cryptic relatedness and population stratification. Fixed-effect meta-analysis across the five cohorts for MSE showed no evidence of substantial excess of statistics deviating from the null expectations as assessed by an overall genomic control value of 1.018, indicating that stratification has been accounted for (Fig. 1). Genomic control values for each population prior to meta-analysis are given in Table 1.
Figure 1.

The quantile–quantile plot for the five-cohort meta-analysis after individual study adjustment for genetic ancestry using principal components and genomic control.
The Manhattan plot of the discovery meta-analysis showed several genomic regions as potential risk loci (Fig. 2). Seven genome-wide significant SNPs were located close to one another (45177852-45169413bp) on chromosome 10 (Supplementary Material, Table S1). Complete discovery meta-analysis P-values and related information for all SNPs that passed quality control are available online in the Stambolian_RE_meta_chr#.csv files (where # = 1–22). The most significant P-value was determined for rs12571148 on chromosome 10 (β = 0.298, P-value = 2.02 × 10−8). Nineteen nearby SNPs on chromosome 10 showed suggestive associations (P-values ranging from 5.18 × 10−8 to 1 × 10−7). An additional 83 SNPs located in 12 different chromosomal regions had P-values of ≤1 × 10−5 (Supplementary Material, Table S1).
Figure 2.

Genome-wide Manhattan plot of genotyped and imputed SNPs for meta-analysis of RE in five discovery samples. Gray line represents the genome-wide significance level P = 5 × 10−8.
For replication, SNPs from the discovery meta-analysis with P ≤ 1 × 10−5 were ranked, and clustering within linkage disequilibrium blocks was examined. A total of 22 SNPs (Table 2) were chosen for replication in a total of 19 763 samples from BMES, CROATIA-Korcula, CROATIA-Split, CROATIA-Vis, ORCADES, DCCT, RS1, RS2, RS3 and Erasmus Rucphen Family Study (ERF) (see supplementary data for replication study descriptions). These consisted of one to four SNPs from each of the 11 high LD regions displaying significant or suggestive association in the discovery meta-analysis. Two SNPs on chromosome 16, rs4581716 and rs10500355, both located in close vicinity to each other within the RBFOX1 gene gave strong signals in the replication analysis (replication, P = 7.34 × 10−5) (Table 2). The three SNPs at that locus that were genome-wide significant in the discovery meta-analysis (Supplementary Material, Table S1) did not show evidence of replication (all replication, P > 0.3, Table 2). In the combined meta-analysis of the discovery and replication sample sets, rs10500355 in the RBFOX1 gene on chromosome 16 achieved genome-wide significance (rs10500355, P = 3.9 × 10−9, β = −0.111) (Fig. 3) and the highly correlated SNP rs4581716 (r2 = 0.63) was nearly significant (P = 1.6 × 10−7, β = 0.098) (Table 2, Fig. 2). Both SNPs, rs4581716 and rs10500355, are located in the same intron of RBFOX1, between exon 1E and 8, 1212 bp apart from each other (Fig. 4). Previously, no diseases have been reported to be associated with either SNP. A search for evolutionary conservation using the ECR browser (http://ecrbrowser.dcode.org/, last accessed date on March 13, 2013) (27) around these SNPs found modest conservation around rs4581716 from human to dog and high conservation around rs10500355 from human to rodent and chicken. Additionally, a PAX6 binding site was found 13 bp from rs10500355 (http://genome.ucsc.edu/, last accessed date on March 13, 2013). MultiTF analysis (http://multitf.dcode.org/, last accessed date on March 13, 2013) demonstrated GATA3 and OCT4 binding at rs10500355, while sTRAP (http://trap.molgen.mpg.de/cgi-bin/trap_form.cgi, last accessed date on March 13, 2013) analysis found significant binding of HNF1a (P-value for WT: 0.282; P-value for minor allele: 0.044). Enhancers/promoter analysis on these highly conserved regions using FPROM, FirstEF, NNPP and Promoter 2.0 did not find any promoters or enhancers in this region. The ENCODE database (http://genome.ucsc.edu/ENCODE/, last accessed date on March 13, 2013) lists a DNaseI Hypersensitivity cluster 100 bp upstream of rs1050035. This cluster is annotated as present in two cell lines analyzed by the ENCODE consortium: H7-hESC (embryonic stem cells) and WERI-RB-1 (retinoblastoma). There is also a small H3K4Me3 mark 450 bp upstream of rs1050035 in the NHLF cell line (lung fibroblast).
Table 2.
Details of 22 SNPs used in the replication phase including rank based on the most significant P-value from the discovery phase
| Rank | SNP | Chra | Bpb | Discovery—5 samples | Pd | Directione (A-K-F-OT-M) | Replication—10 samples |
Direction (B-CK-CS-CV-D-E-O-R1-R2-R3) | Meta-analysis |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β (95% CI)c | β (95% CI) | P | β (95% CI) | P | |||||||
| 7 | rs1343654 | 2 | 11005484 | 0.22 (0.137, 0.303) | 1.81 × 10−7 | +++?+ | 0.028 (−0.015,0.07) | 2.04 × 10−1 | +−+++−++++ | 0.069 (0.0314, 0.107) | 5.68 × 10−5 |
| 20 | rs13094898 | 3 | 58290143 | −0.232 (−0.333, −0.131) | 6.78 × 10−6 | −−−−− | −0.063 (−0.132, 0.006) | 7.46 × 10−2 | −−−−+−−−−+ | −0.121 (−0.177,−0.065) | 6.63 × 10−5 |
| 14 | rs11715445 | 3 | 58343887 | 0.243 (0.14, 0.345) | 3.20 × 10−6 | +++++ | 0.066 (−0.003, 0.135) | 6.31 × 10−2 | ++++−+++++ | 0.125 (0.068, 0.181) | 5.95 × 10−5 |
