

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) latent genomes are tethered to host histones to form a minichromosome also known as an “episome.” Histones, which are core components of chromatin, are heavily modified by various histone-targeting enzymes. Posttranslational modifications of histones significantly influence accessibility of transcriptional factors and thus have profound effects on gene expression. Recent studies showed that epigenetic marks on the KSHV episome are well organized, exemplified by the absence of histone H3 lysine 9 (H3K9) methylation, a heterochromatic histone mark, from immediate early and latent gene promoters in naturally infected cells. The present study revealed a mechanistic insight into KSHV epigenome regulation via a complex consisting of LANA and the H3K9me1/2 histone demethylase JMJD1A/KDM3A. This complex was isolated from HeLa cell nuclear extracts stably expressing LANA and was verified by coimmunoprecipitation analyses and with purified proteins. LANA recruitment sites on the KSHV genome inversely correlated with H3K9me2 histone marks in naturally infected cells, and methylation of H3K9 significantly inhibited LANA binding to the histone H3 tail. Chromatin immunoprecipitation coupled with KSHV tiling arrays identified the recruitment sites of the complex, while depletion of LANA expression or overexpression of a KDM3A binding-deficient mutant decreased KDM3A recruitment to the KSHV genome. Finally, ablation of KDM3A expression from latently KSHV-infected cells significantly inhibited KSHV gene expression, leading to decreased KSHV replication during reactivation. Taken together, our results suggest that LANA may play a role in regulation of epigenetic marks on the KSHV genome, which is in part through association with the histone demethylase KDM3A.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also designated human herpesvirus 8 (HHV-8), has been linked to Kaposi's sarcoma (KS) as well as primary effusion lymphoma (PEL) (or body cavity B lymphoma [BCBL]) (1) and a subset of multicentric Castleman's disease (MCD) (2). KS has emerged as the major malignancy in HIV-infected AIDS patients coinfected with KSHV. Similar to other oncogenic herpesviruses, malignant transformation requires that KSHV must enter a latent state. During this period, all but a few viral genes, such as LANA (ORF73), which is involved in viral latency and/or cell transformation, are silenced. LANA is an essential factor for establishing KSHV latency and is required for maintaining KSHV episomes in infected cells (3–7). KSHV LANA, first identified in 1997, is highly expressed in all KSHV-associated disorders, such as KS, MCD, and PEL (8–12). LANA is a DNA binding protein, which binds to the KSHV origin of DNA replication, located at the terminal repeat sequence of the KSHV genome (3, 6). LANA can serve both as a transcriptional activator and as a repressor, depending on the context of promoters and cell lines used (13–15). Accordingly, LANA associates with a broad range of transcriptional regulators, such as RBP-Jκ, CBP, Daxx, BRD2, RB, p53, and Sp-1 (16–22). LANA-null KSHV, generated from a KSHV bacterial artificial chromosome (BAC) system, exhibited a highly lytic phenotype, indicating, in general, that LANA has a role in silencing of the KSHV genome, which may be, in part, through inhibition of the strong transactivator K-Rta (22–24). Several chromatin modifiers and histone binding proteins have also been identified as LANA-interacting proteins, including Brd4, Daxx, SUV39H1, and the methyl CpG binding protein MeCP2 (19, 25–27), suggesting that LANA may have a role in the epigenetic regulation of the KSHV genome during latency.
Previous studies, using a KSHV viral promoter chip, revealed that the KSHV genome is rapidly associated with acetylated histones after reactivation triggered by K-Rta induction (28). More recent reports revealed a landscape of active and repressive histone marks along the entire latent KSHV genome (29, 30). H3K27me3 is the dominant repressive mark, which covers significant portions of the KSHV genome, whereas H3K9me3, another repressive mark, is clustered in mainly two regions corresponding primarily to late genes (28–30). Importantly, both active (H3K4me3) and repressive (H3K27me3) histone marks coexist in certain viral promoters, resembling stem cell signatures of histone marks (30). This “bivalent” state of such chromatin is postulated to signify the “readiness” of target genes to be transcribed upon reactivation. The major groups of enzymes catalyzing such histone modifications are histone lysine methyltransferases and demethylases; however, the mechanisms as to how these enzymes are recruited to the KSHV genome and their roles in viral gene expression remain to be determined.
Histone methylation has been known for more than 40 years and was considered an irreversible modification until recently. However, the identification of lysine demethylases (KDMs) completely overturned this dogma, and over 20 lysine demethylases have been identified so far (31). Each member of the KDMs demethylates specific lysine residues of basic histone tails in either a mono-, di-, or trimethylated state. KDM3A was the first identified histone demethylase from the jumonji family, which is known to demethylate the putative transcriptional repressive marks histone H3 mono- and dimethylated lysine 9 (H3K9me1/2) (32). Most histone-targeting KDMs contain histone binding domains such as a plant homeodomain (PHD) finger or Tudor domain; however, a few members of the KDMs require a DNA/histone binding protein for anchoring to the targeted methyl marks on their target genomic loci. KDM3A is one of the histone demethylases which lacks a defined histone binding domain and may require assistance from DNA/histone binding proteins (33). One of the partners of KDM3A is hypoxia-inducible factor 1α (HIF-1α). KDM3A recruitment is important for demethylating repressive marks on a subset of HIF-1α-inducible genes (34), thus creating massive transcriptional reorganization through epigenetic reinforcement. In addition to HIF-1α, KDM3A has been shown to interact with other transcriptional factors, such as androgen receptor (32) and Oct-1 (35), for maintaining an open chromatin structure by removing heterochromatic methyl marks.
Similar to cellular gene regulation, the significance of histone demethylases in regulation of herpesvirus gene expression has just begun to be revealed. For example, LSD1, the first identified histone lysine demethylase, is required for herpes simplex virus to reactivate from sensory neurons (36). Epstein-Barr virus (EBV) infection induces KDM6B (jumonji domain-containing 3) protein expression, resulting in B-cell differentiation (37). Similarly, our recent study identified that KDM4A, another jumonji family protein, is associated with the KSHV genome and inhibits heterochromatinization of early-lytic promoters (38).
In this report, we identified that LANA forms a protein complex with KDM3A in naturally infected cells and that KDM3A is recruited to LANA binding sites during latency. Knockdown of KDM3A significantly inhibited KSHV gene expression during reactivation, resulting in decreased KSHV replication. Our study suggests that LANA has a role in organization of the viral epigenome in infected cells to facilitate optimal viral gene expression.
MATERIALS AND METHODS
Cell culture.
Human embryonic kidney 293T (HEK 293T) epithelial cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) in the presence of 5% CO2. The KDM3A knockdown TREx-MH-K-Rta BCBL-1 cell line was generated by infection of TREx-MH-K-Rta BCBL-1 cells (39) with lentiviral particles expressing short hairpin RNA (shRNA) targeting KDM3A (shKDM3A) and grown in RPMI 1640 containing 15% FBS, 50 μg/ml blasticidin, 100 μg/ml hygromycin (Invitrogen), and 1 μg/ml puromycin (Invivogen). The shKDM3A vectors were commercially obtained (Open Biosystems). The control knockdown cells were similarly prepared by transduction of lentivirus generated with an empty vector. Recombinant KSHV.219 (rKSHV.219)-infected Vero cells (40) were maintained in DMEM containing 10% FBS and 1 μg/ml puromycin. rKSHV.219 stocks were produced by induction of lytic replication by treatment with 1 mM sodium butyrate, and cell-free supernatant was used to infect HEK 293T cells. rKSHV-infected HEK 293T cells were then selected with puromycin (1 μg/ml).
