

Background: STIM1 is essential for store-operated Ca2+ entry (SOCE) in endothelial cells.
Results: SOCE-activated AMPKα1-p38β signaling phosphorylates STIM1, which in turn inhibits SOCE in endothelial cells.
Conclusion: SOCE-activated signaling pathway completes a negative feedback loop to regulate SOCE in endothelial cells.
Significance: Selective p38β agonists may represent potential therapeutic agents to reverse the vascular leak syndrome.
Keywords: AMP-activated Kinase (AMPK), Calcium Channels, Endothelial Dysfunction, p38 MAPK, Thrombin, Phosphorylation, Protease-activated Receptor-1, Store-operated Calcium Entry, Stromal Interaction Molecule-1, Vascular Permeability
Abstract
The Ca2+ sensor STIM1 is crucial for activation of store-operated Ca2+ entry (SOCE) through transient receptor potential canonical and Orai channels. STIM1 phosphorylation serves as an “off switch” for SOCE. However, the signaling pathway for STIM1 phosphorylation is unknown. Here, we show that SOCE activates AMP-activated protein kinase (AMPK); its effector p38β mitogen-activated protein kinase (p38β MAPK) phosphorylates STIM1, thus inhibiting SOCE in human lung microvascular endothelial cells. Activation of AMPK using 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) resulted in STIM1 phosphorylation on serine residues and prevented protease-activated receptor-1 (PAR-1)-induced Ca2+ entry. Furthermore, AICAR pretreatment blocked PAR-1-induced increase in the permeability of mouse lung microvessels. Activation of SOCE with thrombin caused phosphorylation of isoform α1 but not α2 of the AMPK catalytic subunit. Moreover, knockdown of AMPKα1 augmented SOCE induced by thrombin. Interestingly, SB203580, a selective inhibitor of p38 MAPK, blocked STIM1 phosphorylation and led to sustained STIM1-puncta formation and Ca2+ entry. Of the three p38 MAPK isoforms expressed in endothelial cells, p38β knockdown prevented PAR-1-mediated STIM1 phosphorylation and potentiated SOCE. In addition, inhibition of the SOCE downstream target CaM kinase kinase β (CaMKKβ) or knockdown of AMPKα1 suppressed PAR-1-mediated phosphorylation of p38β and hence STIM1. Thus, our findings demonstrate that SOCE activates CaMKKβ-AMPKα1-p38β MAPK signaling to phosphorylate STIM1, thereby suppressing endothelial SOCE and permeability responses.
Introduction
Previous studies from our laboratory have demonstrated that an increase in intracellular Ca2+ signaling is critical for protease-activated receptor-1 (PAR-1)2-mediated endothelial hyper-permeability (1). Thrombin-induced increase in intracellular Ca2+ concentration in endothelial cells is dependent on both inositol 1,4,5-triphosphate-induced release of stored Ca2+ and Ca2+ store depletion-mediated Ca2+ entry, termed store-operated Ca2+ entry (SOCE) (1). The channel responsible for mediating Ca2+ entry secondary to ER-stored Ca2+ depletion is termed store-operated Ca2+ entry channels (SOCs) (1, 2). In recent studies, we have shown that transient receptor potential canonical (TRPC) 1 and 4 channels function as SOCs in endothelial cells (3). Other studies have shown that a Ca2+-selective channel (ICRAC), Orai1 channel also contributes to SOCE in endothelial cells (4, 5).
Recent studies have elucidated the mechanism of the ER-localized Ca2+ sensor protein, stromal interacting molecule-1 (STIM1), in activating SOCE through TRPC and Orai1 channels (6–10). ER-store Ca2+ depletion induces clustering of STIM1 at “puncta” on the ER/plasma membrane interface, which in turn binds to and activates SOCs (TRPC and Orai1 channels) (6–10). Many of the molecular details of STIM1-mediated Ca2+ entry (i.e. SOCE) are well understood (6). STIM1 is a multidomain protein containing an EF hand domain at the N terminus projecting into the ER lumen and at the C-terminal ezrin-radixin-moesin (ERM), serine/proline, and lysine-rich cytosolic domains. The ERM domain contains a coiled-coil domain and a highly conserved SOAR (STIM1 Orai activating region) domain (6). The SOAR domain binds to both TRPC and Orai1. STIM1 SOAR domain binding to Orai1 is sufficient to gate Orai1 (6, 7). In the case of TRPC channels, electrostatic interaction between the STIM1 C-terminal Lys domain and TRPC C-terminal acidic residues is required to activate Ca2+ entry through TRPC channels (6, 11). STIM1 is critical for thrombin-induced SOCE by its interaction with TRPC1 and TRPC4 in endothelial cells (3). Studies from another laboratory have shown that STIM1-Orai1 association also mediates SOCE in endothelial cells (4, 5). Regulation of SOCE activity is not as well understood in general and has not been investigated in endothelial cells.
STIM1 was originally identified as a phosphoprotein with multiple serine (Ser) phosphorylation sites (12). Recently, Smyth et al. (13) showed that STIM1-mediated Ca2+ entry was “turned off” by phosphorylation of Ser-486 and Ser-668 residues at the C terminus during mitosis in HeLa cells. Furthermore, they have shown that STIM1 phosphorylation prevented store depletion-induced STIM1 punta at ER-plasma membrane junctions, an event essential for SOCE activation. Another study showed that ERK1/2-mediated phosphorylation of STIM1 at Ser-519 and Ser-575 modulated SOCE in HEK293 cells (14). Thus, we investigated the underlying signaling pathway downstream of PAR-1 in inducing STIM1 phosphorylation at its Ser residues to “turn off” SOCE in endothelial cells.
Sequence analysis for human STIM1, using Group-based prediction system, version 2.1.1 software, revealed the presence of 10 consensus phosphorylation sites (Ser-486, Ser-492, Ser-575, Ser-600, Ser-608, Ser-618, Ser-621, Thr-626, Ser-628, and Ser-668) for p38 MAPK indicating the possibility that p38 MAPK-mediated STIM1 phosphorylation may modulate SOCE in endothelial cells. In recent studies, we have shown that SOCE induced by thrombin resulted in activation of AMPK and its downstream target p38 MAPK in endothelial cells (15). Thus, we addressed the possibility that SOCE-activated AMPK-p38 MAPK signaling axis is involved in inhibiting SOCE in endothelial cells. Our results show that SOCE signal activates AMPKα1 and its downstream target p38β MAPK, which in turn phosphorylates STIM1 to turn off SOCE in endothelial cells.
EXPERIMENTAL PROCEDURES
Materials
Endothelial growth medium (EGM-2) was obtained from Lonza Walkersville, Inc. (Walkersville, MD). Hanks' balanced salt solution (HBSS) and trypsin were from Invitrogen. Fetal bovine serum (FBS) was from Hyclone (Logan, UT). Human α-thrombin was obtained from Enzyme Research Laboratories (South Bend, IN). Protease-activated receptor-1 (PAR-1)-activating peptide (TFFLRNPNDK-NH2) was synthesized as a C-terminal amide (16). Fura-2AM was purchased from Invitrogen. 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) was obtained from Toronto Research Chemical Inc. (Ontario, Canada). SB203580, SB202474, and Evans Blue dye were from Sigma. Antibodies for phospho-AMPK (pAb), AMPK (mAb), AMPKα1 (pAb), and AMPKα2 (pAb) were purchased from Upstate Cell Signaling (Lake Placid, NY). Polyclonal antibodies that specifically react with p38α, -β, and -γ were from Cell Signaling Technologies (Beverly, MA). Anti-STIM1 mAb and anti-phosphoserine pAb were from BD Transduction Laboratories. Anti-STIM1 pAb was from Proteintech Group (Chicago). Anti-Myc pAb, anti-Myc mAb, and anti-phospho-p38 MAPK pAb were purchased from Santa Cruz Biotechnology. Human (h)-specific siRNA to target AMPKα1, AMPKα2, p38α, p38β, p38γ, and scrambled siRNA (sc-siRNA) were from Dharmacon (Lafayette, CO). Myc-tagged-WT-STIM1 (Myc-WT-STIM1) and yellow fluorescent protein (YFP)-WT-STIM1 constructs were prepared as described previously (9, 11). N-terminal GST fusion protein corresponding to full-length human p38β2 activated with MKK6 (active p38β2; catalog no. 14-253) was from EMD Millipore Corp. (Billerica, MA). Kinase assay reagents were purchased from Cell Signaling Technology Inc., (Danvers, MA).
