

Abstract
Sphingomonads comprise a physiologically versatile group within the Alphaproteobacteria that includes strains of interest for biotechnology, human health, and environmental nutrient cycling. In this study, we compared 26 sphingomonad genome sequences to gain insight into their ecology, metabolic versatility, and environmental adaptations. Our multilocus phylogenetic and average amino acid identity (AAI) analyses confirm that Sphingomonas, Sphingobium, Sphingopyxis, and Novosphingobium are well-resolved monophyletic groups with the exception of Sphingomonas sp. strain SKA58, which we propose belongs to the genus Sphingobium. Our pan-genomic analysis of sphingomonads reveals numerous species-specific open reading frames (ORFs) but few signatures of genus-specific cores. The organization and coding potential of the sphingomonad genomes appear to be highly variable, and plasmid-mediated gene transfer and chromosome-plasmid recombination, together with prophage- and transposon-mediated rearrangements, appear to play prominent roles in the genome evolution of this group. We find that many of the sphingomonad genomes encode numerous oxygenases and glycoside hydrolases, which are likely responsible for their ability to degrade various recalcitrant aromatic compounds and polysaccharides, respectively. Many of these enzymes are encoded on megaplasmids, suggesting that they may be readily transferred between species. We also identified enzymes putatively used for the catabolism of sulfonate and nitroaromatic compounds in many of the genomes, suggesting that plant-based compounds or chemical contaminants may be sources of nitrogen and sulfur. Many of these sphingomonads appear to be adapted to oligotrophic environments, but several contain genomic features indicative of host associations. Our work provides a basis for understanding the ecological strategies employed by sphingomonads and their role in environmental nutrient cycling.
INTRODUCTION
Bacteria belonging to the genera Sphingomonas, Sphingobium, Novosphingobium, and Sphingopyxis, collectively referred to as sphingomonads, comprise an assortment of physiologically diverse bacteria within the phylum Alphaproteobacteria (1). Members of this group are broadly distributed in nature and have been isolated from a variety of environments, including human-associated niches (2), chemically contaminated water and sediment (3), activated sludge from wastewater treatment plants (4), seawater (5), and stromatolites (6). Many sphingomonads have been demonstrated to play an important role in nutrient cycling, especially in oligotrophic environments (5). Others reside in various plant- and animal-associated ecosystems and can even be responsible for nosocomial infections in humans (2).
A salient characteristic of many sphingomonads is their ability to degrade a variety of recalcitrant aromatic compounds and polysaccharides (1). A number of sphingomonads have been isolated from environments heavily contaminated with pesticides, herbicides, and other xenobiotics, and some have been shown to use these compounds as a sole carbon source (7, 8). Some sphingomonads have been shown to degrade dioxins (9), diphenyl ethers (7, 10), polyethylene glycol (11), dibenzofurans (12, 13), and many other aromatic and chloroaromatic compounds (8, 14). Moreover, many strains are known to metabolize lignin (15), an aromatic polymer abundant in plant cell walls. Taken together with the ability of many sphingomonads to deconstruct polysaccharides (16, 17), it is clear that these bacteria play important roles in the degradation of diverse recalcitrant compounds in the environment.
To provide insight into the ecology of sphingomonads, we compared the genomes of 26 bacteria belonging to the genera Sphingomonas, Sphingobium, Novosphingobium, and Sphingopyxis. Additionally, we sequenced and analyzed draft genomes of Sphingomonas sp. strain Mn802 and Sphingomonas sp. strain PR090111-T3T-6A, two isolates associated with termites. The primary objectives of this study were to use genomic tools to shed light on the ecological strategies utilized by these bacteria and to broaden our knowledge of the evolutionary processes affecting their genomes. We compared the architecture of all sphingomonad genomes and, through analysis of the sphingomonad pan-genome, identified shared and variably distributed genes. Using targeted comparative analyses of specific genes involved in nutrient uptake and metabolism, our work provides hypotheses regarding the genomic content necessary for sphingomonads to thrive in a diversity of environments. Given the role many sphingomonads have been postulated to play in the cycling of recalcitrant carbon compounds in the environment, we also compared the complement of genes involved in the degradation of polysaccharides and aromatic compounds.
MATERIALS AND METHODS
Termite collection and bacterial isolations.
Termite colonies were sampled from natural populations in South Africa and Puerto Rico (see Table S1 in the supplemental material). Sphingomonas sp. strain Mn802 was isolated from a worker in a nest of the fungus-growing termite Macrotermes natalensis in 2008, and strain PR090111-T3T-6A was isolated from nest material from a Nasutitermes sp. in 2009. For strain Mn802, an underground fungus-growing termite colony of Macrotermes natalensis was excavated in South Africa and workers were collected using sterile forceps. Workers were kept together with fungus material in alcohol-wiped plastic containers until they were processed at the Forestry and Agricultural Biotechnology Institute (FABI) at the University of Pretoria, South Africa. In the laboratory, major workers were ground in 0.5 ml water and a homogenate was plated on 1% carboxymethyl cellulose (CMC) medium (10 g CMC and 20 g agar/liter), and a Sphingomonas isolate was obtained through serial transfer. For strain PR090111-T3T-6A, a colony of a Nasutitermes sp. was sampled from a termitarium in Bosque Estatal Maricao, Puerto Rico, in 2009. A homogenate of nest material in sterile water was plated on chitin medium (4 g chitin, 0.7 g K2HPO4, 0.3 g KH2PO4, 0.5 g MgSO4 · 5H2O, 0.01 g FeSO4 · 7H2O, 0.001 g ZnSO4, 0.0001 g MnCl2, 1 liter water) (18), and colonies on the plates were isolated by serial transfer.
Genome sequencing.
DNA was extracted from two pure cultures of Sphingomonas isolates grown in liquid Luria-Bertani (LB) media using the Qiagen DNeasy blood and tissue kit (Qiagen, CA). Shotgun sequencing libraries were constructed at the DOE Joint Genome Institute (JGI), and all aspects related to library construction and sequencing are available at http://www.jgi.doe.gov/. Whole-genome sequencing was performed for both isolates using a single picotiter plate on a 454 FLX Titanium pyrosequencer (19) and de novo assembled using Newbler, version 2.3. The draft genome of strain Mn802 consists of 25 contigs comprising 3.19 Mb, while the genome of strain PR090111-T3T-6A consists of 26 contigs comprising 3.91 Mb.
Genome annotations.
Proteins from all draft and complete genomes were predicted de novo using Prodigal (20). Protein clusters were constructed through comparison with previously constructed clusters available on the eggNOG Database, version 3.0 (21), using HMMER (22). To do this, all bacterial cluster alignments (bactNOGs) available from eggNOG were downloaded on 28 September 2011, and HMMer3 was used to construct hidden-Markov model profiles for each cluster. For the clusters that lacked alignments on the eggNOG file transfer protocol (FTP), alignments were generated using MUSCLE (23). All sphingomonad proteins predicted were compared to these clusters using hmmscan from HMMer3, with a minimum E value of <1e−5. Proteins were designated as belonging to the same cluster as their top HMMer model hit. Raw data regarding cluster membership can be found in Data Set S1 in the supplemental material.