Plasmids.
The cloning strategy employed throughout these studies involved cloning of fragments into several modified vectors containing a CpoI restriction enzyme site generated within each polylinker. This strategy was described previously (41, 42). Fragments were digested with CpoI, desalted (Qiagen, Valencia, CA), and cloned into CpoI-digested vectors for expression in mammalian cells (pcDNA3-CpoI), lentiviral transduction (pLTRExBGHpA-CpoI), baculoviral expression (pFastBac-CpoI), and glutathione S-transferase (GST) fusion expression (pGEX-2T-CpoI). The resultant clones from all procedures possess an N-terminal Flag, hemagglutinin (HA), green fluorescent protein (GFP), or GST tag. Full-length wild-type LANA and wild-type KDM3A expression vectors were constructed as described above, using cDNA prepared from BCBL-1 cells. These plasmids served as templates for mutagenesis and for construction of truncated LANA and KDM3A fragments by PCR. Oligonucleotide sequences used throughout this study are available upon request. To generate recombinant proteins of Flag-KDM3A and Flag-LANA in the baculovirus protein expression system, KDM3A and LANA were subcloned into pFastBac1. Successive N-terminal deletion mutants encoding KDM3A-I (residues 1 to 250), KDM3A-II (residues 251 to 500), KDM3A-III (residues 501 to 750), KDM3A-IV (residues 751 to 1000), KDM3A-V (residues 1001 to 1322), and KDM3A-JmjC (residues 1059 to 1281) were cloned into a GFP-tagged pcDNA 3.0 or GST-tagged pGEX-2T vector for expression of recombinant protein in mammalian cells and in Escherichia coli, respectively. GST-LANA deletion mutant constructs were prepared essentially as described previously (43). LANA knockdown was achieved by generating a short hairpin RNA for LANA containing the sequence 5′-CACCGCATTTGTGTCTAGTCCTACTCGAAAGTAGGACTAGACACAAATGC-3′, which was cloned into the pENTR H1/TO vector according to the manufacturer's protocol (Invitrogen).
ChIP assay.
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (42). KDM3A knockdown ChIP assays were employed in control or KDM3A knockdown TREx-MH-K-Rta BCBL-1 cell lines. LANA knockdown ChIP assays were performed by using rKSHV.219-infected Vero cells transiently transfected with either control shRNA or LANA shRNA. To examine the effects of the dominant negative deletion protein encompassing residues 191 to 251 of LANA (LANA Δ191–251) on KDM3A occupancy, rKSHV.219-infected HEK 293T cells were transfected with either HA-KDM3A and Flag-LANA (wild type) or HA-KDM3A and Flag–LANA Δ191–251. At 48 h posttransfection, cells were harvested and processed for ChIP analysis. The antibodies used in the ChIP experiments were rabbit anti-LANA IgG (in-house), anti-KDM3A antibody (Proteintech), anti-H3K9me2 antibody (Abcam), anti-Flag antibody (Sigma), anti-HA antibody (Covance), and rabbit nonimmune serum IgG (Alpha Diagnostic International). All immunoprecipitated chromatin DNA was analyzed by SYBR green-based quantitative PCR (qPCR) (Bio-Rad) with primers described previously (43).
KSHV tiling array and ChIP with microarray technology (ChIP-on-chip).
The KSHV tiling array was designed across the KSHV genome sequence (GenBank accession number NC_009333.1) and manufactured by Agilent Technologies. The probes were spotted onto an 8- by 15-K array. In addition, probes specific for the promoter region of 5 human genes were also included in the microarray. This array contains 2,299 probes spanning the entire KSHV genome. Each probe on the array is 60 bases long. Whole ChIP samples and 10 ng of input were linearly amplified by using a WGA Genomeplex whole-genome amplification kit (Sigma). Labeling, hybridization, scanning, and analysis of the arrays were done at the UCD Cancer Center Genomic Shared Resource. The enrichment of LANA or KDM3A binding to the KSHV genome was calculated by the intensity ratio of immunoprecipitated and input DNA for each array spot, and probe signals were further normalized by the blank subtraction normalization method and the Whitehead error model by using Genomic Workbench software.
Immunoprecipitation and Western blot analysis.
Full-length Flag-LANA- and HA-KDM3A-transfected HEK 293T cells were collected in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 6.7], 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1× protease inhibitor cocktail (Roche). Five hundred micrograms of total cell lysates (TCLs) was incubated with anti-Flag M2 agarose beads (Sigma) overnight at 4°C. The interaction was probed by using anti-HA antibody (Covance). BCBL-1 cells were lysed similarly, and 1 mg of TCLs was incubated with either 3 μg of rabbit nonimmune serum IgG or anti-KDM3A rabbit IgG. Immune complexes were captured by protein A- and protein G-Sepharose beads. Beads were washed extensively with RIPA buffer, and the bound proteins were analyzed by immunoblotting with anti-LANA antibody (Advanced Biotechnologies Inc.). LANA interaction sites were mapped by cotransfection of full-length Flag-LANA and GFP-KDM3A deletion mutants in HEK 293T cells. Forty-eight hours after transfection, the cells were harvested and lysed with RIPA buffer. Five hundred micrograms of TCLs was incubated with anti-Flag M2 agarose beads overnight at 4°C. The beads were then washed three times with RIPA lysis buffer, and the interaction was visualized by immunoblotting with anti-GFP antibody (Sigma). Immunoblot analysis for KSHV lytic proteins was done by using anti-K-Rta rabbit antibody (44), anti-K-bZIP (41), anti-ORF57 (Santa Cruz), anti-K8.1 (Santa Cruz), and anti-ORF45 (Santa Cruz). To examine the effects of LANA Δ191-251 on KDM3A recruitment, rKSHV.219-infected HEK 293T cells were transfected with either a control empty Flag vector or a dominant negative Flag-LANA Δ191-251 construct. Twenty-four hours later, the cells were treated with a combination of 20 nM tetradecanoyl phorbol acetate (TPA) and 3 mM sodium butyrate. Total cell lysates were prepared at 0, 4, 8, 24, 56, and 72 h postreactivation.
Purification of recombinant protein.
Spodoptera frugiperda Sf9 cells were maintained in Ex-Cell 420 medium (JRH Biosciences), and recombinant baculoviruses were generated as previously described (42). Recombinant baculovirus bacmid DNA was transfected into Sf9 cells by using FuGENE6 (Roche), and recombinant viruses were subsequently amplified twice. Expression of recombinant proteins was confirmed by immunoblotting with anti-Flag monoclonal antibody (Sigma). Large-scale cultures of Sf9 cells (100 ml) were infected with recombinant baculovirus at a multiplicity of infection (MOI) of 0.1 to 1.0, and cells were harvested 48 h after infection. Recombinant proteins were purified as described previously (41). The purity and amount of protein were measured by SDS-PAGE and Coomassie blue staining, using bovine serum albumin (BSA) as a standard.
Preparation and purification of GST fusion proteins.
