

Abstract
The core promoter is the region in which RNA polymerase II is recruited to the DNA and acts to initiate transcription, but the extent to which the core promoter sequence determines promoter activity levels is largely unknown. Here, we identified several base content and k-mer sequence features of the yeast core promoter sequence that are highly predictive of maximal promoter activity. These features are mainly located in the region 75 bp upstream and 50 bp downstream of the main transcription start site, and their associations hold for both constitutively active promoters and promoters that are induced or repressed in specific conditions. Our results unravel several architectural features of yeast core promoters and suggest that the yeast core promoter sequence downstream of the TATA box (or of similar sequences involved in recruitment of the pre-initiation complex) is a major determinant of maximal promoter activity. We further show that human core promoters also contain features that are indicative of maximal promoter activity; thus, our results emphasize the important role of the core promoter sequence in transcriptional regulation.
INTRODUCTION
The RNA polymerase II (pol-II) core promoter is the region in which pol-II is recruited to the DNA and acts to initiate transcription, a process involving the general transcription factors (GTFs, including the following: TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH) (1).
The first core promoter element identified was the TATA box (1), the binding site of TATA binding protein (TBP; a subunit of TFIID), which is found in all eukaryotes. Most yeast core promoters do not contain a consensus TATA box (2). A recent study showed that almost all yeast promoters contain sequences that differ from the TATA box consensus by up to 2 bases, and that TBP is recruited to such sequences, with lesser affinity (3). In metazoans (e.g. human, drosophila), other core promoter elements (Inr, DPE, etc.) were identified (4), revealing a variety of core promoter architectures. These elements are not found in yeast core promoters. In metazoans, as in yeast, most core promoters do not have a TATA box (4). However, as the TATA box is the best-known element to which the pre-initiation complex (PIC) is recruited, much of our knowledge of transcription initiation is based on TATA box containing core promoters.
A major difference between yeast and metazoan transcription initiation is in transcription start site (TSS) selection. In metazoans, PIC formation over a TATA box results in TSS selection 25–30 bp downstream of it, while in Saccharomyces cerevisiae, selected TSSs are 40–120 bp downstream of the TATA box (1). In both yeast and metazoans, promoter DNA melting was shown to occur ∼20 bp downstream of the TATA box (5), such that the promoter sequence ∼30 bp downstream of the TATA box is at the pol-II active center (6,7). Another study proposed that following PIC formation, the yeast pol-II performs a downstream scan of the melted template strand, searching for TSS sequence signals (5). This scanning model is supported by studies showing that TSS locations can be at varying distances downstream of where the PIC is recruited (e.g. a TATA box), and depend on the sequence at these locations (8–13). Various studies also showed that TFIIB, TFIIF and TFIIH affect TSS selection in a manner depending on the sequence next to the TSS and upstream of it (14–17). Suggested yeast TSS consensus sequences include RRYRR, TCRA (9), YAWR (18) and A(Arich)5NYAWNN(Arich)6 (19).
Ample evidence suggests that variation of the TATA box sequence alters promoter activity levels (20–26). However, the role of downstream yeast core promoter sequence signals that may affect pol-II scanning and TSS selection in determining promoter activity levels is less explored. One study analyzed a set of 95 S. cerevisiae promoters and noted that the sequence 30–10 bp upstream of the TSS was T enriched and A depleted, while the sequence between 8 bp upstream and 15 bp downstream of the TSS was T depleted and A enriched (27). They termed this base content signal the ‘locator’, suggesting that it may affect TSS localization. They showed that the ‘locator’ signal was stronger in 51 ‘strong’ promoters compared with 34 ‘weak’ ones. Their results extend an earlier study that analyzed 17 S. cerevisiae promoters and noted the existence of a pyrimidine-rich stretch ending ∼10 bp upstream of the TSS in the six high-expressing promoters (28). Another study demonstrated the high efficiency of the main TSS of the SNR14 gene, and quantified to what extent did various mutations reduce its efficiency, leading to reduction in mRNA production (12).
Here, we set out to explore the relation between various base content and k-mer features of the yeast core promoter sequence and promoter activity. We found that several features are indicative of the maximal promoter activity, both in promoters that are constitutively active as well as in promoters that are induced or repressed in specific conditions. Most of these features are located within 75 bp upstream and 50 bp downstream of the main TSS, and represent core promoter signals that are downstream of the location to which the PIC is recruited. This suggests that the yeast core promoter sequence can greatly influence promoter activity by affecting pol-II scanning rate and TSS selection. Extending our exploration to human constitutive TSSs, we show that in human, as in yeast, core promoter features are indicative of maximal promoter activity. Taken together, our results suggest that the core promoter is an important determinant of promoter activity.
MATERIALS AND METHODS
Intrinsic nucleosome average occupancy predictions
We predicted the intrinsic nucleosome average occupancy around yeast TSSs using the model published by (29) with the temperature parameter set to 1 and the nucleosome scaling (concentration-related parameter) set to 0.5. Predictions were on 1000 bp flanked sequences around the TSSs, to avoid sequence edge–related errors.
While in vitro–measured average occupancy is intrinsic, it is measured with a nucleosome concentration much lower than in vivo, and therefore does not adequately reflect the intrinsic average occupancy in vivo. Using the above parameters, the mean predicted intrinsic average occupancy over the entire S. cerevisiae genome was slightly >0.6.
Linear model learning
We chose to learn a linear model for several reasons. First, linear models are simple and easy to interpret. Second, for yeast data we have <1 K data points, yet an initial set of >54 K features. This requires efficient feature selection to be performed; otherwise, overfitting the model to the training data is inevitable. Instead of having to perform feature selection before model learning, regularized linear regression algorithms allow to learn relatively sparse models that can avoid, or at least reduce, overfitting.
