

Background: The C2 domain is a Ca2+ sensor that drives the first step in the activation of conventional PKC isozymes.
Results: Hydrophobic interactions drive membrane association and electrostatic interactions drive membrane retention of the C2 domain.
Conclusion: The amplitude and location of conventional PKC signaling is controlled by residues in the C2 domain.
Significance: Point mutations in the C2 domain are associated with disease.
Keywords: Calcium, Calcium-binding Proteins, Calcium Signaling, Protein Kinase C (PKC), Signal Transduction, C2 Domain, Membrane Translocation
Abstract
The cellular activation of conventional protein kinase C (PKC) isozymes is initiated by the binding of their C2 domains to membranes in response to elevations in intracellular Ca2+. Following this C2 domain-mediated membrane recruitment, the C1 domain binds its membrane-embedded ligand diacylglycerol, resulting in activation of PKC. Here we explore the molecular mechanisms by which the C2 domain controls the initial step in the activation of PKC. Using stopped-flow fluorescence spectroscopy to measure association and dissociation rate constants, we show that hydrophobic interactions are the major driving force in the binding of the C2 domain to anionic membranes, whereas electrostatic interactions dominate in membrane retention. Specifically, mutation of select hydrophobic or select basic residues in the Ca2+-binding loops reduces membrane affinity by distinct mechanisms; mutation of hydrophobic residues primarily alters association rate constants, whereas mutation of charged residues affects dissociation rate constants. Live cell imaging reveals that introduction of these mutations into full-length PKCα not only reduces the Ca2+-dependent translocation to plasma membrane but, by impairing the plasma membrane-sensing role of the C2 domain, causes phorbol ester-triggered redistribution of PKCα to other membranes, such as the Golgi. These data underscore the key role of the C2 domain in driving conventional PKC isozymes to the plasma membrane and reveal that not only the amplitude but also the subcellular location of conventional PKC signaling can be tuned by altering the affinity of this module for membranes.
Introduction
The first step in the activation of conventional protein kinase C (PKC) isozymes is a Ca2+-dependent recruitment to the plasma membrane. This step is triggered by the myriad of signals that result in hydrolysis of phosphatidylinositol 4,5-bisphosphate to generate the soluble second messenger inositol trisphosphate, which mobilizes intracellular Ca2+, and the membrane-embedded lipid second messenger diacylglycerol (1, 2). Binding to diacylglycerol is the key “on/off” switch for PKC activation; this phosphatidylserine-assisted interaction provides the energy to expel an autoinhibitory pseudosubstrate segment from the substrate-binding cavity (3). The recognition of this membrane-embedded ligand by a cytosolic enzyme is facilitated for conventional PKC isozymes (α, the alternatively spliced βI and βII, and γ) by Ca2+-dependent recruitment of PKC to membranes via a Ca2+ sensor, the C2 domain. This Ca2+-regulated targeting from three-dimensional space to the membrane allows the enzyme's C1 domain to initiate a two-dimensional search for its membrane-embedded ligand, diacylglycerol; this pretargeting by the C2 domain effectively increases the local concentration of diacylglycerol by three orders of magnitude, thus allowing highly efficient binding and activation (4). Novel PKC isozymes (δ, ϵ, η, and θ) do not have a Ca2+-regulated C2 domain; their activation by diacylglycerol is possible because of a single residue change in their C1B domain that allows them to bind diacylglycerol-containing membranes with 2 orders of magnitude higher affinity than conventional PKC isozymes (5). Thus, for conventional PKC isozymes, canonical second messenger-controlled activation depends on coincident detection of Ca2+ and diacylglycerol that occurs by Ca2+-dependent pretargeting to membranes.
Membrane-targeting modules drive the function of diverse signaling molecules. Such modules include C2, C1, FYVE, and pleckstrin homology domains, which play instrumental roles in signaling by reversibly controlling the localization and activation of signaling molecules in response to second messengers (6–8). Membrane binding of these modules is controlled by a variety of second messengers, including Ca2+ (C2 domains), diacylglycerol (C1 domains), and specific phosphoinositides (FYVE and pleckstrin homology domains). For signaling enzymes, such as PKC, the coordinated use of two membrane-targeting modules allows ultrasensitivity and increased specificity in responding to extracellular signals (9). Precise tuning of the agonist-dependent affinity of signaling enzymes is essential for cellular homeostasis, and dysregulation of this membrane affinity results in pathophysiologies. A case in point is the oncogenic kinase Akt, where a single point mutation in its membrane-targeting pleckstrin homology domain from an acidic to a basic residue results in constitutive membrane localization of Akt and is associated with human breast, colorectal, and ovarian cancers (10). Understanding the mechanisms of membrane binding of these modules could provide therapeutic targets for diseases in which mutations in these domains alter their localization and activity to promote or impair signaling.
Membrane binding acutely controls the amplitude of agonist-dependent signaling by conventional PKC isozymes (11). Live cell imaging studies using genetically encoded reporters to monitor PKC activity have revealed that signals that result in hydrolysis of phosphatidylinositol 4,5-bisphosphate result in two phases of conventional PKC activation: a rapid rise in activity that tracks with the rise in intracellular Ca2+ (t½ of seconds) followed by slow decay in activity that tracks with the decay in diacylglycerol (t½ on the order of minutes at the plasma membrane and tens of minutes at the Golgi) (12). Under some conditions, oscillations in Ca2+ result in oscillations in PKC activity (13), underscoring the role of the C2 domain's membrane attachment in controlling the activity of PKC. Indeed, biophysical studies have previously established that the affinity of PKC for membranes needs to be sufficiently high to provide the energy to release the autoinhibitory pseudosubstrate segment. For conventional PKC isozymes, engagement of both the C1 and C2 domains on membranes is necessary to allow response to physiological agonists (9). Note that phorbol esters, which bind the C1 domain with 2-order of magnitude higher affinity than diacylglycerol (14), are able to engage PKC to membranes with sufficiently high affinity to cause maximal activation. Importantly, the affinity of the C1 and C2 domains for membranes acutely controls the output of agonist-evoked PKC signaling.
Biochemical and biophysical studies over the past 2 decades have provided considerable insight into the structure and function of C2 domains (9, 15–17). The C2 domain is an ∼130 residue segment that folds into two four-stranded β-sheets using one of two different topologies, depending on the protein (15). The domain exists as a Ca2+ sensor in proteins such as conventional PKC isozymes and as a Ca2+-independent module in other proteins, such as the novel PKC isozymes. Ca2+ binding is conferred by the presence of a set of conserved Asp residues in a “mouth” formed by two loops at the tip of the β-sandwich (17–20). Both kinetic and structural studies have established that the C2 domain of PKCα binds two Ca2+ ions (21, 22). Structure-function studies have established that the integrity of these loops and the presence of the conserved Asp residues are necessary for the binding of both the isolated C2 domain and full-length PKC to membranes in vitro and to plasma membrane in cells (21, 23–29). Although Ca2+ binding has been proposed to serve as a simple “electrostatic switch” for C2 domain binding to anionic membranes (30, 31), neutralization of the acidic binding pocket by mutation of key Asp residues (to mimic neutralization upon Ca2+ binding) does not bypass the requirement for Ca2+ to promote membrane interaction (23). Thus, Ca2+ serves a specific structural role in allowing the C2 domain to bind anionic membranes. In addition to the Ca2+-binding site, a lysine-rich cluster within the β3- and β4-sheets of the C2 domain binds phosphatidylinositol 4,5-bisphosphate (27, 32–36). It is this interaction with phosphatidylinositol 4,5-bisphosphate that directs conventional PKC isozymes to the plasma membrane, which is enriched in this lipid (38–41).
Here we dissect the role of electrostatic and hydrophobic interactions in the Ca2+-triggered association of the C2 domain of a conventional PKC with membranes. Stopped-flow fluorescence spectroscopy reveals that hydrophobic interactions dominate in driving the C2 domain to membranes, whereas electrostatic interactions with ionic lipids dominate in retaining the domain on membranes. Altering binding kinetics by perturbation of either binding force through mutation of key charged or hydrophobic residues results in reduced cellular activation, revealing that each force plays a determining role in tuning the biological activity of PKC.