GST-KDM3A deletion mutants and GST-LANA deletion mutants were transformed and expressed in Escherichia coli strain BL21. Briefly, bacterial cells (250 ml) were cultured in Luria broth for each construct. Protein expression was induced with 0.5 mM (final concentration) isopropyl-d-thiogalactopyranoside (IPTG). The cells were harvested and washed once in phosphate-buffered saline (PBS) and then lysed by sonication in PBS containing 1% Triton X-100 and 0.3× Bugbuster lysis buffer (Novagen). After clearing by centrifugation at 7,000 × g for 15 min at 4°C, glutathione-Sepharose beads (200 μl of a 1:1 slurry in PBS) were added to the lysates for affinity purification. After overnight incubation at 4°C with rotation, the beads were washed four times in PBS containing 1% Triton X-100 and 0.3× Bugbuster lysis buffer. The proteins immobilized on the glutathione-agarose beads were quantified by Coomassie blue staining, using BSA as a protein standard.
In vitro interaction assay.
A total of 500 nM (final concentration) full-length Flag-KDM3A or Flag-LANA protein purified from Sf9 cells was incubated in 2× binding buffer (40 mM HEPES [pH 7.9], 120 mM KCl, 2 mM EDTA, 8 mM MgCl2, 2 mM dithiothreitol, 0.04% NP-40, 10% glycerol supplemented with 1 mg/ml BSA, 0.5 mM phenylmethylsulfonyl fluoride, and 1× protease inhibitor cocktail) with its GST-tagged deletion fusion protein counterparts for 1 h at 4°C. The immobilized GST-tagged deletion mutants were washed three times with 1× binding buffer and subjected to SDS-PAGE. The interaction was probed by immunoblotting with anti-Flag antibody.
Reverse transcriptase qPCR (RT-qPCR).
Total RNA was prepared with TRIzol (Invitrogen). cDNAs were synthesized by using SuperScript III first-strand synthesis reagents (Invitrogen). Gene expression was analyzed by qPCR using specific primers designed previously by Fakhari and Dittmer (45). KSHV gene expression was normalized to the cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) signal.
Peptide precipitation assays.
The peptides containing posttranslational modifications at specific residues (H3K9me1, H3K9me2, H3K9me3, H3pS10, H3K36me2, and H3K27me3) or unmodified peptides {histone H3 containing amino acid residues 21 to 44 [H3(1–21)] and H3(21–44)} were commercially obtained (AnaSpec Inc.). One microgram of unmodified or modified biotin-conjugated H3 peptides encompassing either residues 1 to 21 or residues 21 to 44 was incubated with purified Flag-LANA, Flag-EHMT1, or Flag-luciferase in 2× binding buffer (40 mM HEPES [pH 7.9], 120 mM KCl, 2 mM EDTA, 8 mM MgCl2, 2 mM dithiothreitol, 0.04% NP-40, 10% glycerol supplemented with 1 mg/ml BSA, 0.5 mM phenylmethylsulfonyl fluoride, and 1× protease inhibitor cocktail) for 1 h at 4°C in a volume of 80 μl. The amount of Flag-tagged recombinant protein used for the binding assay was 500 nM (final concentration). Each binding mixture was incubated with streptavidin beads (Novagen) overnight to capture biotinylated peptides. The peptide-bound streptavidin beads were sequentially washed 3 times in 1× binding buffer containing a final concentration of 500 mM NaCl. The peptide-bound proteins were eluted into SDS-PAGE sample buffer, and interactions were visualized by immunoblotting with anti-Flag antibody.
Immunofluorescence analyses.
BCBL-1 cells were fixed with 3.7% formaldehyde in PBS for 5 min at room temperature and subsequently treated with 1.0% Triton X-100 followed by 1.0% NP-40 in PBS for 10 min each at room temperature. After washing twice with 0.2% Tween 20 in PBS, cells were smeared onto a coverslip (Fisher). After treatment with blocking solution containing 2% BSA (Fisher) in PBS, cells on the coverslip were incubated with anti-KDM3A rabbit IgG (1:1,000 dilution) and anti-LANA rat IgG (1:1,000 dilution) in PBS–2% BSA for 1 h at room temperature. After washing four times with PBS, Alexa Fluor 488-conjugated anti-rat antibody and Alexa Fluor 555-conjugated goat anti-rabbit IgG F(ab′)2 (1:3,000 dilution) (Molecular Probes) were applied in blocking solution and allowed to react for 1 h at room temperature. After washing twice with Tris-buffered saline–Tween (TBST) and once with PBS, coverslips were air dried and mounted onto a glass slide (Fisher) with Antifade gold reagent (Invitrogen). Cells were imaged by using a wide-field three-dimensional (3D) deconvolution fluorescence microscope (personalDV; Applied Sciences, GE Healthcare) equipped with a 60×, 1.42-numerical-aperture (NA) oil immersion objective lens. 3D image visualization and quantitation were performed by using the VoloCITY digital imaging suite (Improvision, Perkin-Elmer).
RESULTS
LANA interacts with the cellular histone demethylase KDM3A.
Epigenetic gene regulation has profound effects on local gene expression and, hence, may influence the transition of the KSHV latency-lytic switch. Because the KSHV latent genome tightly associates with histones, and these histones are epigenetically decorated (29, 30), we asked whether LANA has a role in changing the local histone modifications of the KSHV genome. In addition, because KSHV LANA tethers KSHV chromatin to the host chromosome and is expressed throughout both phases of the KSHV life cycle, we hypothesized that LANA has a role in coordination of the KSHV epigenome. To explore this idea, we first isolated the LANA protein complex from 8 liters of HeLa cell culture stably expressing three repeats of N-terminally Flag-tagged LANA. The LANA protein complex was immunoprecipitated with Flag M2 agarose beads (Sigma), and LANA-interacting proteins were identified by mass spectrometry analysis. Control cultures, which expressed only the 3×Flag tag, were similarly prepared and subjected to the same procedure for background subtraction. Several potential transcriptional activators were found to be LANA-interacting proteins. These proteins were CCAR1 and THRP3. In addition, LANA also associates with the PRMT5/WDR77 complex, which may have a function in gene silencing through histone H4R3 methylation (46, 47). Importantly, we also identified a histone demethylase, KDM3A, as a LANA-interacting partner along with SF3B1, an RNA splicing factor from the same extracted band (Fig. 1A). Protein identification using MASCOT software identified KDM3A with a confidence interval equal to 100%. Because histone demethylases play a critical role in epigenetic gene regulation, we decided to focus on this interaction.
Fig 1.

