

Abstract
Hepatitis C virus (HCV) infection leading to chronic hepatitis is a major factor in the causation of liver cirrhosis, hepatocellular carcinoma, and liver failure. This process may involve the interplay of various host cell factors, as well as the interaction of these factors with viral RNA and proteins. We report a novel strategy using a sequence-specific biotinylated peptide nucleic acid (PNA)-neamine conjugate targeted to HCV RNA for the in situ capture of subgenomic HCV (+) RNA, along with cellular and viral factors associated with it in MH14 host cells. Using this affinity capture system in conjunction with LC/MS/MS, we have identified 83 cellular factors and three viral proteins (NS5B, NS5A, and NS3–4a protease-helicase) associated with the viral genome. The capture was highly specific. These proteins were not scored with cured MH14 cells devoid of HCV replicons because of the absence of the target sequence in cells for the PNA-neamine probe and also because, unlike oligomeric DNA, cellular proteins have no affinity for PNA. The identified cellular factors belong to different functional groups, including signaling, oncogenic, chaperonin, transcriptional regulators, and RNA helicases as well as DEAD box proteins, ribosomal proteins, translational regulators/factors, and metabolic enzymes, that represent a diverse set of cellular factors associated with the HCV RNA genome. Small interfering RNA-mediated silencing of a diverse class of selected proteins in an HCV replicon cell line either enhanced or inhibited HCV replication/translation, suggesting that these cellular factors have regulatory roles in HCV replication.
The hepatitis C virus, a blood-borne pathogen that causes chronic hepatitis, is the primary reason for liver transplantation in the United States. HCV1 preferentially replicates in liver tissue without any direct cytopathic effect and thus is able to maintain long term, persistent infection. More than 50% of HCV-infected patients do not respond to treatment; instead, the majority of patients develop chronic hepatitis C, which leads to progressive liver fibrosis, cirrhosis, end-stage liver disease, and hepatocellular carcinoma.
The hepatitis C virus is a positive single-stranded RNA virus of a 9.6-kb genome. After its entry into cells, the (+) strand RNA first serves as a messenger RNA for the translation of viral proteins. Newly synthesized HCV replicase (NS5B) then copies the (+) strand RNA genome into the (−) strand RNA, which serves as a template for the production of the viral genome. The conserved 5′- and 3′-nontranslated (5′NTR and 3′NTR) regions of the HCV genome have multiple regulatory elements that are essential for replication of HCV and translation of viral proteins. Although the 5′NTR of HCV contains the internal ribosomal entry site, which is required for cap-independent translation of (+) strand HCV RNA (1–4), it is also the 3′ region of the (−) strand RNA, which functions as the initiation site for replication of the (+) strand HCV RNA genome. The 3′ regions of both (−) and (+) strand HCV RNAs are highly structured and serve as the initiation sites for viral replication (5). Various cellular proteins have been shown to interact with 5′NTR of HCV RNA; these include La autoantigen (6) nuclear factors NF90, NF110, NF45, and RNA helicase A (7), as well as the polypyrimidine tract-binding protein (8–10). Recently, we affinity-captured different cellular proteins interacting with HCV 3′NTR and identified them by LC/MS/MS; some of these proteins were found to be essential for HCV replication as confirmed by siRNA (11).
Another recent study using sequence-specific gene silencing of the RNAi screen has identified 26 human genes encoding proteins that physically interact with HCV RNA or protein and modulate HCV replication (12). A more direct approach would be to capture the replicating HCV RNA genome in situ under physiological conditions and then identify all the cellular and viral factors associated with the viral genome. The structured HCV genome and the interplay of tightly regulated viral and host factors assembled on it should be highly specific within the cells. We present a novel strategy to affinity-capture the replicating HCV RNA and associated cellular and viral proteins in MH14 cells carrying actively replicating HCV replicons. We have identified these proteins by proteomics technology.
EXPERIMENTAL PROCEDURES
MH14 Cells
Cured MH14 and MH14 cells (a kind gifts from Makoto Hijikata, Japan) carrying replicative HCV subgenomic replicons were grown in DMEM (Cellgro) supplemented with 10% fetal calf serum, 100 μg/ml each of penicillin/streptomycin, and 300 μg/ml G418 (13, 14). Cured MH14 cells were prepared by treating MH14 cells with 5,000 IU/ml of α-interferon for 2 weeks. The absence of replicon RNA and viral proteins was checked by Northern blotting, RT-PCR, and Western blotting (14). Cells were grown at 37°C with 5% CO2.
Peptide Nucleic Acid (PNA)
We conjugated a 15-mer PNA targeted to the HCV genome with neamine at the N terminus as described previously (15). The PNA-neamine conjugate contained biotin at the C terminus via the Lys residue (Fig. 1). We obtained the PNA sequence (neamine-TACTCGTGCTTAGGA-Lys-biotin), which is complementary to the N-terminal HCV core coding region downstream of the 5′NTR in the MH14 HCV subgenomic replicon (Fig. 1C), on solid support from Panagene (South Korea) and conjugated it with neamine monomer essentially as described previously (15).
Fig. 1.
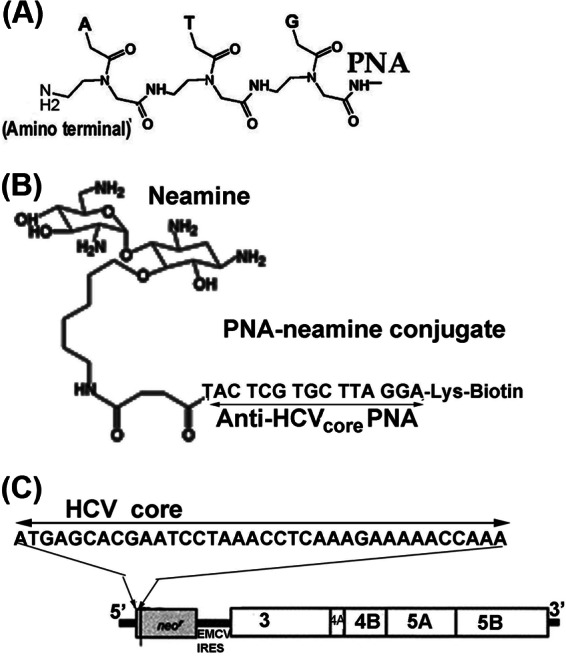
A, structure of PNA. The bases (A, T, G, and C) are linked with polyamide backbone against a sugar phosphate backbone in DNA. B, neamine-PNA-biotin conjugate. PNA targeted to the HCV core coding region is conjugated with neamine at the N terminus and biotin at the C terminus. C, schematic representation of the MH14 HCV subgenomic replicon. The N-terminal HCV core sequence is shown downstream of 5′NTR within the reading frame of neomycin-resistant marker gene.
Preparation of 32P-Labeled RNA Fragment Corresponding to 5′NTR
The HCV 5′NTR flanking the 3′ N-terminal HCV core coding region was amplified by PCR from the pMH14 template using up-primer containing the T-7 promoter (CGG GAG AGC CAT AGT GG) and down-primer complementary to the HCV core coding region (GGT TTT TCT TTG AGG TTT AGG). The PCR product corresponding to domains III and IV of 5′NTR and the 36 nucleotides of the N-terminal coding sequence of the HCV core were transcribed to generate 244-base runoff transcripts, using the T7 transcription kit from Roche Applied Sciences. The RNA transcript was internally labeled by including [α-32P]UTP (3,000 Ci/mmol; Amersham Biosciences) in the reaction solution. Reactions were carried out according to the manufacturer's protocols. The transcripts were purified by phenol/chloroform extraction and ethanol precipitation, dissolved in diethyl pyrocarbonate-treated water, and stored at −80°C. Following treatment with RNase-free DNase I to remove template DNA, the RNA was precipitated with lithium chloride, resuspended in RNase-free water, and used to determine the binding specificity of Nea-PNA-biotin conjugate.
Gel Retardation Assay
The affinity and specificity of the anti-HCV Nea-PNA-biotin conjugate for its target sequence were evaluated by gel electrophoretic mobility shift analysis. The 32P-labeled HCV-5′NTR RNA (20 nm; 10,000 Cerenkov cpm) was incubated with increasing concentrations of neamine-PNA-biotin conjugate in a buffer containing 30 mm Tris-HCl (pH 8.0), 75 mm KCl, 5.5 mm MgCl2, 5 mm DTT, 0.01% Nonidet P-40, and 500 ng of poly r(I-C) in a final volume of 15 μl. After 30 min of incubation at 37°C, samples were subjected to gel electrophoresis on 6% polyacrylamide gel, using the Tris borate buffer system. The RNA-PNA complex was resolved at a constant voltage of 150 V at room temperature for 3 h and subjected to PhosphorImager analysis (GE Healthcare).
Determination of Nonspecific Binding of Cellular Proteins to Biotinylated PNA-neamine Probe Targeted to HCV Genome
Cell extract from cured MH14 cells prepared as described above was used to determine the nonspecific binding of cellular proteins to the PNA probe specific to the HCV RNA genome. For binding experiments, 100 pmol of biotinylated PNA-neamine probe was incubated with 20 μl of cell extracts in binding buffer containing 1× protease inhibitor mixture (mini-EDTA-free, Roche Applied Science), 1 mm DTT, 100 mm NaCl, 20 mm HEPES (pH 7.5), and 20 units/ml SUPERaseIN. The mixture was incubated on ice for 60 min, after which 75 μl of streptavidin-coated paramagnetic bead suspension (Dynal, Invitrogen) was added to the mixture to capture the biotinylated PNA probe. The mixture was further incubated on ice for 30 min with occasional vortexing. Beads were then washed three times with the binding buffer. Elution was done by adding 30 μl of binding buffer and 30 μl of 2× SDS gel loading dye to washed beads and heating at 95°C for 5 min. Following magnetic separation of beads, the supernatant-containing eluted proteins were subjected to SDS-PAGE on 8–16% polyacrylamide. The gel was stained with Sypro Ruby dye (Molecular Probes, Invitrogen) for visualization of protein bands. We also included 10 μm biotin as well as a biotinylated DNA oligonucleotide with sequence identical to the PNA probe as controls.