EXPERIMENTAL PROCEDURES
Reagents
1-Palmitoyl-2-oleoylphosphatidyl-l-serine (PS)4 and 1-palmitoyl-2-oleoylphosphatidylcholine (PC) were purchased from Avanti Polar Lipids. N-(5-(dimethylamino)naphthalene-1-sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (dPE) was purchased from Molecular Probes. PCR primers were prepared by IDT Technologies, Inc. Glutathione-Sepharose 4B was purchased from Amersham Biosciences.
Electrophoresis reagents were from Calbiochem and Bio-Rad. Thrombin was purchased from Calbiochem. The antibody for PKCα was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Phorbol 12,13-dibutyrate (PDBu) and thapsigargin were obtained from Calbiochem. The 1× Hanks' balanced salt solution was purchased from Cellgro. The jetPRIME transfection reagent was from PolyPlus Transfection. All other chemicals were reagent grade.
Cloning, Mutagenesis, and Purification
Rat PKCα DNA was a gift from Y. Nishizuka and Y. Ono (Kobe University, Kobe, Japan). The C2 domain (residues 179–283) was PCR-amplified and subcloned into a pGEX-4T3-GST plasmid using EcoRI and XhoI restriction sites. The C2 domain mutations R216A, R249A, R252A, K268A, W245A, W247A, L191A, and W274A were created using site-directed mutagenesis. The C2 domain of rat PKCα was expressed as a glutathione S-transferase (GST) fusion protein in BL21-pLysS cells as described previously (29). The isolated C2 domain was cleaved from the glutathione-Sepharose 4B-bound fusion protein using thrombin. The concentration of the various C2 domains was determined by UV absorbance using their molar extinction coefficient (20). Human PKCα, a gift from T. Hunter (Salk Institute), was Gateway (GATEWAY Cloning Technology) cloned into pcDNA3-YFP. The mutations R249A and W247A were created using site-directed mutagenesis. mpCFP (13) and GolgiCFP (11) were previously described.
Preparation of Lipid Vesicles
For stopped-flow fluorescence spectroscopy, phospholipids in chloroform were dried under nitrogen and then vacuum-dried for 2 h. The lipids were resuspended in a buffer of varying NaCl concentration (150–200 mm) and 20 mm HEPES, pH 7.4. The resuspended lipid was freeze/thawed five times and extruded through two 0.1-μm polycarbonate filters to prepare phospholipid vesicles of 100-nm diameter. The phosphorus concentration was assayed as described previously to determine the phospholipid concentration (42).
Stopped-flow Fluorescence Spectroscopy
An Applied Photophysics pi*-180 stopped-flow fluorescence spectrophotometer was used to determine the kinetics of binding to lipid vesicles, as described previously (4). For association experiments, purified C2 domain protein (0.2–1.0 μm) was rapidly mixed with equal volumes of increasing concentrations of dansyl-labeled anionic phospholipid vesicles (PS/PC/dPE, 35:60:5 mol ratio) at the indicated Ca2+ and NaCl concentrations, and protein-to-membrane fluorescence resonance energy transfer (FRET) was monitored, as described previously (4). For dissociation experiments, purified C2 domain protein (0.2–1.0 μm) was preincubated for 15–30 min with dansyl-labeled phospholipid vesicles (PS/PC/dPE, 35:60:5 mol ratio) and then rapidly mixed with equal volumes of a 10-fold excess of unlabeled anionic phospholipid vesicles (PS/PC, 40:60 mol ratio) at the indicated Ca2+ and NaCl concentrations, as described previously (20). Traces were fitted to a nonlinear least-squares curve fitting using KaleidaGraph according to the exponential equation,
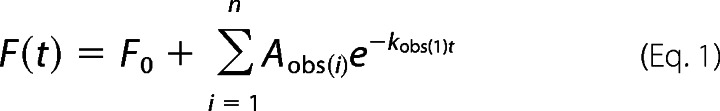 |
where F(t) equals the observed fluorescence at time t, F0 is a fluorescence offset representing the final fluorescence, Aobs(i) equals the amplitude, and the kobs(i) is the observed rate constant for ith of n phases. The experiments were performed under pseudo-first-order conditions; the observed rate constants were plotted as a function of lipid vesicle concentration and fitted with the linear manner equation,
where kon represents the apparent second-order association rate constant, and koff represents the apparent dissociation rate constant. The ratio of koff (determined from FRET association experiments) to kon (determined from FRET trapping experiments) provides the calculated apparent vesicle dissociation constant (Kdcalc).
Fluorescence Spectroscopy to Measure Steady State Ca2+ Binding
Intrinsic Trp emission of the C2 domain (0.5 μm) was monitored on a Jobin Yvon-SPEX FluoroMax-2 fluorescence spectrophotometer using 280-nm excitation light and by recording emission at 345 nm at 25 °C, as described previously (20). The observed fluorescence change (ΔFobs) was plotted as a function of free Ca2+ concentration (x) and fitted to the following modified Hill equation,
where ΔFmax represents the calculated maximum fluorescence change, nH represents the apparent Hill constant, and [Ca2+]½ represents the Ca2+ concentration at the midpoint of the titration, an approximation of the average Ca2+ dissociation constant. Because [Ca2+]½ was considerably greater than the protein concentration, the concentration of total Ca2+ was taken as the free concentration.
Cell Transfection and Imaging
COS-7 cells were maintained in DMEM (Cellgro) with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C. YFP-PKCα-WT, -R249A, or -W247A was co-transfected with mpCFP or GolgiCFP into COS-7 cells using the jetPRIME transfection reagent for 24 h. Cells were washed in 1× Hanks' balanced salt solution and imaged in the dark at room temperature. Imaging experiments were performed on the Zeiss Axiovert microscope (Carl Zeiss MicroImaging, Inc.) using a MicroMax digital camera (Roper-Princeton Instruments) controlled by MetaFluor software (Universal Imaging Corp.) All optical filters were from Chroma Technologies. Data were collected as described previously (13). Cells that were imaged were selected based on equal YFP expression to control for PKCα overexpression. Base-line images were acquired for 3 min before the addition of ligand. Thapsigargin was added at the indicated time to yield a final concentration of 5 μm, and PDBu was added 10 min later at a final concentration of 200 nm. FRET ratios of cells from at least three imaging dishes were normalized to their individual base lines and then averaged.
RESULTS
Design of PKCα C2 Domain Mutants
To study the role of electrostatic and hydrophobic interactions in the kinetics of membrane binding of the C2 domain, we designed eight constructs in which key charged or neutral residues were mutated to Ala (Fig. 1). Specifically, the basic residues Arg-216, Arg-249, and Arg-252 and the hydrophobic residues Leu-191, Trp-245, and Trp-247 were individually mutated to Ala. Residue Arg-216 is located on Ca2+-binding loop 2, and residues Arg-249 and Arg-252 reside on Ca2+-binding loop 3 (22); all three residues have been previously shown to facilitate membrane binding and activation of full-length PKCα (24–26). In addition, electron paramagnetic resonance studies have shown that Arg-249 and Arg-252 are involved in C2 penetration into the membrane (43). The hydrophobic residue Leu-191 is located on Ca2+-binding loop 1, and Trp-245 and Trp-247 are on Ca2+-binding loop 3. Previous studies have established that both Trp-245 and Trp-247 participate in membrane binding and facilitate activation of PKCα (24). We also mutated two residues distal to the Ca2+-binding loops that are not predicted to interact with membranes: Lys-268 and Trp-274.
FIGURE 1.

Structure of the C2 domain of PKCα. A ribbon diagram of the C2 domain of PKCα (based on the structure by Gómez-Fernández and colleagues (22)) shows residues mutated in this study. Basic residues are indicated in blue (Arg-216, Arg-249, and Arg-252 on the Ca2+-binding loops and the distally located Lys-268), and hydrophobic residues are indicated in red (Leu-191, Trp-247, and Trp-245 on the Ca2+-binding loops and the distally located Trp-274).