KDM3A interacts with KSHV LANA in vivo. (A) Generation of stably 3×Flag-LANA-expressing HeLa cells. Immunoblotting was performed with anti-Flag antibody to confirm LANA expression. Cellular GAPDH was probed with anti-GAPDH monoclonal antibody and used as a loading control. (A) Mass spectrometry analysis. The LANA protein complex was isolated from HeLa nuclear extracts. 3×Flag-tagged LANA was stably expressed in HeLa cells, and nuclear extracts were prepared from 8 liters of suspension culture. Stably Flag-expressing control cells were similarly prepared for a background control. Protein complexes were subjected to SDS-PAGE on a 4-to-16% gradient gel, and the gel was stained with Sypro ruby. Some of the interacting partners that were identified by at least three unique peptides and a total of more than 10 reads with a confidence interval equal to 100% (Mascot/Scaffold program) are listed. hnRNPK, heterogeneous nuclear ribonucleoprotein K. (B) Coimmunoprecipitation of KDM3A and LANA in 293T cells (a) or BCBL-1 cells (b). HEK 293T cells were transiently cotransfected with HA-KDM3A and the Flag control or Flag-LANA. Cell lysates (500 μg) were immunoprecipitated with Flag agarose beads and immunoblotted with anti-HA (a). BCBL-1 cell lysates (1 mg) were immunoprecipitated with rabbit IgG control or anti-KDM3A rabbit IgG and immunoblotted with anti-LANA rat IgG (b). W.B., Western blotting. (C) Colocalization. Immunofluorescence staining of anti-LANA (green) and anti-KDM3A (red) in naturally infected BCBL-1 cells shows colocalization (yellow). Images were taken by using a wide-field 3D deconvolution fluorescence microscope, and 3D image visualization and quantitation were performed by using the VoloCITY digital imaging suite.
First, the interaction was confirmed with other methods; KDM3A and LANA expression vectors were constructed from BCBL-1 cDNA and cloned into a pcDNA HA-tagged vector and a Flag-tagged vector, respectively. These constructs were used to perform coimmunoprecipitation assays with HEK 293T cells. After immunoprecipitation with anti-Flag antibody, coprecipitation of KDM3A was probed with anti-HA antibody. The result indeed demonstrated that LANA associated with more than 5% of the input KDM3A in HEK 293T cells (Fig. 1Ba). The interaction was further verified by coimmunoprecipitation and colocalization experiments in naturally infected BCBL-1 cells (Fig. 1Bb and C). Importantly, automated statistical analysis of colocalization showed good correlation (Pearson's coefficient of 0.82 for this image) between red and green fluorescence distributions, indicating specificity of the interaction.
LANA interacts directly with the N-terminal region of KDM3A.
Although an interaction between KDM3A and LANA was observed by using different approaches, it is still possible that the interaction is mediated by histones or other LANA-associated proteins. To exclude this possibility, purified LANA and KDM3A were prepared from recombinant baculovirus-infected cells (Fig. 2A) and used for in vitro binding assays. Deletion mutants of LANA and KDM3A were generated as GST fusion proteins, and a series of GST pulldown assays were performed by incubation with their full-length counterparts. The results revealed that the deletion protein encompassing residues 191 to 251 of LANA (Fig. 2B) and the N-terminal portion (N-250) of KDM3A (Fig. 2C) were responsible for this interaction. Of note, LANA residues 191 to 251 precipitated more than 25% of the input, indicating the high affinity of the interaction. Finally, as LANA is heavily modified in cells, and these posttranslational modifications are known to influence protein interactions, we further confirmed the interaction domain in HEK 293T cells. The GFP-tagged KDM3A deletion plasmids were cotransfected with the full-length LANA expression construct, and the interaction was analyzed by coimmunoprecipitation analysis. The results showed again that the N-terminal portion of KDM3A was responsible for the interaction. Interestingly, the Jumonji C domain of KDM3A also interacted with LANA in cells; this is likely due to indirect interactions through chromatin binding. Taken together, these results clearly demonstrated that LANA associates with KDM3A in vitro and in vivo.
Fig 2.

In vitro interaction between KDM3A and LANA. (A) Flag-KDM3A and Flag-LANA proteins were purified from baculovirus-infected Sf9 cells. Purified proteins were subjected to SDS-PAGE and stained with Coomassie brilliant blue. (B) Schematic diagram of the domains of LANA. Deletion proteins used in this study are depicted in the middle. GST-LANA proteins were subjected to SDS-PAGE and stained with Coomassie (bottom). An in vitro GST pulldown assay was performed by incubation of purified full-length Flag-KDM3A with immobilized LANA-GST deletion mutants in binding buffer. LANA residues 191 to 251 precipitated nearly 25% of the input of full-length KDM3A. (C) Schematic diagram of the KDM3A domains based on the SMART database. Successive N-terminal GST deletion mutants of KDM3A, prepared from bacterial cells, were incubated with full-length baculovirus-purified Flag-LANA for a series of in vitro GST pulldown assays. The interaction was probed with anti-Flag antibody, and GST proteins used in these studies were visualized by Coomassie staining. (D) Confirmation of the interacting domain in HEK 293T cells. The indicated plasmids were cotransfected into HEK 293T cells. The coprecipitated deletion mutant of KDM3A was probed with anti-GFP antibody. Whole-cell lysates (WCL) were similarly probed and used as input controls.
LANA interacts with the nonmethylated histone H3 tail.
A histone binding protein often forms protein complexes with histone methylases or demethylases to generate a binding site(s) on histone tails, which helps the protein complex to anchor the sites. A prime example is the targeting of heterochromatin protein 1 (HP-1) to chromatin through both direct interactions with the lysine methyltransferase SUV39H1 and recognition of SUV39H1-mediated K9 methylation. Recruitment of this complex allows propagation of heterochromatin H3K9me3 methyl marks and, therefore, transcriptional repression (48). To examine if LANA interacts with the histone H3 tail and whether KDM3A facilitates LANA binding to the histone H3 tail, we performed a series of peptide pulldown analyses with purified Flag-LANA. Biotin-labeled peptides were incubated with purified LANA, and the interaction was detected by immunoblotting after pulling down with streptavidin-conjugated beads. Beads alone were used as a background control, and different types of histone modifications were examined for LANA binding. As shown in Fig. 3, LANA but not the luciferase control protein strongly interacted with nonmethylated H3 peptide, which was readily visible even after washing with high-salt buffer (500 mM NaCl). As a positive control, we also prepared full-length EHMT1, an H3K9 methyltransferase which has been shown to bind to H3K9me1/2 marks (49). As expected, the EHMT1 protein strongly interacted with the methylated H3K9me1/2 peptides but had much less affinity for H3K9me3. These experiments establish the specificity of the interaction between LANA and the nonmethylated histone H3 tail. Interestingly, although trimethylation and monomethylation of the H3K9 position inhibited LANA binding, dimethylation at lysine 9 did not. We repeated experiments with different salt concentrations; these experiments again indicated that LANA recognized the H3K9me2 mark but not mono- or trimethylated H3K9 peptides. In addition, the results also demonstrated that LANA specifically recognized the H3 N-terminal tail, because histone H3 containing amino acid residues 21 to 44 [H3(21–44)] precipitated very small amounts of LANA protein (Fig. 3).
Fig 3.

LANA preferentially binds to the nonmethylated N-terminal H3 tail. Peptide pulldown analyses were performed by incubation of 1 μg of biotinylated peptides with purified LANA, luciferase, or EHMT1. Biotinylated peptides were precipitated with a streptavidin bead mixture and washed extensively with binding buffer containing 500 mM NaCl. Interaction with the histone N-terminal tail was probed by immunoblotting with anti-Flag antibody. The EHMT1 protein was used in the peptide pulldown assay as a positive control for H3K9me1/2 binding, while luciferase protein served as a negative control. LANA but not luciferase interacts with the histone H3 N-terminal tail in a posttranslational-modification-specific manner. The band intensities are shown as a percentage of the input.
Identification of LANA/KDM3A recruitment sites on the KSHV genome.