Cellular Uptake and Localization of Nea-PNA Conjugate
The MH14 cells carrying stably replicating HCV subgenomic replicons were grown to 80% confluence in Dulbecco's modified Eagle medium containing 10% FCS. The cells were washed with PBS containing 2% FCS and incubated at 37°C with 2 μm FITC-tagged Nea-PNA conjugate or naked PNA. After 3 h of incubation, the cells were washed, detached, and resuspended in the same buffer. Fluorescent signals per 10,000 cells were then obtained by FACScan. To determine cellular localization of the FITC-labeled PNA-neamine conjugate, the cells were washed with PBS and stained with DAPI and wheat germ agglutinin conjugated with rhodamine to label, respectively, the nuclear DNA (blue) and membrane glycoproteins (red). Uptake of FITC-labeled PNA-neamine shows green fluorescence at 488 nm. The images were acquired on Nikon A1R confocal microscope.
Affinity Capture of HCV (+) Strand RNA-Protein Complex
We gently washed the subconfluent MH14 cells with cold buffer containing 150 mm sucrose, 30 mm HEPES (pH 7.4), 33 mm NH4Cl, 7 mm KCl, and 4.5 mm magnesium acetate. We layered the washed cells with lysolecithin (200 μg/ml) in the wash buffer for 5 min and then aspirated all the solution from the plates as described earlier (16, 17). We then layered the cells with reticulocyte buffer containing 1.6 mm Tris acetate (pH 7.8), 80 mm KCl, 2 mm magnesium acetate, 0.25 mm ATP, 0.1 mm dithiothreitol, and 10 units of RNasin containing 0.5 μm of anti-HCV PNA-neamine-biotin conjugate designed to capture (+) strand HCV RNA-protein complex. After incubation at room temperature for 2 h, the cells were washed once with the same buffer, gently scraped from each plate, and lysed on ice. We centrifuged the lysed cells for 10 min at low speed (7,000 × g). The supernatant (S7 fraction) was incubated on ice with 150 μl of paramagnetic streptavidin beads for 1 h to capture the HCV RNA-protein complex bound to Nea-PNA-biotin conjugate. We washed the beads six times with the reticulocyte buffer containing 500 mm NaCl. The captured (+) strand HCV RNA-protein complex was then eluted from the beads by adding 30 μl of binding buffer and 30 μl of 2× SDS gel loading dye to the washed beads and heating at 95°C for 5 min before magnetic separation of beads from eluted proteins. Aliquots of the samples were subjected to SDS-PAGE on an 8–16% polyacrylamide gel and stained with Sypro Ruby dye (Molecular Probes) as described (11).
Mass Spectrometry, Protein Identification, and Database Search
The RNA-protein complex eluted from the beads was entrapped in polyacrylamide gel and washed three times with 50% methanol containing 10% glacial acetic acid, two times with water, and two times with 50 mm ammonium bicarbonate in 30% acetonitrile. Reduction was done by incubating gel pieces for 30 min at 37°C in solution containing 10 mm DTT, 50 mm ammonium bicarbonate, and 30% acetonitrile. Subsequently, alkylation was done by incubating gel pieces for 30 min at 37°C in solution containing 45 mm iodoacetamide, 50 mm ammonium bicarbonate, and 30% acetonitrile. Gel pieces were then dehydrated by washing two times with 80% acetonitrile and drying at 60°C for 10 min. The dried gel pieces were subjected to trypsin digestion by adding solution containing 50 mm ammonium bicarbonate and 10 ng/ml trypsin (Trypsin Promega Gold MS grade). After overnight incubation at 37°C for digestion, reactions were adjusted to 1% trifluoroacetic acid for extraction of peptides from the gel. The extracted peptides were dried in a Speedvac and resuspended in 10 μl of solvent A (2% acetonitrile, 0.1% formic acid) for LC/MS/MS analysis. In brief, the peptides were first separated by reversed phase liquid chromatography, capillary PepMap100 column (75 μm × 150 mm, 3 μm, 100 Å, C18) (Dionex, Sunnyvale, CA) in a 60-min linear gradient from 10% solvent A to 40% solvent B (95% ACN, 0.1% formic acid). The reversed phase liquid chromatography eluant was directly introduced into a nano-ESI source on an API-US QTOF tandem MS system (Waters). The ESI capillary voltage was set at 3,000 V. The MS spectra (m/z 400–1900) were acquired in the positive ion mode. Argon was used as the collision gas. The collision energy was set within a range between 17 and 55 V, depending on the charge states, and the m/z values of the ions were analyzed. MS/MS spectra were acquired in data-dependent mode, in which the top five most abundant precursors with two to five charges from each MS survey scan were selected for fragmentation. ProteinLynx Global server (PLGS) program version 2.1 was used to convert LC/MS/MS raw data into pkl files. These files were submitted for search by the MASCOT search engine (versions 2.3.0 and 1.9.0) against the NCBInr database (May, 2011, 14261927 entries) with taxonomy limited to human or hepatitis C virus (237,402 or 53,243 entries). The following MASCOT search parameters were used: peptide mass tolerance, 200 ppm; fragment mass tolerance, 0.6 Da; trypsin cleavage with a maximum of two missed cleavages; variable modifications (of S-carbamidomethyl on cysteine and oxidation on methionine). Peptide ion scores >- 40 indicating identity or extensive homology (p < 0.05) were considered significant. Protein identifications were accepted on the basis of at least two identified peptides. The false discovery rate was less than 1.0% at the peptide level and less than 1.0% at the protein level. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped.
siRNA Experiments
Sequence of siRNAs Used
The following sequences were used: DDX6 (sense 5′-CUA UUC CGA GCA ACA UUG Att-3′ and antisense 5′-UCA AUG UUG CUC GGA AUA Gtt-3′ (Sigma-Aldrich)); ADAR1 (sense 5′-GAC UAU CUC UUC AAU GUG Utt-3′ and antisense 5′-ACA CAU UGA AGA GAU AGU Ctt (Sigma-Aldrich)); PA2G4 (sense 5′-CUG AAU UUG AGG UAC AUG Att-3′ and antisense 5′-UCA UGU ACC UCA AAU UCA Gtt-3′ (Sigma-Aldrich)); HSP60 (sense 5′-CAC UGA AUG AUG AAU UAG Att-3′ and antisense 5′-UCA UGU ACC UCA AAU UCA Gtt-3′ (Sigma-Aldrich)); and IGF2BP1 (sense 5′-CAG UAU GUG GGU GCC AUU Att-3′ and antisense 5′-UAA UGG CAC CCA CAU ACU Gtt-3′ (Sigma-Aldrich)).
siRNA targeted to STAU1 had a pool of three different siRNA duplexes as follows: (sense CGA GUA AAG CCU AGA AUC Att and antisense UGA UUC UAG GCU UUA CUC Gtt; sense CUG AGC AAC UGG ACU AUC Utt and antisense AGA UAG UCC AGU UGC UCA Gtt; sense CUA CAC UAC AGG AUA UGA Utt and antisense AUC AUA UCC UGU AGU GUA Gtt (Santa Cruz Biotechnology)).
Primer Sequence Used for RT-PCR
HCV 5′NTR (forward 5′-CGG GAG AGC CAT AGT GG-3′ and reverse 5′-AGT ACC ACA AGG CCT TTC G-3′); actin mRNA (forward 5′-CAG GCA CCA GGG CGT GAT GG-3′ and reverse 5′-AGG CGT ACA GGG ATA GCA CA-3′); glyceraldehyde-3-phosphate dehydrogenase (GADPH) mRNA (forward 5′-CTC TGC TCC TCC TGT TCG AC-3′ and reverse 5′-ATG GGT GGA ATC ATA TTG GAA C-3′).
siRNA Transfection
The MH14 cells (2 × 105/well) carrying replicating HCV replicons were grown in a 6-well plate for 24 h and then transfected with 20 nm siRNAs targeted against STAU1, ADAR1, DDX6, PA2G4, and HSP60 and IGF2G according to the manufacturer's protocol, using siPORT amine as the transfection reagent (Ambion). The transfected cells were further grown for 72 h. One set of cells were washed, lysed, and analyzed for total protein (BCA protein assay; Pierce). An equal quantity of protein from each set was used for Western blot analysis. Another set of cells was processed for the isolation of total mRNA and subsequent RT-PCR analysis for HCV RNA and actin mRNA or GADPH mRNA.
RESULTS
Sequence Specificity of PNA-neamine Conjugate Targeted to Core Coding Region of HCV
We used a new class of DNA mimic, PNA, for in situ capture of the replicating HCV genome and associated proteins from MH14 cells. The chargeless PNA molecule has no sugar phosphate backbone in which purine and pyrimidine bases are linked via peptide bonds (Fig. 1A) (18). Instead, the oligomeric PNA irreversibly binds to the complementary RNA or DNA sequences with very high affinity (19). The cellular uptake of naked PNA is negligible. Earlier, we showed that PNA conjugated with neamine or glucosamine is efficiently taken up by the cells without endosomal entrapment (15, 20). We conjugated 15-mer biotinylated PNAs with neamine at the N terminus and with biotin at the C terminus via a Lys residue (Fig. 1B). The sequence of PNA (neamine-TAC TCG TGC TTA GGA-Lys-biotin) was complementary to the N-terminal coding region of HCV C-protein in the HCV subgenomic replicons stably replicating in MH14 cells (Fig. 1C).
We incubated 20 nm of the 32P-labeled RNA fragment corresponding to domains III and IV of 5′NTR and 36 nucleotides of the N-terminal coding sequence of HCV core with different concentrations of PNA-NeaHCV-Core in binding buffer at room temperature for 15 min. We then analyzed the bound PNA-RNA complex by gel retardation. As shown in Fig. 2A, the binding of PNA-NeaHCV-Core to its target sequence was highly specific, with a binding stoichiometry of 1:1. In the presence of 10 and 15 nm concentrations of PNA-NeaHCV-Core, the respective extents of gel retardation were 50 and 75% of the total 20 nm labeled RNA (Fig. 2A, lanes 2 and 3); at a ratio of 1:1 or higher, all the 32P-labeled RNA was in the form of the PNA-RNA complex (Fig. 2, lanes 4 and 5). The scramble PNA with similar base composition failed to bind the target sequence under similar conditions (Fig. 2B).
Fig. 2.