To identify distinguishing genomic features in the three sphingomonad genera for which multiple genomes are available (Sphingomonas, Sphingobium, and Novosphingobium), we performed an enrichment analysis of the protein clusters represented in these genera. To do this, we used Fisher's exact test (P < 0.01) to identify protein clusters that were overrepresented in one of these genera compared to the other two genera combined (raw data available in Data Set S1 in the supplemental material). For our analysis of highly variable coding potential, protein clusters with highly variable membership across the sphingomonads were identified by calculating the standard deviation of cluster membership across all genomes. The annotations of representative proteins belonging to the clusters with the highest variability (defined here as having a standard deviation of >1.5) were analyzed using the same tools as implemented for whole-genome annotations (see below). Visualization of Venn diagrams was performed using the tool Venny (http://bioinfogp.cnb.csic.es/tools/venny/index.html).
Proteins predicted from the sphingomonad genomes were annotated using the KEGG KAAS server (24) and by comparison with the Clusters of Orthologous Groups (COG) database (25) using RPSBLAST (26) (E value < 1e−5). To identify transposons, we found all proteins with hits to one or more transposon-associated COGs (full list and details in Data Set S2 in the supplemental material). Prophage and phage-like proteins on the chromosomes of the complete sphingomonad genomes were identified using PhiSpy (27) with the associated GenBank files available in the GenBank database (http://www.ncbi.nlm.nih.gov/GenBank/index.html) as the input. Oxygenases were determined by identifying all hits to mono- and dioxygenases in the KEGG Orthology (28) database (full KEGG annotations are available in Data Set S3 in the supplemental material). For carbohydrate-active enzymes (CAZymes) (29), predictions were made as previously described (30) (full annotations for oxygenases and CAZymes are available in Data Set S2). To identify transposons, we found proteins with homology to 27 COG profiles associated with these elements (details and full annotations in Data Set S2).
Phylogenetic analyses.
A multilocus phylogenetic tree was constructed by identifying orthologous protein-coding genes between genomes. This was done by identifying all universally conserved eggNOG clusters and, for each genome, retaining only the protein having the highest HMMER hit to the Markov model for that cluster. This resulted in 268 universally conserved protein-coding genes for each genome. Protein sequences for each conserved cluster were aligned using the E-INS-i algorithm implemented in MAFFT, version 6 (31), and then converted to nucleotide alignments. These were concatenated and used to generate a phylogeny using RAxML (32). The concatenated alignment was partitioned by gene, and GTRGAMMA model parameters were estimated for each partition of the alignment separately. The phylogeny was generated with 100 bootstrap iterations.
For the average amino acid identity (AAI) analyses, we used a strategy similar to one previously described (33). AAI values were calculated for each pair of genomes using the reciprocal best BLASTP hits of their predicted proteins (parameters: X 150, −q −1, −F F, −e 1e−5). The AAI values were then converted into distances by subtracting them from 100. The dendrogram was created by using the resulting pairwise distances as input for the Neighbor package in Phylip (34). Both the multilocus phylogeny and AAI dendrogram were visualized using the interactive Tree of Life (iTOL) (35). The pairwise synteny plots were constructed using the same reciprocal best BLASTP hits as used for the AAI analysis and plotting them using R (36).
Nucleotide sequence accession numbers.
The raw 454 sequences for Sphingomonas sp. strain Mn802 and strain PR090111-T3T-6A are available from the DDBJ Sequence Read Archive (SRA) under accession numbers SRA037852 and SRA037853, respectively, and the assemblies used in this study have been deposited in DDBJ/EMBL/GenBank under the accession numbers AORY00000000 and AORL00000000, respectively.
RESULTS AND DISCUSSION
Phylogenetic analyses of the sphingomonads.
We analyzed the phylogenetic relationships of the sphingomonad genomes using both multilocus sequence analysis (MLSA) and AAI. For the MLSA, we used a concatenated alignment of 268 orthologous genes found to be universally conserved across all genomes. The resulting phylogeny revealed distinct clustering of the four genera, consistent with biochemical and 16S-based phylogenetic analyses distinguishing these groups (37) (Fig. 1). Maximum-likelihood bootstrap support was 100% for all nodes, and the topology of this tree was identical to a dendrogram created by clustering pairwise AAI analysis (Fig. 1), suggesting well-supported phylogenetic relationships. The pairwise AAI values ranged from 55.4% (between Sphingomonas wittichii RW1 and Novosphingobium sp. strain AP12) and 99.5% (between Sphingomonas wittichii RW1 and DP58) (all available in Data Set S4 in the supplemental material). Despite its classification in the genus Sphingomonas, both our MLSA and AAI analysis revealed that strain SKA58 clusters within the genus Sphingobium in the same clade as Sphingobium yanoikuyae XLDN2-5 and strain AP49 (Fig. 1), suggesting that this bacterium should be classified within this genus.
Fig 1.
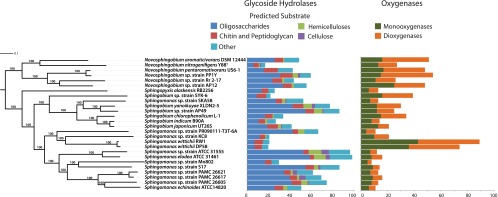
Maximum-likelihood MLSA of 26 sphingomonad genomes constructed from concatenated nucleotide sequences of 268 universally conserved genes, together with the distribution of glycoside hydrolases (GHs) and mono- and dioxygenases identified in the sphingomonad genomes. The topology of this tree is identical to the AAI dendrogram created from the proteins encoded in these genomes. Bootstrap support for the MLSA is given at each node, and the bar indicates the number of nucleotide substitutions per site. GHs were divided into categories based on their predicted substrate as follows: oligosaccharides, GH1, GH2, GH3, GH13, GH15, GH28, GH29, GH31, GH35, GH36, GH39, GH42, GH43, and GH92; hemicelluloses, GH8, GH10, GH26, GH51, GH53, and GH105; chitin and peptidoglycan, GH18, GH20, GH23, GH24, GH25, and GH73; cellulose, GH5, GH6, GH9, and GH44; other, all other GH families.
General genomic organization and selfish genetic elements.
Statistics for the 26 genomes analyzed in this study can be found in Table 1. Considerable variation in the organization of the seven complete genomes was observed (Table 2). For example, Sphingobium chlorophenolicum L-1 and Sphingobium japonicum UT26S contain two chromosomes, while all other complete genomes contain only one. Synteny analysis revealed only a small degree of conserved gene order between the complete genomes (see Fig. S1 in the supplemental material). As noted previously (38), a small degree of shared gene order is present between Sphingobium japonicum UT26S and Sphingobium chlorophenolicum L-1. These genomes appear to have undergone multiple chromosomal rearrangements since their divergence, and neither shares identifiable synteny with strain SYK-6. Also, only a small degree of synteny was observed between the chromosomes of Novosphingobium aromaticivorans DSM 12444 and Novosphingobium sp. strain PP1Y (see Fig. S1).
Table 1.