| 21 | rs10018930 | 4 | 111029634 | −0.173 (−0.249, −0.097) | 7.50 × 10−6 | −−−−− | −0.035 (−0.084, 0.013) | 1.60 × 10−1 | +++−−+−−−+ | −0.075 (−0.115,−0.034) | 3.28 × 10−4 |
| 22 | rs10033229 | 4 | 111030074 | 0.172 (0.096, 0.247) | 8.31 × 10−6 | +++++ | 0.034 (−0.015, 0.083) | 1.68 × 10−1 | −−−++−+++− | 0.076 (0.035, 0.112) | 2.64 × 10−4 |
| 10 | rs13178105 | 5 | 78268128 | −0.247 (−0.347, −0.146) | 1.59 × 10−6 | −−−?− | −0.05 (−0.106,0.006) | 7.93 × 10−2 | +−−−+−−−−− | −0.097 (−0.146,−0.048) | 3.07 × 10−4 |
| 18 | rs583456 | 6 | 24948279 | 0.194 (0.111, 0.277) | 4.65 × 10−6 | +++++ | 0.036 (−0.012, 0.084) | 1.46 × 10−1 | −+++++++−0 | 0.077 (0.036, 0.118) | 7.48 × 10−4 |
| 13 | rs9490548 | 6 | 123090476 | −1.394 (−1.977, −0.811) | 2.75 × 10−6 | ??−?− | 0.011 (−0.33, 0.349) | 9.51 × 10−1 | ????−+?−+− | −0.355 (−0.645, −0.064) | 6.37 × 10−3 |
| 5 | rs7912000 | 10 | 45161041 | −0.289 (−0.396, −0.181) | 1.30 × 10−7 | −−−−− | 0.028 (−0.045, 0.1) | 4.51 × 10−1 | −−−+++−+−+ | −0.077 (−0.137,−0.018) | 2.13 × 10−3 |
| 3 | rs12266496 | 10 | 45169414 | 0.281 (0.180, 0.382) | 4.55 × 10−8 | +++++ | −0.031 (−0.093, 0.031) | 3.33 × 10−1 | −++−−−+−+− | 0.042 (−0.007,0.091) | 1.30 × 10−3 |
| 4 | rs7897547 | 10 | 45177653 | −0.288 (−0.392, −0.184) | 5.18 × 10−8 | −−−−− | 0.025 (−0.045, 0.094) | 4.87 × 10−1 | −−−+++−+−+ | −0.078 (−0.135, −0.021) | 1.30 × 10−3 |
| 2 | rs12771080 | 10 | 45181333 | −0.293 (−0.397, −0.189) | 3.33 × 10−8 | −−−−− | 0.028 (−0.042, 0.097) | 4.37 × 10−1 | −−−+++−+−+ | −0.077 (−0.134,−0.02) | 1.44 × 10−3 |
| 1 | rs12571148 | 10 | 45186816 | 0.298 (0.194, 0.402) | 2.01 × 10−8 | +++++ | −0.024 (−0.093, 0.046) | 5.02 × 10−1 | +++−−−+−+− | 0.081 (0.024, 0.138) | 8.98 × 10−4 |
| 19 | rs10771293 | 12 | 26843918 | −0.149 (−0.213, −0.085) | 5.61 × 10−6 | −−−−− | −0.012 (−0.052, 0.028) | 5.59 × 10−1 | −+−−+−++−− | −0.053 (−0.087,−0.019) | 1.02 × 10−3 |
| 12 | rs1570142 | 14 | 94856261 | −0.16 (−0.226, −0.093) | 2.51 × 10−6 | −−−−− | −0.006 (−0.048, 0.036) | 7.73 × 10−1 | −++−++−−−0 | −0.051 (−0.0866, −0.016) | 2.84 × 10−3 |
| 11 | rs7154685 | 14 | 94867604 | −0.16 (−0.225, −0.094) | 1.82 × 10−6 | −−−−− | −0.002 (−0.042, 0.038) | 9.14 × 10−1 | −++−−++−−+ | −0.047 (−0.08,−0.013) | 4.14 × 10−3 |
| 16 | rs1956704 | 14 | 94870584 | 0.154 (0.088, 0.219) | 3.97 × 10−6 | +++++ | 0 (−0.04, 0.04) | 9.85 × 10−1 | +−−++−−++− | 0.044 (0.01, 0.078) | 5.97 × 10−3 |
| 8 | rs4776318 | 15 | 67074656 | −0.169 (−0.238, −0.101) | 1.16 × 10−6 | −−−−− | −0.028 (−0.07, 0.014) | 1.95 × 10−1 | −−++−−+−−− | −0.069 (−0.104,−0.0334) | 1.75 × 10−4 |
| 15 | rs8025869 | 15 | 67084685 | 0.159 (0.092, 0.227) | 3.61 × 10−6 | +++++ | 0.024 (−0.016, 0.063) | 2.48 × 10−1 | ++−−++−−++ | 0.061 (0.027, 0.096) | 6.83 × 10−4 |
| 6 | rs4581716 | 16 | 7458135 | 0.185 (0.116, 0.255) | 1.79 × 10−7 | +++++ | 0.06 (0.015, 0.104) | 9.00 × 10−3 | −+++++++++ | 0.098 (0.061, 0.136) | 1.64 × 10−7 |
| 17 | rs10500355 | 16 | 7459347 | −0.162 (−0.23, −0.093) | 4.13 × 10−6 | −−−−− | −0.089 (−0.131,−0.044) | 7.34 × 10−5 | −−−+−−−−−− | −0.111 (−0.147,−0.074) | 3.92 × 10−9 |
| 9 | rs8109324 | 19 | 53238740 | 0.184 (0.109, 0.259) | 1.54 × 10−6 | +++++ | −0.01 (−0.056, 0.036) | 6.74 × 10−1 | −−+++−−++− | 0.045 (0.0066, 0.084) | 6.06 × 10−3 |
Studies are listed in order at the top of the Direction columns as AREDS (A), KORA (K), FES (F), OGP-Talana (OT), MESA (M) in the discovery set and BMES (B), Croatia-Korcula (CK), Croatia-Split (CS), Croatia-Vis (CV), DCCT (D), ERF (E), ORCADES(O), RS1 (R1), RS2 (R2), RS3 (R3) in the replication set.
aChr—chromosome location of SNP.
bBP—base pair position of SNP.
cβ (95% CI)—estimate of the effect size of the tested allele and its 95% confidence interval.
dP—significance level of the association test of MSE with this SNP.
dDirection—direction of effect of the tested allele (+ = protective, − = risk, 0 = no effect, ? = no data) in each sample.
Figure 3.

Forest plot of betas for spherical equivalent for top SNP rs10500355.
Figure 4.
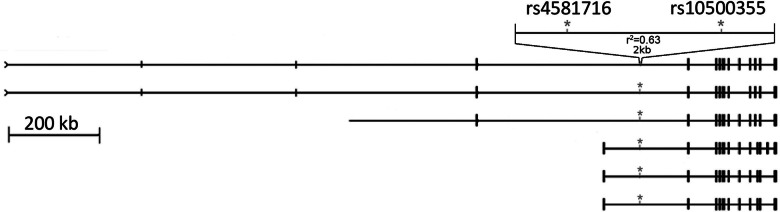
RBFOX1 isoforms and location of rs4581716 and rs10500355.
We also assessed 14 SNPs on chromosome 15q14 reported previously as being associated with RE (23). They have been replicated in a large international meta-analysis consortium (24), but did not show even nominal significance (P ≤ 0.05) in the meta-analysis of the five discovery samples or individually in AREDS, KORA, FES or OGP-TALANA. However, 6 of the 14 SNPs (rs11073058, rs11073059, rs11073060, rs7163001, rs4924134, rs619788) were close to nominal significance in the KORA data (all with P ≤ 0.062). Nominally significant evidence of replication was found in MESA, where 13 of the 14 SNPs had P-values ranging between 0.027 and 0.0066; the top SNP was rs8032019 (P ≤ 0.0066). We were not able to replicate significance for any of the five SNPs previously described as significant at the 15q25 region (22) in our discovery meta-analysis or in any of the individual discovery samples. Supplementary Material, Table S4, lists the SNPs, their heterogeneity test P-values and their association P-values from the meta-analysis of our five discovery datasets. None of the SNPs were significantly heterogeneous across the five populations.