Our linear regression algorithm of choice was the elastic net (30), imposing a combination of L1 and L2 regularization terms. The L1 term contributes to sparseness, while the L2 term contributes to spreading the weight among multiple covariates. For the purpose of learning an elastic net from training data, we used the glmnet software (http://www-stat.stanford.edu/∼tibs/glmnet-matlab/). We chose a mixing ratio of 1:1 between the L1 and the L2 terms (glmnet parameter α = 0.5). To enforce relative sparseness, we limited the number of non-zero effect sizes to 200 (glmnet parameter dfmax = 200). We say that a feature was included in the model if its effect size was non-zero.
glmnet uses least angle regression (31) to generate a grid of solutions on the regularization path of the model coefficients vector, between the 0 model and the non-regularized model. Each solution on the regularization path corresponds to a specific value of the regularization coefficient λ, with λ monotonically decreasing between the 0 model and the non-regularized model (where λ = 0). To select the value of λ, we used a 10-fold cross validation (CV) scheme over the training data. For this purpose, the training set was randomly partitioned (10 different times) to an internal training set and a validation set. For each internal training set, we learned a grid of up to 1000 solutions (glmnet parameter nlambda = 1000) on the regularization path, and took the value of λ of the solution that performed best on the held-out validation set (in terms of the R2 statistic). The final value of λ was taken to be the mean of the 10 selected λ values.
RESULTS
Features of the core promoter sequence greatly differ between S. cerevisiae genes with high and with low maximal expression
In a recent study performed in our lab (to be published elsewhere), 859 native S. cerevisiae promoters were inserted upstream of a YFP reporter gene, and their promoter activity was accurately measured in 10 different conditions. Many of these genes were constitutively expressed in all conditions, and we will refer to them here as constitutive genes. We will refer to all other genes as regulated genes, as each of them is further induced or repressed in a subset of the conditions. This first classification of genes is related to their mode of regulation. We alternatively classified the genes based on their maximal promoter activity. As an approximation to the real maximal promoter activity (in any possible condition), we used the maximal measured promoter activity, denoted by Emax, and classified as follows: low (Emax < 0.1), medium (0.1 ≤ Emax < 0.4), high (0.4 ≤ Emax < 1) and very high (Emax ≥ 1). By definition, this approximation is more accurate for genes with high Emax values, and may be less accurate for genes with low Emax values, as they may be highly expressed in a condition other than the 10 examined. In total, we had 729 genes out of the above 859 that also had their TSS measured by (32) (see Supplementary Table S1). Applying both of the above classification criteria, we partitioned these 729 genes into eight subsets: 171 constitutive–low (both constitutive and low), 104 regulated–low, 190 constitutive–medium, 80 regulated–medium, 122 constitutive–high, 18 regulated–high, 36 constitutive–very high, and 8 regulated–very high.
For each of the above eight gene subsets, we analyzed base content (of mononucleotides and G + C) and hits of the TATA box TATAWAWR consensus (2) in the region between 200 bp upstream and 100 bp downstream of the main TSS (denoted the [−200, 100] region), as mapped by (32).
Strikingly, we found that genes with higher Emax values have core promoter sequences that are significantly (see rank-sum P-values in Supplementary Figure S1) more T rich (Figure 1 and Supplementary Figure S1A) upstream of the main TSS (within the [−70, −10] region), and alternately more A rich (Figure 1 and Supplementary Figure S1B) at and downstream of the main TSS (within the [0, 50] region). A similar result was shown by (27) for a smaller set of genes (see above). Here we also show that this signal is highly similar for both constitutive and regulated genes that have similar levels of Emax.
Figure 1.

Yeast core promoter sequence signals differ between genes with different maximal promoter activity. Mean nucleotide and TATA box content, computed using a sliding window (20 bp long, 10 bp step) over the [−200, 100] region around the main TSS (32), for the eight yeast gene subsets (defined in the main text). Here, the TATA box window content was defined to be the fraction of genes in the subset that had a hit of the TATA box consensus TATAWAWR (2) within the window (a hit was counted if the first T was in the window). Plots are arranged in a table-like fashion. Columns are arranged by the genes’ maximal promoter activity level (Emax). The top three rows are for constitutive genes, while the bottom three rows are for regulated genes. In addition, similar plots were generated for the set of all genes (bottom of the figure). The vertical dashed lines represent the location of the main TSS. The horizontal dotted lines are to assist with the comparison of plots between columns.
Genes with higher Emax values (both constitutive and regulated) also tend to have significantly lower G\C content around their main TSS (Figure 1 and Supplementary Figure S1C). Interestingly, reduced G\C content around the main TSS of genes with high Emax is mainly achieved by significantly reduced G content (Figure 1 and Supplementary Figure S1D) and not C content (Figure1). G\C content is known to be highly correlated with intrinsic nucleosome occupancy (33) that depends only on the DNA sequence and the concentration of histone proteins. This intrinsic occupancy was shown to be well predicted by a thermodynamic model learned based on in vitro nucleosome occupancy data (29). Using this model, we predicted and computed the mean intrinsic nucleosome occupancy around the main TSS (see ‘Materials and Methods’ section) for the eight gene sets defined above, shown in Figure 2A. Indeed, there is similarity between the G\C content tracks (Figure 1) and the predicted intrinsic nucleosome occupancy tracks, suggesting that lower intrinsic nucleosome occupancy around the main TSS contributes to higher levels of maximal promoter activity. This trend is also evident when examining the mean YPD in vivo nucleosome occupancy (29) shown in Figure 2B, although for regulated genes, many of which not induced in YPD (with glucose available), there are significant differences between the YPD in vivo and the predicted intrinsic nucleosome occupancies.
Figure 2.

Nucleosome occupancy of yeast core promoters differs between genes with different maximal promoter activity. (A) Mean predicted nucleosome occupancy in the [−200, 100] region around the main TSS, for the eight yeast gene subsets (defined in the main text). Predictions were made with the model of (29), with parameters that gave a genomic mean nucleosome occupancy of ∼0.6 (indicated by the dotted horizontal lines). Plots are arranged in a table-like fashion. Columns are arranged by the genes’ maximal promoter activity level (Emax). The top row is for constitutive genes, while the bottom row is for regulated genes. The vertical dashed lines represent the location of the main TSS. (B) Mean in vivo (YPD) normalized nucleosome occupancy (29). Y-axis value of 1 (dotted horizontal lines) represents the genomic mean.
Consensus TATA boxes are known to be high-affinity binding sites of TBP, with one and two mismatches leading to weaker binding affinities (3). In accord with (2), when comparing regulated and constitutive genes that have similar Emax values, we found a higher frequency of consensus TATA boxes (Figure 1) in core promoters of regulated genes. Still, for both regulated and constitutive genes, consensus TATA box frequency is higher in genes with higher Emax. This is in support of TBP high-affinity binding contributing to higher expression (26).
These results thus identify several specific core promoter sequence signals that are predictive and may thus affect the maximal promoter activity of yeast promoters.