Membrane Binding Kinetics for PKCα C2 Domain Mutants
To assess the individual contributions of electrostatic versus hydrophobic interactions in the association and dissociation rate constants of the C2 domain, we used stopped-flow fluorescence spectroscopy to measure the binding kinetics of C2 domain mutants. Specifically, we measured FRET between dansyl-labeled lipid vesicles and the four intrinsic tryptophans in the C2 domain as a function of time to obtain the association rate constants (kon). Mutation of any of the three charged residues in the Ca2+ loops to Ala (R216A, R249A, and R252A) had only minimal effects on kon (Fig. 2A and Table 1). In contrast, mutation of the hydrophobic residues in the Ca2+ loops (W245A and W247A) significantly decreased the association rate constants compared with the wild-type domain (Fig. 2A and Table 1). Mutations of the two residues distal to the Ca2+ loops (K268A and W274A) resulted in modest increases in kon. Note that although mutation of individual Trp residues reduced the amplitude of the maximal FRET change (e.g. ∼2-fold for the W247A mutant), this did not affect the kinetic analysis, and data were highly reproducible (S.E. <2%). The much stronger effect of removing hydrophobic residues compared with basic residues on reducing the association rate constant reveals that hydrophobic, rather than electrostatic, interactions of residues within the Ca2+-binding loops are a major driving force in the recruitment of the C2 domain to membranes.
FIGURE 2.

Basic- or hydrophobic-to-neutral mutations on the C2 domain affect its recruitment to and retention on lipid vesicles. A, the binding of purified C2 domain (wild-type, R216A, R249A, R252A, K268A, W245A, W247A, L191A, or W274A) to anionic phospholipid vesicles was measured by FRET association stopped-flow fluorescence spectroscopy. The C2 domain protein (0.2–0.5 μm) was rapidly mixed with equal volumes of increasing concentrations of dansyl-labeled anionic phospholipid vesicles (PS/PC/dPE, 35:60:5 mol ratio) in the presence of 200 μm Ca2+ and 150 mm NaCl, as described under “Experimental Procedures.” The dansyl emission was measured over time, and the kobs was determined using a monophasic fit. The kon was calculated from the slope of the linear plot of kobs versus vesicle concentration. Data represent the weighted average ± S.E. (error bars) of 3–7 independent experiments. B, the binding of purified C2 domain (wild type, R216A, R249A, R252A, K268A, W245A, W247A, L191A, or W274A) to anionic phospholipid vesicles was measured by FRET dissociation stopped-flow analysis. The C2 domain protein (0.2–0.5 μm) was incubated for at least 15 min with dansyl-labeled phospholipid vesicles (PS/PC/dPE, 35:60:5 mol ratio). The protein and labeled lipid were then rapidly mixed with equal volumes of a 10-fold higher concentration of unlabeled anionic phospholipid vesicles (PS/PC, 40:60 mol ratio) in the presence of 200 μm Ca2+ and 150 mm NaCl. The dansyl emission was measured as a function of time, and the koff was determined using a monophasic fit. Data represent the weighted average ± S.E. of 3–7 independent experiments.
TABLE 1.
Effect of C2 domain basic- and hydrophobic-to-neutral residue mutations on equilibrium binding to lipid vesicles
Shown are the Kdcalc values obtained from the kon and koff in Fig. 2. Data represent weighted averages ± S.E. The -fold difference values relative to the Kdcalc of wild-type C2 domain are indicated; error represents S.E.
| C2α mutation | kon (×1010) | koff | Kdcalc | Kd -fold difference from WT |
|---|---|---|---|---|
| m−1s−1 | s−1 | nm | -fold | |
| WT | 2.241 ± 0.009 | 0.71 ± 0.02 | 0.032 ± 0.001 | 1.00 ± 0.04 |
| R216A | 1.87 ± 0.07 | 4.8 ± 0.2 | 0.25 ± 0.01 | 8.1 ± 0.5 |
| R249A | 1.95 ± 0.04 | 13.1 ± 0.2 | 0.67 ± 0.02 | 21.3 ± 0.9 |
| R252A | 2.63 ± 0.02 | 5.03 ± 0.02 | 0.191 ± 0.002 | 6.1 ± 0.2 |
| K268A | 3.40 ± 0.01 | 0.556 ± 0.005 | 0.0126 ± 0.0002 | 0.40 ± 0.01 |
| W245A | 0.496 ± 0.004 | 3.29 ± 0.02 | 0.6640 ± 0.006 | 21.0 ± 0.7 |
| W247A | 0.90 ± 0.02 | 5.9 ± 0.1 | 0.66 ± 0.02 | 20.9 ± 0.9 |
| L191A | 3.10 ± 0.03 | 0.959 ± 0.002 | 0.0309 ± 0.0003 | 0.98 ± 0.03 |
| W274A | 2.71 ± 0.03 | 0.540 ± 0.009 | 0.0208 ± 0.0004 | 0.66 ± 0.02 |
To measure the dissociation rate constants of the various mutants from membranes, we equilibrated the C2 domain with dansyl-labeled lipid vesicles and then used FRET to monitor the dissociation from the labeled vesicles upon the addition of a 10-fold excess of unlabeled vesicles. Mutation of any of the three charged residues in the Ca2+ loops to Ala dramatically increased the koff values compared with that of wild-type C2 domain: R216A, R249A, and R252A had koff values that were higher by 6-, 16-, and 6-fold, respectively (Fig. 2B and Table 1). Note that mutation of Arg-249 to Leu altered the dissociation rate constant by 8-fold (compared with 16-fold for mutation to Ala) (data not shown). The koff for the W245A and W247A mutants also increased by 4-fold and 9-fold, respectively, compared with the wild-type domain. The koff of L191A was similar to that of the wild-type domain (Table 1). Mutation of the distal residues Lys-268 and Trp-274 to Ala caused the koff to decrease slightly (Table 1). These results reveal that both electrostatic and hydrophobic interactions created by specific residues in the Ca2+-binding loops are critical for retaining the C2 domain at the membrane, with Arg-249 playing a particularly strong role in retaining the C2 domain on anionic membranes.
Effect of C2 Domain Mutations on Membrane Equilibrium Binding Constant
Using the experimentally determined values for kon and koff, we calculated the Kdcalc for each C2 domain mutant. The data in Table 1 reveal that the Kdcalc values for the C2 domains in which basic residues in the Ca2+ loops were mutated to Ala were at least 6-fold higher than that for the wild-type C2 domain; the Kdcalc values increased 8-, 21-, and 6-fold for R216A, R249A, and R252A, respectively, compared with wild-type. Mutation of the hydrophobic residues resulted in even greater increases in equilibrium dissociation constants; Kdcalc values for the W245A and W247A mutants increased 21- and 20-fold, respectively, compared with wild-type C2 domain. No significant change in Kdcalc was observed upon mutation of Leu-191 to Ala. Similarly, only modest (0.5-fold) changes in Kdcalc were noted for the K268A and W274A mutants. These results are consistent with previous studies establishing that residues within the Ca2+-binding loops critically determine the equilibrium binding of the C2 domain to anionic membranes. Here, by analysis of individual kinetic constants, we have determined the role of basic versus hydrophobic residues in membrane recruitment and retention. Importantly, we have identified two residues, Arg-249 and Trp-245, whose individual mutation to Ala results in a similar overall change in membrane affinity (21-fold) but by opposing mechanisms; the basic residue affects koff, and the hydrophobic residue primarily affects kon.