Statistics and isolation information for the 26 sphingomonad genomes
| Organism | Genome size (Mbp) | Sequencing status | No. of chromosomes (plasmids) | No. of 16S operons | Isolation source | Reference |
|---|---|---|---|---|---|---|
| Novosphingobium | ||||||
| Novosphingobium aromaticivorans DSM 12444 | 4.2 | Complete | 1 (2) | 3 | Subsurface sediment | 55 |
| Novosphingobium nitrogenifigens Y88T | 4.2 | Draft | 1 | Paper mill wastewater | 56 | |
| Novosphingobium pentaromativorans US6-1 | 5.3 | Draft | 1 | Ulsan Bay sediment | 57 | |
| Novosphingobium sp. strain AP12 | 5.6 | Draft | 1 | Cottonwood rhizosphere | 58 | |
| Novosphingobium sp. strain PP1Y | 5.3 | Complete | 1 (3) | 3 | Seawater | 59 |
| Novosphingobium sp. strain Rr 2-17 | 4.5 | Draft | 1 | Grapevine crown gall | 60 | |
| Sphingopyxis | ||||||
| Sphingopyxis alaskensis RB2256 | 3.4 | Complete | 1 (1) | 1 | Seawater | 46 |
| Sphingobium | ||||||
| Sphingobium chlorophenolicum L-1 | 4.6 | Complete | 2 (1) | 3 | Soil | 38 |
| Sphingobium indicum B90A | 4.1 | Draft | 2 | Sugarcane rhizosphere | 61 | |
| Sphingobium japonicum UT26S | 4.4 | Complete | 2 (3) | 3 | Contaminated soil | 49 |
| Sphingobium sp. strain AP49 | 4.5 | Draft | 1 | Cottonwood rhizosphere | 58 | |
| Sphingobium sp. strain SYK-6 | 4.3 | Complete | 1 (1) | 2 | Pulp mill wastewater | 41 |
| Sphingobium yanoikuyae XLDN2-5 | 5.4 | Draft | 1 | Contaminated soil | 62 | |
| Sphingomonas | ||||||
| Sphingomonas echinoides ATCC 14820 | 4.2 | Draft | 1 | Plate contaminant | 63 | |
| Sphingomonas elodea ATCC 31461 | 4.1 | Draft | 1 | Plant (Elodea) tissue | 64 | |
| Sphingomonas sp. strain KC8 | 4.1 | Draft | 1 | Activated sludge | 4 | |
| Sphingomonas sp. strain Mn802 | 3.2 | Draft | 1 | Termite | This study | |
| Sphingomonas sp. strain PR090111-T3T-6A | 3.9 | Draft | 1 | Termite | This study | |
| Sphingomonas sp. strain S17 | 4.3 | Draft | 1 | Hypersaline lake | 6 | |
| Sphingomonas sp. strain SKA58 | 3.9 | Draft | 4 | Seawater | NA | |
| Sphingomonas sp. strain ATCC 31555 | 4.0 | Draft | 1 | Pond water | 65 | |
| Sphingomonas sp. strain PAMC 26605 | 4.7 | Draft | 1 | Arctic lichen | 66 | |
| Sphingomonas sp. strain PAMC 26617 | 4.7 | Draft | 1 | Arctic lichen | 67 | |
| Sphingomonas sp. strain PAMC 26621 | 4.8 | Draft | 2 | Arctic lichen | 68 | |
| Sphingomonas wittichii DP58 | 5.6 | Draft | 1 | Pimiento rhizosphere | 69 | |
| Sphingomonas wittichii RW1 | 5.9 | Complete | 1 (2) | 2 | Elbe River | 3 |
Table 2.
Size and number of encoded selfish genetic elements and biodegradative enzymes in the 7 complete sphingomonad genomes
| Organism | Size (kb) | No. of: |
|||||
|---|---|---|---|---|---|---|---|
| Putative prophage | Phage-like proteins (% of total proteins) | Transposons | Monooxygenases | Dioxygenases | CAZymes | ||
| Novosphingobium aromaticivorans DSM 12444 | |||||||
| Chromosome | 3,561.6 | 5 | 309 (9.3) | 16 | 13 | 19 | 58 |
| Plasmid pNL1 | 184.5 | 1 | 1 | 1 | 13 | 1 | |
| Plasmid pNL2 | 487.3 | 2 | 3 | 8 | |||
| Novosphingobium sp. strain PP1Y | |||||||
| Chromosome | 3,911.5 | 3 | 113 (3.3) | 24 | 10 | 33 | 36 |
| Plasmid Lpl | 192.1 | 1 | 5 | 2 | |||
| Plasmid Spl | 48.7 | 1 | 1 | ||||
| Plasmid Mpl | 1,161.6 | 17 | 5 | 6 | 41 | ||
| Sphingobium chlorophenolicum L-1 | |||||||
| Chromosome 1 | 3,080.8 | 1 | 107 (3.8) | 15 | 9 | 4 | 30 |
| Chromosome 2 | 1,368.7 | 1 | 74 (6.7) | 8 | 6 | 15 | 17 |
| Plasmid pSPHCH01 | 123.7 | 2 | 1 | ||||
| Sphingobium japonicum UT26S | |||||||
| Chromosome 1 | 3,514.8 | 2 | 95 (2.7) | 37 | 9 | 6 | 28 |
| Chromosome 2 | 681.9 | 7 | 2 | 2 | 22 | ||
| Plasmid pUT1 | 31.8 | 5 | |||||
| Plasmid pUT2 | 5.4 | ||||||
| Plasmid pPCHQ1 | 191.0 | 2 | 6 | 2 | 2 | ||
| Sphingobium sp. strain SYK-6 | |||||||
| Chromosome | 4,199.3 | 1 | 88 (2.3) | 29 | 16 | 23 | 25 |
| Plasmid pSLPG | 148.8 | 6 | 1 | ||||
| Sphingomonas wittichii RW1 | |||||||
| Chromosome | 5,382.26 | 2 | 183 (3.8) | 36 | 42 | 38 | 33 |
| Plasmid pSWIT01 | 310.2 | 2 | 9 | 2 | 1 | ||
| Plasmid pSWIT02 | 222.8 | 1 | 31 | 1 | 6 | 2 | |
| Sphingopyxis alaskensis RB2256 | |||||||
| Chromosome | 3,345.2 | 1 | 62 (2.0) | 36 | 9 | 7 | 33 |
| F plasmid | 28.5 | ||||||
All of the complete genomes, except that of Sphingopyxis alaskensis RB2256, contained megaplasmids (plasmids of >100 kb [39]), consistent with previous suggestions that these replicons are common in sphingomonads (40). Although some megaplasmids were comparable in size to the secondary chromosomes of Sphingobium chlorophenolicum L-1 and Sphingobium japonicum UT26S, these replicons do not encode 16S rRNA operons or translational machinery characteristic of chromosomes (see the Translation category in Fig. S2 in the supplemental material). Furthermore, the metabolic potential encoded on the different replicons is markedly different, with the vast majority of proteins involved in essential processes such as amino acid and coenzyme metabolism encoded on the chromosomes, while hypothetical proteins are more commonly encoded on megaplasmids and plasmids (see Fig. S2).
