

Abstract
EFhd2 is a conserved calcium binding protein, abundant within the central nervous system. Previous studies identified EFhd2 associated with pathological forms of tau proteins in the tauopathy mouse model JNPL3, which expresses the human tauP301L mutant. This association was validated in human tauopathies, such as Alzheimer’s disease (AD). However, the role that EFhd2 may play in tauopathies is still unknown. Here, we show that EFhd2 formed amyloid structures in vitro, a capability that is reduced by calcium ions. Electron microscopy (EM) analyses demonstrated that recombinant EFhd2 formed filamentous structures. EM analyses of sarkosyl insoluble fractions derived from human AD brains also indicated that EFhd2 co-localizes with aggregated tau proteins and formed granular structures. Immunohistological analyses of brain slices demonstrated that EFhd2 co-localizes with pathological tau proteins in AD brains, confirming the co-aggregation of EFhd2 and pathological tau. Furthermore, EFhd2’s coiled-coil domain mediated its self-oligomerization in vitro and its association with tau proteins in JNPL3 mouse brain extracts. The results demonstrate that EFhd2 is a novel amyloid protein associated with pathological tau proteins in AD brain and that calcium binding may regulate the formation of EFhd2’s amyloid structures. Hence, EFhd2 may play an important role in the pathobiology of tau-mediated neurodegeneration.
Keywords: EFhd2, tauopathies, tau, amyloid, calcium, Alzheimer’s disease
INTRODUCTION
EFhd2 is a conserved calcium binding protein that contains two functional EF-hand motifs and is expressed in different tissues, but predominantly in the central nervous system (Vega et al. 2008; Ferrer-Acosta et al. 2012; Hagen et al. 2012). This novel protein was first identified in human CD8 lymphocytes (Vuadens et al. 2004). Further studies suggested that EFhd2 modulates the calcium flux in response to B-cell receptor (BCR) stimulation in B cells and is required for the association between BCR, spleen tyrosine kinase (Syk) and phospholipase Cγ2 (PLCγ2) (Kroczek et al. 2010; Hagen et al. 2012). EFhd2’s Src-homology 3 (SH3) binding site at the N-terminus was required for its localization at the lipid rafts of WEHI231 cells (Kroczek et al. 2010). The lipid raft localization of Syk and PLCγ2 and BCR-induced calcium flux were also affected upon deletion of EFhd2’s SH3 binding site (Kroczek et al. 2010). These studies, however, did not determined if EFhd2’s coiled-coil domain was required to mediate the association of EFhd2 with Syk and PLCγ2. The coiled-coil domain is known to mediate protein-protein interactions in other proteins, such as the filamentous proteins keratin and collagen (McAlinden et al. 2003; Mason and Arndt, 2004). At present, it is still unknown whether EFhd2’s coiled-coil domain mediates interaction of EFhd2 with other proteins or facilitates its own oligomerization.
The novel protein EFhd2 was found associated to tau proteins in the tauopathy mouse model JNPL3 (Vega et al. 2008). The association between endogenous EFhd2 and the transgenic human tauP301L mutant expressed in JNPL3 was detected in terminally ill mice (Vega et al. 2008). This result suggested that EFhd2 was associated to pathological forms of tau. The insolubility of hyperphosphorylated and aggregated tau proteins in the detergent sarkosyl is a biochemical property of pathological tau in JNPL3 mice and human tauopathies (Sahara et al. 2002; Zhukareva et al. 2002). Consistent with these results, EFhd2 co-purified with tau proteins in the sarkosyl-insoluble fraction (Vega et al. 2008). The association between EFhd2 and tau was validated in AD and FTDP-17 human brains, suggesting that EFhd2 may play a role in the pathobiology of tau-mediated neurodegeneration (Vega et al. 2008). However, the physiological and pathological roles of EFhd2 in the central nervous system are still unknown.
The association and co-purification of EFhd2 and tau in the sarkosyl-insoluble fraction indicates that these two proteins form a very strong interaction (Vega et al. 2008). EFhd2 has an isoelectric point of 5.0, indicating that at physiological pH this protein has a net negative charge. Negatively charged molecules have been shown to serve as nucleation factors for the aggregation of tau proteins in vitro (Friedhoff et al. 1998). The coiled-coil domain at the C-terminus of EFhd2 proteins also suggests that this protein may form homo-oligomers or interact with other proteins. Therefore, it is plausible to hypothesize that EFhd2 may function as nucleation factor for the pathological formation of tau filaments observed in tauopathies. This hypothesis implies that EFhd2 may be a novel amyloid protein that forms oligomers that mediate the formation of tau filaments. Amyloid-forming proteins have been directly associated to several diseases collectively known as proteinopathies, including AD, Parkinson’s disease, Huntington’s, prion diseases and others (Li et al. 2009). These proteins undergo similar conformational changes that lead to the formation of insoluble aggregates characterized by cross-β structures (Li et al. 2009). In order to gain insights in support of this hypothesis, the capacity of EFhd2 to form amyloid structures, filaments and its association with neurofibrillary tangles in AD was investigated.
METHODS
Recombinant proteins
His EFhd2 purification
Recombinant EFhd2 proteins used in these experiments had an N-terminus 6X Histidine tag. EFhd2 WT, N-terminus truncation and C-terminus truncation were overexpressed in E. coli bacteria and purified as described in Ferrer-Acosta et al. (2012).
Flag EFhd2 protein purification
Recombinant EFhd2 WT protein was cloned into an N-terminus FLAG tag vector. FLAG-EFhd2 WT construct was transformed in E.coli BL21. These cells were grown in an overnight preculture grown at 37°C and a larger culture was made the following day starting at an optical density (OD) of 0.2. Once an OD of 0.6 was reached, 0.4 mM IPTG was added and induction was carried for 1 hr, shaking at 37°C. To prepare bacterial lysate, cells were collected, frozen at −80°C and resuspended in 50 mM Tris Base, 150 mM NaCl pH 7.4, 1x protease inhibitor cocktail (SIGMA) and 0.5 mM PMSF. This pellet was sonicated and centrifuged at 15,000 × g for 10 min at 4°C. Bacterial supernatant was added Anti-FLAG M2 (SIGMA) FLAG-antibody conjugated beads (50μL of beads per 1 mL of bacterial lysate) overnight at 4°C. FLAG-EFhd2 was eluted from affinity column using the 3XFlag®tag peptide (SIGMA) at a concentration of 150 μg/mL. Buffer exchanges were made using a 3,000 MWCO centrifugal filter unit (Centricon, Millipore).