T richness upstream of the main TSS is a predictor of the maximal promoter activity
We next sought to test the extent to which core promoter sequence features can predict Emax levels. To this end, we focused on the T-richness signal upstream of the main TSS (Figure 1). For each of our sliding windows (20 bp long, 10 bp step) within the [−80, −1] region, we computed per promoter, the T content to A\T content ratio in that window, and took the maximum over these windows to be a T-richness feature value. Taking the T content to A\T content ratio and not the T content itself normalizes out differences in A\T content between constitutive and regulated genes with similar Emax. For each pair of gene subsets (out of the above eight), we then computed an AUC score, quantifying the extent to which our T-richness feature separates between genes of one subset and the other subset. A score of 0.5 is attained by random, while a score of 1 represents a perfect separation (classification). An advantage of reporting AUC scores over P-values (of rank-sum tests, for instance) is that P-values are highly sensitive to the sizes of the compared gene subsets, while the AUC scores are not. The computed AUC scores are shown in Figure 3 in a triangular color matrix. Each intersection of a row and a column holds the AUC score for how well the T-richness feature separates between the subset at the right of the row and the one at the head of the column. For each AUC score, we also tested whether it is significantly different than 0.5 by computing an empirical P-value based on 10 000 random permutations of the feature values. AUC scores with a non-significant P-value (controlled for allowing a false discovery rate of 0.05) were marked by ‘x'.
Figure 3.

T richness upstream of the main TSS is a predictor of maximal activity of yeast promoters. AUC scores for seperating genes of one yeast gene subset from another, based on a feature of T richness upstream of the main TSS (top right, see also main text). AUC scores are shown in a triangular color matrix, where each cell holds the score for separating the gene subset at the head of the column from the gene subset to the right of the row. Scores not significantly different than 0.5 (based on an empirical P-value, controlled for allowing an FDR of 0.05) are marked with ‘x’. For four of the scores, the degree of separation of the two gene subsets is further exemplified by comparing the distribution of feature values of the two subsets. Con = Constitutive; Reg = Regulated.
We found two important observations. First, the T-richness feature does not significantly separate between gene subset pairs (constitutive versus regulated) of the same Emax level. Second, in most cases, the T-richness feature can significantly separate between gene subsets that have different levels of Emax, and the measure of separation (AUC) increases as the difference in Emax levels increases. There are four exceptions to this rule (constitutive–medium versus Constitutive–low, regulated–medium versus regulated–low, regulated–high versus regulated–medium and regulated–very high versus regulated–high), where separation is weak and not significant.
Taken together, these results demonstrate that the T-richness feature defined above is predictive of the Emax level of a gene.
Prediction of maximal promoter activity from core promoter sequence features
Encouraged by our above results, we sought to learn a quantitative model that predicts a gene’s Emax from features of its core promoter sequence, with two goals in mind: to provide a lower bound on how predictive the core promoter region is of the maximal promoter activity, and to elucidate core promoter sequence features that may have a role in determining it.
For each of the 729 genes, we computed a large set of base content and k-mer (k = 1, … ,4) counts and existence features over different windows within the [−200, 50] region. For each 4-mer feature, we computed another version of it with up to 1 mismatch of the 4-mer allowed. Adhering to a 10-fold CV scheme, we defined 10 pairs of training and test gene sets in the following way: we randomly partitioned the 729 genes into 10 subsets of (about) the same size. Each time we took one of the 10 subsets to be the test set, while the training set consisted of all other genes. For each of the 10 training sets, we first pruned sparse features (that had a support of <5% of the genes in the training set), and used the remaining features (>54 K) to learn a linear model that predicts Emax based on a small subset of them (see ‘Materials and Methods’ section). This model was then used to predict the Emax values of the genes in the respective test set. Model performance was evaluated by three measures: the R2 statistic (quantifying the proportion of variance in the data that is explained by the model), the Pearson correlation, r, and the Spearman correlation, ρ.
The mean (over the 10 models) performance measures are shown in Figure 4A (bar plot). Most importantly, the mean test R2 is 0.254, indicating that the core promoter sequence itself can explain at least 25.4% of the variance in the maximal promoter activity of yeast promoters. The difference between the mean test Pearson correlation (0.527) and the mean test Spearman correlation (0.425) indicates a small bias of the models to better predict high Emax values. This can be expected in cases such as ours, where the distribution of response values is greatly skewed (Supplementary Figure S3). A comparison of mean model performance on the training data versus the test data (Figure 4A, bar plot) shows that there is a degree of overfitting to the training data. This too is expected, as each model was learned on a large set (>54 K) of features. We will therefore focus on features that were included in at least 5 of the 10 models (48 such features), as they are likely to represent real signals. We term these features, robust features.
Figure 4.

Core promoter sequence features explain 25% of the variance in maximal promoter activity in yeast. Results of learning linear models (in a 10-fold CV scheme) that predict Emax from features of the [−200, 50] region (relative to the main TSS). (A) The bar plot shows mean model performance measures (R2; Pearson correlation, r; Spearman correlation, ρ), with error bars indicating ±1 standard deviation. The mean test R2 of 0.254 indicates that core promoter features explained 25.4% of the Emax variance of held-out genes. For each gene, the dot plot shows log Emax versus log test prediction (each gene appeared in the test set for one of the 10 models). The log transformation was applied after shifting both response and test prediction values by a constant such that all values became positive (the response was the Emax values centered around 0). The table enumerates 48 robust features (included in at least 5 of the 10 models). Each feature involves a sequence element (base content, k-mer) within a specific window. Features were manually classified (leftmost column), and sorted within each class by their mean effect size (color coded in the rightmost column). (B) Illustration of the classes of sequence features (with their location relative to the main TSS) that predict higher or lower Emax.
A table detailing the robust features is included in Figure 4A. For each feature (row) we show its sequence element (e.g. the 4-mer ‘TTTT’) and the promoter window (relative to the main TSS) where it was computed. Notably, most features could be related to one of the core promoter sequence signals that we discussed above. Based on that, we partitioned the features to several classes (class definitions appear in the left column), and sorted them within each class by their mean effect size (computed over the 10 models, color coded in the right column).
