

Abstract
Tuberculosis remains one of the top three leading causes of morbidity and mortality worldwide, complicated by the emergence of drug-resistant Mycobacterium tuberculosis strains and high rates of HIV coinfection. It is important to develop new antimycobacterial drugs and immunomodulatory therapeutics and compounds that enhance antituberculous immunity. Dipterinyl calcium pentahydrate (DCP), a calcium-complexed pterin compound, has previously been shown to inhibit human breast cancer cells and hepatitis B virus (HBV). DCP inhibitory effects were attributed to induction of apoptosis and/or increased production of interleukin 12 (IL-12) and granulocyte-macrophage colony-stimulating factor (GM-CSF). In this study, we tested the ability of DCP to mediate inhibition of intracellular mycobacteria within human monocytes. DCP treatment of infected monocytes resulted in a significant reduction in viability of intracellular but not extracellular Mycobacterium bovis BCG. The antimicrobial activity of DCP was comparable to that of pyrazinamide (PZA), one of the first-line antituberculosis drugs currently used. DCP potentiated monocyte antimycobacterial activity by induction of the cysteine-cysteine (C-C) chemokine macrophage inflammatory protein 1β (MIP-1β) and inducible nitric oxide synthase 2. Addition of human anti-MIP-1β neutralizing antibody or a specific inhibitor of the l-arginase-nitric oxide pathway (NG-monomethyl l-arginine [l-NMMA] monoacetate) reversed the inhibitory effects of DCP on intracellular mycobacterial growth. These findings indicate that DCP induced mycobacterial killing via MIP-1β- and nitric oxide-dependent effects. Hence, DCP acts as an immunoregulatory compound enhancing the antimycobacterial activity of human monocytes.
INTRODUCTION
Tuberculosis (TB), caused by Mycobacterium tuberculosis, remains a global health crisis because the disease claims approximately 2 million lives annually, more than any other single bacterial disease. The usual route of M. tuberculosis infection is via the lung, where alveolar macrophages are the primary host targets for initial pathogen replication. Although innate and adaptive immune responses prevent the development of TB disease in about 90% of those infected, the latent state of M. tuberculosis infection in these individuals can result in TB reactivation disease when immunity is compromised, such as after coinfection with human immunodeficiency virus (HIV) (1–3).
The health burden of TB is exacerbated by the increasing emergence of multidrug-resistant (MDR) and extremely resistant (XDR) strains of M. tuberculosis (4–7). An estimated 440,000 cases of MDR-TB were reported to the World Health Organization (WHO) in 2008. MDR-TB is caused by M. tuberculosis resistant to at least the two first-line anti-TB drugs, isoniazid (PZA) and rifampin (RIF); XDR-TB is resistant to these two first-line anti-TB medications as well as any fluoroquinolone and aminoglycoside. Most recently, M. tuberculosis strains resistant to all currently available antituberculosis drugs have been reported (8, 9). The rising prevalence of drug-resistant tuberculosis and association with the HIV pandemic underscores the need for new antimycobacterial drugs and/or immunomodulatory therapeutics and compounds that enhance antituberculous immunity.
We employed an in vitro mycobacterial growth inhibition assay to assess the effects of a calcium-complexed pterin compound called dipterinyl calcium pentahydrate (DCP) on intracellular mycobacterial growth in human monocytes. Pterins are derivatives of pteridines, natural compounds involved in the biosynthesis of vitamins and cofactors required for various enzyme activities, containing a 2-amino-4-oxo heterocyclic structural backbone. For example, tetrahydrobiopterin (BH4), synthesized via de novo and salvage pathways from GTP and 7,8-dihydrobiopterin, respectively, is a cofactor of mammalian nitric oxide (NO) synthases (10–13). Neopterin, a pterin formed from dihydroneopterin triphosphate in the BH4 biosynthesis pathway, increases the inducible isoform of NO synthase (iNOS or NOS2) in rat vascular smooth muscle cells (14). iNOS catalyzes the production of NO in a variety of mammalian cells by metabolic conversion of l-arginine to l-citruline, producing reactive nitrogen intermediates (RNI) with important microbicidal effects against many human infectious diseases (15–18).
DCP previously has been shown to mediate antitumor and anti-hepatitis B virus (HBV) effects in mice (19–21). Ca2+-mediated induction of apoptosis in MDA-MB231 human breast tumor cells was proposed to be involved in DCP-induced antitumor effects. Moreover, DCP has immunomodulatory effects in vivo. DCP treatment of nude mice inoculated with MDA-MB231 tumor cells was shown to induce significant increases in plasma interleukin 12 (IL-12) (22). The anti-HBV effects of DCP were associated with increased granulocyte-macrophage colony-stimulating factor (GM-CSF) levels (19). Both IL-12 and GM-CSF, produced by a myriad of activated immune cell types, are important immunomodulators leading to the expansion and maturation of dendritic cells, differentiation of monocytes into macrophages, and increased macrophage activity (23, 24). IL-12 and GM-CSF are important for the induction of protective TB immunity. Mice lacking IL-12 or GM-CSF are unable to control growth of M. tuberculosis and succumb to infection (25, 26).
The purpose of the present study was to determine whether DCP could mediate antimycobacterial effects through either direct or immunomodulatory mechanisms similar to those associated with its antitumor and anti-HBV activities. We report herein that DCP significantly inhibited intracellular mycobacterial growth in human monocytes by enhancing production of the cysteine-cysteine (C-C) chemokine macrophage inflammatory protein 1β (MIP-1β) and the iNOS-NO effector pathway.
MATERIALS AND METHODS
Dipterinyl calcium pentahydrate and anti-TB drugs.
DCP (C6H4N5O)2Ca · 5H2O; molecular weight [MW], 454.4) was obtained from SanRx Pharmaceuticals, Inc., La Jolla, CA. It is a yellowish compound synthesized by mixing calcium and pterin, as previously described (20). Briefly, pure pterin, a derivative of pteridine with a 2-amino-4-oxo structure, was dissolved in distilled H2O and 0.1 N sodium hydroxide and CaCl2 · 2H2O was added, with constant stirring. The resulting yellowish precipitate was collected and dried. The molecular and X-ray crystallographic structures of DCP were reported previously (19, 20). The DCP suspension used in this study was prepared by suspending the compound in endotoxin-free deionized and distilled water (Lonza BioWhittaker; 17-724Q) and stored at −80°C until required for use in experiments.
The anti-TB drugs PZA (Sigma; P7136) and RIF (Sigma; R8883) were purchased from Sigma-Aldrich (St. Louis, MO).
Media and reagents.
Antibiotic-free RPMI 1640 (Gibco 11875) supplemented with 10% heat-inactivated human AB serum (Sigma; H4522) and 1% l-glutamine (Lonza BioWhittaker; 17-605E) (designated complete RPMI 1640 medium) was used for cell culture and dilution of stock DCP, PZA, and RIF for treatment of Mycobacterium bovis BCG- or M. tuberculosis H37Rv-infected monocytes. Anti-human MIP-1α (ab10381), anti-human MIP-1β (ab9675), and anti-human RANTES (ab9679) were obtained from Abcam Inc., Cambridge, MA, and used at concentrations previously shown to neutralize the bioactivities of MIP-1α/CCL3, MIP-1β/CCL4, and RANTES/CCL5, respectively. Rabbit polyclonal IgG (ab27478) was used as a control antibody. Anti-human gamma interferon (IFN-γ) (BD Biosciences 551221; clone NIB42) and anti-human tumor necrosis factor alpha (TNF-α) (BD Biosciences 551220; clone MAb1) were used to neutralize the bioactivity of IFN-γ and TNF-α, respectively. NG-monomethyl l-arginine (l-NMMA) monoacetate was purchased from Enzo Life Sciences (Plymouth Meeting, PA). Mycobacterium bovis BCG Connaught strain (ATCC 35745; TMC 1030) was grown in suspension with constant gentle rotation in roller bottles containing Middlebrook 7H9 broth (BD Difco; 271310) supplemented with 10% Middlebrook albumin, dextrose, and catalase (ADC) enrichment (BD Biosciences; 211887) and 0.05% Tween 80 (Sigma-Aldrich). When the bacterial density reached log-phase growth, mycobacteria were harvested and stored in phosphate-buffered saline (PBS) at −80°C. The concentration, expressed as CFU/ml, was quantified on Middlebrook 7H10 agar (BD Difco; 262710) plates. M. tuberculosis H37Rv (catalog number NR-123) was obtained from BEI Resources, Manassas, VA. Before being used for infection, bacteria were thawed and sonicated for l min in a cup sonicator (W385; Heat Systems Ultrasonics, Farmingdale, NY) to obtain a single-cell suspension and diluted appropriately in antibiotic-free complete RPMI 1640 medium.
