

Abstract
Rolofylline is a potent, selective adenosine A1 receptor antagonist that was under development for the treatment of patients with acute congestive heart failure and renal impairment. Rolofylline is metabolized primarily to the pharmacologically active M1-trans and M1-cis metabolites (metabolites) by cytochrome P450 (CYP) 3A4. The aim of this investigation was to provide a pharmacokinetic (PK) model for rolofylline and metabolites following intravenous administration to healthy volunteers. Data included for this investigation came from a randomized, double-blind, dose-escalation trial in four groups of healthy volunteers (N = 36) where single doses of rolofylline, spanning 1 to 60 mg ,were infused over 1–2 h. The rolofylline and metabolite data were analyzed simultaneously using NONMEM. The simultaneous PK model comprised, in part, a two-compartment linear PK model for rolofylline, with estimates of clearance and volume of distribution at steady-state of 24.4 L/h and 239 L, respectively. In addition, the final PK model contained provisions for both conversion of rolofylline to metabolites and stereochemical conversion of M1-trans to M1-cis. Accordingly, the final model captured known aspects of rolofylline metabolism and was capable of simultaneously describing the PK of rolofylline and metabolites in healthy volunteers.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-012-9443-5) contains supplementary material, which is available to authorized users.
KEY WORDS: adenosine A1 receptor antagonist, pharmacokinetics, rolofylline
INTRODUCTION
Rolofylline is a potent, selective adenosine A1 receptor antagonist (1). It is reported that rolofylline blocks the re-absorption of sodium by the proximal tubules without triggering tubuloglomerular feedback (TGF), a compensatory mechanism to avoid excessive sodium and water loss (2). TGF promotes the release of adenosine, and adenosine binding to A1 receptors is associated with vasoconstriction of the afferent arteriole, decreased renal blood flow, and enhanced sodium re-absorption by the proximal tubule; this results in a decrease in glomerular filtration rate, diminished renal function, and sodium and water retention. Blocking adenosine A1 receptors via a selective adenosine receptor antagonist may limit sodium re-absorption by the proximal tubules without triggering TGF and promotes vasodilatation of the afferent arteriole of the glomerulus (3). Accordingly, rolofylline was under development for the treatment of patients with acute congestive heart failure and renal function impairment (1,4–7).
Rolofylline is eliminated primarily by oxidative metabolism to M1-trans and M1-cis metabolites. As with rolofylline, both M1-trans and M1-cis metabolites are eliminated primarily by metabolism (by oxidation and glucuronidation, respectively) and are not excreted unchanged in urine (1,8). Preclinical data indicate that M1-trans and M1-cis undergo stereochemical interconversion, and that the conversion of M1-trans to M1-cis is faster than the conversion of M1-cis to M1-trans in humans. The structures of rolofylline and both M1-trans and M1-cis metabolites are given in Fig. 1.
Fig. 1.
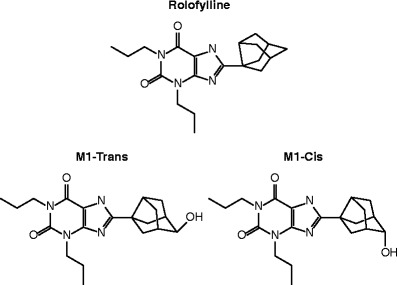
Structure of rolofylline and both M1-trans and M1-cis metabolites
In vitro data show that rolofylline and its M1-trans and M1-cis metabolites have similar affinities to the human adenosine A1 receptor expressed in CHO cells. In rats, intravenous (IV) treatment with M1-trans or M1-cis alone resulted in diuretic and natriuretic effects similar to those induced by rolofylline. In addition, both M1-trans and M1-cis had similar reno-protective effects compared with rolofylline in an acute rat renal injury model (Merck & Co., Inc., Whitehouse Station, NJ, USA and Kyowa Hakko Kirin Co., Ltd., Tokyo, Japan; data on file).
An understanding of the pharmacokinetics of all three analytes becomes important given the potential contribution of rolofylline, M1-trans, and M1-cis to the overall pharmacologic activity of rolofylline administration. Noncompartmental analyses of both rolofylline and metabolites following administration of single (9) and multiple (8) IV doses of rolofylline in healthy volunteers indicate that exposures of M1-trans were comparable to those for rolofylline, while M1-cis circulated at lower levels. Additionally, the terminal phases of rolofylline and metabolite concentration–time profiles appeared similar (with accompanying apparent terminal half-lives as obtained by noncompartmental analysis of approximately 15, 12, and 14 h for rolofylline, M1-trans, and M1-cis, respectively), suggesting the metabolites were formation-rate limited. While these analyses are valuable in characterizing the pharmacokinetics of rolofylline and metabolites independently, a complementary compartmental analysis provides a platform for explicitly capturing the relationship between the pharmacokinetics of rolofylline and metabolites. A model that links parent and metabolite pharmacokinetics (“simultaneous model”) potentially facilitates the estimation of the impact of new clinical scenarios (such as intrinsic and extrinsic factors, changes in dosing regimen, etc.) on both the relative and total levels of circulating active analytes (10). In addition, explicit provisions to link the disposition of parent to metabolite pharmacokinetics become important for formation-limited kinetics (11).