Robust features 1–11 (serial numbers appear in the gray-shaded column) are of T-rich k-mers in windows within the [−75, −1] region, and have positive effects (toward higher predicted Emax values), in accord with our above observations. Notably, most predictive are T-rich k-mers within the [−20, −11] region, as they are included in both robust features 1 and 2, and their effect size is the sum of effects of the two features. Robust feature 12 is also of a T-rich 4-mer, but at a small window further upstream.
Robust features 13–19 are of T-less k-mers within the [−100, −1] region. Robust features 20–22 are of the maximum A-content to A\T-content ratio, taken over a sliding window (size 10 bp, step 5 bp), within the [−75, −16] region. Robust features 13–22 all represent T-poor elements upstream of the main TSS, and have negative effects.
Robust features 23–29 likely involve TBP binding signals, as they include k-mers that are part of the consensus TATA box, up to 1 mismatch (3), and are upstream of the main TSS. Interestingly, such signals within the 100 bp upstream of the main TSS (robust features 23–26) have positive effects, while those further upstream (robust features 27–29) have negative effects, suggesting that TBP binding far upstream of the main TSS is less suited for achieving high promoter activity. This is in accord with the pol-II scanning model, as TBP binding far upstream of the main TSS would require longer pol-II scanning, and perhaps even increase the chance of pol-II falling off before initiation.
Robust features 30–41 are in windows overlapping the main TSS or slightly downstream of it. These features have positive effects and include A content (robust feature 33), as well as A-rich k-mers that also contain pyrimidines, and capture signals that have some resemblance to the TSS motif of (19).
Robust feature 42 is the occurrence of the ‘AATG’ 4-mer within the 50 bp downstream of the main TSS, and has a positive effect. This feature may in fact represent a translation-related signal, suggesting that a short 5′UTR contributes to higher translation rates (recall that the promoter activity measures were based on YFP measurements), perhaps in relation with the ribosome scanning of the mRNA for the first AUG (34). To further assess this result, we compared 5′UTR lengths (32) of four gene subsets: constitutive–high, constitutive–low, regulated–high and regulated–low. Figure 5 shows the cumulative distributions of 5′UTR lengths for these four subsets. For constitutive–high genes alone, 5′UTRs were found to be significantly shorter than those of the other subsets (rank-sum P-values < 10−5). This suggests that 5′UTR length may indeed have an effect on expression for constitutive genes, but less so for regulated genes. Our result extends previous results (35,36) that showed that constitutive genes tend to have shorter 5′UTRs than other genes.
Figure 5.
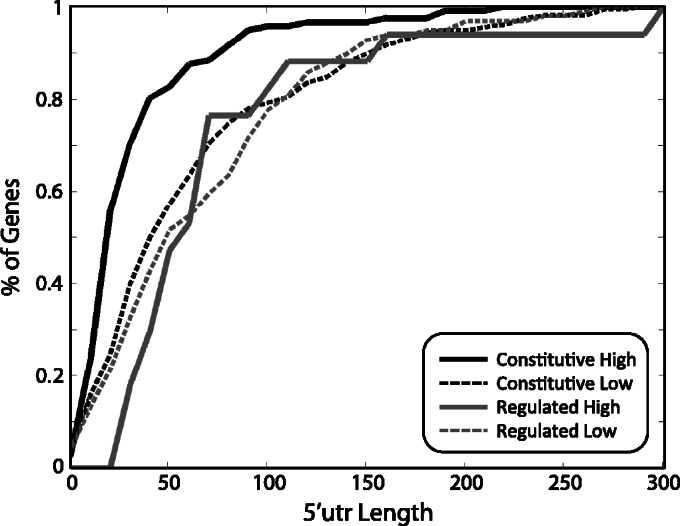
5′UTR length may affect expression in constitutive genes. Cumulative distributions of 5′UTR lengths (32) for four gene subsets. 5′UTRs of constitutive–high genes tend to be shorter than those of the other three subsets (rank-sum P-values < 10−5).
Robust features 43–48 are of G\C content and of G\C-rich 4-mers in windows around the main TSS, and have negative effects, in accord with above observations.
Thus, by using a quantitative modeling approach, our results demonstrate that core promoter sequence features can explain a large fraction of the variance in maximal promoter activities of held-out genes. We were also able to suggest specific sequence features in different parts of the core promoter as having a role in determining the maximal promoter activity. These features fall into several classes, as illustrated in Figure 4B.
The effects of the complexity and location of sequence features on prediction
The linear modeling results reported in the previous section were based on features of the core promoter region with sequence complexity ranging from base content and up to 4-mers. An interesting question that arises is whether low sequence complexity features suffice to get similar performance. To test this, we repeated our linear model learning scheme several times, starting with only base content features, and gradually added features of higher sequence complexity, up to features of 5-mers. The mean (over 10 models) test R2 is reported in Figure 6A for each sequence complexity threshold, demonstrating that using base content features alone can explain (on average) >20% of the variance in the test data. Still, increasing sequence complexity up to 4-mers further contributed to test performance, suggesting that higher complexity sequence features of the core promoter region indeed play an additional non-redundant role in determining the maximal promoter activity.
Figure 6.

A comparison of mean test R2 of linear models learned with varying complexity/location constraints on the sequence features. Here, the same 10-fold cross validated linear model learning scheme, described in the main text and in the Figure 4 legend, was applied. (A) Comparing the mean test R2 of models with increasing complexity of the sequence features allowed to be used. Using base content features alone can result in mean test R2 of >0.2, but <0.254, which can only be attained when higher order features (up to 4-mers) are also allowed to be used. (B) Comparing the mean test R2 of models where the sequence features allowed to be used are constrained to be within different 100 and 50 bp windows (around the main TSS). Each window is represented by a rectangle over its positions, color coded according to its mean test R2. The highest mean test R2 of 0.194 is attained for features within the [−75, 24] or within the [−50, 49] windows.
Another interesting question is which regions of the core promoter are more predictive of the maximal promoter activity. To shed light on this, we again repeated our linear model learning scheme multiple times (using features of up to 4-mers), each time using only features that fall within a certain window (of length 100 or 50 bp) over the core promoter region. For each such window, its resulting mean test R2 is shown in Figure 6B, showing that it suffices to take features within the [−50, 49] or the [−75, 24] regions to explain 19.4% of the test variance. Indeed, most of the robust features that were found to have high (absolute) effects (Figure 4A) were computed over windows that fall within the [−75, 49] region around the TSS.
Core promoter sequence is also indicative of maximal promoter activity in human
Our results above show that core promoter sequence is indicative of maximal promoter activity in yeast. A natural question that arises is whether these results also hold for other organisms.