Isolation of PBMCs and monocytes.
Human peripheral blood mononuclear cells (PBMCs) were obtained from healthy tuberculin skin test-negative donors by leukapheresis. Monocytes were purified from PBMCs by plastic adherence based on the unique adhesion properties of monocytes/macrophages among PBMC populations (27–29). Briefly, 1.5 × 105 or 1.5 × 106 PBMCs were plated in each well of 96-well round-bottom microtiter plates (Corning Inc.; Costar 3799). Nonadherent cells were washed off with complete RMPI 1640 medium after 2 h or overnight incubation at 37°C and 5% CO2. In general, approximately 10% of the initial number of PBMCs plated were retained as adherent monocytes (>90% CD14+ [data not shown]), which were then cultured for one more day before infection with M. bovis BCG or M. tuberculosis H37Rv.
Preparation of murine BM-derived macrophages.
Bone marrow (BM) cells were isolated from femora of wild-type C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME). To generate BM-derived macrophages (BMDMϕ), BM cells (2.5 × 104 cells/well) were cultured in 10% fetal bovine serum–Dulbecco modified Eagle medium (FBS-DMEM) (D10 medium) for 7 days with 20 ng/ml murine recombinant M-CSF (PeproTech; 315-02). After 7 days of culture, BMDMϕ formed a confluent monolayer, and the density of Mϕ was estimated to be 2.5 × 105 cells per well in 96-well plates (Corning Inc.; Costar 3595). BMDMϕ were infected with M. bovis BCG Connaught strain at a multiplicity of infection (MOI) of 3:1 (bacteria/BMDMϕ) in antibiotic-free D10 medium at 37°C in 5% CO2. After overnight culture, infected BMDMϕ were washed with antibiotic-free D10 medium to remove the extracellular M. bovis BCG before treatment with DCP.
Assay of DCP-mediated inhibition of intracellular mycobacterial growth.
Two different concentrations of adherent human monocytes were used (∼1.5 × 104 and ∼1.5 × 105/well in 96-well plates, designated low and high density, respectively). The assay was performed, with slight modifications, as previously described (29–31). The lower-density adherent monocytes were used in most experiments to assess the inhibitory effects of DCP. The higher-density adherent monocytes were used in some inhibition assays as well as in experiments harvesting total cellular RNA to investigate the molecular mechanisms involved in DCP-mediated inhibition of intracellular mycobacteria. Briefly, adherent monocytes were used at day 2 after in vitro culture in 96-well, round-bottom plates (Corning Inc.; Costar 3799). Monocytes were infected overnight with mycobacteria at an MOI of 0.3 or 3, and then extracellular bacilli were washed away. Infected cells were treated or not with DCP or control anti-TB drugs at 37°C and 5% CO2 for 72 h.
Determination of viable residual intracellular mycobacteria.
Following the 3 days of various treatments, monocytes were lysed with a final concentration of 0.2% saponin (Sigma S7900) in RPMI 1640 medium. After cell lysis, 1 μCi [5,6-3H]uridine (PerkinElmer Wallac Inc., Boston, MA; catalog number NET-367) in 7H9 Middlebrook broth supplemented with 10% ADC enrichment medium (BD Biosciences; 211887) was added per well, and plates were incubated for another 72 h in a 37°C, 5% CO2 incubator. Then BCG cells were harvested onto fiberglass filtermat, 90 by 120 mm (PerkinElmer Wallac Inc.; catalog number 1450-421), using Tomtec Mach III cell harvester 96 (Tomtec Inc., Hamden, CT). Filtermats were air dried, and mycobacterial incorporation of the tritiated uridine was quantified as disintegrations per minute (dpm) in a Microbeta scintillation counter (PerkinElmer Wallac Inc.). Also, 10-fold dilutions of the lysates were plated in duplicate on Middlebrook 7H10 agar plates. The numbers of colonies were counted after 3 weeks of incubation at 37°C. The percentages of mycobacterial growth inhibition were determined using the following formula: percent inhibition = 100 − [100 × (dpm or CFU in the DCP-treated cultures/dpm or CFU in the untreated cultures)].
BD cytometric bead array assay.
Macrophage inflammatory protein 1α (MIP-1α; also known as CCL3), MIP-1β (also known as CCL4), regulated upon activation of normal T cell expressed and secreted protein (RANTES; also known as CCL5), IFN-γ-inducible 10-kDa protein (IP-10; also known as CXCL10), and monokine induced by IFN-γ (MIG; also known as CXCL9) were simultaneously quantified in each sample with the cytokine bead array (CBA) human soluble protein flex set (IP-10, number 558280; MIG, number 558286; MIP-1α, number 558325; MIP-1β, number 558288; and RANTES, number 558324; BD Biosciences) according to the manufacturer's procedure. Incubation time was 3 h. The fluorescence intensity of CBA beads was measured on a FACSCalibur flow cytometer (BD Biosciences) and analyzed with FCAP Array software (32).
Total cellular RNA isolation and quantitative reverse transcription-PCR (RT-PCR).
Total RNA from human monocytes that had been left uninfected and untreated, left uninfected and treated with DCP, infected with M. bovis BCG alone, and infected with M. bovis BCG and treated with DCP were isolated using an RNeasy Plus minikit with genomic DNA (gDNA) eliminator columns (Qiagen; 74134) according to the manufacturer's instructions. The quantity of RNA obtained was assessed using a NanoDrop ND-2000 instrument (Nano Technologies). Fifty nanograms of total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems; catalog number 4374966) according to the manufacturer's instructions. The specific cDNAs were amplified with NOS2 (Hs00167248_m1), indoleamine 2,3-dioxygenase 1 (IDO1) (Hs00984148_m1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Hs99999905_m1) TaqMan gene expression assays in a 7500 Plus real-time PCR system (Applied Biosystems). The reactions were performed using the following conditions: an initial single cycle of 50°C for 2 min, followed by 95°C for 10 min, and then 45 cycles of 95°C for 15 s, followed by 60°C for 1 min. The relative level of mRNA expression, normalized against GAPDH, was calculated by the threshold cycle (2−ΔΔCT) method (33, 34).
Measurement of NO production in murine macrophages.
Concentrations of nitrite (NO2−) produced by DCP-treated, BCG-infected murine BMDMϕ were measured by using the Griess reagent system (part number TB229; Promega Corporation, Madison, WI). A serially diluted sodium NO2− standard (0 to 100 μM) was used to generate a standard curve to quantify the amount of NO2− in culture supernatants according to the manufacturer's protocol (35, 36). Cell-free medium was used as a blank for the assay.
Statistical analysis.
Statistical analyses of experimental data, employing nonparametric tests as indicated below, were performed using GraphPad Prism (GraphPad Software, San Diego, CA). For unpaired and paired comparisons, Mann-Whitney U tests and Wilcoxon matched-pairs signed-rank tests, respectively, were applied.
RESULTS
DCP-mediated inhibition of intracellular mycobacteria in human monocytes.
We first determined the ability of DCP to inhibit the growth of M. bovis BCG within human monocytes as noted in Materials and Methods. As shown in Fig. 1A, DCP inhibited intracellular M. bovis BCG growth in a dose-dependent manner. DCP treatment at 1,600 μM was able to suppress growth of intracellular M. bovis BCG by 72.65% ± 5.52% compared with the untreated control cultures. We then compared the in vitro activity of DCP with two routinely used first-line anti-TB drugs, RIF and PZA, which were used at achievable serum concentrations of 10 μg/ml and 50 μg/ml, respectively (Fig. 1B and C). The antimicrobial activity of DCP was comparable to that of PZA. As expected, RIF showed greater activity than PZA, inhibiting growth of intracellular BCG by 99% ± 0.05%.
Fig 1.

DCP mediates inhibition of intracellular mycobacteria growth in a dose-dependent manner. PBMCs were plated at a density of 1.5 × 106 cells per well in round-bottom 96-well plates and incubated overnight at 37°C. After overnight incubation, nonadherent cells were washed off, and the enriched monocytes (∼1.5 × 105 cells/well or ∼10% of the initial PBMC numbers = high monocyte density) were cultured for one more day before infection with BCG at a multiplicity of infection of 3 bacteria/cell. Extracellular BCG was washed off, and infected cells were treated with DCP at the indicated doses, 50 μg/ml PZA, or 10 μg/ml RIF for 72 h. Data are percent inhibition of intracellular BCG growth calculated as described in Materials and Methods. (A) Treatment with the highest dose of DCP resulted in >70% decreases in viability of intracellular mycobacteria. (B and C) Activity of DCP compared with current anti-TB drugs, PZA and RIF. Data shown are means ± standard errors of the means of results obtained with monocytes from 4 different volunteers. P values were obtained by using Mann-Whitney U tests.