Cell Culture
Human lung microvascular endothelial cells (HLMVECs) were from Lonza (Walkersville). Mouse (C57BL6) lung endothelial cells (mLECs) were isolated and cultured as described previously (16, 17). HLMVECs were grown in EGM-2MV supplemented with 15% FBS, and mLECs were grown in EGM-2 supplemented with 5% FBS (growth media from Lonza). Both cell types were used between passages 4 and 6.
Reverse Transcription-PCR
Total RNA from ECs were isolated using TRIzol reagent. RT was performed using oligo(dT) primers and superscript RT (Invitrogen) following the manufacturer's instructions. Human p38 isoforms and GAPDH were amplified using the following primer sets: p38α (sense, 5′-GATCAGTTGAAGCTCATTTTAA-3′, and antisense, 5′-CACTTGAATAATATTTGGAGAGT-3′); p38β (sense, 5′-AGCCATATCTGGCAAGAAGCTGGA-3′, and antisense, 5′-AAGTGTCCGAGTCCAAGTCCACAT-3′); p38γ (sense, 5′-TTGAATTGGATGCGCTACACGCAG-3′, and antisense, 5′-AGGGCTTGCATTGGTCAGGATAGA-3′); p38δ (sense, 5′-TGTGCAGAAGCTGAACGACAAAGC-3′, and antisense, 5′-TGCCATGCAAGATGAGTCCCTACA-3′), and GAPDH (sense, 5′-TATCGTGGAAGGACTCATGACC-3′, and antisense, 5′-TACATGGCAACTGTGAGGGG-3′). RT product (2 μl) was amplified in a 20-μl volume containing 100 pmol of primers and 2.5 units of TaqDNA polymerase. Reaction conditions were as follows: 95 °C for 2 min, 95 °C for 1 min, 62 °C for 30 s, 72 °C for 1 min for 30 cycles, and then 72 °C for 7 min. The amplified products were electrophoresed on 1.5% agarose gels in TAE buffer and visualized by ethidium bromide staining.
Cytosolic Ca2+ ([Ca2+]i) Measurement
The cytoplasmic Ca2+ concentration ([Ca2+]i) in ECs was measured using the Ca2+-sensitive fluorescent dye Fura-2/AM (3). Cells were grown to confluence on gelatin-coated glass coverslips and then washed two times with serum-free medium and incubated for 2 h at 37 °C in culture medium containing 1% FBS. Cells were washed once and loaded with 3 μm Fura-2/AM for 30 min. After loading, cells were washed with HBSS, and the coverslips were transferred on a perfusion chamber at 37 °C and imaged using a semi-motorized microscope (Axio Observer D1; Carl Zeiss GmbH, Jena, Germany) equipped with an AxioCam HSm camera (Carl Zeiss) and a Fluar ×40 oil immersion objective. Light was provided by the DG-4 wavelength switcher (Princeton Scientific Instruments, Monmouth Junction, NJ). A dual excitation at 340 and 380 nm was used, and emission was collected at 520 nm. The AxioVision physiology software module was used to acquire the images at 1-s intervals, and the data were analyzed off-line. In each experiment, 20–30 cells were selected to measure change in [Ca2+]i.
Transendothelial Electrical Resistance
The real time change in endothelial monolayer resistance (TER) was measured to assess endothelial barrier function. Before the experiment, confluent endothelial monolayer was kept in 1% FBS containing medium for 2 h and then a thrombin-induced real time change in TER was measured. Data are presented in resistance normalized to its value at time 0 (18).
Assessment of Lung Microvessel Permeability in Mice
C57BL6J mice obtained from Charles Rivers Laboratories (Wilmington, MA) were housed in the University of Illinois Animal Care Facility and used according to approved animal protocols. Mice (22–25 g) were anesthetized with (2.5% sevoflurane in room air) for insertion of an indwelling jugular catheter and then were allowed to recover for 30 min. Mice were then injected with AICAR (500 mg/kg, intraperitoneal) or saline. At 195 min after AICAR or saline administration, mice received 100 μl of Evans blue dye conjugated with albumin (EBA) (20 mg/kg) through the jugular vein. At the end of 4 h, the mice were sacrificed and lungs harvested. Thirty min before sacrificing, mice received either saline (100 μl) or PAR-1 peptide (100 μl (1 mg/kg)) through the jugular vein. The EBA presence in lung tissue was measured as described previously (19).
siRNA Transfection
ECs grown to ∼70% confluence on gelatin-coated culture dishes were transfected with target siRNAs or sc-siRNA using DharmaFECT transfection reagent as per the manufacturer's instructions. At 72 h after transfection, cells were used for Ca2+ measurements or harvested for Western blot analysis.
Immunoprecipitation
ECs grown to confluence challenged with agonists were washed three times with phosphate-buffered saline at 4 °C and lysed in lysis buffer as in Ref. 17. Lysate protein (300 μg) was subjected to immunoprecipitation. Insoluble material was removed by centrifugation (13,000 × g for 15 min) before overnight immunoprecipitation with 1 μg/ml antibody at 4 °C. Protein A/G-agarose beads were added to each sample and incubated for 1 h at 4 °C. Immunoprecipitates were gently washed three times with wash buffer (Tris-buffered saline containing 0.05% Triton X-100, 1 mm Na3VO4, 1 mm NaF, 2 μg/ml leupeptin, 2 μg/ml pepstatin A, 2 μg/ml aprotinin, and 44 μg/ml phenylmethylsulfonyl fluoride). Immunoprecipitated proteins were resolved on SDS-PAGE and immunoblotted with appropriate antibodies.
Immunoblotting
EC lysates or immunoprecipitates were resolved by SDS-PAGE on a 10% separating gel under reducing conditions and transferred to Duralose membrane. Membranes were blocked with 5% dry milk in 10 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.05% Tween 20 for 1 h. Membranes were incubated with the indicated primary antibody (diluted in blocking buffer) overnight. After three washes, membranes were incubated with horseradish peroxidase-conjugated secondary antibody. Protein bands were detected by enhanced chemiluminescence.
In Vitro Kinase Assay
Briefly, HEK293 cells in 100-mm dishes were transfected with Myc-WT-STIM1 expression construct (1 μg/ml) (17). At 48 h after transfection, total cell lysate (800 μg of protein) was immunoprecipitated using anti-Myc mAb. The precipitated proteins (substrate) were washed twice and incubated with N-terminal GST fusion-active p38β2 (0.25 μg) in the presence or absence of SB203580 (10 μm) in kinase buffer (50 mm Tris-HCl, 100 mm NaCl, 25 mm β-glycerophosphate, 25 mm MgCl2, pH 7.4, protease and phosphatase inhibitor mixtures, and 200 μm ATP) for 15 or 30 min at 30 °C (20, 21). The reaction was terminated by adding 2× SDS-PAGE sample buffer. Samples were boiled for 5 min; the proteins were separated on 10% SDS-PAGE and immunoblotted using anti-phospho-Ser pAb, anti-Myc pAb, or anti-STIM1 mAb, or anti-phospho-p38 pAb. We also performed in vitro kinase assay as described above for STIM1 immunoprecipitated from unstimulated (control) HLMVECs using anti-STIM1 pAb.
Confocal Imaging
ECs were transfected with the YFP-WT-STIM1 (2 μg/ml) construct as described previously (3). 48 h after transfection, cells were washed and placed in HBSS, and then confocal live cell images of the YFP-tagged fluorescent protein were acquired near the surface of the cell using a 514-nm laser excitation/530-nm LP emission filter with the pinhole set to achieve 1 Airy unit (∼0.5-μm optical sections). Image configurations acquired before and after thrombin stimulation were not changed, and the cells were maintained at 37 °C.
Statistical Analysis
Comparisons were made with a two-tailed Student's t test. Experimental values were reported as mean ± S.E. Differences in mean values between two or more groups were determined by one-way analysis of variance. A p value <0.05 was considered statistically significant.
RESULTS
STIM1 Phosphorylation Inhibits SOCE in Endothelial Cells
To study the relationship between STIM1 phosphorylation and SOCE in endothelial cells, we treated HLMVECs with thrombin for different time periods, and then cells were used to examine STIM1 phosphorylation at its serine residues. After thrombin treatment, cell lysates were immunoprecipitated with anti-STIM1 mAb, and the precipitate was immunoblotted with anti-phospho-Ser pAb. Here, we observed that thrombin stimulation caused a time-dependent phosphorylation of STIM1, which reached a maximum of ∼8-fold over basal within 10 min (Fig. 1A). At 30 and 60 min after thrombin treatment, STIM1 phosphorylation was significantly reduced (Fig. 1A). These results indicate that thrombin stimulation caused STIM1 phosphorylation in endothelial cells.
FIGURE 1.