To explore this, we examined mRNA expression data (37) of 10 human cell lines (GM12878, GM12892, H1-hESC, HCT116, HeLa-S3, HepG2, HSMM, HUVEC, K562 and MCF7), available at TSS resolution (for each gene, an FPKM mRNA abundance measure is reported for different TSSs, see also Supplementary Information). For each cell line, there were between two and four replicates of mRNA expression measurements, and for each TSS we conservatively took the minimum replicate value as its mRNA expression level. We then chose only TSSs that were expressed in all cell lines, and were the most highly expressed out of all other TSSs of the same gene. This left us with a set of 8025 TSSs that are constitutively expressed in the above 10 cell lines (see Supplementary Table S2). We further defined two subsets of these TSSs, based on their maximal mRNA expression (the maximal FPKM over the 10 different cell lines): 1035 TSSs with high maximal mRNA expression (maximal FPKM ≥ 100) and 1218 TSSs with low maximal expression (maximal FPKM < 5). The high maximal expression subset is, by definition, a subset of TSSs with high maximal promoter activity. The low maximal expression subset is an approximation of a subset with low maximal promoter activity, as some of its TSSs may be highly expressed in other cell lines or conditions, or alternatively, their mRNA products may be strongly downregulated posttranscriptionally.
Similar to our analysis in yeast (see above), we analyzed various sequence signals within the [−200, 100] region around the TSSs, including base content (mononucleotides and G + C), CpG and GpC content, as well as the percent of TSSs with TATA box hits, or with hits of 6-mers of the SP1 transcription factor motif consensus (GGGCGG or its reverse complement CCGCCC). For all of these sequence signals (Figure 7A) there were significant differences (see rank-sum P-values in Supplementary Figure S4) between the set of high maximal expression TSSs (Figure 7A, left column) and the set of low maximal expression TSSs (Figure 7A, middle column).
Figure 7.

Human core promoter sequence signals differ between constitutive TSSs with different maximal expression. (A) Mean nucleotide, k-mers and TATA box content, computed using a sliding window (20 bp long, 10 bp step) over the [−200, 100] region around TSSs that are constitutively expressesed in 10 different human cell lines (37). Plots are arranged in three columns: constitutive TSSs with high maximal expression in the left, constitutive TSSs with low maximal expression in the middle and all constitutive TSSs in the right. The vertical dashed lines represent the location of the TSS. The horizontal dotted lines are to assist with the comparison of plots between columns. SP1 - the GGGCGG\CCGCCC 6-mers of the SP1 TF binding motif consensus. (B) Fifty of the above constitutive TSSs were of RPs, and they were divided into two subsets of 25 higher expressed and 25 lower expressed (based on their maximal expression). Mean nucleotides content around the TSSs is shown for the two subsets.
High maximal expression TSSs tend to have significantly lower A and T content around the TSS than low maximal expression TSSs (Supplementary Figure S4A and B), and, conversely, higher C, G, G\C, GpC and CpG content around the TSS (Supplementary Figure S4C–G). While human core promoters are known to have high G\C, GpC and CpG content compared with flanking regions (38,39), here we show that their core promoter content is indicative of the maximal promoter activity. Accordingly, features of G\C, GpC and CpG richness around the TSS were found to significantly separate between the high and the low maximal expression TSSs, with AUC scores of 0.623, 0.64 and 0.682, respectively (Supplementary Figure S5). Recently, one study showed that high G\C and CpG content promote nucleosome depletion in mammalian promoters, both in vivo and in vitro (40). Thus, higher G\C and CpG content around the TSS would lower its nucleosome occupancy, making it more accessible for PIC formation and hence more highly expressed, in line with our results. Importantly, our focus here on constitutive core promoters removes the possibility that the differences between the high and the low maximal expression TSSs are due to differences between constitutive and tissue-specific core promoters (for instance, constitutive human core promoters are known to be CpG richer (39)).
Among several TF motifs that are known to be enriched in core promoters of constitutive genes, SP1 motifs are the most abundant (38). SP1 consensus 6-mers (GGGCGG or its reverse complement CCGCCC) in the 100 bp upstream of the TSS are found in 3355 of the 8025 (41.8%) constitutive core promoters, and are significantly depleted in the low maximal expression subset (found in 304 out of 1218, 25%, P < 10−39), suggesting that their existence may contribute to higher levels of maximal promoter activity. Other TF motifs known to be enriched in core promoters include those of NF-Y (the CAAT-box) and ETS (38). Similar to the SP1 consensus 6-mers, both NF-Y consensus 5-mers (CCAAT or its reverse complement ATTGG) and ETS consensus 6-mers (CCGGAA or its reverse complement TTCCGG) occur more around high maximal expression TSSs than around low maximal expression TSSs (Supplementary Figure S6).
While 10–24% of human genes were estimated to have a core promoter containing a TATA box, core promoters of constitutively expressed human genes were shown to be mostly TATA-less (41). In line with this, consensus TATA boxes (TATAWAWR) that are distanced 50–20 bp upstream of the TSS are found in only 60 of the 8025 constitutive core promoters (0.75%). Albeit the small numbers, they are significantly enriched in the high maximal expression subset (found in 29 out of 1035, 2.8%, P < 10−11), and depleted in the low maximal expression subset (found in 3 out of 1218, 0.25%, P-value 0.013), suggesting that the TATA box may contribute to higher levels of maximal promoter activity, as in yeast (see above).
While for most of the above sequence elements, the mean signal of the high maximal expression TSSs (Figure 7A, left column) seems relatively similar to that of all constitutive TSSs (Figure 7A, right column), there are significant differences between the two sets (see Supplementary Figure S4B–E). Most notably, high maximal expression TSSs tend to have higher C content immediately upstream of the TSS, and lower G content at and downstream of the TSS, compared with the average constitutive TSS.