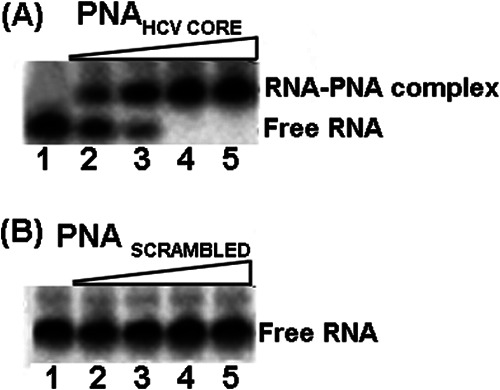
Specificity of PNA-neamine conjugate to its target sequence. A, 15-mer PNA-neamine conjugate complementary to the N-terminal HCV core of HCV RNA was incubated with 32P-labeled target RNA and analyzed by gel retardation assay. Lane 1, 3 nm of 32P-labeled 120-base-long RNA. Lanes 2–5 represent incubation of the labeled RNA with, respectively, 1.5, 2.0, 3, and 5 nm of PNA. B, 15-mer scrambled PNA-neamine conjugate incubated with 3 nm of labeled RNA. Lanes 1–5 represent the labeled RNA with, respectively, 0, 2, 5, 10, and 15 nm of scrambled PNA.
Cellular Proteins Do Not Bind to Sequence-specific PNA-Nea-HCV-Core Conjugate
We determined whether cellular proteins could recognize the base sequences on the PNA-NeaHCV-Core probe and bind to it nonspecifically. We incubated the biotinylated PNA-nea conjugate with cell lysate from cured MH14 cells devoid of HCV subgenomic replicons. As a control, we also incubated the cell lysate separately with biotin as well as with a biotinylated oligonucleotide DNA probe with nucleotide base sequence corresponding to the PNA-NeaHCV-Core probe. The biotin and biotinylated probes were then captured from the incubation mixture by paramagnetic streptavidin beads. After the beads were washed, cellular proteins associated with the HCV RNA bound with the biotinylated PNA-neamine probe were resolved on SDS-PAGE. The gel was stained with Sypro Ruby. As shown in Fig. 3, many cellular proteins were captured on the oligonucleotide DNA probe (lane 1). In contrast, fewer numbers of cellular proteins captured on the biotinylated PNA probe (Fig. 3, lane 2) were due to their affinity for biotin (lane 3). The LC/MS/MS analysis of these proteins identified many of them as biotin-binding metabolic enzymes (supplemental Table 1).
Fig. 3.
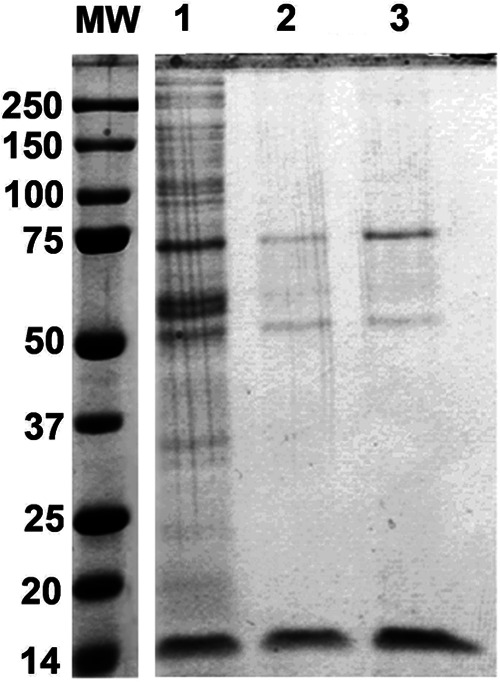
Binding affinity of cellular proteins to oligomeric PNA and DNA. The lysate from cured MH14 cells (HCV-negative) was incubated with 10 μm of biotinylated 15-mer PNA targeted to the HCV core coding region or biotinylated 15-mer oligo DNA containing an identical sequence. Biotin (10 μm) was included as control. After 30 min of incubation, the biotinylated PNA and DNA probes were captured on paramagnetic streptavidin beads and washed with buffer containing 0.5 m NaCl. Protein bands were visualized by staining the gel with Sypro Ruby. Lane 1, cellular proteins bound to oligomeric biotinylated DNA; lane 2, cellular proteins bound to oligomeric biotinylated PNA; lane 3, cellular protein bound to biotin.
PNA-neamine Conjugate Is Efficiently Taken Up by the Cells and Distributed in Both Cytosol and Nucleus
For uptake studies, we prepared fluorescein-tagged conjugate of a 15-mer PNA-neamine. The fluorescein probe was attached to the PNA moiety of the conjugate (15). The fluorescein-labeled conjugate was dissolved in water, and its concentration was determined by absorption of fluorescein at 490 nm (ε = 67,000) and absorption of PNA at 260 nm (ε = 171,200). Similar molar concentrations obtained by these two methods established their accuracy and indicated the absence of free fluorescein in the preparation. Using this fluorescein-tagged PNA-neamine conjugate, we did a series of experiments to determine the uptake efficiency of the conjugate using flow cytometry. The fluorescein-tagged naked (unconjugated) PNA was used as a control. The results, shown in Fig. 4, A and B, indicate that the PNA-neamine conjugate supplemented in the medium is efficiently taken up by Huh7 cells. At a 2 μm concentration of the conjugate, nearly 80% of cells were fluorescence-positive within 3 h of incubation. Uptake of fluorescein-tagged naked PNA at 5 μm concentration was negligible. We also determined the localization of the conjugate in the cells. As shown in Fig. 4C, the conjugate is uniformly distributed in the cytosol and also localized in the nucleus upon prolonged incubation.
Fig. 4.

Cellular uptake of PNA-neamine conjugates by Huh7 cells. MH14 cells grown in a monolayer were incubated at room temperature for 6 h with fluorescein-tagged 5 μm of naked PNA (A) or 2 μm of PNA-neamine (B) conjugate. The cells were then washed, detached, and resuspended at 2 × 106 cells/ml in PBS containing 2% FCS. Uptake was monitored by FACScan. Although PNA is neutral and has no charge, it is not taken up by the cell, although its neamine conjugate is efficiently internalized. C, localization of PNA-neamine conjugate in MH14 cells. Cells were incubated with 2 μm of fluorescein-tagged PNA-neamine conjugate. After 6 and 12 h of incubation, cells were washed and stained first with wheat germ agglutinin conjugated with rhodamine to label the membrane glycoproteins (red) to visualize the membrane boundary and then with DAPI to stain the nuclear DNA (blue). Uptake of PNA-neamine shows green fluorescence measured at 488 nm. Images were obtained Nikon A1R confocal microscope.
In Situ Capture of HCV (+) Strand RNA-Protein Complex
We used both MH14 cells (HCV-positive) carrying stably replicating HCV subgenomic replicons and cured MH14 cells (HCV-negative) in our experiments on affinity capture by PNA-NeaHCV-Core conjugate. The biotinylated PNA-neamine conjugate complementary to nucleotide sequence 342–356 of HCV (+) strand RNA was incubated with MH14 cells to capture the HCV RNA protein complex in situ. We also incubated the conjugate probe with cured MH14 cells that were devoid of HCV subgenomic replicons. The cells were washed and lysed. The biotin-PNA-neamine conjugate internalized in the cells was captured from the lysate by paramagnetic streptavidin beads. After washing the beads, the PNA-bound RNA-protein complex released in Laemmli gel loading buffer was either resolved by SDS-PAGE, followed by gel staining with Sypro ruby, or subjected to LC/MS/MS.
The PNA-NeaHCV-Core conjugate efficiently penetrated the cells and captured the HCV-RNA protein complex in situ from MH14 cells (Fig. 5). Proteins bands associated with the captured HCV (+) RNA genome from MH14 cells could be seen in the gel (Fig. 5, lane 3). Binding of the RNA-protein complex to the PNA probe was tight enough to withstand washing with 0.5 m salt, as only few protein bands could be seen in the washes (Fig. 5A, lanes 4 and 5). In contrast, the affinity capture lane from cured MH14 cells devoid of HCV replicons showed few protein bands in the gel (Fig. 5B, lane 3). LC/MS/MS analysis of these proteins identified 23 cellular factors associated with biotinylated PNA-streptavidin complex (supplemental Table 1). Among these, eight were biotin-binding metabolic enzymes, including pyruvate carboxylase, acetyl-CoA carboxylase, methylcrotonyl-CoA carboxylase, and propionyl-CoA carboxylase. These proteins were excluded from the list of cellular factors identified as being associated with HCV (+) strand RNA genome.
Fig. 5.

Affinity capture of HCV (+) RNA-protein complex from MH-14 cells (positive control) and cured MH14 cells (negative control). The biotinylated PNA-Nea-HCV-Core conjugate complementary to nucleotide sequence 342–356 of the HCV (+) strand RNA genome was incubated with MH14 cells (HCV-positive) (A) or cured MH14 cells (HCV-negative) (B). The conjugate that penetrated the cells and bound to its target sequence was captured from cell lysate on paramagnetic streptavidin beads. The beads were washed, suspended in SDS gel loading buffer, and heated at 90°C for 5 min. Following magnetic separation of beads, the eluate was resolved by SDS-PAGE and visualized by staining the gel with Sypro Ruby. Lane 1, cell lysate; lane 2, cell lysate supernatant flow-through following affinity capture; lane 3, affinity-captured proteins bound to biotinylated anti-HCV PNA-neamine conjugate; lanes 4 and 5, bead washes with 0.5 m NaCl in reticulocyte buffer.
Identification of Cellular or Viral Proteins Associated with HCV (+) Strand RNA Genome
For identification of specific cellular and viral proteins associated with the HCV RNA genome, the RNA-protein complex released in Laemmli gel loading buffer was processed for LC/MS/MS analysis to achieve the highest level of confidence in our identification. We used LC/MS/MS tandem mass spectrometric detection. The LC/MS/MS approach has three distinct advantages. It separates the tryptic peptides before mass spectrometric analysis, provides sequence information for fragmented peptides, and identifies proteins in protein mixtures. These proteins are listed in Table I with their accession numbers obtained from the protein database (NCBI). We identified three HCV proteins (NS3-4a protease-helicase, NS5A, and NS5B) and 83 cellular proteins in the affinity capture. Many of the cellular proteins belong to transcriptional regulators, as do the far upstream element-binding protein 1 (FBP1), hnRNP-U, hnRNP C-like 1, nuclear co-repressor KAP-1, the La ribonucleoprotein domain family, and the subunit 1 isoform of CCR4-NOT transcription complex. FBP1, a transactivator of c-myc gene transcription, has been shown to be expressed in hepatocellular carcinoma cells and enhance HCV replication (21).