Previous studies showed that LANA alters the landscape of histone modifications near LANA foci (50), suggesting that LANA may have a function as an “epigenetic modifier” as part of a protein complex. This prompted us to investigate the local histone modification at LANA binding sites. Previously, we determined LANA binding sites on the KSHV genome with KSHV tiling arrays, which consisted of 60-mer oligonucleotides covering the entire KSHV genome. In this study, we used a polyclonal anti-LANA rabbit IgG generated in our laboratory and repeated experiments similar to those previously described (43). Rabbit IgG has a higher affinity for protein A/G than rat IgG, which may allow increased sensitivity of our analyses. The ChIP-on-KSHV-chip with the anti-LANA rabbit IgG showed that there was a significant number of recruitment sites when a ratio of ChIP/input of over 2-fold was considered enrichment. There appeared to be several preferential recruitment sites on the KSHV genome, including previously identified loci such as the LANA promoter region, K1 promoter region, ORF75 promoter region, K15 regions, and ORF57 regions (Fig. 4A). Of note, LANA was largely eliminated at late gene cluster regions such as the ORF64 coding region. These LANA recruitment sites on the KSHV genome were confirmed by quantitative PCR analyses with specific primer pairs. The results were in good agreement with the tiling array data, which showed that LANA was recruited to the promoter and 5′ coding region of LANA but not the ORF64 coding region (Fig. 4B). These results were also consistent with previous reports (43, 51, 52).
Fig 4.

LANA and KDM3A recruitment sites on the KSHV genome. (A) LANA and KDM3A occupancy on the KSHV genome was examined by tiling array analyses. Co-occupying regions with more than 5-fold enrichment of both proteins are marked with orange shading. (B) Confirmation of ChIP-on-chip analyses. qPCR was performed on the samples with independent ChIP analyses. LANA and KDM3A were recruited to LANA loci but not the ORF64 coding region, which inversely correlated with the histone H3K9me2 mark. (C) Effects of LANA knockdown on KDM3A recruitment. Efficacy of LANA knockdown was examined by immunoblotting (a), and effects on viral copy number were monitored before ChIP analyses (b). Occupancy of LANA and KDM3A were examined by ChIP analysis followed by qPCR using primers for the LANA promoter and ORF64 coding region (c).
Identification of KDM3A binding sites on the KSHV genome.
With LANA binding sites on the KSHV genome now in our hands, recruitment of KDM3A was examined with KSHV tiling arrays. Corecruitment of LANA and KDM3A was observed mostly at the LANA promoter and LANA coding regions. In addition to the latent gene cluster region, a sharp peak was also observed downstream of the K-Rta coding region. Results of tiling array analyses were confirmed by qPCR (Fig. 4B). Consistent with the presence of KDM3A, H3K9me2 marks were depleted at the LANA region, and an elevated H3K9me2 level was observed at the ORF64 coding region (Fig. 4B). To further confirm that LANA is important for recruitment of KDM3A to LANA binding sites, LANA expression was transiently knocked down by introducing shRNA against LANA into latently rKSHV.219-infected Vero cells (Fig. 4Ca). We first confirmed that transient knockdown of LANA did not affect overall viral copy numbers in cells, which was examined by qPCR of viral genomic DNA (Fig. 4Cb). As shown in Fig. 4Cc, knockdown of LANA decreased KDM3A recruitment at the LANA promoter, indicating that LANA plays a role in the recruitment of KDM3A at this locus. Of note, we did not see increased lytic gene expression by the transient knockdown of LANA in our setting; this indicated that the decreased KDM3A recruitment was not due to alterations in chromatin configuration by the induction of lytic replication via reduced expression of LANA.
Virological significance of KDM3A.
As a next task, we addressed the significance of KDM3A in the KSHV life cycle. In addition, we asked if KDM3A is important for demethylation of H3K9me2 marks at LANA binding sites. To examine this, KDM3A expression was knocked down from K-Rta-inducible BCBL-1 cells. In these cells, K-Rta expression is driven by a tetracycline-inducible cytomegalovirus (CMV) promoter. Use of this cell line allowed us to purely study the effects of KDM3A during reactivation by eliminating effects of endogenous K-Rta promoter activity and, hence, K-Rta expression, which has profound effects on viral reactivation. Knockdown of KDM3A expression was first confirmed by immunoblotting (Fig. 5A), and ChIP assays were performed to examine and confirm the recruitment of KDM3A. As expected, shKDM3A significantly decreased KDM3A recruitment on the KSHV genome, which led to a slight increase in H3K9me2 levels (Fig. 5B). The fact that knockdown of KDM3A increased levels of H3K9me2 marks at the ORF64 coding region indicated that recruitment of KDM3A may not be entirely dependent on LANA and has global effects on H3K9me2 marks on the KSHV genome despite the existence of other demethylases that target H3K9me2. Nonetheless, the results suggested that KDM3A is actively demethylating H3K9me2 on the KSHV genome in vivo. Next, effects on KSHV gene expression during a course of reactivation were examined. KSHV reactivation was triggered by exogenous K-Rta expression for 4 h by addition of doxycycline to the culture medium. Total RNA was harvested at several different time points after reactivation, and the dynamics of viral gene expression were examined by a KSHV PCR array. As shown in Fig. 6Aa, knockdown of KDM3A inhibited the majority of viral gene expression at 56 h after induction of K-Rta. Curiously, effects on viral gene expression were dependent on the viral gene examined, and some viral genes, such as ORF48 and ORF59, showed increased gene expression levels when KDM3A was knocked down. To further confirm the results, a few viral genes were selected for relative gene expression analysis by qPCR with a more detailed time course. The results again showed that knockdown of KDM3A, in general, exhibited decreased viral gene expression levels, which is consistent with the diminished viral protein expression level (Fig. 6B). Consistent with this, the overall level of viral replication was decreased, as shown by viral copy numbers in BCBL-1 cells after 96 h of induction (Fig. 6C). It appeared that a 4-h pulse induction of K-Rta was able to activate viral gene expression, but it was not sufficient for a robust induction of viral DNA replication.
Fig 5.

Histone H3K9me2 demethylation of the KSHV genome by KDM3A. (A) KDM3A knockdown from K-Rta-inducible TREx-MH-K-Rta BCBL-1 cell lines. Immunoblot analysis was performed with the indicated antibodies. GAPDH was used as a loading control. (B) ChIP assays. ChIP assays were performed with the indicated antibodies with either shControl or shKDM3A stable TREx-MH-K-Rta BCBL-1 cell lines. Immunoprecipitated DNA was analyzed by qPCR analysis using specific primer pairs for the indicated loci.
Fig 6.

Effects of KDM3A knockdown on the KSHV life cycle. (A) Viral gene expression. KSHV reactivation was triggered by incubation with doxycycline for 4 h. Total RNA was harvested at the indicated time points, and KSHV PCR array analysis (a) or individual RT-qPCR (b) was performed to monitor viral gene expression. Cellular GAPDH and actin were used for internal controls, and the mean normalized expression (MNE) level is shown. For array analyses, individual gene expression levels were normalized to the lowest expression level for each column, and fold induction of viral gene expression is shown as a heat map. (B) Viral protein expression. A series of immunoblot analyses was performed to examine the expression of KSHV immediate early, early, and late genes at the protein level. The indicated antibodies were used to probe viral protein expression. Cellular GAPDH was used as a loading control. (C) Viral replication. The intracellular viral DNA copy number was measured by qPCR. The actin coding region was used as an internal control.