We previously observed that the density of macrophage culture conditions can influence their abilities to support and/or control intracellular mycobacterial growth (29). Therefore, we next studied whether DCP could induce macrophages to inhibit intracellular mycobacterial growth when cultured at a 10-fold-lower density. Figure 2 clearly confirms that DCP can induce inhibition of intracellular mycobacteria within human monocytes plated at suboptimal densities, suggesting that DCP could have antimycobacterial effects under various physiologic conditions encountered in vivo.
Fig 2.
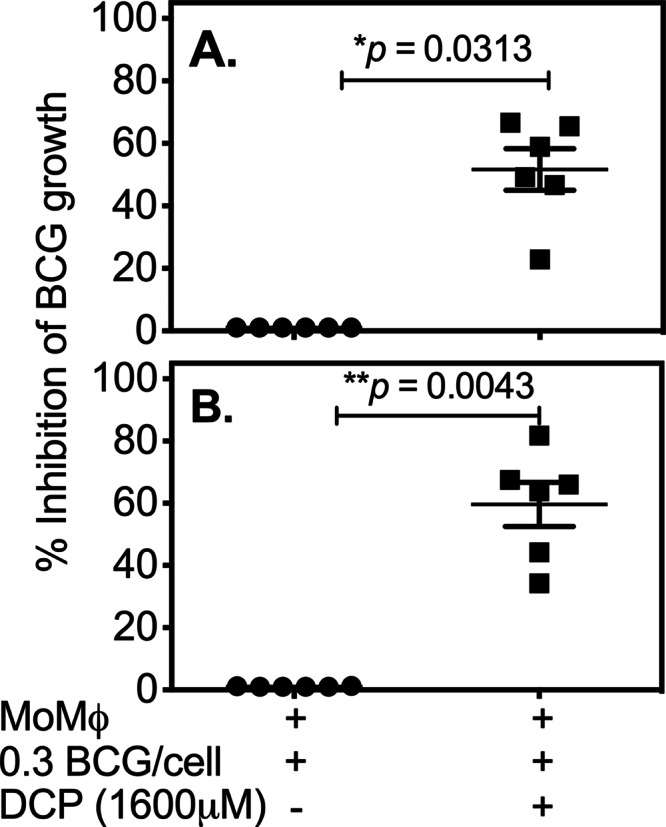
DCP inhibits intracellular mycobacterial growth in monocytes grown at lower densities. PBMCs from 6 different volunteers were plated at a density of 1.5 × 105 cells per well in a 96-well plate and incubated overnight at 37°C in a 5% CO2 incubator. After overnight incubation, nonadherent cells were washed off, and the enriched monocytes (∼1.5 × 104 cells/well or ∼10% of the initial PBMC numbers = low monocyte density) were cultured for one more day before infection with BCG at a multiplicity of infection of 0.3 bacteria/cell. Extracellular (nonphagocytosed) bacilli were washed off, and infected cells were treated with 1,600 μM DCP for 72 h. Data shown are percent inhibition of intracellular M. bovis BCG growth derived from two separate experiments: experiment 1 (A) and experiment 2 (B). P values were obtained by using Mann-Whitney U tests.
DCP does not have cytolytic effects on macrophages or mycobacteria.
To rule out the possibility that the observed mycobacterial inhibitory activity of DCP was the result of direct lytic effects on human monocytes, we cultured adherent cells in the presence or absence of DCP. After 3 days of incubation, cells were gently detached, and viable cells were counted using 0.4% (wt/vol) trypan blue solution. Data shown in Table 1 and Fig. 3A demonstrate that similar cell numbers were recovered from cultures in wells with and without DCP. Therefore, the inhibition of intracellular mycobacteria was not simply due to a direct cytolytic effect of DCP on human monocytes.
Table 1.
Determination of cell viability in cultures with and without DCP
| Volunteer and well no. | No. of viable cells after incubation with: |
|
|---|---|---|
| Complete RPMI medium | DCP (1,600 μM) | |
| Volunteer 1 | ||
| Well 1 | 2.40 × 104 | 1.20 × 104 |
| Well 2 | 1.80 × 104 | 1.20 × 104 |
| Well 3 | 1.80 × 104 | 1.80 × 104 |
| Well 4 | 0.60 × 104 | 1.20 × 104 |
| Mean ± SEM (×104) | 1.65 ± 0.38 | 1.35 ± 0.15a |
| Volunteer 2 | ||
| Well 1 | 1.80 × 104 | 1.80 × 104 |
| Well 2 | 0.60 × 104 | 4.80 × 104 |
| Well 3 | 4.20 × 104 | 3.00 × 104 |
| Well 4 | 1.80 × 104 | 2.40 × 104 |
| Mean ± SEM (×104) | 2.10 ± 0.76 | 3.00 ± 0.65a |
Not significantly different from value for control group; P = 0.49 (Student t test).
Fig 3.

DCP has no cytolytic effect on monocytes and no direct inhibitory effect on mycobacteria. (A) Monocytes (∼1.5 × 104 cells/well or ∼10% of the initial PBMC numbers), enriched by plastic overnight adherence in round-bottom 96-well plates (Table 1), were incubated with antibiotic-free complete RPMI medium alone or with 1,600 μM DCP for 72 h. At the end of 3 days, cells were gently detached from wells using a 1:1 mixture of 10 mM EDTA in PBS (pH 7.4) and RPMI 1640 medium containing 20% FBS. The cell viability was determined by using 0.4% trypan blue exclusion. Data shown are absolute numbers of viable cells recovered after incubation with medium alone or DCP. There were no differences in the mean cell numbers cultured with and without DCP, indicating that DCP did not have cytolytic effects on monocytes during the 72-h treatment duration. Each bar represents the mean of two different volunteers (P = 0.7792; t test). (B) A total of 450,000 CFU of BCG was cultured in cell-free Middlebrook 7H9 broth in the absence or presence of DCP (1,600 μM) in round-bottom 96-well plates for 3 days. On day 3, BCG was pulsed with [3H]uridine for 3 more days, and incorporation of tritiated uridine was measured. Data shown are averages of disintegrations per minute obtained from 4 separate experiments. There was no difference in the quantity of BCG in cultures with and without DCP. Each bar represents the mean of 4 separate experiments (P = 0.2000; Mann-Whitney U test). n.s., not significant.
To determine whether DCP possessed any direct inhibitory effects on mycobacteria, we studied the effects of DCP on extracellular BCG. BCG was incubated in 100 μl Middlebrook 7H9 broth media with or without DCP at 37°C for 3 days. Residual viable BCG bacilli were estimated by incorporation of tritiated-uridine. As demonstrated in Fig. 3B, similar amounts of bacilli survived in cultures with or without DCP for 72 h, indicating that DCP did not have direct effects on mycobacterial replication. These data confirm that the antimycobacterial inhibitory effects of DCP observed in the intracellular mycobacterial-growth inhibition assays shown in Fig. 1 and 2 were due to enhancement of macrophage microbicidal activities which resulted in better control of intracellular mycobacterial growth.
DCP enhances C-C chemokine MIP-1β production by mycobacterium-infected monocytes.
Monocytes and macrophages are endowed with microbicidal effector programs, which are activated through receptors upon recognition of danger signals. These specialized effector programs establish an intracellular environment that limits survival of invading microbes. Infection of macrophages with M. tuberculosis or M. bovis BCG induces release of effector proteins, including proinflammatory and anti-inflammatory cytokines, chemokines with autocrine activating and chemotactic effects, reactive oxygen species, and RNI (17, 18, 37–39). We first sought to determine whether treatment of M. bovis BCG-infected human monocytes with DCP could enhance the production of proinflammatory cytokines and chemokines. We analyzed the secretion of human IL-1β, TNF-α, IL-12p70, IFN-γ, GM-CSF, IL-6, and IL-17A, which activate intracellular microbicidal activities of monocytes/macrophages and other cells or inhibit the anti-inflammatory cytokine IL-10. As measured by CBA, the above cytokines were not detected in the supernatants of mycobacterium-infected monocytes after treatment with DCP (data not shown).