Out of the above defined set of 8025 constitutive TSSs, 50 were TSSs of ribosomal proteins (RPs). Human RPs share a common core promoter architecture (42,43), and most of them are similarly expressed across tissues (44), suggesting that they are jointly regulated. Many of the 50 constitutive RP TSSs were very highly expressed, with 46 of them included in the above high maximal expression TSSs subset. We partitioned these 50 TSSs to the 25 with higher maximal expression and the 25 with lower maximal expression, and compared their mean base content around the TSS (Figure 7B). Here, too, we found significant differences between the two subsets. Most notably, RP TSSs with higher maximal expression tend to be C richer and G poorer in the [−10, 9] window around the TSS (rank-sum P-values 0.02 and 0.032, respectively). Accordingly, C content in that window separates the higher maximal expression RP TSSs from the lower ones with an AUC score of 0.69. Mammalian RP core promoters were shown to have a polypyrimidine initiator (43). Our results suggest that the pyrimidine content of this initiator element may affect its efficiency.
Finally, following the same 10-fold CV linear model learning scheme as with the yeast data (see above), we learned 10 linear models that predict the maximal expression (log of the maximal FPKM measure) of the 8025 constitutive human TSSs from base content and k-mer features of their core promoters. Mean model performance measures (R2, Pearson correlation r, Spearman correlation ρ) and a table detailing 58 robust features (that were included in at least 9 out of the 10 models) are shown in Supplementary Figure S7. The linear models could only explain, on average, 7% of the variation in the maximal expression of held-out test TSSs (with corresponding test mean r = 0.268 and ρ = 0.238), but consistently so (low standard deviation between models). The low predictive power of these linear models is likely due to the great complexity and diversity of human core promoter architectures. In accord with the results above, features of CpG containing k-mers were found to be the most significant predictors of higher maximal expression, and features of TATA box, NF-Y, ETS and SP1 consensus k-mers were also found to contribute to higher maximal expression.
Taken together, our results show that various human core promoter sequence features are predictive of maximal promoter activity, suggesting that they likely have a causal role in their determination.
DISCUSSION
Possible functional roles of different yeast core promoter features
In this work, we studied the relation between the yeast core promoter sequence and maximal promoter activity. Using a linear modeling framework, we were able to highlight a concise set of yeast core promoter features that may play a role in determining maximal promoter activity, out of a large initial set of base content and k-mer features.
The majority of these features are located between 75 bp upstream and 50 bp downstream of the main TSS, mostly downstream of TBP binding signals (see Figure 1). Following our base content analysis shown in Figure 1, we assigned these features into 3 major classes (Figure 4): features of T-rich or T-poor elements upstream of the main TSS, features of TSS-related elements (mostly A rich) at and downstream of the main TSS and features of G\C-rich elements around the main TSS.
We showed that core promoters that have high maximal promoter activity tend to be T rich upstream of the main TSS and A rich at and downstream of the main TSS (Figures 1, 3 and 4), and moreover, that this is true for both constitutive and regulated genes (Figures 1 and 3). This suggests that the T-richness followed by A-richness signals do not affect the regulation of PIC recruitment (that differs between constitutive and regulated genes), and because they are physically downstream of where the PIC is formed, they probably represent signals that affect pol-II scanning and TSS selection. This is in line with past evidence that pol-II is subjected to additional rate-limiting steps following its recruitment (45).
High maximal promoter activity core promoters also tend to have lower G\C content around their main TSS (Figure 1). Again, this is true for both constitutive and regulated genes, although they differ in their overall G\C-content landscape, suggesting that here too the effect is on pol-II scanning and TSS selection. As G\C content and the intrinsic nucleosome occupancy are highly correlated ((33), Figures 1 and 2A), this suggests that lower intrinsic occupancy of the +1 nucleosome (Figure 2A) over the TSS contributes to higher maximal promoter activity.
Two major differences between core promoters of constitutive and regulated genes are evident (Figure 2). First, constitutive core promoters encode for an intrinsic nucleosome-free region (NFR) upstream of their main TSS, while most regulated core promoters encode for an intrinsic NFR at the main TSS. Second, the in vivo nucleosome occupancy in rich media growth conditions and the intrinsic nucleosome occupancy of constitutive core promoters are highly similar, while for many regulated core promoters (especially with medium and high Emax), they are not, with their in vivo NFR situated upstream of the TSS. Recently, one study showed that in yeast cells grown in rich medium, TSSs of ‘TATA-less' genes (most of which are constitutive genes) are tightly located around the 5′ edge of the +1 nucleosome, while TSSs of ‘TATA-containing' genes (most of which are regulated genes) are more freely dispersed downstream into the +1 nucleosome location (3). This study suggested that in the core promoters of ‘TATA-containing' genes there may be competition between the PIC and the +1 nucleosome, where the PIC formation is coupled with +1 nucleosome eviction that removes an impediment to pol-II scanning. This hypothesis would be more adequate had the +1 nucleosome occupancy over the TSS been intrinsic (which is often not the case, as shown in Figure 2). Instead, we suggest the following explanation. In many of the regulated genes, repression or downregulation is achieved by remodeling of the +1 nucleosome, shifting it from its intrinsically favored location, downstream of the TSS, to a more upstream location where the TSS is occupied. This mode of repression was recently demonstrated for genes that are either repressed on carbon starvation or repressed in rich medium (and induced on carbon starvation) (46). The shift of the +1 nucleosome away from its intrinsically favored location is transient (47), still allowing PIC recruitment at lower rates as the +1 nucleosome shifts back to its intrinsically favored position, in which the TSS is not occupied. Such remodeling does not occur in constitutive genes, and thus, their in vivo nucleosome occupancy around the TSS is highly similar to the intrinsic one. Under such a model of repression by +1 nucleosome remodeling, the +1 nucleosome is not evicted, and in both constitutive and regulated genes, the PIC is recruited to the core promoter when the +1 nucleosome is at its intrinsically favored position. Consequently, for both constitutive and regulated genes, lower intrinsic nucleosome occupancy over the TSS can be expected to result in fewer impediments on the scanning pol-II and contribute to higher maximal promoter activity. This is in line with what we observed.
Is yeast core promoter T richness followed by A richness a TSS locator signal?
