

Abstract
Reactive oxygen species (ROS) can be both beneficial and deleterious. Under normal physiological conditions, ROS production is tightly regulated, and ROS participate in both pathogen defense and cellular signaling. However, insufficient ROS detoxification or ROS overproduction generates oxidative stress, resulting in cellular damage. Oxidative stress has been linked to various inflammatory diseases. Inflammation is an essential response in the protection against injurious insults and thus important at the onset of wound healing. However, hampered resolution of inflammation can result in a chronic, exaggerated response with additional tissue damage. In the pathogenesis of several inflammatory skin conditions, e.g., sunburn and psoriasis, inflammatory-mediated tissue damage is central. The prolonged release of excess ROS in the skin can aggravate inflammatory injury and promote chronic inflammation. The cellular redox balance is therefore tightly regulated by several (enzymatic) antioxidants and pro-oxidants; however, in case of chronic inflammation, the antioxidant system may be depleted, and prolonged oxidative stress occurs. Due to the central role of ROS in inflammatory pathologies, restoring the redox balance forms an innovative therapeutic target in the development of new strategies for treating inflammatory skin conditions. Nevertheless, the clinical use of antioxidant-related therapies is still in its infancy.
Keywords: ROS, oxidative stress, inflammation, antioxidant, skin
1. Introduction
The primary function of healthy skin is to form a physical and chemical barrier between the external environment and the organism’s internal milieu to defend against injurious insults. Harmful stimuli such as trauma, pathogens or irritants evoke a complex response known as inflammation (1). Inflammation protects organisms against pathogenic invaders and cleans up damaged cells after injury to prevent further tissue damage. Hereto the injurious agents are identified and eliminated and wound healing is initiated for re-establishment of tissue homeostasis. The strength and duration of the inflammatory response depends on stimulus and context, however, and the initial steps are stereotyped as part of the innate immune response [1]. The five classical signs of acute inflammation are: pain, heat, redness, swelling, and functional loss. These signs can be explained by the different phases that the inflammatory response generally follows: (1) dilation of capillaries to increase blood flow; (2) vasopermeabilization; (3) leukocyte recruitment; (4) elimination of pathogens or injurious stimuli; and 5) resolution of inflammation. Unfortunately, inflammation can also become chronic and destructive, and accumulating evidence demonstrate a contribution of chronic inflammation to the development of diseases like Alzheimer’s disease, atherosclerosis, and type 2 diabetes [2].
In most cases, inflammation of skin represents a beneficial and protective process after injury or infection [3]. However, the skin can also be subjected to excessive inflammatory responses resulting in chronic inflammation, auto-inflammation and auto-immunity [4]. The epidermal layer of the skin is composed of predominantly keratinocytes, a few Langerhans cells—which are specialized dendritic cells—and some pigment-producing melanocytes [3,4]. Keratinocytes produce different keratins that generate the toughness of the epidermis [5]. Embedded in the connective tissue of the underlying dermis different types of immune cells can be found, such as macrophages, T cells and mast cells [4]. The connective tissue is composed of an extracellular matrix produced and secreted by fibroblasts, the principal cell type of the dermis [6].
Under homeostatic conditions, the skin surface is colonized by a diversity of microorganisms. However, a dynamic, healthy equilibrium between the epidermis and the microorganismal population is regulated by production of antibiotic and antifungal compounds by dermal sebocytes as well as the microorganisms themselves. Additionally, keratinocytes also produce antibacterial substances constitutively and after infection or injury [7]. Some of these keratinocyte-derived antimicrobics and cytokines influence the immunological properties of dendritic cells and T cells [3,4]. Thus, the skin balances between ensuring an efficient pathogen defense and immunosurveillance and to reduce excessive immune responses that can lead to disease [1].
At the molecular level, inflammation is activated by the inflammasome that is a cytosolic multiprotein complex regulating caspase-1 activation. Caspase-1 subsequently activates the pro-inflammatory cytokines IL-1β and IL-18 by proteolytic cleavage. The inflammasome is composed of the danger sensor protein NALP and the caspase-1 recruiter protein ASC [8]. Dependent on the activation stimuli, different inflammasomes are formed as a response to danger signals as each inflammasome has unique roles in pathogen recognition [8].
Signal transduction by IL-1β and IL-18 results in a series of phosphorylation and ubiquitination events that ultimately leads to activation of nuclear factor (NF)-κB and p38 mitogen-activated protein kinase (MAPK) pathways, which cooperatively induce the expression of IL-1 target genes, including IL-6 [9]. Pathogen-mediated activation of the inflammasome also induces a specific form of caspase-1-dependent cell death, pyroptosis, that results in osmotic swelling and plasma membrane rupture [10,11]. This induces a strong inflammatory response via the release of pro-inflammatory cytokines and spreading of pro-inflammatory molecules [12].
In contrast to immune cells, human keratinocytes constitutively express inflammasome proteins and are a potent source of the pro-inflammatory cytokines pro-IL1α and pro-IL1β [13–15], which are activated and released upon UV exposure [13,16]. This implies important roles for keratinocytes as non-professional immune cells following sunburn, and in innate immune responses dependent on the inflammasome and IL1β. Keratinocytes are thus important in the inflammatory response under both physiological and pathological conditions [13,17].
Notably, it has been reported that the NLRP3 inflammasome assembly is stimulated in a ROS-sensitive manner by ROS generation [18,19]. The predominant source of ROS as response to danger stimuli are mitochondria, which also control inflammation via release of mitochondrial DNA [20,21]. Excessive release of ROS by mitochondria, activated leukocytes and endothelial cells in chronic inflammatory conditions can ultimately result in severe cell and tissue damage and can further promote and aggravate inflammatory injury. In many inflammatory diseases, currently available intervention strategies fail or are of limited success, warranting the need for novel strategies to treat chronic inflammatory conditions. The prolonged presence of oxidative stress is postulated to promote and fuel these deleterious inflammatory processes and may form a novel target for treatment of chronic inflammatory conditions.
2. Reactive Species Mediate Cellular Signaling
ROS are free radicals generated from molecular oxygen, such as superoxide anion (O2•−), hydroxyl radical (HO•), and non-radical species including hydrogen peroxide (H2O2). Today, it is well accepted that ROS mediate and modulate signaling processes [22,23]. In moderate concentrations, ROS induce a cascade of cell signaling networks, triggering a ROS wave that propagates throughout tissues carrying signals across large distances [24]. They thereby act as important regulatory mediators in different signaling pathways and processes, including cell proliferation, differentiation, and apoptosis [25,26].
Today H2O2 is recognized as a major ROS signaling molecule [27] that can mediate oxidation of protein thiols [28], and it is widely accepted that ROS can elicit cellular effects by covalently modifying amino acids and subsequently affecting protein activity. Redox-regulated proteins sense the changes in cellular redox state by different mechanisms. Several transcription factors, including nuclear factor κB (NF-κB), hypoxia inducible factor-1 (HIF-1), and p53, contain redox-sensitive cysteine residues in their DNA binding sites [29]. Oxidative modifications of these residues affect DNA binding and subsequently regulate gene transcription of redox sensitive genes [26,30,31]. Additionally, the DNA binding properties of these transcriptional regulators are further indirectly affected by redox-sensitive proteins, like apurinic/apyrimidinic endonuclease/redox effector factor-1 (APE/Ref-1) protein and histone deacetylases [32,33].
Oxidative posttranslational modifications form a major redox-regulated mechanism of protein function [34], targeting multiple types of amino acids with various susceptibilities and subsequent structural and functional consequences [35]. For instance, reversible oxidation of cysteines and oxidative nitration of tyrosines regulate the activity of various kinases involved in signaling pathways, including c-Jun N-terminal kinases (JNK), mitogen-activated protein kinase (MAPK), and protein kinase C [26]. Altered kinase activities due to oxidative modifications affect downstream signaling pathways and consequently transcription factor activation and contribute to additional redox-regulated gene transcription [36]. Effects of some of these redox-mediated alterations of signaling are exemplified below.
Based on the chemical characteristics, only a few amino acids can undergo oxidative modification, namely cysteine, methionine, and tyrosine [37]. Selective oxidation, reduction or chemical modification of these sensor thiols results in a change in protein activity and signal transduction in response to redox fluctuations [38]. However, the reactivity and modification of a particular cysteine residue is highly dependent on the microenvironment [28,39].
The side chain of a cysteine residue contains a terminal thiol (–SH) as a functional group allowing multiple oxidation states and is a common event in redox signaling [27,40–43]. Oxidation of these residues result in reactive sulfenic acid (–SOH) that can form disulfide bonds (RS–SR’) with nearby cysteines or undergo further oxidation to sulfinic acid (–SO2H) and sulfonic acid (–SO3H) [44]. Most of these modifications are generated by diffusible small molecules like H2O2 and are reversible by reducing systems like thioredoxin and peroxiredoxin with the exception of the sulfinic and sulfonic states [27]. For instance, reversible inhibition of protein tyrosine phosphatase (PTP) activity and subsequently regulation of cellular tyrosine phosphorylation and downstream signaling is mediated by H2O2-mediated oxidation of the catalytic cysteine to the sulfenic form [45,46].
Disulfide bond formation is important for protein structure and function [47], and recently its role as signaling event has been demonstrated [48,49]. Some cysteine-containing proteins also form intra- and intermolecular disulfide bonds, resulting in conformational changes and altered function due to ROS-mediated oxidation. For instance, ROS-mediated cysteine oxidation of PTPs is accompanied by intramolecular disulfide bond formation to protect against further oxidation [50–53]. Moreover, the accompanying conformational change might ease access for reducing enzymes and subsequent reactivation of the enzyme [54]. Cysteine redox switches in enzymes have been extensively discussed elsewhere [55].