Effect of C2 Domain Mutations on Ionic Strength Sensitivity for Membrane Recruitment and Retention
We next addressed how mutating charged or hydrophobic residues in the C2 domain affected the ionic strength sensitivity of membrane recruitment. Increasing the ionic strength from 150 to 175 mm NaCl resulted in a modest (∼20%) increase in the kon of the wild-type C2 domain; this increase was reversed upon further increase to 200 mm NaCl (Fig. 3A). This same trend was observed for the R216A mutant. The increase observed in kon as the ionic strength was increased from 150 to 175 mm probably reflects strengthening of hydrophobic interactions; however, an even higher ionic strength could screen electrostatic interactions such as those present between anionic phospholipid headgroups and the C2 domain, resulting in a lowering of kon. In contrast, increasing ionic strength markedly enhanced kon for the domains in which Trp-245 or Trp-247 were mutated to Ala. Most strikingly, altering the ionic strength from 150 to 200 mm NaCl resulted in an increase in kon for the W247A mutant to a level comparable with that for the wild-type domain (Fig. 3A). Thus, increasing ionic strength rescues the reduction in kon resulting from removal of key hydrophobic residues. One explanation consistent with this result is that increasing ionic strength enhances the remaining hydrophobic interactions. Thus, removal of a key hydrophobic residue (e.g. Trp-247) reduces kon. However, strengthening of remaining hydrophobic interactions (e.g. those mediated by Trp-245) by increasing ionic strength increases on rate constants. Only minor effects of altering ionic strength were observed for the association rate constants of the L191A (Fig. 3A), W274A, and K268A domains (not shown). These data underscore the key role of hydrophobic interactions in driving the association of the C2 domain with membranes.
FIGURE 3.

Ionic strength sensitivity of membrane binding kinetics of C2 domain mutants. A, the binding of purified C2 domain (wild type, R216A, R249A, R252A, W245A, W247A, or L191A) to anionic phospholipid vesicles was measured by FRET association stopped-flow analysis as described in the legend to Fig. 2A. The conditions were the same except for increasing concentrations of NaCl as indicated. Data represent the weighted average ± S.E. (error bars) of 3–7 independent experiments. B, the binding of purified C2 domain (wild type, R216A, R249A, R252A, K268A, W245A, W247A, L191A, or W274A) to anionic phospholipid vesicles was measured by FRET dissociation stopped-flow analysis as described in Fig. 2B. The conditions were the same except for increasing concentrations of NaCl as indicated. Data represent the weighted average ± S.E. of 3–7 independent experiments. C, the Kdcalc values were obtained from the kon and koff obtained from FRET association and dissociation experiments in A and B. Data represent the weighted average ± S.E. of 3–7 independent experiments.
We next examined how ionic strength controlled the membrane dissociation rate constants for the C2 domain mutants. The koff of the wild type C2 domain increased as ionic strength increased, with an almost 3-fold higher koff measured in the presence of 200 mm NaCl compared with 150 mm NaCl (Fig. 3B). This 3-fold increase in koff in the presence of 200 mm NaCl compared with 150 mm NaCl was observed for all mutants except W245A. For this domain, altering ionic strength had no significant effect on koff. This suggests that for this mutant, increased hydrophobic interactions compensate for reduced electrostatic interactions, whereas for all other mutants and for the wild-type domain, electrostatic interactions dominate in membrane retention.
Effect of Ionic Strength on Equilibrium Constants for C2 Domain Mutations
We next examined the effect of ionic strength increase on Kdcalc values obtained from the kon and koff, as above. Normalizing the Kdcalc values to the values obtained at 150 mm NaCl revealed that the relative ionic strength sensitivity of R216A, R249A, R252A, and L191A followed the same trend as that of the wild-type C2 domain (Fig. 3C). In contrast, the binding affinity of W245A and W247A to lipid vesicles was insensitive to changes in ionic strength under the conditions examined (Fig. 3C). Examination of the individual contributions of the kon and koff to the Kdcalc revealed that the changes in recruitment and retention cancel each other out so that the apparent lipid vesicle binding affinity is insensitive to increases in ionic strength. Thus, without these hydrophobic residues, the equilibrium binding of the C2 domain to anionic membranes is insensitive to ionic strength changes, whereas wild-type C2 domain and the charged-to-neutral residue mutations are sensitive to increasing ionic strength.
Kinetic Constants for Double Mutations in C2 Domain
To examine whether the charged residues cooperate in promoting the binding of the C2 domain to anionic membranes, we introduced double mutations of basic residues in the C2 domain of PKCα: R216A/R249A, R249A/R252A, and R216A/R252A. Although previous studies have examined the contribution of R249A/R252A, R216A, and R249A to equilibrium binding, the combined and individual contributions of these residues to kon and koff have not been reported (24–26). We measured the binding kinetics of these domains using both the FRET association and FRET dissociation experiments. The kon values for R216A/R252A and R249A/R252A were comparable with that of the wild-type C2 domain (Fig. 4A and Table 2). In contrast, kon for the R216A/R249A mutant decreased ∼2-fold compared with wild-type C2 domain (Fig. 4A and Table 2). These data suggest that either Arg-216 or Arg-249 is sufficient for unimpaired association of the C2 domain to membranes, but loss of both charges impairs membrane association.
FIGURE 4.

Double charged-to-neutral residue mutations on the C2 domain affect its recruitment to and retention at lipid vesicles. The binding of purified C2 domains (wild-type, R216A/R249A, R216A/R252A, or R249A/R252A) to anionic phospholipid vesicles was measured by FRET association stopped-flow analysis, and the kon was determined as described in the legend to Fig. 2A. Data represent the weighted average ± S.E. (error bars) of 3–7 independent experiments. B, the binding of purified C2 domains (wild type, R216A/R249A, R216A/R252A, or R249A/R252A) to anionic phospholipid vesicles was measured by FRET dissociation stopped-flow analysis, and the koff was determined as described in the legend to Fig. 2B. Data represent the weighted average ± S.E. of 3–7 independent experiments.
TABLE 2.
Effect of C2 domain double charged-to-neutral residue mutations on equilibrium binding to lipid vesicles
Kdcalc values were obtained from the kinetic rate constants in Fig. 4. Data represent the weighted average ± S.E. of 3–7 independent experiments. The -fold difference values relative to the Kdcalc of wild-type C2 domain are indicated; error represents S.E.
| C2α mutations | kon (×1010) | koff | Kdcalc | Kd -fold difference from WT |
|---|---|---|---|---|
| m−1s−1 | s−1 | nm | -fold | |
| WT | 2.241 ± 0.009 | 0.71 ± 0.02 | 0.032 ± 0.001 | 1.00 ± 0.04 |
| R216A/R249A | 0.9 ± 0.1 | 42 ± 4 | 4.7 ± 0.7 | 148 ± 22 |
| R216A/R252A | 2.5 ± 0.1 | 30.3 ± 0.7 | 1.28 ± 0.05 | 41 ± 2 |
| R249A/R252A | 2.6 ± 0.2 | 32.0 ± 0.4 | 1.16 ± 0.09 | 37 ± 3 |
In contrast to modest effects on association rate constants, koff for the R216A/R249A, R216A/R252A, and R249A/R252A domains increased by 50-, 37-, and 38-fold relative to wild-type C2 domain (Fig. 4B and Table 2). These results confirm the critical role that these charged residues play in retaining the C2 domain at lipid vesicles. The overall increase in equilibrium dissociation constant for the double mutants R216A/R249A and R216A/R252A (148- and 41-fold higher than wild-type C2 domain) were as expected for the independent contribution of each residue. However, the R249A/R252A mutation resulted in only a 37-fold increase relative to the wild-type C2 domain rather than ∼120-fold, as would be expected had the two residues contributed independently (Table 2). Thus, Arg-249 and Arg-216 contribute independently to the binding of the C2 domain to membranes, whereas Arg-252 and Arg-249 have redundant function.