DISCUSSION
While a heritable component to RE has been recognized for several decades, the genetic determinants of RE have been generally elusive. Our analysis adds an additional large GWAS to two prior European GWASs on RE and identifies a new genome-wide significant locus associated with RE susceptibility at chromosome 16p13.3. While the genome-wide significant SNPs on chromosome 10 from the discovery meta-analysis did not replicate in our replication meta-analysis, this region should be examined in a larger set of independent samples. To our knowledge, this region on chromosome 10 (10q11.21) has not been seen in other published association studies of MSE. The Solouki et al. (23) GWAS on MSE found suggestive evidence of association (P < 1 × 10−6) at 10p12.3, but this was 23 Mb from our discovery step signal at 10q11.21). Moreover, the Rotterdam RS1 data used as the discovery set in the Solouki et al. paper (23) was one of our replication datasets and did not show any evidence of association to our 10q11.21 replication SNPs. However, Nallasamy et al. (28) mapped high-grade myopia in a Hutterite family to a locus at 10q21.1 using linkage analysis (LOD = 3.22). Their linkage peak is located ∼10 Mb distal of our association signal. It remains to be seen whether the two signals might represent the same locus.
In this GWAS of RE in Caucasians, combined analysis showed the strongest association with two highly correlated variants, namely rs10500355 and rs4581716 on 16p13.3 in the RBFOX1 gene, with the former reaching genome-wide significance. Details of both SNPs are summarized in Supplementary Material, Table S6. RBFOX1 (RNA-Binding Fox-1 homolog) codes a tissue-specific alternative splicing regulator expressed in many different tissues including the eye (http://genome.ucsc.edu/, last accessed date on March 13, 2013) (29,30). Some observations in humans indicated that mutations of this gene might possibly be implicated in rare forms of ataxia, retardation, epilepsy or autism (31–34). Zhang et al. (35) analyzed RBFOX1 gene targets and predicted that six myopia genes were targets of RBFOX1. These genes include SHQ1 and IGF1R, both differentially expressed in a chick model of myopia (36,37); FGFR-1, differentially expressed in a tree shrew model for myopia (38); and HGF (39,40) and ZNF644 (41), in which mutations have been detected in myopic patients by exome sequencing. HGF has also been shown to induce expression of egr-1/ZENK, a modifier of myopia development (39,40). In the five datasets used in our discovery meta-analysis, some evidence of association (P < 0.01) was observed in or near four of these five genes (FGFR1, IGF1R, ZNF644, SHQ1 but not HGF) in either the meta-analysis or at least one of the discovery datasets (Supplementary Material, Figures S6–S12). In the case of IGF1R, the association P-value of an intronic SNP rs11635251 is 0.0035 in the meta-analysis and 0.0011 in the AREDS data. While none of these results are genome-wide significant, they provide further support to the prior evidence that this pathway may be involved in RE development in humans.
Two recent publications (31,42) describe the RBFOX1 protein as a regulator of both neuronal alternative splicing and coordinative transcription, both of which are required for normal neurological development. Dysfunction of this regulation leads to neurological disease. From other resources, there is additional evidence why dysregulation of the RBFOX1 gene may lead to abnormal development of refraction. First, the gene is expressed in the retina according to the NCBI UniGene (http://www.ncbi.nlm.nih.gov/unigene) database and in the human retina and RPE/choroid/sclera according to our RNA-Seq data (unpublished). Second, the RBFOX1 protein has been shown to interact with a number of genes that have been related to human myopia and animal models of myopia (see above). Third, our two most significant SNPs are located in a highly conserved region and very close to a DNase1 hypersensitivity site and may be either in linkage disequilibrium with another SNP that influences this site or directly influence this site themselves. It is our hypothesis that RBFOX1 expression is altered to an extent that the growth of the eye is affected resulting in an RE phenotype. It is also possible that binding of Oct4 and/or GATA3 at rs10500355 that is predicted by MultiTF analysis may influence the transcription of RBFOX. However, neither GATA3 nor Oct4 has been reported to be associated with RE.
No statistically significant evidence of association was found in any of the discovery datasets or the discovery meta-analysis (Supplementary Material, Table S4) between RE and the previously reported associated SNPs at the 15q25 (22) region, although power calculations based on the published effect sizes suggested that we should have had adequate power for replication (Supplementary Material, Table S5). Moreover, no evidence of association with RE was observed in the discovery meta-analysis (Supplementary Material, Table S4) or in the AREDS, KORA, OGP-Talana or FES samples with SNPs at 15q14 that also were previously described as being significantly associated (23). The replication of the 15q14 association by a larger meta-analysis (24) suggests that our failure to replicate this region may be a matter of insufficient power (given that the published effect sizes may be inflated) or of differing LD patterns across the studies.
In conclusion, our GWAS increases the number of genome-wide significant loci associated with MSE to three in Caucasians. Although this study brings together over 7000 individuals in the discovery cohorts and is comparable in size with the two previous GWASs on RE in Caucasians, there is an evident need for meta-analyses even larger than the present one in order to produce a more extensive list of associated variants. Moreover, other types of genetic variation (copy number variants, rare variants including de novo mutations) will likely further expand the list of causative genetic loci. Studies directed at these types of genetic variations should also be conducted in order to improve our understanding of RE susceptibility.
MATERIALS AND METHODS
Populations
The five GWASs meta-analyzed in the discovery study included 1869 subjects aged 35–84 years from the Cooperative Health Research in the Region of Augsburg Study (KORA F3, Southern Germany), 1877 subjects aged 55–81 from the Age-related Eye Study (AREDS), 1389 unrelated subjects aged 28–84 from the FES, 1462 subjects aged 46–86 from the Multi-Ethnic Study of Atherosclerosis (MESA) study and 683 subjects aged 18–88 from the OGP-Talana study in Sardinia, resulting in a total sample size of 7280. All individuals were of European ancestry. Approval was obtained by the local ethics committees for all studies and an informed consent was obtained from the study participants. Validation samples were obtained from Australia, Croatia (three independent studies), Canada, Netherlands (four independent samples) and Scotland for a total of 19 673 individuals. Key features of these populations are found in the Supplementary Material.
Study design
The study was divided into three stages: a discovery stage, a validation stage and a combined analysis.
Discovery stage
A GWAS of spherical equivalent using the Illumina HumanOmni2.5 chip (2.5 million SNPs) was performed on the AREDS and KORA Study (‘Cooperative Health Research in the Region of Augsburg’). The results from these two datasets were then combined into a ‘discovery’ meta-analysis with total GWAS results from three other datasets (genotyped on other array platforms) drawn from the following studies: the FES, the OGP-Talana study and the MESA.