Finally, the significance of the interaction between KDM3A and LANA was examined by utilizing a dominant negative LANA mutant, LANA Δ191–251, which has a deletion in the KDM3A binding domain. The LANA mutant was first transfected into rKSHV-infected HEK 293T cells to prevent recruitment of KDM3A at LANA recruitment sites. Reactivation was then induced by addition of TPA and sodium butyrate. KSHV reactivation was monitored by viral protein expression. The results showed that in the presence of mutant LANA, viral protein expression was significantly inhibited (Fig. 7A). Recruitment of KDM3A was then examined by ChIP analyses after cotransfection of KDM3A and wild-type or mutant LANA. Equal amounts of KDM3A expression were first determined by immunoblotting (Fig. 7Ba). Results of the ChIP analyses showed that KDM3A recruitment was inhibited in the presence of the LANA mutant (Fig. 7Bb). Although the former experiment could not rule out that the observed inhibition of viral gene expression was due to other effects, such as an abundance of mutant LANA, the latter experiment at least pointed out that wild-type but not mutant LANA was able to recruit KDM3A and that mutant expression modulated KSHV reactivation. Taken together, these results suggest that KDM3A plays an important role in effective viral reactivation, which may be, in part, through the physical interaction with LANA.
Fig 7.

Effects of the dominant negative LANA Δ191–251 mutant on the viral life cycle and KDM3A recruitment. (A) Viral gene expression. rKSHV.219-infected HEK 293T cells were transfected with either the empty Flag vector alone or Flag–LANA Δ191–251. At 24 h posttransfection, cells were treated with a combination of TPA (20 nM) and sodium butyrate (3 mM), and lysates were subjected to immunoblotting analyses at the indicated time points. Immunoblotting was performed by using the indicated antibodies. (B) ChIP assays. (a) Immunoblot analysis of rKSHV-infected HEK 239T cells after transient transfection of the indicated plasmids. (b) Transfected cells were used for ChIP analyses using anti-Flag M2 antibody or anti-HA antibody. Wt, wild type.
DISCUSSION
KSHV establishes latency in infected individuals, in which most genes are silenced, except for a few latent genes. A KSHV recombinant which lacks LANA demonstrated a “highly lytic” phenotype, and similar observations were also made for rhesus rhadinovirus (RRV) (53, 54). These studies, at least, indicated that LANA protein has a role in inhibition of lytic gene expression. Our studies, however, indicated that LANA might have functions not only for establishment and maintenance of latency but also for effective reactivation via inhibition of heterochromatin formation (Fig. 8). The function may allow prompt reactivation by keeping an open chromatin configuration on certain regions of the KSHV genome where the LANA/KDM3A complex resides.
Fig 8.
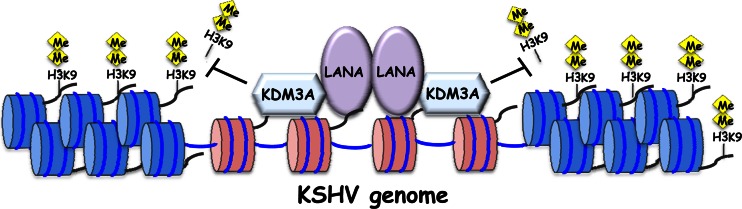
Hypothetical model of KDM3A and LANA interactions. During latency, the recruitment of KDM3A through LANA interactions maintains a euchromatin state (histones marked in red) on a subset of viral genes by preventing possession of the repressive H3K9me1/2 histone marks that form heterochromatin (histones marked in blue). Depletion of these repressive marks sets the stage for efficient lytic reactivation.
Our large-scale immunoprecipitation analyses with cells stably expressing LANA identified several interacting proteins. Interestingly, LANA interacted with both transcriptional coactivators (CCAR1 and THRP3) as well as corepressors (PRMT5/WDR77). In the mass spectrometry analyses, we also identified previously known LANA-interacting proteins, which include several ribosome-associated proteins, PRMT1, HDAC1, and histones (43, 55, 56). Although this method does not distinguish direct binding from indirect binding via other biomolecules, such as protein, DNA, or RNA, the result does indicate that these proteins exist in proximity to LANA and thus are likely to influence LANA's function. Accordingly, identification of multiple RNA binding proteins as LANA-interacting proteins suggests a possibility that LANA binds to RNA. We examined the possibility with in vitro RNA binding assays and found that LANA is indeed an RNA binding protein (M. Campbell, S. Huerta, P.-C. Chang, R. Davis, C. G. Tepper, K. Y. Kim, B. Schevchenko, D.-H. Wang, C. Izumiya, H.-J. Kung, and Y. Izumiya, unpublished data). Our studies (43) as well as others (52) independently studied LANA recruitment sites on both KSHV and human genomes. These studies revealed multiple recruitment sites with different occupancies, which may be attributed to different modes of binding, such as direct DNA binding, histone binding, as well as the tethering of local noncoding RNA; this may reflect the signal intensities of LANA recruitment observed by ChIP-on-KSHV-chip analyses (Fig. 4A).
Previously, we showed recruitment of KDM4A onto the KSHV genome and the concomitant loss of H3K9me3 marks at the KDM4A-recruited sites (38). Importantly, KDM4A can catalyze histone demethylation from H3K9me3 only to the monomethyl form. In principle, histone demethylases that catalyze demethylation of H3K9me1 to a nonmethylated state are required for H3K9 acetylation for gene activation and, hence, lytic reactivation. Substrates of KDM3A are H3K9me2 and -me1; thus, LANA/KDM3A may cooperate with KDM4A to generate a nonmethyl status. In fact, cooperation among histone demethylases for effective gene expression has been reported (57).
To our surprise, we found that LANA recognized the histone H3 tail in a posttranslational-modification-dependent manner. In silico analysis, however, did not identify any known consensus histone binding motifs, such as the Tudor domain or PHD finger, in the LANA protein sequence. LANA is known to bind histones through an acidic patch on H2B (56), which may facilitate the initial recruitment of the LANA/KDM3A complex proximal to histone H3, thus enabling KDM3A to demethylate local H3K9me1/2 marks. Removal of these methyl marks may allow a tighter association between the LANA/KDM3A protein complex and histones. It is important to note that the association between LANA and the histone H3 tail was very tight, which was resistant to 500 mM NaCl present in the washing buffer. This indicates the specificity of LANA's interaction with residues 1 to 21 of histone H3. It is tempting to speculate that there may be a possible positive-feedback loop between KDM3A and LANA in which recruitment of KDM3A may allow LANA to bind more tightly on the KSHV genome by removing H3K9 methylation. Importantly, our results are also in good agreement with previous work which showed that LANA was depleted from the H3K9me3 marks (50).