Thioflavin-S staining in vitro
EFhd2 WT Thioflavin S binding assay was performed using various protein concentrations of purified, recombinant His-EFhd2 WT protein (7.5μM, 30μM and 40μM) in 50 mM PIPES, 150 mM NaCl, 1mM DTT, pH 7.4. Reactions were incubated at 37°C with or without 3.3μM heparin, in a 50μL final volume, for 20 hrs. After incubation, 0.5mM Thioflavin-S (SIGMA) was added to the samples and analyzed in a fluorimeter (Cary Eclipse fluorimeter, Agilent Technologies) with a temperature-controlled cell holder set at 37°C. Excitation of Thioflavin S (Thio-S) was made at 440 nm and emission was read from 450–800nm, having a λmax at 550nm. A slit width of 5nm was used and an average of 5 scans per sample was made. To study the effect of calcium on EFhd2’s cross-β structure transition, the same reactions were prepared with recombinant EFhd2 WT (40μM), 1mM DTT, with or without 1mM CaCl2 and with no heparin added. These reactions were incubated for 20 hrs at 37°C and Thio-S binding signal was measured using the same fluorimeter parameters.
Transmission electron microscopy
In vitro EFhd2 filament formation
Recombinant, purified EFhd2 (40μM) in HEPES buffer (10mM HEPES, 100mM NaCl, pH 7.4) with 1mM DTT, 3.3μM Heparin and 1mM CaCl2 was incubated for 17 hrs. at 37°C, and negatively stained for TEM observation with the following procedure: 10μL of the protein solution were placed on a 300 mesh copper grid with a carbon support film for 2 minutes. The excess solution was removed with a filter paper, and the grid was gently rinsed with distilled de-ionized water (ddH2O). Coating for the negative staining was performed using 1.5μL of a 2% uranyl acetate (UA) solution in dd H2O for 40 seconds. The excess UA solution was then removed.
Sarkosyl-insoluble fraction preparation
The sarkosyl-insoluble preparation was performed as described in Vega et al. (2008). Briefly, about 0.08g of human brain frontal cortex tissue was homogenized in 400μL of Buffer A (20mM Tris base pH 7.4, 150mM NaCl, 1mM EDTA, 1mM EGTA, 1mM PMSF, 5mM Sodium Pyrophosphate, 10mM β-glycerolphosphate, 30mM sodium fluoride). The preparation was ultra-centrifuged at 156,565 × g for 15 min. at 4°C. The pellet obtained was added to 400μL of Buffer B (20mM Tris base pH 7.4, 800mM NaCl, 1mM EDTA, 1mM EGTA, 1mM PMSF, 5mM Sodium Pyrophosphate, 10mM β-glycerolphosphate, 30mM Sodium Fluoride and 10% sucrose) and re-homogenized. A second ultra-centrifugation was made at 156,565 × g for 15 min. at 4°C. The supernatant was collected, and 1% sarkosyl was added, mixed, and incubated for 1 hr. at 37°C. A final ultra-centrifugation was made at 156,565 × g for 30 min. at 4°C. The supernatant was removed and the pellet formed was re-suspended in 50μL of TBS (called the P3, sarkosyl-insoluble brain fraction).
Human brain sarkosyl-insoluble fractions for immunogold
10 μL of the sarkosyl-insoluble human brain fraction was placed on a 300 mesh copper grid with a carbon support film for 2 minutes. The excess solution was removed using a filter paper, and the grid was rinsed with ddH2O. A blocking solution (25μL) of TBS with 0.04% purified bovine serum albumin (BSA) and 2% horse serum was added for 1 hr. at room temperature in a humid chamber. Anti-EFhd2 primary antibody (1:10), anti-Tau13 primary antibody (1:50), were diluted in the blocking solution and added for 2 hrs. to the grids at room temperature. Primary antibodies solution was removed with a filter paper, and grids were washed with TBS. Secondary antibodies were diluted in the blocking solution 1:20, added and incubated for 1.5 hrs. at room temperature. Grids were rinsed with TBS and finally with ddH2O. Excess liquid was removed with a filter paper and grids were negatively stained with 1.5 μL of 2% UA for 40 sec. UA excess was removed with a filter. Control grids were also prepared without the primary antibodies, or without UA staining (to allow easier identification of the gold nanoparticles). Replicates from two separates protein preparations were observed.
TEM observations
TEM examinations were performed using a Carl Zeiss LEO 922 TEM operated at 200 kV and equipped with an in-column energy filter.
Human brain lysis and western blot analysis
Human frontal cortex from normal aging (average age 74.5 y. o.; n=17) and Alzheimer’s disease (average age 75.4 y. o.; n=17) were obtained from UPENN Pathology Core Facilities. The samples selected were pathologically confirmed as Alzheimer’s disease cases with abundant senile plaque pathology and Braak & Braak stage of IV or more for neurofibrillary pathology. Age-matched cases without history of dementia or detectable tau pathology were used as controls. Human frontal cortex from normal aging and Alzheimer’s were homogenized in five volumes of Buffer A (20 mM Tris base, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 5 mM sodium pyrophosphate, 30 mM β-glycerol phosphate, and 30 mM sodium fluoride) and the protein extract centrifuged at 21,000 × g for 10 min. The supernatant was transferred to a clean tube and used for western blot analyses. To determine protein concentration, Bradford assay was performed by detecting change in absorbance using a spectrophotometer (DU730 Beckman Coulter). All samples were resolved in SDS-PAGE and transferred to pure nitrocellulose membrane (0.45μm BIO-RAD). The membrane was blocked using 5% dry-milk solution in 1x TBS-T (2.5mM Tris-Base, 15mM NaCl, 30mM KCl and 0.02% Tween 20 detergent). The nitrocellulose membrane was incubated with a predetermined concentration of primary antibody overnight at 4°C. The membrane was washed three times with 1x TBS-T and incubated in secondary antibody for 1 hr. at room temperature. The recognized protein bands were visualized by enhanced chemiluminescence reactions detected on X-ray film (cat. #4741008379, Fisher Scientific). OD was expressed as a ratio of EFhd2 and GAPDH (loading control) using a densitometer, from western-blots in films, (BIO RAD GS-800) and Quantity One software (System ID: 3DoF758E). All p-values for column graphs were obtained using one-tail student’s T-test (**p<0.01).