STIM1 phosphorylation inhibits SOCE in endothelial cells. A, thrombin-induced STIM1 phosphorylation was measured. HLMVECs exposed to thrombin (50 nm) for different time periods were lysed; lysates were immunoprecipitated (IP) with STIM1 mAb, and the immunoprecipitate was immunoblotted (IB) with anti-phospho-Ser pAb (top row). Total cell lysates were immunoblotted with anti-STIM1 mAb (bottom row). Phosphoprotein bands were quantified by densitometry and expressed as relative to control (bottom panel). Results shown are mean ± S.E. of four experiments. B, HLMVECs were preincubated either with thrombin (50 nm) (top panel) or PAR-1 peptide (40 μm) (bottom panel) for different time periods. After the specified time points, cells were used to measure TG (1 μm)-induced store Ca2+ release and Ca2+ entry (see details under “Experimental Procedures”). The experiment was repeated three times with similar results.
Next, we investigated whether STIM1 phosphorylation inhibits SOCE in HLMVECs. We challenged HLMVECs with thrombin or PAR-1 peptide for different time intervals. The cells were then washed, loaded with Fura-2AM for 30 min, and then thapsigargin (TG)-induced ER-stored Ca2+ release and Ca2+ release-activated Ca2+ entry (SOCE) were measured. In control cells (not pretreated with thrombin or PAR-1 peptide), we observed a normal TG-induced store release and SOCE (Fig. 1B). In cells pretreated with thrombin or PAR-1 peptide for 10 min, TG failed to induce either store release or SOCE (Fig. 1B). Interestingly, TG response was partially rescued in 30-min thrombin- or -PAR-1 peptide-pretreated cells (Fig. 1B), whereas in TG response was largely rescued in 60-min thrombin or -PAR-1 peptide-pretreated cells (Fig. 1B). These results are in agreement with the time course of thrombin-induced STIM1 phosphorylation. Thus, STIM1 phosphorylation in its Ser residues may inhibit SOCE in HLMVECs.
Pharmacological Activation of AMPK Induces STIM1 Phosphorylation and Prevents PAR-1-induced Ca2+ Entry and Lung Microvessel Permeability
Studies from our laboratory (15) and others (22) showed that Ca2+ entry signal activates AMPK in endothelial cells. Therefore, we tested whether AMPK signaling is involved in modulating SOCE in endothelial cells. AMPK is a serine/threonine (Ser/Thr) protein kinase composed of a catalytic α-subunit and regulatory β- and γ-subunits (23, 24). Thr-172 phosphorylation in the α-subunit is essential for catalytic function of AMPK (23, 24). We pretreated HLMVECs with the AMPK activator AICAR (23) and measured Thr-172 phosphorylation of AMPK α-subunit (AMPKα). We observed a dose-dependent increase in the phosphorylation of AMPKα (Fig. 2A), with an optimal increase (∼2.5-fold) in cells treated with 1 or 2 mm (Fig. 2A). To study the effect of AMPK activation on STIM1 phosphorylation, we measured STIM1 phosphorylation in its Ser residues in control and AICAR-treated HLMVECs. In this experiment, cells were lysed; lysates were immunoprecipitated with anti-phospho-Ser pAb, and the immunoprecipitate was immunoblotted with anti-STIM1 mAb. We observed that STIM1 phosphorylation was increased significantly in AICAR-pretreated cells compared with control cells (Fig. 2B).
FIGURE 2.
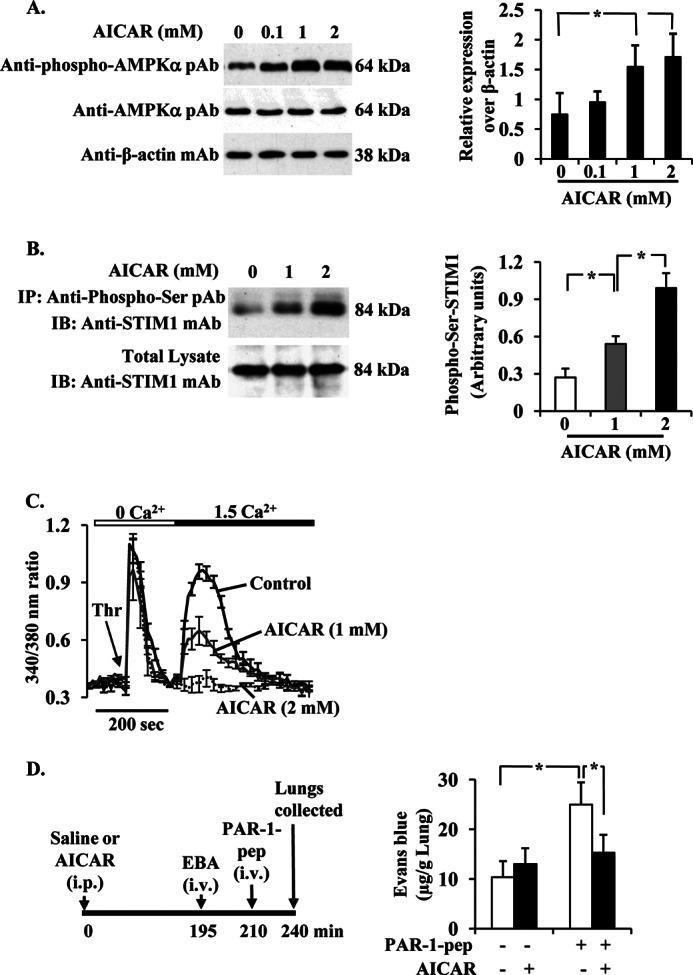
AMPK activation abrogates PAR-1-induced Ca2+ entry in ECs and lung microvessel permeability. A, AICAR induces phosphorylation of AMPKα. HLMVECs grown to 80% confluence were pretreated with AICAR (0, 0.1, 1, and 2 mm) for 2 h in 1% serum-containing medium. Cells were then lysed and immunoblotted with anti-phospho-AMPKα mAb (top), anti-AMPKα pAb (middle), and anti-β-actin mAb. A representative blot is shown from four independent experiments. The protein bands were quantified by densitometry relative to β-actin (right panel). *, significantly different from cells not treated with AICAR. B, AICAR induces STIM1 phosphorylation. HLMVECs exposed to the indicated concentrations of AICAR for 2 h as above. Cells were lysed; lysates were immunoprecipitated (IP) with anti-phospho-Ser pAb, and then the precipitate was immunoblotted (IB) with anti-STIM1 mAb. Results shown are from representative of four experiments. The quantified results are shown in the right panel. *, significantly different from control. C, AICAR inhibits thrombin-induced Ca2+ entry. HLMVECs grown on coverslips and pretreated with AICAR (1 or 2 mm) for 2 h were used to measure Ca2+ entry. Fura-2-loaded cells placed in Ca2+- and Mg2+-free HBSS were stimulated with thrombin (50 nm). After return of [Ca2+]i to base-line levels, CaCl2 (1.5 mm) was added to extracellular medium to induce Ca2+ entry. Arrow indicates time at which cells were stimulated with thrombin (Thr). Results shown are mean ± S.E. of four experiments. D, AICAR pretreatment abrogates PAR-1-induced lung vascular permeability increase. C57BL/6 mice either injected with AICAR (500 mg/kg, intraperitoneally) or saline were used to measure PAR-1-peptide-induced EBA uptake in lungs. Top panel shows experimental design. Results are mean ± S.E. of changes in lung EBA after PAR-1 agonist peptide administration (n = 6; in each group). * indicates the significance between the treatment groups and the respective control groups (p < 0.05).
Next, we measured thrombin-induced Ca2+ entry (i.e. SOCE) in cells treated with or without AICAR. AICAR pretreatment had no significant effect on thrombin-induced store Ca2+ release (Fig. 2C), whereas thrombin-induced Ca2+ entry was blocked in AICAR-treated cells (Fig. 2C). These results indicate that the AMPK signal may play a role in the reversal of SOCE in ECs. We also investigated the in vivo relevance of AMPK signaling in regulating lung microvessel permeability. In this study, we injected mice (C57BL6J) with AICAR (500 mg/kg i.p.), and the control mice received saline. At 4 h after AICAR or saline injection, mice were used to assess PAR-1 agonist peptide-induced lung microvessel permeability by measuring EBA uptake in lungs (see details under “Experimental Procedures”). PAR-1 peptide induced an ∼2-fold increase in EBA uptake over control (Fig. 2D). AICAR alone had no effect on basal EBA uptake in lungs (Fig. 2D), whereas AICAR pretreatment markedly reduced PAR-1-induced EBA uptake in lungs (Fig. 2D). These results collectively suggest that AMPK-mediated STIM1 phosphorylation may play a role in reversing lung vascular permeability responses.