With every mechanistic provision included in the simultaneous model comes an increasing level of complexity, including the question of structural identifiability; structural identifiability pertains to the ability to uniquely estimate the parameters of a model given ideal, error-free data as an input. One can perform structural identifiability analysis (SIA) on a candidate model with a variety of techniques (12,13). The freely available DAISY software tool (http://www.dei.unipd.it/wdyn/?IDsezione=4364) that is used in the current evaluation utilizes a differential algebra approach (14). The robust algorithm as implemented in DAISY has been used for a growing set of SIA examples (15–17) including pharmacokinetic modeling (18). To maintain structural identifiability, simultaneous models usually contain some degree of simplification of the true underlying process and/or reparameterization. This is especially true in the common case where individual metabolites are not available to administer separately, and information regarding the disposition of the metabolites is gleaned following administration and conversion of the parent molecule (10,19). The additional complexity that results from treating parent and metabolite simultaneously may be a contributor to the relatively low frequency of this approach in the wider literature (18).
The aim of this investigation was to develop a pharmacokinetic model capable of describing the plasma concentration–time profile of rolofylline, M1-trans, and M1-cis simultaneously (simultaneous PK model) and to thereby demonstrate consistency of observed PK data with known features of the biotransformation pathway of rolofylline. The simultaneous PK model was based on single rising-dose data obtained in healthy volunteers following single IV infusions from 1 to 60 mg of rolofylline in Study KW-3902 IV-EU01.
MATERIALS AND METHODS
Clinical Protocol
Model development was based on data collected in a randomized, double-blind, dose-escalation design with single doses of IV rolofylline in four groups of healthy volunteers, with the exception of the first dose given to group 1 (1 mg rolofylline IV and placebo) and the second 2.5-g dose given to group 2, which were open-label (Study KW-3902 IV-EU01). In KW-3902 IV-EU01 single doses of 1, 2.5, 5, 10, 20, 30, 40, 50, and 60 mg rolofylline were given IV. The rate for volume administration was kept constant for all groups (1 ml/min). For doses less than or equal to 30 mg, the infusion time was 1 h; administration of 40 to 60 mg lasted 2 h. The actual dose regimen was as follows:
1 and 2.5 mg rolofylline or placebo over 60 min each.
2.5 mg, followed by 5 and 10 mg rolofylline or placebo over 60 min each.
20 and 30 mg rolofylline or placebo over 60 min each.
40, 50, and 60 mg rolofylline or placebo over 120 min each.
Thirty-seven healthy white male subjects with a mean age of 29 years (range = 18–42 years), a mean height of 175 cm (range = 161–191 cm), and a mean weight of 73.3 kg (range = 60.2–90.0 kg) were enrolled in this study; in total, 36 subjects received study medication in different periods. The number of subjects entered into the analysis by treatment is indicated in Table I.
Table I.
Number of Subjects Analyzed for Rolofylline Pharmacokinetics by Group Following Single-Dose IV Infusion Administration at the Indicated Dose Level
| Group | Rolofylline dose (mg) | Number of subjects analyzed |
|---|---|---|
| 1 | 1.0 | 5 |
| 2.5 | 4a | |
| 2 | 2.5 | 8 |
| 5.0 | 6 | |
| 10 | 6 | |
| 3 | 20 | 6 |
| 30 | 6 | |
| 4 | 40 | 5 |
| 50 | 6 | |
| 60 | 6 |
aOne subject received less than 1/10 of the 2.5-mg dose
The study was conducted in accordance with principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agencies, and all subjects provided written informed consent.
To determine the plasma concentrations of rolofylline and M1-cis and M1-trans metabolites following the administration of rolofylline, blood samples were drawn at 0 (before start of infusion), 0.5, 1.17, 1.5, 2, 3, 5, 9, 13, 25, 37, and 49 h after start of infusion on the profile day for groups 1, 2 and 3 and at 0 (before start of infusion), 1, 2.17, 2.5, 3, 4, 6, 10, 14, 26, 38, and 50 h after start of infusion on the profile day for group 4.