As mentioned above, (27) suggested that yeast core promoter T richness followed by A richness is a signal that plays a role in TSS localization, and termed it the TSS ‘locator’. Because most genes have multiple alternative TSSs (of varying intensities, see (12) for example), this suggests that a stronger ‘locator’ signal would lead toward focused transcription initiation, at one strong TSS, while a weak ‘locator’ signal would lead toward dispersed transcription initiation, at multiple weak TSSs. To assess this, we used data of (48) that measured multiple TSS instances per gene (some instances being different measurements of the same TSS) for many of the S. cerevisiae genes. Each measured TSS instance is in fact a sample from the unknown TSS distribution of the respective gene, and the number of samples depended on the expression level of the gene in cells grown in rich media conditions. We therefore limited our analysis to a comparison of constitutive genes with either high or very high maximal promoter activity (as above defined), as they are highly expressed in rich medium. Still, to avoid cases that were significantly undersampled, we used only genes that had at least 10 measured TSS instances. This left us with 31 (out of 36) constitutive–very high genes, and with 77 (out of 122) constitutive–high genes. For each gene we computed the proportion of TSS instances that were within 20 bp of the main TSS (the one with most instances), indicating how focused is transcription initiation of that gene. In Figure 8, we show the histograms of these values for the constitutive–very high and for the constitutive–high genes. It is evident that transcription initiation of constitutive–very high genes tends to be more focused than that of constitutive–high genes (rank-sum P-value 0.0077). This provides some support for the above TSS ‘locator’ hypothesis, as the T-richness followed by A-richness signal is stronger in constitutive–very high genes (Figure 1).
Figure 8.
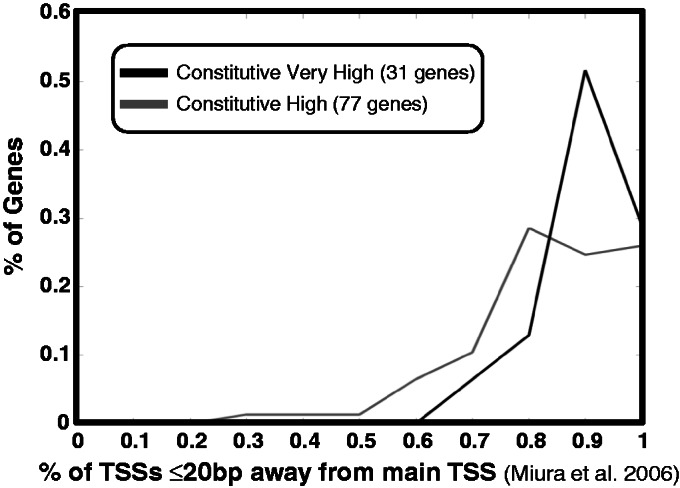
Transcription initiation in constitutive genes with high expression is more localized than in constitutive genes with lower expression. Histograms of a measure indicating how focused (less dispersed) is transcription initiation of a gene (the proportion of TSS instances that are within 20 bp of the main TSS, according to the data of (48)). This measure was computed for constitutive–very high and for constitutive–high genes that had at least 10 TSS instances measured, and was found to be significanly higher in constitutive–very high genes (rank-sum P-value 0.0077).
In this study, we show that core promoter sequence is predictive of maximal promoter activity, and suggest various sequence features that play a role in determining it in yeast as well as in human. Our results also highlight open questions on how the core promoter sequence affects promoter activity. In yeast, we do not yet know how the core promoter sequence determines the TSS distribution and how this TSS distribution affects promoter activity. In human, we still do not know the relation between the multitude of possible configurations of core promoter elements and promoter activity. We intend to pursue these questions in future studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2, Supplementary Figures 1–7 and Supplementary Information.
FUNDING
The European Research Council (ERC) and the US National Institutes of Health (NIH) (to E.S.). E.S. is the incumbent of the Soretta and Henry Shapiro career development chair. Funding for open access charge: ERC; the US NIH.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Ann. Rev. Biochem. 2003;72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- 2.Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004;116:699–709. doi: 10.1016/s0092-8674(04)00205-3. [DOI] [PubMed] [Google Scholar]
- 3.Rhee HS, Pugh BF. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012;483:295–301. doi: 10.1038/nature10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juven-Gershon T, Kadonaga JT. Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev. Biol. 2010;339:225–229. doi: 10.1016/j.ydbio.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giardina C, Lis JT. DNA melting on yeast RNA polymerase II promoters. Science. 1993;261:759–762. doi: 10.1126/science.8342041. [DOI] [PubMed] [Google Scholar]
- 6.Bushnell DA, Westover KD, Davis RE, Kornberg RD. Structural basis of transcription: an RNA polymerase II-TFIIB cocrystal at 4.5 Angstroms. Science. 2004;303:983–988. doi: 10.1126/science.1090838. [DOI] [PubMed] [Google Scholar]
- 7.Miller G, Hahn S. A DNA-tethered cleavage probe reveals the path for promoter DNA in the yeast preinitiation complex. Nat. Struct. Mol. Biol. 2006;13:603–610. doi: 10.1038/nsmb1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen W, Struhl K. Yeast mRNA initiation sites are determined primarily by specific sequences, not by the distance from the TATA element. EMBO J. 1985;4:3273–3280. doi: 10.1002/j.1460-2075.1985.tb04077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hahn S, Hoar ET, Guarente L. Each of three “TATA elements” specifies a subset of the transcription initiation sites at the CYC-1 promoter of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA. 1985;82:8562–8566. doi: 10.1073/pnas.82.24.8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagawa F, Fink GR. The relationship between the “TATA” sequence and transcription initiation sites at the HIS4 gene of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA. 1985;82:8557–8561. doi: 10.1073/pnas.82.24.8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McNeil JB, Smith M. Transcription initiation of the Saccharomyces cerevisiae iso-1-cytochrome c gene. Multiple, independent T-A-T-A sequences. J. Mol. Biol. 1986;187:363–378. doi: 10.1016/0022-2836(86)90439-0. [DOI] [PubMed] [Google Scholar]
- 12.Kuehner JN, Brow DA. Quantitative analysis of in vivo initiator selection by yeast RNA polymerase II supports a scanning model. J. Biol. Chem. 2006;281:14119–14128. doi: 10.1074/jbc.M601937200. [DOI] [PubMed] [Google Scholar]