Our studies pointed out a relationship between KSHV replication and hypoxia. It seems likely that KSHV is well prepared for hypoxic conditions of the host and takes advantage of such an environment for viral replication. Previous studies showed that KSHV reactivation could be triggered by hypoxic conditions, which is initiated by K-Rta activation (58). Importantly, HIF transcriptional factors are stabilized in KSHV-infected cells by multiple mechanisms, which in turn induce viral replication as well as cell proliferation (59–61). In this report, we found that KDM3A, one of the HIF-1α transcriptional target genes, is a LANA binding partner and, together with LANA, may generate a suitable environment for KSHV gene expression. Interestingly, Cai et al. demonstrated that LANA physically interacts with HIF-1α at the N-terminal region of LANA (residues 46 to 89) and that the interaction between LANA and HIF-1α is important for nuclear accumulation of HIF-1α (61). As HIF-1α functionally and physically associates with KDM3A (34, 62), it is possible that LANA, HIF-1α, and KDM3A can form a ternary complex under hypoxic conditions. In accord with this, the KDM3A recruitment sites that we identified significantly overlap HIF-1α binding sites (63).
Our finding that KDM3A is important for KSHV gene expression suggests one possible molecular mechanism of hypoxia-mediated KSHV reactivation. Accordingly, we examined whether KDM3A alone was able to reactivate KSHV from recombinant KSHV-infected Vero cells; however, the results turned out to be negative (38) (data not shown). This may be due to the fact that the K-Rta promoter is silenced mainly by the histone H3K27me3 mark but not H3K9me2 marks (29, 30); thus, KDM3A's role in the KSHV life cycle is limited to the preparation of the KSHV genome for efficient gene expression by inhibiting “heterochromatin” formation on certain loci of the KSHV genome.
Initially, to our surprise, we found that KDM3A plays a role in maintaining ongoing viral gene expression more than the activation of promoters (Fig. 6A). Knockdown of KDM3A decreased viral gene expression quickly at the 56-h time point, but subtle differences were observed at the 24-h time point compared to control cells. This may indicate that other KSHV lytic products, such as K-Rta, may utilize KDM3A to sustain viral gene expression. Accordingly, transient reporter analysis showed that KDM3A was able to enhance K-Rta transactivation activity (data not shown). In this experiment, we induced KSHV reactivation by induction of exogenous K-Rta through an inducible CMV promoter; therefore, it may also mask initial effects of viral reactivation. Alternatively, other lysine demethylases, such as KDM4A, which demethylates H3K9me3 to H3K9me2, are required to see a significant phenotype in initial viral gene expression. This idea is also supported by our previous studies that showed inhibition of viral gene expression by ablation of KDM4A expression (38).
Based on the facts that cancer cells showed elevated expression levels of KDM3A, which lead to upregulation of cell proliferation (64–66), and KDM3A is a critical epigenetic modifier for robust gene expression under conditions of hypoxia (34, 62), inhibition of KDM3A may have cancer therapeutic potential for KSHV-associated malignancies via inhibition of cell growth as well as viral replication.
ACKNOWLEDGMENTS
We thank our collaborators, Clifford G. Tepper and Ryan Davis (University of California, Davis), for technical support for tiling arrays. We also thank Frank Chuang (University of California, Davis) for technical assistance in 3D deconvolution fluorescence microscopy and analyses of 3D images.
This work was supported by grants from the National Institutes of Health (R01-CA147791 to Y.I. and P01-DE19085 to Y.I. and H.-J.K.). This work was also supported partly by University of California, Davis, startup funds and a UCD Committee on Research award to Y.I.
Footnotes
Published ahead of print 10 April 2013
REFERENCES
- 1. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 2. Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hu J, Garber AC, Renne R. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 76:11677–11687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kelley-Clarke B, De Leon-Vazquez E, Slain K, Barbera AJ, Kaye KM. 2009. Role of Kaposi's sarcoma-associated herpesvirus C-terminal LANA chromosome binding in episome persistence. J. Virol. 83:4326–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kelley-Clarke B, Ballestas ME, Komatsu T, Kaye KM. 2007. Kaposi's sarcoma herpesvirus C-terminal LANA concentrates at pericentromeric and peri-telomeric regions of a subset of mitotic chromosomes. Virology 357:149–157 [DOI] [PubMed] [Google Scholar]
- 6. Komatsu T, Ballestas ME, Barbera AJ, Kelley-Clarke B, Kaye KM. 2004. KSHV LANA1 binds DNA as an oligomer and residues N-terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 319:225–236 [DOI] [PubMed] [Google Scholar]
- 7. Ye FC, Zhou FC, Yoo SM, Xie JP, Browning PJ, Gao SJ. 2004. Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J. Virol. 78:11121–11129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120:939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat. Rev. Cancer 10:707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verma SC, Lan K, Robertson E. 2007. Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 312:101–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schulz TF. 2001. KSHV/HHV8-associated lymphoproliferations in the AIDS setting. Eur. J. Cancer 37:1217–1226 [DOI] [PubMed] [Google Scholar]
- 12. Kedes DH, Lagunoff M, Renne R, Ganem D. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi's sarcoma-associated herpesvirus. J. Clin. Invest. 100:2606–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Renne R, Barry C, Dittmer D, Compitello N, Brown PO, Ganem D. 2001. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:458–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. An FQ, Compitello N, Horwitz E, Sramkoski M, Knudsen ES, Renne R. 2005. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J. Biol. Chem. 280:3862–3874 [DOI] [PubMed] [Google Scholar]
- 15. Jeong JH, Orvis J, Kim JW, McMurtrey CP, Renne R, Dittmer DP. 2004. Regulation and autoregulation of the promoter for the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 279:16822–16831 [DOI] [PubMed] [Google Scholar]
- 16. Verma SC, Borah S, Robertson ES. 2004. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 78:10348–10359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Friborg J, Jr, Kong W, Hottiger MO, Nabel GJ. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889–894 [DOI] [PubMed] [Google Scholar]
- 18. Radkov SA, Kellam P, Boshoff C. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6:1121–1127 [DOI] [PubMed] [Google Scholar]
- 19. Murakami Y, Yamagoe S, Noguchi K, Takebe Y, Takahashi N, Uehara Y, Fukazawa H. 2006. Ets-1-dependent expression of vascular endothelial growth factor receptors is activated by latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus through interaction with Daxx. J. Biol. Chem. 281:28113–28121 [DOI] [PubMed] [Google Scholar]
- 20. Platt GM, Simpson GR, Mittnacht S, Schulz TF. 1999. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 73:9789–9795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lim C, Gwack Y, Hwang S, Kim S, Choe J. 2001. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 276:31016–31022 [DOI] [PubMed] [Google Scholar]