Immunohistochemistry of human brain slices
Paraffin-embedded human pre-frontal cortex slices of AD and normal-aging control patients were deparaffinized using Histoclear™ (National Diagnostics) for 5 min., 3 times. Tissue slices were incubated in the following ethanol gradient for 5 min.: 100%, 100%, 95%, and 70%. The slices were incubated in ddH2O for 2 min. For antigen retrieval, slices were microwaved in ddH2O at 1,100W for 15 min. Brain slices samples were allowed to cool and incubated in a 0.3% H2O2 solution made in PBS for 20 min. After washing in PBS, blocking was made with 5% FBS in PBS for 20 min. at room temperature. Primary antibodies anti- EFhd2 (1:200) and anti-PHF-1 (1:200) were diluted in blocking solution and incubated at 25°C for 2 hrs. in a humidity chamber. Three washes of 5 min. in PBS were made and secondary antibodies were added 1:2000 in blocking solution. Anti-goat conjugated with FITC was used to label EFhd2, and anti-mouse conjugated with Alexa 568 was used for PHF-1 detection. Brain slices were incubated with these secondary antibodies for 1 hr. at 25°C. After 3 washes in PBS, TOPRO-3® iodide stain was added on a 1:2000 dilution in blocking solution for 30 min. at 25°C. Slices were washed 3 times in PBS for 5 min. and Vectashield mounting media was used to lay the coverslip. A Zeiss LSM 510 META on an Axiovision Z1 microscope was used to examine the samples.
Protein-protein interaction assay using mouse brain lysates
Animals
Lewis et al. (2000) developed the tauopathy mouse model JNPL3, which expresses the cDNA of the human tau 0N4R isoform bearing a common missense (P301L) mutation found in FTDP-17. The expression of the integrated tau gene is under the control of the mouse prion promoter. In hemizygotes JNPL3, the expression of the integrated human tau gene is equivalent to that of endogenous tau (Lewis et. al. 2000). Phenotypically, the JNPL3 hemizygotes animal showed motor and behavioral deficits as early as 6-months of age, while non-transgenic (NT) littermates are normal (Lewis et. al. 2000). Ultrastructural neuronal studies of JNPL3 brain slices revealed that intracellular NFT are composed of straight and twisted ribbon tau filaments similar to those observed in FTDP-17 and CBD (Lewis et. al. 2000). Mice used in these experiments were quickly euthanized using a CO2 filled chamber to minimize pain and discomfort, under the IACUC approved protocol (#00212-08-08-2012) for the use of transgenic animals.
Interaction assay
Age-matched 11-month-old JNPL3 and non-transgenic mouse brains (from male mice) were homogenized in Buffer A and centrifuged at 15,000 × g for 10 min., at 4°C. Supernatant was collected and Bradford assay was used to determine total protein concentration. 1mg of total protein brain lysates with 5mM imidazole were incubated with 30μg of recombinant His-EFhd2 WT, NT, CC or no protein (as control) in a 100μL final reaction volume. Reactions were left rotating at 4°C for 18 hrs. After incubation, 10μL of packed Histidine-tag affinity nickel beads (His-select, SIGMA) were added to each reaction. These samples were left rotating with nickel beads for 4 hrs. at 4°C. Nickel beads were eventually washed 3 times with 0.6mL buffer A, and eluted with 25μL of 2XSDS loading buffer. 5μL of eluted proteins were loaded on a 12% acrylamide SDS-PAGE for western blotting. Anti-His antibody (1:3000) was used to confirm pulldown and elution of His tagged proteins, and Tau13 (1:12500) was added to confirm tau pulldown with recombinant EFhd2 WT and mutants.
Indirect ELISA
Recombinant protein FLAG-EFhd2 WT (4μg total protein) was coupled to the bottom of a 96-well plate overnight at room temperature in 100μL of a 50mM sodium carbonate solution. Wells were washed three times with 200μL PBS-T (140mM NaCl; 10mM Na2PO4; 0.05% Tween 20; pH 7.4), blocked for 1 hr. with 100μl of 3% BSA in PBS-T (blocking solution) and washed 3 times. Wells were incubated for 1 hr. with recombinant His-EFhd2 WT, His-EFhd2 NT, His-EFhd2 CC or GST induced-bacterial lysate (8μg total protein) in blocking solution. After washing the wells, 100μL of Anti-His antibody (SIGMA, cat # H1029) (1:2000) in blocking solution was added for 1 hr. to determine interaction of His-tagged proteins with the bound Flag EFhd2 WT. Wells were washed 3 times and incubated with Anti- mouse IgG antibody (1:2000) (Millipore cat# AP181P) in blocking solution. The binding of secondary antibodies was determined using 100μL of BioRad HRP Substrate Kit (cat#172-1064). The reaction was stopped after 5 min. with 100μL of a 2% oxalic acid solution. The colorimetric reaction was measured with a Molecular Devices SpectraMax 340 microplate reader at 415 nm. Measurements were corrected by subtracting the background absorbance measured from reactions where the induced bacterial lysate was incubated in wells without coupled FLAG-EFhd2. A Student’s paired T-test analysis was performed to determine statistical significance between the columns (p****<0.0001).