S-glutathionylation is one of the most common S-thionylation reactions inside the cell and is generated from the reaction between cysteine sulfenic acid and a reduced thiol like GSH. This forms a mixed glutathione disulfide (GSSG) that prevents further irreversible oxidation [56]. Hereby, cells can effectively and reversibly respond to redox input, and glutathionylation indeed regulates most cellular pathways [30]. For instance, activation of the master regulator of several antioxidant genes, NF-E2-related factor 2 (Nrf2), is physically confined to binding partner Keap1 in the cytosol [57]. Keap1 glutathionylation leads to dissociation of the Nrf2-Keap1 complex, allowing Nrf2 to translocate to the nucleus and activate expression of target genes [58]. S-glutathionylation has also been found to control NF-κB pathway activation on different levels [30]. Several members of this pathway are inhibited by S-glutathionylation [59], and NF-κB itself is subjected to redox-regulation [36,60]. Oxidative modifications may also modulate protein–protein interactions and thereby affect the stability of protein complexes and activity of the protein partners involved [61]. For instance, NF-κB is sequestered by IκB in the cytosol. ROS activate IκB kinases that phosphorylate IκB, leading to its degradation by the proteasome and exposure of the nuclear localization sequence of NF-κB. Next, NF-κB translocates to the nucleus, and activates transcription of various target genes [62]. Moderate ROS levels promote NF-κB activation and cell survival, whereas high levels of ROS inactivate NF-κB, leading to cell death [26,36].
S-nitrosylation is the nonenzymatic adduction of NO to a thiol group (-SNO), and is reversible through the action of the protein denitrosylation system mediated by reduced glutathione (GSH) or the thioredoxin system [63,64]. Redox effects of NO are mediated by S-nitrosylation [65] that is thought to exert its regulatory effects through direct modification of protein function via conformational changes or through the protection of thiol groups against further oxidation [66]. However, the exact mechanisms underlying S-nitrosylation-mediated regulation is still not fully explored. S-nitrosylation plays a dual role as it can act as a protective modification and as an intermediate to further oxidation [67] as well as intermediate for formation of secondary posttranslational modifications such as ubiquitination [68]. Also, S-nitrosylation mediates the anti-inflammatory effects of NO in the cardiovascular system, as S-nitrosylation of N-ethylmaleimide factor suppresses vascular inflammation as well as inhibits NF-κB-dependent expression of pro-inflammatory cytokines and adhesion molecules [65].
Mitochondrial ROS production is also a participant in redox signaling networks [69,70]. The respiratory chain generates O2•− that is converted to H2O2, and both species can act as redox signals, but with different properties and interactions [27,71]. Membrane-impermeable O2•− is restricted to the mitochondrial matrix where it is thought to act as a redox signal within the mitochondria [72,73]. O2•− itself is not particularly reactive with most biomolecules except for iron–sulfur proteins, NO, and quinones [74–76]. Mitochondrial aconitase is an Fe-S cluster containing protein that reacts with O2•− to release H2O2 and ferrous iron [77]. ROS-mediated damage to the Fe–S cluster leads to aconitase inactivation and a cellular metabolic shift from ATP production to fat storage due to coenzyme A buildup [78]. This feedback loop has been suggested to be an antioxidant defense mechanism by reducing respiration and subsequently mitochondrial ROS formation [72,78]. Indeed, mitochondrial oxidative stress has been suspected to contribute to metabolic syndrome [78].
Also, mitochondrial ROS production is thought to play a role in oxygen sensing via HIF-1 [79,80] under both hypoxic [81] and non-hypoxic conditions [82]. Additionally, NADPH oxidases contribute for ROS formation and redox-dependent HIF-1 stabilization [83]. HIF-1 is a heterodimeric transcription factor that activates numerous genes involved in hypoxic adaptation via its binding to a hypoxia response element (HRE) in gene promoter regions [84]. Under normoxic conditions, the HIF1α subunit is tagged at the oxygen-dependent degradation domain for ubiquitination and subsequent proteasomal degradation by oxygen- and iron-dependent prolyl hydroxylases. Under hypoxia, the hydroxylation of HIF-1α subunit is inhibited, thereby enabling the formation of the active HIF transcription factor and subsequent induction of target gene transcription [79]. This is achieved through ROS-mediated oxidation of ferrous iron that is a co-activator of a prolyl hydroxylase function [85]. Moreover, a recent study describes perinuclear accumulation of mitochondria due to an altered microtubule-dependent transport under hypoxia [86]. As a result, increased levels of ROS in the nucleus caused oxidative modifications of DNA bases in the HRE of the VEGF promoter that is important for facilitated assembly of the HIF-1 transcriptional complex and thus increased VEGF gene expression [80,86].
Together, these examples illustrate that ROS-mediated posttranslational modifications act as key regulatory mediators in different signaling pathways and processes, including cell proliferation, differentiation, and apoptosis [25,26]. Paradoxically, ROS-signaling also promotes pathways protecting against oxidative stress to restore an imbalanced ratio between cellular oxidants and antioxidants [87]. Also, from these examples it becomes clear that the redox system affects protein function and biological activity either indirectly at the level of protein expression or stability or directly through posttranslational modifications [26]. An exhaustive collection of literature further points towards the importance of well-regulated ROS-mediated signaling as ROS has been linked to diverse groups of diseases, ranging from cardiovascular problems to neurodegeneration, see e.g., [88]. However, despite the ever-increasing number of studies, it is evident that ROS-mediated signaling and importantly, the regulation and specificity hereof, is still poorly defined and warrants for more investigation [27].
3. ROS and (Chronic) Inflammation of the Skin
Excess levels of ROS due to overproduction or because of insufficient scavenging generate oxidative stress, leading to injurious effects via: (1) oxidative modification and damage of biomolecules, altering lipid/protein/DNA structure and function; (2) further irreversible oxidation of reactive protein thiol groups which is hallmark of oxidative stress [89]; and (3) dysregulation of cell signaling pathways [90], triggering downstream signaling cascades leading to altered cytokine release and exacerbation of inflammation [91]. Combined, excess ROS lead to pathological changes in cells and tissues, as exemplified by inflammatory skin conditions like psoriasis.
Inflammatory skin diseases range from acute rashes with itching and redness to chronic conditions like dermatitis (eczema) and psoriasis. Acute skin inflammation may develop following exposure to, e.g., UV radiation (sunburns), allergens, physical wounding, or contact with chemical irritants, and is resolved within two weeks with only minor tissue destruction. In contrast, chronic skin inflammation results from a sustained, exaggerated inflammatory response, negatively affecting skin health.
3.1. Sunburn
Acute dermal overexposure to UV radiation causes sunburn and is an inflammatory response with increased prostaglandin and pro-inflammatory cytokine production, causing erythema, vasodilation, and leukocyte infiltration as predominant features [92,93].
Oxidative stress is thought to play a central role in the cellular response following UV exposure. Solar UV radiation is classified as UV-A (320–400 nm), UV-B (290–320 nm) and UV-C (100–290 nm) [94]. However, skin will only be exposed to UV-A and UV-B, as UV-C and partly UV-B radiation is absorbed by the ozone layer. UV-A photons are absorbed by endogenous UV-absorbing chromophores (e.g., riboflavin, quinines, tryptophan, and porphyrins) that subsequently transfer this energy to molecular oxygen, resulting in the formation of O2−•. Superoxide anions are then converted to H2O2 which in the presence of redox-active transition metals can be converted into HO• [95]. UV-B radiation is mainly absorbed by the epidermis and is primarily responsible for sun burn, whereas UV-A penetrates deeply into the skin. UV-A and UV-B radiation have different targets and outcomes. DNA is a prominent target of UV-B radiation, resulting in the formation pyrimidine and purine photoproducts and DNA strand breaks [96]. Additionally, UV-B-derived HO• can also damage the DNA, inhibiting normal cell function [97].
The effects of UV-A radiation exposure are a result of both direct and indirect damage to biomolecules and the subsequent physiological consequences hereof. Firstly, UV radiation damages skin lipids [98] and lipid peroxidation leads to increased production of prostaglandins, promoting inflammation in the skin [99]. Additionally, after UV exposure, keratinocytes and other skin-related cells upregulate pro-inflammatory cytokine production, e.g., IL-1, IL-6 and TNFα, and induce the expression of vascular adhesion molecules. TNFα is considered central to mediating UV-induced inflammation [100].
Furthermore, UV radiation of sunlight has been demonstrated to generate ROS leading to oxidative stress in skin due to depletion of endogenous antioxidant enzymes [101–103]. Chronically sun-exposed skin demonstrated no difference to sun-protected skin with respect to expression levels of Cu/Zn-dependent superoxide dehydrogenase (SOD)1, Mn-dependent SOD2, and catalase; however, sun exposure induced a marked increase in heme oxygenase expression [98]. ROS generated by UV radiation primarily cause damage to DNA through oxidative modifications and mutations, but also by inducing expression of different genes, such as matrix metalloproteinases and collagenases, thereby affecting collagen integrity and skin aging [104,105]. Additionally, UV-mediated ROS generation also indirectly affects cellular function and survival via its effect on cell signaling pathways [106]. For instance, activation of MAPK proteins occurs after UV exposure, suggesting that it may be responsible for executing the effects of UV-induced oxidative stress [93,98].