Assay Methods
Plasma samples were analyzed for rolofylline, M1-trans, and M1-cis concentrations at the contract laboratory Hoechst Marion Roussel (Frankfurt, Germany). The analytes were extracted from heparinized human plasma at pH 8 using diethyl ether. Urine samples were processed by dilution (20-fold) prior to analysis (since levels of the three analytes were below or just above the limit of quantitation at the highest dose group, the determination of the samples of the lower doses was canceled to avoid unnecessary bioanalytical activities). The analytes were then separated and detected by a high-performance liquid chromatograph tandem mass spectrometric system (HPLC–MS/MS). The HPLC was carried out isocratically on a C-18 column using 20 mM ammonium format at pH 3 and methanol. [2H7]-rolofylline was used as internal standard to rolofylline. The analog internal standards KF 16082 and KF 18267 were used as internal standard for the M1-trans and M1-cis metabolites. Analyte concentrations were determined from the peak area ratios of the analyte to its respective internal standard. The dynamic range for the assay of rolofylline and its metabolites in plasma was 0.5–500 ng/mL when 0.25 mL of plasma was processed. The assays were selective and specific for analytes in human biological fluids, and there was no significant interference observed from endogenous components in control human biological fluids. The limit of quantification (LOQ) for rolofylline, M1-cis and M1-trans in plasma was 0.5 ng/mL for all three analytes. Accuracy ranged from 92.9–103.7%, 86.8–106.5%, and 90.1–100.1% for rolofylline, M1-trans, and M1-cis respectively; precision ranged from 0.2 to 10.5%, 0.8–10.5%, and 4.6–10.8% for rolofylline, M1-trans, and M1-cis respectively.
Modeling Methods
The pharmacokinetics of rolofylline and both M1-trans and M1-cis metabolites were analyzed using NONMEM Version VI level 2.0 (GloboMax, Hanover, MD) using the general linear model (ADVAN 7) and first-order conditional estimation with interaction (20). The SIA was performed using the DAISY software tool Version 1.5 as described elsewhere (14,18). Concentration data below LOQ (223 of 1,914 post-dose observations or 65, 52, and 106 observations corresponding to rolofylline, M1-trans, and M1-cis, respectively) were treated as missing. The inter-individual variability (IIV) was captured using exponential random-effects terms. The residual variability was estimated using proportional or both proportional and additive error models independently for rolofylline, M1-trans, and M1-cis. Visual predictive checks (VPC) were generated for rolofylline and the M1-trans and M1-cis metabolites. Two sets of VPCs were generated. The first set of VPCs depicted the collection of subjects receiving a 1 h infusion (corresponding to dose levels up to and including 30 mg); observed plasma concentrations were dose-normalized to 10 mg for this depiction. The second set of VPCs were generated for patients receiving 2-h infusions (dose levels of 40 mg and higher), and observed concentrations were dose-normalized to the 50-mg dose level. The VPCs were generated using 1,000 simulations.
To provide an estimate of prediction error (PE), we determined the ratio of the areas under the curve (AUC0–∞) and maximum plasma concentrations (Cmax) based on individual predicted concentration profiles to those based upon observed plasma concentrations for rolofylline and metabolites. Noncompartmental analysis of individual predicted and observed data was conducted in WinNonlin Version 5.2.1 (Pharsight Corporation, Mountain View, CA). Since there was a relatively limited amount of data above the limit of quantitation following administration of the 1-mg rolofylline dose, calculation of the PE was limited to data following administration of doses 2.5 mg and higher.
Initial exploratory pharmacokinetic modeling proceeded in three sequential steps, with data from an additional analyte added for each round. The objective function value and diagnostic plots were used to guide model development. At the conclusion of a given round, a pharmacokinetic model based on the present set of analytes was selected and brought forward to the subsequent round. Round 1 included the rolofylline concentration data only. For round 2, both rolofylline and M1-trans concentration data were included; however, the pharmacokinetic parameters that influenced the concentration–time profile of rolofylline were held fixed to those of the model selected in round 1. In round 3, rolofylline, M1-trans, and M1-cis data were included; however, the pharmacokinetic parameters that influenced the pharmacokinetics of both rolofylline and M1-trans were held fixed to the best parameterization of round 2. Once the three initial rounds of model building were completed, the parameters of the model from round 3 were simultaneously estimated. This was considered the final model. Since the final model was dependent on the results of each sequential round of exploratory model building, the specific details regarding the parameterization for each round are included in the RESULTS AND DISCUSSION.