Table I. Cellular and viral proteins associated with HCV (+) RNA genome.
Most of the host cell proteins listed had a minimum of two peptides matching. The exceptions were Apobec-1 complementation factor (gi|6996658), NICE-4 protein (gi|11990132), nuclear co-repressor KAP-1 (gi|1699027), RAS-p21 activating protein (gi|5031703), and chaperonin T-complex polypeptide (gi|5453603), each of which had a single peptide match (supplemental file 1).
| Protein descriptiona | NCBI accession no. | Protein score | Molecular mass | No. of unique peptides | Protein sequence coverage |
|---|---|---|---|---|---|
| Da | % | ||||
| HCV proteins | |||||
| 1 NS3-4a protease-helicase | gi|365813424 | 32 | 75,350 | 3 | 6.7 |
| 2 NS5A | gi|110225779 | 29 | 49,170 | 4 | 13.4 |
| 3 NS5B | gi|329750779 | 34 | 65,010 | 7 | 19.9 |
| Host cell proteins | |||||
| 1 ES/130 | gi|3299885 | 155 | 108,619 | 3 | 6 |
| 2 DDX-5 (growth-regulated nuclear 68 protein) | gi|226021 | 117 | 66,881 | 2 | 6.4 |
| 3 5′–3′- Exoribonuclease 2 | gi|18860916 | 187 | 108,513 | 2 | 6.1 |
| 4 60 S ribosomal protein L4 | gi|16579885 | 120 | 47,667 | 3 | 13.8 |
| 5 Adenosine deaminase acting on RNA 1-A (ADAR1) | gi|915284 | 81 | 135,910 | 2 | 2.7 |
| 6 A-kinase anchor protein 8 | gi|5031579 | 186 | 76,061 | 2 | 4.5 |
| 7 Apobec-1 complementation factor | gi|6996658 | 49 | 64,245 | 1 | 2.2 |
| 8 Arginyl-tRNA synthetase | gi|1217668 | 113 | 74,930 | 2 | 4.1 |
| 9 ATP synthase subunit α | gi|4757810 | 625 | 59,714 | 7 | 17.9 |
| 10 ATP synthase subunit β | gi|32189394 | 708 | 56,525 | 9 | 23.3 |
| 11 Autoantigen | gi|533202 | 164 | 131,776 | 2 | 3.7 |
| 12 C protein | gi|306875 | 245 | 31,947 | 3 | 16.9 |
| 13 C9orf10a | gi|8118021 | 72 | 116,627 | 3 | 4.6 |
| 14 Calnexin | gi|7709904 | 117 | 31,166 | 2 | 8.1 |
| 15 Caprin-1 isoform 1 | gi|42558250 | 189 | 78,318 | 3 | 10.7 |
| 16 CCR4-NOT transcription complex subunit 1 isoform a | gi|42716275 | 68 | 266,768 | 2 | 4.4 |
| 17 Cell cycle protein p38–2G4 homolog | gi|2697005 | 114 | 43,785 | 2 | 6.1 |
| 18 Chaperonin (HSP60) | gi|306890 | 347 | 60,986 | 5 | 17.1 |
| 19 Clathrin heavy chain 1 | gi|4758012 | 206 | 191,493 | 3 | 2.9 |
| 20 Cytochrome b-c1 complex subunit 2 (UBQRC2) | gi|50592988 | 245 | 48,413 | 3 | 13.7 |
| 21 Dead box, X isoform (DDX3) | gi|2580550 | 368 | 73,226 | 5 | 9.5 |
| 22 DEAH box polypeptide 30 (DDX-30) | gi|119585237 | 579 | 140,404 | 7 | 9.1 |
| 23 DDX6 (Rck/p54) | gi|458727 | 73 | 53,183 | 2 | 7.6 |
| 24 Developmentally regulated GTP-binding protein 1 (DRG1) | gi|4758796 | 85 | 40,517 | 2 | 7.6 |
| 25 DNA-binding protein B | gi|181486 | 374 | 39,954 | 5 | 33.2 |
| 26 EIF-3 p110 subunit | gi|1931584 | 169 | 105,276 | 2 | 3.8 |
| 27 EIF3A protein | gi|32449796 | 506 | 96,804 | 6 | 12.6 |
| 28 Elongation factor 1-γ | gi|4503481 | 130 | 50,087 | 2 | 5.9 |
| 29 Elongation factor Tu | gi|704416 | 258 | 49,509 | 3 | 12.2 |
| 30 Endoplasmic reticulum lipid raft-associated 2 isoform 1 (Erlin-2) | gi|6005721 | 180 | 37,815 | 2 | 11.5 |
| 31 Eukaryotic translation initiation factor 2 subunit 3 | gi|4503507 | 204 | 51,077 | 3 | 8.9 |
| 32 Eukaryotic translation initiation factor 3 subunit C | gi|4503525 | 446 | 88,106 | 6 | 11.8 |
| 33 Eukaryotic translation initiation factor 3 subunit E | gi|4503521 | 625 | 52,187 | 8 | 21.8 |
| 34 Eukaryotic translation initiation factor 3 subunit L | gi|7705433 | 434 | 66,684 | 6 | 14.7 |
| 35 Eukaryotic translation initiation factor 3 subunit M | gi|23397429 | 139 | 42,476 | 2 | 8.3 |
| 36 Far upstream element-binding protein 1 | gi|17402900 | 80 | 67,518 | 2 | 4.3 |
| 37 Glutaminyl-tRNA synthetase | gi|31958 | 276 | 162,923 | 3 | 9.7 |
| 38 Heat shock 70-kDa protein 5 | gi|16507237 | 556 | 72,288 | 8 | 20.2 |
| 39 Heat shock cognate 71-kDa protein isoform 1 | gi|5729877 | 340 | 70,854 | 5 | 10.2 |
| 40 HNRNA U-like 1, isoform CRA_c | gi|119577432 | 173 | 84,447 | 2 | 5.1 |
| 41 hnRNP A/B isoform a | gi|55956919 | 150 | 35,945 | 2 | 9.6 |
| 42 hnRNP H | gi|5031753 | 258 | 49,198 | 3 | 18.3 |
| 43 hnRNP K | gi|460789 | 169 | 51,040 | 2 | 6 |
| 44 hnRNP M4 protein | gi|187281 | 380 | 77,555 | 9 | 13.2 |
| 45 hnRNP U protein | gi|32358 | 469 | 88,890 | 7 | 13.8 |
| 46 hnRNP U-like protein 2 | gi|118601081 | 135 | 72,378 | 2 | 7.2 |
| 47 IGF2BP1 | gi|119615099 | 190 | 47,883 | 3 | 9.8 |
| 48 IGF-II mRNA-binding protein 2 | gi|4191610 | 266 | 65,950 | 4 | 11.9 |
| 49 KIAA0115 | gi|473947 | 75 | 50,680 | 2 | 5.5 |
| 50 KIAA1352 protein | gi|7243085 | 312 | 138,273 | 5 | 5.7 |
| 51 KIAA1401 protein (tumor suppressor) (TSR 1) | gi|7243183 | 340 | 96,987 | 5 | 9 |
| 52 Lanosterol 14-α demethylase | gi|1698484 | 140 | 56,742 | 2 | 7.8 |
| 53 La-related protein 7 (LARP-7) | gi|109809739 | 162 | 66,857 | 2 | 5 |
| 54 Mammary tumor-associated protein INT6 | gi|2695641 | 625 | 52,044 | 8 | 21.8 |
| 55 M-phase phosphoprotein 4 | gi|1770458 | 161 | 66,703 | 2 | 6.4 |
| 56 mRNA-binding protein CRDBP | gi|7141072 | 494 | 63,417 | 6 | 17.3 |
| 57 Na+/K+-ATPase α subunit | gi|179212 | 158 | 81,680 | 3 | 6.2 |
| 58 Na+/K+-transporting ATPase subunit α-1 isoform a | gi|21361181 | 201 | 112,824 | 3 | 7.2 |
| 59 NF45 protein | gi|532313 | 230 | 44,669 | 4 | 13.5 |
| 60 NICE-4 protein (UBAP2L) | gi|11990132 | 50 | 43,252 | 1 | 3.7 |
| 61 Nodal modulator 2 isoform 2 (NOMO 2) | gi|27734709 | 258 | 134,107 | 4 | 7.8 |
| 62 Nuclear corepressor KAP-1 | gi|1699027 | 42 | 88,479 | 1 | 2.4 |
| 63 NY-REN-2 antigen | gi|5360085 | 146 | 61,282 | 2 | 4.7 |
| 64 ORF1 | gi|483915 | 174 | 40,076 | 3 | 11.2 |
| 65 P1.11659_4 (stomatin-like protein 2) | gi|2984585 | 190 | 38,725 | 2 | 9.2 |
| 66 PABPC4 protein | gi|41388837 | 266 | 69,563 | 4 | 8.7 |
| 67 Polyadenylate-binding protein II | gi|693937 | 154 | 58,481 | 2 | 9.4 |
| 68 Prt1 homolog (eIF3 p110) | gi|1778051 | 409 | 98,820 | 6 | 11.3 |
| 69 Putative helicase MOV-10 | gi|14211540 | 353 | 113,599 | 4 | 10.9 |
| 70 Putative RNA-binding protein KOC | gi|2105469 | 432 | 63,681 | 4 | 15.2 |
| 71 Ras GTPase-activating-like protein IQGAP2 | gi|116089337 | 111 | 122,764 | 2 | 4 |
| 72 RAS-p21 activating protein (G3BP1) | gi|5031703 | 73 | 52,132 | 1 | 3.4 |
| 73 RcNSEP1 | gi|82802829 | 206 | 35,435 | 3 | 31 |
| 74 Regulator of nonsense transcript stability | gi|1575536 | 294 | 123,039 | 4 | 7.2 |
| 75 Ribophorin-1 | gi|4506675 | 277 | 68,527 | 4 | 9.9 |
| 76 Staufen protein | gi|4335947 | 361 | 54,985 | 4 | 13.3 |
| 77 T-cluster binding protein | gi|2281006 | 160 | 29,631 | 2 | 12.8 |
| 78 T-complex protein 1 subunit β isoform 1 (TCP1) | gi|5453603 | 41 | 57,452 | 1 | 2.2 |
| 79 Translation initiation factor eIF3 p44 subunit | gi|3264859 | 276 | 35,674 | 4 | 13.4 |
| 80 Translational initiation factor β subunit | gi|182067 | 293 | 38,376 | 5 | 20.4 |
| 81 Valosin-containing protein (VCP) | gi|6005942 | 307 | 89,266 | 5 | 9.4 |
| 82 Vigilin | gi|4885409 | 222 | 141,352 | 2 | 4.7 |
| 83 YTHDF1 protein | gi|13277546 | 185 | 59,658 | 2 | 5.5 |
a The results are based on two separate experiments. The listed proteins are those scored in both experiments. The cellular proteins from cured MH14 cells (HCV-negative) associated with biotinylated PNA-streptavidin complex are shown in the supplemental Table I.