Thus, UV-induced oxidative damage is due to either immediate damage from UV radiation or indirectly from activated immune cells and dysregulated cellular signaling. An important aspect of UV damage is the depletion of the antioxidant defenses, which leaves the skin vulnerable to additional ROS damage [107]. This redox imbalance may also have systemic effects due to the wide-ranging effects of ROS, thereby rendering the body more prone to other ROS-mediated pathologies. This is further supported by several studies demonstrating an association between psoriasis and the prevalence and incidence of diabetes [108–110], a condition with ROS-mediated pathology due to increased ROS production and impaired antioxidant defense [111].
3.2. Psoriasis
Psoriasis is a chronic inflammatory skin disease manifested by red, thickened skin and skin scales due to keratinocyte hyperproliferation and is caused by a multitude of factors, including genetic, immunological, and environmental factors [112]. The pathogenesis of psoriasis includes complex interactions between skin and immune cells in concert with growth factors and pro-inflammatory chemo- and cytokines [113,114]; however, the exact underlying mechanisms still remain to be elucidated.
Oxidative stress is believed to be a key factor in the pathogenesis of psoriasis [115], as studies have suggested the involvement of increased ROS levels in psoriasis pathogenesis [101,116]. Increased ROS generation by infiltrated leukocytes into psoriatic lesions [117] is accompanied by substantial biomolecular damage, like psoriatic skin lesions containing oxidized LDL [118,119]. Indeed, a relationship between psoriasis severity, lipoprotein levels and oxidative damage has been proposed [120]. Notably, decreased antioxidant levels have been found together with increased levels of lipid peroxidation markers in blood of psoriasis patients [121,122]. Also, serum levels of catalase were elevated in psoriatic patients [123] and increased activity of superoxide dismutase (SOD) [124], and expression levels of peroxiredoxin (Prdx)2 and glutathione peroxidase (GPx)6 have been found in psoriatic skin lesions [125,126]. It is tempting to speculate that increased compensatory antioxidant levels counteract the skewed redox balance.
Besides direct damaging effects of unregulated ROS production, dysregulation of several pro-inflammatory pathways, like MAPK, NF-κB, and JAK-STAT, has been considered to contribute to psoriasis etiology [127]. Several members of the MAPK signaling pathways, like ERK1/2, JNK, and MAPK, were activated in psoriatic skin, further supporting this notion [128–130].
NF-κB, another redox-sensitive transcription involved in cellular processes like inflammation, cell proliferation and survival, has recently been demonstrated to be upregulated and active in psoriatic skin [129,131,132]. Dysregulation of NF-κB-mediated signaling may further exaggerate disease severity, as NF-κB-mediated upregulation of pro-inflammatory cytokines activates NF-κB via a positive feedback loop [133]. Importantly, inhibition of NF-κB nuclear translocation and DNA binding activity dampens the inflammatory component of psoriasis [134–136]. Many of the cytokines involved in psoriasis pathology induce keratinocyte proliferation and these signals are via ROS-mediated action transmitted to transcription factors and associated proliferation pathways [137,138]. Together, these observations strongly support a misbalanced oxidant/antioxidant balance as the major culprit in sustaining the inflammatory component in the pathology of psoriasis.
3.3. Burn Injury
A skin burn is a posttraumatic inflammatory condition accompanied by local as well as distant effects, leading to exaggerated inflammation, tissue damage, and infection. After skin burn, molecular signals, including inflammatory mediators and oxidants, are released at the injury site, further contributing to tissue damage and ischemic tissue necrosis [139,140]. Liquid from burn injury blisters contains a substantial amount of keratinocyte-derived pro-inflammatory cytokine IL-1β [141]. Moreover, induction of the inflammatory phase and attraction of immune cells leads to ROS production, exacerbating tissue damage [142]. Also, burns are often accompanied by secondary tissue damage distant to the site of injury [143–145] that is thought to be mediated by ROS and activated immune cells/neutrophils [146,147]. Besides local effects, burns also initiate systemic inflammatory reactions via ROS production, leading to distant oxidative damage to lipids and proteins [144,148]. Additionally, this systemic inflammatory response contributes to secondary damage as neutrophils have been demonstrated in various organs distant from the burn site within hours after injury and may lead to exaggerated oxidative stress and damage [145]. Notably, the intensity of the systemic inflammatory response and subsequently ROS generation correlates with the severity of the burn injury [149,150]. This pathological ROS production may affect cell signaling networks at the site of injury, thereby damaging cells and biomolecules. Also, ROS-mediated lipid peroxidation in skin is an important cause of cellular membrane dysfunction and subsequently cell death after burn injury [151,152]. A correlation has been described between the lipid peroxidation load and the degree of complications [153–156].
Importantly, these pathological effects seem to be caused by an overwhelming of the antioxidant systems, as decreased antioxidant scavenging capacity and reduced levels of SOD, GSH, and bilirubin have been reported after burns [148,156–159], likely due to massive consumption of these antioxidants as an attempt to counteract the oxidative stress. Thus, a tight control of the cellular redox balance is crucial, as excessive ROS production or decreased activity of ROS detoxifying enzymes results in aberrant wound healing [160] caused by exacerbated tissue damage due to macromolecular damage or by responses via stress-induced pathways.
4. Maintaining the Redox Balance in the Skin
Free radicals are formed in the skin following exposure to environmental stimuli and immune reactions. In addition, ROS generation in skin occurs naturally as part of normal cellular metabolism, like mitochondrial respiration. These ROS are normally rapidly neutralized by non-enzymatic and enzymatic antioxidants, thereby maintaining the oxidant/antioxidant balance and thus tissue homeostasis [101] (Figure 1).
Figure 1.
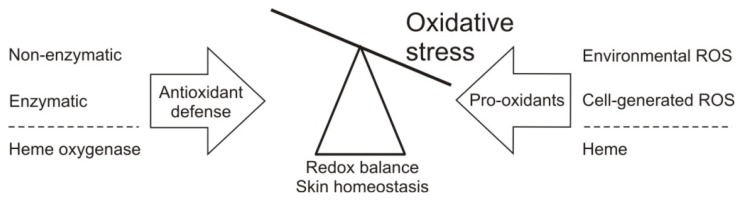
Redox balance maintenance in skin. ROS in the skin originate from normal cellular metabolism, e.g., mitochondrial respiration, and enzymatic activity. Besides, exogenous ROS are generated following physical insults, like UV light or persistent presence of leukocytes, facilitating chronic inflammatory skin conditions. To regulate ROS levels, the skin is rich in enzymatic and non-enzymatic antioxidant defense systems, thereby maintaining physiological homeostasis. In addition to the classical antioxidant defense, the cytoprotective enzyme heme oxygenase exhibits antioxidant properties via its degradation of pro-oxidant heme and generation of its antioxidant effector molecule bilirubin.
However, if the cytoprotective antioxidant factors get overwhelmed or are depleted, as may occur in hyperglycemic patients, the redox balance gets skewed towards oxidative stress, and may aggravate inflammatory injury.
4.1. Enzymatic Sources of Oxidants in the Skin
A major ROS source is the mitochondrial electron transport chain, leaking electrons during respiration. These electrons contribute to O2•− generation that is released into both the mitochondrial intermembranal space and matrix [69]. Matrix O2•− is converted to H2O2 by mitochondrial Mn-dependent SOD2 and can easily reach the cytosol by diffusion, whereas O2•− from the intermembranal space exits the mitochondria via voltage-dependent anion channels and is converted to H2O2 by Cu/Zn-dependent SOD1 in the cytosol [161].
Various enzyme systems produce ROS secondary to their main enzymatic function, including xanthine oxidoreductase [162], lipid peroxidases [163], cytochrome P450 enzymes [164], and nitric oxide synthase [165]. Nitric oxide (NO) synthase produces the free radical NO, which, under normal conditions acts protective, but which under oxidative circumstances gets converted into peroxynitrite, which is highly damaging [166]. Moreover, under certain conditions, like loss of cofactor tetrahydrobiopterine, NO synthase uncouples and produces O2•− instead of NO [167] leading to oxidative stress affecting cardiovascular performance [168].
The most important enzyme responsible for ROS production is the membrane–bound enzyme complex NAPDH oxidase (Nox). The Nox family consists of 7 members, Nox1-5 and Duox1-2, bearing a catalytic Nox or Duox domain, respectively. Furthermore, Nox isoforms contain a stabilizing domain (p22phox) and several regulatory subunits [169]. They display differential expression, regulation, and subcellular localization, and produce different ROS products [170]. Nox1, 2 and 5 are the key sources of O2−•, whereas Nox4 mainly produces H2O2 from molecular oxygen using NADPH as electron donor [171]. Nox-dependent ROS generation is activated by a wide range of chemical, physical, environmental, and biological factors [172]. The functions of the different Nox isoforms depend on their cellular localization and mode of activation [173] and have been thoroughly reviewed [174]. In keratinocytes, Nox1 is the only Nox expressed despite the detection of mRNAs encoding Nox1, Nox2, and Nox4 [175]. Keratinocyte Nox activity has been suggested to induce VEGF expression [176], MAPK activation [177], and cell growth [178]. Upon inflammation, infiltration of activated macrophages and neutrophils and their Nox2 and myeloperoxidase activities will exacerbate oxidative stress and prolong the inflammatory state [179].
4.2. Non-Enzymatic Sources of Oxidants in the Skin
Heme is crucial as the functional group of various hemoproteins, such as hemoglobin, cytochromes, peroxidases, and catalases [180]. In addition, heme can act as signaling molecule [181]. However, large amounts of free heme may be injurious to cells since heme is an iron chelate with the potential to catalyze iron-dependent reactions leading to ROS generation and membrane peroxidation [182]. Heme has indeed been demonstrated to catalyze ROS formation via the Fenton reaction [183] through its iron-dependent reaction with H2O2 [184,185].