MATERIALS
Antibodies
Antibodies used for Western Blot of His-EFhd2 and tau interaction assay were anti-His (SIGMA, 1:3000, cat # H1029) and Tau13 (Abcam, 1:12500, cat # ab24634). For the immunogold of human brain fractions, goat anti-EFhd2 (Novus, 1:10, cat # NB100-1477) and mouse anti-Tau13 (Abcam, 1:50, cat # ab24634) were used. Secondary antibodies used for these experiments were 6 nm gold-conjugated donkey polyclonal antibody to goat IgG (Abcam, 1:20, cat # ab105268) and 12 nm gold-conjugated donkey polyclonal antibody to mouse IgG (Abcam,1:20, cat # ab 105277). For immunohistochemistry and western blot, primary anti-EFhd2 (Novus, 1:200, cat # NB100-1477), anti-PHF-1 (1:200, kindly provided by Dr. P. Davies) and anti-GAPDH (1:10000; Advanced Immunochemical) were used. Secondary antibodies used were anti-goat FITC (SIGMA, 1:2000, cat # F7367) and anti-mouse Alexa Fluor®568 (Invitrogen 1:2000, cat # A11031) for immunohistochemistry, and peroxidase-conjugated goat anti-rabbit (1:2000), goat anti-mouse (1:2000) antibodies, and rabbit anti-goat (1:2000) (Chemicon, Temecula, CA, USA) for western blot. Nuclear staining of cells was made with TOPRO®-3 iodide 642/661nm (Invitrogen, cat # T3605). For the indirect ELISA assay, anti-His (SIGMA, 1:2000, cat # H1029) was used.
RESULTS
One of the first methods developed to detect amyloids was the use of fluorescent dyes, such as Congo red and Thioflavin S (ThioS), which have affinity to the crossβ-sheet structures formed by amyloids, protofibrils and fibrils (Li et al. 2009). In order to test EFhd2’s capacity to form amyloid structures, different concentrations of recombinant EFhd2 were incubated in the presence of ThioS under conditions that have been previously used to detect tau filament formation (Adams et al. 2010) (Fig. 1A). The results showed that ThioS fluorescence increased in a concentration-dependent manner, indicating that there is an accumulation of cross-β structures as EFhd2 protein concentration increases (Fig. 1A). The highest intensity of ThioS signal was detected in the sample containing 40μM EFhd2 (Fig. 1A). In these experiments the buffer contained heparin, a negatively charged nucleation factor used for the polymerization of tau proteins in vitro (Fig. 1A). However, EFhd2 has a net negative charge at physiological pH. Thus, heparin may not be required for the formation of EFhd2 amyloid structures. In addition, EFhd2 is also a calcium binding protein (Vega et al. 2008; Ferrer-Acosta et al. 2012). Therefore, the role that heparin and calcium ions play in the formation of EFhd2 amyloid structures was studied. The results show that heparin is not necessary for the formation of EFhd2 cross-β structures, indicating that EFhd2 does not require a nucleation factor or inducer to form these amyloid structures (Fig. 1B). In contrast, the addition of calcium reduced the ability of EFhd2 to form amyloid structures (Fig. 1B). Thus, EFhd2 proteins can form self-seeded amyloid structures at physiological pH and temperature, a capability that is lessened by the presence of calcium ions. These results suggested that EFhd2 may form filaments that share similar properties and morphology of those formed by amyloidogenic and pathological proteins such as tau (Goedert et al. 1996).
Figure 1. EFhd2 forms amyloid structures in vitro.

A, Various concentrations of purified, recombinant His EFhd2 WT (7.5, 30, 40μM) were incubated at 37°C with heparin and DTT (dash, dotted, solid line, respectively). After 20 hrs, samples were added Thioflavin-S (ThioS) and intensity was read in a fluorimeter at Exc.440 nm and Em.550 nm. ThioS stain has a high affinity for cross-β containing structures such as amyloids. EFhd2 showed affinity for this stain and an increase in ThioS binding was observed in a concentration-dependent manner. Baseline ThioS signal of buffer with heparin and DTT is shown (gray line) for comparison with EFhd2-containing reactions. B, Recombinant His EFhd2 WT (40μM) can also bind ThioS stain when incubated without heparin. Formation of cross-β structures by His EFhd2 WT was affected upon the addition of calcium, as it shows a reduction in the binding of ThioS (solid line, minus calcium; dashed lines, plus 1mM CaCl2). Baseline ThioS signal of buffers with heparin and DTT minus (gray line) and plus (dotted line) 1mM CaCl2 are shown for comparison with protein-containing reactions.
In order to determine if the ThioS affinity for EFhd2 was due to the formation of filaments similar to tau proteins, recombinant EFhd2 was examined using electron microscopy techniques (Fig. 2). The results obtained indicate that EFhd2 proteins can form filaments (Fig. 2). The filaments observed had lengths ranging from 50 to 500 nm (Fig. 2A–C). This result showed that EFhd2 can self-oligomerize to form filamentous structures. Thus, EFhd2’s coiled-coil domain on its C-terminus could mediate the formation of self-oligomers. To start testing this hypothesis, protein-protein interaction assays were performed to determine whether the coiled-coil domain mediated EFhd2’s self-oligomerization. Indirect ELISA assays were performed using differentially epitope-tagged recombinant EFhd2 proteins, as illustrated (Fig. 2D). FLAG EFhd2 WT was coupled to the bottom of an ELISA microplate and His-tagged EFhd2 (WT), N-terminus (ΔNT) and C-terminus (ΔCC) truncation mutants were incubated with the coupled FLAG EFhd2 WT protein. The results showed that His EFhd2WT and His EFhd2ΔNT mutant interacted with FLAG EFhd2WT. However, the association was significantly reduced when the coiled-coil domain was deleted (Fig. 2E, His EFhd2ΔCC). It is important to mention that previous structural analyses using circular dichroism indicated that N-terminal and C-Terminal truncations did not disrupt the secondary structure of EFhd2 (Ferrer-Acosta et al. 2012). The results show that EFhd2 is a novel amyloid protein that may use its coiled-coil domain to self-oligomerize.
Figure 2. EFhd2 can self-oligomerize and form filamentous structures in vitro.