As expected, many of the identified cellular factors interacting with the HCV (+) strand RNA genome are translation factors, such as elongation factors (eEF2 and EFTu) and initiation factors (eIF3 subunits CELM, eIF3-p44, eIF2 subunit 3, and eIF3-p110). Among these, the initiation factors eIF3 and eIF2 complexed with GTP and tRNA-Met have been shown to be recruited by the HCV IRES to assemble an 80 S ribosome translation complex (22–24). The signaling group of proteins was also found to be associated with the HCV genome. This included cell cycle protein PA-2G4, UBAP2L (NICE-4), and NOMO2. Among these, PA-2G4, which is involved in RNA processing and signaling, has been shown to be associated with HCV IRES (25), whereas UBAP2L (NICE-4) has been identified as one of the nine gene products associated with the progression of hepatocellular carcinoma (26). Another group of proteins that we identified belongs to chaperon proteins, ribosomal proteins, RNA helicases, DEAD box proteins, oncogenic proteins, and various RNA-binding proteins and metabolic enzymes. Together, they represent a diverse set of cellular factors associated with the HCV RNA genome.
Bioinformatics Analysis of the Identified Proteins
Ingenuity pathway analysis of the identified proteins indicated their association with 12 different molecular and cellular functions (Fig. 6B). Although 15% of the proteins were involved in replication of RNA viruses, 6% of them were directly linked with HCV replication. Another major group of proteins was associated with different cancers, including digestive organ tumors. The canonical pathway analysis has matched the identified protein associated with 24 different pathways with significant −log (p) value (Fig. 6C). The highest −log(p) values were for the protein ubiquitination pathway, endothelial nitric-oxide synthase signaling, eukaryotic initiation factor 2 (eIF2) signaling, and regulation of eIF4 and p70 S6K signaling involved in the initiation of protein synthesis. Ingenuity pathway analysis also demonstrated that four major disease and disorder developments were significantly associated with our affinity-captured proteins. The majority of them are related to cancer (46%), infectious disease (30%), reproductive system disease (15%), and hepatic system disease (7%) (Fig. 6A).
Fig. 6.

Bioinformatics analysis of the identified proteins. The affinity-captured proteins identified by LC/MS/MS were matched against the Ingenuity pathway database of disease and disorder (A), molecular function (B), and canonical pathways (C) that were most significant to the set of identified proteins. The canonical pathway of the identified proteins with a p value for each pathway is indicated by the bar and is expressed as −1 times the log of the p value. The line indicated with the arrow represents the ratio of the number of genes in a given pathway that meet the cutoff criterion (p ≥ 0.05) divided by the total number of genes that make up that pathway due to chance alone. The percent of total protein matched in each category in the dataset of disease and disorder and molecular function is indicated. The gene symbol of each protein that matched against molecular function (B) and individual pathway (C) is also shown.
Silencing of Diverse Class of Identified Cellular Factors Modulates HCV Replication and Translation
Using siRNA, we silenced two RNA editing factors (ADAR1 and Stau1), an RNA helicase (DDX6), a cell cycle signaling protein (PA2G4), a molecular chaperone (HSP60), and a regulator of mRNA stability, translation, and turnover (IGF2BP1) to examine their effect on HCV replication and expression of the viral protein NS5A. Three of the cellular factors (DDX6, PA2G4, and IGF2BP1) were positive controls as they have been implicated on HCV replication (25, 27) or translation (28), although others (Stau1, ADR1, and HSP60) were considered as the novel targets. The siRNA was delivered into MH14 cells, which carry actively replicating HCV replicons. Fig. 7 shows that expression of all the siRNA targeted genes was reduced by more than 95% as demonstrated by Western blot analysis (lane 4). We also determined HCV RNA replicon level by RT-PCR and viral protein expression level by Western blotting for HCV NS5A. Although we noted a direct correlation between reduction in HCV RNA replication and reduced level of expression of Stau1, DDX6, and HSP60 in MH14 cells, there was an inverse relation between HCV replication and reduced expression of ADAR1 and PA2G4. We found that down-regulation of IGF2BP1 significantly reduced HCV translation as judged by Western blotting of NS5A but had no effect on HCV replication. The down-regulation of ADAR1 and PA2G4 significantly enhanced both HCV replicon RNA and expression of the viral protein NS5A. ADAR1, which catalyzes the deamination of adenosine in double-stranded RNA (29), has been suggested to be involved in IFN-α-mediated clearance of HCV RNA (30). The down-regulation of ADAR1 resulted in 2- and 3-fold stimulation of HCV RNA replication and viral protein expression, respectively. PA2G4, a proliferation-associated signaling protein, has been shown to induce cell cycle arrest in the G2/M phase of the cell cycle (31). Down-regulation of PA2G4 significantly enhanced both HCV RNA replication and viral protein expression in MH14 cells. Stau1, a double-stranded RNA-binding protein, must interact with influenza virus protein NS1 for efficient virus replication (32). We found that down-regulation of Stau1 reduced HCV replication to a nearly undetectable level, indicating its involvement in HCV replication. DDX6 (Rck/p54), one of the several RNA helicases associated with the HCV RNA genome, is overexpressed in human hepatocytes from patients with chronic hepatitis C (33). We found that down-regulation of DDX6 resulted in more than 90% inhibition of both HCV replication and expression of viral protein. HSP60 is a molecular chaperone required for cell survival during stress, which it achieves by restraining p53 function (34). Down-regulation of HSP60 in MH14 cells resulted in more than 90% reduction in HCV replication and 75% reduction in the expression of viral protein NS5A. IGF2BP1, which is a regulator of RNA stability, turnover, and translation, has been shown associated with HCV RNA and involved in IRES-mediated HCV translation (28). We found that down-regulation of IGF2BP1 had no effect on HCV replication, but it caused ∼60% reduction in the expression of viral protein NS5A suggesting its specific role in regulation of translation of HCV proteins.
Fig. 7.

Modulation of HCV replication/translation by siRNA-mediated down-regulation of Stau1, ADAR1, DDX6, PA2G4, HSP60, and IGF2BP1. The control MH14 cells carrying replicating HCV replicon were grown for 24 h (lane 1) and then transfected with 20 nm of siRNA duplexes targeting individual cellular factors (lanes 4–6) or with control siRNA duplexes (lane 3). A mock transfection was also done using only transfection reagent (lane 2). Cells were grown for 72 h after transfection, and total protein and RNA were isolated. A shows controls (lanes 1–3) as well as siRNA-mediated down-regulation of targeted host cell proteins (lanes 4–6) as assessed by Western blotting, inhibition, or stimulation of HCV replication by RT-PCR (5′NTR region of the HCV genome) and HCV translation by Western blotting of HCV NS5A. B–D, respectively, show the protein bands of targeted cellular proteins, corresponding RT-PCR of HCV RNA, and expression of HCV NS5A quantified by Quantity One software (Bio-Rad).
DISCUSSION
Earlier, we used in vitro transcribed HCV 3′NTR annealed with biotinylated oligo-DNA as bait to capture interacting cellular proteins from cell lysate (11). Although this strategy identified many cellular proteins interacting with HCV 3′NTR, some proteins interacted with the oligo-DNA probe alone; they were also identified and subtracted from the list as nonspecific binders.
In this study, we devised a novel strategy to capture the replicating HCV (+) strand RNA genome in situ and identified associated cellular or viral factors. This strategy, which uses a sequence-specific biotinylated PNA conjugated with polycationic neamine moiety of neomycin, has four advantages as follows: 1) PNA, being an unnatural DNA mimic with no sugar phosphate backbone, is not recognized by cellular proteins and does not have any affinity for them, so that background signal due to nonspecific protein binding was eliminated; 2) binding of PNA to its target is stoichiometric and irreversible under physiological conditions; 3) the PNA-neamine conjugate in either free form or bound to its target sequence is highly stable and completely resistant to cellular nucleases and proteases; and 4) most significantly, PNA-neamine conjugate efficiently penetrates the cells and binds to its target RNA in the cytosol, which can then be quantitatively recovered from the cell lysate. We used this strategy specifically to capture the HCV (+) strand RNA genome within MH14 cells carrying a stably replicating HCV subgenomic replicon. Following incubation with the conjugate, the cells were lysed, and the biotinylated PNA-neamine molecules in the cell lysate were recovered by immobilizing them on paramagnetic streptavidin beads. This approach to capturing the HCV genomic RNA-protein complex has positively identified many of the cellular and viral factors associated with the viral genome.
A recent report identified various cellular factors that affect HCV infection and replication in cultured hepatoma cells (12). Also, a combination of proteomics and computational modeling has identified novel host proteins that function as key regulators of HCV-associated metabolic changes (35). Consistent with these observations, we have identified 83 cellular proteins and three viral proteins (NS5B, NS5A, and NS3–4a helicase) that are associated with replicating HCV (+) strand RNA genome in cultured hepatoma cells. As expected, some ribosomal proteins and translation factors were associated with HCV (+) strand RNA. Besides these, several metabolic enzymes were also scored among the host factors associated with the viral genome.