After injury, large amounts of free heme are released from hemoproteins and aggravate tissue damage [182,186]. High local accumulation of heme can overwhelm the cellular ROS detoxification systems and prolongs oxidative and inflammatory stress [186–188]. Additionally, several studies have indicated that free heme also possesses pro-inflammatory properties [189–192], whereas low concentrations of free heme contribute to resolution of inflammation by downregulating inflammatory mediators, probably via induction of heme oxygenase (HO) activity [193–195]. Heme has therefore been suggested to act as a molecular switch due to its opposite, concentration-dependent effects [196].
Furthermore, free redox active metals, e.g., copper, zinc, and iron provide catalytic function to diverse (anti)oxidant enzymes [197], and redox cycling between Cu+/Cu2+ and Fe2+/Fe3+ is an integral part of the mitochondrial electron transport chain and ATP generation [198]. Transition metal homeostasis is regulated on both cellular and systemic levels and metal overload due to defective metal transporters is a central feature of several human diseases, like neurodegeneration [199]. Moreover, unregulated interaction of these metals with molecular oxygen also facilitates excessive ROS generation, predominantly via Fenton chemistry [200].
4.3. Antioxidant Systems in the Skin
Normally, the skin is constantly exposed to free radicals from the internal and external environment, challenging the functionality of the skin. Is our skin well enough prepared to withstand exogenous and endogenous ROS, and could targeting the redox status of the skin be a strategy to combat inflammatory skin conditions?
Under physiological conditions, ROS buildup in the skin is limited by numerous antioxidant defense systems, including both non-enzymatic and enzymatic mechanisms that either scavenge generated ROS before it can cause damage or prevent its formation (Figure 1, Table 1). In contrast to non-enzymatic antioxidants the enzymatic counterparts are not consumed and have a high affinity and reaction rate when scavenging ROS. Furthermore, the efficiency of the dietary non-enzymatic antioxidants depends on bioavailability as well as conversion into the active form upon ingestion [201] and antioxidant enzymes may therefore confer more efficient protection against acute oxidative and inflammatory stress [201].
Table 1.
Enzymes and factors involved in antioxidant defense in the skin.
| Antioxidants | Examples | Target |
|---|---|---|
| Enzymatic | Superoxide dismutase | Superoxide |
| Catalase | Hydrogen peroxide | |
| Glutathione peroxidase | Hydrogen peroxide, lipid peroxides | |
| Peroxiredoxin | Hydrogen peroxide | |
| Heme oxygenase | Heme | |
|
| ||
| Non-enzymatic | Bilirubin | Lipid peroxides |
| Vitamin C | Superoxide, hydroxyl radical, reactive nitrogen species, trace metals | |
| Vitamin E | Lipid peroxides | |
|
| ||
| Metal-binding proteins | Ferritin | Free iron |
The antioxidant defense system in skin is mainly comprised of the abundantly expressed antioxidant enzymes catalase, SOD, GPx, and Prdx [202].
SOD exists as three isoforms: cytosolic Cu/Zn-dependent SOD1, mitochondrial Mn-dependent SOD2, and extracellular SOD3 and catalyzes the conversion of O2−• radicals into H2O2 using NAPDH as cofactor [203]. Excessive amounts of H2O2 are harmful to cells and rapid scavenging hereof is thus important [204]. This task is performed by either catalase, Prdx, or by GPx and the glutathione system that reduces the H2O2 to oxygen and water.
Mammalian cells may express 5 GPx isoforms [205], with GPx1 being the most prominent isoform in skin cells, reducing H2O2 and a range of organic peroxides [206]. GPx1 expression and activity is upregulated by ROS [207], and GPx1 and SOD display similar expression patterns [208], thereby securing an effective detoxification of H2O2 generated by SODs and avoiding the generation of HO• radicals. Moreover, GPx1 has been linked to the regulation of acute oxidative stress [209], as GPx1 knockout (KO) mice are highly susceptible to oxidative injury induced by paraquat and H2O2 [207] and GPx1 KO fibroblasts show increased sensitivity to oxidant-induced apoptosis [209].
Besides catalase and GPx, the six members of the Prdx family all catalyze the reduction of H2O2 and a wide spectrum of organic peroxides and peroxynitrite using GSH together with thioredoxin (Prdx1-5) or ascorbate (Prdx6), respectively [210]. Prdx6 is important in the cellular stress response, as Prdx6 overexpressing cells are protected from ROS-induced toxicity [211,212] and Prdx6 overexpressing keratinocytes are less sensitive towards photo-damage by UV radiation [213]. On the contrary, Prdx6 knockdown increases sensitivity towards oxidative stress [214–216].
Additionally, the NADPH/NADP+ ratio is an important index reflecting the cellular redox status. NADPH, the principal intracellular reductant, is generated from NADP+ by different groups of enzymes: (1) cytosolic glucose-6-phosphate dehydrogenase (G6PD) and 6-gluconate phosphate dehydrogenase; (2) cytosolic and mitochondrial isocitrate dehydrogenases; (3) cytosolic and mitochondrial malic enzymes; and (4) mitochondrial transhydrogenase [217]. NADPH is a central component in the cellular antioxidant system, as NADPH is required by glutathione reductase to reduce glutathione disulfide to GSH, an obligate co-substrate for GPxs [218]. Also, NAPDH is required by several pro-oxidant and antioxidant enzymes, including Nox and catalase [219]. G6PD is a major contributor to NADPH generation that despite its status as housekeeping enzyme is subjected to tissue-specific regulation by various factors, including oxidative stress, nutrients and hormones [220]. Notably, studies have shown that abrogation of G6PD activity dramatically increases cellular sensitivity to oxidative stress in vitro [221,222]. However, G6PD plays a dual role in the (anti)oxidative system as G6PD activity also provides Nox with NADPH. Obese and hyperglycemic rats demonstrated significantly higher Nox-derived O2−• production due to increased G6PD activity and subsequently elevated NADPH levels to fuel this overproduction [223]. The resulting oxidative stress induced pathological changes of the heart and aorta and reduced cardiovascular function [223]. Together, this places G6PD and NAPDH centrally in the maintenance of a balanced ratio between pro- and antioxidants.
Besides the classical ROS-detoxifying enzymes discussed earlier, the HO system also exhibits potent antioxidant functions [224]. HO enzymes comprise the inducible HO-1 and the predominantly constitutively expressed HO-2 isoforms [225] and catalyze the degradation of heme into carbon monoxide (CO), iron, and biliverdin. Biliverdin is rapidly converted to bilirubin by biliverdin reductase [226,227]. Because heme is a redox active molecule that is cytotoxic in high concentrations [228], a direct beneficial effect of HO activity on the cellular redox balance can be ascribed to its active heme detoxification.
Numerous studies have linked the HO system to the regulation of various (patho)physiological processes, including cellular adaptation to oxidative stress and promotion of inflammatory resolution [229]. The expression of HO-1 is induced by various ROS-producing stresses [230], including heme and heavy metals [231], UV light [98,232], and H2O2 [233]. Also, oxidative stress-mediated induction of HIF-1 stimulates HO-1 activity in many tissues [234]. Actually, HO-1 induction is considered a marker of cellular oxidative stress and is involved in the protective response against oxidative damage [235].
Moreover, HO-1 overexpression counteracts the cytotoxic effects caused by high concentrations of free heme [181,190,236,237], whereas inhibition of HO activity intensifies oxidative cellular and tissue damage [238–241]. Preclinical and epidemiological evidence indicate that the cytoprotective and oxidative effects of the HO system are mediated via the generated effector molecules CO, bilirubin/biliverdin and by co-induced ferritin [For a recent review, see 230].
Both biliverdin and bilirubin generated from HO-mediated heme degradation are strong antioxidants [242,243], and bilirubin ameliorates oxidative stress in different diseases, including atherosclerosis, diabetes mellitus, and inflammatory and autoimmune diseases (Recently reviewed in [244]). For instance, bilirubin protects cells against high H2O2 concentrations [242,245,246] and nanomolar concentrations of bilirubin suppress Nox activity in vitro, thereby reducing ROS levels and subsequent tissue damage [247–249]. Thus, the involvement of biliverdin and bilirubin in antioxidant defense is evident [250].
Ferrous iron (Fe2+) is released during heme degradation by HO enzymes and can participate in ROS-generating Fenton chemistry, leading to cellular damage [251]. Ferritin is a ubiquitous iron-binding protein that can accommodate up to 4,500 iron atoms per molecule [252,253]. Notably, ferritin expression is not only regulated by cellular iron levels but also by oxidative stress via Nrf2 binding to an antioxidant responsive element (ARE) in the ferritin promoter [254], leading to co-induction with HO-1 [196]. Thus, the HO system contributes significantly to the cellular antioxidant defense by degrading redox active heme, thereby generating ROS-targeting effector molecules that further contributing to counteracting oxidative stress.
Numerous studies using cellular and animal models have demonstrated protective effects of vitamin C, a water-soluble compound, and vitamin E, a fat-soluble compound, against cytoplasmic oxidative damage and lipid peroxidation, respectively, in the etiology of atherosclerosis. Disappointingly, clinical studies have turned out less promising [255].
Together, under normal physiological conditions, ROS-mediated reactions in the skin are well balanced and protect the cells against oxidative stress. However, under pathological conditions like inflammation excessive damaging ROS formation may occur due to overwhelming of the antioxidant systems, contributing to a worsened clinical outcome.
