

Abstract
Human La protein has been implicated in facilitating the internal initiation of translation as well as replication of hepatitis C virus (HCV) RNA. Previously, we demonstrated that La interacts with the HCV internal ribosome entry site (IRES) around the GCAC motif near the initiator AUG within stem-loop IV by its RNA recognition motif (RRM) (residues 112 to 184) and influences HCV translation. In this study, we have deciphered the role of this interaction in HCV replication in a hepatocellular carcinoma cell culture system. We incorporated mutation of the GCAC motif in an HCV monocistronic subgenomic replicon and a pJFH1 construct which altered the binding of La and checked HCV RNA replication by reverse transcriptase PCR (RT-PCR). The mutation drastically affected HCV replication. Furthermore, to address whether the decrease in replication is a consequence of translation inhibition or not, we incorporated the same mutation into a bicistronic replicon and observed a substantial decrease in HCV RNA levels. Interestingly, La overexpression rescued this inhibition of replication. More importantly, we observed that the mutation reduced the association between La and NS5B. The effect of the GCAC mutation on the translation-to-replication switch, which is regulated by the interplay between NS3 and La, was further investigated. Additionally, our analyses of point mutations in the GCAC motif revealed distinct roles of each nucleotide in HCV replication and translation. Finally, we showed that a specific interaction of the GCAC motif with human La protein is crucial for linking 5′ and 3′ ends of the HCV genome. Taken together, our results demonstrate the mechanism of regulation of HCV replication by interaction of the cis-acting element GCAC within the HCV IRES with human La protein.
INTRODUCTION
Hepatitis C virus (HCV), a major etiologic agent of posttransfusion and sporadic non-A, non-B hepatitis (NANBH), belongs to the Hepacivirus genus of the Flaviviridae family (1, 2). The HCV genome consists of a 9.6-kb single-stranded positive-sense RNA containing an open reading frame (ORF) encoding an ∼3,000-residue polyprotein precursor flanked at both ends by highly structured and conserved untranslated regions (UTRs) (1–4). HCV translation is mediated by an internal ribosome entry site (IRES) located mostly within the 341-nucleotide (nt) 5′ UTR and extending to 30 to 40 nucleotides downstream of the initiator AUG (iAUG) codon. Sequence elements that are directly involved in HCV replication are located mostly in the 3′ UTR (1, 4–6). The HCV 3′ UTR varies between 200 and 235 nt in length, including a short variable region, a poly(U/UC) tract (with an average length of 80 nt), and an invariant 98-nt X-tail region (7–9). Initiation of HCV replication takes place by formation of a ribonucleoprotein (RNP) complex at the 3′ UTR of the viral genome. The newly synthesized negative-strand RNA then serves as a template for the production of the plus-strand copies of viral RNA (10, 11).
Some reports have revealed the presence of cis-acting elements in the HCV 5′ UTR that affect viral replication. For example, the sequence upstream of the IRES is essential for viral RNA replication, IRES sequences are required for high-level RNA replication, and stem-loop domain II of the IRES is crucial for replication (12). Several studies have also revealed that the 5′ UTR is capable of binding to a liver-specific microRNA, mir-122, resulting in enhanced HCV replication (13, 14).
Apart from cis-acting elements present in HCV RNA, HCV nonstructural proteins (except NS2) are required for efficient HCV replication. Intracellular changes resulting from HCV replication are not very well understood. Development of a selectable and efficiently replicating HCV subgenomic replicon in the human hepatoma cell line Huh7 has allowed investigation of viral persistence and pathogenesis mechanisms in the context of HCV replication in cell culture (10, 11, 15). A significant number of studies have focused on the cellular and viral proteins that directly form RNP complexes with the 5′ and 3′ UTRs (7, 9, 11, 16). Several trans-acting proteins, such as polypyrimidine tract binding protein (PTB) (17) and poly(rC) binding protein 2 (PCBP-2) (18), interact with the 5′ and/or 3′ UTR. These interactions may be important for HCV replication, although the precise roles are not yet known.
Human La protein is a multifunctional phosphoprotein originally identified as an autoantigen in patients with autoimmune disorders. It is an RNA binding protein of the RNA recognition motif (RRM) family. La protein is located predominantly in the nucleus and functions in the maturation of RNA polymerase III transcripts and the unwinding of double-stranded RNA (19, 20). La protein associates with U1 RNA (21), telomerase RNA (22), adenovirus VA RNA (23), and the HIV trans-activation response (TAR) element (24). Other targets of La include the 5′ UTRs of poliovirus (25), hepatitis C virus (26, 27), encephalomyocarditis virus (28), and Bip mRNA (29). La protein binds to the HCV 5′ UTR near the initiator AUG and stimulates IRES-mediated translation (26). La protein has three putative RRMs and a basic region followed by an acidic region at the C terminus (30). Our previous studies revealed that only the RRM at residues 112 to 184 binds HCV RNA around the region encompassing the GCAC motif (which is relatively conserved across HCV genotypes) located in the stem region within stem-loop IV (SLIV) near the iAUG. Mutations in the GCAC motif alter La binding to the HCV IRES and have drastic effects on HCV translation both in vitro and ex vivo (31, 32).
Here, we investigated the possible role of La interaction with the GCAC motif in HCV replication. We have demonstrated that a GCAC motif mutation near the iAUG within SLIV, which alters the primary sequence while retaining the overall secondary structure, has a drastic effect on HCV replication in the context of a monocistronic subgenomic replicon and an infectious JFH1 RNA. To further address whether reduced La protein interactions with the GCAC motif affect HCV replication as a consequence of translation inhibition, we mutated the bicistronic replicon pSGR-JFH1/Luc. Interestingly, we found that HCV RNA levels decreased in the pSGR-JFH1/Luc mutant as well. Furthermore, we found that this inhibition of replication can be rescued by La overexpression. We also found that the mutation reduced the interaction between La and NS5B. Additionally, we demonstrated that this mutation affected the translation-to-replication switch regulated by the interplay between NS3 protease and human La protein. Furthermore, we showed that specific interaction of La with the GCAC motif promotes linkage between both ends of the HCV genome, indicating a plausible mechanism for regulating HCV replication by long-range RNA-RNA interactions. To our knowledge, this study constitutes the first report demonstrating the importance of the cis-acting element GCAC located within the HCV IRES in HCV replication.
MATERIALS AND METHODS
Cell culture.
Huh7 and Huh7.5 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U of penicillin/ml, and 100 μg of streptomycin sulfate/ml. Cells were maintained at 37°C in 5% CO2.
Materials.
Hemin was purchased from Merck Millipore. Puromycin and cycloheximide were purchased from Sigma.
Plasmids.
The HCV monocistronic replicon construct pFKi383hygubiNS3-3′5.1 (33), the full-length pJFH1 construct (34), and the bicistronic replicon construct pSGR-JFH1/Luc (35) used in this study were generous gifts from Ralf Bartenschlager and Takaji Wakita.
Site-directed mutagenesis.
Mutated replicons were generated by using a QuikChange Lightning multisite-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's protocol.
Transient transfection.
Huh7 cells (80% confluent) were transiently transfected with HCV mono- or bicistronic replicon RNA (2 μg) by using Lipofectamine 2000 reagent (Invitrogen) in antibiotic-free medium. Cells were harvested by using Tri reagent (Sigma) for RNA isolation, followed by reverse transcriptase PCR (RT-PCR). Cell lysates were prepared by using passive lysis buffer (Promega) to measure luciferase activities, using luciferase assay reagent (Promega).
Semiquantitative RT-PCR.
Total RNA was extracted by using Tri reagent (Sigma). One microgram of total RNA was reverse transcribed by using RT reaction mix (containing 1× reverse transcription buffer [Thermo Scientific], 1 mM deoxynucleoside triphosphate [dNTP] mix [Thermo Scientific], 40 U of RNase inhibitor [Promega], and 40 U of RevertAid Moloney murine leukemia virus [M-MuLV] RT [Thermo Scientific]), using an HCV 5′ primer (forward) or an HCV 3′ primer (reverse) to detect the HCV negative or positive RNA strand, respectively. Equal volumes (2 μl) of resulting cDNAs were used for PCR amplification corresponding to the HCV IRES by using PCR mix (containing 1× Taq polymerase buffer [New England BioLabs], 0.2 mM dNTP mix, and Taq polymerase [1 U/20 μl; New England BioLabs]) and the primers listed below. The PCR products were analyzed by running on a 2% agarose gel. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. Serial dilutions of the cDNA samples (1:2, 1:4, 1:8, and 1:16) were used to detect the exponential phase of the PCR. The following primers were used: HCV 5′ (forward) primer 5′-TGCGGAACCGGTGAGTACA-3′, HCV 3′ (reverse) primer 5′-CTTAAGGTTTAGGATTCGTGCTCAT-3′, GAPDH 5′ (forward) primer 5′-CATGAGAAGTATGACAACAGCCT-3′, and GAPDH 3′ (reverse) primer 5′-AGTCCTTCCACGATACCAAAGT-3′.
Quantitative RT-PCR.