Validation stage
The most significant results from this discovery meta-analysis were then replicated by meta-analysis of association results from the following studies: Croatia-Korcula, Croatia-Split, Croatia-Vis, ORCADES, The Diabetes Control and Complications Trial (DCCT), the Blue Mountains Eye Study, three cohorts from the Rotterdam Study (RS1, RS2, RS3) and ERF. All SNPs requested from all datasets for which the principal investigators of the respective studies entered into collaboration are included in the replication analysis. Finally, a fixed-effect meta-analysis of the discovery and replication results was performed on all SNPs used in the replication analysis (i.e. all the SNPs in Table 2 that were chosen from the discovery meta-analysis).
SNPs were chosen from the discovery meta-analysis ranked by meta-analysis P-value (≤1 × 10−5), but only a subset of such SNPs were chosen per region to reduce the number of SNPs used in the replication analysis. Twelve regions were identified (two on chromosome 10) which contained at least one SNP with meta-analysis P-value ≤1 × 10−5. From these regions, two lists of SNPs were chosen. The first list contained the most significant SNP from each location (one SNP each on chromosomes 2, 3, 4, 5, 6, 10, 12, 14, 15, 16 and 19). The second list included the next most significant SNPs from each location that met our significance threshold for replication. In regions where multiple SNPs met the significance threshold, replication SNPs were chosen to span the region. Thus, no additional SNPs were chosen for chromosomes 2, 5, 12 and 19, one additional SNP each was picked from the chromosomes 3, 4, 6, 15 and 16 regions, two additional SNPs on chromosomes 14 and 4 on chromosome 10. The regions on chromosomes 10 and 14 had multiple SNPs chosen for this replication because they had a large number of SNPs in a small region that were close to genome-wide significance (see Table 2 for P-values of these SNPs in the Discovery meta-analysis and Supplementary Material, Figure S12). Fixed-effect meta-analyses were performed on the resulting set of 22 SNPs with METAL (43) using the standard error and effect size for each population and a two-sided test. Genomic control was performed for each study and then again for the final meta-analysis.
Replication of 15q Loci: Replication of previously reported associations on 15q14 and 15q25 were performed in PLINK and METAL using a two-sided test. Heterogeneity scores were calculated in METAL, using a chi-square test with n − 1 degrees of freedom where n is the number of studies being analyzed. For the 15q14 locus, we had 70% power to detect a variant in the AREDS sample alone (at P = 0.025). For the 15q25 locus, we had 85% power to detect a signal in the AREDS sample. In the combined discovery datasets, our expected power was 0.9999 and 1 for these two candidate regions, respectively (see Supplementary Material, Table S5). Power calculations assumed an additive quantitative trait loci variance based on the reported effect size from the original publications of 0.005 for 15q14 (23) and 0.007 for 15q25 (22), based on average minor-allele frequencies across all the original SNPs of 0.4 and assuming complete LD (D′ = 1) between the causal allele and tested SNPs.
Combined analysis
Finally, a meta-analysis of the discovery and replication datasets was performed on the SNPs carried over to the validation stage.
Quality control of discovery datasets
AREDS and KORA
Individuals with chromosome abnormalities and sex discrepancies were removed. Cryptic relatedness was estimated by calculating pairwise identical by descent (IBD) coefficients using the software PLINK (version 1.07; http://pngu.mgh.harvard.edu/~purcell/plink, last accessed date on March 13, 2013) (44) and pseudo-kinship coefficients using EMMAX (45) for all pairs of individuals in the study. Estimates of the average proportion of alleles shared IBD (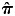 ) were almost identical using both methods. For each pair with a kinship coefficient of ≥0.125, one member of the pair was dropped based on the genotyping rate and trait phenotype, preferring to retain the person with a higher genotyping rate and more extreme phenotype. Population stratification was assessed using a subset of ∼40 000 independent, polymorphic (MAF > 0.01), autosomal markers from the Human OMNI 2.5 panel. We utilized the principal components method in the EIGENSTRAT (46) software and implemented in the EIGENSOFT (47) package (version 3.0; http://genepath.med.harvard.edu/~reich/Software.htm, last accessed date on March 13, 2013) for population stratification testing. We investigated possible batch effects by testing each batch against the others using Fisher's exact test to account for possible small numbers in some cells and the loop-assoc function in PLINK. We also used this function to look for patterns of missingness per batch. As AREDS was a multicenter study, we also tested for differences between collection sites. Samples were dropped for poor performance on the array (genotyping rate of <98%). We used Hardy–Weinberg test statistics generated using PLINK to identify those SNPs which were poor performers in both populations. An SNP was also removed from a population if its call rate was <99%, its minor-allele frequency was <0.01 or if its distribution departed significantly from Hardy–Weinberg expectations (HWE, P < 1 × 10−4) in a single population, as recommended by the GENEVA consortium (48). We additionally dropped SNPs in both populations, where HWE P < 1 × 10−4 in one population and HWE P < 1 × 10−3 in the other. This was because the two populations were genotyped at the same time in the same laboratory, with samples from both populations represented on every plate. If an SNP did not look good in one population, it is reasonable to exclude it if the other population is approaching the cutoff for exclusion. SNPs were also excluded if they showed more than one genotype inconsistency between: (1) HapMap control samples and the consensus genotype in the HapMap database (http://hapmap.ncbi.nlm.nih.gov/, last accessed date on March 13, 2013) or (2) investigator-provided duplicate samples. The final marker list contained 2 182 680 high-quality SNPs with a minor-allele frequency of ≥0.01, a genotype call rate >99%, and whose distribution was consistent with HWEs (P > 1 × 10−4).
) were almost identical using both methods. For each pair with a kinship coefficient of ≥0.125, one member of the pair was dropped based on the genotyping rate and trait phenotype, preferring to retain the person with a higher genotyping rate and more extreme phenotype. Population stratification was assessed using a subset of ∼40 000 independent, polymorphic (MAF > 0.01), autosomal markers from the Human OMNI 2.5 panel. We utilized the principal components method in the EIGENSTRAT (46) software and implemented in the EIGENSOFT (47) package (version 3.0; http://genepath.med.harvard.edu/~reich/Software.htm, last accessed date on March 13, 2013) for population stratification testing. We investigated possible batch effects by testing each batch against the others using Fisher's exact test to account for possible small numbers in some cells and the loop-assoc function in PLINK. We also used this function to look for patterns of missingness per batch. As AREDS was a multicenter study, we also tested for differences between collection sites. Samples were dropped for poor performance on the array (genotyping rate of <98%). We used Hardy–Weinberg test statistics generated using PLINK to identify those SNPs which were poor performers in both populations. An SNP was also removed from a population if its call rate was <99%, its minor-allele frequency was <0.01 or if its distribution departed significantly from Hardy–Weinberg expectations (HWE, P < 1 × 10−4) in a single population, as recommended by the GENEVA consortium (48). We additionally dropped SNPs in both populations, where HWE P < 1 × 10−4 in one population and HWE P < 1 × 10−3 in the other. This was because the two populations were genotyped at the same time in the same laboratory, with samples from both populations represented on every plate. If an SNP did not look good in one population, it is reasonable to exclude it if the other population is approaching the cutoff for exclusion. SNPs were also excluded if they showed more than one genotype inconsistency between: (1) HapMap control samples and the consensus genotype in the HapMap database (http://hapmap.ncbi.nlm.nih.gov/, last accessed date on March 13, 2013) or (2) investigator-provided duplicate samples. The final marker list contained 2 182 680 high-quality SNPs with a minor-allele frequency of ≥0.01, a genotype call rate >99%, and whose distribution was consistent with HWEs (P > 1 × 10−4).