Both M. tuberculosis and M. bovis BCG can stimulate human monocytes and macrophages to produce cysteine-cysteine (C-C) family chemokines, including monocyte chemotactic protein-1 (MCP-1), RANTES, MIP-1α, and MIP-1β (40–42). In addition, the C-X-C chemokines IP-10 and MIG are involved in IFN-γ-induced responses important for mycobacterial immunity (43–45). Based on these reports, we next investigated the levels of these C-C and C-X-C chemokines by CBA in the same culture supernatants as used (see above) to study cytokine responses. Shown in Fig. 4A to E are the amounts of chemokines produced by infected monocytes treated with DCP or not. BCG infection alone induced monocytes to produce IP-10, MIG, RANTES, and MIP-1β but not MIP-1α (Fig. 4A to E). In contrast, treatment of infected monocytes with DCP suppressed the synthesis of IP-10 and MIG (Fig. 4A and B) but did not affect production of RANTES or MIP-1α (Fig. 4C and D). Importantly, MIP-1β was significantly increased following DCP treatment of BCG-infected monocytes. These results suggested that DCP-induced MIP-1β may be uniquely important for the mycobacterial inhibitory effects observed in Fig. 1 and 2.
Fig 4.

DCP enhances MIP-1β production by mycobacterium-infected monocytes. Monocytes, enriched from PBMCs through plastic adherence (∼1.5 × 104 cells/well or ∼10% of the initial PBMC numbers), were infected overnight with BCG (MOI = 0.3). Extracellular bacteria were gently washed off, and BCG-infected cells were treated with 1,600 μM DCP for 72 h at 37°C. After 72 h of culture, supernatants were collected and quantities of IP-10 (A; P = 0.0043), MIG (B; P = 0.0022), CCL5 (C; P = 0.9372), MIP-1α (D; P = 0.1823), and MIP-1β (E; P = 0.0313) were measured using CBA. (F) DCP-mediated inhibition of intracellular BCG was reversed with 10 μg/ml human anti-MIP-1β (*, P = 0.0151; **, P = 0.0022). (G and H) Additional experiments showed that anti-MIP-1β can reverse DCP-mediated inhibition of BCG by both tritiated-uridine uptake (G) and CFU (H) endpoints, but anti-MIP-1β had no effects on BCG-infected monocytes not treated with DCP (*, P = 0.0159; **, P = 0.0079). Data shown are means ± standard errors of the means of results obtained with monocytes from 6 different volunteers (A to F) and 5 different volunteers (G and H). P values were obtained by using Mann-Whitney U tests.
We next examined the functional impact of MIP-1β in DCP-mediated inhibition of intracellular mycobacteria. Mycobacterium-infected monocytes were treated with DCP in the presence or absence of neutralizing anti-MIP-1β antibody, and the effects on intracellular mycobacterial growth were compared. Figure 4F shows that the ability of DCP-treated monocytes to inhibit intracellular mycobacterial growth was prevented by anti-MIP-1β antibody. Thus, the antimycobacterial activity of DCP treatment required the production of the C-C chemokine MIP-1β. It is important to note that blockade of MIP-1β induced by BCG infection alone did not affect intracellular mycobacterial growth (Fig. 4G and 4H). The latter results suggest either (i) that the higher levels of MIP-1β induced by DCP treatment are required for mycobacterial inhibitory effects or (ii) that although MIP-1β is required for DCP-mediated inhibitory effects, additional factors induced by DCP are necessary as well.
DCP enhances induction of iNOS expression by mycobacterium-infected monocytes.
We next considered the possibility that the endogenously produced MIP-1β could be involved in the induction of iNOS, previously shown to be an important antimycobacterial effector response that produces microbicidal NO (18). Villalta et al. (46) and Aliberti and colleagues (47) demonstrated that C-C chemokines could induce NO production in human macrophages infected with Trypanosoma cruzi, with trypanocidal effects. We found that neutralization of IFN-γ and TNF-α, cytokines commonly involved in iNOS induction, did not affect the DCP-induced inhibition of intracellular mycobacteria (data not shown). In addition, we were unable to detect DCP-mediated induction of NO by Griess reaction or iNOS protein by Western blotting in human macrophages (data not shown). However, multiple authors have shown that these endpoints are difficult to measure in cultures of human monocytes/macrophages (48, 49). Therefore, we next evaluated iNOS mRNA expression using real-time RT-PCR as described in Materials and Methods. Figure 5A illustrates the expression levels of iNOS mRNA in uninfected or BCG-infected human monocytes before and after treatment with DCP. In both uninfected and infected monocytes, DCP induced increased production of iNOS mRNA. In addition, DCP induced significantly higher levels of iNOS in mycobacterium-infected monocytes than in uninfected monocytes. Murine BMDMϕ were used to directly demonstrate that DCP induces NO production (Fig. 5B).
Fig 5.

DCP enhances iNOS expression in monocytes infected with BCG. (A) Uninfected or BCG-infected human monocytes (∼1.5 × 105 cells/well or ∼10% of the initial PBMC numbers = high monocyte density), enriched from PBMCs through overnight plastic adherence, were cultured with or without DCP at 37°C. After 12 h of culture, total cellular RNA was isolated and iNOS mRNA expression measured by quantitative RT-PCR. Data shown are means ± standard errors of the means of fold increases in iNOS expression in monocytes obtained from 6 different volunteers. Significant differences in mean iNOS mRNA were detected in the following comparisons: uninfected without DCP versus uninfected with DCP (**, P = 0.0050), uninfected with DCP versus BCG infected with DCP (*, P = 0.0260), and BCG infected without DCP versus BCG infected with DCP (**, P = 0.0022). (B) Direct evidence that DCP induces NO production in M. bovis BCG-infected murine BMDMϕ (*, P = 0.031). (C) Effects of addition of anti-MIP-1β on iNOS induction in BCG-infected cells treated with DCP or not (*, P = 0.0158). Rab, rabbit control antibody. P values were obtained by using the Mann-Whitney U test (A), Wilcoxon signed-rank test (B), and paired Student t test (C).
Because MIP-1β was shown (see above) to be important for DCP-induced intracellular inhibition of mycobacterial growth, we next examined the effect of addition of anti-MIP-1β on NOS2 induction in infected monocytes. BCG-infected monocytes were treated with DCP or not in the presence of anti-MIP-1β neutralizing antibody or control rabbit polyclonal IgG, and iNOS mRNA was measured by real-time RT-PCR. Blocking endogenous MIP-1β, which was enhanced in the presence of DCP (Fig. 4E), significantly reduced DCP-mediated iNOS induction (Fig. 5C). These data indicate that MIP-1β produced by DCP-treated infected monocytes is involved in the induction of iNOS and are in agreement with previous reports that MIP-1β activates human macrophages to inhibit intracellular pathogens via the iNOS-NO pathway (42, 46, 47).
To confirm a role for the iNOS-NO effector pathway in DCP-mediated inhibition of intracellular mycobacteria, we assessed the functional significance of increased iNOS mRNA expression by inhibiting NO production with l-NMMA (50). As shown in Fig. 6A, l-NMMA significantly inhibited the antimycobacterial effect of DCP, confirming the role for NO in DCP-mediated inhibitory activity in human monocytes. The results of a second experiment are shown in Fig. 6B (uptake of tritiated uridine) and Fig. 6C (CFU counts). l-NMMA reversed the antimycobacterial activity of DCP treatment as measured by both the tritiated-uridine and CFU endpoints. The latter results also demonstrate that l-NMMA had no effect on mycobacterial growth in monocytes not treated with DCP, suggesting that infection alone does induce functional iNOS responses.
Fig 6.

iNOS inhibition reverses the DCP-mediated antimycobacterial effects. (A) The addition of the iNOS inhibitor (l-NMMA monoacetate) markedly reversed the DCP-mediated inhibition of intracellular M. bovis BCG growth. **, P = 0.0022 for untreated infected monocytes versus DCP-treated infected monocytes; **, P = 0.0022 for DCP-treated infected monocytes versus DCP-treated infected monocytes plus 1 mM l-NMMA monoacetate. n.s., P = 0.0649. P values were obtained using Mann-Whitney U tests. (B and C) Effects of addition of l-NMMA to infected monocytes without DCP. Monocytes were obtained from PBMCs of 6 different donors (**, P = 0.0079). (D) Uninfected or BCG-infected human monocytes were cultured with or without DCP at 37°C. After 12 h of culture, total cellular RNA was isolated and IDO1 mRNA expression measured by real-time RT-PCR. Data shown are means ± standard errors of the means of fold gene expression in monocytes obtained from 6 different volunteers. DCP moderately induced IDO1 expression in uninfected macrophages. In vitro infection by M. bovis BCG induced monocytes to express greater amounts of IDO1, which was suppressed by treatment with DCP. Significant differences in mean IDO1 levels were detected in the following comparisons: uninfected without DCP versus uninfected with DCP (**, P = 0.0037), uninfected with DCP versus BCG infected without DCP (*, P = 0.0152) and BCG infected without DCP versus BCG infected with DCP (*, P = 0.0411). P values were obtained using Mann-Whitney U tests.