Total RNA was isolated by using Tri reagent (Sigma) and reverse transcribed by avian myoblastosis virus (AMV) RT (Promega), using an HCV 5′ primer (forward) or an HCV 3′ primer (reverse) to detect the negative or positive RNA strand, respectively. cDNA (diluted 1:10) was used for PCR amplification by using a real-time assay mixture (Finnzymes) according to the manufacturer's instructions. The data were analyzed by using an ABI Prism real-time PCR machine, using the comparative ΔΔCT method (36). GAPDH was used as an internal control.
Tagged cDNA RT-PCR.
Total RNA was isolated by using Tri reagent (Sigma) and reverse transcribed with AMV RT (Promega), using a tagged HCV 5′ primer. Two rounds of PCR were done by using Hot Start Taq DNA polymerase (Genei). The first round of PCR was performed on cDNA using the tag-only primer and the HCV 3′ primer. The resulting PCR product was diluted 1:100 and used for the second round of PCR using a set of internal primers. The PCR product was run on a 2% agarose gel. GAPDH was used as an internal control.
Immunoprecipitation.
Huh7 cells (80% confluent) were transiently transfected with HCV monocistronic replicon RNA by using Lipofectamine 2000 reagent (Invitrogen) in antibiotic-free medium. After 24 h, cells were lysed by using buffer containing 100 mM KCl, 5 mM MgCl2, 10 mM HEPES (pH 7.0), 0.05% Nonidet P-40, 1 mM dithiothreitol (DTT), 100 U/ml RNase inhibitor, and 25 μl/ml protease inhibitor cocktail. Supernatants were incubated with anti-La antibody. Immunocomplexes were immobilized on protein G-Sepharose beads, followed by proteinase K and DNase treatments. Finally, phenol-chloroform-extracted and alcohol-precipitated RNA was used for RT-PCR. The same protocol was used for experiments using the bicistronic replicon RNA by using anti-NS5B antibody. Antiactin was used as a nonspecific control.
In vitro transcription.
mRNAs were transcribed in vitro from linearized plasmid constructs under T7 promoters in runoff transcription reactions using T7 RNA polymerase (Promega). Standard transcription reactions were performed according to the manufacturer's instructions.
In vitro translation.
In vitro-transcribed mRNAs were translated in vitro by using micrococcal nuclease-treated rabbit reticulocyte lysate (RRL), amino acid mix, and an RNase inhibitor (Promega), according to the manufacturer's instructions. Luciferase activity was measured by using luciferase assay reagent (Promega).
Preparation of cell extracts for Western blotting.
Huh7 cells were harvested and then lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 0.25% sodium deoxycholate, 1% Nonidet P-40, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM sodium orthovanadate, 1 mM sodium fluoride, and 12 μl/ml protease inhibitor cocktail) for 30 min at 4°C, followed by centrifugation at 12,000 × g for 10 min to pellet cellular debris. Supernatants were used for Western blotting.
Western blotting.
Protein concentrations in lysates were assayed by a Bradford assay (Bio-Rad), and equal protein amounts in cell extracts were separated by SDS–10% PAGE and transferred onto a nitrocellulose membrane (Pall Life Sciences). Samples were then analyzed by immunodetection using rabbit anti-La or anti-NS5B antibody followed by a secondary antibody (horseradish peroxidase-conjugated anti-rabbit IgG; Sigma). The blots were developed by using ECL techniques (Amersham Pharmacia Biotech). Mouse monoclonal antiactin antibody (Sigma) was used to verify equal loading of cell extracts.
Coimmunoprecipitation assay.
Transfected Huh7 cells were washed with cold phosphate-buffered saline (PBS) and lysed by using RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 12 mM deoxycholate sodium salt, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 0.2 mM sodium vanadate, and 1 mM phenylmethylsulfonyl fluoride). Cell lysates were centrifuged at 12,000 × g for 10 min at 4°C, and the supernatants were precleared with protein G-agarose at 4°C for 3 h. In parallel, anti-La (or anti-NS5B) antibody was incubated with protein G-agarose overnight in 1 ml of PBS at 4°C. This was followed by incubation of antibody conjugated with protein G-agarose with the precleared lysates at 4°C for 4 h. Precipitates were washed three times with RIPA buffer, and bound proteins were fractionated by SDS–10% PAGE and subjected to Western blot analysis using anti-NS5B (or anti-La) antibody.
Purification of La protein.
Escherichia coli BL21(DE3) cells were transformed with a bacterial expression vector encoding La (or mutant La). Transformed colonies were inoculated into 100 ml of LB medium containing 80 μg/ml ampicillin and grown at 37°C in an incubator shaker set at 200 rpm until the A600 reached 0.6. The culture was induced with 0.6 mM isopropyl-β-d-thiogalactopyranoside for 4 h at 37°C. The crude extracts were mixed with a Ni-nitrilotriacetic acid (NTA) agarose slurry (Qiagen) and rocked at 4°C for 2 h. The lysate was loaded onto a column and washed with 50 ml of wash buffer (50 mM Tris [pH 7.4], 300 mM NaCl, and 40 mM imidazole), and the bound proteins were eluted with 500 μl of elution buffer containing 500 mM imidazole. The eluted proteins were dialyzed at 4°C for 6 h in dialysis buffer (50 mM Tris [pH 7.4], 100 mM KCl, 7 mM β-mercaptoethanol, and 20% glycerol), aliquoted, and stored at −70°C
Two-cycle RNase protection assay.
A two-cycle RNase protection assay was performed as previously described (37). Briefly, total RNA was self-hybridized overnight at 60°C in the absence of radiolabeled probe, followed by RNase treatment for 1 h in digestion mix (500 mM NaCl, 10 mM Tris-HCl [pH 7.5], 5 mM EDTA, 4.5 μg of RNase A/ml, and 350 U of RNase T1/ml). Samples were subjected to proteinase K treatment followed by phenol-chloroform extraction and ethanol precipitation with 5 μg of tRNA. RNA samples were then suspended in hybridization buffer containing 75 fmol of radiolabeled probe and subjected to a standard RNase protection procedure.
UV cross-linking assay.
α-32P-labeled RNA probes were allowed to form complexes with recombinant La protein at 30°C for 20 min in RNA binding buffer (containing 5 mM HEPES [pH 7.6], 25 mM KCl, 2 mM MgCl2, 3.8% glycerol, 2 mM DTT, and 0.1 mM EDTA) and next irradiated with a UV lamp for 15 min. The reaction mixtures were treated with 30 μg of RNase A at 37°C for 30 min, followed by SDS-PAGE analysis and phosphorimaging.
5′-3′ coprecipitation assay.
A 5′-3′ coprecipitation assay was performed as previously described (38), with slight modifications. Briefly, 2 μg of in vitro-transcribed biotinylated 5′-UTR RNA was bound to 50 μl of streptavidin beads for 30 min at room temperature. The 32P-labeled 3′-UTR RNA probe was incubated with recombinant La or bovine serum albumin (BSA) in the presence of 10 μg of tRNA. The RNA-protein complex was then incubated with the 5′-UTR RNA-coated streptavidin beads at 4°C for 2 h. The beads were collected and washed. Precipitated RNA was analyzed on a denaturing 5% Tris-borate PAGE gel and autoradiographed.
Statistical analysis.
All graphs represent means ± standard deviations (SD). Student's t test was performed to determine statistical significance, and the criterion for statistical significance was a P value of <0.05 (* represents a P value of <0.05, ** represents a P value of <0.005, and *** represents a P value of <0.0005).
RESULTS
Mutation of the GCAC motif in the SLIV region affects HCV RNA functions.
Previously, it was shown that the GCAC sequence motif (which is relatively conserved across HCV genotypes) near the initiator AUG in the SLIV region of the HCV internal ribosomal entry site (IRES) is important for IRES-mediated translation. Consequently, mutation of the GCAC motif reduces IRES activity drastically (32). However, the role of this GCAC motif in HCV replication is unknown. Thus, to investigate whether the GCAC motif influences HCV replication, we introduced a mutation into SLIV in an HCV monocistronic replicon (33) in such a way as to alter the primary nucleotide sequence of the GCAC motif, while a compensatory mutation in the other strand maintained the secondary structure and GC content (Fig. 1A).
Fig 1.

Effect of mutation of the GCAC motif in SLIV on HCV RNA functions. (A) Schematic representation of the HCV 5′ UTR showing the position of the mutation incorporated into the SLIV region of the HCV monocistronic subgenomic replicon (33). Nucleotide positions of initiator AUG (iAUG) and the GCAC motif are also marked. (B) Huh7 cells were transfected with either in vitro-transcribed wild-type or mutated HCV monocistronic subgenomic RNA. The cells were harvested at 24 h posttransfection, and HCV RNA (negative-strand) levels were quantified by using semiquantitative RT-PCR. (C) Serial dilutions of the cDNA samples (1:2, 1:4, 1:8, and 1:16) were made. Amplified PCR products were analyzed by electrophoresis on a 2% agarose gel. GAPDH was used as an internal control. “U” denotes undiluted cDNA sample. (D to F) Tagged cDNA RT-PCR (D), quantitative RT-PCR (E), and Western blot for NS5B using anti-NS5B antibody (F) for the experiment performed in panel B. Actin was used as a control to ensure equal loading. (G) Huh7.5 cells were transfected with wild-type or mutated HCV JFH1 RNA. The cells were harvested at 24 h posttransfection, and HCV RNA (negative-strand) levels were quantified by using quantitative RT-PCR. (H) For the experiment performed in panel G, Western blotting for NS5B was done by using anti-NS5B antibody. Actin was used as a control to ensure equal loading.