RESULTS AND DISCUSSION
Model development was driven by known aspects of the metabolism of rolofylline and metabolites. Accordingly, the simultaneous PK model was to contain the provisions as itemized the introduction for (1) conversion of rolofylline to both M1-trans and M1-cis metabolites and (2) conversion of M1-trans to M1-cis. Counterbalancing the aim of capturing the fate of rolofylline and metabolites were considerations of structural identifiability and the need to obtain convergence during opimization; these considerations were driven minimally from the fact that the system had three outputs (i.e., rolofylline, M1-trans, and M1-cis concentration time histories) with one input (i.e., only rolofylline was dosed). To address these considerations, from the “MATERIALS AND METHODS”, selection of the final model proceeded through three rounds of model building, each of which considered an additional analyte in the optimization. As described in more detail below, in moving from the second round (i.e., the round in which rolofylline and M1-trans only were considered) to the third round (i.e., the round in which all analytes were considered), fractionation of rolofylline to M1-trans versus M1-cis was expressed in terms of the apparent fraction of rolofylline that was metabolized to M1-cis (FM); SIA was used to ensure that the final model could be characterized by a unique set of model parameters.
In the first round of optimization, only rolofylline pharmacokinetic data were included; from Fig. 2a, a two-compartment model with first-order elimination from the central compartment was the only structural model considered for the first round. Selection of this structural model was based upon inspection of rolofylline concentration profiles following single- and multiple-dose administration (8,9), and the presence of a biphasic fall from levels following the end of infusion. For round 2, both rolofylline and M1-trans concentration data were included, however the pharmacokinetic parameters which influenced the concentration–time profile of rolofylline were held fixed to those of the model selected in round 1 (i.e., parameters V1, V2, CL1 and CL2 of Fig. 2b were held fixed to the values of the previous round). Both a one- and two-compartment model, each with first-order elimination, were considered for M1-trans for the structural model. The two-compartment model was selected for M1-trans based on improved model diagnostics, both from inspection of diagnostic plots and a drop of 355.694 in the objective function value relative to the one-compartment model. In round 3, rolofylline, M1-trans, and M1-cis data were considered; however, the model parameters that influenced the pharmacokinetics of both rolofylline and M1-trans were held fixed to the optimal parameterization of round 2. Figure 2 panels c and d illustrate the two structural models considered in round 3. Both variations included conversion of M1-trans to M1-cis metabolite and a single-compartment model for M1-cis with first-order elimination. The second of two structural models considered in round 3 also permitted conversion of rolofylline to M1-cis and was parameterized in terms FM as described previously. Relative to rolofylline and M1-trans, less data were available above the limit of quantitation for M1-cis to inform a more complicated model than a single-compartment model; we did not explore anything beyond a one-compartment model for M1-cis. Attempts to fit the first of two candidate models to M1-cis data (i.e., the model of Fig. 2c with V1, V2, V3, V4, CL1, CL2, CL3, and CL4 held fixed to values of the previous round) did not result in a successful covariance step. In contrast, attempts with the second of two candidate models (i.e., the model of Fig. 2d again with V1, V2, V3, V4, CL1, CL2, CL3, and CL4 held fixed to values of the previous round) converged with both a successful covariance step and a favorable change of 172.314 in the objective function relative to the model of Fig. 2c.
Fig. 2.
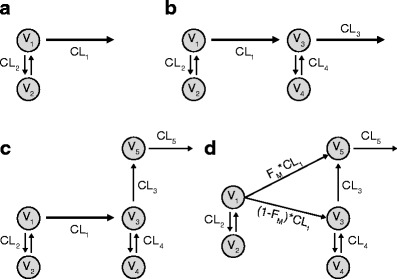
The pharmacokinetic models considered during three rounds of exploratory pharmacokinetic modeling of rolofylline and metabolite data. Compartments 1, 3, and 5 represent the first compartments for rolofylline, M1-trans, and M1-cis, respectively. a In round 1, only rolofylline data were considered. b The model from round 1 was brought forward to round 2; the pharmacokinetic parameters which influenced the concentration–time profile of rolofylline were held fixed to those from round 1, and parameters which influenced the pharmacokinetics of M1-trans were optimized. c and d For round 3, the model selected from round 2 was brought forward. The model parameters which influenced the pharmacokinetics of both rolofylline and M1-trans were held fixed to the optimal parameterization of round 2, and parameters which influenced the pharmacokinetics of M1-cis were optimized. Two structural models were considered for M1-cis; d the second of the two models had a provision for conversion of rolofylline to M1-cis, while c the first did not. The depiction of panel d is the final model brought forward; a definition of the model terms in panel d (i.e., V, CL, and FM) is provided in Table I
The model of Fig. 2d was selected for one final round of fitting, where all model parameters were re-estimated. The SIA analysis in DAISY confirmed that the model of Fig. 2d was globally identifiable (14,18). The parameter estimates following this final optimization are in Table II. The fixed effects were generally estimated with reasonable precision (RSE = 100 × SE/ Estimate <35%). The typical individual estimates for rolofylline CL and Vss were 24.4 L/h and 239 L, respectively. Random effects were not estimable for the distributional clearance for both rolofylline and M1-trans and the volume term associated with M1-cis, and were accordingly fixed to zero.