Effects of Ca2+ Concentration on C2 Domain Kinetics and Equilibrium Constants
Surprisingly, the charged-to-neutral mutations of R216A, R249A, and R252A showed only a modest change in their recruitment to the membrane. Yet, previous studies have shown that electrostatic interactions play a critical role in the membrane recruitment of C2 domains, including that of PKCα (4, 20, 31). To examine the effect of Ca2+ concentration on the membrane recruitment and retention of the C2 domain, we measured binding kinetics at subsaturating (low) and saturating (high) Ca2+ concentrations (20 and 200 μm, respectively) using the FRET association and FRET dissociation experiments. Increasing Ca2+ concentrations from 20 to 200 μm resulted in an ∼2-fold increase in kon (Fig. 5A and Table 3). However, no difference in association kinetics was observed at 150 mm NaCl compared with 200 mm NaCl. No binding was detected in the presence of 2 μm Ca2+ (data not shown). Dissociation rate constants were also sensitive to Ca2+ concentration; increasing the Ca2+ concentration decreased the koff for the wild-type C2 domain ∼5-fold (Fig. 5B and Table 3). Thus, the overall equilibrium dissociation constant increased 7-fold as the Ca2+ concentration was decreased (Fig. 5C and Table 3). Increasing ionic strength had the same relative effect on koff and Kdcalc (increasing both ∼3-fold) at both low and high Ca2+ concentrations. These results support a previous study showing that Ca2+ binding both increases the association rate constant and decreases the dissociation rate constant (4) and reveal that ionic strength effects at the concentrations tested are independent of Ca2+ concentration.
FIGURE 5.

Increasing Ca2+ affects the C2 domain recruitment to and retention at lipid vesicles. A, the binding of purified wild-type C2 domain and anionic phospholipid vesicles in the presence of 20 μm (low) or 200 μm (high) Ca2+ was measured by FRET association stopped-flow analysis, and kon was determined as described in the legend to Fig. 2A. Data represent the weighted average ± S.E. of 3–7 independent experiments. B, the binding of purified wild-type C2 domain and anionic phospholipid vesicles in the presence of 20 or 200 μm Ca2+ was measured by FRET dissociation stopped-flow analysis, and koff was measured as described in the legend to Fig. 2B. The weighted averages are calculated from three or four independent experiments. C, data for kon obtained in A and koff obtained in B were used to calculate Kdcalc.
TABLE 3.
Effect of Ca2+ on the membrane binding affinity of the C2 domain
Shown is a summary of kinetic constants for the binding of purified C2 domain to anionic lipids, in the presence of 150 mm NaCl, described in the legend to Fig. 5. Data represent weighted averages ± S.E.
| Ca2+ | kon (×1010) | koff | Kdcalc |
|---|---|---|---|
| μm | m−1s−1 | s−1 | nm |
| 20 | 1.012 ± 0.009 | 3.88 ± 0.06 | 0.23 ± 0.02 |
| 200 | 2.241 ± 0.009 | 0.71 ± 0.02 | 0.031 ± 0.007 |
Effects of C2 Domain Mutations on Ca2+ Binding
Because Ca2+ binding drives both the recruitment and retention of the C2 domain to anionic membranes (4), we next addressed how mutations in the C2 domain affected Ca2+ binding. To do this, we monitored Trp quenching of the C2 domain as a function of increasing Ca2+ concentration in the presence or absence of anionic lipid vesicles. In the absence of lipid, half-maximal binding of Ca2+ ([Ca2+]½) to the C2 domain was observed with 32 ± 4 μm Ca2+ (Table 4), as reported previously (21). Mutation of Arg-216 did not significantly affect Ca2+ binding. In contrast, mutation of Arg-249 or Arg-252 resulted in a large increase in the concentration of Ca2+ required for half-maximal binding (44- and 12-fold, respectively). This strong reduction in Ca2+ affinity was abrogated in the presence of saturating concentrations of anionic lipid vesicles (Table 4). Curiously, mutation of Trp-245 had only a modest (2-fold) effect on [Ca2+]½ in the absence of vesicles but caused a dramatic 22-fold reduction in affinity in the presence of saturating lipid concentrations. In contrast, mutation of Trp-247 increased [Ca2+]½ ∼8-fold both in the absence and presence of lipid vesicles. These data reveal that both Arg and Trp residues play key roles in binding Ca2+, with some residues having more dominant roles in the membrane-bound state (e.g. Trp-245), some being more dominant in the membrane-free state (e.g. Arg-249 and Arg-252), and others contributing to binding independently of membrane binding (e.g. Trp-247).
TABLE 4.
Effect of C2 domain basic- and hydrophobic-to-neutral residue mutations on Ca2+ binding affinity
The binding of Ca2+ to purified C2 domains (wild type, R216A, R249A, R252A, K268A, W245A, W247A, L191A, or W274A) was measured by monitoring Trp quenching as a function of increasing concentration of Ca2+, as described under “Experimental Procedures.” The [Ca2+]½ values indicate the weighted averages ± S.E. of at least three independent experiments.
| C2α mutation | [Ca2+]1/2 | [Ca2+]1/2 with lipid vesicles |
|---|---|---|
| μm | μm | |
| WT | 32 ± 4 | 25.6 ± 0.3 |
| R216A | 38 ± 2 | 46.0 ± 0.8 |
| R249A | 1409 ± 69 | 71 ± 3 |
| R252A | 395 ± 27 | 27.6 ± 0.3 |
| W245A | 74 ± 4 | 567 ± 17 |
| W247A | 263 ± 12 | 188 ± 4 |
Mutations in the C2 Domain of PKCα Alter Membrane Translocation in Cells
This study identified two mutations, which, by different mechanisms, reduce the membrane affinity of the C2 domain of PKCα by the same factor, 21-fold. Although it is well established that C2 domain mutations that impair the binding of the isolated domain to membranes also impair the binding of the full-length protein (38), we asked whether mutations that altered binding by different mechanisms (either reducing the membrane association rate constant or increasing the membrane dissociation rate constant) had similar or different effects on the translocation kinetics of full-length PKCα in cells. Specifically, we measured the increase in FRET upon the translocation of YFP-tagged wild-type or mutant PKCα to plasma membrane-targeted CFP in COS7 cells following stimulation with thapsigargin to elevate intracellular Ca2+. This rise in Ca2+ triggered the engagement of the C2 domain of PKCα to membranes; the subsequent addition of PDBu resulted in maximal translocation of PKC by engaging the C1 domain. Real-time imaging of FRET changes was used to assess the membrane interaction of PKC. Fig. 6A shows that mutation of either Trp-247 or Arg-249 to Ala markedly reduced the thapsigargin-induced translocation of PKCα to the plasma membrane. Furthermore, the maximal translocation induced by phorbol esters was also lower for these mutants compared with wild-type PKCα. The latter observation was intriguing and suggested that these mutants may have an intrinsically higher affinity for a different membrane, notably Golgi, the preferred binding surface for PKC isozymes that do not use the C2 domain for membrane interactions (11). Thus, we assessed how these mutations affected Golgi translocation by targeting CFP to the Golgi using the N-terminal 33 amino acids of endothelial nitric-oxide synthase and monitoring translocation of YFP-PKCα to this membrane (11). Fig. 6B shows that thapsigargin resulted in similar translocation of the mutants and wild-type PKCα to Golgi; however, subsequent phorbol ester treatment caused considerably greater translocation of the C2 domain mutants to the Golgi compared with the modest translocation for wild-type PKCα. These data not only indicate that the mutations that reduce either the kon or increase the koff of the C2 domains for PKC reduce membrane translocation but, importantly, reveal that impairment of the C2 domain removes the plasma membrane-binding properties of PKC and drives it to the Golgi instead (Fig. 7). These data are consistent with several elegant studies showing that the affinity of the C2 domain for PIP2 at the plasma membrane recruits conventional PKC isozymes to the plasma membrane, sequestering it away from the diacylglycerol-rich Golgi (38, 44). Here, we show that mutations that impair the C2 domain not only reduce membrane binding but also result in relocalization of the conventional PKCα to Golgi.
FIGURE 6.

Effects of basic- and hydrophobic-to-neutral residue mutations on the translocation of PKCα in cells. PKC translocation in COS7 cells expressing YFP-tagged PKCα-WT (red trace), PKCα-W247A (green trace), or PKCα-R249A (blue trace) was monitored by measuring FRET changes induced by translocation of PKC to CFP targeted to plasma membrane (A) and Golgi (B). Thapsigargin was added at the indicated time to elevate intracellular Ca2+, and PDBu was added to maximally translocate PKC. The traces were analyzed as described under “Experimental Procedures”; data represent the mean ± S.E. (error bars) of at least three independent experiments.