Indoleamine 2,3-dioxygenase-1 (IDO1) is induced in macrophages and dendritic cells by both IFN-γ/signal transducer and activator of transcription-1 (Stat-1) and lipopolysaccharide (LPS)/Toll-like receptor signaling pathways (51, 52). It is the main inducible intracellular enzyme that catabolizes the essential amino acid l-tryptophan through the kynurenine pathway. l-Tryptophan depletion by IDO1 activity has been shown to be an important factor for restricting the intracellular growth of pathogens that depend on exogenous tryptophan, such as Toxoplasma gondii and mycobacteria (53–55). Therefore, we next investigated whether growth inhibition of intracellular mycobacteria in DCP-treated cultures involved increased IDO degradation of l-tryptophan and the restriction of this essential amino acid for pathogen replication. Figure 6D shows that IDO1 mRNA measured by quantitative RT-PCR was increased by both BCG infection alone and DCP treatment alone. However, DCP treatment of BCG-infected monocytes inhibited IDO1 mRNA expression compared with untreated but BCG-infected monocytes. These results demonstrate that the inhibition of intracellular mycobacterial growth mediated by DCP is caused not by the depletion of l-tryptophan but rather by the iNOS induction mechanism described above.
Finally, we studied the in vitro effects of DCP on intracellular growth of virulent M. tuberculosis (H37Rv strain). As shown in Fig. 7, DCP treatment also significantly reduced the intracellular growth of virulent M. tuberculosis. These data further suggest that DCP or other compounds with similar activity could be useful for treatment of TB infection and/or disease.
Fig 7.
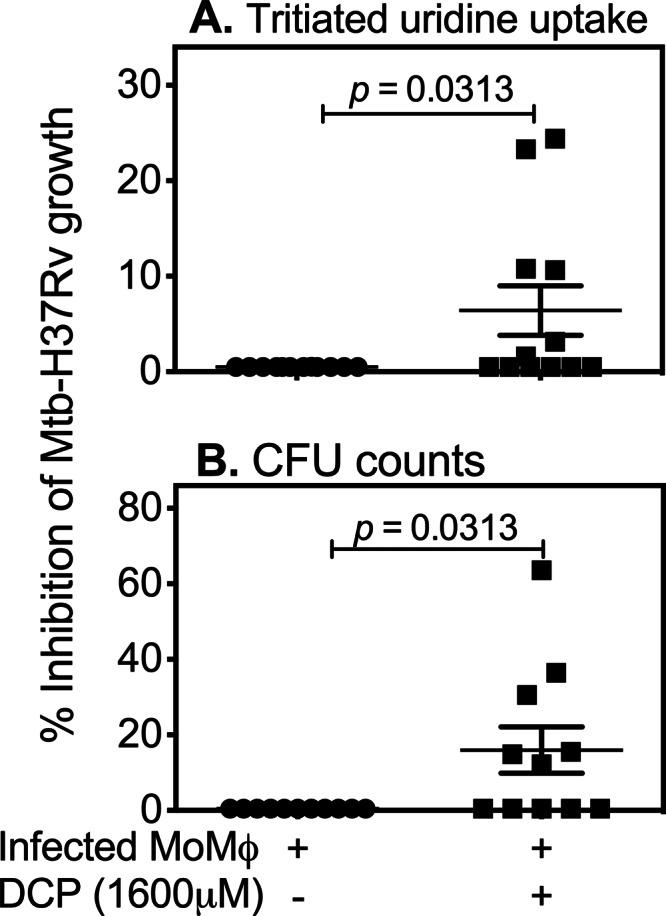
DCP activity against M. tuberculosis H37Rv. PBMCs were plated at a density of 1.5 × 106 cells per well in round-bottom 96-well plates and incubated overnight at 37°C. After 2 h of incubation, nonadherent cells were washed off, and the enriched monocytes (∼1.5 × 105 cells/well or ∼10% of the initial PBMC numbers = high monocyte density) were cultured for one more day before infection with M. tuberculosis H37Rv (Mtb-H37Rv) at a multiplicity of infection of 3 bacteria/cell. Extracellular (nonphogocytosed) bacilli were washed off, and infected cells were treated with 1,600 μM DCP for 72 h, after which cultures were lysed and residual bacteria quantitated by determination of uptake of tritiated uridine and CFU counts. Data shown are percent inhibition of intracellular mycobacterial growth calculated from uptake of tritiated uridine (A) and CFU counts (B). Each black square or circle represents an individual volunteer. P values were obtained by Wilcoxon signed-rank tests.
DISCUSSION
The increasing incidence of drug-resistant M. tuberculosis strains has compromised efforts toward both the treatment and prevention of TB (4, 56). The ability of the WHO and its member countries to control the enormous burden of TB will require the development of not only new-generation vaccines but also novel anti-TB drugs and/or immunomodulatory therapeutics and compounds that can enhance the antituberculosis activity of human macrophages.
Based on the demonstration that DCP induces protective effects against tumor growth and hepatitis B virus in vivo (19, 22), we tested the hypothesis that DCP could exert microbicidal activity against mycobacteria within infected human monocytes/macrophages. Our study demonstrates that DCP induces a potent inhibitory effect against mycobacteria, resulting in a 65 to 75% decrease in viability of bacilli within human monocytes at 72 h posttreatment (Fig. 1 and 2). Though the activity of DCP was lower than that of RIF, DCP and PZA exhibited similar in vitro activities against intracellular M. bovis BCG (Fig. 1B and C), further demonstrating the potential clinical relevance of our work. PZA is a first-line agent used in combination with isoniazid, RIF, and ethambutol for short-course (6-month) treatment of drug-sensitive TB and frequently for MDR-TB (35, 57, 58).
DCP had no lytic effects on mycobacterium-infected macrophages (Fig. 3A) and no direct inhibitory effects on mycobacterial growth (Fig. 3B). Similar numbers of viable monocytes were recovered after 3 days of culture in the absence or presence of the highest doses (1.6 mM) of DCP. Moreover, a possible direct effect of DCP on mycobacteria was ruled out by culturing extracellular M. bovis BCG with DCP. These observations demonstrated that DCP activates the antimycobacterial effector functions of human macrophages.
The antimicrobial effects of RIF are attributed to its interaction with and inhibition of the bacterial DNA-dependent β-subunit of RNA polymerase (59, 60). RIF has bactericidal activity in vitro against slow-growing and intermittently growing M. tuberculosis. PZA, an analog of nicotinamide, is a prodrug requiring conversion into its active form, pyrazinoic acid (POA), by the bacterial pyrazinamidase (61). It is preferentially active against nonreplicating persisting bacilli in vivo during active inflammation. The biochemical basis for antituberculosis activity of PZA was recently reported to be the inhibition of trans-translation in M. tuberculosis (62). Shi and colleagues showed that POA binds ribosomal protein S1, which is involved in protein translation and the ribosome-sparing process of trans-translation. Interestingly, PZA does not act directly on M. bovis because of the lack of a pncA gene in this species. Therefore, our results further suggest that PZA activates human macrophages to better control intracellular mycobacteria, which may explain the clinical observation that PZA seems more important for the efficacy of the first-line multidrug treatment regimens than would be predicted based on PZA MICs alone.
Mononuclear phagocytes, including macrophages, play an important role in innate immunity against M. tuberculosis and therefore are considered the first and essential line of defense against TB (38, 63). Whereas resting monocytes/macrophages fail to control replication of mycobacteria and other intracellular pathogens, activated macrophages can suppress growth of intracellular bacilli. The suppression of growth is accomplished via various effector mechanisms, such as biosynthesis of inflammatory cytokines (e.g., TNF-α and IL-1β), chemokine-mediated recruitment of neutrophils/lymphocytes, activation of intracellular microbicidal activities, or direct activation of the production of reactive oxygen species and/or reactive nitrogen intermediates (NO and/or its derivatives) (18, 37, 42, 64).
Our results indicate that the impairment of intracellular mycobacterial growth observed in human monocytes treated with DCP can be attributed at least in part to the production of MIP-1β by infected cells. Neutralization of MIP-1β reversed the DCP-mediated inhibitory effects on intracellular mycobacteria (Fig. 4F). As illustrated in Fig. 4G and 4H, the microbicidal role of MIP-1β we observed is in agreement with the findings reported by Saukkonen and colleagues, who demonstrated that MIP-1β suppresses growth of mycobacteria within macrophages (42).