A–C Transmission electron micrographs of EFhd2 TEM showed numerous filamentous structures (A), with various sizes, ranging from 50–500 nm in length (B, C). TEM micrographs were made at a 200 kV voltage. D An indirect ELISA assay was used to study whether the N- or C- terminus of EFhd2 was required for self-oligomerization. Recombinant Flag-EFhd2 WT proteins were bound to the bottom of a 96-well plate and incubated with recombinant His EFhd2 WT, truncations His EFhd2 ΔNT, His EFhd2 ΔCC. These wells were washed and added anti-His antibody to determine interaction of the His-tagged proteins with the bound Flag EFhd2 WT. Anti-His signal was significantly increased for His EFhd2 WT and ΔNT, compared to the ΔCC (which has no coiled-coil domain) or the Glutathione-S transferase (GST) negative control (T-Test, ****p < 0.0001, WT/ΔCC interaction; ****p < 0.0001, WT/GST interaction and p= n.s. for WT/ΔNT).
EFhd2 co-purification with sarkosyl-insoluble tau indicates that the association between them is resistant to high salt and detergent treatments, suggesting that EFhd2 may co-aggregate with pathological tau proteins in tauopathies, such as AD (Vega et al. 2008). To test this hypothesis, sarkosyl insoluble tau was obtained from human normal aging controls and AD cases, and prepared for immunogold labeling (Fig. 3). The co-localization of EFhd2 and tau proteins was confirmed by TEM (Fig. 3). The results showed that EFhd2 and tau co-aggregate in filamentous structures present in the sarkosyl insoluble fraction of AD cases (Fig. 3A–B), but not in normal aging controls (Fig. 3E–F). In addition to the normal aging control, the detected signal was specific to the presence of tau and EFhd2 proteins, since the gold nanoparticles were not observed in AD samples treated with secondary antibodies alone (data not shown). Additionally, granules containing either tau or EFhd2 were detected in the sarksosyl insoluble fraction (Fig. 3C–D). These granules could represent early stages of aggregation or independent pathological lesions. The co-aggregation of EFhd2 and tau proteins on filamentous structures in the sarkosyl insoluble fraction indicates that EFhd2 is associated with pathological tau in human tauopathy.
Figure 3. EFhd2 co-aggregates with tau in the sarkosyl-insoluble fraction of AD brain.
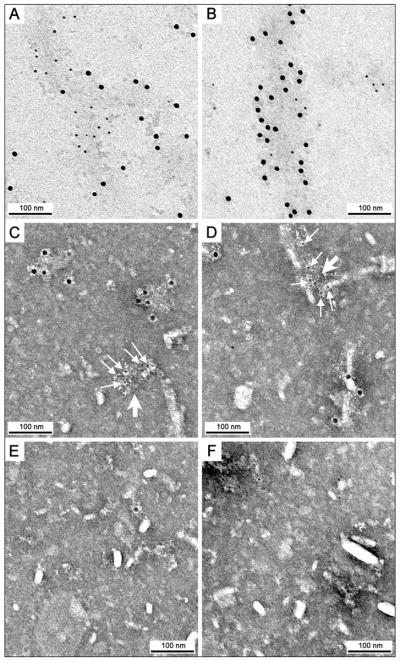
A–D TEM micrographs of a sarkosyl-insoluble fraction from an AD patient brain with immunogold labeling against EFhd2 (6 nm) and tau (12 nm). A, B: Unstained grid, showing the co-localization of EFhd2 with Tau in filaments. C, D: Stained grid, showing independent aggregates of EFhd2 and tau (small arrows point 6 nm EFhd2 gold particles, large arrows point the granular aggregates), grid stained with UA. E–F TEM micrographs of a normal aging patient brain sarkosyl-insoluble fraction with immunogold labeling against EFhd2 (6 nm) and tau (12 nm), grid stained with UA. The overall absence of gold particles indicates that normal aging patients do not present these protein aggregates. TEM micrographs were made at a 200 kV voltage.
To further corroborate the association between EFhd2 and pathological tau, an immunohistological analysis of frontal cortex slices from normal aging (N) and AD cases was performed (Fig. 4). Pathological tau proteins were detected with PHF-1 antibody, which recognizes hyperphosphorylated (pS396/pS404) and aggregated tau proteins (Fig. 4C–G). The pathological tau was readily detected in AD cases (Fig. 4G), but not in normal aging controls (Fig. 4C). Consistent with the immunogold TEM results, EFhd2 co-localized with PHF-1, providing further evidence that EFhd2 is associated with pathological tau in PHF-1 positive cells (Fig. 4F–H). It is important to mention that EFhd2 is preferentially detected in brain slices from AD cases (Fig. 4F) and a very weak signal is detected in normal aging controls (Fig. 4B). Previous results suggested that EFhd2 is overexpressed in AD brains when compared to normal aging controls (Vega et al. 2008). In order to confirm that EFhd2 is overexpressed in AD brain, frontal cortices were subjected to western blot analyses (Fig. 4I). The results indicate that EFhd2 is significantly overexpressed in AD in comparison with normal aging controls (Fig. 4I–J). In most cases, the overexpression of EFhd2 coincided with the detection of PHF-1 immunoreactive tau. The overexpression of EFhd2 can explain the differential detection level between AD and normal aging controls in immunohistochemistry, which could be attributable to the anti-EFhd2 antibody detection limits. Together, these results indicate that EFhd2 is a novel amyloid protein that aggregates with pathological tau proteins and is overexpressed in AD brain.
Figure 4. EFhd2 co-localizes with PHF-1 positive cells in human brain.

A–H Immunohistochemistry of human frontal brain slices was made to study EFhd2 co-localization with pathology-related tau. A–D: Normal patient brain slice (N), was labeled with nuclear stain (blue, A), anti-EFhd2 (green, B) and anti-PHF-1(red, C). Braak and Braak stage of this patient was 0. E–H: Alzheimer’s disease (AD) brain labeled with nuclear stain (blue, E), anti-EFhd2 (green, F) and anti-PHF-1(red, G). Braak and Braak stage of this patient was 6. Co-localization of both proteins (yellow in merge panel) is clearly observed in AD (panel H) and in few of the normal aged patient’s PHF-1 positive cells (panel D). AD brains showed a very strong co-localization of these proteins in cells (E–H, inset). I–J: Western blot (panel I) and densitometry analyses (panel J) were done to detect EFhd2 protein levels in normal aging and AD. Frontal cortices from 11 normal aging and AD cases were analyzed. The detected proteins are illustrated (panel I). The optical density of the detected EFhd2 protein expression in normal aging and AD cases was determined (panel J). GAPDH was used as a loading control. Statistical analysis indicated a significant difference in the expression of EFhd2 between normal aging and AD cases (**p<0.01).