To study the effect of this mutation on HCV replication, Huh7 cells were transiently transfected with in vitro-transcribed wild-type or mutated HCV monocistronic replicon RNA. Total RNA was isolated at 24 h posttransfection, and semiquantitative RT-PCR was performed (Fig. 1B). The mutated replicon showed a drastic reduction in HCV negative-strand synthesis compared to the wild-type replicon, suggesting the importance of the GCAC motif in replication.
PCR quantification can be performed only if the PCR product is measured within the exponential phase of the PCR, where the amount of amplified target is directly proportional to the amount of input target. To determine the exponential phase of the PCR empirically, serial dilutions of cDNA samples were made, and PCR amplification was performed over the same number of cycles. This assay also suggested that negative-strand RNA levels were low in the case of the mutated replicon (Fig. 1C).
Additionally, to discriminate negative-strand RNAs among a vast excess of positive-strand RNAs, RT-PCR using tagged primers was performed to selectively measure the level of negative-strand synthesis (39). Mutation of the GCAC motif caused an ∼75% reduction in HCV replication (Fig. 1D). Quantitative RT-PCR further confirmed these observations (Fig. 1E). In parallel, a Western blot analysis was performed where we could not detect NS5B in the case of the mutated replicon (Fig. 1F), which showed a drastic decrease in NS5B production from the mutated RNA.
Similarly, the effect of the mutation on HCV RNA replication in an infectious cell culture system was determined by transient transfection of Huh7.5 cells with JFH1 RNA. HCV negative-strand levels were measured at 24 h posttransfection by quantitative RT-PCR (Fig. 1G). The mutated RNA displayed a significant reduction (P < 0.005) in RNA replication as well as a considerable decrease in the production of NS5B protein, as seen in the Western blot analysis (Fig. 1H).
In the above-mentioned experiments, we normalized the transfection efficiency by performing transfection of Huh7 or Huh7.5 cells with the wild-type or mutated viral genome along with a Renilla luciferase reporter construct. At 24 h posttransfection, luciferase assays were performed, and there was no difference in transfection efficiencies (Fig. 2A and B).
Fig 2.

Effect of mutation of the GCAC motif on HCV RNA stability. (A) Huh7 cells were cotransfected with either in vitro-transcribed wild-type or mutated HCV monocistronic subgenomic RNA and a Renilla luciferase reporter construct. At 24 h posttransfection, a luciferase assay was performed, and the results are represented graphically (“ns” represents values that are not significant). (B) Huh7.5 cells were cotransfected with either in vitro-transcribed wild-type or mutated HCV JFH1 RNA and a Renilla luciferase reporter construct. At 24 h posttransfection, a luciferase assay was performed, and the results are represented graphically. (C) Integrity of in vitro-transcribed wild-type and mutated HCV monocistronic RNAs was checked in a 0.8% formaldehyde agarose gel. (D and E) Huh7 cells were transfected with wild-type or mutated HCV monocistronic subgenomic RNA and treated simultaneously with hemin (100 μM). Total RNA was isolated at 12 h and 24 h posttransfection, and the level of input positive-strand HCV was checked by quantitative RT-PCR using primers corresponding to either the 5′ UTR (D) or the 3′ UTR (E). GAPDH was used as an internal control for normalization [HCV(+) denotes HCV positive strand].
To rule out the possibility that low HCV RNA levels might be due to a decrease in the stability/integrity of the mutated viral genome rather than a defect in replication, we checked the integrity of wild-type and mutated RNAs in vitro using formaldehyde agarose gel electrophoresis (Fig. 2C).
Treatment with the replication inhibitor hemin, an iron donor, increases intracellular iron levels. Elevated iron levels inactivate NS5B (viral RNA-dependent RNA polymerase [RdRp]) by allowing iron to bind specifically and with high affinity to NS5B's Mg2+ binding pocket and thus suppressing new viral RNA synthesis without affecting translation (40, 41). We specifically determined the ex vivo stability of both the transfected RNAs (positive sense) by blocking (using hemin) the formation of subsequent HCV RNA strands as a result of replication of input RNA. For this purpose, Huh7 cells were transfected with wild-type or mutated RNA and treated simultaneously with hemin (100 μM). Total RNA was isolated at two time points (12 and 24 h posttransfection), and quantitative RT-PCR was performed by using primers corresponding to the 5′ and 3′ UTRs to determine the levels of positive-strand RNA (Fig. 2D and E). We did not find any change in RNA levels, suggesting that both wild-type and mutated RNAs are equally stable, even after transfection. The relative decrease in the RNA level at two subsequent time points might be the consequence of degradation of input RNA in the absence of replication.
Taken together, our results suggest that mutation in the cis-acting element GCAC in SLIV reduces HCV RNA replication and translation without affecting its stability.
Mutation of the GCAC motif reduces La interaction with HCV RNA.
Human La protein interaction with the GCAC motif near the initiator AUG within the HCV IRES is critical for internal initiation of translation (31, 32). To investigate whether this interaction is also important for HCV replication, Huh7 cells were transfected with in vitro-transcribed wild-type or mutated replicon RNA (positive sense) and simultaneously treated with hemin (100 μM) to block negative-strand synthesis. Cells were harvested at 24 h posttransfection, and ribonucleoprotein (RNP) complexes were immunoprecipitated by using anti-La antibody. The difference in the association of La with input positive-sense wild-type and mutated RNAs was measured by quantitative RT-PCR using RNA isolated from the immunoprecipitated complex. Interestingly, we found a significant reduction (P < 0.005) in HCV positive-sense RNA levels in complex from mutated RNA compared to the wild type (Fig. 3A). Western blotting was performed to show equal precipitation of La by using anti-La antibody (Fig. 3B). This observation suggested that mutation of the GCAC motif reduced La binding with positive-sense (input) RNA and hence decreased negative-strand synthesis (as shown in Fig. 1). This was confirmed by immunoprecipitation followed by semiquantitative (data not shown) and tagged cDNA RT-PCR of HCV negative-strand RNA (Fig. 3C).
Fig 3.

Effect of mutation of the GCAC motif on La interaction with HCV RNA. (A) Huh7 cells were transfected with in vitro-transcribed (positive-sense) wild-type or mutant monocistronic subgenomic RNA and simultaneously treated with hemin (100 μM). Cells were harvested at 24 h posttransfection, and ribonucleoprotein (RNP) complexes were immunoprecipitated by using anti-La antibody, followed by quantitative RT-PCR of the positive-sense RNA extracted from the immunoprecipitated complex. (B) Western blotting was done to verify equal precipitation of La. (C) Huh7 cells were transfected with wild-type or mutated HCV monocistronic subgenomic RNA. The cells were harvested at 24 h posttransfection, and RNA-protein complexes were precipitated by using anti-La antibody, followed by tagged cDNA RT-PCR for HCV negative-strand RNA. IP, immunoprecipitation. (D) La overexpression can rescue the inhibition of HCV RNA replication. Huh7 cells were cotransfected with wild-type or mutant HCV monocistronic subgenomic RNA along with a plasmid construct expressing either La or P4La (mutant La). The cells were harvested at 24 h posttransfection, and negative-strand HCV RNA levels were quantified by quantitative RT-PCR (“C” denotes control, “Wt” denotes wild-type RNA, “Mut” denotes mutated RNA, and “ns” represents values that are not significant). (E) Western blotting was done by using anti-NS5B antibody to check NS5B production levels. (F) La and P4La overexpression was checked by Western blot analysis using anti-La antibody.
We further investigated whether La overexpression could rescue the observed RNA replication inhibition. Huh7 cells were transiently cotransfected with wild-type or mutated RNA and the plasmid construct expressing La. HCV RNA levels were quantified by using quantitative RT-PCR (Fig. 3D). Interestingly, La overexpression rescued the inhibition of replication and increased HCV negative-strand RNA levels significantly (P < 0.05). P4La, a mutant La protein which interacts inefficiently with the HCV IRES (42), was used as a negative control. P4La overexpression decreased HCV replication of both wild-type and mutated RNAs slightly, perhaps due to competition between endogenous La and P4La supplied by overexpression to bind to the GCAC motif. Similar observations were made at the protein level during Western blot analysis (Fig. 3E). La/P4La overexpression was verified by semiquantitative RT-PCR (data not shown) and Western blot analysis (Fig. 3F). Taken together, our results suggest that mutation of the GCAC motif decreases La interaction with HCV positive-strand RNA and consequently affects negative-strand synthesis. This effect of the GCAC mutation can be rescued by supplying exogenous La by overexpression of the La gene.
Mutation of the GCAC motif affects HCV replication independent of translation.