Table II.
Final Parameter Estimates for Simultaneous Pharmacokinetic Model
| Variable | Description | Estimate (RSE) | Between-subject variability estimate (RSE) |
|---|---|---|---|
| V1 | Volume central compartment, parent (L) | 37.8 (3.15) | 12.3 (29.8) |
| CL1 | Parent to metabolite clearance (L/h) | 24.4 (4.39) | 21.4 (22.7) |
| V2 | Volume peripheral compartment, parent (L) | 201 (5.08) | 22.0 (51.0) |
| CL2 | Distributional clearance, parent (L/h) | 13.2 (3.47) | – |
| V3 | Volume central compartment, M1-trans (L) | 26.1 (9.16) | 53.9 (24.1) |
| CL3 | Interconversion clearance (L/h) | 19.6 (6.89) | 42.3 (21.1) |
| V4 | Volume peripheral compartment, M1-trans (L) | 41.7 (7.29) | 32.9 (30.7) |
| CL4 | Distributional clearance, M1-trans (L/h) | 28.4 (11.7) | – |
| V5 | Volume central compartment, M1-cis (L) | 3.78 (34.13) | – |
| CL5 | Clearance M1-cis (L/h) | 91.6 (6.77) | 39.1 (27.8) |
| FM | Fraction parent metabolized to M1-cis | 0.194 (11.8) | 41.1 (33.4) |
| Proportional residual error term, parent | 26.1 ( 10.6) | ||
| Additive residual error term, parent | – | ||
| Proportional residual error term, M1-trans | 17.4 (17.3) | ||
| Additive residual error term, M1-trans | 0.217 (177) | ||
| Proportional residual error term, M1-cis | 15.0 (20.4) | ||
| Additive residual error term, M1-cis | 0.614 (39.0) |
Coefficient of variation expressed as a percentage
RSE relative standard error, expressed as a percentage
Figure 3 depicts model diagnostic plots for rolofylline, M1-trans, and M1-cis concentrations for the simultaneous PK model (additional representations of both mean of observed and population mean predicted plasma concentrations as functions of time and by analyte and dose are provided in Supplemental Figure S-1). By inspection, the simultaneous PK model provides a reasonable fit, although there is some tendency for underprediction of relatively high plasma concentrations. The VPC results are depicted in Fig. 4 for subjects receiving both the 1- and 2-h rolofylline infusions. From inspection of the VPC representations of Fig. 4, observed plasma concentrations of rolofylline, M1-trans, and M1-cis were generally within the 10% to 90% intervals; however, there was a relatively increased frequency of peak M1-trans levels (i.e., prior to ∼5 h post-dose) outside of the upper 90% interval. Conversely, the upper 90% interval for M1-cis appears somewhat elevated relative to observed peak levels, suggesting some tendency for the model to over predict variability for M1-cis. We next calculated the PE based on AUC0–∞ and Cmax estimates from noncompartmental analysis of both individual predicted and observed concentrations as a complementary diagnostic to the VPC, and calculated the intra-individual ratio of AUC0-∞ and Cmax based upon predicted and observed data for each individual. The geometric mean (coefficient of variation, CV%) of the intra-individual ratios (AUC0–∞ based on individual predicted/AUC0–∞ based on observed) were 1.01 (15.9%), 1.03 (13.0%), and 1.07 (16.7%) for rolofylline, M1-trans, and M1-cis, respectively. The geometric mean (CV%) of the intra-individual ratios (Cmax based on individual predicted/Cmax based upon observed) were 0.92 (16.4%), 0.98 (12.5%), and 0.97 (10.0%) for rolofylline, M1-trans, and M1-cis, respectively. Additionally, the apparent terminal half-life estimated from noncompartmental analysis of population predicted profiles was approximately 17 h for all analytes, which is consistent with formation-rate limited kinetics for the M1-trans and M1-cis metabolites.
Fig. 3.