Some of the identified RNA-binding proteins, including H1, M4, and K, belong to the hnRNP group. The hnRNP group of proteins is important in regulating HCV translations or replication. Among these, hnRNP K physically interacts with HCV core protein (36), although hnRNP C-like protein, which binds large pyrimidine-rich region HCV 3′NTRs, may function in the initiation and/or regulation of HCV RNA replication (37). The hnRNP U acts as a basic transcriptional regulator that represses basic transcription driven by several viral and cellular promoters (38). It has also been found to be associated with HCV IRES-binding proteins and is involved in IRES-dependent down-regulation of HCV translation (25). The hnRNP M4 interacts with cell membrane receptors to trigger signaling pathways that promote metastasis (39).
Other RNA binders that score in proteomics include the following: KOC (KH homology), a cancer-related autoantigen that is overexpressed in cancer cells (40); nuclear factor 45 (NF45), which has been shown to be part of HCV replication machineries involved in the regulation of viral translation and RNA replication (7); high density lipoprotein-binding protein (Vigilin), known as a marker gene product of HCV-associated hepatocellular carcinoma (41); and IGF2BP1, which not only affects mRNA nuclear export, localization, stability, and translation but is associated with HCV replicon RNA and enhances IRES-mediated HCV translation (28). T-cluster-binding protein was also scored as HCV RNA binder, which is one of the IFN-stimulated gene products specifically expressed in chronic hepatitis C (42).
RNA and DNA helicases are another important group of proteins found to be associated with HVC (+) strand RNA. Among these, DDX3 has been implicated in several processes that regulate gene expression. This protein has been the prime target for many viruses, including HCV, HBV, and HIV, which interact with DDX3 and modulate its function (43). HCV core protein specifically interacts with DDX3 and may be involved in regulating host cell mRNA translation (44). DDX6 (Rck/p54) is a cellular RNA helicase with ATP-dependent RNA-unwinding activity (45) that has been suggested to function as a proto-oncogene. It is overexpressed in human hepatocytes from patients with chronic hepatitis C (33) and in some colorectal cancers (46); also, its helicase activity is essential for efficient HCV replication (27). The RNA helicase DDX5 (growth-related nuclear 68 protein), which interacts with the C-terminal region of HCV NS5B, has been suggested to be part of the HCV replicase complex (47), whereas Ras-GTPase-activating protein-binding protein 1 (G3BP1) interacts with both HCV NS5B and the 5′ end of the HCV minus-strand RNA, suggesting that it is part of HCV replication complex (48). Another RNA helicase DDX30 that we scored has been shown to have an inhibitory effect on HIV-1 packaging and to reduce viral infectivity (49). A similar role for this helicase may be predicted in the HCV life cycle, wherein sequestering of DDX30 by the HCV genome may reduce its restrictive function.
The signaling group of proteins found to be associated with the HCV genome included the cell cycle protein PA-2G4 homolog, which is associated with HCV IRES (25); UBAP2L (NICE-4), which has been identified as one of the nine gene products associated with the progression of HCC (26); and nodal modulator 2 isoform (NOMO 2), which participates in the nodal signaling pathway during vertebrate development and is one of the signature proteins for vascular invasion of hepatitis C virus-related HCCs (50).
The cellular proteins belonging to the RNA editing group were also found to be associated with HCV genomic RNA. One such factor is an adenosine deaminase that acts on double-stranded RNA (ADAR1) and is induced by INF-α. The ADAR1 catalyzes deamination of adenosine in double-stranded RNA (29), which is then targeted for degradation by specific cellular RNase. It has been suggested that the inosine editing function of ADAR1 is involved in successful in vitro clearance of HCV RNA by IFN-α; this protein promises to offer a new therapeutic strategy for viral infections (30). Another RNA editing factor identified was Staufen, a double-stranded RNA-binding protein involved in mRNA transport and localization. This protein is overexpressed in HIV-1-infected cells and incorporated into the package of HIV-1 virions (51). It has also been shown to interact with influenza virus protein NS1 and is required for efficient virus replication (32). A similar role of Staufen in HCV replication may be suggested as we found that its down-regulation in MH14 cells carrying replicating HCV replicons drastically reduced HCV replication (Fig. 7). It is possible that Staufen may be involved in HCV RNA dimerization to regulate the molecular transition from the synthesis of (+) strand and (−) strand viral RNA to viral RNA translation. The APOBEC1 complementation factor that we have identified is an essential component of an editing complex involved in introducing site-specific deamination of cytosine in mammalian apolipoprotein B mRNA. Although the human liver is deficient in APOBEC1 expression, its interacting partner, APOBEC1 complementation factor, is abundantly expressed and suppresses apoptosis in liver cells (52, 53). However, hepatitis C virus triggers the expression of APOBEC1 in hepatocytes and chronic hepatic inflammation caused by HCV infection (54).The cytidine deaminase activity expression induced by APOBEC family members may function as a genome mutator that generates somatic mutation in targeted host genes, thus contributing to tumor genesis (55).
As expected, we also found an array of transcriptional regulators to be associated with the HCV genome. The FUSE-binding protein (FBP) is a transcription transactivator of the c-myc gene (56, 57). We have demonstrated that FBP interacts with HCV NS5A and significantly enhances HCV replication while strongly inhibiting translation of viral proteins (21). FBP is not expressed in normal somatic cells but is overexpressed in HCC and required for tumor growth (58). Another protein that negatively regulates cellular transcription is the La-related protein LARP7, which was associated with viral RNA. LARP7 binds to 7SK RNA and reverses its antagonistic effect on P-TEFb-mediated stimulation of transcription elongation by RNA polymerase II (59). The RNAi-mediated silencing of LARP7 stimulated polymerase II transcription of cellular as well as viral (HIV-1) genes. Another protein in this group was KAP-1, which interacts with STAT1 and negatively regulates interferon (IFN)/STAT1-mediated interferon-regulatory factor-1 (IRF-1) gene expression (60). Therefore, KAP1 could be one target host protein of HCV infection, which controls IRF-1 activation. CNOT1 is one of the nine components of the CCR4-NOT complex, which functions as a global regulator of gene expression (61) and is required for HCV infection (62).
Cellular factors involved in a variety of transport functions were also affinity-captured with the HCV RNA genome. These are UBQRC2, a subunit of the mitochondrial respiratory chain protein ubiquinol-cytochrome-c reductase complex III, which is involved in electron transfer from ubiquinol to cytochrome c (63); nucleoporin-like protein, which is required for the export of mRNAs containing poly(A) tails from the nucleus into the cytoplasm (64), as well as docking of HIV-1 Vpr at the nuclear envelope (65); and valosin-containing protein (VCP), also known as transitional endoplasmic reticulum ATPase (TER ATPase) or p97, which is an enzyme involved in vesicle transport and fusion, 26 S proteasome function, assembly of peroxisomes, and various cellular events that are regulated during mitosis (66). These cellular factors also include Caprin 1, a cell cycle-associated phosphoprotein required for normal progression through the G1-S phase of the cell cycle (67). Caparin 1 forms a complex with G3BP1 (68), which has been shown to interact with both HCV NS5B and the 5′ end of the HCV (minus) strand RNA (48).
Oncogenic proteins were another group of cellular factors associated with HCV RNA. Among them was the developmentally regulated GTP-binding protein (DRG1), which is critical in cell growth, associated with stem cell leukemia/T-cell acute lymphoblastic leukemia 1 (SCL/TAL1), and stimulates the co-transforming activity of c-Myc and Ras (68). Its abnormal expression triggers the disruption of normal growth control. ELAC2 has been shown to be associated with prostate cancer as mutation in the gene increases the risk of prostate cancer (69). Another oncogenic protein, KIAA1401, is also a transcriptional system regulator 1 (TSR1) and is required during maturation of the 40 S ribosomal subunit in the nucleolus; it also is associated with breast and thymus cancer (70). Endoplasmic reticulum lipid raft-associated 2 isoform 1 (ERLIN2) has been identified as one of the most potently transforming oncogenes expressed in breast cancer (71). Autoantigen is a nuclear protein specifically expressed in cancer cells during the S and G2 phase. Its N-terminal region of SG2NA (amino acids 1–391) acts as a strong transcriptional activator in both yeast and mammalian cells (72).
We have also scored various chaperon proteins associated with the viral genome. Most notable among them are HSP70 protein 5, HSP70 protein 8 isoform 1, and HSP60. These heat shock proteins (HSPs), designated as chaperones, are required for cell survival during stress and for protein folding, degradation, and reactivation of misfolded proteins (73). HSP70 is highly expressed in hepatocellular carcinoma as compared with its level in normal or benign livers (74). HSP70 physically interacts with NS5A and has been implicated in HCV IRES-mediated translation (75). The chaperonin T-complex polypeptide (TCP1), also known as TCP1 ring complex (TRiC), participates in HCV RNA replication and virion production, possibly through its interaction with NS5B (76). Another molecular chaperone, calnexin, which has a major role in controlling the quality of folding of HCV glycoproteins (77), was also affinity-captured with HCV RNA.
We have also found several translation factors, ribosomal proteins, and metabolic enzymes associated with HCV genomic RNA. Indeed, the information we have generated regarding the identity of cellular factors associated with replicating HCV (+) RNA subgenomic replicons will provide a strong basis for numerous hypothesis-driven studies on the interactions of cellular factors with viral RNA and proteins, as well as the functions of these factors in establishing chronic HCV infection and promoting its progression to LC and HCC. The major challenges are to determine the hierarchical importance of these interactions between HCV and host cell factors, to delineate how these interactions affect patients infected with HCV, and to determine which of these interactions may be potential targets for therapeutic intervention.
Supplementary Material
Footnotes
* This work was supported, in whole or in part, by National Institutes of Health Grant AI073703 from NIAID and Grant DK083560 from NIDDK.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- HCV
- hepatitis C virus
- 3′NTR
- 3′-nontranslated region of HCV RNA genome
- 5′NTR
- 5′-nontranslated region of HCV RNA genome
- PNA
- peptide nucleic acid
- NS5A
- nonstructural protein 5A of hepatitis C virus
- NS5B
- nonstructural protein 5B of hepatitis C virus
- HCC
- hepatocellular carcinoma
- IRES
- internal ribosomal entry site
- PNA-nea
- PNA-neamine
- FBP
- Fuse-binding protein
- hnRNP
- heterogeneous nuclear ribonucleoprotein.