To investigate the structural requirements for the association between EFhd2 and tau proteins, an in vitro protein-protein interaction assay was performed using His EFhd2 WT and truncation mutants. Equal amounts of these recombinant proteins were incubated with equal protein concentrations of either non-transgenic (NT) or JNPL3 (TG) mouse brain extracts (Fig. 5). After incubation, the recombinant proteins bound to nickel beads were intensively washed and the bound proteins were resolved by SDS-PAGE for western blot analysis, using human specific anti-tau (Tau-13) antibody and anti-EFhd2 antibody. As expected, human tau was not detected in the samples incubated with NT brain lysates since the antibody used specifically recognizes human tau. Tau proteins, however, were readily detected in the wild type and N-terminal truncated (ΔNT) EFhd2 samples incubated with JNPL3 brain extract (Fig. 5). The detected association was specific since tau proteins were barely detected in the beads alone negative control (Fig. 5, BA). Interestingly, the 64kDa tau species was detected (Fig. 5A, arrow), which represents hypephosphorylated tau proteins (Lewis et al. 2000; Sahara et al. 2002). This result is consistent with our previous immunoprecipitation results that showed that EFhd2 co-immunoprecipitated tau’s 64kDa species (Vega et al. 2008). The lower molecular weight tau protein bands detected could be truncation fragments or tau proteins phosphorylated at different levels. Also, the reduction in the abundance of tau associated with the N-teminus EFhd2 truncated mutant could be explained by the small difference in the amount of recombinant protein available or a subtle conformational change in the EFhd2 NT protein that reduced the binding affinity between both proteins. In contrast, tau was detected at background levels when JNPL3 brain extract was incubated with the coiled-coil truncated (ΔCC) EFhd2 mutant. Circular dichroism analyses indicated that deletion of the N-terminus and C-terminus did not disturb the secondary structure of EFhd2 (Ferrer-Acosta et al. 2012). Thus, these results suggest that the association between EFhd2 and tau proteins requires the presence of EFhd2’s coiled-coil domain.
Figure 5. EFhd2’s coiled-coil domain mediates the association with tau proteins.
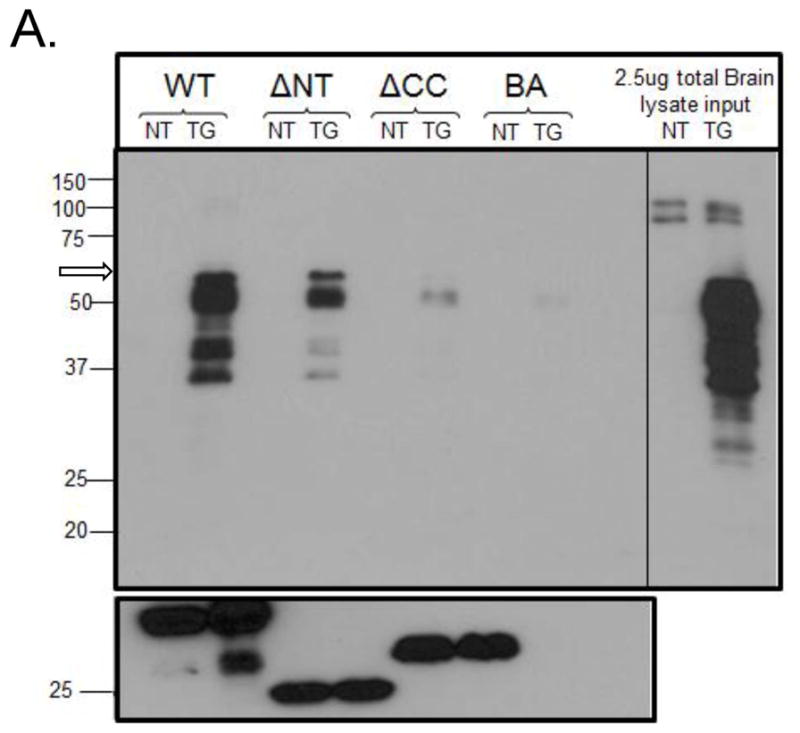
Recombinant His EFhd2WT (WT), His EFhd2 ΔNT (NT) and His EFhd2 ΔCC (ΔCC) proteins were incubated with 11month-old P301L (TG) or an age-matched non-transgenic mouse (NT) whole brain lysate. A control incubated without recombinant proteins was also added (BA). After incubation, proteins were pulled down by their His tag using a nickel column, including the negative control (BA). Proteins bound to the columns were resolved on an SDS-PAGE and an immunoblot against tau (using anti-Tau13) and His (anti-His) was performed. 2.5μg of total protein from TG and NT brain lysates (representing 0.25% total protein input in this experiment) is shown in the right panel. An open arrow (left panel, over 50kDa band) points the migration of the 64kDa band representing hyperphosphorylated tau proteins.