We identified type IV secretion system components encoded in the majority of the genomes (see Table S2 in the supplemental material), raising the possibility for natural transformation or plasmid-mediated gene transfer as mechanisms for gene exchange. Components of the type IV apparatus are encoded on the megaplasmid of each megaplasmid-containing bacterium except Novosphingobium aromaticivorans DSM 12444, suggesting that these replicons may encode their own mechanisms for transfer. Experiments demonstrating the transfer of the 184.5-kb megaplasmid pNL1 of Novosphingobium aromaticivorans DSM 12444 to other sphingomonads have previously been reported (40), and these experiments also showed rapid recombination between the transferred plasmid and the chromosome of the new host. Moreover, ∼120 open reading frames (ORFs) with shared synteny and >98% amino acid identity are found on plasmid pSLGB of Sphingobium sp. strain SYK-6, the main chromosome of Sphingobium japonicum UT26S, and plasmid pSPHCH01 of Sphingobium chlorophenolicum L-1 (previously reported in references 38 and 41), suggesting recent transfer events. The transfer of essential genes to plasmids has been postulated to give rise to secondary chromosomes in other bacteria (42), providing a potential explanation for the multichromosomal arrangement in Sphingobium chlorophenolicum L-1 and Sphingobium japonicum UT26S.
Other selfish genetic elements, such as prophage and transposons, were also prevalent features of the sphingomonads. The program PhiSpy (27) identified several prophage encoded on the chromosomes (Table 2), with the highest number predicted from Novosphingobium aromaticivorans DSM 12444: 5 putative prophage are predicted to contribute 9.3% of all proteins encoded by this bacterium. Novosphingobium sp. strain PP1Y encoded 3 prophage contributing 113 protein-coding genes to its chromosome. One or 2 putative prophage were also identified in all chromosomes of the complete genomes, with the exception of the smaller chromosome of Sphingobium japonicum UT26S (Table 2). COG-based annotation also identified a highly variable number of transposons in the sphingomonads, with between 2 and 65 of these elements encoded in the complete and draft genomes (Table 2; see also Data Set S2 in the supplemental material). The largest numbers were found in Sphingomonas sp. strain PAMC 26621, Novosphingobium sp. strain Rr 2-17, and Sphingobium yanoikuyae XLDN2-5 (63 to 65 each), while the fewest were identified in the two termite-associated Sphingomonas strains (Mn802 and PR090111-T3T-6A).
Pan-genomics of the sphingomonads.
To investigate the pan-genome of the sphingomonads, we used Prodigal (20) to identify a total of 110,698 proteins. Comparison of proteins encoded in the sphingomonad genomes to the eggNOG Database (21), which contains previously generated protein clusters from bacterial genomes, revealed a total of 23,087 protein clusters, including 268 that were universally conserved between all genomes and 492 that were conserved between the complete genomes. These genes consisted primarily of ribosomal proteins and proteins involved in replication, DNA repair, amino acid metabolism, transcription, and other basic metabolic functions (see Fig. S3 in the supplemental material).
Although numerous genus-specific protein clusters were identified in our analyses (Fig. 2A), the majority of these were represented only in one or a few genomes within the genera (Fig. 2B), indicating that they do not represent features that consistently distinguish the sphingomonad genera. Note that because the genome of Sphingopyxis alaskensis RB2256 is the only representative of its genus, it was not possible to ascertain whether the protein clusters unique to this bacterium are representative of other Sphingopyxis isolates. To investigate consistent differences in coding potential between sphingomonad genera for which multiple genomes are available, we identified protein clusters that were overrepresented in any of the genera Sphingomonas, Sphingobium, and Novosphingobium compared to the other two combined (Fisher's exact test, P < 0.01). Using this approach, we identified 87 protein clusters overrepresented in Sphingomonas, 163 in Sphingobium, and 145 in Novosphingobium (details are in Data Set S1 in the supplemental material). Interestingly, most of these clusters have no COG annotation, have a general or unknown functional prediction, or are associated with DNA processing (see Fig. S4 in the supplemental material), suggesting that they encode selfish genetic elements rather than conserved metabolic processes.
Fig 2.

(A) Venn diagram showing the number of shared and unique protein clusters identified in the 4 sphingomonad genera. (B) Distribution of protein clusters identified in the complete and draft sphingomonad genomes. The x axis represents the number of genomes encoding a member of a protein cluster, and the y axis represents the total number of clusters having that many genomes represented.
Consistent with intergenus findings, the majority of protein clusters identified as highly variable are predicted to encode transposases, phage-related proteins, or proteins that could not be annotated (Fig. 3). Some of these variably distributed proteins encode glycoside hydrolases (GHs) and oxygenases, suggesting that sphingomonad genomes may be particularly affected by adaptations related to changes in biodegradative capabilities. Clustering analysis of the variable protein clusters revealed that many were represented in specific genera or species, suggesting that shared ancestry may be responsible for their distribution (Fig. 3). Sphingomonas wittichii strains RW1 and DP58, for example, both encoded multiple proteins belonging to clusters annotated as outer membrane receptors, dehydrogenases, and transposases (Fig. 3). Overall, however, our pan-genomic analysis revealed no large-scale differences in coding potential that distinguish the four sphingomonad genera.
Fig 3.

Heat map representing the sphingomonad protein clusters with the most variable distributions across the 26 genomes. The dendrogram was constructed using the pairwise Pearson's r correlation calculated for the protein cluster membership distributions (raw values in Data Set S1 in the supplemental material).
Aromatic compound and polysaccharide degradation.
To investigate the capacity of sphingomonads to degrade aromatic compounds, we analyzed the encoded mono- and dioxygenases, enzymes that catalyze the ring cleavage step critical to aromatic compound degradation (43). The distribution of mono- and dioxygenases varied by an order of magnitude across the sphingomonad phylogeny, with between 4 and 43 monooxygenases and 3 and 46 dioxygenases encoded (Fig. 1; see also Table S3 in the supplemental material). Both Sphingomonas wittichii RW1 and DP58 encoded the largest complement of oxygenases, although Sphingobium chlorophenolicum L-1, Sphingobium sp. strain SYK-6, and all Novosphingobium species except for Novosphingobium nitrogenifigens Y88T also encoded a large complement of these enzymes (see Table S3). The most abundant monooxygenase families identified included nitronate monooxygenase (NMO, K000459), vanillate monooxygenase (VanA, K03862), and alkanesulfonate monooxygenase (SsuD, K04091), while the most abundant dioxygenase families included taurine dioxygenase (TauD, K03119) and 4-hydroxyphenylpyruvate dioxygenase (HppD, K00457) (Table 3). Both Spingomonas wittichii RW1 and DP58 contained the largest number of predicted vanillate monooxygenases (24 and 19, respectively) as well as multiple dioxin dioxygenases (HcaA homologs, K05708), with genes for the latter being generally absent from the other sphingomonad genomes (see Data Set S2 in the supplemental material).
Table 3.