The role of the La interaction with the GCAC motif in HCV replication might be dependent on or independent of translation. To investigate this, an HCV bicistronic replicon, pSGR-JFH1/Luc, where luciferase translation is driven by the HCV IRES and nonstructural HCV proteins are produced by the encephalomyocarditis virus (EMCV) IRES, was used (35). We mutated the GCAC motif in the HCV IRES of this bicistronic replicon without affecting the EMCV IRES, and semiquantitative RT-PCR was performed at 24 h and 48 h posttransfection. At the 24-h time point, we observed an ∼50% reduction in HCV negative-strand synthesis of the GCAC-mutated RNA compared to the wild type (Fig. 4A), providing evidence that the GCAC motif has a distinct role in replication independent of translation. Quantitative RT-PCR further corroborated this result (data not shown). At 48 h posttransfection, we found only a marginal increase in HCV RNA levels in cells transfected with wild-type RNA but a considerable decrease in cells transfected with mutated RNA, further suggesting that the mutated RNA does not replicate efficiently and that the GCAC motif is an essential cis-acting element required for efficient HCV replication.
Fig 4.

Mutation of the GCAC motif affects HCV replication independent of translation. (A) Huh7 cells were transfected with in vitro-transcribed wild-type or mutant pSGR-JFH1/Luc RNA. The cells were harvested at 24 h and 48 h posttransfection, and HCV RNA levels were quantified by using semiquantitative RT-PCR. The schematic above the panel represents the pSGR-JFH1/Luc replicon (35). (B) Luciferase levels were also assayed. (C) Effect of La silencing on HCV replication. Huh7 cells were cotransfected with wild-type or mutant pSGR-JFH1/Luc RNA and siLa (50 nM). The cells were harvested at 24 h and 48 h posttransfection, and HCV RNA levels were quantified by using semiquantitative RT-PCR. (D) Western blotting was done for La by using anti-La antibody to check silencing. Actin was used as a control to ensure equal loading. (E) A luciferase assay was also performed, and the results are represented graphically. (F) Western blotting for NS3 was done using by anti-NS3 antibody to check EMCV IRES-mediated translation. Actin was used as a control to ensure equal loading.
To investigate the effect of the GCAC motif mutation on HCV translation, in vitro-transcribed wild-type and mutated pSGR-JFH1/Luc RNAs were in vitro translated in RRL. A drastic reduction in the internal initiation of translation of mutated RNA was observed (data not shown). This was further confirmed under ex vivo conditions. Huh7 cells were transiently transfected with equal amounts of wild-type or mutated RNA. Cells were lysed at 24 and 48 h posttransfection, and extracts were assayed for luciferase activity. Consistent with the in vitro results, translation efficiency was reduced drastically (P < 0.0005) for the RNA with a mutated GCAC motif (Fig. 4B).
Furthermore, we investigated the effect of La silencing on replication of wild-type and mutated pSGR-JFH1/Luc RNAs. Huh7 cells were transiently cotransfected with equal amounts of wild-type or mutated RNA and small interfering RNA targeting the La gene (siLa) (50 nM). HCV RNA levels were quantified at 24 h and 48 h posttransfection (Fig. 4C). Although we observed a decrease in the replication of both wild-type and mutated RNAs after La silencing, this decrease was greater for RNA with the mutated GCAC motif. La silencing was verified by Western blot analysis (Fig. 4D). A similar observation was found when translation was tested in a luciferase-based translation assay (Fig. 4E).
To investigate if the GCAC mutation in the HCV IRES has any effect on the EMCV IRES activity of the pSGR-JFH1/Luc bicistronic RNA, we performed Western blot analyses to detect the viral proteins NS3 (Fig. 4F) and NS5B (data not shown) in the presence of hemin (100 μM). As expected, no change in the translation level was observed for both wild-type and mutated RNAs.
Therefore, the results suggest that although viral proteins are being synthesized equally well in wild-type and mutated RNAs, replication is still inhibited, indicating that this replication inhibition might be independent of translation inhibition.
GCAC facilitates interaction between La and viral RdRp.
Host and viral proteins interact with each other directly via protein-protein interactions or indirectly via RNA to form a functional replication complex on positive-sense viral genomic RNA. Formation of these complexes is a prerequisite for replication (43, 44). To investigate whether the GCAC motif located in the positive strand of the HCV IRES plays any role in mediating the interaction of La with viral RdRp (NS5B), we performed coimmunoprecipitation assays. Huh7 cells were transfected with in vitro-transcribed positive-sense wild-type or GCAC motif-mutated bicistronic subgenomic replicon RNA and treated with hemin (100 μM). Immunoprecipitation was performed at 24 h posttransfection by using anti-La antibody, and Western blot analysis was performed to detect NS5B. Although equal amounts of La were immunoprecipitated in the cases of both wild-type and mutated RNAs, less La-associated NS5B was immunoprecipitated by the mutated RNA than by the wild type (Fig. 5A).
Fig 5.
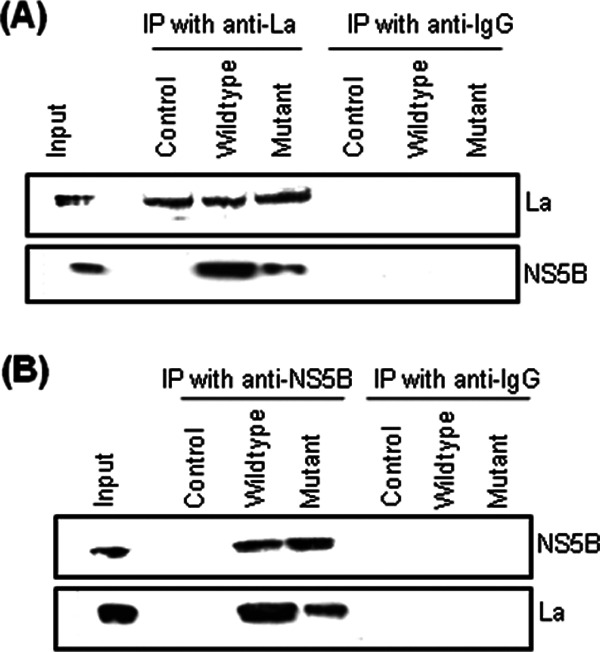
Mutation of the GCAC motif affects interactions between La and NS5B. Huh7 cells were transfected with in vitro-transcribed wild-type or mutated pSGR-JFH1/Luc RNA along with treatment with hemin (100 μM). At 24 h posttransfection, immunoprecipitation was done by using anti-La antibody (A) or anti-NS5B antibody (B), followed by Western blot analysis for La and NS5B. Anti-IgG antibody was used as a negative control for immunoprecipitation. “Control” indicates no transfection.
In a similar experiment, we performed immunoprecipitation with anti-NS5B antibody, followed by Western blot analysis for La. As in the above-described experiment, we found less La associated with NS5B when the GCAC motif was mutated (Fig. 5B).
These results suggest that the mutation in the GCAC motif of the HCV positive-strand RNA affects an interaction between La and NS5B, which may be crucial for initiation of negative-strand synthesis.
Exogenous La can rescue HCV replication inhibition but not translation inhibition.
Since mutation of the GCAC motif inhibits replication by affecting interactions between La and viral polymerase, we were interested in investigating the effect of exogenous La on replication inhibition. To address this, Huh7 cells were transiently cotransfected with equal amounts of wild-type or mutated pSGR-JFH1/Luc RNA and a construct encoding La or P4La. HCV RNA levels were quantified at 24 h posttransfection by using semiquantitative RT-PCR. As described above (Fig. 3D), we found that exogenous La rescued replication of the mutated RNA (Fig. 6A).
Fig 6.
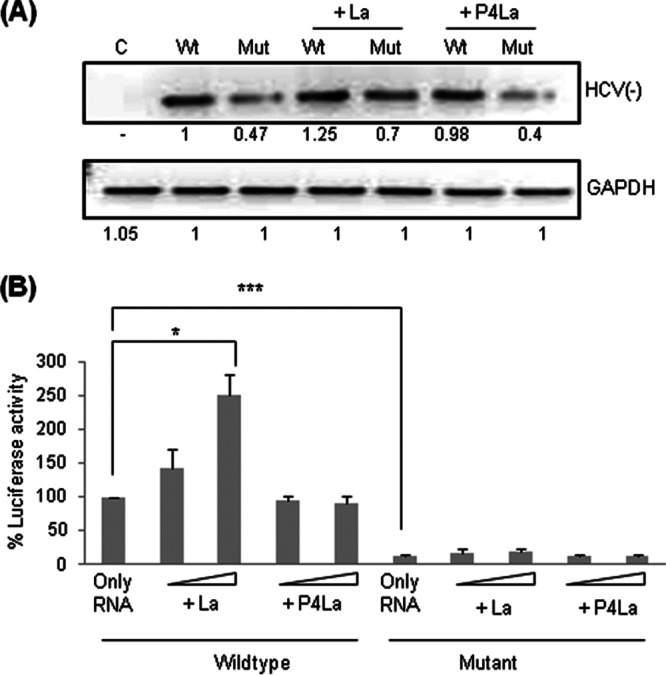
Effect of exogenous La on HCV replication and translation. (A) Huh7 cells were cotransfected with wild-type or mutated pSGR-JFH1/Luc RNA and a construct expressing La/P4La (mutant La). The cells were harvested at 24 h posttransfection, and replication was checked by semiquantitative RT-PCR (“C” denotes control, “Wt” denotes wild type, and “Mut” denotes mutant). (B) A luciferase assay was also performed to check HCV IRES-mediated translation.