REFERENCES
- 1. Tsukiyama-Kohara K., Iizuka N., Kohara M., Nomoto A. (1992) Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 66, 1476–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang C., Sarnow P., Siddiqui A. (1993) Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J. Virol. 67, 3338–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rijnbrand R. C., Lemon S. M. (2000) Internal ribosome entry site-mediated translation in hepatitis C virus replication. Curr. Top. Microbiol. Immunol. 242, 85–116 [DOI] [PubMed] [Google Scholar]
- 4. Reed K. E., Rice C. M. (2000) Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top. Microbiol. Immunol. 242, 55–84 [DOI] [PubMed] [Google Scholar]
- 5. Oh J. W., Sheu G. T., Lai M. M. (2000) Template requirement and initiation site selection by hepatitis C virus polymerase on a minimal viral RNA template. J. Biol. Chem. 275, 17710–17717 [DOI] [PubMed] [Google Scholar]
- 6. Ali N., Siddiqui A. (1997) The La antigen binds 5′-noncoding region of the hepatitis C virus RNA in the context of the initiator AUG codon and stimulates internal ribosome entry site-mediated translation. Proc. Natl. Acad. Sci. U.S.A. 94, 2249–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Isken O., Baroth M., Grassmann C. W., Weinlich S., Ostareck D. H., Ostareck-Lederer A., Behrens S. E. (2007) Nuclear factors are involved in hepatitis C virus RNA replication. RNA 13, 1675–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ali N., Siddiqui A. (1995) Interaction of polypyrimidine tract-binding protein with the 5′-noncoding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J. Virol. 69, 6367–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anwar A., Ali N., Tanveer R., Siddiqui A. (2000) Demonstration of functional requirement of polypyrimidine tract-binding protein by SELEX RNA during hepatitis C virus internal ribosome entry site-mediated translation initiation. J. Biol. Chem. 275, 34231–34235 [DOI] [PubMed] [Google Scholar]
- 10. Paek K. Y., Kim C. S., Park S. M., Kim J. H., Jang S. K. (2008) RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J. Virol. 82, 12082–12093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harris D., Zhang Z., Chaubey B., Pandey V. N. (2006) Identification of cellular factors associated with the 3′-nontranslated region of the HCV genome by RNA affinity capture and mass spectrometry. Mol. Cell. Proteomics 5, 1006–1018 [DOI] [PubMed] [Google Scholar]
- 12. Randall G., Panis M., Cooper J. D., Tellinghuisen T. L., Sukhodolets K. E., Pfeffer S., Landthaler M., Landgraf P., Kan S., Lindenbach B. D., Chien M., Weir D. B., Russo J. J., Ju J., Brownstein M. J., Sheridan R., Sander C., Zavolan M., Tuschl T., Rice C. M. (2007) Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U.S.A. 104, 12884–12889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miyanari Y., Hijikata M., Yamaji M., Hosaka M., Takahashi H., Shimotohno K. (2003) Hepatitis C virus nonstructural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 278, 50301–50308 [DOI] [PubMed] [Google Scholar]
- 14. Murata T., Ohshima T., Yamaji M., Hosaka M., Miyanari Y., Hijikata M., Shimotohno K. (2005) Suppression of hepatitis C virus replicon by TGF-β. Virology 331, 407–417 [DOI] [PubMed] [Google Scholar]
- 15. Riguet E., Tripathi S., Chaubey B., Désiré J., Pandey V. N., Décout J. (2004) A peptide nucleic acid-neamine conjugate that targets and cleaves HIV-1 TAR RNA inhibits viral replication. J. Med. Chem. 47, 4806–4809 [DOI] [PubMed] [Google Scholar]
- 16. Ali N., Tardif K. D., Siddiqui A. (2002) Cell-free replication of the hepatitis C virus subgenomic replicon. J. Virol. 76, 12001–12007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Waris G., Sarker S., Siddiqui A. (2004) Two-step affinity purification of the hepatitis C virus ribonucleoprotein complex. RNA 10, 321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. 18. Kim S. K., Nielsen P. E., Egholm M., Buchardt O., Berg R. H., Norden B. (1993) Right-handed triplex formed between peptide nucleic acid PNA-T-8 and poly(dA) shown by linear and circular dichroism spectroscopy. J. Am. Chem. Soc. 115, 6477–6481 [Google Scholar]
- 19. Cherny D. Y., Belotserkovskii B. P., Frank-Kamenetskii M. D., Egholm M., Buchardt O., Berg R. H., Nielsen P. E. (1993) DNA unwinding upon strand-displacement binding of a thymine-substituted polyamide to double-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 90, 1667–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Das I., Désiré J., Manvar D., Baussanne I., Pandey V. N., Décout J. L. (2012) A peptide nucleic acid-aminosugar conjugate targeting transactivation response element of HIV-1 RNA genome shows a high bioavailability in human cells and strongly inhibits tat-mediated transactivation of HIV-1 transcription. J. Med. Chem. 55, 6021–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Z., Harris D., Pandey V. N. (2008) The FUSE-binding protein is a cellular factor required for efficient replication of hepatitis C virus. J. Virol. 82, 5761–5773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pestova T. V., Shatsky I. N., Fletcher S. P., Jackson R. J., Hellen C. U. (1998) A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 12, 67–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ji H., Fraser C. S., Yu Y., Leary J., Doudna J. A. (2004) Coordinated assembly of human translation initiation complexes by the hepatitis C virus internal ribosome entry site RNA. Proc. Natl. Acad. Sci. 101, 16990–16995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Otto G. A., Puglisi J. D. (2004) The pathway of hepatitis C virus internal ribosome entry site-mediated translation initiation. Cell 119, 369–380 [DOI] [PubMed] [Google Scholar]
- 25. Pacheco A., Reigadas S., Martínez-Salas E. (2008) Riboproteomic analysis of polypeptides interacting with the internal ribosome-entry site element of foot-and-mouth disease viral RNA. Proteomics 8, 4782–4790 [DOI] [PubMed] [Google Scholar]
- 26. Wurmbach E., Chen Y. B., Khitrov G., Zhang W., Roayaie S., Schwartz M., Fiel I., Thung S., Mazzaferro V., Bruix J., Bottinger E., Friedman S., Waxman S., Llovet J. M. (2007) Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 45, 938–947 [DOI] [PubMed] [Google Scholar]
- 27. Jangra R. K., Yi M., Lemon S. M. (2010) DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J. Virol. 84, 6810–6824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weinlich S., Hüttelmaier S., Schierhorn A., Behrens S. E., Ostareck-Lederer A., Ostareck D. H. (2009) IGF2BP1 enhances HCV IRES-mediated translation initiation via the 3′UTR. RNA 15, 1528–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saunders L. R, Barber G. N. (2003) The dsRNA-binding protein family: critical roles, diverse cellular functions. FASEB J. 17, 961–983 [DOI] [PubMed] [Google Scholar]
- 30. Taylor D. R., Puig M., Darnell M. E., Mihalik K., Feinstone S. M. (2005) New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J. Virol. 79, 6291–6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y., Akinmade D., Hamburger A. W. (2005) The ErbB3-binding protein Ebp1 interacts with Sin3A to repress E2F1 and AR-mediated transcription. Nucleic Acids Res. 33, 6024–6033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Lucas S., Peredo J., Marión R. M., Sánchez C., Ortín J. (2010) Human Staufen1 protein interacts with influenza virus ribonucleoproteins and is required for efficient virus multiplication. J. Virol. 84, 7603–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miyaji K., Nakagawa Y., Matsumoto K., Yoshida H., Morikawa H., Hongou Y., Arisaka Y., Kojima H., Inoue T., Hirata I., Katsu K., Akao Y. (2003) Overexpression of a DEAD box/RNA helicase protein, rck/p54, in human hepatocytes from patients with hepatitis C virus-related chronic hepatitis and its implication in hepatocellular carcinogenesis. J. Viral. Hepat. 10, 241–248 [DOI] [PubMed] [Google Scholar]
- 34. Ghosh J. C., Dohi T., Kang B. H., Altieri D. C. (2008) Hsp60 regulation of tumor cell apoptosis. J. Biol. Chem. 283, 5188–5194 [DOI] [PubMed] [Google Scholar]
- 35. Diamond D. L., Syder A. J., Jacobs J. M., Sorensen C. M., Walters K. A., Proll S. C., McDermott J. E., Gritsenko M. A., Zhang Q., Zhao R., Metz T. O., Camp D. G., 2nd, Waters K. M., Smith R. D., Rice C. M., Katze M. G. (2010) Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 6, e1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hsieh T. Y., Matsumoto M., Chou H. C., Schneider R., Hwang S. B., Lee A. S., Lai M. M. (1998) Hepatitis C virus Core protein interacts with heterogeneous nuclear ribonucleoprotein K. J. Biol. Chem. 273, 17651–17659 [DOI] [PubMed] [Google Scholar]
- 37. Gontarek R. R., Gutshall L. L., Herold K. M., Tsai J., Sathe G. M., Mao J., Prescott C., Del Vecchio A. M. (1999) hnRNP C and polypyrimidine tract-binding protein specifically interact with the pyrimidine-rich region within the 3′NTR of the HCV RNA genome. Nucleic Acids Res. 27, 1457–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gabler S., Schütt H., Groitl P., Wolf H., Shenk T., Dobner T. (1998) E1B 55-kilodalton-associated protein: a cellular protein with RNA-binding activity implicated in nucleocytoplasmic transport of adenovirus and cellular mRNAs. J. Virol. 72, 7960–7971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bajenova O., Stolper E., Gapon S., Sundina N., Zimmer R., Thomas P. (2003) Surface expression of heterogeneous nuclear RNA-binding protein M4 on Kupffer cell relates to its function as a carcinoembryonic antigen receptor. Exp. Cell Res. 291, 228–241 [DOI] [PubMed] [Google Scholar]