AMPKα1 Negatively Regulates Thrombin-induced Ca2+ Entry in HLMVECs
Two isoforms of the catalytic subunits (α1 and α2) have been identified (23). Endothelial cells express both α1 and α2 subunits (25–28). The above results showed that pharmacological activation of AMPK induced STIM1 phosphorylation and suppressed SOCE, so we addressed which isoform of the α-subunits of AMPK were involved in regulating SOCE in HLMVECs. We measured phosphorylation of AMPK catalytic α1- and α2-subunits in response to thrombin. Interestingly, we observed thrombin induced a time-dependent robust increase in phosphorylation of AMPKα1 but not AMPKα2 (Fig. 3, A–C). The maximum level of phosphorylation of AMPKα1 was seen 10 min after thrombin and then gradually decreased at 30 and 60 min (Fig. 3, A, 1st lane, and B). We did not observe significant changes in the level of phosphorylation of AMPKα2 in response to thrombin (Fig. 3, A, 2nd lane, and C). Because we observed an activation of AMPKα1 after thrombin stimulation, we then silenced endogenous expression of AMPKα1 or AMPKα2 by transfecting siRNA specific to AMPKα1 or AMPKα2 in HLMVECs. In AMPKα1-siRNA-transfected cells, AMPKα1 protein expression was markedly suppressed compared with control or Sc-siRNA transfected cells (Fig. 3D). Similarly, we observed AMPKα2 expression was markedly reduced in AMPKα2-siRNA-transfected cells (Fig. 3D). Next, we determined thrombin-induced Ca2+ entry in Sc-siRNA-, AMPKα1-siRNA-, or AMPKα2-siRNA-transfected cells. In AMPKα1-transfected cells, we observed an augmented thrombin-induced Ca2+ entry (Fig. 3E), whereas the thrombin-induced Ca2+ entry was not altered in cells transfected with AMPKα2-siRNA compared with Sc-siRNA-transfected cells (Fig. 3E). These results indicate that AMPKα1 may regulate SOCE in HLMVECs.
FIGURE 3.

PAR-1-activated AMPKα1 regulates SOCE in HLMVECs. A, HLMVECs were challenged with thrombin (50 nm) for different time intervals at 37 °C. After thrombin stimulation, cells were lysed and immunoprecipitated (IP) with anti-phospho-AMPKα mAb. The precipitated proteins were immunoblotted (IB) with anti-AMPKα1 pAb (1st lane) or AMPKα2 pAb (2nd lane). Total cell lysates were immunoblotted with anti-AMPKα1 pAb (3rd lane) or AMPKα2 pAb (4th lane). Results are shown as the mean ± S.E. of four independent experiments for AMPKα1 (B) or AMPKα2 (C). * indicates the significance compared with cells not treated with thrombin. D, HLMVECs transfected with Sc-siRNA, AMPKα1-siRNA, or AMPKα1-siRNA (see details under “Experimental Procedures”) were lysed and immunoblotted with anti-AMPKα1 pAb (left, top panel) or anti-AMPKα2 pAb (left, bottom panel). The membrane was stripped and probed with anti-β-actin mAb as loading control. In right panels, AMPKα1 and AMPKα2 proteins were quantified by densitometry relative to β-actin. Results shown are mean ± S.E. of four experiments. *, significantly different compared with control or Sc-siRNA transfected cells. E, HLMVECs transfected with Sc-siRNA, AMPKα1-siRNA (100 or 200 nm), or AMPKα2-siRNA (100 or 200 nm) were used to measure thrombin (Thr)-induced Ca2+ store release and Ca2+ entry as described in Fig. 2C. Arrow indicates the time when the cells are challenged with thrombin (Thr). Results shown are mean ± S.E. of four experiments.
p38 MAPK Inhibition Augments SOCE and Prevents SOCE-mediated Phosphorylation of STIM1 in HLMVECs
Because AMPK lies upstream of p38 MAPK (15), we investigated whether AMPK modulates SOCE by activating its downstream target p38 MAPK. In this study, we initially performed an in vitro kinase assay to determine whether p38 MAPK directly phosphorylates STIM1 using recombinant active p38 MAPK (20, 21). In this assay, we determined STIM1 phosphorylation by Western using the anti-phospho-Ser pAb. We observed that active p38β2 caused phosphorylation of STIM1 at its serine residues (Fig. 4, A and B). The phosphorylation observed was prevented by SB203580, a p38 MAPK-selective inhibitor (Fig. 4, A and B) (29–31) indicating that STIM1 is a direct target of p38 MAPK. Next, we studied the role of p38 MAPK in regulating SOCE in HLMVECs. In this experiment, HLMVECs were treated with vehicle (0.01% DMSO) or SB203580, and then thrombin-induced STIM1 phosphorylation was measured. Thrombin stimulation increased a time-dependent phosphorylation of STIM1 with a maximum level at 10 min and a return to basal level 60 min after thrombin stimulation (Fig. 4C). In cells preincubated with SB203580, thrombin-induced STIM1 phosphorylation was suppressed (Fig. 4C). To understand the functional relevance, we determined thrombin-induced Ca2+ entry in control (vehicle-treated) cells, in SB203580-treated cells or in SB202474 (a negative control compound)-treated cells. We observed normal thrombin-induced Ca2+ entry in vehicle- or SB202474-treated cells (Fig. 4D), whereas in cells preincubated with the p38 MAPK inhibitor (SB203580), we observed a sustained increase in Ca2+ entry (Fig. 4D), indicating that p38 MAPK signaling is required for the reversal of SOCE.
FIGURE 4.

p38 MAPK downstream of AMPK signaling controls SOCE via phosphorylation of STIM1. A and B, phosphorylation of STIM1 by active p38β2 was determined using in vitro kinase assay (see details under “Experimental Procedures”). A, Myc-STIM1 was ectopically expressed in HEK293 cells and immunoprecipitated (IP) using anti-Myc; mAb was used as substrate for active p38β2. The assay was performed in the presence (+) and absence (−) of p38 inhibitor, SB203580 (10 μm). Lane 1, active p38β2 was not included in the kinase mixture; lane 7, control A/G beads were incubated with Myc-STIM1 expressing HEK cell lysates included in the kinase assay mixture loaded. Equal volume of assay mixture was immunoblotted (IB) with anti-phospho-Ser pAb, anti-Myc pAb, anti-STIM1 mAb, or anti-phospho-p38 pAb (left panel). Phosphoprotein bands were quantified by densitometry and expressed as relative to Myc-STIM1 (right panel). Results shown are mean ± S.E. of three independent experiments. *, significantly different from SB203580 treatment. B, unstimulated HLMVECs were immunoprecipitated using anti-STIM1 pAb, and the immunoprecipitate was used as substrate for active p38β2. The assay was performed as above. Equal volume of assay mixture was immunoblotted with anti-phospho-Ser pAb, anti-STIM1 mAb, or anti-phospho-p38 pAb (left panel). Results shown are mean ± S.E. of four independent experiments (right panel). *, significantly different from SB203580 treatment. Note that active p38β2-mediated STIM1 phosphorylation was detectable by anti-phospho-Ser pAb. C, HLMVECs pretreated with vehicle (DMSO, 0.01%) or SB203580 (10 μm) for 30 min were used to measure thrombin-induced phosphorylation of STIM1 as above in Fig. 1A. Phosphoprotein bands were quantified by densitometry and expressed as relative to control (right panel). Results shown are mean ± S.E. of three experiments. *, significantly different from cells not stimulated with thrombin or significant difference between control and SB203580-treated cells. D, HLMVECs pretreated with SB203580 (10 μm) or SB202474 (10 μm) were used to measure thrombin-induced Ca2+ entry as described above. Arrow indicates time of addition of thrombin (Thr). Results shown are mean ± S.E. of four independent experiments. E, HLMVECs grown to ∼70% confluence on glass-bottomed 35-mm dishes were transfected with YFP-WT-STIM1 expression construct. At 48 h after transfection, cells pretreated with SB203580 (10 μm) for 30 min were washed and placed in HBSS, and then PAR-1 peptide-induced STIM1 puncta formation was monitored in real time using a confocal microscope. STIM1 puncta were seen both in vehicle or SB203580-pretreated cells, whereas an increase or sustained puncta formation was observed in SB203580-treated cells. The images acquired from representative experiments are shown (top panels). STIM1 puncta formed after PAR-1 peptide addition was quantified, and results shown are mean ± S.E. (bottom panel). n = 4 from each group; *, significantly different from vehicle.