- 22. Lan K, Kuppers DA, Robertson ES. 2005. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 79:3468–3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu F, Day L, Gao SJ, Lieberman PM. 2006. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J. Virol. 80:5273–5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lan K, Kuppers DA, Verma SC, Robertson ES. 2004. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J. Virol. 78:6585–6594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76:11596–11604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ottinger M, Christalla T, Nathan K, Brinkmann MM, Viejo-Borbolla A, Schulz TF. 2006. Kaposi's sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 80:10772–10786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakakibara S, Ueda K, Nishimura K, Do E, Ohsaki E, Okuno T, Yamanishi K. 2004. Accumulation of heterochromatin components on the terminal repeat sequence of Kaposi's sarcoma-associated herpesvirus mediated by the latency-associated nuclear antigen. J. Virol. 78:7299–7310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ellison TJ, Izumiya Y, Izumiya C, Luciw PA, Kung HJ. 2009. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi's sarcoma-associated herpesvirus. Virology 387:76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gunther T, Grundhoff A. 2010. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 6:e1000935 doi: 10.1371/journal.ppat.1000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 6:e1001013 doi: 10.1371/journal.ppat.1001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pedersen MT, Helin K. 2010. Histone demethylases in development and disease. Trends Cell Biol. 20:662–671 [DOI] [PubMed] [Google Scholar]
- 32. Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. 2006. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125:483–495 [DOI] [PubMed] [Google Scholar]
- 33. Shi Y, Whetstine JR. 2007. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 25:1–14 [DOI] [PubMed] [Google Scholar]
- 34. Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ. 2010. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol. Cell. Biol. 30:344–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shakya A, Kang J, Chumley J, Williams MA, Tantin D. 2011. Oct1 is a switchable, bipotential stabilizer of repressed and inducible transcriptional states. J. Biol. Chem. 286:450–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. 2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 15:1312–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anderton JA, Bose S, Vockerodt M, Vrzalikova K, Wei W, Kuo M, Helin K, Christensen J, Rowe M, Murray PG, Woodman CB. 2011. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin's lymphoma. Oncogene 30:2037–2043 [DOI] [PubMed] [Google Scholar]
- 38. Chang PC, Fitzgerald LD, Hsia DA, Izumiya Y, Wu CY, Hsieh WP, Lin SF, Campbell M, Lam KS, Luciw PA, Tepper CG, Kung HJ. 2011. Histone demethylase JMJD2A regulates Kaposi's sarcoma-associated herpesvirus replication and is targeted by a viral transcriptional factor. J. Virol. 85:3283–3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 77:4205–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240 [DOI] [PubMed] [Google Scholar]
- 41. Izumiya Y, Ellison TJ, Yeh ET, Jung JU, Luciw PA, Kung HJ. 2005. Kaposi's sarcoma-associated herpesvirus K-bZIP represses gene transcription via SUMO modification. J. Virol. 79:9912–9925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Izumiya Y, Izumiya C, Hsia D, Ellison TJ, Luciw PA, Kung HJ. 2009. NF-kappaB serves as a cellular sensor of Kaposi's sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jkappa coactivator. J. Virol. 83:4435–4446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Campbell M, Chang PC, Huerta S, Izumiya C, Davis R, Tepper CG, Kim KY, Shevchenko B, Wang DH, Jung JU, Luciw PA, Kung HJ, Izumiya Y. 2012. Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J. Biol. Chem. 287:5806–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Izumiya Y, Izumiya C, Van Geelen A, Wang DH, Lam KS, Luciw PA, Kung HJ. 2007. Kaposi's sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J. Virol. 81:1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fakhari FD, Dittmer DP. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213–6223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu X, Hoang S, Mayo MW, Bekiranov S. 2010. Application of machine learning methods to histone methylation ChIP-Seq data reveals H4R3me2 globally represses gene expression. BMC Bioinformatics 11:396 doi: 10.1186/1471-2105-11-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Antonysamy S, Bonday Z, Campbell RM, Doyle B, Druzina Z, Gheyi T, Han B, Jungheim LN, Qian Y, Rauch C, Russell M, Sauder JM, Wasserman SR, Weichert K, Willard FS, Zhang A, Emtage S. 2012. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U. S. A. 109:17960–17965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stewart MD, Li J, Wong J. 2005. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol. Cell. Biol. 25:2525–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, Cheng X. 2008. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat. Struct. Mol. Biol. 15:245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stuber G, Mattsson K, Flaberg E, Kati E, Markasz L, Sheldon JA, Klein G, Schulz TF, Szekely L. 2007. HHV-8 encoded LANA-1 alters the higher organization of the cell nucleus. Mol. Cancer 6:28 doi: 10.1186/1476-4598-6-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Campbell M, Izumiya Y. 2012. Post-translational modifications of Kaposi's sarcoma-associated herpesvirus regulatory proteins—SUMO and KSHV. Front. Microbiol. 3:31 doi: 10.3389/fmicb.2012.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lu F, Tsai K, Chen HS, Wikramasinghe P, Davuluri RV, Showe L, Domsic J, Marmorstein R, Lieberman PM. 2012. Identification of host-chromosome binding sites and candidate gene targets for Kaposi's sarcoma-associated herpesvirus LANA. J. Virol. 86:5752–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wen KW, Dittmer DP, Damania B. 2009. Disruption of LANA in rhesus rhadinovirus generates a highly lytic recombinant virus. J. Virol. 83:9786–9802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li Q, Zhou F, Ye F, Gao SJ. 2008. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 379:234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen W, Dittmer DP. 2011. Ribosomal protein S6 interacts with the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 85:9495–9505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861 [DOI] [PubMed] [Google Scholar]
- 57. Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, Metzger E, Schule R. 2007. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 9:347–353 [DOI] [PubMed] [Google Scholar]
- 58. Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R. 2001. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 97:3244–3250 [DOI] [PubMed] [Google Scholar]
- 59. Shin YC, Joo CH, Gack MU, Lee HR, Jung JU. 2008. Kaposi's sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 68:1751–1759 [DOI] [PubMed] [Google Scholar]
- 60. Carroll PA, Kenerson HL, Yeung RS, Lagunoff M. 2006. Latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J. Virol. 80:10802–10812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cai Q, Murakami M, Si H, Robertson ES. 2007. A potential alpha-helix motif in the amino terminus of LANA encoded by Kaposi's sarcoma-associated herpesvirus is critical for nuclear accumulation of HIF-1alpha in normoxia. J. Virol. 81:10413–10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mimura I, Nangaku M, Kanki Y, Tsutsumi S, Inoue T, Kohro T, Yamamoto S, Fujita T, Shimamura T, Suehiro J, Taguchi A, Kobayashi M, Tanimura K, Inagaki T, Tanaka T, Hamakubo T, Sakai J, Aburatani H, Kodama T, Wada Y. 2012. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol. Cell. Biol. 32:3018–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Veeranna RP, Haque M, Davis DA, Yang M, Yarchoan R. 2012. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen induction by hypoxia and hypoxia-inducible factors. J. Virol. 86:1097–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cho HS, Toyokawa G, Daigo Y, Hayami S, Masuda K, Ikawa N, Yamane Y, Maejima K, Tsunoda T, Field HI, Kelly JD, Neal DE, Ponder BA, Maehara Y, Nakamura Y, Hamamoto R. 2012. The JmjC domain-containing histone demethylase KDM3A is a positive regulator of the G(1)/S transition in cancer cells via transcriptional regulation of the HOXA1 gene. Int. J. Cancer 131:E179–E189 doi: 10.1002/ijc.26501 [DOI] [PubMed] [Google Scholar]
- 65. Guo X, Shi M, Sun L, Wang Y, Gui Y, Cai Z, Duan X. 2011. The expression of histone demethylase JMJD1A in renal cell carcinoma. Neoplasma 58:153–157 [DOI] [PubMed] [Google Scholar]
- 66. Bjorkman M, Ostling P, Harma V, Virtanen J, Mpindi JP, Rantala J, Mirtti T, Vesterinen T, Lundin M, Sankila A, Rannikko A, Kaivanto E, Kohonen P, Kallioniemi O, Nees M. 2012. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene 31:3444–3456 [DOI] [PubMed] [Google Scholar]