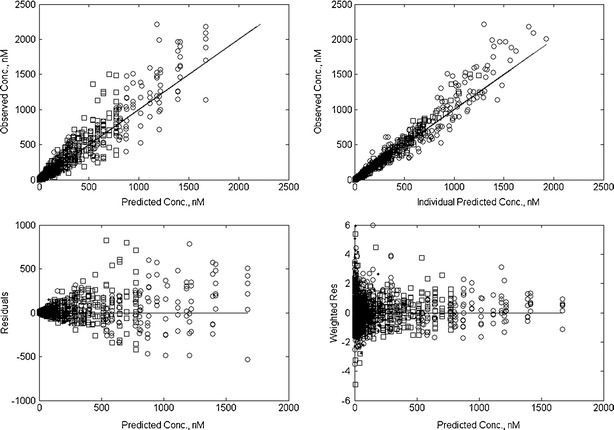
Diagnostic plots following fit of the simultaneous PK model to observed rolofylline (circles), M1-trans (squares), and M1-cis (dots) plasma concentrations. Progressing clockwise from the top-left corner, the model diagnostic plots are comprised of a observed versus population predicted concentrations, b observed versus individual predicted concentrations, c residual and d weighted residual plots
Fig. 4.
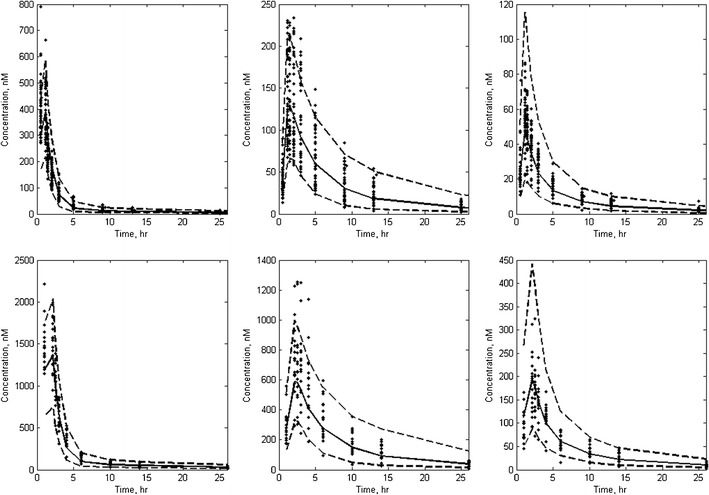
Visual predictive check for rolofylline (left-most panels), M1-trans (center panels) and M1-cis (right-most panels). Concentration profiles corresponding to 1 and 2 h infusion times were dose-normalized to the 10- and 50-mg dose levels, respectively, and depicted on the top and bottom panels, respectively. Observed data are indicated as points; the solid and dashed lines represent median and both 10% and 90% intervals of the model-predicted concentrations. For clarity, only data up to 25 h post-start of infusion are depicted in this representation
Two caveats are important in interpretation of parameter estimates. First, although several known aspects of the biotransformation pathway for rolofylline and metabolites are captured by the simultaneous model, additional routes of elimination are active for rolofylline and metabolites in vivo (e.g. hydroxylation of M1-trans metabolite) that are not explicitly captured in the simultaneous model. Addition of additional elimination routes for either rolofylline or M1-trans in addition to those included in the final model (i.e., first-order elimination to the environment for rolofylline and M1-trans) resulted in nonidentifiable systems in both instances; similarly including an oxidative pathway for elimination of M1-trans instead of interconversion resulted in an a system that was not identifiable. This impacts the physiological interpretability of parameter estimates associated with the simultaneous model (10,21). Second, the treatment of data below the LOQ as missing may have adversely affected parameter bias and precision and model selection (22,23); with this caveat, selection of a more sophisticated method for treatment of below LOQ data would result in additional model and computational complexity with potentially limited practical improvement in parameter estimates (24).
As mentioned in the “INTRODUCTION”, results from both in vitro binding experiments and preclinical in vivo experiments suggested that the pharmacological activity of rolofylline administration would be attributable to the combined action of rolofylline, M1-trans, and M1-cis. The overall contribution of rolofylline and metabolites to the observed pharmacodynamic effect is expected to be a function of the exposures and potencies of these analytes (25). A historical evaluation of the contribution of an active metabolite to the overall pharmacological activity of tesofensine was captured via a competitive interaction PK/PD model that included provisions for the relative exposures of parent and metabolite to the overall activity observed in mice (26) and again in the clinic (27). Although model diagnostics pointed to challenges in capturing peak levels of rolofylline and metabolites, collectively the model diagnostics were generally favorable for use of the simultaneous model in estimation of overall exposures. The simultaneous model provided both a framework for understanding the disposition of rolofylline and metabolites and a potential bridge to understanding the impact of new clinical scenarios on relative exposures and the time course of the pharmacodynamic effect.