The detailed intracellular signaling pathways by which DCP induces production of MIP-1β by infected monocytes are unknown. There are lines of evidence indicating that many of the processes involved in killing of intracellular mycobacteria are regulated by Ca2+. For instance, activation of cytokine production and preformed granule secretion is regulated by Ca2+ (65, 66). Even more relevant to our work, Méndez-Samperio and colleagues demonstrated that in vitro, M. bovis BCG stimulates human monocytes to produce C-C chemokines through mobilization of intracellular Ca2+ and influx of extracellular Ca2+ (40). Therefore, the mechanism of DCP effects observed in our work could at least partially involve Ca2+ fluxes directly altered by the Ca2+ complexed within DCP. However, further investigations are required to determine the specific signaling events triggered by DCP that result in MIP-1β production.
The influence and biological activities of chemokines are wide-ranging and involve more than simple recruitment of circulating leukocytes. In addition to their chemotatic role, chemokines can stimulate a cascade of proinflammatory events, lead to macrophage activation, and enhance T-cell activation and proliferation (67, 68). In human macrophages, C-C chemokines were reported to induce NO production and killing of Trypanosoma cruzi, an intracellular parasite and the causative agent of Chagas' disease (46). Taking into account the importance of NO as an effector molecule involved in the control of mycobacteria both in vitro and in vivo (69, 70), it was of relevance to investigate whether DCP-induced MIP-1β and/or DCP alone could induce iNOS and thus production of NO. Quantitative RT-PCR results demonstrated that DCP can stimulate increased production of iNOS mRNA in mycobacterium-infected monocytes (Fig. 5A). Additional experiments with murine macrophages confirmed that DCP induced NO production in mycobacterium-infected macrophages (Fig. 5B). We then established that DCP-induced MIP-1β production was required for DCP-induced upregulation of the monocyte/macrophage iNOS pathway by showing that the addition of anti-MIP-1β antibody prevented induction of iNOS mRNA by DCP. As shown in Fig. 5C, neutralization of MIP-1β significantly reduced iNOS mRNA in infected cells treated with DCP, demonstrating that MIP-1β is involved in the induction of iNOS.
We confirmed the importance of the iNOS-NO pathway in DCP-mediated inhibition of intracellular mycobacteria by adding the l-arginine analogue that serves as an iNOS inhibitor, l-NMMA monoacetate (50), to DCP-treated infected monocytes. Figure 6A clearly demonstrates that inhibition of iNOS activity by l-NMMA prevented the DCP-mediated suppression of intracellular mycobacterial growth. These data verify that DCP induction of the iNOS-NO effector pathway in human monocytes significantly contributes to suppression of intracellular mycobacteria. The mechanism(s) by which NO and/or RNI kill mycobacteria is unclear but has been suggested to involve disruption of bacterial DNA, proteins, signaling, and/or induction of monocyte/macrophage apoptosis (15, 71). Furthermore, given that previously reported activities of DCP include the induction of apoptosis (20), there may be a connection between DCP-mediated NO production and apoptosis of infected cells. Since M. tuberculosis promotes its replication by inhibiting the apoptosis of infected macrophages (reviewed in reference 72), it would be worth investigating in future studies whether apoptosis is restored in mycobacterium-infected cells by DCP treatment.
iNOS is regulated at both the transcriptional and posttranscriptional levels by a number of signal transduction pathways and molecules, such as Jak-1/Stat-α/IRF-1; IκB/nuclear factor kappa B (NF-κB); enzyme activity cofactors, protein kinases, and phosphatases; and tyrosine phosphatases (11, 18, 37). The induction of iNOS mRNA expression is the main regulatory step controlling iNOS activity, and the iNOS promoter is activated by multiple different transcription factors (reviewed in reference 73). Bacterial LPS, IL-1β, TNF-α, and oxidative stress have been shown to induce iNOS expression by activating the transcription factor NF-κB. Furthermore, the interaction of C-C chemokines with their cognate Gαi-protein-coupled receptors can activate phosphatidylinositol 3-kinase (PI3K), which, in turn, stimulates NF-κB production and thus can lead to activation of the iNOS promoter (reviewed in references 73 and 74). Therefore, DCP induction of MIP-1β may result in both NF-κB and iNOS promoter activation, important for the observed intracellular inhibitory effects on mycobacteria. We have shown that MIP-1β is necessary for the inhibitory effects of DCP and that neutralization of MIP-1β alone inhibited iNOS induction (Fig. 5B). These results suggest that the inhibitory effects of DCP involve in part the induction of iNOS via the MIP-1β signaling pathway. However, the exact molecular details require further detailed dissection.
Various laboratories have shown that different human cell types are capable of controlling growth of parasites (55, 75), bacteria (76, 77), and viruses (78, 79) via IDO1-dependent antimicrobial effects. In fact, IDO1 induction was thought to be at least partially responsible for the anticancer and antiviral effects of DCP (19, 20, 22). Catabolism of local l-tryptophan into kynurenine and its metabolites has been implicated as the mechanism of IDO1-mediated inhibition of microbial replication (55). However, in recent years evidence has emerged indicating that degradation of l-tryptophan by IDO1 also can downregulate T cell responses in a wide range of disease states (52, 80). Müller and coworkers addressed these seemingly contradictory roles of IDO1 by determining the relative concentrations of l-tryptophan required for bacterial and T cell growth (81). The levels of l-tryptophan required to support bacterial growth were 10- to 40-fold higher than the levels necessary for optimal T cell activation and proliferation (81). Hence, IDO1 can exert predominant antimicrobial effects during the acute phase of a rapidly replicating infection. In contrast, at later time points of infection, especially with a chronic, slow-growing pathogen, the immunoregulatory effects of IDO1 may predominate. In our model system, accumulation of IDO1 mRNA in mycobacterium-infected monocytes was suppressed by DCP treatment (Fig. 6D). Therefore, IDO1 does not appear to be involved in the DCP-mediated inhibitory effects on intracellular mycobacteria. However, DCP may be able to reduce the immunoregulatory effects of IDO1 during TB infection, providing further benefits during treatment of TB disease. The latter possibility should be tested in future experiments.
In conclusion, this study shows that DCP has significant effects on intracellular mycobacterial growth. The findings presented herein favor the concept that DCP mediates its effects via the C-C chemokine MIP-1β and iNOS-NO effector pathways. It will be interesting to investigate whether macrophage activation with DCP would enhance the killing of M. tuberculosis with current anti-TB drugs, such as PZA, which is preferentially used with several new drug candidates for optimal efficiency (82). Further studies are warranted to evaluate DCP effects on drug-resistant and drug-sensitive virulent strains of M. tuberculosis in vivo, used alone and in combination with PZA. In addition, further molecular dissection of the pathways involved in DCP-mediated enhancement of intracellular control of mycobacteria is important to pursue.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01-A1-48391 (to D.F.H., Principal Investigator), a VA Office of Research and Development Merit Review Award (to R.F.S.), and grant NO1-AI-50036 (to P.M.).
P.M. holds stock and stock options, in SanRx Pharmaceuticals, Inc., which has been assigned patent rights to DCP.
We thank Dietmar Fuchs (Innsbruck Medical University, Austria) for his helpful comments and suggestions regarding this work.