- 40. Müeller-Pillasch F., Lacher U., Wallrapp C., Micha A., Zimmerhackl F., Hameister H., Varga G., Friess H., Büchler M., Beger H. G., Vila M. R., Adler G., Gress T. M. (1997) Cloning of a gene highly overexpressed in cancer coding for a novel KH-domain containing protein. Oncogene 14, 2729–2733 [DOI] [PubMed] [Google Scholar]
- 41. Jacobs J. M., Diamond D. L., Chan E. Y., Gritsenko M. A., Qian W., Stastna M., Baas T., Camp D. G., 2nd, Carithers R. L., Jr., Smith R. D., Katze M. G. (2005). Proteome analysis of liver cells expressing a full-length hepatitis C virus (HCV) replicon and biopsy specimens of post-transplantation liver from HCV-infected patients. J. Virol. 79, 7558–7569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Helbig K. J., Lau D. T., Semendric L., Harley H. A., Beard M. R. (2005) Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology 42, 702–710 [DOI] [PubMed] [Google Scholar]
- 43. Schröder M. (2010) Human DEAD-box protein 3 has multiple functions in gene regulation and cell cycle control and is a prime target for viral manipulation. Biochem. Pharmacol. 79, 297–306 [DOI] [PubMed] [Google Scholar]
- 44. Mamiya N., Worman H. J. (1999) Hepatitis C virus core protein binds to a DEAD box RNA helicase. J. Biol. Chem. 274, 15751–15756 [DOI] [PubMed] [Google Scholar]
- 45. Weston A., Sommerville J. (2006) Xp54 and related (DDX6-like) RNA helicases: roles in messenger RNP assembly, translation regulation, and RNA degradation. Nucleic Acids Res. 34, 3082–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hashimoto K., Nakagawa Y., Morikawa H., Niki M., Egashira Y., Hirata I., Katsu K., Akao Y. (2001) Co-overexpression of DEAD box protein Rck/p54 and c-Myc protein in human colorectal adenomas and the relevance of their expression in cultured cell lines. Carcinogenesis 22, 1965–1970 [DOI] [PubMed] [Google Scholar]
- 47. Goh P. Y., Tan Y. J., Lim S. P., Tan Y. H., Lim S. G., Fuller-Pace F., Hong W. (2004) Cellular RNA helicase p68 relocalization and interaction with the hepatitis C virus (HCV) NS5B protein and the potential role of p68 in HCV RNA replication. J. Virol. 78, 5288–5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yi Z., Pan T., Wu X., Song W., Wang S., Xu Y., Rice C. M., Macdonald M. R., Yuan Z. (2011) Hepatitis C virus co-opts ras-GTPase-activating protein-binding protein 1 for its genome replication. J. Virol. 85, 6996–7004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou Y., Ma J., Bushan Roy B., Wu J. Y., Pan Q., Rong L., Liang C. (2008) The packaging of human immunodeficiency virus type 1 RNA is restricted by overexpression of an RNA helicase DHX30. Virology 372, 97–106 [DOI] [PubMed] [Google Scholar]
- 50. Mínguez B., Hoshida Y., Villanueva A., Toffanin S., Cabellos L., Thung S., Mandeli J., Sia D., April C., Fan J. B., Lachenmayer A., Savic R., Roayaie S., Mazzaferro V., Bruix J., Schwartz M., Friedman S. L., Llovet J. M. (2011). Gene-expression signature of vascular invasion in hepatocellular carcinoma. J. Hepatol. 55, 1325–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mouland A. J., Mercier J., Luo M., Bernier L., DesGroseillers L., Cohen E. A. (2000) The double-stranded RNA-binding protein Staufen is incorporated in human immunodeficiency virus type 1: evidence for a role in genomic RNA encapsidation. J. Virol. 74, 5441–5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Blanc V., Henderson J. O., Newberry E. P., Kennedy S., Luo J., Davidson N. O. (2005) Targeted deletion of the murine apobec-1 complementation factor (acf) gene results in embryonic lethality. Mol. Cell. Biol. 25, 7260–7269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pazienza V., Clément S., Pugnale P., Conzelmann S., Pascarella S., Mangia A., Negro F. (2009) Gene expression profile of Huh-7 cells expressing hepatitis C virus genotype 1b or 3a core proteins. Liver Int. 29, 661–669 [DOI] [PubMed] [Google Scholar]
- 54. Kou T., Marusawa H., Kinoshita K., Endo Y., Okazaki I. M., Ueda Y., Kodama Y., Haga H., Ikai I., Chiba T. (2007) Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int. J. Cancer 120, 469–476 [DOI] [PubMed] [Google Scholar]
- 55. Marusawa H. (2008) Aberrant AID expression and human cancer development. Int. J. Biochem. Cell Biol. 40, 1399–1402 [DOI] [PubMed] [Google Scholar]
- 56. Avigan M. I., Strober B., Levens D. (1990) A far upstream element stimulates c-Myc expression in undifferentiated leukemia cells. J. Biol. Chem. 265, 18538–18545 [PubMed] [Google Scholar]
- 57. Duncan R., Bazar L., Michelotti G., Tomonaga T., Krutzsch H., Avigan M., Levens D. (1994) A sequence-specific, single-strand binding protein activates the far upstream element of c-Myc and defines a new DNA-binding motif. Genes Dev. 8, 465–480 [DOI] [PubMed] [Google Scholar]
- 58. Rabenhorst U., Beinoraviciute-Kellner R., Brezniceanu M. L., Joos S., Devens F., Lichter P., Rieker R. J., Trojan J., Chung H. J., Levens D. L., Zörnig M. (2009) Overexpression of the far upstream element-binding protein 1 in hepatocellular carcinoma is required for tumor growth. Hepatology 50, 1121–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Markert A., Grimm M., Martinez J., Wiesner J., Meyerhans A., Meyuhas O., Sickmann A., Fischer U. (2008) The La-related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes. EMBO Rep. 9, 569–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kamitani S., Ohbayashi N., Ikeda O., Togi S., Muromoto R., Sekine Y., Ohta K., Ishiyama H., Matsuda T. (2008) KAP1 regulates type I interferon/STAT1-mediated IRF-1 gene expression. Biochem. Biophys. Res. Commun. 370, 366–370 [DOI] [PubMed] [Google Scholar]
- 61. Collart M. A., Timmers H. T. (2004) The eukaryotic Ccr4-not complex: A regulatory platform integrating mRNA metabolism with cellular signaling pathways? Prog. Nucleic Acid Res. Mol. Biol. 77, 289–322 [DOI] [PubMed] [Google Scholar]
- 62. Li Q., Brass A. L., Ng A., Hu Z., Xavier R. J., Liang T. J., Elledge S. J. (2009) A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. U.S.A. 106, 16410–16415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schägger H., Brandt U., Gencic S., von Jagow G. (1995) Ubiquinol-cytochrome-c reductase from human and bovine mitochondria. Methods Enzymol. 260, 82–96 [DOI] [PubMed] [Google Scholar]
- 64. Strahm Y., Fahrenkrog B., Zenklusen D., Rychner E., Kantor J., Rosbach M., Stutz F. (1999) The RNA export factor Gle1p is located on the cytoplasmic fibrils of the NPC and physically interacts with the FG-nucleoporin Rip1p, the DEAD-box protein Rat8p/Dbp5p, and a new protein Ymr 255p. EMBO J. 18, 5761–5777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Farjot G., Sergeant A., Mikaélian I. (1999) A new nucleoporin-like protein interacts with both HIV-1 Rev nuclear export signal and CRM-1. J. Biol. Chem. 274, 17309–17317 [DOI] [PubMed] [Google Scholar]
- 66. Guinto J. B., Ritson G. P., Taylor J. P., Forman M. S. (2007) Valosin-containing protein and the pathogenesis of frontotemporal dementia associated with inclusion body myopathy. Acta Neuropathol. 114, 55–61 [DOI] [PubMed] [Google Scholar]
- 67. Solomon S., Xu Y., Wang B., David M. D., Schubert P., Kennedy D., Schrader J. W. (2007) Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2α, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol. Cell. Biol. 27, 2324–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mahajan M. A., Park S. T., Sun X. H. (1996) Association of a novel GTP-binding protein, DRG, with TAL oncogenic proteins. Oncogene 12, 2343–2350 [PubMed] [Google Scholar]
- 69. Rebbeck T. R., Walker A. H., Zeigler-Johnson C., Weisburg S., Martin A. M., Nathanson K. L., Wein A. J., Malkowicz S. B. (2000) Association of HPC2/ELAC2 genotypes and prostate cancer. Am. J. Hum. Genet. 67, 1014–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Higo K., Ushijima T., Oyabu A., Ye C., Yagyu S., Takahashi H., Matsuyama M. (2000) Generation of a polymorphic marker linked to thymoma susceptibility gene of rat 1 by genetically directed representational difference analysis. Exp. Anim. 49, 189–195 [DOI] [PubMed] [Google Scholar]
- 71. Yang Z. Q., Liu G., Bollig-Fischer A., Giroux C. N., Ethier S. P. (2010) Transforming properties of 8p11-12-amplified genes in human breast cancer. Cancer Res. 70, 8487–8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhu W., Chan E. K., Li J., Hemmerich P., Tan E. M. (2001) Transcription activating property of autoantigen SG2NA and modulating effect of WD-40 repeats. Exp. Cell Res. 269, 312–321 [DOI] [PubMed] [Google Scholar]
- 73. Parsell D. A., Lindquist S. (1993) The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu. Rev. Genet. 27, 437-496 [DOI] [PubMed] [Google Scholar]
- 74. Di Tommaso L., Franchi G., Park Y. N., Fiamengo B., Destro A., Morenghi E., Montorsi M., Torzilli G., Tommasini M., Terracciano L., Tornillo L., Vecchione R., Roncalli M. (2007) Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology 45, 725-734 [DOI] [PubMed] [Google Scholar]
- 75. Gonzalez O., Fontanes V., Raychaudhuri S., Loo R., Loo J., Arumugaswami V., Sun R., Dasgupta A., French S. W. (2009) The heat shock protein inhibitor Quercetin attenuates hepatitis C virus production. Hepatology 50, 1756–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Inoue Y., Aizaki H., Hara H., Matsuda M., Ando T., Shimoji T., Murakami K., Masaki T., Shoji I., Homma S., Matsuura Y., Miyamura T., Wakita T., Suzuki T. (2011) Chaperonin TRiC/CCT participates in replication of hepatitis C virus genome via interaction with the viral NS5B protein. Virology 410, 38–47 [DOI] [PubMed] [Google Scholar]
- 77. Dubuisson J., Rice C. M. (1996) Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J. Virol. 70, 778–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.