Studies have demonstrated that ER-stored Ca2+ release-mediated assembly of STIM1 into puncta at the ER/plasma membrane interface is required for Ca2+ entry (6). Also, studies have shown that STIM1 phosphorylation prevents store depletion-induced STIM1 puncta formation and SOCE (13). To address whether inhibition of p38 MAPK may influence the STIM1 puncta formation, we expressed YFP-WT-STIM1 in HLMVECs and used a confocal microscope to observe STIM1 puncta after PAR-1-agonist peptide stimulation. In cells treated with 0.01% DMSO (vehicle), we noted that PAR-1 peptide stimulation caused STIM1 puncta formation in a time-dependent manner (Fig. 4E, top panel). STIM1 puncta appeared within 20 s of PAR-1 peptide addition and were seen for 400 s. By contrast, in cells incubated with SB203580 for 30 min, we observed a sustained increase in puncta formation over a period of 500 s (Fig. 4E, bottom panel). These results indicate that p38 MAPK-mediated STIM1 phosphorylation may regulate SOCE in endothelial cells.
p38β MAPK Signaling Is Required to Reverse PAR-1-induced SOCE
Next, we investigated which isoform of p38 MAPK activation downstream of AMPK is involved in regulating SOCE in endothelial cells. Because p38 MAPK has four isoforms (α, β, γ, and δ) (29–31), we first determined the expression profiles of p38 MAPK isoforms in HLMVECs and mouse (m) LECs. RT-PCR confirmed that the mRNA for p38α, p38β, and p38γ were present in HLMVECs, but mLECs express the mRNA for only α and β isoforms (Fig. 5A).
FIGURE 5.

p38β MAPK regulates SOCE in HLMVECs. A, RT-PCR analysis of mRNA expression for p38 MAPK isoforms in HLMVECs and mLECs. Total RNA from HLMVECs and mLECs was isolated, and RT-PCR was performed to determine the expression of transcripts for p38 MAPK (α, β, γ, and δ) and GAPDH. B, HLMVECs were transfected with Sc-siRNA or siRNA specific to p38 MAPK isoforms (p38α, p38β, and p38γ). At 72 h after transfection, cells were used to determine expression of p38α, p38β, and p38γ by immunoblot. C–E, HLMVECs were transfected with Sc-siRNA or siRNA specific to p38α (C), β (D), or γ (E). At 72 h after transfection, cells were used to determine thrombin-induced Ca2+ entry as described above. Note the sustained Ca2+ entry in p38β-siRNA transfected cells (D), whereas in p38α-siRNA transfected cells the Ca2+ entry was blocked (C). Experiments were repeated at least three times, and the results shown are mean ± S.E. F, HLMVECs transfected with Sc-siRNA, p38α-siRNA, or p38β-siRNA were immunoblotted with anti-STIM1 mAb. Note that in p38α-siRNA-transfected cells STIM1 expression was reduced.
In the next set of experiments, we knocked down individual p38 isoforms using siRNA to address the specific role of p38 isoforms in regulating SOCE in HLMVECs. In these experiments, cells were transfected with control siRNA (Sc-siRNA), si-RNA specific to human p38α, human p38β, or human p38γ. At 72 h after transfection, we examined the protein expression by Western blot. p38 MAPK levels were markedly suppressed in target siRNA-transfected cells compared with Sc-siRNA-transfected cells or control cells (Fig. 5B). We then determined Ca2+ entry secondary to thrombin-induced Ca2+ store depletion. In p38α knockdown cells, both Ca2+ entry (Fig. 5C) and STIM1 expression (Fig. 5F) were blocked, whereas p38γ knockdown had no significant effect on thrombin-induced Ca2+ entry (Fig. 5E). Interestingly, p38β knockdown had no effect on STIM1 expression (Fig. 5F), but thrombin-induced Ca2+ entry was sustained (Fig. 5D). These results suggest that p38β may regulate SOCE presumably by phosphorylating STIM1, whereas the p38α signal may be required for the expression of STIM1 in endothelial cells.
Ca2+ Entry-CaMKKβ-AMPKα1 Axis Signaling Is Required to Activate p38β MAPK and STIM1 Phosphorylation
We have shown that Ca2+ entry through TRPC channels activates the CaMKKβ-AMPK-p38 signaling pathway in human pulmonary artery endothelial cells (15). In this study, we exposed HLMVECs to the CaMKKβ-selective inhibitor STO-609 and then measured thrombin-induced phosphorylation of AMPKα1. We observed that STO-609 treatment blocked thrombin-induced phosphorylation of AMPKα1 (Fig. 6A) indicating that CaMKKβ is essential to activate AMPKα1 in HLMVECs. Next, we investigated whether SOCE signaling activates p38β in HLMVECs. In this experiment, we measured thrombin-induced phosphorylation of p38β in the presence and absence of SOCE blocker Gd3+ (3, 4). We observed a time-dependent increase in phosphorylation of p38β in control HLMVECs challenged with thrombin, whereas a marked reduction in thrombin-induced p38β phosphorylation was observed in cells treated with Gd3+ (Fig. 6B). These results show an obligatory role for SOCE in signaling thrombin-induced p38β activation in HLMVECs.
FIGURE 6.
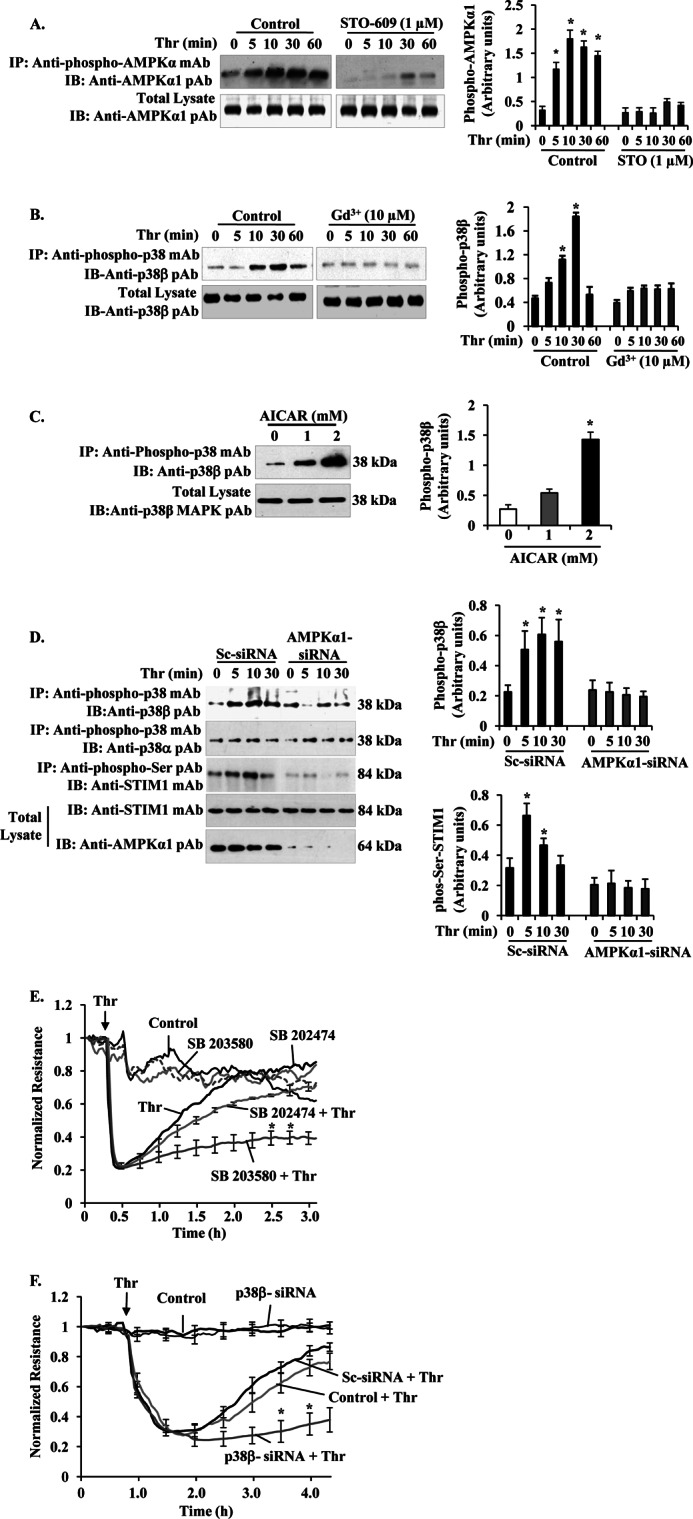
Ca2+ entry-CaMKKβ-AMPKα1-p38β MAPK axis signaling mediates STIM1 phosphorylation to inhibit SOCE and endothelial permeability. A, HLMVECs were pretreated with or without CaMKKβ inhibitor STO-609 (1 μm) for 30 min, and then thrombin-induced AMPKα1 phosphorylation was measured as described in Fig. 3A. The experiment was repeated four times, and the results shown are mean ± S.E. (right panel). *, significantly different from cells not stimulated with thrombin. B, HLMVECs, thrombin-induced p38β phosphorylation was measured in the absence and presence of Gd3+ (10 μm). After thrombin treatment, cell lysates were immunoprecipitated (IP) with anti-phospho-p38 mAb, and the precipitate was immunoblotted (IB) with anti-p38β pAb to determine p38β phosphorylation (top panel). Total cell lysates were blotted with anti-p38β mAb (bottom panel). A representative blot is shown from four independent experiments. Phosphoprotein bands were quantified by densitometry and are expressed in arbitrary units. *, significantly different from cells not stimulated with thrombin. Note that impaired thrombin-induced p38β phosphorylation in cells treated with Gd3+ to inhibit Ca2+ entry. C, HLMVECs pretreated with AICAR (0, 1, and 2 mm) were lysed and immunoprecipitated with anti-phospho-p38 mAb. The precipitated proteins were immunoblotted with anti-p38β MAPK pAb. Total cell lysates were immunoblotted with anti-p38β MAPK pAb. D, HLMVECs transfected with either Sc-siRNA or AMPKα1siRNA (200 nm) as described in Fig. 3D were stimulated with thrombin (50 nm) for different time intervals at 37 °C. After thrombin treatment, cells were lysed and immunoprecipitated (IP) with anti-phospho-p38 mAb. The precipitated proteins were immunoblotted (IB) with anti-p38β pAb (top row) or anti-p38α pAb (2nd row). Total cell lysates were immunoprecipitated with anti-phospho-Ser pAb, and the precipitate was immunoblotted with STIM1 mAb (3rd row). Total cell lysates were immunoblotted with anti-STIM1 mAb (4th row) and anti-AMPKα1 pAb (bottom row). Results shown are the mean ± S.E. of four independent experiments (right panels). *, significantly different from control cells. E, HLMVECs were grown to