Selected catabolic enzymes identified in the sphingomonad genomesa
| Organism | No. of enzymes |
|||||||
|---|---|---|---|---|---|---|---|---|
| Monooxygenases |
Dioxygenases |
Cellulase families |
||||||
| NMO (K00459) | VanA (K03862) | SsuD (K04091) | TauD (K03119) | HppD (K00457) | GH5 | GH6 | GH9 | |
| Novosphingobium | ||||||||
| Novosphingobium aromaticivorans DSM 12444 | 4 | 4 | 1 | 2 | 2 | |||
| Novosphingobium nitrogenifigens Y88T | 3 | 1 | 2 | 3 | 1 | |||
| Novosphingobium pentaromativorans US6-1 | 6 | 5 | 2 | 2 | 2 | |||
| Novosphingobium sp. strain AP12 | 5 | 16 | 17 | 1 | ||||
| Novosphingobium sp. strain PP1Y | 3 | 2 | 1 | 1 | 2 | 1 | 1 | |
| Novosphingobium sp. strain Rr 2-17 | 3 | 2 | 5 | 1 | ||||
| Sphingopyxis | ||||||||
| Sphingopyxis alaskensis RB2256 | 6 | 1 | 1 | 1 | ||||
| Sphingobium | ||||||||
| Sphingobium chlorophenolicum L-1 | 7 | 1 | 1 | 6 | 1 | |||
| Sphingobium indicum B90A | 5 | 1 | 1 | |||||
| Sphingobium japonicum UT26S | 7 | 2 | 1 | 1 | ||||
| Sphingobium sp. strain AP49 | 4 | 1 | 2 | 4 | 1 | |||
| Sphingobium sp. strain SYK-6 | 2 | 7 | 7 | 1 | ||||
| Sphingobium yanoikuyae XLDN2-5 | 4 | 2 | 4 | 2 | 2 | |||
| Sphingomonas | ||||||||
| Sphingomonas echinoides ATCC 14820 | 2 | 1 | 1 | |||||
| Sphingomonas elodea ATCC 31461 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | |
| Sphingomonas sp. strain KC8 | 4 | 1 | 2 | 1 | ||||
| Sphingomonas sp. strain Mn802 | 2 | 1 | 1 | |||||
| Sphingomonas sp. strain PR090111-T3T-6A | 3 | 1 | 1 | |||||
| Sphingomonas sp. strain S17 | 3 | 2 | 1 | 2 | ||||
| Sphingomonas sp. strain SKA58 | 3 | 1 | 1 | 1 | ||||
| Sphingomonas sp. strain ATCC 31555 | 2 | 1 | 1 | 2 | 2 | |||
| Sphingomonas sp. strain PAMC 26605 | 3 | 3 | 1 | 1 | 1 | |||
| Sphingomonas sp. strain PAMC 26617 | 2 | 2 | 1 | 2 | 4 | 2 | ||
| Sphingomonas sp. strain PAMC 26621 | 2 | 2 | 1 | 2 | 2 | |||
| Sphingomonas wittichii DP58 | 4 | 19 | 5 | 8 | 1 | |||
| Sphingomonas wittichii RW1 | 5 | 24 | 4 | 7 | 1 | |||
NMO, nitranoate monooxygenase; VanA, vanillate monooxygenase; SsuD, alkanesulfonate monooxygenase; TauD, taurine dioxygenase; HppD, 4-hydroxyphenylpyruvate dioxygenase.
The abundance of tauD and ssuD homologs in many of the genomes suggests that sulfonated compounds may be sources of both carbon and sulfur for many of the sphingomonads. Bacteria have been shown to use a variety of organosulfur compounds as carbon and sulfur sources, and these compounds are abundant in soil and aquatic environments, as well as in environmental pollutants and fossil fuels (44). Novosphingobium sp. strain AP12, isolated from the rhizosphere of the cottonwood tree (Populus deltoides), for example, contained a particularly large amount of these genes (16 ssuD homologs and 17 tauD homologs). Homologs to the sulfonate transport apparatus (ssuABC) were also identified in strain AP12 as well as all of the soil- or rhizosphere-associated Sphingobium species (S. chlorophenolicum L-1, S. japonicum UT26S, strain AP49, and S. yanoikuyae XLDN2-5) (see Table S2 in the supplemental material). Interestingly, S. japonicum UT26S contained all ssuABC homologs but no ssuD dioxygenase, suggesting that it may degrade sulfur-containing compounds using alternative enzymes (Table 3; see also Table S2 and Data Set S3 in the supplemental material).
In contrast to ssuD and tauD, the presence of nitronate monooxygenase (NMO, K00457) gene homologs in the sphingomonad genomes was more consistent, with all genomes coding for between 2 and 7 copies. The largest complement of these enzymes was found in Sphingobium chlorophenolicum L-1 and Sphingobium japonicum UT26S. NMO preferentially acts on nitroalkanes, raising the possibility that these compounds may be naturally occurring sources of both nitrogen and carbon to some sphingomonads.
Many of the sphingomonads contained a diversity of GHs, suggesting adaptations to degrade plant polysaccharides. Sphingomonas elodea ATCC 31461 and Sphingomonas sp. strain ATCC 31555 contained the largest complement of these enzymes (99 and 97, respectively), including cellulases and hemicellulases (Fig. 1). The genomes with the most predicted GHs also encoded representatives from the cellulase families GH5, GH6, and GH9 (Table 3), in support of their ability to hydrolyze cellulose. The genomes of these also encode a diversity of other GHs, carbohydrate esterases, and polysaccharide lyases that likely target pectins, xylans, mannans, and other plant cell wall polysaccharides (see Table S3 in the supplemental material). The isolates with the largest complement of hemicellulases and cellulases, Sphingomonas elodea ATCC 31461, Sphingomonas sp. ATCC 31555, Sphingobium yanoikuyae XLDN2-5, and Sphingobium sp. strain AP49, were isolated from environments rich in plant biomass, such as plant tissue, soil, and pond water, suggesting sphingomonad participation in the degradation of environmental plant biomass.
Among the sphingomonads with the fewest GHs (20 to 22 each) were strains encoding a large number of oxygenases (e.g., Sphingomonas wittichii strains RW1 and DP58 and Sphingobium sp. strain SYK-6 [Fig. 1; see also Table S3 in the supplemental material]), suggesting specialization in the degradation of aromatic compounds and an absence of the capacity to deconstruct recalcitrant polysaccharides. However, although Novosphingobium nitrogenifigens Y88T encoded only 17 GHs, it also lacked a substantial coding potential for oxygenases compared to other Novosphingobium strains (Fig. 1; see also Table S3), suggesting that the degradation of a wide diversity of recalcitrant carbon compounds may be less important for this strain than for other sphingomonads.
Analysis of the seven complete sphingomonad genomes revealed that many oxygenases and GHs are encoded on plasmids or megaplasmids (Table 2). In Novosphingobium sp. strain PP1Y, more GHs were encoded on the megaplasmid than the chromosome (41 versus 36), and the plasmids of Sphingomonas wittichii RW1 and Novosphingobium aromaticivorans DSM 12444 contain several dioxygenases. Moreover, the smaller of the two chromosomes in Sphingobium chlorophenolicum L-1 has been shown to encode the genes necessary to degrade the aromatic pesticide pentachlorophenol, and evidence suggests that three genes involved have arisen through multiple horizontal gene transfer events (38). Together with the high incidence of chromosomal rearrangements and plasmid-mediated gene transfers observed, this is consistent with the assertion that plasmids and other selfish genetic elements are likely responsible for frequent exchange of biodegradative capabilities in sphingomonads (14).