FIGURE 7.

Model showing spatial regulation of PKC by the C2 domain. Upon agonist stimulation, the binding of Ca2+ (gray circle) to the PIP2-sensing C2 domain (yellow) results in the association of conventional PKC with the PIP2-enriched plasma membrane; this C2-dependent membrane association is driven primarily by hydrophobic interactions, and retention is driven primarily by electrostatic interactions. Once at the membrane, the C1 domain (orange) finds its membrane-embedded ligand, diacylglycerol (DAG), resulting in a high affinity membrane interaction that releases the autoinhibitory pseudosubstrate segment (green rectangle) from the kinase domain (cyan circle) and thus activation (top right species of PKC). Impairing the membrane-binding properties of the C2 domain results in loss of plasma membrane recognition and redirection of conventional PKC, via the now dominant C1 domain, to diacylglycerol/phorbol esters in the much more abundant membrane surface of Golgi (bottom right species of PKC); this binding pattern mimics that of novel PKC isozymes, which do not have a Ca2+-binding C2 domain. Note that the C1b domain of novel PKC isozymes binds diacylglycerol with 2-order of magnitude higher affinity than the C1b domain of conventional PKC isozymes, so pretargeting to membranes is not necessary for novel isozymes to respond to agonist-evoked increases in diacylglycerol. Although the impaired C2 domain mutants of PKC are redirected to Golgi, their affinity for diacylglycerol (but not phorbol esters) may be too low for significant activation following agonist stimulation.
DISCUSSION
Ca2+-dependent binding of the C2 domain to membranes provides the first step in the agonist-evoked activation of conventional PKC isozymes. Here we used stopped-flow kinetics to dissect the role of hydrophobic versus electrostatic interactions in the recruitment and retention of the C2 domain of PKCα to anionic membranes. Analysis of on and off rate constants for the equilibrium binding of the C2 domain as a function of mutation of specific residues reveals that basic and hydrophobic residues have distinct roles in the membrane interaction of this domain; hydrophobic residues drive the initial recruitment to the membrane, whereas electrostatic interactions involving basic residues play a key role in retaining the C2 domain at the membrane. The tuning of membrane kinetics by loss of basic or hydrophobic residues also tunes the translocation of PKC in cells; imaging of PKCα translocation in real time reveals that perturbation of either of these mechanisms reduces the magnitude of Ca2+-triggered as well as PDBu-stimulated translocation in addition to driving PKCα to the Golgi instead of the plasma membrane.
Membrane Association Is Controlled by Hydrophobic Residues in the Ca2+ Binding Loops
Mutation of two highly conserved Trp residues in the Ca2+-binding loops of the C2 domain revealed that they play a key role in driving the membrane association of the C2 domain. In contrast, mutation of highly conserved Arg residues in the Ca2+-binding loops has no significant effect on membrane association. Specifically, mutation of either Trp-245 or Trp-247 to Ala reduced kon 4- and 2-fold relative to the wild-type domain (Fig. 2A and Table 1). Mutation of another conserved hydrophobic residue, Leu-191, located on Ca2+-binding loop 2 had no significant effect on either the on rate constant or off rate constant, suggesting that this residue does not have a significant role in C2 domain membrane binding (Fig. 2A and Table 1). Trp-245 and Trp-247 are located on Ca2+-binding loop 3 and have been previously proposed to penetrate into the membrane (22, 24, 25). Our data suggest that such membrane penetration plays a key role in allowing the Ca2+-bound C2 domain to interact with membranes.
We note that the magnitude of the membrane association constants measured is very high, perhaps exceeding the rate constant for protein diffusion in solution. This enhanced association rate constant probably results from the reduction in dimensionality of the C2 domain binding anionic lipids in a two-dimensional plane, as discussed (45).
If hydrophobic interactions dominate in the association of the C2 domain with anionic membranes, then increasing ionic strength would be expected to strengthen such interactions. Consistent with this, raising ionic strength from 150 to 175 mm NaCl resulted in an ∼20% increase in kon of the wild-type domain for binding to anionic membranes (Fig. 3A). In fact, this modest increase in ionic strength resulted in an increased kon for all Ca2+-binding loop mutants except the L191A (Fig. 3A); the reduced kon of the L191A mutant suggests that this Leu contributes to the hydrophobic interactions that are strengthened at 175 mm NaCl. Note that at even higher ionic strength (200 mm NaCl), kon did not further increase (Fig. 3A). Thus, disruption of electrostatic interactions began to compensate for the increased hydrophobic interactions as ionic strength was increased further. However, the kon for W245A was increased even further at this higher ionic strength. One possibility is that loss of the Trp at position 245 may unmask other hydrophobic residues (e.g. the key Trp at position 247) so that membrane penetration is significantly strengthened at this higher ionic strength, dominating over electrostatic interactions. In summary, these data support an important role of hydrophobic interactions in the association of the C2 domain with membranes.
Membrane Retention Is Primarily Controlled by Basic Residues in the Ca2+ Binding Loops
In contrast to the conserved Trp in the Ca2+-binding loops, replacement of the conserved Arg with Ala revealed no significant role in driving the membrane association of the C2 domain (Fig. 2A and Table 1). Rather, these residues play a key role in retaining the domain on membranes. Loss of Arg at position 249 resulted in a dramatic reduction in residency time, as assessed by the 16-fold increase in koff; loss of Arg at position 216 or 252 resulted in a 6-fold increase in koff (Fig. 2B and Table 1). All three residues have previously been shown to decrease membrane binding affinity, translocation to the membrane, and enzymatic activity (26, 46). Our current data reveal that this reduced affinity is caused primarily by reduced residency time of the domain on anionic membranes.
Membrane retention was also sensitive to loss of Trp-245 or Trp-247 (Fig. 2B and Table 1). As discussed above, the membrane penetration of these residues has previously been proposed to control the C2 domain membrane association. However, of all of the residues examined, Arg-249 has the most profound effect on the membrane residency time of the C2 domain.
The finding that mutation of Arg-249 to an Ala has the greatest impact on the membrane residency time of the C2 domain suggests a particularly important role of this residue in stabilizing the domain-phospholipid complex. Arg-249 has previously been proposed to interact with phospholipid acyl chains via the guanidinium group, which has been suggested to interact with the sn-2 ester carbonyl of phospholipid acyl chains (22, 47). Mutation of either Arg-216 or Arg-252 to an Ala also significantly decreased the retention of the C2 domain on the membrane. Similar to Arg-249, the guanidinium group of Arg-216 may also interact with the sn-2 ester carbonyl of the fatty acyl chain (22). Electron paramagnetic resonance studies indicate that Arg-249 penetrates deeper into the bilayer than Arg-252 (22, 43); thus, Arg-249 may interact at or near the interface with the hydrocarbon core, whereas Arg-252 is probably located closer to the bilayer surface. Taken together, these data suggest that Arg-216, Arg-252, and, most importantly, Arg-249 play an important role in stabilizing the C2 domain-lipid complex.
Concurrent mutation of both Arg-216 and Arg-249 caused a 2-order of magnitude increase in the equilibrium dissociation constant of the domain, resulting primarily from a large (50-fold) decrease in residency time at the membrane (Fig. 4B and Table 2). This double mutation also resulted in reduced membrane association, with a 2-fold decrease in kon (Fig. 4A and Table 2). These data underscore the key role of the guanidinium groups in membrane interactions. The overall binding affinity of the double mutants R216A/R252A and R249A/R252A was significantly decreased relative to the wild-type domain; however, the change was less than expected, suggesting that Arg-252 may be able to somehow compensate for the loss of the guanidinium group of Arg-216 and Arg-249.