confluence on gold electrodes (see details under “Experimental Procedures”). Cells were washed and incubated with 1% serum-containing medium for 2 h and then incubated 30 min with or without the indicated concentrations of 10 μm SB203580 or SB202474 before the addition of 50 nm thrombin (Thr). Note that in SB203580-treated cells, thrombin produced a marked decrease in TER, but the TER recovery to basal level was delayed compared with control cells treated with thrombin or cells pretreated with SB202474 followed by thrombin addition. The arrow indicates the time at which the cells were challenged with thrombin (Thr) or medium. F, HLMVECs transfected with 200 nm Sc-siRNA or p38 β-siRNA were used to measure thrombin-induced TER changes. Note the delay in thrombin-induced decrease in TER recovery to basal level in p38β-siRNA transfected cells indicating hyper-permeability associated with prolonged SOCE. The arrow indicates the time at which the cells were challenged with thrombin (Thr) or medium. Results shown are the mean ± S.E. of four independent experiments. *, significantly different from control cells treated with thrombin.
To further elucidate whether AMPK mediates the p38β activation which in turn phosphorylates STIM1 to inhibit SOCE, we measured phosphorylation of p38β in the AMPK activator, AICAR-exposed HLMVECs. AICAR induced an increase in p38β phosphorylation ∼3-fold over basal (Fig. 6C). We showed above (Fig. 3E) that AMPKα1 knockdown enhanced SOCE in HLMVECs; thus, we examined whether AMPKα1 signaling is required to activate p38β. We suppressed AMPKα1 expression using AMPKα1-siRNA in HLMVECs and then measured thrombin-induced p38β and STIM1 phosphorylation. We observed that thrombin induced phosphorylation of p38β in control siRNA (Sc-siRNA)-transfected cells, whereas this response was markedly reduced in AMPKα1-siRNA transfected cells (Fig. 6D, 1st lane). In contrast, p38α phosphorylation did not change in either Sc-siRNA- or AMPKα1-siRNA-transfected cells (Fig. 6D, 2nd lane). Interestingly, thrombin-induced STIM1 phosphorylation was markedly reduced in AMPKα1 knockdown cells compared with control cells (Fig. 6D, 3rd lane). These results suggest that AMPKα1 signaling is essential for the activation of p38β and subsequent STIM1 phosphorylation, thereby regulating SOCE in endothelial cells.
To address the functional relevance of p38β activity downstream of AMPKα1 and thereby regulating vascular endothelial barrier function, we determined thrombin-mediated changes in TER (18). In control HLMVECs, thrombin addition caused an ∼70% maximum decrease in TER and the TER return to basal 2 h after thrombin challenge (Fig. 6E). In cells incubated with the p38 inhibitor (SB203580), thrombin caused a similar decrease in TER, but TER return to basal did not occur (Fig. 6E). In the control compound (SB202474), the incubated cells thrombin-induced response was similar to control cells. In another set of experiments, we knocked down p38β in HLMVECs and measured TER. In control cells or in cells transfected with Sc-siRNA, thrombin produced an ∼60% decrease in TER, and the TER recovered to basal levels within 2 h of thrombin addition (Fig. 6F), whereas in p38β siRNA-transfected cells, thrombin produced a similar decrease in TER, but TER recovery to basal levels did not occur (Fig. 6F). Thus, these results collectively support the conclusion that SOCE-mediated CaMKKβ-AMPKα1-p38β signaling serves as a turn off switch for SOCE, reversing the permeability responses.
DISCUSSION
SOCE in nonexcitable cells regulates many cellular processes, including cell migration, apoptosis, and induction of inflammatory genes. We have shown that SOCE induced by thrombin ligation of PAR-1 mediates vascular barrier dysfunction and amplifies the expression of inflammatory genes in endothelial cells (3, 15, 16, 32, 33). Recent studies have shown that STIM1 is crucial for the activation of SOCE in endothelial cells (3–5). However, the downstream signaling pathways involved in terminating SOCE are unknown. Further evidence suggests that SOC function may be inhibited by phosphorylation of STIM1 on its C-terminal serine residues (13). The deduced STIM1 sequence revealed the presence of 10 putative phosphorylation sites for p38 MAPK. In recent studies, we showed that SOCE activates the CaMKKβ-AMPK-p38 MAPK signaling pathway in endothelial cells (15). Thus, we tested the postulate that PAR-1-induced SOCE signaling is essential for Ser phosphorylation of STIM1 and subsequent inhibition of SOCE in endothelial cells. Using an antibody that reacts with phospho-Ser, we observed phosphorylation of STIM1 in response to PAR-1-induced SOCE. Interestingly, we observed a negative correlation between the time course of PAR-1-induced STIM1 phosphorylation and SOCE activity indicating that STIM1 phosphorylation serves as an “off switch” for SOCE in endothelial cells.
AMPK is an energy sensor activated by an increase in AMP levels in cells (23). AMPK activation requires phosphorylation of Thr-172 in the activation loop of the α-subunit (23, 24). The upstream-activating enzyme AMPK kinase or the closely related tumor suppressor kinase LKB1 can phosphorylate the catalytic α-subunit of AMPK in an AMP-dependent manner (23, 34). AMPK can also be activated in an AMP-independent manner, which involves CaMKKβ (22, 23). An increase in intracellular Ca2+ is required for CaMKKβ activation (22). The pharmacological AMPK activator, AICAR, has been shown to phosphorylate AMPK subunit α and activate AMPK both in vitro and in vivo (23, 34). AICAR is metabolized to 5-aminoimidazole-4-carboxamide ribonucleoside, which mimics all effects of AMP on AMPK systems (23, 34). Zhao et al., (35) have shown that AICAR-mediated AMPK activation reduced endotoxin-induced acute lung injury in mice. Another study showed that administration of AMPK activator metformin (anti-diabetic drug) increased the survival rate of endotoxemic mice (36). Creighton et al. (27) have demonstrated that AMPKα1 signaling promotes the endothelial barrier repair process. More importantly, studies have shown that AICAR-induced AMPK activation targets its downstream effector p38 MAPK (37). Based on the existing evidence, we tested the hypothesis that pharmacological activation of AMPK would block PAR-1-mediated SOCE by phosphorylating STIM1 in endothelial cells. In support of this hypothesis, we observed that AICAR pretreatment induced AMPK subunit α phosphorylation in HLMVECs. Also, AICAR pretreatment induced STIM1 phosphorylation and blocked thrombin-induced Ca2+ entry in HLMVECs. To address the in vivo physiological relevance of AMPK signaling, we pretreated mice with AICAR and then determined PAR-1-induced lung vascular permeability by measuring EBA uptake in lungs. We observed that AICAR pretreatment markedly reduced PAR-1-induced increase in lung vascular permeability indicating that AICAR-induced STIM1 phosphorylation may contribute to the inhibition of PAR-1-mediated lung vascular permeability.