CONCLUSION
The proposed simultaneous pharmacokinetic model adequately describes the concentration–time profile of rolofylline and both M1-trans and M1-cis metabolites following single rising doses in healthy volunteers. The final model captures features of the biotransformation pathway of rolofylline, including the conversion of rolofylline to M1-trans and M1-cis metabolites, as well as conversion of M1-trans to M1-cis. Since the simultaneous model is capable of providing insight regarding the relative exposures of rolofylline and active metabolites, it acts as a key component in understanding the impact of new clinical scenarios upon overall activity.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
(PPTX 81.4 kb)
Acknowledgments
We wish to acknowledge Dr. Maria Pia Saccomani, University of Padova, for helpful discussion on regarding SIA and use of the DAISY software to accomplish this analysis.
References
- 1.Slawsky MT, Givertz MM. Rolofylline: a selective adenosine 1 receptor antagonist for the treatment of heart failure. Expert Opin Pharmacother. 2009;10(2):311–322. doi: 10.1517/14656560802682213. [DOI] [PubMed] [Google Scholar]
- 2.Briggs JP, Schnermann J. The tubuloglomerular feedback mechanism—functional and biochemical aspects. Annu Rev Physiol. 1987;49:251–273. doi: 10.1146/annurev.ph.49.030187.001343. [DOI] [PubMed] [Google Scholar]
- 3.Welch WJ. Adenosine A(1) receptor antagonists in the kidney: effects in fluid-retaining disorders. Curr Opin Pharmacol. 2002;2:165–170. doi: 10.1016/S1471-4892(02)00134-0. [DOI] [PubMed] [Google Scholar]
- 4.Cotter G, Dittrich HC, Weatherley BD, Bloomfield DM, O’Connor CM, Metra M, et al. The PROTECT pilot study: a randomized, placebo-controlled, dose-finding study of the adenosine A(1) receptor antagonist rolofylline in patients with acute heart failure and renal impairment. J Card Fail. 2008;14:631–640. doi: 10.1016/j.cardfail.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Dittrich HC, Gupta DK, Hack TC, Dowling T, Callahan J, Thomson S. The effect of KW-3902, an adenosine A(1) receptor antagonist, on renal function and renal plasma flow in ambulatory patients with heart failure and renal impairment. J Card Fail. 2007;13:609–617. doi: 10.1016/j.cardfail.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 6.Givertz MM, Massie BM, Fields TK, Pearson LL, Dittrich HC. The effects of KW-3902, an adenosine A1-receptor antagonist, on diuresis and renal function in patients with acute decompensated heart failure and renal impairment or diuretic resistance. J Am Coll Cardiol. 2007;50:1551–1560. doi: 10.1016/j.jacc.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 7.Weatherley BD, Cotter G, Dittrich HC, Delucca P, Mansoor GA, Bloomfield DM, et al. Design and rationale of the PROTECT study: a placebo-controlled randomized study of the selective A1 adenosine receptor antagonist rolofylline for patients hospitalized with acute decompensated heart failure and volume overload to assess treatment effect on congestion and renal function. J Card Fail. 2010;16:25–35. doi: 10.1016/j.cardfail.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 8.Stroh M, Dishy V, Radziszewski W, Hwang E, Lazarus-Shipitofsky N, Dittrich H, et al. The effects of multiple doses of rolofylline on the single-dose pharmacokinetics of midazolam in healthy subjects. Am J Ther. 2010;17:53–60. doi: 10.1097/MJT.0b013e3181c12313. [DOI] [PubMed] [Google Scholar]
- 9.Radziszewski W, Lai E, Shipitofsky NL, Stroh M, Dishy V, Han LL, et al. A single supratherapeutic dose of rolofylline does not prolong the QTcF interval in healthy volunteers. Am J Ther. 2010;17:8–16. doi: 10.1097/MJT.0b013e3181c3cbdb. [DOI] [PubMed] [Google Scholar]
- 10.Duffull SB, Chabaud S, Nony P, Laveille C, Girard P, Aarons L. A pharmacokinetic simulation model for ivabradine in healthy volunteers. Eur J Pharm Sci. 2000;10:285–294. doi: 10.1016/S0928-0987(00)00086-5. [DOI] [PubMed] [Google Scholar]