We have previously reported that Ca2+ binding controls both the association and dissociation rate constants of the C2 domain of PKCβII for anionic membranes (4), accounting for the dramatic effect of this second messenger in increasing the equilibrium binding of the C2 domain to membranes (4, 24, 25, 31). Consistent with our previous study, we show that Ca2+ binding also increases both the recruitment and retention of the C2 domain of PKCα at the membrane. Also supporting previous studies (24), analysis of the apparent affinity of the various C2 domain mutants for Ca2+ reveals that Arg-249 and, to a lesser extent, Arg-252 play dominant roles in allowing Ca2+ binding to the free C2 domain, interactions that become dispensable in the presence of lipid. In contrast, Trp-245 does not influence binding of Ca2+ to the free domain but dramatically stabilizes the binding of Ca2+ to the lipid-bound domain. These data support the requirement for Ca2+ in tethering the C2 domain to membranes and delineate residues that promote binding of Ca2+ to the soluble form versus those that stabilize binding to the membrane-bound form.
Mutations in C2 Domain Modulate PKCα Translocation in Cells
Using a FRET-based translocation assay, we show that introduction into full-length PKCα of mutations that impair the association or dissociation of the C2 domain with membranes significantly reduce the Ca2+-dependent translocation of PKCα to the plasma membrane. Specifically, mutation of either Arg-249 or Trp-247 to Ala results in a comparable decrease in thapsigargin-induced redistribution of PKC from the cytosol to the plasma membrane compared with wild-type enzyme. In the isolated C2 domain, replacement of either of these residues with Ala decreases the membrane affinity of the C2 domain by the same factor (21-fold), but by different mechanisms. In the context of the full-length protein, mutation of either residue results in a comparable reduction in plasma membrane translocation. These data reveal that altering the affinity of the C2 domain for membranes, either by modulating kon or koff, significantly impairs Ca2+-dependent translocation of PKC to plasma membrane.
Surprisingly, the phorbol ester-dependent translocation to the plasma membrane was significantly reduced for PKCα mutants with impaired C2 domains, compared with the wild-type enzyme. One possibility is that by impairing the plasma membrane-sensing properties of PKC (conferred by PIP2 recognition by the C2 domain), the PKCα mutants are relocalized to other membranes (e.g. Golgi) via now dominant C1 domain interactions. Consistent with this, phorbol ester-dependent translocation to Golgi was greatly enhanced in mutants of PKCα in which the C2 domain was impaired. Thus, the C2 domain not only regulates the degree of membrane translocation of PKC but also the membrane location. Our data support a model in which impairing the Ca2+-binding ability of the C2 domain (the plasma membrane-directing unit) of conventional PKCs allows the C1 domain (the diacylglycerol-sensing module) to become the dominant membrane-binding module and redirects PKC to the most diacylglycerol-enriched membrane, which is the Golgi (Fig. 7). These data validate the biophysical work of the Falke and Gómez-Fernández groups proposing that the C2 domain drives conventional PKCs to the PIP2-enriched plasma membrane (38, 44).
CONCLUSION
The Ca2+-controlled targeting of the C2 domain to the plasma membrane plays a key role in allowing conventional PKC isozymes to find their membrane-embedded allosteric activator, diacylglycerol. Mutation of residues in the membrane-interfacing Ca2+-binding loops to impair either association or dissociation kinetics alters the magnitude and localization of Ca2+-induced translocation of PKC in cells. The recent identification of mutations in the C2 domain of conventional PKC isozymes in human cancers suggests that resulting alterations in PKC signaling could contribute to cancer (48, 49). Of note, an Arg to His mutation in PKCγ at the residue corresponding to Arg-252 in PKCα has been identified in pancreatic cancer (37). Whether this mutation conferred a survival advantage to these tumor cells remains to be established.
Acknowledgments
We are indebted to Dr. Maya Kunkel for critical help with manuscript preparation, Dr. Eric Nalefski for advice, and Dr. Patricia Jennings for use of the stopped-flow fluorescence spectrometer.
This work was supported, in whole or in part, by National Institutes of Health Grant GM43154 (to A. C. N.).
- PS
- 1-palmitoyl-2-oleoylphosphatidyl-l-serine
- PC
- 1-palmitoyl-2-oleoylphosphatidylcholine
- dPE
- N-(5-dimethylaminonapthalene-1-sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine
- PDBu
- phorbol 12,13-dibutyrate.
REFERENCES
- 1. Newton A. C. (2010) Protein kinase C. Poised to signal. Am. J. Physiol. Endocrinol. Metab. 298, E395–E402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Griner E. M., Kazanietz M. G. (2007) Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 7, 281–294 [DOI] [PubMed] [Google Scholar]
- 3. Orr J. W., Keranen L. M., Newton A. C. (1992) Reversible exposure of the pseudosubstrate domain of protein kinase C by phosphatidylserine and diacylglycerol. J. Biol. Chem. 267, 15263–15266 [PubMed] [Google Scholar]
- 4. Nalefski E. A., Newton A. C. (2001) Membrane binding kinetics of protein kinase C βII mediated by the C2 domain. Biochemistry 40, 13216–13229 [DOI] [PubMed] [Google Scholar]
- 5. Dries D. R., Gallegos L. L., Newton A. C. (2007) A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J. Biol. Chem. 282, 826–830 [DOI] [PubMed] [Google Scholar]
- 6. Hurley J. H. (2006) Membrane binding domains. Biochim. Biophys. Acta 1761, 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Newton A. C. (2009) Lipid activation of protein kinases. J. Lipid Res. 50, S266–S271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pawson T., Nash P. (2003) Assembly of cell regulatory systems through protein interaction domains. Science 300, 445–452 [DOI] [PubMed] [Google Scholar]
- 9. Newton A. C., Johnson J. E. (1998) Protein kinase C. A paradigm for regulation of protein function by two membrane-targeting modules. Biochim. Biophys. Acta 1376, 155–172 [DOI] [PubMed] [Google Scholar]
- 10. Carpten J. D., Faber A. L., Horn C., Donoho G. P., Briggs S. L., Robbins C. M., Hostetter G., Boguslawski S., Moses T. Y., Savage S., Uhlik M., Lin A., Du J., Qian Y. W., Zeckner D. J., Tucker-Kellogg G., Touchman J., Patel K., Mousses S., Bittner M., Schevitz R., Lai M. H., Blanchard K. L., Thomas J. E. (2007) A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444 [DOI] [PubMed] [Google Scholar]
- 11. Gallegos L. L., Newton A. C. (2008) Spatiotemporal dynamics of lipid signaling. Protein kinase C as a paradigm. IUBMB Life 60, 782–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gallegos L. L., Kunkel M. T., Newton A. C. (2006) Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J. Biol. Chem. 281, 30947–30956 [DOI] [PubMed] [Google Scholar]
- 13. Violin J. D., Zhang J., Tsien R. Y., Newton A. C. (2003) A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J. Cell Biol. 161, 899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mosior M., Newton A. C. (1996) Calcium-independent binding to interfacial phorbol esters causes protein kinase C to associate with membranes in the absence of acidic lipids. Biochemistry 35, 1612–1623 [DOI] [PubMed] [Google Scholar]
- 15. Nalefski E. A., Falke J. J. (1996) The C2 domain calcium-binding motif. Structural and functional diversity. Protein Sci. 5, 2375–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malmberg N. J., Falke J. J. (2005) Use of EPR power saturation to analyze the membrane-docking geometries of peripheral proteins. Applications to C2 domains. Annu. Rev. Biophys. Biomol. Struct. 34, 71–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cho W., Stahelin R. V. (2006) Membrane binding and subcellular targeting of C2 domains. Biochim. Biophys. Acta 1761, 838–849 [DOI] [PubMed] [Google Scholar]