Footnotes
Published ahead of print 18 March 2013
REFERENCES
- 1. Boom WH, Canaday DH, Fulton SA, Gehring AJ, Rojas RE, Torres M. 2003. Human immunity to M. tuberculosis: T cell subsets and antigen processing. Tuberculosis (Edinb.) 83:98–106 [DOI] [PubMed] [Google Scholar]
- 2. Kaufmann SH. 2001. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 1:20–30 [DOI] [PubMed] [Google Scholar]
- 3. Kaufmann SH, Hussey G, Lambert PH. 2010. New vaccines for tuberculosis. Lancet 375:2110–2119 [DOI] [PubMed] [Google Scholar]
- 4. Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, van Soolingen D, Jensen P, Bayona J. 2010. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet 375:1830–1843 [DOI] [PubMed] [Google Scholar]
- 5. Wells CD. 2010. Global impact of multidrug-resistant pulmonary tuberculosis among HIV-infected and other immunocompromised hosts: epidemiology, diagnosis, and strategies for management. Curr. Infect. Dis. Rep. 12:192–197 [DOI] [PubMed] [Google Scholar]
- 6. WHO 2008. Anti-tuberculosis drug resistance in the world: fourth global report. WHO/HTM/TB/2008.394. World Health Organization, Geneva, Switzerland: http://www.who.int/tb/publications/2008/en/index.html [Google Scholar]
- 7. WHO 2009. Global Tuberculosis Control 2009: Epidemiology. Strategy. Financing. 13th annual report on global tuberculosis control. WHO/HTM/TB/2009.411. World Health Organization, Geneva, Switzerland: http://www.who.int/tb/publications/2008/en/index.html [Google Scholar]
- 8. Migliori GB, De Iaco G, Besozzi G, Centis R, Cirillo DM. 2007. First tuberculosis cases in Italy resistant to all tested drugs. Euro Surveill. 12:E070517 070511 [DOI] [PubMed] [Google Scholar]
- 9. Velayati AA, Masjedi MR, Farnia P, Tabarsi P, Ghanavi J, Ziazarifi AH, Hoffner SE. 2009. Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest 136:420–425 [DOI] [PubMed] [Google Scholar]
- 10. Förstermann U, Gath I, Schwarz P, Closs EI, Kleinert H. 1995. Isoforms of nitric oxide synthase. Properties, cellular distribution and expressional control. Biochem. Pharmacol. 50:1321–1332 [DOI] [PubMed] [Google Scholar]
- 11. Jorens PG, Matthys KE, Bult H. 1995. Modulation of nitric oxide synthase activity in macrophages. Mediators Inflamm. 4:75–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmidt TS, Alp NJ. 2007. Mechanisms for the role of tetrahydrobiopterin in endothelial function and vascular disease. Clin. Sci. 113:47–63 [DOI] [PubMed] [Google Scholar]
- 13. Tzeng E, Billiar TR, Robbins PD, Loftus M, Stuehr DJ. 1995. Expression of human inducible nitric oxide synthase in a tetrahydrobiopterin (H4B)-deficient cell line: H4B promotes assembly of enzyme subunits into an active dimer. Proc. Natl. Acad. Sci. U. S. A. 92:11771–11775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schobersberger W, Hoffmann G, Grote J, Wachter H, Fuchs D. 1995. Induction of inducible nitric oxide synthase expression by neopterin in vascular smooth muscle cells. FEBS Lett. 377:461–464 [DOI] [PubMed] [Google Scholar]
- 15. Chan ED, Chan J, Schluger NW. 2001. What is the role of nitric oxide in murine and human host defense against tuberculosis? Current knowledge. Am. J. Respir. Cell Mol. Biol. 25:606–612 [DOI] [PubMed] [Google Scholar]
- 16. Kröncke KD, Fehsel K, Kolb-Bachofen V. 1998. Inducible nitric oxide synthase in human diseases. Clin. Exp. Immunol. 113:147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kwon OJ. 1997. The role of nitric oxide in the immune response of tuberculosis. J. Korean Med. Sci. 12:481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. MacMicking J, Xie QW, Nathan C. 1997. Nitric oxide and macrophage function. Annu. Rev. Immunol. 15:323–350 [DOI] [PubMed] [Google Scholar]
- 19. Moheno P, Morrey J, Fuchs D. 2010. Effect of dipterinyl calcium pentahydrate on hepatitis B virus replication in transgenic mice. J. Transl. Med. 8:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moheno P, Pfleiderer W, DiPasquale AG, Rheingold AL, Fuchs D. 2008. Cytokine and IDO metabolite changes effected by calcium pterin during inhibition of MDA-MB-231 xenograph tumors in nude mice. Int. J. Pharm. 355:238–248 [DOI] [PubMed] [Google Scholar]
- 21. Moheno PB. 2004. Calcium pterin as an antitumor agent. Int. J. Pharm. 271:293–300 [DOI] [PubMed] [Google Scholar]
- 22. Moheno P, Pfleiderer W, Fuchs D. 2009. Plasma cytokine concentration changes induced by the antitumor agents dipterinyl calcium pentahydrate (DCP) and related calcium pterins. Immunobiology 214:135–141 [DOI] [PubMed] [Google Scholar]
- 23. Hornell TM, Beresford GW, Bushey A, Boss JM, Mellins ED. 2003. Regulation of the class II MHC pathway in primary human monocytes by granulocyte-macrophage colony-stimulating factor. J. Immunol. 171:2374–2383 [DOI] [PubMed] [Google Scholar]
- 24. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. 2001. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 15:557–567 [DOI] [PubMed] [Google Scholar]
- 25. Cooper AM, Kipnis A, Turner J, Magram J, Ferrante J, Orme IM. 2002. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J. Immunol. 168:1322–1327 [DOI] [PubMed] [Google Scholar]
- 26. Gonzalez-Juarrero M, Hattle JM, Izzo A, Junqueira-Kipnis AP, Shim TS, Trapnell BC, Cooper AM, Orme IM. 2005. Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infection. J. Leukoc. Biol. 77:914–922 [DOI] [PubMed] [Google Scholar]
- 27. Bennett S, Breit SN. 1994. Variables in the isolation and culture of human monocytes that are of particular relevance to studies of HIV. J. Leukoc. Biol. 56:236–240 [DOI] [PubMed] [Google Scholar]
- 28. Johnson WD, Jr, Mei B, Cohn ZA. 1977. The separation, long-term cultivation, and maturation of the human monocyte. J. Exp. Med. 146:1613–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Worku S, Hoft DF. 2003. Differential effects of control and antigen-specific T cells on intracellular mycobacterial growth. Infect. Immun. 71:1763–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoft DF, Worku S, Kampmann B, Whalen CC, Ellner JJ, Hirsch CS, Brown RB, Larkin R, Li Q, Yun H, Silver RF. 2002. Investigation of the relationships between immune-mediated inhibition of mycobacterial growth and other potential surrogate markers of protective Mycobacterium tuberculosis immunity. J. Infect. Dis. 186:1448–1457 [DOI] [PubMed] [Google Scholar]
- 31. Worku S, Hoft DF. 2000. In vitro measurement of protective mycobacterial immunity: antigen-specific expansion of T cells capable of inhibiting intracellular growth of Bacille Calmette-Guerin. Clin. Infect. Dis. 30(Suppl 3):S257–S261 [DOI] [PubMed] [Google Scholar]
- 32. Varro R, Chen R, Sepulveda H, Apgar J. 2007. Bead-based multianalyte flow immunoassays: the cytometric bead array system. Methods Mol. Biol. 378:125–152 [DOI] [PubMed] [Google Scholar]
- 33. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 34. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 35. Campos-Neto A, Ovendale P, Bement T, Koppi TA, Fanslow WC, Rossi MA, Alderson MR. 1998. CD40 ligand is not essential for the development of cell-mediated immunity and resistance to Mycobacterium tuberculosis. J. Immunol. 160:2037–2041 [PubMed] [Google Scholar]
- 36. Chae SY, Lee M, Kim SW, Bae YH. 2004. Protection of insulin secreting cells from nitric oxide induced cellular damage by crosslinked hemoglobin. Biomaterials 25:843–850 [DOI] [PubMed] [Google Scholar]
- 37. Bogdan C, Rollinghoff M, Diefenbach A. 2000. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr. Opin. Immunol. 12:64–76 [DOI] [PubMed] [Google Scholar]
- 38. Fenton MJ, Vermeulen MW. 1996. Immunopathology of tuberculosis: roles of macrophages and monocytes. Infect. Immun. 64:683–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gordon S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3:23–35 [DOI] [PubMed] [Google Scholar]
- 40. Méndez-Samperio P, Vazquez A, Ayala H. 2003. Infection of human monocytes with Mycobacterium bovis BCG induces production of CC-chemokines. J. Infect. 47:139–147 [DOI] [PubMed] [Google Scholar]
- 41. Sadek MI, Sada E, Toossi Z, Schwander SK, Rich EA. 1998. Chemokines induced by infection of mononuclear phagocytes with mycobacteria and present in lung alveoli during active pulmonary tuberculosis. Am. J. Respir. Cell Mol. Biol. 19:513–521 [DOI] [PubMed] [Google Scholar]