5. Oxidative Stress as Therapeutic Target in Inflammatory Skin Conditions
The skin is constantly subjected to ROS formation via UV irradiation, environmental exposure, and cellular metabolism and is rich in enzymatic and non-enzymatic antioxidant systems keeping the ROS levels at homeostatic levels. However, inflammatory conditions in combination with an overwhelmed antioxidant system leads to pathological levels of ROS and oxidative stress, further exacerbating disease state. Attenuation of ROS levels and restoring the redox balance could then normalize the inflammatory response.
A potential therapeutic approach to restore antioxidant levels in the skin could therefore be achieved through (1) reducing the ROS production; (2) increasing endogenous antioxidant enzymatic defenses; or (3) enhancing the non-enzymatic antioxidant defenses via dietary or pharmacological approaches.
5.1. Reduction of ROS Production
5.1.1. Nox Inhibition
Increased ROS production and oxidative stress contribute to cellular and tissue injury during the progress to chronic inflammatory skin diseases. Recent studies suggest that Nox-generated ROS contribute to cellular and tissue damage by fuelling the acute inflammatory response [174,256–259]. For instance, studies indicate that Nox-generated ROS contribute to TNFα-mediated activation of NF-κB and vascular adhesion molecule expression [256,260–262]. Indeed, the central role of Nox in a multitude of diseases suggests it to be a putative therapeutic target, and several Nox-targeting inhibitors have been developed [263,264] with the main focus on the macrophage-specific Nox2 due to its involvement in several inflammatory conditions [173]. However, a recent study demonstrated that genetic Nox deficiency enhanced inflammatory responses after LPS challenge in vivo, suggesting that Nox-generated ROS in certain settings also have anti-inflammatory functions [265,266]. These contradictive and highly condition-specific outcomes of Nox-generated ROS may in the future challenge the use of Nox as therapeutic target to counteract redox imbalances.
5.1.2. Metal Scavenging Proteins
Redox active metals obstruct cellular signaling pathways by ROS-dependent and independent mechanisms. Both enzymatic and non-enzymatic antioxidants protect against metal-mediated ROS generation by (i) chelating redox-active metals; (ii) maintaining the metal redox state and preventing Fenton chemistry; and (iii) scavenging of metal-mediated ROS [200].
Excess free iron released after insults can catalyze ROS formation via Fenton reaction and cause tissue damage [228]. Furthermore, under in vivo stress conditions, an excess of superoxide releases iron from iron-binding molecules, including ferritin [267], contributing to further iron overload that can have deleterious effects [268,269]. Thus, the application of suitable metal chelators may contribute to a reduction in metal-induced ROS formation.
Chelation therapy is medical treatment for heavy metal poisoning and scavenging of redox active metals. Normally, labile cellular iron not contained by ferroproteins is scavenged by ferritin, thereby neutralizing the pro-inflammatory and pro-oxidative potential of free iron and preventing immune-mediated inflammatory conditions [270]. The bacterial siderophore Deferoxamine is an iron chelator frequently used to treat acute iron overload and related complications [271]. Also, plant-derived polyphenolic compounds (so-called botanicals) can be effective metal chelators [272]. For instance, the antioxidative effect of the plant-derived flavanoid quercetin is predominantly due to its chelating of redox-active iron [273].
5.2. Increasing Enzymatic Antioxidant Defenses
5.2.1. Antioxidant Upregulation
As mentioned earlier, superoxide scavenging is performed by the three mammalian SOD enzymes. The primary location of SOD3 is the extracellular matrix and on cell surfaces, whereas SOD1 and SOD2 are intracellularly located [274]. SOD3 is expressed in the epidermis and dermis of skin [275], but the role of SOD3 in this tissue is less clear. SODs have been suggested to be involved in the defense against UV-mediated ROS, as SOD enzyme expression and activity are affected by UV exposure [276–278]; however, SOD3 needs significantly higher UV doses before being activated than SOD1 and SOD2.
Numerous studies have investigated the antioxidative effects of elevated SOD levels in tissues by different means. For instance, intramuscular injections of SOD1 were successfully applied as anti-fibrotic therapy in treating cutaneous radiation-induced fibrosis in humans [279] and similar promising results were obtained with SOD2 in a porcine model of radiation-induced fibrosis [280].
Additionally, cutaneous SOD2 gene therapy reduced superoxide levels and normalized wound healing in mice with chemically-induced diabetes [281]. Similarly, diabetic transgenic SOD2 mice also demonstrated reduced superoxide levels and improved wound healing after ischemic stress compared to wild type controls [282].
Notably, chemically-induced contact dermatitis was alleviated in transgenic mice overexpressing SOD3 under the control of the keratin14 promoter, including a reduction in the levels of ROS and pro-inflammatory cytokines as well a reduced immune cell infiltration [283,284]. By contrast, SOD3 KO mice display exaggerated IL23-mediated psoriasis-like skin inflammation, including increased immune cell infiltration and higher levels of pro-inflammatory cytokines compared to WT controls [285,286], suggesting a role for SOD3 in cutaneous inflammation. Also, SOD3 expression is reduced in psoriasis patients compared to healthy subjects [284], further supporting this.
Moreover, SOD3 is suggested to play a role in pulmonary, arthritic, and neurological conditions [287]. Importantly, increasing SOD3 levels in various experimental disease models, e.g., chemically induced diabetes, hypertension, and inflammatory arthritis, reduced oxidative stress and improved disease state [288], thereby placing SOD3 as a central therapeutic target. Many studies have focused on SOD3 therapy in targeting ROS-mediated cardiovascular effects; however, the outcome has been variable [288]. Importantly, this discrepancy may relate to the use of predominantly rats in these studies despite that fact that rat SOD3 differs structurally and chemically from other mammalian SOD3 forms, resulting in a lower SOD3 concentration in rat vascular tissues [289], probably creating a therapeutic window that would not be present in other mammals, e.g., dogs, that did not show a therapeutic benefit from SOD3 gene therapy [290].
Thus, it is clear that increasing the enzymatic antioxidant defense by exogenous applications like injection or gene transfer may be a future therapeutic approach for multiple disorders with an inflammatory component; however, more research in terms of tissue- and condition-specific effects is warranted about the role of the SOD enzymes in skin inflammation.
5.2.2. Synthetic Antioxidants
Low-molecular mass synthetic compounds exhibiting catalytic activity, thus operating as enzyme mimics, have been developed as putative antioxidant therapy. The first mimetics were SOD-like because SOD is the first line of antioxidant defense, and today different classes of SOD mimetics based on metalloporphyrins, manganese (Mn) cyclic polyamines, and salen Mn derivatives have been developed [291]. Fortunately, their chemical and biophysical properties not only make them potent SOD mimics but also allow them to neutralize other types of ROS, including peroxynitrite and H2O2, and can thus also be considered catalase/peroxidase mimetics [292]. This broad specificity allows for modulation of the cellular redox environment and thus confers advantages over non-enzymatic antioxidants [293].
Moreover, these compounds have been shown to be effective in reducing oxidative stress in different in vitro cytotoxicity models involving ROS production [293,294] as well as in numerous in vivo models [292]. Notably, many of these compounds are not only functionally protective but also reduce oxidative damage to biomolecules [295–300] and the salen Mn compounds confer protection against mitochondrial damage [299].
Importantly, these mimetics display anti-inflammatory potential as H2O2 mediates activation of inflammatory genes as mentioned earlier, and therefore conditions with an inflammatory component may be the main target for such strategy. Also, reduced levels of activated macrophages were reported following treatment with mimetics in a radiation-induced lung injury model, however, no effects on pro-inflammatory cytokine levels were detected [300]. Also, a recent study demonstrated beneficial effects of systemic mimetic treatment on wound healing after skin irradiation by reducing oxidative damage [301].
Thus, these compounds do show promising prospects for therapeutic strategies in treating ROS-mediated complications in a wide range of conditions. Unfortunately, comparative studies on the different synthetic antioxidant classes in in vitro and in vivo settings are still very sparse and so far none of the antioxidant mimetics has been approved for clinical use.
5.2.3. Induction of the HO System
HO-1 is a stress-responsive enzyme with both antioxidant and cytoprotective effects mediated by the generated effector molecules biliverdin/bilirubin and CO, placing HO-1 as a central player in protection and homeostatic re-establishment after a wide range of pathological insults [302]. Numerous studies have demonstrated significant therapeutic effects of upregulation of HO-1 expression and/or activity as well as effector molecule administration in multiple pathological inflammatory conditions [230,302]. Also, ferritin is co-induced with HO-1 induction, which may provide a beneficial side effect, as ferritin scavenges free iron and thereby contributes to a restoration of the redox balance.
Recently, strategies to employ HO-1 as therapeutic target have been considered. This is further substantiated by HO-1 gene deficiency cases [303,304] and by the fact that promoter polymorphisms of the HMOX1 gene affect HO-1 protein expression levels and, subsequently, the severity of pathological human conditions [305]. Being an inducible enzyme, several synthetic molecules, including porphyrins [306] and heme arginate [307] have been developed and identified as possible HO-1 inducers. Heme arginate has for years been used as a clinical approach to treat porphyria, a disorder caused by non-functional heme metabolism [308]. Lately, a focus has been shed on the expanding number of plant-derived dietary compounds, so-called phytochemicals, with HO-1 inducing effects [309], like curcumin [310–312] or quercetin [313,314]. However, the effects of these pharmacological non-toxic, low-cost compounds still need to be studied in more detail before clinical use.