Framingham eye study
Quality control was carried out in several stages. Samples were chosen based on pedigree information and genotyping quality. Samples with a genotypic call rate below 98% were excluded from the analysis. The mean call rate for analyzed samples was 99.2% (SD = 0.4%). The final marker list contained 436 494 high-quality SNPs with a minor-allele frequency of ≥0.01, a Mendelian error rate below 2% across all pedigrees, a genotype call rate above 95%, and whose distribution was consistent with HWEs (P > 1 × 10−4).
MESA
SNPs with MAF ≤0.02 or HWE, P-value ≤0.001 were removed from the analysis. Genotyping was performed using the Affymetrix Genome-Wide Human SNP Array 6.0. IMPUTE version 2.1.0 was used to perform imputation for the MESA Caucasian participants (chromosomes 1–22) using HapMap Phase I and II - CEU as the reference panel (release #24 - NCBI Build 36 (dbSNP b126)). SNPs with genotype call rate less than 0.95, MAF ≤0.02, HWE P-value ≤0.001, or the average of the observed divided by expected variance ratio of any SNP ≤0.3 (indicating deviation from Hardy–Weinberg equilibrium and poor quality of imputation, sometimes denoted as Rsq) were removed from the analysis. Association tests were performed by SNPTEST v2 (49).
OGP-Talana
Quality control of the SNP data was performed using the GenABEL software package in R. Samples with overall SNP call rate <93%, with a minor allele frequency of <0.01, with Hardy–Weinberg P-value > 10−6, showing excess heterozygosity, or being classified as outliers by allelic identity-by-state (IBS) clustering analysis, were excluded.
Genotype imputation of data
All AREDS and KORA high-quality SNPs were filtered by those present on the Illumina Omni2.5 array to produce a reduced set of genotypes for imputing to the HapMap. Imputation to the HapMap-II reference panel (CEU population release 22, NCBI build 36) was performed in MACH (43,50) in two stages. Stage 1 was the model parameter estimation stage which used a random sample of 300 individuals from each population, using the greedy option which only uses the reference haplotypes (supplied here from the HapMap) and 100 Markov Chain iterations. Stage 2 is the actual imputation stage and uses the model parameters estimated in stage 1 to speed up the imputation of the genotypes.
Genotype imputation of the FFES data to the HapMap-II reference panel (CEU population release 22, NCBI build 36) was carried out in a two-step process using the Markov Chain Haplotyping (MACH version 1.0.16.a) software. First, crossover and error-rate maps were built using 400 unrelated individuals (200 male and 200 female) sampled from Framingham Heart Study (FHS) subjects. Second, genotype imputations of ∼2.5 million autosomal HapMap-II SNPs were carried out on the entire FHS dataset using parameters estimated from step 1.
For MESA, IMPUTE version 2.1.0 was used to perform imputation for the Caucasian participants (chromosomes 1–22) using HapMap phase I and II–CEU as the reference panel (release #24 - NCBI Build 36 (dbSNP b126)).
For OGP-Talana, using the phase II CEU HapMap individuals (release 22, NCBI build 36) as reference panel for imputation, genotypes were imputed for nearly 2.5 milion SNPs using MACH. SNPs imputed with Rsq <0.3 were excluded.
Data analysis
Genetic association was estimated by fitting a linear regression model. The dependent variable was the spherical equivalent refraction, averaged between the eyes (or Mean Spherical Equivalent [MSE]). A general additive genetic model was used to code the SNP effect (i.e. SNPs were coded according to the number of minor alleles [0,1,2] for each person); covariates included age; sex; and an ordinal variable representing education level. For AREDS, KORA and FES, this was accomplished using the PLINK (version 1.07) statistical software (http://pngu.mgh.harvard.edu/~purcell/plink, last accessed date on March 13, 2013) (44). For AREDS and FES analyses, the values for each individual of the first three most significant principal components of the EIGENSTRAT analysis were also included along with the covariates listed above. For MESA, these association tests were performed by SNPTEST v2. For OGP-Talana, all regression models were run using the ProbABEL package from the GenABEL suite which adjusts jointly for cryptic relatedness and population stratification.
Bioinformatics
Bioinformatic analysis of the SNPs in RBFOX1 gene began with gathering general annotated information from dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/, last accessed date on March 13, 2013) and HapMap (http://hapmap.ncbi.nlm.nih.gov/, last accessed date on March 13, 2013) (51,52). The region was viewed on UCSC Genome Browser (http://genome.ucsc.edu/, last accessed date on March 13, 2013) in order to evaluate annotations for regulatory elements, other SNPs in close proximity, conservation, transcription factor binding sites (TFBSs) and disease connection (53). Conservation of the SNP area was viewed in more detail using the Evolutionary Conserved Region Browser (http://ecrbrowser.dcode.org/, last accessed date on March 13, 2013) (54) which aligns genomes from different species to visually compare the evolutionary conservation. MultiTF (http://multitf.dcode.org/, last accessed date on March 13, 2013) and the TRAP web tool (http://trap.molgen.mpg.de/cgi-bin/home.cgi, last accessed date on March 13, 2013) (55) were used to find TFBSs in the conserved regions. TRAP is different from the other TFBS programs because it looks at affinity of binding to an area rather than sequence matching. Another tool, sTRAP, analyzes two slightly different sequences in order to compare the difference in binding affinities. Sequences with and without the effect allele were loaded into sTRAP, while using TRANSFAC and Jaspar TFBS databases. Promoter and enhancer prediction tools were utilized to determine whether the SNP region contains a promoter/enhancer. Promoter/enhancer prediction tools used included FPROM from Softberry (http://linux1.softberry.com/berry.phtml?topic=fprom&group=programs&subgroup=promoter, last accessed date on March 13, 2013), FirstEF from Cold Spring Harbor Laboratory (http://rulai.cshl.org/tools/FirstEF/, last accessed date on March 13, 2013) Promoter 2.0 from Denmark Technical University (http://www.cbs.dtu.dk/services/Promoter/, last accessed date on March 13, 2013) and Neural Network Promoter Prediction developed by Berkeley Drosophila Genome Project (http://www.fruitfly.org/seq_tools/promoter.html, last accessed date on March 13, 2013).