Adaptations to the environment.
All of the sphingomonad genomes contained between 1 and 4 copies of the 16S rRNA gene, with the majority containing one or two copies (Table 1). Previous work has shown that low 16S rRNA copy number is a characteristic commonly associated with bacteria that inhabit a specialized niche and do not rapidly respond to changing environmental conditions (45). Together with the specific sets of oxygenases and GHs encoded by most of the sphingomonads, it seems likely that many of these bacteria are specialized heterotrophs capable of utilizing components of refractory detritus or xenobiotics as carbon, sulfur, and phosphorus sources. This has previously been suggested for Sphingopyxis alaskensis RB2256 (46–48), Sphingobium japonicum UT26S (49), and sphingomonads residing in subsurface sediment, such as Novosphingobium aromaticivorans DSM 12444 (50). This is likely a successful ecological strategy in oligotrophic environments, where few alternative nutrient sources would be available and where other microbes would likely not possess the enzymatic machinery necessary to compete for their use. Thus, rather than remaining dormant for long periods and doubling rapidly when environmental conditions become favorable, sphingomonads may be adapted to specialize in the utilization of particular recalcitrant compounds in oligotrophic conditions.
Adaptation to oligotrophic conditions has already been shown for the marine bacterium Sphingopyxis alaskensis RB2256, an important contributor to nutrient cycling in marine ecosystems (46, 47, 51). This ultramicrobacterium, which maintains an extremely small cell volume (<0.08 μm3), has adapted to persist in seawater containing extremely low concentrations of amino acids and other nutrients (5, 47). Previous analyses have suggested that this genome has not undergone the same degree of genomic reduction as has been observed in other marine ultramicrobacteria (47, 52). This species has the second-smallest genome of all of the sphingomonads analyzed in this study (smallest among the complete genomes) and is the only complete sphingomonad genome lacking a megaplasmid, suggesting that it may in fact have undergone a considerable amount of genomic streamlining compared to other sphingomonads.
Despite the apparent adaptations of many sphingomonads to oligotrophic conditions, many of these bacteria can be found in host-associated environments that are likely more nutrient rich. Many of the species analyzed in this study were isolated from environments including termites, lichen, plant tissue, or the rhizosphere (Table 1). Type IV secretion systems, implicated in host-microbe interactions in other Alphaproteobacteria (53), were identified in most of the sphingomonad genomes (see Table S2 in the supplemental material), raising the possibility that these systems are used during host interaction, in addition to their likely role in DNA transfer. Many of these associations may, however, be short term and not involve host-microbe specificity. For example, some of the sphingomonads isolated from the rhizosphere, such as Novosphingobium sp. strain AP49 and Sphingobium sp. strain AP12, contain genes for organosulfur and nitroaromatic compound utilization, indicating that they may subsist on recalcitrant compounds or secondary metabolites produced by potential plant associates. These bacteria may therefore degrade these compounds in the soil or similar environments, without the necessity for any specific association with metazoan hosts.
Two Sphingomonas species sequenced as part of this study, strain Mn802 and strain PR090111-T3T-6A, were isolated from termites (see Table S1 in the supplemental material). Strain PR090111-T3T-6A encodes 80 CAZymes, suggesting that it may play a role in the degradation of the copious quantities of plant biomass ingested by its potential termite host. Strain Mn802, however, encodes relatively few CAZymes and has the smallest genome size of all the sphingomonads, suggesting that it may have reduced biodegradative capabilities and subsist on easily accessible nutrients readily available in the termite environment. Sphingomonads have previously been isolated from termites (54), indicating that members of this group may have a consistent but as-yet-uncharacterized termite association. If this is correct, our analyses suggest that termite-associated sphingomonads do not all partake in the breakdown of recalcitrant plant material but rather may perform alternative functions in the associations.
Conclusions.
Consistent with previous biochemical characterizations and analyses of 16S rRNA genes (37), the genera Sphingomonas, Sphingobium, Novosphingobium, and Sphingopyxis form well-resolved phylogenetic clades at the whole-genome level, although Sphingomonas sp. strain SKA58 appears to belong in the genus Sphingobium. A large number of species-specific genes were present in the sphingomonad genomes, and few genomic features can reliably distinguish the genera. Selfish genetic elements appear to be prominent forces shaping this genome evolution, and megaplasmids, prophage, transposons, and frequent chromosomal rearrangements are prevalent features in the group. Many sphingomonads appear to successfully occupy oligotrophic niches and utilize recalcitrant compounds as sources of carbon, nitrogen, and sulfur. Species with few oxygenases or GHs may have evolved to live in more nutrient-rich environments, including host associations, while others may have undergone genomic streamlining to adapt to extreme oligotrophic conditions. As more information on the distribution and catabolic activities of the sphingomonads emerges, insights into their ecology will greatly advance our understanding of their impact on global nutrient cycles and potential applications to biotechnology.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494), supporting F.O.A., S.M.A., A.V., B.R.M., R.A.S., C.R.C., G.S., and M.P., and by a STENO stipend awarded by the Danish Agency for Science, Technology and Innovation to M.P. The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.
We thank W. de Beer, M. Wingfield, and the Forestry and Agricultural Biotechnology Institute, University of Pretoria, for assistance and three anonymous reviewers for helpful suggestions to improve this work.
Footnotes
Published ahead of print 5 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00518-13.