5.3. Increasing Non-Enzymatic Antioxidant Defenses
5.3.1. Oral and Topical Administration
Epidemiological studies have suggested that consumption of antioxidant-rich food is associated with lower disease rates and preventive protection of cardiovascular disease [315]. Thus, the supplementation of non-enzymatic, dietary antioxidants could be a feasible way of restoring redox homeostasis and reduce ROS-associated diseases.
However, clinical studies employing antioxidant supplement therapy have been inconclusive. For instance, the antioxidant compound N-acetyl cysteine (NAC) has been successfully used in the treatment of idiopathic pulmonary fibrosis [316–319], an inflammatory condition with etiology linked to Nox4-mediated ROS generation [320]; however, this therapeutic effect may not purely be ascribed to direct antioxidant effects of NAC itself but to its link to cellular glutathione replenishment [321]. Also, the combined use of NAC together with other commonly used treatment protocols should be carefully dissected and considered for each patient situation, as a recent study demonstrated increased mortality and severe treatment-related adverse effects when employing NAC in a three-drug regimen [322].
Frequently studied dietary and naturally occurring antioxidants such as carotenoids, flavonoids, and several vitamins have been implicated as promoters of skin health and rejuvenation [323]. External factors like chronic sun exposure, smoking, and pollution are significant contributors to skin aging, and both vitamins C and E have been demonstrated to have differential UVB photoprotective effects when applied both topically and orally [324,325]. Moreover, oral combination therapies of vitamins C and E resulted in a dramatically increased photoprotective effect compared to monotherapies [326]. Also oral intake of β-carotene or provitamin A reduces UV-induced erythema formation in different clinical studies; however, this effect is highly dose- and time-dependent [327–329]. Notably, β-carotene has been demonstrated in vitro to quench UV-induced radical formation and lipid peroxidation [330,331] and to reduce mitochondrial mutagenesis after UV exposure of skin fibroblasts [332].
Polyphenols are plant-derived micronutrients such as green tea polyphenols and curcumin that also have gained more attention in skin research during the last decade due to their antioxidant properties and their potential beneficial effects on cancer, neurodegenerative and cardiovascular diseases that all have been linked to oxidative stress [333]. Various polyphenols have been reported to be photoprotectors [334]. Oral as well as topical application of green tea polyphenols to mice protected in a time-dependent manner against UV-induced cutaneous edema, depletion of the epidermal antioxidant defense, and cyclooxygenase induction [335,336]. Studies using animal models have also demonstrated anti-inflammatory activities of administration of green tea polyphenols, predominantly mediated by the major green tea component epigallocatechin-3-gallate [337,338]. Also in humans topical application of green tea polyphenols dose-dependently reduced erythema formation and sunburn cells [337]. Sunburn cells are keratinocytes undergoing apoptosis due to irreversible DNA damage [339]. However, the underlying mechanism of these compounds is still not well understood but has been suggested to be mediated via effects on signal transduction pathways [340].
Another powerful antioxidant involved in skin antioxidant defense is coenzyme Q10 (CoQ10) [341]. However, being part of the respiratory chain, CoQ10 also contributes to ROS formation, as discussed earlier. In skin, the CoQ10 level is 10 times higher in the epidermis compared to the dermis [341]. Being the outermost skin layer, the epidermis is directly exposed to UV irradiation and it is known that UV irradiation depletes antioxidants in the skin [107]. The epidermis may thus be an optimal target for CoQ10 administration. Dietary CoQ10 supplementation to rats increased the levels of CoQ10 and its homologs in tissues and mitochondria therein [342]. This was accompanied by a reductive shift in plasma aminothiol status and decreased oxidative damage to mitochondrial proteins in skeletal muscle [342]. In contrast, mice on CoQ10 enriched diets did not show any effect on the systemic redox balance, nor the lifespan, despite a buildup of CoQ10 in tissues [343]. Notably, human epidermal keratinocytes isolated from topically CoQ10-treated skin demonstrated improved mitochondrial function and protection against UV-induced mitochondrial damage compared to non-treated controls [344]. Other studies demonstrated that CoQ10 stabilizes mitochondrial function, improves cell viability, and attenuates oxidative effects in human skin [345,346].
In summary, despite their great availability and use, the effects of dietary supplements on skin and general health still remain controversial. However, several things that could be crucial for treatment outcome must be considered. Firstly, these bioactive compounds have to be prepared and taken up by the target organ. The stability of these compounds will ultimately determine efficiency, and the use of vitamin C in creams has proven difficult due to a low stability in the presence of oxygen [347]. To account for this and to facilitate uptake, more stable derivates are often used though several of these compounds are not efficiently converted into the active form of the antioxidant [348]. Another issue is the bioavailability of the oral supplements. Dietary supplements have to pass through the gastrointestinal tract, enter circulation, and reach the target tissue and selected cell types and/or cellular compartments, which may be important for the effective dose to be given. Also, toxic effects due to e.g., cross-reactions and organ-specific reactions to certain compounds should be taken into account [349]. Additionally, route of administration is important, as e.g., curcumin is effective to skin when applied topically, but only to the colon when applied orally [340]. Importantly, antioxidant supplementation may interfere with the endogenous antioxidant response normally initiated after exercise and subsequently interferes with ROS signaling [350]. Other clinical studies on vitamins A, C and E, coenzyme Q10, carotene, and plant-derived flavanoids have turned out rather disappointing, as no significant effects these dietary supplements on general health have been detected [351,352]. More importantly, the long-term effects of many of these supplements are yet to be assessed, but one study linked increased mortality to long-term intake of antioxidant supplements [353].
Interestingly, the oxidated targeted site may determine the success of antioxidant therapy. Although both GSH and bilirubin are potent endogenous antioxidants, they protect against distinct targets. GSH mainly protects against hydrophilic proteins, whereas bilirubin protects against lipid peroxidation [354]. Recently, we demonstrated that iatrogenic induction of mild hyperbilirubinemia ameliorated the serum antioxidant status and vascular function in diabetic patients [355]. Thus, more in-depth studies are still needed to gain more knowledge about in vivo and in vitro obtained data on the diverse group of potential beneficial antioxidants. Knowledge concerning dose-response, optimal administration, cellular and compartmental targeting and translational studies are still urgently needed.
5.3.2. Targeted Antioxidant Delivery
Mitochondria are central organelles in cell survival and function [356] and increasing attention has been paid to the role of mitochondrial dysfunction in aging, apoptosis, neurodegeneration, and cancer [357]. Despite already being equipped with antioxidant systems, great interest has been paid to developing supplementary approaches to further protect the mitochondria from ROS-mediated damage. For instance, CoQ10 and related ubiquinones have been used as therapy to decrease mitochondrial damage in Parkinson’s disease patients [358,359]. However, delivery issues limited the therapeutic effect as oral administration only resulted in a limited mitochondrial uptake of ubiquinones [360–362]. Conjugation of a lipophilic triphenylphosphonium cation to ubiquinones led to the development of MitoQ, which selectively is taken up by and accumulated within mitochondria [363]. Moreover, MitoQ efficiently prevented oxidative stress in isolated mitochondria, as well as in the ischemic heart [364–366]. Later, other effective compounds such as SkQ1 and Trolex were developed and characterized [367–369]. Lately, nanotechnology has been employed in targeted mitochondrial delivery. Different drugs employed in cancer, Alzheimer’s disease, and obesity demonstrated improvement in the drug therapeutic index after nanoparticle-mediated delivery compared to a non-targeted carrier or to the free form of the therapeutics [370].
Notably, a recent report described induction and subsequently mitochondrial translocation of HO-1 following gastric mucosal injury, resulting in prevention of mitochondrial oxidative stress and pathology [371], suggesting that HO-1 induction may represent another mitochondrial targeting strategy. Together, this targeted approach thus holds big potential for future treatment strategies.
4. Concluding Remarks
ROS are crucial for cellular functions and provide essential protective mechanisms. However, ROS and oxidative stress have also been linked to various disease states, including inflammation, diabetes mellitus, cardiovascular diseases, and aging [88]. A well-balanced redox homeostasis is important, as oxidative stress either by increased levels of ROS and/or by depleted antioxidant systems may dysregulate protein function due to oxidative modifications and further aggravate inflammatory conditions. Targeting oxidative stress in inflammatory skin conditions may ameliorate disease outcome by dampening inflammation and improving recovery. However, although promising, efficacy and clinical studies employing antioxidant therapy are still in its infancy.
Acknowledgments
This work was supported by grants from the Dutch Burns Foundation (#09.110) and TASENE (NWO-WOTRO; W02.29.101).