Similarly, luciferase assays performed ex vivo demonstrated a dose-dependent increase in IRES activity for wild-type RNA but not for mutated RNA (Fig. 6B). This result was also confirmed under in vitro conditions by incubating wild-type and mutated pSGR-JFH1/Luc RNAs in the presence of increasing concentrations of purified recombinant La protein in the in vitro translation reaction mix. Consistent with the ex vivo results, in vitro data also revealed a dose-dependent stimulation of HCV IRES-mediated translation for wild-type but not for mutated RNA (data not shown). These observations suggest that exogenous La supplementation can rescue replication inhibition but not IRES-mediated inhibition caused by GCAC motif mutation.
Mutation of the GCAC motif in the SLIV region affects the translation-to-replication switch.
Previously, it was demonstrated that NS3 (viral factor) and human La protein (host factor) share similar binding sites in the SLIV region near the GCAC motif, and interplay between NS3 protease and human La protein regulates the translation-to-replication switch of HCV RNA (45). To investigate the effect of the GCAC motif mutation on the translation-to-replication switch, wild-type or mutated pSGR-JFH1/Luc RNA was transiently cotransfected with either an NS3pro-encoding construct or vector DNA. At different time points (0, 6, 12, 18, and 24 h posttransfection), HCV negative-strand RNA was measured by semiquantitative RT-PCR (Fig. 7A and B) and tagged cDNA RT-PCR (data not shown). GAPDH was used as an internal control. Overexpression of NS3pro caused a greater increase in HCV negative-strand wild-type RNA levels at the 18-h time than mutated RNA levels. Vector DNA was used as a control.
Fig 7.

Effect of mutation of the GCAC motif on the translation-to-replication switch. (A and B) Huh7 cells were cotransfected with wild-type (A) or mutant (B) pSGR-JFH1/Luc RNA and a construct expressing NS3pro or the vector. Total RNA was isolated at different time points, and HCV RNA was quantified by using semiquantitative RT-PCR (left). GAPDH was used as an internal control (right). (C and D) For the experiments detailed in panels A and B, luciferase assays were performed to detect translation levels. The percent luciferase activities were plotted for each reaction at different time points, taking the 6-h time point as 100% (control) (“ns” represents values that are not significant). (E and F) The ratio of percent replication to translation levels in vector-transfected control cells was compared with that of cells expressing NS3pro and is graphically represented by plotting the ratio of percent replication to translation on the y axis and time on the x axis. (G) For the experiments performed in panels A and B, the RNA protein complex was immunoprecipitated at 18 h posttransfection by using anti-NS5B antibody followed by semiquantitative RT-PCR for HCV negative-strand RNA. (H) Huh7 cells were cotransfected with in vitro-transcribed wild-type or mutated pSGR-JFH1/Luc RNA and a construct expressing NS3. Immunoprecipitation was done at 18 h posttransfection by using anti-La antibody, followed by Western blotting for NS3 by using anti-NS3 antibody. Western blotting for La was done to verify equal precipitation. Anti-IgG antibody was used as a negative control for immunoprecipitation. “Control” represents no transfection. − and + represent “without RNase A treatment” and “with RNase A treatment,” respectively.
In parallel, luciferase assays were performed at the same time points (starting at 6 h posttransfection) to assess the rate of IRES-mediated translation (Fig. 7C and D). Ratios of HCV RNA amplified (representing replication) to luciferase synthesis (representing translation) at each time point showed that wild-type HCV negative-strand RNA levels at 18 h posttransfection were ∼3.6-fold higher than those of the vector control (Fig. 7E). This ratio was found to be only ∼1.7-fold higher than that of the vector control for mutated RNA (Fig. 7F).
To further confirm our observation, the replication complex was immunoprecipitated by using anti-NS5B antibody at 18 h posttransfection, and HCV negative-strand RNA isolated from the complex was quantified by semiquantitative RT-PCR (Fig. 7G). Our results clearly showed an increase in negative-strand synthesis in the immunoprecipitated complex from the Huh7 cells transfected with the plasmid construct overexpressing NS3pro.
Moreover, we investigated the effect of mutation on the interaction between La, NS3, and HCV RNA during the translation-to-replication switch by coimmunoprecipitation assays. Less NS3 associated with La was immunoprecipitated by mutated RNA than by the wild type, suggesting the importance of the GCAC motif in mediating the interaction of La and NS3 with HCV RNA (Fig. 7H).
To further validate the above-described observations, we used translation inhibitors, i.e., puromycin and cycloheximide, which are known to induce dissociation or freezing of ribosomes, respectively, and hence have contrasting effects on negative-strand synthesis (46, 47). Huh7 cells were transfected with wild-type or mutated pSGR-JFH1/Luc RNA, followed by treatment with puromycin or cycloheximide at 12 h posttransfection. HCV negative-strand RNA was detected at different time points by using a two-cycle RNase protection assay (37). Our results clearly showed that puromycin treatment caused a greater increase in HCV negative-strand wild-type RNA synthesis at the 18-h time point than that of mutated RNA (Fig. 8A). In contrast, cycloheximide treatment caused a gradual decrease in both wild-type and mutated HCV negative-strand RNA levels (Fig. 8B). Controls were given no drug treatment (data not shown).
Fig 8.

The GCAC motif participates directly in enhancing HCV replication. (A and B) Huh7 cells were transfected with in vitro-transcribed wild-type or mutated pSGR-JFH1/Luc RNA, followed by treatment with puromycin (A) or cycloheximide (B) (12 h posttransfection) and detection of HCV negative-strand RNA at different time points (as indicated) by using a two-cycle RNase protection assay (the arrow above the 12-h time point indicates the time point of addition of puromycin or cycloheximide). (C and D) Luciferase assays were performed to check HCV IRES-mediated translation.
In parallel, we performed luciferase assays at different time points to determine the effect of puromycin and cycloheximide on HCV IRES-mediated translation (Fig. 8C and D). These observations clearly suggested the importance of the GCAC motif in regulating the translation-to-replication switch in the HCV life cycle by “directly” participating in negative-strand synthesis.
Site-directed mutagenesis reveals differential roles of individual GCAC motif nucleotides in HCV translation and replication.
To further investigate the role of the GCAC motif in replication at the single-nucleotide level, different point mutations (G345A, A347C, or C348G) were introduced into the pSGR-JFH1/Luc replicon, and HCV RNA levels produced by these mutated replicons were checked by semiquantitative RT-PCR. The G345A and C348G mutations caused a considerable reduction in HCV RNA levels, whereas the A347C mutation did not, suggesting that these nucleotides are not equally important for RNA replication (Fig. 9A).
Fig 9.
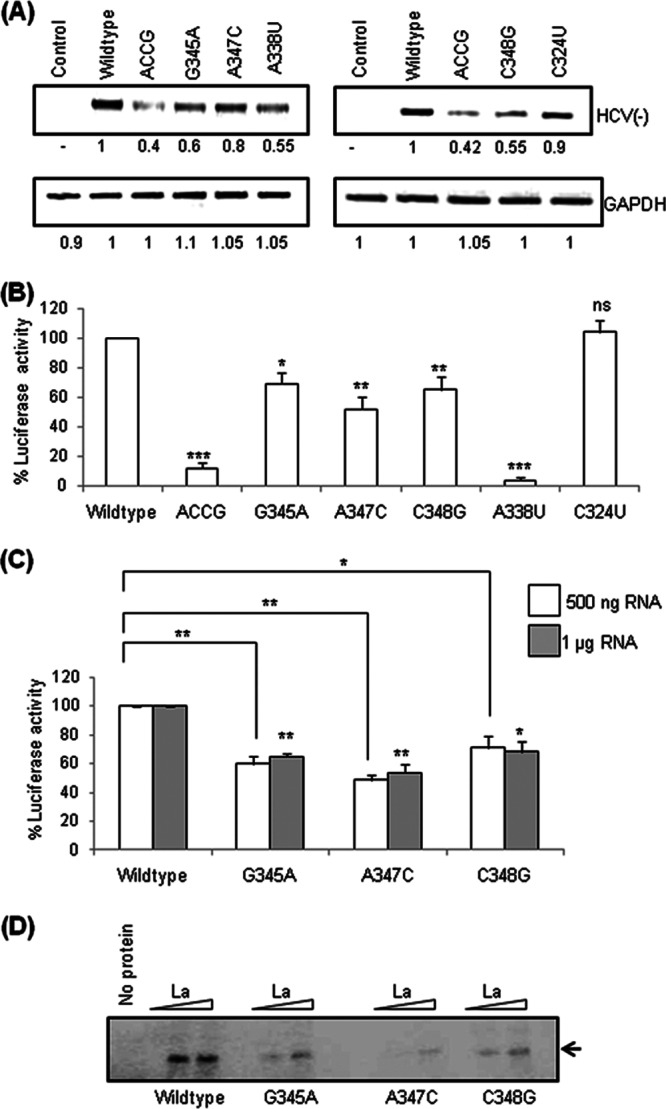
Effect of point mutations of the GCAC motif on HCV replication and translation. (A) Huh7 cells were transfected with in vitro-transcribed wild-type or mutated pSGR-JFH1/Luc RNA. The cells were harvested at 24 h posttransfection, and HCV negative-strand RNA levels were quantified by using semiquantitative RT-PCR. GAPDH was used as an internal control (ACCG indicates the same mutant represented in Fig. 1A, whereas G345A, A347C, C348G, A338U, and C324U represent point mutants). (B) One microgram of wild-type and various mutated pSGR-JFH1/Luc RNAs was translated in RRL, and percent luciferase activities were plotted (“ns” represents data that are not significant). (C) Wild-type and mutated (G345A, A347C, and C348G) HCV IRES-Luc RNAs (42) were translated in vitro in RRL, and percent luciferase activities were plotted. (D) In vitro binding of α-32P-labeled wild-type and mutated HCV IRES RNAs with increasing concentrations of recombinant La was performed by using a UV cross-linking assay. No protein was used as a negative control.