- 11.Klein CE, Gupta E, Reid JM, Atherton PJ, Sloan JA, Pitot HC, et al. Population pharmacokinetic model for irinotecan and two of its metabolites, SN-38 and SN-38 glucuronide. Clin Pharmacol Ther. 2002;72:638–647. doi: 10.1067/mcp.2002.129502. [DOI] [PubMed] [Google Scholar]
- 12.Chis OT, Banga JR, Balsa-Canto E. Structural identifiability of systems biology models: a critical comparison of methods. PLoS One. 2011;6:e27755. doi: 10.1371/journal.pone.0027755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yates JW, Jones RD, Walker M, Cheung SY. Structural identifiability and indistinguishability of compartmental models. Expert Opin Drug Metab Toxicol. 2009;5:295–302. doi: 10.1517/17425250902773426. [DOI] [PubMed] [Google Scholar]
- 14.Bellu G, Saccomani MP, Audoly S, D’Angio L. DAISY: a new software tool to test global identifiability of biological and physiological systems. Comput Methods Programs Biomed. 2007;88:52–61. doi: 10.1016/j.cmpb.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roper RT, Pia SM, Vicini P. Cellular signaling identifiability analysis: a case study. J Theor Biol. 2010;264:528–537. doi: 10.1016/j.jtbi.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 16.Saccomani MP, Audoly S, Bellu G, D’Angio L. Examples of testing global identifiability of biological and biomedical models with the DAISY software. Comput Biol Med. 2010;40:402–407. doi: 10.1016/j.compbiomed.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saccomani MP. An effective automatic procedure for testing parameter identifiability of HIV/AIDS models. Bull Math Biol. 2011;73:1734–1753. doi: 10.1007/s11538-010-9588-2. [DOI] [PubMed] [Google Scholar]
- 18.Ogungbenro K, Aarons L. Structural identifiability analysis of pharmacokinetic models using DAISY: semi-mechanistic gastric emptying models for 13C-octanoic acid. J Pharmacokinet Pharmacodyn. 2011;38:279–292. doi: 10.1007/s10928-011-9193-5. [DOI] [PubMed] [Google Scholar]
- 19.Bertrand J, Laffont CM, Mentre F, Chenel M, Comets E. Development of a complex parent-metabolite joint population pharmacokinetic model. AAPS J. 2011;13:390–404. doi: 10.1208/s12248-011-9282-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boeckmann AJ, Sheiner LB, Beal SL. NONMEM Users Guide-Part V, Introductory Guide. University of California San Francisco: NONMEM Project Group; 1994.
- 21.Evans ND, Godfrey KR, Chapman MJ, Chappell MJ, Aarons L, Duffull SB. An identifiability analysis of a parent-metabolite pharmacokinetic model for ivabradine. J Pharmacokinet Pharmacodyn. 2001;28:93–105. doi: 10.1023/A:1011521819898. [DOI] [PubMed] [Google Scholar]
- 22.Duval V, Karlsson MO. Impact of omission or replacement of data below the limit of quantification on parameter estimates in a two-compartment model. Pharm Res. 2002;19:1835–1840. doi: 10.1023/A:1021441407898. [DOI] [PubMed] [Google Scholar]
- 23.Byon W, Fletcher CV, Brundage RC. Impact of censoring data below an arbitrary quantification limit on structural model misspecification. J Pharmacokinet Pharmacodyn. 2008;35:101–116. doi: 10.1007/s10928-007-9078-9. [DOI] [PubMed] [Google Scholar]
- 24.Xu XS, Dunne A, Kimko H, Nandy P, Vermeulen A. Impact of low percentage of data below the quantification limit on parameter estimates of pharmacokinetic models. J Pharmacokinet Pharmacodyn. 2011;38:423–432. doi: 10.1007/s10928-011-9201-9. [DOI] [PubMed] [Google Scholar]
- 25.Holford NHG, Sheiner LB. Kinetics of pharmacologic response. Pharmacol Ther. 1982;16:143–166. doi: 10.1016/0163-7258(82)90051-1. [DOI] [PubMed] [Google Scholar]
- 26.Lehr T, Staab A, Tillmann C, Nielsen EO, Trommeshauser D, Schaefer HG, et al. Contribution of the active metabolite M1 to the pharmacological activity of tesofensine in vivo: a pharmacokinetic–pharmacodynamic modelling approach. Br J Pharmacol. 2008;153:164–174. doi: 10.1038/sj.bjp.0707539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehr T, Staab A, Trommeshauser D, Schaefer HG, Kloft C. Quantitative pharmacology approach in Alzheimer’s Disease: efficacy modeling of early clinical data to predict clinical outcome of tesofensine. AAPS J. 2010;12:117–129. doi: 10.1208/s12248-009-9164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PPTX 81.4 kb)