REFERENCES
- 1. Balkwill DL, Fredrickson J, Romine M. 2006. The prokaryotes: a handbook on the biology of bacteria, p 605–629 Springer, Singapore [Google Scholar]
- 2. Ryan MP, Adley CC. 2010. Sphingomonas paucimobilis: a persistent Gram-negative nosocomial infectious organism. J. Hosp. Infect. 75:153–157 [DOI] [PubMed] [Google Scholar]
- 3. Miller TR, Delcher AL, Salzberg SL, Saunders E, Detter JC, Halden RU. 2010. Genome sequence of the dioxin-mineralizing bacterium Sphingomonas wittichii RW1. J. Bacteriol. 192:6101–6102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu A, He J, Chu KH, Yu CP. 2011. Genome sequence of the 17β-estradiol-utilizing bacterium Sphingomonas strain KC8. J. Bacteriol. 193:4266–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eguchi M, Ostrowski M, Fegatella F, Bowman J, Nichols D, Nishino T, Cavicchioli R. 2001. Sphingomonas alaskensis strain AFO1, an abundant oligotrophic ultramicrobacterium from the North Pacific. Appl. Environ. Microbiol. 67:4945–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Farias ME, Revale S, Mancini E, Ordonez O, Turjanski A, Cortez N, Vazquez MP. 2011. Genome sequence of Sphingomonas sp. S17, isolated from an alkaline, hyperarsenic, and hypersaline volcano-associated lake at high altitude in the Argentinean Puna. J. Bacteriol. 193:3686–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coronado E, Roggo C, Johnson DR, van der Meer JR. 2012. Genome-wide analysis of salicylate and dibenzofuran metabolism in Sphingomonas wittichii RW1. Front. Microbiol. 3:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rentz JA, Alvarez PJ, Schnoor JL. 2008. Benzo(a)pyrene degradation by Sphingomonas yanoikuyae JAR02. Environ. Pollut. 151:669–677 [DOI] [PubMed] [Google Scholar]
- 9. Wittich RM, Wilkes H, Sinnwell V, Francke W, Fortnagel P. 1992. Metabolism of dibenzo-p-dioxin by Sphingomonas sp. strain RW1. Appl. Environ. Microbiol. 58:1005–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schmidt S, Wittich RM, Erdmann D, Wilkes H, Francke W, Fortnagel P. 1992. Biodegradation of diphenyl ether and its monohalogenated derivatives by Sphingomonas sp. strain SS3. Appl. Environ. Microbiol. 58:2744–2750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sugimoto M, Tanabe M, Hataya M, Enokibara S, Duine JA, Kawai F. 2001. The first step in polyethylene glycol degradation by sphingomonads proceeds via a flavoprotein alcohol dehydrogenase containing flavin adenine dinucleotide. J. Bacteriol. 183:6694–6698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harms H, Wilkes H, Wittich R, Fortnagel P. 1995. Metabolism of hydroxydibenzofurans, methoxydibenzofurans, acetoxydibenzofurans, and nitrodibenzofurans by Sphingomonas sp. strain HH69. Appl. Environ. Microbiol. 61:2499–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilkes H, Wittich R, Timmis KN, Fortnagel P, Francke W. 1996. Degradation of chlorinated dibenzofurans and dibenzo-p-dioxins by Sphingomonas sp. strain RW1. Appl. Environ. Microbiol. 62:367–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stolz A. 2009. Molecular characteristics of xenobiotic-degrading sphingomonads. Appl. Microbiol. Biotechnol. 81:793–811 [DOI] [PubMed] [Google Scholar]
- 15. Masai E, Katayama Y, Nishikawa S, Fukuda M. 1999. Characterization of Sphingomonas paucimobilis SYK-6 genes involved in degradation of lignin-related compounds. J. Ind. Microbiol. Biotechnol. 23:364–373 [DOI] [PubMed] [Google Scholar]
- 16. Sutherland IW, Kennedy L. 1996. Polysaccharide lyases from gellan-producing Sphingomonas spp. Microbiology 142(Part 4):867–872 [DOI] [PubMed] [Google Scholar]
- 17. Hashimoto W, Miyake O, Momma K, Kawai S, Murata K. 2000. Molecular identification of oligoalginate lyase of Sphingomonas sp. strain A1 as one of the enzymes required for complete depolymerization of alginate. J. Bacteriol. 182:4572–4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hsu SC, Lockwood JL. 1975. Powdered chitin agar as a selective medium for enumeration of actinomycetes in water and soil. Appl. Microbiol. 29:422–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, Jensen LJ, von Mering C, Bork P. 2012. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 40:D284–D289 doi: 10.1093/nar/gkr1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14:755–763 [DOI] [PubMed] [Google Scholar]
- 23. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35:W182–W185 doi: 10.1093/nar/gkm321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29:22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akhter S, Aziz RK, Edwards RA. 2012. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 40:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. 2008. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36:D480–D484 doi: 10.1093/nar/gkm882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37:D233–D238 doi: 10.1093/nar/gkn663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suen G, Scott JJ, Aylward FO, Adams SM, Tringe SG, Pinto-Tomas AA, Foster CE, Pauly M, Weimer PJ, Barry KW, Goodwin LA, Bouffard P, Li L, Osterberger J, Harkins TT, Slater SC, Donohue TJ, Currie CR. 2010. An insect herbivore microbiome with high plant biomass-degrading capacity. PLoS Genet. 6:e1001129 doi: 10.1371/journal.pgen.1001129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Katoh K, Toh H. 2010. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26:1899–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690 [DOI] [PubMed] [Google Scholar]
- 33. Konstantinidis KT, Tiedje JM. 2005. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187:6258–6264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Felsenstein J. 1989. PHYLIP—Phylogeny Inference Package (version 3.2). Cladistics 5:164–166 [Google Scholar]
- 35. Letunic I, Bork P. 2007. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128 [DOI] [PubMed] [Google Scholar]
- 36. R Development Core Team 2008. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 37. Takeuchi M, Hamana K, Hiraishi A. 2001. Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int. J. Syst. Evol. Microbiol. 51:1405–1417 [DOI] [PubMed] [Google Scholar]
- 38. Copley SD, Rokicki J, Turner P, Daligault H, Nolan M, Land M. 2012. The whole genome sequence of Sphingobium chlorophenolicum L-1: insights into the evolution of the pentachlorophenol degradation pathway. Genome Biol. Evol. 4:184–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwartz E. 2009. Microbial megaplasmids. Springer-Verlag, Berlin, Germany [Google Scholar]
- 40. Basta T, Keck A, Klein J, Stolz A. 2004. Detection and characterization of conjugative degradative plasmids in xenobiotic-degrading Sphingomonas strains. J. Bacteriol. 186:3862–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Masai E, Kamimura N, Kasai D, Oguchi A, Ankai A, Fukui S, Takahashi M, Yashiro I, Sasaki H, Harada T, Nakamura S, Katano Y, Narita-Yamada S, Nakazawa H, Hara H, Katayama Y, Fukuda M, Yamazaki S, Fujita N. 2012. Complete genome sequence of Sphingobium sp. strain SYK-6, a degrader of lignin-derived biaryls and monoaryls. J. Bacteriol. 194:534–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Slater SC, Goldman BS, Goodner B, Setubal JC, Farrand SK, Nester EW, Burr TJ, Banta L, Dickerman AW, Paulsen I, Otten L, Suen G, Welch R, Almeida NF, Arnold F, Burton OT, Du Z, Ewing A, Godsy E, Heisel S, Houmiel KL, Jhaveri J, Lu J, Miller NM, Norton S, Chen Q, Phoolcharoen W, Ohlin V, Ondrusek D, Pride N, Stricklin SL, Sun J, Wheeler C, Wilson L, Zhu H, Wood DW. 2009. Genome sequences of three Agrobacterium biovars help elucidate the evolution of multichromosome genomes in bacteria. J. Bacteriol. 191:2501–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harayama S, Kok M, Neidle EL. 1992. Functional and evolutionary relationships among diverse oxygenases. Annu. Rev. Microbiol. 46:565–601 [DOI] [PubMed] [Google Scholar]