DISCUSSION
There are still unanswered questions about the polymerization of tau and its aggregation in the course of tau-mediated neurodegeneration. At first, neurofibrillary tangles (NFTs) were considered as the neurotoxic lesion that induced neuronal death. It is now accepted by the research community that these ultrastructures of tau aggregates may serve a neuroprotective role rather than inducing neurodegeneration (SantaCruz et al. 2005; Castellani et al. 2008; Lee et al. 2011). However, it remains controversial if tau dimers, oligomers or filaments are the particles that induce the neurodegeneration process (Lee et al. 2011). In vitro studies indicate that tau oligomers can induce intracellular aggregation of tau proteins, suggesting that amyloid structures could serve as nucleation factors for the conversion of “aggregation-prone” tau molecules (Lasagna-Reeves et al. 2012; Mandelkow and Mandelkow 2012). But, what promotes the formation of these toxic molecules in vivo? Hyperphosphorylation of tau proteins disrupt its association with the microtubules (Lee et al. 2001; Lee et al. 2011). Tau polymerization assays indicate that phosphorylation enhances the formation of tau filaments in vitro, in comparison to unphosphorylated tau (Rankin et al. 2007). Additionally, it has been shown that negatively charged molecules, such as heparin and arachidonic acid, enhance tau filament formation (Wilson and Binder, 1997; Friedhoff et al. 1998). Other studies suggest that posttranslational modifications, such as truncation and acetylation, may play an important role inducing tau oligomer formation (García-Sierra et al. 2008; Cohen et al. 2011; Kolarova et al. 2012). Thus, the mechanism that induces tau oligomer or filament formation still remains poorly understood.
The results presented here show that EFhd2 is a novel amyloid protein that is associated to aggregated tau in AD brain. To our knowledge, this is the second amyloid protein associated with tau. Alpha synuclein, an amyloid protein aggregated in Parkinson’s disease, has been shown to induce fibrillization of tau into intraneuronal filamentous inclusions characteristic of neurodegenerative diseases. This co-aggregation has been observed in rare cases of PD with dementia and in vitro (Giasson et al. 2003; Clinton et al. 2012). Recently, a known calcium-binding protein, S100A6, was shown to form fibrils and enhance the aggregation of SOD-1 in vitro, a protein that aggregates in the motor neuron disorder Amyotrophic lateral sclerosis (ALS) (Botelho et al. 2012). Interestingly, S100A6 protein’s capacity to form fibrils was also affected by calcium (Botelho et al. 2012). So far, there is no proof of an in vivo interaction between these two proteins or its association to a diseased-state. However, EFhd2 has been found associated to human tau in AD and FTDP17 cases.
EFhd2 is a protein whose calcium-binding capacity may regulate its physiological function (Dütting et al. 2011; Ferrer-Acosta et al. 2012). Interestingly, neuronal calcium dysregulation has been proposed as a molecular signal involved in the process of neurodegeneration (Mattson et al. 2007). The influx and efflux of calcium ions mediates changes in the proteome, such as activation of proteases and kinases, which have been implicated in the pathobiology of tau-mediated neurodegeneration (Wang et al. 2007; Reinecke et al. 2011). Thus, it is plausible to suggest that an increase in neuronal calcium ion concentration may promote the association of EFhd2 with tau proteins, favoring a shift from a physiological to a pathological function. Conversely, our study shows that calcium ions are not required for the formation of EFhd2 filaments or amyloid structures. Therefore, the calcium binding capacity of EFhd2 proteins may be related to its physiological function in the nervous system, which suggests that factors that disrupt EFhd2 calcium binding capacity may induce a shift of this protein into a pathological molecule. Consistently, our results indicated that EFhd2 is associated to pathological tau in terminally ill JNPL3 mice, but it was not detected to associate with tau proteins in JNPL3 mice that lacked tau pathology (Vega et al. 2008). These results suggest that EFhd2 may undergo a molecular switch between its physiological function and a pathobiological role throughout tau-mediated neurodegeneration.
Does EFhd2 play a neuroprotective or neurodegenerative role in tau-mediated neurodegeneration? EFhd2 association with NFTs in AD brains could be indicative of a neuroprotective role. EFhd2 has a negative net charge at physiological pH due to its low isoelectric point of 5. Thus, EFhd2 may serve as nucleation factor that enhances the transition from tau oligomers to filaments, and finally to NFTs. EFhd2 could enhance the kinetics of filament formation driving tau oligomers to a less toxic conformation in affected neurons. EFhd2 granules were observed in sarkosyl insoluble fractions obtained from AD brain, suggesting that this novel amyloid protein may form nucleation centers to induce the formation of less toxic tau aggregates. Alternatively, EFhd2, as a novel amyloid protein, may play a central role in the pathobiology of tau-mediated neurodegeneration. Posttranslational modifications may promote EFhd2 to switch from a physiological to a pathological role in vivo. An induced conformational change may convert EFhd2 into a neurotoxic molecule, suggesting a gain-of-toxicity effect. In contrast, EFhd2 may perform an essential physiological role in the central nervous system. An induced conformational change as a consequence of the neurodegeneration process may promote a loss-of-function effect, depriving neurons of EFhd2’s essential physiological function. Further experiments are needed in order to discriminate between these two possible roles of EFhd2 in tauopathy. Nevertheless, the results reported here show, for the first time, the presence of a novel amyloid protein intimately associated to tau in AD brain.
Acknowledgments
This work was supported, in part, by NIH-NINDS grant 1SC1NS066988 to IEV. YFA and ENRC were supported by NIH-NIGMS training grant 5R25GM061151. JV was supported by NIH-NIGMS training grant 8R25NS080687. The grants ISI0 RR-13705-01 and DBI-0923132 support the Confocal Microscopy Facility at the University of Puerto Rico (CIF-UPR). NSF and NASA for their support to the Institute for Functional Nanomaterials (IFN) Nanoscopy Facility. The contribution by GSB was supported by Alzheimer’s Association grant number 4079 and the Owens Family Foundation.