Eyebrowse (http://eyebrowse.cit.nih.gov/, last accessed date on March 13, 2013) is a visualization tool for transcript information from NEIBank, an online collection of transcripts found in eye tissues (56) and was used to verify expression of transcripts in the eye.
CpG islands were searched for in the area of interest using the CpG island tool in the Sequence Manipulation Suite (http://www.bioinformatics.org/sms2/cpg_islands.html), last accessed date on March 13, 2013 (57).
SUPPLEMENTARY MATERIAL
FUNDING
This work was funded in part by the Intramural Research Program of the National Human Genome Research Institute (J.E.B.W., R.W., C.L.S.) and the National Eye Institute (M.C., E.Y.C.), National Institutes of Health and NIH RO1EY020483 (D.S., J.E.B.W.) and K08EY022943 (R.W.). The KORA Study is supported by funds from Helmholtz Center Munich and the German Federal Ministry of Education and Research (BMBF). The Multi-Ethnic Study of Atherosclerosis (MESA) and MESA SNP Health Association Resource (SHARe) are conducted and supported by the National Heart, Lung and Blood Institute (NHLBI) in collaboration with MESA investigators. Support is provided by grants and contracts N01 HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169 and RR-024156. Funding for SHARe genotyping was provided by NHLBI Contract N02-HL-6-4278. Funding for the collection of RE data was supported by the Intramural Research Program of the National Eye Institute (ZIAEY000403). Support was also provided by the National Center for Research Resources, Grant UL1RR033176, and is now at the National Center for Advancing Translational Sciences, Grant UL1TR000124. The Blue Mountains Eye Study was supported by the Australian National Health & Medical Research Council (NHMRC) project grants (IDs 974159, 991407, 211069 and 457349) and Centre for Clinical Research Excellence (CCRE) in Translational Clinical Research in Eye Diseases, CCRE in TCR-Eye (ID 529923). The Blue Mountains Eye Study GWAS and genotyping costs were supported by Australian NHMRC project grants (IDs 512423, 475604, 529912 and 590204), and the Wellcome Trust, UK, as part of Wellcome Trust Case Control Consortium 2 (grant IDs 085475/B/08/Z and 085475/08/Z). E.G.H. (631096), P.N.B. (1028444) and J.W. (358702 and 632909) are supported by the NHMRC fellowship scheme. The Center for Eye Research Australia receives Operational Infrastructure Support from the Victorian government. OGP-Talana was supported by grants from the Italian Ministry of Education, University and Research (MIUR) No. 5571/DSPAR/2002 and (FIRB) D.M no. 718/Ric/2005. The DCCT Research Group is sponsored through research contracts from the National Institute of Diabetes, Endocrinology and Metabolic Diseases of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, N01-DK-6-2204, R01-DK-077510) and the National Institutes of Health. A.D.P. holds a Canada Research Chair in the Genetics of Complex Diseases. The Rotterdam Study and ERF were supported by the Netherlands Organization of Scientific Research (NWO) (Vidi 91796357); Erasmus Medical Center and Erasmus University, Rotterdam, The Netherlands; Netherlands Organization for Health Research and Development (ZonMw); UitZicht; the Research Institute for Diseases in the Elderly; the Ministry of Education, Culture and Science; the Ministry for Health, Welfare and Sports; the European Commission (DG XII); the Municipality of Rotterdam; the Netherlands Genomics Initiative/NWO; Center for Medical Systems Biology of NGI; Lijf en Leven; M.D. Fonds; Henkes Stichting; Stichting Nederlands Oogheelkundig Onderzoek; Swart van Essen; Bevordering van Volkskracht; Blindenhulp; Landelijke Stichting voor Blinden en Slechtzienden; Rotterdamse Vereniging voor Blindenbelangen; OOG; Algemene Nederlandse Vereniging ter Voorkoming van Blindheid; the Rotterdam Eye Hospital Research Foundation and Topcon Europe. The Croatian studies were funded by grants from the Medical Research Council (UK), from the Republic of Croatia Ministry of Science, Education and Sports (108-1080315-0302). We acknowledge the Wellcome Trust Clinical facility (Edinburgh) for the genotyping of the CROATIA-Vis study, an EU framework 6 project EUROSPAN (contract no LSHG-CT-2006-018947) for the genotyping of the CROATIA-Korcula study that was performed at the Helmholtz Zentrum Munchen (Munich, Germany). ORCADES was supported by the Chief Scientist Office of the Scottish Government, the Royal Society, the Medical Research Council Human Genetics Unit and the European Union framework program 6 EUROSPAN project (contract no. LSHG-CT-2006-018947).
Supplementary Material
ACKNOWLEDGEMENTS
We thank numerous clinicians, clinical staff, patients and their families for their participation in and dedication to the project; the Ogliastra population and all the individuals who participated in this study. We are very grateful to the municipal administrators for their collaboration to the project and for economic and logistic support. We are extremely appreciative of the support and wisdom provided by Dr Hemin Chin of the National Eye Institute. The Center for Inherited Disease Research, fully funded through a federal contract (HHSN268200782096C) from National Institutes of Health to The Johns Hopkins University, performed genotyping of the AREDS and KORA cohorts. Clinical data and DNA from the DCCT/EDIC study will be made available through the National Institute of Diabetes and Digestive and Kidney Diseases repository at https://www.niddkrepository.org/niddk/home.do, last accessed date on March 13, 2013. This manuscript was not prepared under the auspices of and does not represent analyses or conclusions of the NIDDK Central Repositories, or the NIH. Rotterdam Study and ERF thank Ada Hooghart, Corina Brussee, Riet Bernaerts-Biskop, Patricia van Hilten, Pascal Arp, Jeanette Vergeer, Marijn Verkerk and Sander Bervoets. We acknowledge the Wellcome Trust Clinical facility (Edinburgh) for DNA extraction for the ORCADES study and Peter Lichner and the Helmholtz Zentrum Munchen genotyping staff (Munich, Germany) for genotyping.
Conflict of Interest Statement. None declared.