Abbreviations
- APE/Ref-1
apurinic/apyrimidinic endonuclease/redox effector factor-1
- ARE
antioxidant responsive element
- CO
carbon monoxide
- CoQ10
coenzyme Q10
- ERK
extracellular signal-regulated protein kinases
- G6PD
glucose-6-phosphate dehydrogenase
- GPx
glutathione peroxidase
- GSH
reduced glutathione
- H2O2
hydrogen peroxide
- HO•
hydroxyl radical
- HIF-1
hypoxia inducible factor-1
- HO
heme oxygenase
- HRE
hypoxia response element
- JNK
c-Jun N-terminal kinases
- KO
knockout
- LDL
low density lipoprotein
- MAPK
mitogen-activated protein kinase
- Mn
manganese
- NAC
N-acetyl cysteine
- NF-κB
nuclear factor-κB
- Nox
NADPH oxidase
- Nrf2
NF-E2-related factor 2
- O2−•
superoxide anion
- Prdx
peroxiredoxin
- PTP
protein tyrosine phosphatase
- ROS
reactive oxygen species
- SOD
superoxide dismutase.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Contassot E., Beer H.D., French L.E. Interleukin-1, inflammasomes, autoinflammation and the skin. Swiss Med. Wkly. 2012;142 doi: 10.4414/smw.2012.13590. [DOI] [PubMed] [Google Scholar]
- 2.Martinon F., Mayor A., Tschopp J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 3.Feldmeyer L., Werner S., French L.E., Beer H.D. Interleukin-1, inflammasomes and the skin. Eur. J. Cell Biol. 2010;89:638–644. doi: 10.1016/j.ejcb.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Nestle F.O., Di Meglio P., Qin J.Z., Nickoloff B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009;9:679–691. doi: 10.1038/nri2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuchs E., Raghavan S. Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet. 2002;3:199–209. doi: 10.1038/nrg758. [DOI] [PubMed] [Google Scholar]
- 6.Sorrell J.M., Caplan A.I. Fibroblast heterogeneity: More than skin deep. J. Cell Sci. 2004;117:667–675. doi: 10.1242/jcs.01005. [DOI] [PubMed] [Google Scholar]
- 7.Glaser R., Harder J., Lange H., Bartels J., Christophers E., Schroder J.M. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat. Immunol. 2005;6:57–64. doi: 10.1038/ni1142. [DOI] [PubMed] [Google Scholar]
- 8.Masters S.L. Specific inflammasomes in complex diseases. Clin. Immunol. 2012 doi: 10.1016/j.clim.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Weber A., Wasiliew P., Kracht M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010;3 doi: 10.1126/scisignal.3105cm1. [DOI] [PubMed] [Google Scholar]
- 10.Bergsbaken T., Fink S.L., Cookson B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeretssian G., Labbe K., Saleh M. Molecular regulation of inflammation and cell death. Cytokine. 2008;43:380–390. doi: 10.1016/j.cyto.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 12.Miao E.A., Leaf I.A., Treuting P.M., Mao D.P., Dors M., Sarkar A., Warren S.E., Wewers M.D., Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldmeyer L., Keller M., Niklaus G., Hohl D., Werner S., Beer H.D. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr. Biol. 2007;17:1140–1145. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe H., Gaide O., Petrilli V., Martinon F., Contassot E., Roques S., Kummer J.A., Tschopp J., French L.E. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J. Investig. Dermatol. 2007;127:1956–1963. doi: 10.1038/sj.jid.5700819. [DOI] [PubMed] [Google Scholar]
- 15.Kupper T.S., Ballard D.W., Chua A.O., McGuire J.S., Flood P.M., Horowitz M.C., Langdon R., Lightfoot L., Gubler U. Human keratinocytes contain mRNA indistinguishable from monocyte interleukin 1 alpha and beta mRNA. Keratinocyte epidermal cell-derived thymocyte-activating factor is identical to interleukin 1. J. Exp. Med. 1986;164:2095–2100. doi: 10.1084/jem.164.6.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondo S., Sauder D.N., Kono T., Galley K.A., McKenzie R.C. Differential modulation of interleukin-1 alpha (IL-1 alpha) and interleukin-1 beta (IL-1 beta) in human epidermal keratinocytes by UVB. Exp. Dermatol. 1994;3:29–39. doi: 10.1111/j.1600-0625.1994.tb00263.x. [DOI] [PubMed] [Google Scholar]
- 17.Barker J.N., Mitra R.S., Griffiths C.E., Dixit V.M., Nickoloff B.J. Keratinocytes as initiators of inflammation. Lancet. 1991;337:211–214. doi: 10.1016/0140-6736(91)92168-2. [DOI] [PubMed] [Google Scholar]
- 18.Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 19.Tschopp J., Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev.. Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 20.Nakahira K., Haspel J.A., Rathinam V.A., Lee S.J., Dolinay T., Lam H.C., Englert J.A., Rabinovitch M., Cernadas M., Kim H.P., et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada K., Crother T.R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V.K., Wolf A.J., Vergnes L., Ojcius D.M., et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Droge W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 23.Forman H.J., Fukuto J.M., Miller T., Zhang H., Rinna A., Levy S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch. Biochem. Biophys. 2008;477:183–195. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niethammer P., Grabher C., Look A.T., Mitchison T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamata H., Hirata H. Redox regulation of cellular signalling. Cell. Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 26.Trachootham D., Lu W., Ogasawara M.A., Nilsa R.D., Huang P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finkel T. Signal transduction by reactive oxygen species. J. Cell. Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhee S.G., Chang T.S., Bae Y.S., Lee S.R., Kang S.W. Cellular regulation by hydrogen peroxide. J. Am. Soc. Nephrol. 2003;14:S211–S215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- 29.Haddad J.J. Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell. Signal. 2002;14:879–897. doi: 10.1016/s0898-6568(02)00053-0. [DOI] [PubMed] [Google Scholar]
- 30.Pastore A., Piemonte F. S-Glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci. 2012;46:279–292. doi: 10.1016/j.ejps.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 31.Jones D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fritz G., Grosch S., Tomicic M., Kaina B. APE/Ref-1 and the mammalian response to genotoxic stress. Toxicology. 2003;193:67–78. doi: 10.1016/s0300-483x(03)00290-7. [DOI] [PubMed] [Google Scholar]
- 33.Rahman I., Marwick J., Kirkham P. Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem. Pharm. 2004;68:1255–1267. doi: 10.1016/j.bcp.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 34.England K., Cotter T.G. Direct oxidative modifications of signalling proteins in mammalian cells and their effects on apoptosis. Redox Rep. 2005;10:237–245. doi: 10.1179/135100005X70224. [DOI] [PubMed] [Google Scholar]
- 35.Bourdon E., Blache D. The importance of proteins in defense against oxidation. Antioxid. Redox Signal. 2001;3:293–311. doi: 10.1089/152308601300185241. [DOI] [PubMed] [Google Scholar]
- 36.Pantano C., Reynaert N.L., van der Vliet A., Janssen-Heininger Y.M. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid. Redox Signal. 2006;8:1791–1806. doi: 10.1089/ars.2006.8.1791. [DOI] [PubMed] [Google Scholar]
- 37.Shao D., Oka S., Brady C.D., Haendeler J., Eaton P., Sadoshima J. Redox modification of cell signaling in the cardiovascular system. J. Mol. Cell. Cardiol. 2012;52:550–558. doi: 10.1016/j.yjmcc.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacob C., Giles G.I., Giles N.M., Sies H. Sulfur and selenium: The role of oxidation state in protein structure and function. Angew. Chem. 2003;42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 39.Martyniuk C.J., Fang B., Koomen J.M., Gavin T., Zhang L., Barber D.S., Lopachin R.M. Molecular mechanism of glyceraldehyde-3-phosphate dehydrogenase inactivation by alpha, beta-unsaturated carbonyl derivatives. Chem. Res. Toxicol. 2011;24:2302–2311. doi: 10.1021/tx200437y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Go Y.M., Duong D.M., Peng J., Jones D.P. Protein cysteines map to functional networks according to steady-state level of oxidation. J. Proteomics Bioinform. 2011;4:196–209. doi: 10.4172/jpb.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forman H.J., Fukuto J.M., Torres M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol. Cell Physiol. 2004;287:C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 42.Winterbourn C.C., Hampton M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Paulsen C.E., Carroll K.S. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung H.S., Wang S.B., Venkatraman V., Murray C.I., Van Eyk J.E. Cysteine oxidative posttranslational modifications: Emerging regulation in the cardiovascular system. Circ. Res. 2013;112:382–392. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denu J.M., Dixon J.E. Protein tyrosine phosphatases: Mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol. 1998;2:633–641. doi: 10.1016/s1367-5931(98)80095-1. [DOI] [PubMed] [Google Scholar]
- 46.Ostman A., Frijhoff J., Sandin A., Bohmer F.D. Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 2011;150:345–356. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 47.Tu B.P., Weissman J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell. Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frand A.R., Cuozzo J.W., Kaiser C.A. Pathways for protein disulphide bond formation. Trends Cell. Biol. 2000;10:203–210. doi: 10.1016/s0962-8924(00)01745-1. [DOI] [PubMed] [Google Scholar]
- 49.Jones D.P., Go Y.M., Anderson C.L., Ziegler T.R., Kinkade J.M., Jr, Kirlin W.G. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J. 2004;18:1246–1248. doi: 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- 50.Chen C.Y., Willard D., Rudolph J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry. 2009;48:1399–1409. doi: 10.1021/bi801973z. [DOI] [PubMed] [Google Scholar]
- 51.Caselli A., Marzocchini R., Camici G., Manao G., Moneti G., Pieraccini G., Ramponi G. The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J. Biol. Chem. 1998;273:32554–32560. doi: 10.1074/jbc.273.49.32554. [DOI] [PubMed] [Google Scholar]