We further studied whether the various GCAC point mutations affected translation and replication equally or not. To study this, translation of RNAs containing point mutations was checked in vitro (Fig. 9B and C). Interestingly, it was observed that the A347C mutation had a greater inhibitory effect on translation than did the G345A and C348G mutations. Previously, it was shown that the A338U mutation renders RNA translationally incompetent (48), whereas the C324U mutation does not affect translation (32). Therefore, RNAs bearing the A338U and C324U mutations were used as translation controls. Additionally, using a UV cross-linking assay, we investigated the binding of recombinant La protein with the wild-type and mutated RNAs. The results of the in vitro binding studies of point mutants (Fig. 9D) corroborate the translation data (Fig. 9B and C), suggesting that it could be because of reduced and differential affinities of La for mutated RNAs. Taken together, our observations suggested differential effects of GCAC motif nucleotides on replication and translation.
La protein promotes interaction of 5′ and 3′ ends of the HCV genome via the GCAC motif.
Previously reported observations have shown that La protein binds both the 5′ and 3′ UTRs of the HCV genome (26, 49, 50). La also can undergo dimerization through its C terminus (20). It is therefore plausible that La protein could act as a protein bridge to mediate long-range RNA-RNA interactions between the 5′ and 3′ ends of the HCV genome for efficient RNA replication. To test this possibility, a 5′-3′ coprecipitation assay was used, wherein radiolabeled 3′-UTR RNA was precipitated by using the biotin-labeled 5′ UTR in the presence of La protein (Fig. 10A). We observed a significant increase (P < 0.0001) in the 5′-UTR–3′-UTR interaction in the presence of La protein (Fig. 10B). In contrast, in control reaction mixtures where La protein was absent or the unrelated protein BSA was used, very little coprecipitation was observed. Similarly, P4La (mutant La) could not coprecipitate 5′ and 3′ UTRs (data not shown). Moreover, increasing amounts of the unlabeled 5′- or 3′-UTR RNA could compete away the interaction between 5′ and 3′ UTRs (Fig. 10C), demonstrating that La linkage to both ends of the HCV RNA is a specific interaction.
Fig 10.
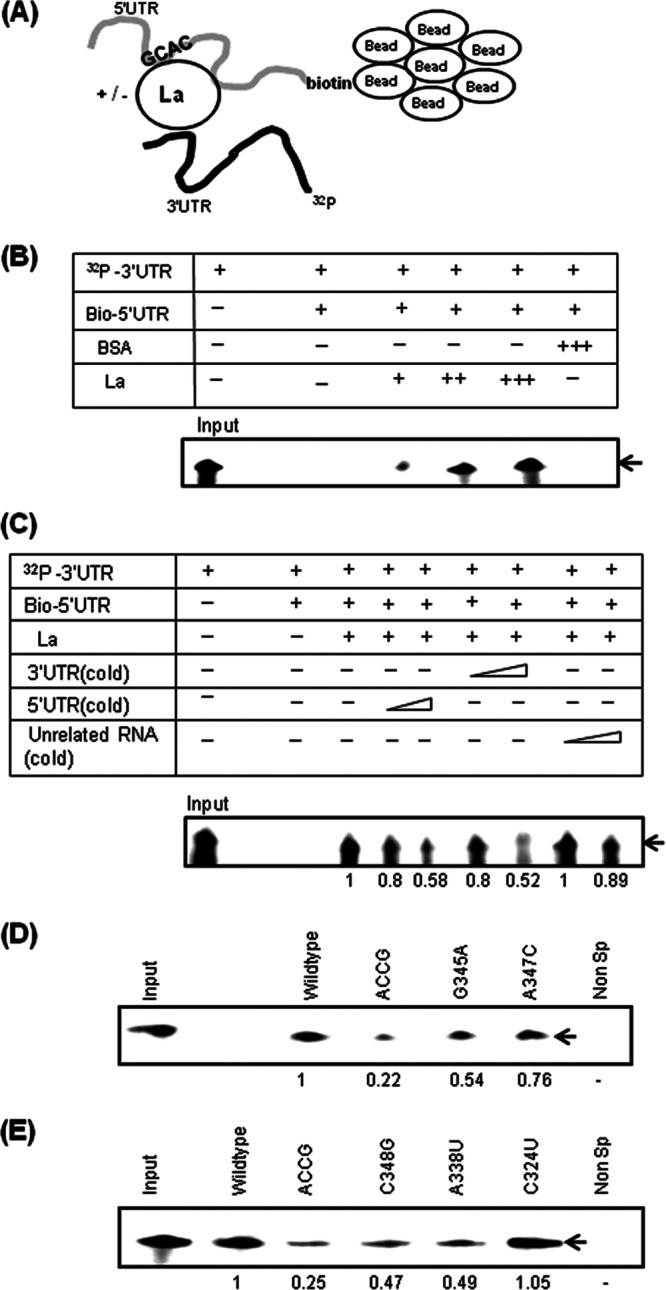
La protein interaction with the GCAC motif promotes 5′-to-3′ linkage. (A) Schematic representation of the 5′-3′ coprecipitation assay carried out with a biotinylated 5′ UTR and a 32P-labeled 3′ UTR. The location of the GCAC motif is also indicated. (B) Autoradiograph of precipitated RNAs in the absence or presence of increasing amounts of La (25, 50, and 100 ng). BSA (100 ng) was used as an unrelated protein control. (C) Increasing amounts of unlabeled (cold) 5′ UTR and 3′ UTR competed away the binding of the 32P-labeled 3′ UTR to the biotinylated 5′ UTR (Bio-5′UTR). (D and E) The same assay was performed with wild-type and various indicated mutated 5′-UTR RNAs in the presence of La (50 ng). Nonspecific RNA (unrelated nonviral RNA) that replaced the 5′ UTR in the assay mixture was used as a control.
Since the GCAC motif plays an important role in HCV replication, it was therefore likely that La promotes this interaction between the 5′ and 3′ UTRs via the GCAC motif. To test this hypothesis, we performed a 5′-3′ coprecipitation assay with mutated RNAs; RNAs bearing an ACCG, G345A, C348G, or A338U mutation formed significantly fewer 5′-3′ interactions, whereas the A347C and C324U mutations did not affect these interactions (Fig. 10D and E). Interestingly, coprecipitation assays of GCAC mutations gave results similar to those obtained by RT-PCR assays (Fig. 9A), suggesting that La mediating specific interactions between the GCAC motif (within the 5′ UTR) and the 3′ UTR is an important mechanism required for efficient HCV RNA replication.
DISCUSSION
Hepatitis C virus translation and replication are facilitated by several host and viral protein factors. One such host cell protein that facilitates HCV RNA translation as well as replication is the human La protein (11, 26, 27). Previous publications reported that the La protein binds to the GCAC motif of the SLIV region of the HCV IRES and thereby aids in ribosome assembly and translation initiation (31, 32). Apart from La and other proteins, such as NF90/NFAR, Rck/p54, and LSm1-7 (43, 44), RNA elements in the 5′ UTR are known to be crucial for RNA replication. However, the mechanistic details of replication enhancement by cis-acting RNA elements are not fully understood (12). The full mechanism of HCV RNA replication, therefore, is still under investigation.
In this study, we have explored whether the GCAC motif plays any role in HCV replication. Mutation of this region reduced the replication abilities of both subgenomic and full-length infectious JFH1 RNAs. Considering the fact that mutation of a critical RNA structural element might alter its stability, the loss of HCV RNA in a mutated genome could be simply the result of altered stability and not of decreased replication. Our observations using a replication inhibitor, hemin (40, 41), suggested that mutation of the GCAC motif does not affect RNA stability.
Furthermore, we found that an important reason behind the low level of negative-strand synthesis in the case of the mutant was the reduction in La protein association with the positive-sense mutated RNA. Therefore, an overexpression of La allowed an increase in the level of replication of the mutant RNA significantly. Moreover, our observations from the experiments performed with the bicistronic replicon clearly indicated the possibility of the existence of an independent role of the La protein interaction with the GCAC motif in replication. Here, we also found that La overexpression could rescue replication inhibition but not translation inhibition. It is possible that, apart from binding the La, the GCAC motif is involved in some other tertiary RNA-RNA interactions in the HCV genome that might be critical for internal initiation of translation (but not replication). It seems that, in the case of the mutated bicistronic replicon RNA, these interactions are not restored even after supplying La exogenously, and thus, IRES-mediated translation could not be rescued, although replication was restored to some extent.