- 42. Saukkonen JJ, Bazydlo B, Thomas M, Strieter RM, Keane J, Kornfeld H. 2002. Beta-chemokines are induced by Mycobacterium tuberculosis and inhibit its growth. Infect. Immun. 70:1684–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qiu L, Huang D, Chen CY, Wang R, Shen L, Shen Y, Hunt R, Estep J, Haynes BF, Jacobs WR, Jr, Letvin N, Du G, Chen ZW. 2008. Severe tuberculosis induces unbalanced up-regulation of gene networks and overexpression of IL-22, MIP-1alpha, CCL27, IP-10, CCR4, CCR5, CXCR3, PD1, PDL2, IL-3, IFN-beta, TIM1, and TLR2 but low antigen-specific cellular responses. J. Infect. Dis. 198:1514–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sauty A, Dziejman M, Taha RA, Iarossi AS, Neote K, Garcia-Zepeda EA, Hamid Q, Luster AD. 1999. The T cell-specific CXC chemokines IP-10, Mig, and I-TAC are expressed by activated human bronchial epithelial cells. J. Immunol. 162:3549–3558 [PubMed] [Google Scholar]
- 45. Seiler P, Aichele P, Bandermann S, Hauser AE, Lu B, Gerard NP, Gerard C, Ehlers S, Mollenkopf HJ, Kaufmann SH. 2003. Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur. J. Immunol. 33:2676–2686 [DOI] [PubMed] [Google Scholar]
- 46. Villalta F, Zhang Y, Bibb KE, Kappes JC, Lima MF. 1998. The cysteine-cysteine family of chemokines RANTES, MIP-1alpha, and MIP-1beta induce trypanocidal activity in human macrophages via nitric oxide. Infect. Immun. 66:4690–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aliberti JC, Machado FS, Souto JT, Campanelli AP, Teixeira MM, Gazzinelli RT, Silva JS. 1999. β-Chemokines enhance parasite uptake and promote nitric oxide-dependent microbiostatic activity in murine inflammatory macrophages infected with Trypanosoma cruzi. Infect. Immun. 67:4819–4826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Coers W, Timens W, Kempinga C, Klok PA, Moshage H. 1998. Specificity of antibodies to nitric oxide synthase isoforms in human, guinea pig, rat, and mouse tissues. J. Histochem. Cytochem. 46:1385–1392 [DOI] [PubMed] [Google Scholar]
- 49. Denis M. 1994. Human monocytes/macrophages: NO or no NO? J. Leukoc. Biol. 55:682–684 [DOI] [PubMed] [Google Scholar]
- 50. Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. 1990. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 101:746–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fujigaki S, Saito K, Sekikawa K, Tone S, Takikawa O, Fujii H, Wada H, Noma A, Seishima M. 2001. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Eur. J. Immunol. 31:2313–2318 [DOI] [PubMed] [Google Scholar]
- 52. Mellor AL, Munn DH. 2004. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol. 4:762–774 [DOI] [PubMed] [Google Scholar]
- 53. Gupta SL, Carlin JM, Pyati P, Dai W, Pfefferkorn ER, Murphy MJ., Jr 1994. Antiparasitic and antiproliferative effects of indoleamine 2,3-dioxygenase enzyme expression in human fibroblasts. Infect. Immun. 62:2277–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hayashi T, Rao SP, Takabayashi K, Van Uden JH, Kornbluth RS, Baird SM, Taylor MW, Carson DA, Catanzaro A, Raz E. 2001. Enhancement of innate immunity against Mycobacterium avium infection by immunostimulatory DNA is mediated by indoleamine 2,3-dioxygenase. Infect. Immun. 69:6156–6164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pfefferkorn ER. 1984. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. U. S. A. 81:908–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hafkin J, Gammino VM, Amon JJ. 2010. Drug-resistant tuberculosis in sub-Saharan Africa. Curr. Infect. Dis. Rep. 12:36–45 [DOI] [PubMed] [Google Scholar]
- 57. Bass JB, Jr, Farer LS, Hopewell PC, O'Brien R, Jacobs RF, Ruben F, Snider DE, Jr, Thornton G. 1994. Treatment of tuberculosis and tuberculosis infection in adults and children. American Thoracic Society and The Centers for Disease Control and Prevention. Am. J. Respir. Crit. Care Med. 149:1359–1374 [DOI] [PubMed] [Google Scholar]
- 58. Davidson PT, Le HQ. 1992. Drug treatment of tuberculosis—1992. Drugs 43:651–673 [DOI] [PubMed] [Google Scholar]
- 59. Calvori C, Frontali L, Leoni L, Tecce G. 1965. Effect of rifamycin on protein synthesis. Nature 207:417–418 [DOI] [PubMed] [Google Scholar]
- 60. Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. 2001. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104:901–912 [DOI] [PubMed] [Google Scholar]
- 61. Scorpio A, Zhang Y. 1996. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 2:662–667 [DOI] [PubMed] [Google Scholar]
- 62. Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE, III, Wang H, Zhang W, Zhang Y. 2011. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333:1630–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Edwards D, Kirkpatrick CH. 1986. The immunology of mycobacterial diseases. Am. Rev. Respir. Dis. 134:1062–1071 [DOI] [PubMed] [Google Scholar]
- 64. Novak EJ, Masewicz SA, Liu AW, Lernmark A, Kwok WW, Nepom GT. 2001. Activated human epitope-specific T cells identified by class II tetramers reside within a CD4high, proliferating subset. Int. Immunol. 13:799–806 [DOI] [PubMed] [Google Scholar]
- 65. Tapper H. 1996. The secretion of preformed granules by macrophages and neutrophils. J. Leukoc. Biol. 59:613–622 [DOI] [PubMed] [Google Scholar]
- 66. Watanabe N, Suzuki J, Kobayashi Y. 1996. Role of calcium in tumor necrosis factor-alpha production by activated macrophages. J. Biochem. 120:1190–1195 [DOI] [PubMed] [Google Scholar]
- 67. Taub DD, Ortaldo JR, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. 1996. Beta chemokines costimulate lymphocyte cytolysis, proliferation, and lymphokine production. J. Leukoc. Biol. 59:81–89 [DOI] [PubMed] [Google Scholar]
- 68. Taub DD, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. 1996. Chemokines and T lymphocyte activation. I. Beta chemokines costimulate human T lymphocyte activation in vitro. J. Immunol. 156:2095–2103 [PubMed] [Google Scholar]
- 69. Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. 1995. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect. Immun. 63:736–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nozaki Y, Hasegawa Y, Ichiyama S, Nakashima I, Shimokata K. 1997. Mechanism of nitric oxide-dependent killing of Mycobacterium bovis BCG in human alveolar macrophages. Infect. Immun. 65:3644–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Herbst S, Schaible UE, Schneider BE. 2011. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS One 6:e19105 doi:10.1371/journal.pone.0019105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Briken V, Miller JL. 2008. Living on the edge: inhibition of host cell apoptosis by Mycobacterium tuberculosis. Future Microbiol. 3:415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. 2010. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide 23:75–93 [DOI] [PubMed] [Google Scholar]
- 74. Wu Y, Yoder A. 2009. Chemokine coreceptor signaling in HIV-1 infection and pathogenesis. PLoS Pathog. 5:e1000520 doi:10.1371/journal.ppat.1000520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nagineni CN, Pardhasaradhi K, Martins MC, Detrick B, Hooks JJ. 1996. Mechanisms of interferon-induced inhibition of Toxoplasma gondii replication in human retinal pigment epithelial cells. Infect. Immun. 64:4188–4196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Byrne GI, Lehmann LK, Landry GJ. 1986. Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect. Immun. 53:347–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. MacKenzie CR, Hadding U, Daubener W. 1998. Interferon-gamma-induced activation of indoleamine 2,3-dioxygenase in cord blood monocyte-derived macrophages inhibits the growth of group B streptococci. J. Infect. Dis. 178:875–878 [DOI] [PubMed] [Google Scholar]
- 78. Adams O, Besken K, Oberdorfer C, MacKenzie CR, Russing D, Daubener W. 2004. Inhibition of human herpes simplex virus type 2 by interferon gamma and tumor necrosis factor alpha is mediated by indoleamine 2,3-dioxygenase. Microb. Infect. 6:806–812 [DOI] [PubMed] [Google Scholar]
- 79. Bodaghi B, Goureau O, Zipeto D, Laurent L, Virelizier JL, Michelson S. 1999. Role of IFN-gamma-induced indoleamine 2,3 dioxygenase and inducible nitric oxide synthase in the replication of human cytomegalovirus in retinal pigment epithelial cells. J. Immunol. 162:957–964 [PubMed] [Google Scholar]
- 80. Favre D, Mold J, Hunt PW, Kanwar B, Loke P, Seu L, Barbour JD, Lowe MM, Jayawardene A, Aweeka F, Huang Y, Douek DC, Brenchley JM, Martin JN, Hecht FM, Deeks SG, McCune JM. 2010. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci. Transl. Med. 2:32ra36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Müller A, Heseler K, Schmidt SK, Spekker K, Mackenzie CR, Daubener W. 2009. The missing link between indoleamine 2,3-dioxygenase mediated antibacterial and immunoregulatory effects. J. Cell. Mol. Med. 13:1125–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yew WW, Cynamon M, Zhang Y. 2011. Emerging drugs for the treatment of tuberculosis. Expert Opin. Emerg. Drugs 16:1–21 [DOI] [PubMed] [Google Scholar]