REFERENCES
- 1.Pizzarello L., Abiose A., Ffytche T., Duerksen R., Thulasiraj R., Taylor H., Faal H., Rao G., Kocur I., Resnikoff S. VISION 2020: the right to sight: a global initiative to eliminate avoidable blindness. Arch. Ophthalmol. 2004;122:615–620. doi: 10.1001/archopht.122.4.615. [DOI] [PubMed] [Google Scholar]
- 2.Lin L.L., Shih Y.F., Hsiao C.K., Chen C.J. Prevalence of myopia in Taiwanese schoolchildren: 1983 to 2000. Ann. Acad. Med. Singapore. 2004;33:27–33. [PubMed] [Google Scholar]
- 3.He M., Zeng J., Liu Y., Xu J., Pokharel G.P., Ellwein L.B. Refractive error and visual impairment in urban children in southern china. Invest. Ophthalmol. Vis. Sci. 2004;45:793–799. doi: 10.1167/iovs.03-1051. [DOI] [PubMed] [Google Scholar]
- 4.Holden B.A., Fricke T.R., Ho S.M., Wong R., Schlenther G., Cronje S., Burnett A., Papas E., Naidoo K.S., Frick K.D. Global vision impairment due to uncorrected presbyopia. Arch. Ophthalmol. 2008;126:1731–1739. doi: 10.1001/archopht.126.12.1731. [DOI] [PubMed] [Google Scholar]
- 5.Dirani M., Tong L., Gazzard G., Zhang X., Chia A., Young T.L., Rose K.A., Mitchell P., Saw S.M. Outdoor activity and myopia in Singapore teenage children. Br. J. Ophthalmol. 2009;93:997–1000. doi: 10.1136/bjo.2008.150979. [DOI] [PubMed] [Google Scholar]
- 6.Ip J.M., Rose K.A., Morgan I.G., Burlutsky G., Mitchell P. Myopia and the urban environment: findings in a sample of 12-year-old Australian school children. Invest. Ophthalmol. Vis. Sci. 2008;49:3858–3863. doi: 10.1167/iovs.07-1451. [DOI] [PubMed] [Google Scholar]
- 7.Morgan I., Rose K. How genetic is school myopia? Prog. Retin. Eye Res. 2005;24:1–38. doi: 10.1016/j.preteyeres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Rose K.A., Morgan I.G., Ip J., Kifley A., Huynh S., Smith W., Mitchell P. Outdoor activity reduces the prevalence of myopia in children. Ophthalmology. 2008;115:1279–1285. doi: 10.1016/j.ophtha.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 9.Saw S.M., Hong C.Y., Chia K.S., Stone R.A., Tan D. Nearwork and myopia in young children. Lancet. 2001;357:390. doi: 10.1016/S0140-6736(05)71520-8. [DOI] [PubMed] [Google Scholar]
- 10.Peet J.A., Cotch M.F., Wojciechowski R., Bailey-Wilson J.E., Stambolian D. Heritability and familial aggregation of refractive error in the Old Order Amish. Invest. Ophthalmol. Vis. Sci. 2007;48:4002–4006. doi: 10.1167/iovs.06-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wojciechowski R., Congdon N., Bowie H., Munoz B., Gilbert D., West S.K. Heritability of refractive error and familial aggregation of myopia in an elderly American population. Invest. Ophthalmol. Vis. Sci. 2005;46:1588–1592. doi: 10.1167/iovs.04-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein A.P., Suktitipat B., Duggal P., Lee K.E., Klein R., Bailey-Wilson J.E., Klein B.E. Heritability analysis of spherical equivalent, axial length, corneal curvature, and anterior chamber depth in the Beaver Dam Eye Study. Arch. Ophthalmol. 2009;127:649–655. doi: 10.1001/archophthalmol.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyhne N., Sjolie A.K., Kyvik K.O., Green A. The importance of genes and environment for ocular refraction and its determiners: a population based study among 20–45 year old twins. Br. J. Ophthalmol. 2001;85:1470–1476. doi: 10.1136/bjo.85.12.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammond C.J., Snieder H., Gilbert C.E., Spector T.D. Genes and environment in refractive error: the twin eye study. Invest. Ophthalmol. Vis. Sci. 2001;42:1232–1236. [PubMed] [Google Scholar]
- 15.Biino G., Palmas M.A., Corona C., Prodi D., Fanciulli M., Sulis R., Serra A., Fossarello M., Pirastu M. Ocular refraction: heritability and genome-wide search for eye morphometry traits in an isolated Sardinian population. Hum. Genet. 2005;116:152–159. doi: 10.1007/s00439-004-1231-6. [DOI] [PubMed] [Google Scholar]
- 16.Dirani M., Chamberlain M., Shekar S.N., Islam A.F., Garoufalis P., Chen C.Y., Guymer R.H., Baird P.N. Heritability of refractive error and ocular biometrics: the genes in Myopia (GEM) twin study. Invest. Ophthalmol. Vis. Sci. 2006;47:4756–4761. doi: 10.1167/iovs.06-0270. [DOI] [PubMed] [Google Scholar]
- 17.Lee K.E., Klein B.E., Klein R., Fine J.P. Aggregation of refractive error and 5-year changes in refractive error among families in the Beaver Dam Eye Study. Arch. Ophthalmol. 2001;119:1679–1685. doi: 10.1001/archopht.119.11.1679. [DOI] [PubMed] [Google Scholar]
- 18.Wojciechowski R. Nature and nurture: the complex genetics of myopia and refractive error. Clin. Genet. 2011;79:301–320. doi: 10.1111/j.1399-0004.2010.01592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y., Qu J., Zhang D., Zhao P., Zhang Q., Tam P.O., Sun L., Zuo X., Zhou X., Xiao X., et al. Genetic variants at 13q12.12 are associated with high myopia in the Han Chinese population. Am. J. Hum. Genet. 2011;88:805–813. doi: 10.1016/j.ajhg.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z., Qu J., Xu X., Zhou X., Zou H., Wang N., Li T., Hu X., Zhao Q., Chen P., et al. A genome-wide association study reveals association between common variants in an intergenic region of 4q25 and high-grade myopia in the Chinese Han population. Hum. Mol. Genet. 2011;20:2861–2868. doi: 10.1093/hmg/ddr169. [DOI] [PubMed] [Google Scholar]
- 21.Li Y.J., Goh L., Khor C.C., Fan Q., Yu M., Han S., Sim X., Ong R.T., Wong T.Y., Vithana E.N., et al. Genome-wide association studies reveal genetic variants in CTNND2 for high myopia in Singapore Chinese. Ophthalmology. 2011;118:368–375. doi: 10.1016/j.ophtha.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hysi P.G., Young T.L., Mackey D.A., Andrew T., Fernandez-Medarde A., Solouki A.M., Hewitt A.W., Macgregor S., Vingerling J.R., Li Y.J., et al. A genome-wide association study for myopia and refractive error identifies a susceptibility locus at 15q25. Nat. Genet. 2010;42:902–905. doi: 10.1038/ng.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solouki A.M., Verhoeven V.J., van Duijn C.M., Verkerk A.J., Ikram M.K., Hysi P.G., Despriet D.D., van Koolwijk L.M., Ho L., Ramdas W.D., et al. A genome-wide association study identifies a susceptibility locus for refractive errors and myopia at 15q14. Nat. Genet. 2010;42:897–901. doi: 10.1038/ng.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verhoeven V.J., Hysi P.G., Saw S.M., Vitart V., Mirshahi A., Guggenheim J.A., Cotch M.F., Yamashiro K., Baird P.N., Mackey D.A., et al. Large scale international replication and meta-analysis study confirms association of the 15q14 locus with myopia. The CREAM consortium. Hum Genet. 2012;131:1467–1480. doi: 10.1007/s00439-012-1176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakanishi H., Yamada R., Gotoh N., Hayashi H., Yamashiro K., Shimada N., Ohno-Matsui K., Mochizuki M., Saito M., Iida T., et al. A genome-wide association analysis identified a novel susceptible locus for pathological myopia at 11q24.1. PLoS Genet. 2009;5:e1000660. doi: 10.1371/journal.pgen.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan Q., Barathi V.A., Cheng C.Y., Zhou X., Meguro A., Nakata I., Khor C.C., Goh L.K., Li Y.J., Lim W., et al. Genetic variants on chromosome 1q41 influence ocular axial length and high myopia. PLoS Genet. 2012;8:e1002753. doi: 10.1371/journal.pgen.1002753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ovcharenko I., Nobrega M.A., Loots G.G., Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32:W280–W286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nallasamy S., Paluru P.C., Devoto M., Wasserman N.F., Zhou J., Young T.L. Genetic linkage study of high-grade myopia in a Hutterite population from South Dakota. Mol. Vis. 2007;13:229–236. [PMC free article] [PubMed] [Google Scholar]