Because endothelial cells express both the catalytic α1- and α2-subunits of AMPK (25–28), we compared the extent of phosphorylation of the α1- and α2-subunits in response to thrombin. We observed that thrombin induced phosphorylation of the α1-subunit but not the α2-subunit of AMPK in HLMVECs. To determine the functional role of SOCE-mediated AMPKα1 phosphorylation, we transfected HLMVECs with siRNA specific to AMPKα1 and then measured SOCE in response to thrombin. We observed that in AMPKα1-depleted cells, thrombin-induced SOCE was augmented, raising the possibility that AMPKα1 activation is required for STIM1 phosphorylation and hence inhibition of SOCE in endothelial cells.
Li. et al. (38) have shown that activated AMPK interacts with the scaffold protein TAB1, and the resulting complex associates with the p38 MAPK, which in turn promotes p38 MAPK autophosphorylation in ischemic hearts. We have shown that inhibition of AMPK in endothelial cells prevented thrombin-induced p38 MAPK activation, demonstrating that AMPK lies upstream of p38 MAPK (15). Thus, we exposed HLMVECs to a specific inhibitor of p38 MAPK (SB203580, which inhibits both p38α and p38β isoforms (29–31)) and measured thrombin-induced STIM1 phosphorylation and Ca2+ entry. We observed that p38 inhibition suppressed thrombin-induced STIM1 phosphorylation and enhanced thrombin-induced Ca2+ entry in HLMVECs. Because ER-store Ca2+ release-mediated assembly of STIM1 into puncta at ER/plasma membrane activates Ca2+ entry (6, 13), we studied the effect of p38 MAPK inhibition on PAR-1-induced STIM1 puncta formation. We observed that p38 MAPK inhibition prolonged the STIM1 puncta formation upon PAR-1 activation indicating the possibility that STIM1 phosphorylation may regulate SOCE in endothelial cells. It is also possible that STIM1 phosphorylation may induce the dissociation of SOC components or undetermined STIM1-binding proteins from STIM1 to limit SOCE in endothelial cells.
There are four p38 MAPK isoforms (MAPK14 (p38α), MAPK11 (p38β), MAPK12 (p38γ), and MAPK13 (p38δ)) expressed in mammalian cells (29–31). Gene knock-out and pharmacological studies suggest that p38α signaling is essential for development, transcriptional regulation of genes, and inflammatory responses (29–31, 39–41). Recent emerging studies suggest that p38β signaling may play a critical role in cell survival and the reversal of inflammatory responses (42). p38δ and p38γ isoforms have been shown to regulate transcription of genes (43). Because we observed endothelial expression of α, β, and γ isoforms of p38 MAPK, we attempted to identify the isoform(s) activated downstream of AMPK in response to thrombin-induced SOCE in HLMVECs. We suppressed the expression of each p38 isoform by gene silencing. In this study, we observed that knockdown of p38α suppressed STIM1 expression and thrombin-induced SOCE in HLMVECs indicating that p38α signaling may regulate the expression of STIM1 rather than SOCE function in HLMVECs. Knockdown of p38γ had no significant effect on thrombin-induced SOCE in HLMVECs. Interestingly, knockdown of p38β had no effect on STIM1 expression but enhanced thrombin-induced SOCE in HLMVECs. Consistent with the enhanced SOCE, the thrombin-induced increase in permeability was also augmented in p38β-depleted HLMVECs.
It is known that Ca2+ entry signaling activates CaMKKβ to induce AMPKα phosphorylation in endothelial cells (15, 22). In this study, we observed that pretreatment with CaMKKβ inhibitor STO-609 suppressed thrombin-induced AMPKα1 phosphorylation indicating that CaMKKβ is essential to activate AMPKα1 in endothelial cells. To determine whether AMPKα1 is essential for SOCE-mediated p38β activation and subsequent STIM1 phosphorylation, we silenced AMPKα1 expression by transfection of siRNA specific to AMPKα1 and measured phosphorylation of p38β and STIM1 in response to thrombin in HLMVECs. We observed that AMPKα1 knockdown markedly reduced thrombin-induced phosphorylation of p38β and STIM1. In another experiment, we observed that AICAR treatment also resulted in p38β phosphorylation in HLMVECs. Moreover, we measured phosphorylation of p38β in the presence of SOCE inhibitor Gd3+ (3, 4) and observed markedly reduced phosphorylation of p38β in response to thrombin. These findings demonstrate that the SOCE-induced CaMKKβ-AMPKα1-p38β signaling pathway is vital in the mechanism of reversal of SOCE and permeability responses.
In summary, we have shown that PAR-1-mediated SOCE (i.e. Ca2+ entry through SOC) results in activation of the CaMKKβ-AMPKα1-p38β signaling axis (Fig. 7), which is essential for phosphorylation of ER-localized STIM1 to turn off SOCE in endothelial cells. Thus, Ca2+ entry-dependent phosphorylation of STIM1 via the CaMKKβ-AMPKα1-p38β axis provides an important negative feedback signal to terminate SOCE and thereby regulates vascular permeability responses.
FIGURE 7.

Signaling pathway downstream of SOCE involved in turning off SOCE in endothelial cells. PAR-1-induced ER store Ca2+ depletion via phospholipase C (PLC)-inositol 1,4,5-triphosphate (IP3) activates SOCE. SOCE signal activates CaMKKβ-AMPKα1-p38β MAPK signal axis, which in turn phosphorylates STIM1 to terminate SOCE in endothelial cells. DAG, diacylglycerol.
This work was supported, in whole or in part, by National Institutes of Health Grants GM058531 and P01HL077806. This work was also supported by a postdoctoral fellowship from Midwest Affiliate American Heart Association (to P. C. S.).
- PAR-1
- protease-activated receptor-1
- SOCE
- store-operated Ca2+ entry
- STIM1
- stromal interaction molecule-1
- AMPK
- AMP-activated protein kinase
- p38 MAPK
- p38 mitogen-activated protein kinase
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- HLMVEC
- human lung microvascular endothelial cell
- Sc-siRNA
- scrambled siRNA
- CaMKKβ
- CaM kinase kinase β
- TER
- transendothelial electrical resistance
- EC
- endothelial cell
- TRPC
- transient receptor potential canonical
- pAb
- polyclonal antibody
- TG
- thapsigargin
- ER
- endoplasmic reticulum
- mLEC
- mouse lung endothelial cell
- SOC
- store-operated Ca2+ entry channels
- HBSS
- Hanks' balanced salt solution
- h
- human
- EBA
- Evans blue dye conjugated with albumin.