The regulatory functions of IRES elements of HCV and picornaviruses are mediated through their interactions with many host proteins (18, 43, 51). For example, PTB interacts with multiple pyrimidine tracts in the HCV 5′ UTR and positively regulates HCV IRES translation (16). In the current study, we observed that the cis-acting element GCAC in the HCV IRES regulates the interaction between the host factor La and the viral polymerase NS5B. While levels of NS5B synthesized by transfected (positive-sense) wild-type and mutated pSGR-JFH1/Luc RNAs were equal, the association of NS5B with La (and vice versa) in an immunoprecipitated complex was reduced in mutated RNA compared to the wild type. Although we have not deciphered whether the interaction of NS5B and La in an RNP complex is a direct protein-protein interaction or is mediated indirectly via HCV RNA, it is still very clear that mutation of the GCAC motif affects this interaction. Therefore, we suggest that the GCAC motif mutation might block the initiation of negative-strand synthesis by affecting the binding of La protein to the GCAC motif and thus reduces the recruitment of NS5B at the replication site.
Recently, it was demonstrated that La protein and NS3 protease share a binding region near the initiator AUG in SLIV. It has been speculated that as soon as viral positive-strand RNA enters a cell, La binds to the GCAC motif and facilitates synthesis of viral proteins by IRES-mediated translation. However, once a threshold level of NS3 protein is synthesized, NS3 inhibits the La-mediated enhancement of translation by dislodging La protein from SLIV. This in turn allows HCV RNA to undergo replication (45). It is possible that NS3 does not dislodge La protein completely but instead competes with La until equilibrium is attained, as both proteins have similar Kd (dissociation constant) values for binding to the HCV IRES. Therefore, it appears that this interaction of La with the GCAC motif switches its role from translation to replication in the presence of NS3 by initiating the assembly of a functional replication complex on the positive strand involving NS5B and 5′- and 3′-UTR RNA elements. This hypothesis was supported by our data where the mutation of the GCAC motif (which affects negative-strand synthesis) reduces La and NS3 interactions. Therefore, apart from the role of NS3 in translation inhibition, this study suggests a possible role of NS3 in replication initiation, perhaps as a part of the replication complex formed by RNA circularization (as described below and as shown in Fig. 11).
Fig 11.
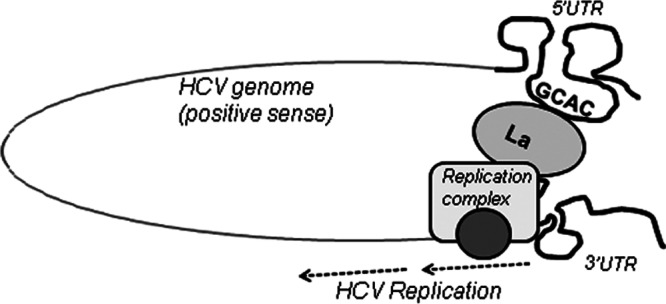
Model for initiation of HCV replication. La interaction with the GCAC motif within HCV IRES promotes 5′-to-3′ communication in favor of HCV negative-strand synthesis by assisting in the assembly of the replication complex.
It is well known that extensive evolution of HCV quasispecies takes place during persistent infection, which may lead to escape from host immune effector mechanisms through changes in B- and T-cell epitopes (52). Our studies also provide support for the possibility that replication can be regulated by incorporating point mutations in HCV RNA to promote long-term persistence of HCV infection in humans. Moreover, this kind of regulation might be a general phenomenon in the HCV life cycle due to the high level of conservation of the GCAC motif across most genotypes.
Genome circularization by long-range RNA-RNA interactions between the functional domains of the 5′ and 3′ ends is a common feature of many positive-stranded RNA viruses. However, specific details of circularization mechanisms likely vary from family to family, e.g., RNA-RNA interactions for flaviviruses (53), RNA-protein-protein-RNA interactions for picornaviruses (51), and cRNA-protein-protein-RNA interactions for coronaviruses and alphaviruses (54). The HCV genome contains well-defined RNA structural elements in its UTRs. Previous studies demonstrated that the protein-independent interaction of domain IIId and short stem-loop 5BSL3.2 is essential for replication (55). Also, previously reported data indicated the participation of different cellular factors in inducing interactions between the HCV 5′ and 3′ UTRs (38, 56). Although many studies have shown the interaction of La protein with both 5′ and 3′ UTRs (26, 49, 50), our study is the first to demonstrate that La binds to the 5′ and 3′ ends via the highly conserved GCAC motif and that this binding may aid in HCV genome circularization. As the GCAC motif plays an essential role in La and NS5B associations, it is possible that 5′-to-3′ cross-communication through La positively regulates HCV replication by providing a suitable platform for the viral polymerase NS5B to localize at the appropriate site to form a functional replication complex (Fig. 11).
Since La has RNA chaperone activity, it is also possible that this activity could be important for viral genome replication in various ways, e.g., by assisting in refolding of local RNA elements into different conformations and/or annealing complementary sequences in the 5′ and 3′ UTRs. There may be many other factors involved in HCV RNA replication that may interact along with La, NS5B, and NS3 at the GCAC motif. Future studies should focus on identifying the protein partners of this complex. Furthermore, it would be interesting to investigate whether this RNA-protein complex interacts with any other part of the HCV RNA to enhance the replication process.
Taken together, our study demonstrates that human La protein plays a critical role in regulating HCV replication by interacting with the GCAC motif. Our results firmly support the existence of La-dependent 5′-to-3′ communication in the HCV genome that involves RNA elements essential for viral translation and replication. Particularly, this study highlights the role of the GCAC motif within the IRES in HCV RNA replication in addition to its role in internal initiation of translation. Therefore, we may also consider this motif as a potent antiviral target. The interaction of La and GCAC was targeted previously to inhibit primarily IRES-mediated translation (42). However, this interaction can also be targeted specifically to block viral RNA replication. Since the La protein is less abundant in the cytoplasm of the host cell than in the nucleus, blocking of this protein might have drastic effects on viral RNA functions. These findings may also have important consequences for understanding the HCV replicative cycle and the genetic variability of the virus.
ACKNOWLEDGMENTS
We are grateful to Ralf Bartenschlager and Takaji Wakita for sharing plasmid constructs. We thank Charles M. Rice for providing Huh7.5 cells. We also thank Rebecca Cerio for critical reading of the manuscript and our laboratory members for their helpful discussions.
This work was supported by a grant from the Department of Biotechnology, India, to S.D. A.K. and U.R. were supported by predoctoral fellowships from the Council of Scientific and Industrial Research, India.