- 13.Sugihara F, Kasahara K, Kokubo T. Highly redundant function of multiple AT-rich sequences as core promoter elements in the TATA-less RPS5 promoter of Saccharomyces cerevisiae. Nucleic Acids Res. 2011;39:59–75. doi: 10.1093/nar/gkq741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faitar SL, Brodie SA, Ponticelli AS. Promoter-specific shifts in transcription initiation conferred by yeast TFIIB mutations are determined by the sequence in the immediate vicinity of the start sites. Mol. Cell. Biol. 2001;21:4427–4440. doi: 10.1128/MCB.21.14.4427-4440.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khaperskyy DA, Ammerman ML, Majovski RC, Ponticelli AS. Functions of Saccharomyces cerevisiae TFIIF during transcription start site utilization. Mol. Cell. Biol. 2008;28:3757–3766. doi: 10.1128/MCB.02272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fishburn J, Hahn S. Architecture of the yeast RNA polymerase II open complex and regulation of activity by TFIIF. Mol. Cell. Biol. 2012;32:12–25. doi: 10.1128/MCB.06242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goel S, Krishnamurthy S, Hampsey M. Mechanism of start site selection by RNA polymerase II: interplay between TFIIB and Ssl2/XPB helicase subunit of TFIIH. J. Biol. Chem. 2012;287:557–567. doi: 10.1074/jbc.M111.281576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furter-Graves EM, Hall BD. DNA sequence elements required for transcription initiation of the Schizosaccharomyces pombe ADH gene in Saccharomyces cerevisiae. Mol. Gen. Genet. 1990;223:407–416. doi: 10.1007/BF00264447. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z, Dietrich FS. Mapping of transcription start sites in Saccharomyces cerevisiae using 5′ SAGE. Nucleic Acids Res. 2005;33:2838–2851. doi: 10.1093/nar/gki583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen W, Struhl K. Saturation mutagenesis of a yeast his3 “TATA element”: genetic evidence for a specific TATA-binding protein. Proc. Natl Acad. Sci. USA. 1988;85:2691–2695. doi: 10.1073/pnas.85.8.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wobbe CR, Struhl K. Yeast and human TATA-binding proteins have nearly identical DNA sequence requirements for transcription in vitro. Molecular and cellular biology. 1990;10:3859–3867. doi: 10.1128/mcb.10.8.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahadevan S, Struhl K. Tc, an unusual promoter element required for constitutive transcription of the yeast HIS3 gene. Mol. Cell. Biol. 1990;10:4447–4455. doi: 10.1128/mcb.10.9.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singer VL, Wobbe CR, Struhl K. A wide variety of DNA sequences can functionally replace a yeast TATA element for transcriptional activation. Genes Dev. 1990;4:636–645. doi: 10.1101/gad.4.4.636. [DOI] [PubMed] [Google Scholar]
- 24.Yean D, Gralla J. Transcription reinitiation rate: a special role for the TATA box. Mol. Cell. Biol. 1997;17:3809–3816. doi: 10.1128/mcb.17.7.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blake WJ, Balázsi G, Kohanski MA, Isaacs FJ, Murphy KF, Kuang Y, Cantor CR, Walt DR, Collins JJ. Phenotypic consequences of promoter-mediated transcriptional noise. Mol. Cell. 2006;24:853–865. doi: 10.1016/j.molcel.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Mogno I, Vallania F, Mitra RD, Cohen BA. TATA is a modular component of synthetic promoters. Genome Res. 2010;20:1391–1397. doi: 10.1101/gr.106732.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maicas E, Friesen JD. A sequence pattern that occurs at the transcription initiation region of yeast RNA polymerase II promoters. Nucleic Acids Res. 1990;18:3387–3393. doi: 10.1093/nar/18.11.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobson MJ, Tuite MF, Roberts NA, Kingsman AJ, Kingsman SM, Perkins RE, Conroy SC, Fothergill LA. Conservation of high efficiency promoter sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 1982;10: 2625–2637. doi: 10.1093/nar/10.8.2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou H, Hastie T. Regularization and variable selection via the elastic net. J. R Stat. Soc. 2005;67:301–320. [Google Scholar]
- 31.Efron B, Hastie T, Johnstone I, Tibshirani R. Least angle regression. Ann. Stat. 2004;32:407–451. [Google Scholar]
- 32.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tillo D, Hughes TR. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics. 2009;10:442. doi: 10.1186/1471-2105-10-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson RJ, Hellen CUT, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM. A high-resolution map of transcription in the yeast genome. Proc. Natl Acad. Sci. USA. 2006;103:5320–5325. doi: 10.1073/pnas.0601091103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Z, Li W-H. Evolution of 5′ untranslated region length and gene expression reprogramming in yeasts. Mol. Biol. Evol. 2012;29:81–89. doi: 10.1093/molbev/msr143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.FitzGerald PC, Shlyakhtenko A, Mir AA, Vinson C. Clustering of DNA sequences in human promoters. Genome Res. 2004;14:1562–1574. doi: 10.1101/gr.1953904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandelin A, Carninci P, Lenhard B, Ponjavic J, Hayashizaki Y, Hume DA. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat. Rev. Genet. 2007;8:424–436. doi: 10.1038/nrg2026. [DOI] [PubMed] [Google Scholar]
- 40.Fenouil R, Cauchy P, Koch F, Descostes N, Cabeza JZ, Innocenti C, Ferrier P, Spicuglia S, Gut M, Gut I, et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012;22:2399–2408. doi: 10.1101/gr.138776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang C, Bolotin E, Jiang T, Sladek FM, Martinez E. Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene. 2007;389:52–65. doi: 10.1016/j.gene.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshihama M, Uechi T, Asakawa S, Kawasaki K, Kato S, Higa S, Maeda N, Minoshima S, Tanaka T, Shimizu N, et al. The human ribosomal protein genes: sequencing and comparative analysis of 73 genes. Genome Res. 2002;12:379–390. doi: 10.1101/gr.214202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perry RP. The architecture of mammalian ribosomal protein promoters. BMC Evol. Biol. 2005;5:15. doi: 10.1186/1471-2148-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishii K, Washio T, Uechi T, Yoshihama M, Kenmochi N, Tomita M. Characteristics and clustering of human ribosomal protein genes. BMC Genomics. 2006;7:37. doi: 10.1186/1471-2164-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Venters BJ, Pugh BF. A canonical promoter organization of the transcription machinery and its regulators in the Saccharomyces genome. Genome Res. 2009;19:360–371. doi: 10.1101/gr.084970.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L, Ma H, Pugh BF. Stable and dynamic nucleosome states during a meiotic developmental process. Genome Res. 2011;21:875–884. doi: 10.1101/gr.117465.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell. 2012;149:1461–1473. doi: 10.1016/j.cell.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miura F, Kawaguchi N, Sese J, Toyoda A, Hattori M, Morishita S, Ito T. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proc. Natl Acad. Sci. USA. 2006;103:17846–17851. doi: 10.1073/pnas.0605645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.