- 29.Bitel C.L., Nathan R., Wong P., Kuppasani S., Matsushita M., Kanazawa H., Frederikse P.H. Evidence that 'brain-specific’ FOX-1, FOX-2, and nPTB alternatively spliced isoforms are produced in the lens. Curr. Eye. Res. 2011;36:321–327. [PubMed] [Google Scholar]
- 30.Nakahata S., Kawamoto S. Tissue-dependent isoforms of mammalian Fox-1 homologs are associated with tissue-specific splicing activities. Nucleic Acids Res. 2005;33:2078–2089. doi: 10.1093/nar/gki338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gehman L.T., Stoilov P., Maguire J., Damianov A., Lin C.H., Shiue L., Ares M., Jr, Mody I., Black D.L. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat. Genet. 2011;43:706–711. doi: 10.1038/ng.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhalla K., Phillips H.A., Crawford J., McKenzie O.L., Mulley J.C., Eyre H., Gardner A.E., Kremmidiotis G., Callen D.F. The de novo chromosome 16 translocations of two patients with abnormal phenotypes (mental retardation and epilepsy) disrupt the A2BP1 gene. J. Hum. Genet. 2004;49:308–311. doi: 10.1007/s10038-004-0145-4. [DOI] [PubMed] [Google Scholar]
- 33.Martin C.L., Duvall J.A., Ilkin Y., Simon J.S., Arreaza M.G., Wilkes K., Alvarez-Retuerto A., Whichello A., Powell C.M., Rao K., et al. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am. J. Med. Genet. B. 2007;144B:869–876. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- 34.Voineagu I., Wang X., Johnston P., Lowe J.K., Tian Y., Horvath S., Mill J., Cantor R.M., Blencowe B.J., Geschwind D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C., Zhang Z., Castle J., Sun S., Johnson J., Krainer A.R., Zhang M.Q. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008;22:2550–2563. doi: 10.1101/gad.1703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schippert R., Schaeffel F., Feldkaemper M.P. Microarray analysis of retinal gene expression in chicks during imposed myopic defocus. Mol. Vis. 2008;14:1589–1599. [PMC free article] [PubMed] [Google Scholar]
- 37.Penha A.M., Schaeffel F., Feldkaemper M. Insulin, insulin-like growth factor-1, insulin receptor, and insulin-like growth factor-1 receptor expression in the chick eye and their regulation with imposed myopic or hyperopic defocus. Mol. Vis. 2011;17:1436–1448. [PMC free article] [PubMed] [Google Scholar]
- 38.Gentle A., McBrien N.A. Retinoscleral control of scleral remodelling in refractive development: a role for endogenous FGF-2? Cytokine. 2002;18:344–348. doi: 10.1006/cyto.2002.1046. [DOI] [PubMed] [Google Scholar]
- 39.Veerappan S., Pertile K.K., Islam A.F., Schache M., Chen C.Y., Mitchell P., Dirani M., Baird P.N. Role of the hepatocyte growth factor gene in refractive error. Ophthalmology. 2010;117:239–245. doi: 10.1016/j.ophtha.2009.07.002. e231–232. [DOI] [PubMed] [Google Scholar]
- 40.Yanovitch T., Li Y.J., Metlapally R., Abbott D., Viet K.N., Young T.L. Hepatocyte growth factor and myopia: genetic association analyses in a Caucasian population. Mol. Vis. 2009;15:1028–1035. [PMC free article] [PubMed] [Google Scholar]
- 41.Shi Y., Li Y., Zhang D., Zhang H., Lu F., Liu X., He F., Gong B., Cai L., Li R., et al. Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet. 2011;7:e1002084. doi: 10.1371/journal.pgen.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fogel B.L., Wexler E., Wahnich A., Friedrich T., Vijayendran C., Gao F., Parikshak N., Konopka G., Geschwind D.H. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum. Mol. Genet. 2012;21:4171–4186. doi: 10.1093/hmg/dds240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanna S., Jackson A.U., Nagaraja R., Willer C.J., Chen W.M., Bonnycastle L.L., Shen H., Timpson N., Lettre G., Usala G., et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat. Genet. 2008;40:198–203. doi: 10.1038/ng.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang H.M., Sul J.H., Service S.K., Zaitlen N.A., Kong S.Y., Freimer N.B., Sabatti C., Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 47.Patterson N., Price A.L., Reich D. Population structure and eigen analysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laurie C.C., Doheny K.F., Mirel D.B., Pugh E.W., Bierut L.J., Bhangale T., Boehm F., Caporaso N.E., Cornelis M.C., Edenberg H.J., et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet. Epidemiol. 2010;34:591–602. doi: 10.1002/gepi.20516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 50.Willer C.J., Sanna S., Jackson A.U., Scuteri A., Bonnycastle L.L., Clarke R., Heath S.C., Timpson N.J., Najjar S.S., Stringham H.M., et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smigielski E.M., Sirotkin K., Ward M., Sherry S.T. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000;28:352–355. doi: 10.1093/nar/28.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 53.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ovcharenko I., Loots G.G., Giardine B.M., Hou M., Ma J., Hardison R.C., Stubbs L., Miller W. Mulan: multiple-sequence local alignment and visualization for studying function and evolution. Genome Res. 2005;15:184–194. doi: 10.1101/gr.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas-Chollier M., Hufton A., Heinig M., O'Keeffe S., Masri N.E., Roider H.G., Manke T., Vingron M. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nat. Prot. 2011;6:1860–1869. doi: 10.1038/nprot.2011.409. [DOI] [PubMed] [Google Scholar]
- 56.Wistow G., Peterson K., Gao J., Buchoff P., Jaworski C., Bowes-Rickman C., Ebright J.N., Hauser M.A., Hoover D. NEIBank: genomics and bioinformatics resources for vision research. Mol. Vis. 2008;14:1327–1337. [PMC free article] [PubMed] [Google Scholar]
- 57.Stothard P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques. 2000;28:1102–1104. doi: 10.2144/00286ir01. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.