- 44. Kertesz MA. 2000. Riding the sulfur cycle-metabolism of sulfonates and sulfate esters in gram-negative bacteria. FEMS Microbiol. Rev. 24:135–175 [DOI] [PubMed] [Google Scholar]
- 45. Klappenbach JA, Dunbar JM, Schmidt TM. 2000. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66:1328–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lauro FM, McDougald D, Thomas T, Williams TJ, Egan S, Rice S, DeMaere MZ, Ting L, Ertan H, Johnson J, Ferriera S, Lapidus A, Anderson I, Kyrpides N, Munk AC, Detter C, Han CS, Brown MV, Robb FT, Kjelleberg S, Cavicchioli R. 2009. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. U. S. A. 106:15527–15533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Williams TJ, Ertan H, Ting L, Cavicchioli R. 2009. Carbon and nitrogen substrate utilization in the marine bacterium Sphingopyxis alaskensis strain RB2256. ISME J. 3:1036–1052 [DOI] [PubMed] [Google Scholar]
- 48. Fegatella F, Lim J, Kjelleberg S, Cavicchioli R. 1998. Implications of rRNA operon copy number and ribosome content in the marine oligotrophic ultramicrobacterium Sphingomonas sp. strain RB2256. Appl. Environ. Microbiol. 64:4433–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nagata Y, Natsui S, Endo R, Ohtsubo Y, Ichikawa N, Ankai A, Oguchi A, Fukui S, Fujita N, Tsuda M. 2011. Genomic organization and genomic structural rearrangements of Sphingobium japonicum UT26, an archetypal gamma-hexachlorocyclohexane-degrading bacterium. Enzyme Microb. Technol. 49:499–508 [DOI] [PubMed] [Google Scholar]
- 50. Fredrickson JK, Balkwill DL, Romine MF, Shi T. 1999. Ecology, physiology, and phylogeny of deep subsurface Sphingomonas sp. J. Ind. Microbiol. Biotechnol. 23:273–283 [DOI] [PubMed] [Google Scholar]
- 51. Cavicchioli R, Ostrowski M, Fegatella F, Goodchild A, Guixa-Boixereu N. 2003. Life under nutrient limitation in oligotrophic marine environments: an eco/physiological perspective of Sphingopyxis alaskensis (formerly Sphingomonas alaskensis). Microb. Ecol. 45:203–217 [DOI] [PubMed] [Google Scholar]
- 52. Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M, Rappe MS, Short JM, Carrington JC, Mathur EJ. 2005. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309:1242–1245 [DOI] [PubMed] [Google Scholar]
- 53. Hubber A, Vergunst AC, Sullivan JT, Hooykaas PJ, Ronson CW. 2004. Symbiotic phenotypes and translocated effector proteins of the Mesorhizobium loti strain R7A VirB/D4 type IV secretion system. Mol. Microbiol. 54:561–574 [DOI] [PubMed] [Google Scholar]
- 54. Wenzel M, Schonig I, Berchtold M, Kampfer P, Konig H. 2002. Aerobic and facultatively anaerobic cellulolytic bacteria from the gut of the termite Zootermopsis angusticollis. J. Appl. Microbiol. 92:32–40 [DOI] [PubMed] [Google Scholar]
- 55. Balkwill DL, Drake GR, Reeves RH, Fredrickson JK, White DC, Ringelberg DB, Chandler DP, Romine MF, Kennedy DW, Spadoni CM. 1997. Taxonomic study of aromatic-degrading bacteria from deep-terrestrial-subsurface sediments and description of Sphingomonas aromaticivorans sp. nov., Sphingomonas subterranea sp. nov., and Sphingomonas stygia sp. nov. Int. J. Syst. Bacteriol. 47:191–201 [DOI] [PubMed] [Google Scholar]
- 56. Strabala TJ, Macdonald L, Liu V, Smit AM. 2012. Draft genome sequence of Novosphingobium nitrogenifigens Y88T. J. Bacteriol. 194:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Luo YR, Kang SG, Kim SJ, Kim MR, Li N, Lee JH, Kwon KK. 2012. Genome sequence of benzo(a) pyrene-degrading bacterium Novosphingobium pentaromativorans US6-1. J. Bacteriol. 194:907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brown SD, Utturkar SM, Klingeman DM, Johnson CM, Martin SL, Land ML, Lu TY, Schadt CW, Doktycz MJ, Pelletier DA. 2012. Twenty-one genome sequences from Pseudomonas species and 19 genome sequences from diverse bacteria isolated from the rhizosphere and endosphere of Populus deltoides. J. Bacteriol. 194:5991–5993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. D'Argenio V, Petrillo M, Cantiello P, Naso B, Cozzuto L, Notomista E, Paolella G, Di Donato A, Salvatore F. 2011. De novo sequencing and assembly of the whole genome of Novosphingobium sp. strain PP1Y. J. Bacteriol. 193:4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gan HM, Chew TH, Hudson AO, Savka MA. 2012. Genome sequence of Novosphingobium sp. strain Rr 2-17, a nopaline crown gall-associated bacterium isolated from Vitis vinifera L. grapevine. J. Bacteriol. 194:5137–5138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Anand S, Sangwan N, Lata P, Kaur J, Dua A, Singh AK, Verma M, Kaur J, Khurana JP, Khurana P, Mathur S, Lal R. 2012. Genome sequence of Sphingobium indicum B90A, a hexachlorocyclohexane-degrading bacterium. J. Bacteriol. 194:4471–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gai Z, Wang X, Tang H, Tai C, Tao F, Wu G, Xu P. 2011. Genome sequence of Sphingobium yanoikuyae XLDN2-5, an efficient carbazole-degrading strain. J. Bacteriol. 193:6404–6405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shin SC, Kim SJ, Ahn do H, Lee JK, Park H. 2012. Draft genome sequence of Sphingomonas echinoides ATCC 14820. J. Bacteriol. 194:1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gai Z, Wang X, Zhang X, Su F, Wang X, Tang H, Tai C, Tao F, Ma C, Xu P. 2011. Genome sequence of Sphingomonas elodea ATCC 31461, a highly productive industrial strain of gellan gum. J. Bacteriol. 193:7015–7016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang X, Tao F, Gai Z, Tang H, Xu P. 2012. Genome sequence of the welan gum-producing strain Sphingomonas sp. ATCC 31555. J. Bacteriol. 194:5989–5990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shin SC, Ahn do H, Lee JK, Kim SJ, Hong SG, Kim EH, Park H. 2012. Genome sequence of Sphingomonas sp. strain PAMC 26605, isolated from arctic lichen (Ochrolechia sp.). J. Bacteriol. 194:1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee J, Shin SC, Kim SJ, Kim BK, Hong SG, Kim EH, Park H, Lee H. 2012. Draft genome sequence of a Sphingomonas sp., an endosymbiotic bacterium isolated from an arctic lichen Umbilicaria sp. J. Bacteriol. 194:3010–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee H, Shin SC, Lee J, Kim SJ, Kim BK, Hong SG, Kim EH, Park H. 2012. Genome sequence of Sphingomonas sp. strain PAMC 26621, an arctic-lichen-associated bacterium isolated from a Cetraria sp. J. Bacteriol. 194:3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ma Z, Shen X, Hu H, Wang W, Peng H, Xu P, Zhang X. 2012. Genome sequence of Sphingomonas wittichii DP58, the first reported phenazine-1-carboxylic acid-degrading strain. J. Bacteriol. 194:3535–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.