REFERENCES
- 1. Tiruppathi C., Ahmmed G. U., Vogel S. M., Malik A. B. (2006) Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13, 693–708 [DOI] [PubMed] [Google Scholar]
- 2. Nilius B., Droogmans G. (2001) Ion channels and their functional role in vascular endothelium. Physiol. Rev. 81, 1415–1459 [DOI] [PubMed] [Google Scholar]
- 3. Sundivakkam P. C., Freichel M., Singh V., Yuan J. P., Vogel S. M., Flockerzi V., Malik A. B., Tiruppathi C. (2012) The Ca2+ sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca2+ entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol. Pharmacol. 81, 510–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abdullaev I. F., Bisaillon J. M., Potier M., Gonzalez J. C., Motiani R. K., Trebak M. (2008) Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 103, 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li J., Cubbon R. M., Wilson L. A., Amer M. S., McKeown L., Hou B., Majeed Y., Tumova S., Seymour V. A., Taylor H., Stacey M., O'Regan D., Foster R., Porter K. E., Kearney M. T., Beech D. J. (2011) Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 108, 1190–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee K. P., Yuan J. P., Hong J. H., So I., Worley P. F., Muallem S. (2010) An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs. FEBS Lett. 584, 2022–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., Cahalan M. D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang G. N., Zeng W., Kim J. Y., Yuan J. P., Han L., Muallem S., Worley P. F. (2006) STIM1 carboxy terminus activates native SOC, ICRAC and TRPC1 channels. Nat. Cell Biol. 8, 1003–1010 [DOI] [PubMed] [Google Scholar]
- 9. Kim M. S., Zeng W., Yuan J. P., Shin D. M., Worley P. F., Muallem S. (2009) Native store-operated Ca2+ influx requires the channel function of Orai1 and TRPC1. J. Biol. Chem. 284, 9733–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan J. P., Zeng W., Dorwart M. R., Choi Y. J., Worley P. F., Muallem S. (2009) SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zeng W., Yuan J. P., Kim M. S., Choi Y. J., Huang G. N., Worley P. F., Muallem S. (2008) STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 32, 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manji S. S., Parker N. J., Williams R. T., van Stekelenburg L., Pearson R. B., Dziadek M., Smith P. J. (2000) STIM1: a novel phosphoprotein located at the cell surface. Biochim. Biophys. Acta 1481, 147–155 [DOI] [PubMed] [Google Scholar]
- 13. Smyth J. T., Petranka J. G., Boyles R. R., DeHaven W. I., Fukushima M., Johnson K. L., Williams J. G., Putney J. W., Jr. (2009) Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat. Cell Biol. 11, 1465–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pozo-Guisado E., Campbell D. G., Deak M., Alvarez-Barrientos A., Morrice N. A., Alvarez I. S., Alessi D. R., Martín-Romero F. J. (2010) Phosphorylation of STIM1 at ERK1/2 target sites modulates store-operated calcium entry. J. Cell Sci. 123, 3084–3093 [DOI] [PubMed] [Google Scholar]
- 15. Bair A. M., Thippegowda P. B., Freichel M., Cheng N., Ye R. D., Vogel S. M., Yu Y., Flockerzi V., Malik A. B., Tiruppathi C. (2009) Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-κB activation in endothelial cells through AMP-activated protein kinase and protein kinase Cδ. J. Biol. Chem. 284, 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tiruppathi C., Freichel M., Vogel S. M., Paria B. C., Mehta D., Flockerzi V., Malik A. B. (2002) Impairment of store-operated Ca2+ entry in TRPC4−/− mice interferes with increase in lung microvascular permeability. Circ. Res. 91, 70–76 [DOI] [PubMed] [Google Scholar]
- 17. Sundivakkam P. C., Kwiatek A. M., Sharma T. T., Minshall R. D., Malik A. B., Tiruppathi C. (2009) Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am. J. Physiol. Cell Physiol. 296, C403–C413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tiruppathi C., Malik A. B., Del Vecchio P. J., Keese C. R., Giaever I. (1992) Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc. Natl. Acad. Sci. U.S.A. 89, 7919–7923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaneider N. C., Leger A. J., Agarwal A., Nguyen N., Perides G., Derian C., Covic L., Kuliopulos A. (2007) Role reversal for the receptor PAR-1 in sepsis-induced vascular damage. Nat. Immunol. 8, 1303–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Chiara G., Marcocci M. E., Torcia M., Lucibello M., Rosini P., Bonini P., Higashimoto Y., Damonte G., Armirotti A., Amodei S., Palamara A. T., Russo T., Garaci E., Cozzolino F. (2006) Bcl-2 phosphorylation by p38 MAPK. Identification of target sites and biologic consequences. J. Biol. Chem. 281, 21353–21361 [DOI] [PubMed] [Google Scholar]
- 21. Thornton T. M., Pedraza-Alva G., Deng B., Wood C. D., Aronshtam A., Clements J. L., Sabio G., Davis R. J., Matthews D. E., Doble B., Rincon M. (2008) Phosphorylation by p38 MAPK as an alternative pathway for GSK3β inactivation. Science 320, 667–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stahmann N., Woods A., Carling D., Heller R. (2006) Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase β. Mol. Cell. Biol. 26, 5933–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hardie D. G. (2003) Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 144, 5179–5183 [DOI] [PubMed] [Google Scholar]
- 24. Hawley S. A., Davison M., Woods A., Davies S. P., Beri R. K., Carling D., Hardie D. G. (1996) Characterization of the AMP-activated protein kinase kinase from rat liver, and identification of threonine-172 as the major site at which it phosphorylates and activates AMP-activated protein kinase. J. Biol. Chem. 271, 27879–27887 [DOI] [PubMed] [Google Scholar]
- 25. Thors B., Halldórsson H., Thorgeirsson G. (2011) eNOS activation mediated by AMPK after stimulation of endothelial cells with histamine or thrombin is dependent on LKB1. Biochim. Biophys. Acta 1813, 322–331 [DOI] [PubMed] [Google Scholar]
- 26. Liu C., Liang B., Wang Q., Wu J., Zou M. H. (2010) Activation of the AMP-activated protein kinase αI alleviates endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins BCL-2 and survivin. J. Biol. Chem. 285, 15346–15355 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27. Creighton J., Jian M., Sayner S., Alexeyev M., Insel P. A. (2011) Adenosine monophosphate-activated kinase α1 promotes endothelial barrier repair. FASEB J. 25, 3356–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fisslthaler B., Fleming I. (2009) Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ. Res. 105, 114–127 [DOI] [PubMed] [Google Scholar]
- 29. Kumar S., Boehm J., Lee J. C. (2003) p38 MAP kinases: Key signaling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2, 717–726 [DOI] [PubMed] [Google Scholar]
- 30. Coulthard L. R., White D. E., Jones D. L., McDermott M. F., Burchill S. A. (2009) p38 (MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 15, 369–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wagner E. F., Nebreda A. R. (2009) Signaling integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9, 537–549 [DOI] [PubMed] [Google Scholar]
- 32. Paria B. C., Bair A. M., Xue J., Yu Y., Malik A. B., Tiruppathi C. (2006) Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-κB activation in endothelial cells. J. Biol. Chem. 281, 20715–20727 [DOI] [PubMed] [Google Scholar]
- 33. Paria B. C., Vogel S. M., Ahmmed G. U., Alamgir S., Shroff J., Malik A. B., Tiruppathi C. (2004) Tumor necrosis factor-α-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 287, L1303–L1313 [DOI] [PubMed] [Google Scholar]
- 34. Towler M. C., Hardie D. G. (2007) AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 100, 328–341 [DOI] [PubMed] [Google Scholar]
- 35. Zhao X., Zmijewski J. W., Lorne E., Liu G., Park Y. J., Tsuruta Y., Abraham E. (2008) Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L497–L504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsoyi K., Jang H. J., Nizamutdinova I. T., Kim Y. M., Lee Y. S., Kim H. J., Seo H. G., Lee J. H., Chang K. C. (2011) Metformin inhibits HMGB1 release in LPS-treated RAW 264.7 cells and increases survival rate of Endotoxemic mice. Br. J. Pharmacol. 162, 1498–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lemieux K., Konrad D., Klip A., Marette A. (2003) The AMP-activated protein kinase activator AICAR does not induce GLUT4 translocation to transverse tubules but stimulates glucose uptake and p38 mitogen-activated protein kinases α and β in skeletal muscle. FASEB J. 17, 1658–1665 [DOI] [PubMed] [Google Scholar]
- 38. Li J., Miller E. J., Ninomiya-Tsuji J., Russell R. R., 3rd, Young L. H. (2005) AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ. Res. 97, 872–879 [DOI] [PubMed] [Google Scholar]
- 39. Cuenda A., Rousseau S. (2007) p38 MAP-kinases pathway regulation, function, and role in human diseases. Biochim. Biophys. Acta 1773, 1358–1375 [DOI] [PubMed] [Google Scholar]
- 40. Adams R. H., Porras A., Alonso G., Jones M., Vintersten K., Panelli S., Valladares A., Perez L., Klein R., Nebreda A. R. (2000) Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell 6, 109–116 [PubMed] [Google Scholar]
- 41. Beardmore V. A., Hinton H. J., Eftychi C., Apostolaki M., Armaka M., Darragh J., McIlrath J., Carr J. M., Armit L. J., Clacher C., Malone L., Kollias G., Arthur J. S. (2005) Generation and characterization of p38β (MAPK11) gene-targeted mice. Mol. Cell. Biol. 25, 10454–10464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferrari G., Terushkin V., Wolff M. J., Zhang X., Valacca C., Poggio P., Pintucci G., Mignatti P. (2012) TGF-β1 induces endothelial cell apoptosis by shifting VEGF activation of p38(MAPK) from the prosurvival p38β to proapoptotic p38α. Mol. Cancer Res. 10, 605–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Risco A., del Fresno C., Mambol A., Alsina-Beauchamp D., MacKenzie K. F., Yang H. T., Barber D. F., Morcelle C., Arthur J. S., Ley S. C., Ardavin C., Cuenda A. (2012) p38γ and p38δ kinases regulate the Toll-like receptor 4 (TLR4)-induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc. Natl. Acad. Sci. U.S.A. 109, 11200–11205 [DOI] [PMC free article] [PubMed] [Google Scholar]