Abbreviations used
- AD
Alzheimer’s Disease
- TEM
Transmission electron microscopy
- ELISA
Enzyme Linked Immunosorbent Assay
- FTDP-17
Frontotemporal dementia with parkinsonism linked to chromosome 17
- EFhd2
EF-hand domain containing protein 2
- SDS PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- EM
Electron microscopy
- OD
optical density
- WT
wild type
- N-terminus
amino terminus
- C-terminus
carboxy terminus
- ΔNT
N-terminus truncation
- ΔCC
C-terminus truncation
- BA
beads alone
- BCR
B cell receptor
- PLCγ
phospholipase C gamma
- UA
uranyl acetate
- TBS
Tris base saline
- SDS
sodium dodecyl sulfate
- MWCO
molecular weight cut off
- ddH2O
distilled, deionized water
- g
gravitational force
- PBS-T
Phosphate buffer saline with 0.02% Tween 20 detergent
- GST
Glutathione-S-Transferase
- His
6X Histidine tag
- y.o.
years old
- FBS
fetal bovine serum
- PBS-T
phosphate buffer saline with 0.05% Tween 20
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- Adams SJ, DeTure MA, McBride M, Dickson DW, Petrucelli L. Three repeat isoforms of tau inhibit assembly of four repeat tau filaments. PLoS One. 2010;5:e10810. doi: 10.1371/journal.pone.0010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho HM, Leal SS, Cardoso I, Yanamandra K, Morozova-Roche LA, Fritz G, Gomes CM. S100A6 amyloid fibril formation is calcium-modulated and enhances superoxide dismutase-1 (SOD1) aggregation. J Biol Chem. 2012 doi: 10.1074/jbc.M112.396416. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani RJ, Nunomura A, Lee H, Perry G, Smith MA. Phosphorylated tau: toxic, protective, or none of the above. J Alzheimers Dis. 2008;14:377–383. doi: 10.3233/jad-2008-14404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30:7281–7289. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dütting S, Brachs S, Mielenz D. Fraternal twins: Swiprosin-1/EFhd2 and Swiprosin-2/EFhd1, two homologous EF-hand containing calcium binding adaptor proteins with distinct functions. Cell Commun Signal. 2011;9:2–14. doi: 10.1186/1478-811X-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Acosta Y, Rodríguez-Cruz EN, Vaquer-Alicea AC, Vega IE. Functional and structural analysis of the conserved EFhd2 protein. Protein Pept Let. 2012 doi: 10.2174/0929866511320050011. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedhoff P, von Bergen M, Mandelkow EM, Davies P, Mandelkow E. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci USA. 1998;95:15712–15717. doi: 10.1073/pnas.95.26.15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sierra F, Mondragón-Rodríguez S, Basurto-Islas G. Truncation of tau protein and its pathological significance in Alzheimer’s disease. J Alzheimers Dis. 2008;14:401–409. doi: 10.3233/jad-2008-14407. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- Hagen S, Brachs S, Kroczek C, Fürnrohr BG, Lang C, Mielenz D. The B cell receptor-induced calcium flux involves a calcium mediated positive feedback loop. Cell Calcium. 2012;51:411–417. doi: 10.1016/j.ceca.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Kolarova M, García-Sierra F, Bartos A, Ricny J, Ripova D. Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis. 2012:731526. doi: 10.1155/2012/731526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroczek C, Lang C, Brachs S, Grohmann M, Dütting S, Schweizer A, Nitschke L, Feller SM, Jäck HM, Mielenz D. Swiprosin-1/EFhd2 controls B cell receptor signaling through the assembly of the B cell receptor, Syk, and phospholipase C gamma2 in membrane rafts. J Immunol. 2010;184:3665–3676. doi: 10.4049/jimmunol.0903642. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Muñoz MJ, Kiritoshi T, Neugebauer V, Jackson GR, Kayed R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. doi: 10.1038/srep00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VMY, Goedert M, Trojanowski JQ. Neurodegenrative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lee VM, Brunden KR, Hutton M, Trojanowski JQ. Developing therapeutic approaches to tau, selected kinases, and related neuronal protein targets. Cold Spring Harb Perspect Med. 2011;1:a006437. doi: 10.1101/cshperspect.a006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Li H, Rahimi F, Sinha S, Murakami K. Amyloids and protein aggregation: analytical methods. In: Meyers RA, editor. Encyclopedia of Analytical Chemistry. John Wiley and Sons; UK: 2009. pp. 1–32. [Google Scholar]
- Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JM, Arndt KM. Coiled coil domains: stability, specificity and biological implications. Chem Bio Chem. 2004;5:170–176. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- McAlinden A, Smith TA, Sandell LJ, Ficheux D, Parry DAD, Hulmes DJS. α-Helical coiled-coil oligomerization domains are almost ubiquitous in the collagen superfamily. J Biol Chem. 2003;43:42200–42207. doi: 10.1074/jbc.M302429200. [DOI] [PubMed] [Google Scholar]
- Rankin CA, Sun Q, Gamblin TC. Tau phosphorylation by GSK-3beta promotes tangle-like filament morphology. Mol Neurodegener. 2007;28:12. doi: 10.1186/1750-1326-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke JB, DeVos SL, McGrath JP, Shepard AM, Goncharoff DK, Tait DN, Fleming SR, Vincent MP, Steinhilb ML. Implicating calpain in tau-mediated toxicity in vivo. PLoS One. 2011;6:e23865. doi: 10.1371/journal.pone.0023865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara N, Lewis J, DeTure M, McGowan E, Dickson DW, Hutton M, Yen SH. Assembly of tau in transgenic animals expressing P301L tau: alteration of phosphorylation and solubility. J Neurochem. 2002;83:1498–1508. doi: 10.1046/j.1471-4159.2002.01241.x. [DOI] [PubMed] [Google Scholar]
- SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A. [Google Scholar]
- Vega IE, Traverso EE, Ferrer-Acosta Y, Matos E, Colon M, Gonzalez J, Dickson D, Hutton M, Lewis J, Yen SH. A novel calcium-binding protein is associated with tau proteins in tauopathy. J Neurochem. 2008;106:96–106. doi: 10.1111/j.1471-4159.2008.05339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuadens F, Rufer N, Kress A, Corthésy P, Schneider P, Tissot JD. Identification of swiprosin 1 in human lymphocytes. Proteomics. 2004;4:2216–2220. doi: 10.1002/pmic.200300779. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. Am J Pathol. 1997;150:2181–95. [PMC free article] [PubMed] [Google Scholar]
- Zhukareva V, Shah K, Uryu K, Braak H, Del Tredici K, Sundarraj S, Clark C, Trojanowski JQ, Lee VM. Biochemical analysis of tau proteins in argyrophilic grain disease, Alzheimer’s disease, and Pick’s disease: a comparative study. Am J Pathol. 2002;161:1135–1141. doi: 10.1016/s0002-9440(10)64390-8. [DOI] [PMC free article] [PubMed] [Google Scholar]