Footnotes
Published ahead of print 3 April 2013
REFERENCES
- 1. Suzuki T, Aizaki H, Murakami K, Shoji I, Wakita T. 2007. Molecular biology of hepatitis C virus. J. Gastroenterol. 42:411–423 [DOI] [PubMed] [Google Scholar]
- 2. Suzuki T, Ishii K, Aizaki H, Wakita T. 2007. Hepatitis C viral life cycle. Adv. Drug Deliv. Rev. 59:1200–1212 [DOI] [PubMed] [Google Scholar]
- 3. Brown EA, Zhang H, Ping LH, Lemon SM. 1992. Secondary structure of the 5′ nontranslated regions of hepatitis C virus and pestivirus genomic RNAs. Nucleic Acids Res. 20:5041–5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362 [DOI] [PubMed] [Google Scholar]
- 5. Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. 1992. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 66:1476–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yi M, Lemon SM. 2003. 3′ nontranslated RNA signals required for replication of hepatitis C virus RNA. J. Virol. 77:3557–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ito T, Lai MM. 1999. An internal polypyrimidine-tract-binding protein-binding site in the hepatitis C virus RNA attenuates translation, which is relieved by the 3′-untranslated sequence. Virology 254:288–296 [DOI] [PubMed] [Google Scholar]
- 8. Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463 [DOI] [PubMed] [Google Scholar]
- 9. Tanaka T, Kato N, Cho MJ, Shimotohno K. 1995. A novel sequence found at the 3′ terminus of hepatitis C virus genome. Biochem. Biophys. Res. Commun. 215:744–749 [DOI] [PubMed] [Google Scholar]
- 10. Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974 [DOI] [PubMed] [Google Scholar]
- 11. Domitrovich AM, Diebel KW, Ali N, Sarker S, Siddiqui A. 2005. Role of La autoantigen and polypyridimine tract binding protein in HCV replication. Virology 335:72–86 [DOI] [PubMed] [Google Scholar]
- 12. Friebe P, Lohmann V, Krieger N, Bartenschlager R. 2001. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J. Virol. 75:12047–12057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581 [DOI] [PubMed] [Google Scholar]
- 14. Shimakami T, Yamane D, Jangra RK, Kempf BJ, Sapniel C, Barton DJ, Lemon SM. 2012. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. U. S. A. 109:941–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 16. Ali N, Siddiqui A. 1995. Interaction of polypyrimidine tract-binding protein with the 5′ noncoding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J. Virol. 69:6367–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chung RT, Kaplan LM. 1999. Heterogeneous nuclear ribonucleoprotein I (hnRNP-I/PTB) selectively binds the conserved 3′ terminus of hepatitis C viral RNA. Biochem. Biophys. Res. Commun. 254:351–362 [DOI] [PubMed] [Google Scholar]
- 18. Fukushi S, Okada M, Kageyama T, Hoshino FB, Nagai K, Katayama K. 2001. Interaction of poly(rC)-binding protein 2 with the 5′-terminal stem loop of the hepatitis C-virus genome. Virus Res. 73:67–69 [DOI] [PubMed] [Google Scholar]
- 19. Tan EM. 1989. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv. Immunol. 44:93–151 [DOI] [PubMed] [Google Scholar]
- 20. Wolin SL, Cedervall T. 2002. The La protein. Annu. Rev. Biochem. 71:375–403 [DOI] [PubMed] [Google Scholar]
- 21. Madore SJ, Wieban ED, Pederson T. 1984. Eukaryotic small ribonucleoproteins. Anti-La human autoantibodies react with U1 RNA-protein complexes. J. Biol. Chem. 259:1923–1933 [PubMed] [Google Scholar]
- 22. Ford LP, Shay JW, Wright WE. 2001. The La antigen associates with the human telomerase ribonucleoprotein and influences telomere length in vivo. RNA 7:1068–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Francoeur AM, Mathews MB. 1982. Interaction between VA RNA and the lupus antigen La: formation of a ribonucleoprotein particle in vitro. Proc. Natl. Acad. Sci. U. S. A. 79:6772–6776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang YN, Kenan DJ, Keene JD, Gatignol A, Jeang KT. 1994. Direct interactions between autoantigen La and human immunodeficiency virus leader RNA. J. Virol. 68:7008–7020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meerovitch K, Svitkin YV, Lee HS, Lejbkowicz F, Kenan DJ, Chan EK, Agol VI, Keene JD, Sonenberg N. 1993. La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J. Virol. 67:3798–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ali N, Siddiqui A. 1997. The La antigen binds 5′ noncoding region of the hepatitis C virus RNA in the context of the initiator AUG codon and stimulates internal ribosome entry site-mediated translation. Proc. Natl. Acad. Sci. U. S. A. 94:2249–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ali N, Pruijn GJM, Kenan DJ, Keene JD, Siddiqui A. 2000. Human La antigen is required for the hepatitis C virus internal ribosome entry site-mediated translation. J. Biol. Chem. 275:27531–27540 [DOI] [PubMed] [Google Scholar]
- 28. Kim YK, Jang SK. 1999. La protein is required for efficient translation driven by encephalomyocarditis virus internal ribosomal entry site. J. Gen. Virol. 80:3159–3166 [DOI] [PubMed] [Google Scholar]
- 29. Kim YK, Back SH, Rho J, Lee SH, Jang SK. 2001. La autoantigen enhances translation of BiP mRNA. Nucleic Acids Res. 29:5009–5016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goodier JL, Fan H, Maraia RJ. 1997. A carboxy-terminal basic region controls RNA polymerase III transcription factor activity of human La protein. Mol. Cell. Biol. 17:5823–5832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pudi R, Abhiman S, Srinivasan N, Das S. 2003. Hepatitis C virus internal ribosome entry site-mediated translation is stimulated by specific interaction of independent regions of human La autoantigen. J. Biol. Chem. 278:12231–12240 [DOI] [PubMed] [Google Scholar]
- 32. Pudi R, Srinivasan P, Das S. 2004. La protein binding at the GCAC site near the initiator AUG facilitates the ribosomal assembly on the hepatitis C virus RNA to influence internal ribosome entry site-mediated translation. J. Biol. Chem. 279:29879–29888 [DOI] [PubMed] [Google Scholar]
- 33. Frese M, Barth K, Kaul A, Lohmann V, Schwarzle V, Bartenschlager R. 2003. Hepatitis C virus RNA replication is resistant to tumour necrosis factor-alpha. J. Gen. Virol. 84:1253–1259 [DOI] [PubMed] [Google Scholar]
- 34. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich H-G, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kato T, Date T, Miyamoto M, Sugiyama M, Tanaka Y, Orito E, Ohno T, Sugihara K, Hasegawa I, Fujiwara K, Ito K, Ozasa A, Mizokami M, Wakita T. 2005. Detection of anti-hepatitis C virus effects of interferon and ribavirin by a sensitive replicon system. J. Clin. Microbiol. 43:5679–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 37. Novak JE, Kirkegaard K. 1991. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive strand encapsidation and the ratio of positive to negative strands in infected cells. J. Virol. 65:3384–3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Isken O, Baroth M, Grassmann CW, Weinlich S, Ostareck DH, Ostareck-Lederer A, Behrens SE. 2007. Nuclear factors are involved in hepatitis C virus RNA replication. RNA 13:1675–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Craggs JK, Ball JK, Thomson BJ, Irving WL, Grabowska AM. 2001. Development of a strand-specific RT-PCR based assay to detect the replicative form of hepatitis C virus RNA. J. Virol. Methods 94:111–120 [DOI] [PubMed] [Google Scholar]
- 40. Fillebeen C, Rivas-Estilla AM, Bisaillon M, Ponka P, Muckenthaler M, Hentze MW, Koromilas AE, Pantopoulous K. 2005. Iron inactivates the RNA polymerase NS5B and suppresses subgenomic replication of hepatitis C virus. J. Biol. Chem. 280:9049–9057 [DOI] [PubMed] [Google Scholar]
- 41. Bartolomei G, Cevik RE, Marcello A. 2011. Modulation of hepatitis C virus replication by iron and hepcidin in Huh7 hepatocytes. J. Gen. Virol. 92:2072–2081 [DOI] [PubMed] [Google Scholar]
- 42. Mondal T, Ray U, Manna AK, Gupta R, Roy S, Das S. 2008. Structural determinant of human La protein critical for internal initiation of translation of hepatitis C virus RNA. J. Virol. 82:11927–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Isken O, Grassmann CW, Sarisky RT, Kann M, Zhang S, Grosse F, Kao PN, Behrens SE. 2003. Members of the NF90/NFAR protein group are involved in the life cycle of a positive-strand RNA virus. EMBO J. 22:5655–5665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scheller N, Mina LB, Galão RP, Chari A, Giménez-Barcons M, Noueiry A, Fischer U, Meyerhans A, Diez J. 2009. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc. Natl. Acad. Sci. U. S. A. 106:13517–13522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ray U, Das S. 2011. Interplay between NS3 protease and human La protein regulates translation-replication switch of hepatitis C virus. Sci. Rep. 1:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barton DJ, Morasco BJ, Flanegan JB. 1999. Translating ribosomes inhibit poliovirus negative-strand RNA synthesis. J. Virol. 73:10104–10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gamarnik AV, Andino R. 1998. Switch from translation to RNA replication in a positive stranded RNA virus. Genes Dev. 12:2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shirasaki T, Honda M, Mizuno H, Shimakami T, Okada H, Sakai Y, Murakami S, Wakita T, Kaneko S. 2010. La protein required for internal ribosome entry site-directed translation is a potential therapeutic target for hepatitis C virus replication. J. Infect. Dis. 202:75–85 [DOI] [PubMed] [Google Scholar]
- 49. Spångberg K, Goobar-Larsson L, Wahren-Herlenius M, Schwartz S. 1999. The La protein from human liver cells interacts specifically with the U-rich region in the hepatitis C virus 3′ untranslated region. J. Hum. Virol. 2:296–307 [PubMed] [Google Scholar]
- 50. Spångberg K, Wiklund L, Schwartz S. 2001. Binding of the La autoantigen to the hepatitis C virus 3′ untranslated region protects the RNA from rapid degradation in vitro. J. Gen. Virol. 82:113–120 [DOI] [PubMed] [Google Scholar]
- 51. Herold J, Andino R. 2001. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol. Cell 7:581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kato N, Sekiya H, Ootsuyama Y, Nakazawa T, Hijikata M, Ohkoshi S, Shimotohno K. 1993. Humoral immune response to hypervariable region 1 of the putative envelope glycoprotein (gp70) of hepatitis C virus. J. Virol. 67:3923–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hahn CS, Hahn YS, Rice CM, Lee E, Dalgarno L, Strauss EG, Strauss JH. 1987. Conserved elements in the 3′ untranslated region of flavivirus RNAs and potential cyclization sequences. J. Mol. Biol. 198:33–41 [DOI] [PubMed] [Google Scholar]
- 54. Frolov I, Hardy R, Rice CM. 2001. cis-Acting RNA elements at the 5′ end of Sindbis virus genome RNA regulate minus- and plus-strand RNA synthesis. RNA 7:1638–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Romero-López C, Berzal-Herranz A. 2009. A long-range RNA-RNA interaction between the 5′ and 3′ ends of the HCV genome. RNA 15:1740–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang L, Jeng KS, Lai MM. 2011. Poly(C)-binding protein 2 interacts with sequences required for viral replication in the hepatitis C virus (HCV) 5′ untranslated region and directs HCV RNA replication through circularizing the viral genome. J. Virol. 85:7954–7964 [DOI] [PMC free article] [PubMed] [Google Scholar]