- 18. Sutton R. B., Davletov B. A., Berghuis A. M., Südhof T. C., Sprang S. R. (1995) Structure of the first C2 domain of synaptotagmin I. A novel Ca2+/phospholipid-binding fold. Cell 80, 929–938 [DOI] [PubMed] [Google Scholar]
- 19. Newton A. C. (1995) Protein kinase C. Seeing two domains. Curr. Biol. 5, 973–976 [DOI] [PubMed] [Google Scholar]
- 20. Nalefski E. A., Slazas M. M., Falke J. J. (1997) Ca2+-signaling cycle of a membrane-docking C2 domain. Biochemistry 36, 12011–12018 [DOI] [PubMed] [Google Scholar]
- 21. Kohout S. C., Corbalán-García S., Torrecillas A., Goméz-Fernandéz J. C., Falke J. J. (2002) C2 domains of protein kinase C isoforms α, β, and γ. Activation parameters and calcium stoichiometries of the membrane-bound state. Biochemistry 41, 11411–11424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verdaguer N., Corbalan-Garcia S., Ochoa W. F., Fita I., Gómez-Fernández J. C. (1999) Ca2+ bridges the C2 membrane-binding domain of protein kinase Calpha directly to phosphatidylserine. EMBO J. 18, 6329–6338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edwards A. S., Newton A. C. (1997) Regulation of protein kinase C βII by its C2 domain. Biochemistry 36, 15615–15623 [DOI] [PubMed] [Google Scholar]
- 24. Medkova M., Cho W. (1998) Mutagenesis of the C2 domain of protein kinase C-α. Differential roles of Ca2+ ligands and membrane binding residues. J. Biol. Chem. 273, 17544–17552 [DOI] [PubMed] [Google Scholar]
- 25. Stahelin R. V., Cho W. (2001) Roles of calcium ions in the membrane binding of C2 domains. Biochem. J. 359, 679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Conesa-Zamora P., Lopez-Andreo M. J., Gómez-Fernández J. C., Corbalán-García S. (2001) Identification of the phosphatidylserine binding site in the C2 domain that is important for PKC α activation and in vivo cell localization. Biochemistry 40, 13898–13905 [DOI] [PubMed] [Google Scholar]
- 27. Corbalán-García S., Rodríguez-Alfaro J. A., Gómez-Fernández J. C. (1999) Determination of the calcium-binding sites of the C2 domain of protein kinase Cα that are critical for its translocation to the plasma membrane. Biochem. J. 337, 513–521 [PMC free article] [PubMed] [Google Scholar]
- 28. García-García J., Corbalán-García S., Gómez-Fernández J. C. (1999) Effect of calcium and phosphatidic acid binding on the C2 domain of PKC α as studied by Fourier transform infrared spectroscopy. Biochemistry 38, 9667–9675 [DOI] [PubMed] [Google Scholar]
- 29. Johnson J. E., Giorgione J., Newton A. C. (2000) The C1 and C2 domains of protein kinase C are independent membrane targeting modules, with specificity for phosphatidylserine conferred by the C1 domain. Biochemistry 39, 11360–11369 [DOI] [PubMed] [Google Scholar]
- 30. Shao X., Li C., Fernandez I., Zhang X., Südhof T. C., Rizo J. (1997) Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch. Neuron 18, 133–142 [DOI] [PubMed] [Google Scholar]
- 31. Murray D., Honig B. (2002) Electrostatic control of the membrane targeting of C2 domains. Mol. Cell 9, 145–154 [DOI] [PubMed] [Google Scholar]
- 32. Rodríguez-Alfaro J. A., Gomez-Fernandez J. C., Corbalan-Garcia S. (2004) Role of the lysine-rich cluster of the C2 domain in the phosphatidylserine-dependent activation of PKCα. J. Mol. Biol. 335, 1117–1129 [DOI] [PubMed] [Google Scholar]
- 33. Manna D., Bhardwaj N., Vora M. S., Stahelin R. V., Lu H., Cho W. (2008) Differential roles of phosphatidylserine, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 in plasma membrane targeting of C2 domains. Molecular dynamics simulation, membrane binding, and cell translocation studies of the PKCα C2 domain. J. Biol. Chem. 283, 26047–26058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lai C. L., Landgraf K. E., Voth G. A., Falke J. J. (2010) Membrane docking geometry and target lipid stoichiometry of membrane-bound PKCα C2 domain. A combined molecular dynamics and experimental study. J. Mol. Biol. 402, 301–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Landgraf K. E., Malmberg N. J., Falke J. J. (2008) Effect of PIP2 binding on the membrane docking geometry of PKC α C2 domain. An EPR site-directed spin-labeling and relaxation study. Biochemistry 47, 8301–8316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ausili A., Corbalán-García S., Gómez-Fernández J. C., Marsh D. (2011) Membrane docking of the C2 domain from protein kinase Cα as seen by polarized ATR-IR. The role of PIP2. Biochim. Biophys. Acta 1808, 684–695 [DOI] [PubMed] [Google Scholar]
- 37. Jones S., Zhang X., Parsons D. W., Lin J. C., Leary R. J., Angenendt P., Mankoo P., Carter H., Kamiyama H., Jimeno A., Hong S. M., Fu B., Lin M. T., Calhoun E. S., Kamiyama M., Walter K., Nikolskaya T., Nikolsky Y., Hartigan J., Smith D. R., Hidalgo M., Leach S. D., Klein A. P., Jaffee E. M., Goggins M., Maitra A., Iacobuzio-Donahue C., Eshleman J. R., Kern S. E., Hruban R. H., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V. E., Kinzler K. W. (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Evans J. H., Murray D., Leslie C. C., Falke J. J. (2006) Specific translocation of protein kinase Cα to the plasma membrane requires both Ca2+ and PIP2 recognition by its C2 domain. Mol. Biol. Cell 17, 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guerrero-Valero M., Ferrer-Orta C., Querol-Audí J., Marin-Vicente C., Fita I., Gómez-Fernandez J. C., Verdaguer N., Corbalán-García S. (2009) Structural and mechanistic insights into the association of PKCα-C2 domain to PtdIns(4,5)P2. Proc. Natl. Acad. Sci. U.S.A. 106, 6603–6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Corbalán-García S., Guerrero-Valero M., Marín-Vicente C., Gómez-Fernández J. C. (2007) The C2 domains of classical/conventional PKCs are specific PtdIns(4,5)P(2)-sensing domains. Biochem. Soc Trans. 35, 1046–1048 [DOI] [PubMed] [Google Scholar]
- 41. Marín-Vicente C., Gómez-Fernández J. C., Corbalán-García S. (2005) The ATP-dependent membrane localization of protein kinase Cα is regulated by Ca2+ influx and phosphatidylinositol 4,5-bisphosphate in differentiated PC12 cells. Mol. Biol. Cell 16, 2848–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bartlett G. R. (1959) Phosphorus assay in column chromatography. J. Biol. Chem. 234, 466–468 [PubMed] [Google Scholar]
- 43. Kohout S. C., Corbalán-García S., Gómez-Fernández J. C., Falke J. J. (2003) C2 domain of protein kinase C α. Elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry 42, 1254–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Corbalán-García S., García-García J., Rodríguez-Alfaro J. A., Gómez-Fernández J. C. (2003) A new phosphatidylinositol 4,5-bisphosphate-binding site located in the C2 domain of protein kinase Cα. J. Biol. Chem. 278, 4972–4980 [DOI] [PubMed] [Google Scholar]
- 45. Astumian R. D., Chock P. B. (1985) Interfacial reaction dynamics. J. Phys. Chem. 89, 3477–3482 [Google Scholar]
- 46. Bolsover S. R., Gomez-Fernandez J. C., Corbalan-Garcia S. (2003) Role of the Ca2+/phosphatidylserine binding region of the C2 domain in the translocation of protein kinase Cα to the plasma membrane. J. Biol. Chem. 278, 10282–10290 [DOI] [PubMed] [Google Scholar]
- 47. Ochoa W. F., Corbalán-Garcia S., Eritja R., Rodríguez-Alfaro J. A., Gómez-Fernández J. C., Fita I., Verdaguer N. (2002) Additional binding sites for anionic phospholipids and calcium ions in the crystal structures of complexes of the C2 domain of protein kinase Cα. J. Mol. Biol. 320, 277–291 [DOI] [PubMed] [Google Scholar]
- 48. Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Forbes S. A., Bindal N., Bamford S., Cole C., Kok C. Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A., Teague J. W., Campbell P. J., Stratton M. R., Futreal P. A. (2011) COSMIC. Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]