- 52.Lee S.R., Yang K.S., Kwon J., Lee C., Jeong W., Rhee S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 53.Sohn J., Rudolph J. Catalytic and chemical competence of regulation of cdc25 phosphatase by oxidation/reduction. Biochemistry. 2003;42:10060–10070. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 54.Tonks N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 55.Klomsiri C., Karplus P.A., Poole L.B. Cysteine-based redox switches in enzymes. Antioxid. Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shackelford R.E., Heinloth A.N., Heard S.C., Paules R.S. Cellular and molecular targets of protein S-glutathiolation. Antioxid. Redox Signal. 2005;7:940–950. doi: 10.1089/ars.2005.7.940. [DOI] [PubMed] [Google Scholar]
- 57.Shlomai J. Redox control of protein-DNA interactions: From molecular mechanisms to significance in signal transduction, gene expression, and DNA replication. Antioxid. Redox Signal. 2010;13:1429–1476. doi: 10.1089/ars.2009.3029. [DOI] [PubMed] [Google Scholar]
- 58.Dinkova-Kostova A.T., Holtzclaw W.D., Cole R.N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shelton M.D., Mieyal J.J. Regulation by reversible S-glutathionylation: Molecular targets implicated in inflammatory diseases. Mol. Cells. 2008;25:332–346. [PMC free article] [PubMed] [Google Scholar]
- 60.Qanungo S., Starke D.W., Pai H.V., Mieyal J.J., Nieminen A.L. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J. Biol. Chem. 2007;282:18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 61.Cross J.V., Templeton D.J. Regulation of signal transduction through protein cysteine oxidation. Antioxid. Redox Signal. 2006;8:1819–1827. doi: 10.1089/ars.2006.8.1819. [DOI] [PubMed] [Google Scholar]
- 62.Gilmore T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 63.Benhar M., Forrester M.T., Stamler J.S. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 2009;10:721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 64.Benhar M., Forrester M.T., Hess D.T., Stamler J.S. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lima B., Forrester M.T., Hess D.T., Stamler J.S. S-nitrosylation in cardiovascular signaling. Circ. Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Evangelista A.M., Kohr M.J., Murphy E. S-Nitrosylation: Specificity, occupancy, and interaction with other post-translational modifications. Antioxid. Redox Signal. 2013 doi: 10.1089/ars.2012.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Becker K., Savvides S.N., Keese M., Schirmer R.H., Karplus P.A. Enzyme inactivation through sulfhydryl oxidation by physiologic NO-carriers. Nat. Struct. Biol. 1998;5:267–271. doi: 10.1038/nsb0498-267. [DOI] [PubMed] [Google Scholar]
- 68.Kim S., Wing S.S., Ponka P. S-nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol. Cell. Biol. 2004;24:330–337. doi: 10.1128/MCB.24.1.330-337.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Collins Y., Chouchani E.T., James A.M., Menger K.E., Cocheme H.M., Murphy M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012;125:801–806. doi: 10.1242/jcs.098475. [DOI] [PubMed] [Google Scholar]
- 71.Murphy M.P., Holmgren A., Larsson N.G., Halliwell B., Chang C.J., Kalyanaraman B., Rhee S.G., Thornalley P.J., Partridge L., Gems D., et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011;13:361–366. doi: 10.1016/j.cmet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoehn K.L., Salmon A.B., Hohnen-Behrens C., Turner N., Hoy A.J., Maghzal G.J., Stocker R., van Remmen H., Kraegen E.W., Cooney G.J., et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. USA. 2009;106:17787–17792. doi: 10.1073/pnas.0902380106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou L., Aon M.A., Almas T., Cortassa S., Winslow R.L., O’Rourke B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput. Biol. 2010;6:e1000657. doi: 10.1371/journal.pcbi.1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Winterbourn C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 75.Hausladen A., Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem. 1994;269:29405–29408. [PubMed] [Google Scholar]
- 76.Fridovich I. Superoxide anion radical (O2 −•), superoxide dismutases, and related matters. J. Biol. Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 77.Vasquez-Vivar J., Kalyanaraman B., Kennedy M.C. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J. Biol. Chem. 2000;275:14064–14069. doi: 10.1074/jbc.275.19.14064. [DOI] [PubMed] [Google Scholar]
- 78.James A.M., Collins Y., Logan A., Murphy M.P. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol. Metab. 2012;23:429–434. doi: 10.1016/j.tem.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 79.Kietzmann T., Gorlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin. Cell Dev. Biol. 2005;16:474–486. doi: 10.1016/j.semcdb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 80.Murphy M.P. Modulating mitochondrial intracellular location as a redox signal. Sci. Signal. 2012;5 doi: 10.1126/scisignal.2003386. [DOI] [PubMed] [Google Scholar]
- 81.Chandel N.S., Maltepe E., Goldwasser E., Mathieu C.E., Simon M.C., Schumacker P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patten D.A., Lafleur V.N., Robitaille G.A., Chan D.A., Giaccia A.J., Richard D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: The essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell. 2010;21:3247–3257. doi: 10.1091/mbc.E10-01-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ushio-Fukai M., Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266:37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsai Y.P., Wu K.J. Hypoxia-regulated target genes implicated in tumor metastasis. J. Biomed. Sci. 2012;19 doi: 10.1186/1423-0127-19-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Acker T., Fandrey J., Acker H. The good, the bad and the ugly in oxygen-sensing: ROS, cytochromes and prolyl-hydroxylases. Cardiovasc. Res. 2006;71:195–207. doi: 10.1016/j.cardiores.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 86.Al-Mehdi A.B., Pastukh V.M., Swiger B.M., Reed D.J., Patel M.R., Bardwell G.C., Pastukh V.V., Alexeyev M.F., Gillespie M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012;5 doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Valko M., Leibfritz D., Moncol J., Cronin M.T., Mazur M., Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 88.Alfadda A.A., Sallam R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012;2012 doi: 10.1155/2012/936486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mailloux R.J., Harper M.E. Mitochondrial proticity and ROS signaling: Lessons from the uncoupling proteins. Trends Endocrinol. Metab. 2012;23:451–458. doi: 10.1016/j.tem.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 90.Lenaz G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol. 2012;942:93–136. doi: 10.1007/978-94-007-2869-1_5. [DOI] [PubMed] [Google Scholar]
- 91.Garcia-Bailo B., El-Sohemy A., Haddad P.S., Arora P., Benzaied F., Karmali M., Badawi A. Vitamins D, C, and E in the prevention of type 2 diabetes mellitus: Modulation of inflammation and oxidative stress. Biologics. 2011;5:7–19. doi: 10.2147/BTT.S14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nasti T.H., Timares L. Inflammasome activation of IL-1 family mediators in response to cutaneous photodamage. Photochem. Photobiol. 2012;88:1111–1125. doi: 10.1111/j.1751-1097.2012.01182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Muthusamy V., Piva T.J. The UV response of the skin: A review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch. Dermatol. Res. 2010;302:5–17. doi: 10.1007/s00403-009-0994-y. [DOI] [PubMed] [Google Scholar]
- 94.Diaz J.H., Nesbitt L.T., Jr Sun exposure behavior and protection: Recommendations for travelers. J. Travel Med. 2013;20:108–118. doi: 10.1111/j.1708-8305.2012.00667.x. [DOI] [PubMed] [Google Scholar]
- 95.Aroun A., Zhong J.L., Tyrrell R.M., Pourzand C. Iron, oxidative stress and the example of solar ultraviolet A radiation. Photochem. Photobiol. Sci. 2012;11:118–134. doi: 10.1039/c1pp05204g. [DOI] [PubMed] [Google Scholar]
- 96.Rastogi R.P., Richa, Kumar A., Tyagi M.B., Sinha R.P. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J. Nucleic Acids. 2010;2010 doi: 10.4061/2010/592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Valko M., Rhodes C.J., Moncol J., Izakovic M., Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 98.Akasaka E., Takekoshi S., Horikoshi Y., Toriumi K., Ikoma N., Mabuchi T., Tamiya S., Matsuyama T., Ozawa A. Protein oxidative damage and heme oxygenase in sunlight-exposed human skin: Roles of MAPK responses to oxidative stress. Tokai J. Exp. Clin. Med. 2010;35:152–164. [PubMed] [Google Scholar]
- 99.Hruza L.L., Pentland A.P. Mechanisms of UV-induced inflammation. J. Investig. Dermatol. 1993;100:35S–41S. doi: 10.1111/1523-1747.ep12355240. [DOI] [PubMed] [Google Scholar]
- 100.Clydesdale G.J., Dandie G.W., Muller H.K. Ultraviolet light induced injury: Immunological and inflammatory effects. Immunol. Cell Biol. 2001;79:547–568. doi: 10.1046/j.1440-1711.2001.01047.x. [DOI] [PubMed] [Google Scholar]
- 101.Bickers D.R., Athar M. Oxidative stress in the pathogenesis of skin disease. J. Investig. Dermatol. 2006;126:2565–2575. doi: 10.1038/sj.jid.5700340. [DOI] [PubMed] [Google Scholar]
- 102.Vile G.F., Tanew-Ilitschew A., Tyrrell R.M. Activation of NF-kappa B in human skin fibroblasts by the oxidative stress generated by UVA radiation. Photochem. Photobiol. 1995;62:463–468. doi: 10.1111/j.1751-1097.1995.tb02369.x. [DOI] [PubMed] [Google Scholar]
- 103.Vile G.F., Tyrrell R.M. UVA radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen. Free Radic. Biol. Med. 1995;18:721–730. doi: 10.1016/0891-5849(94)00192-m. [DOI] [PubMed] [Google Scholar]
- 104.Scharffetter K., Wlaschek M., Hogg A., Bolsen K., Schothorst A., Goerz G., Krieg T., Plewig G. UVA irradiation induces collagenase in human dermal fibroblasts in vitro and in vivo. Arch. Dermatol. Res. 1991;283:506–511. doi: 10.1007/BF00371923. [DOI] [PubMed] [Google Scholar]
- 105.Wlaschek M., Briviba K., Stricklin G.P., Sies H., Scharffetter-Kochanek K. Singlet oxygen may mediate the ultraviolet A-induced synthesis of interstitial collagenase. J. Investig. Dermatol. 1995;104:194–198. doi: 10.1111/1523-1747.ep12612751. [DOI] [PubMed] [Google Scholar]
- 106.Ryter S.W., Tyrrell R.M. Singlet molecular oxygen ((1)O2): A possible effector of eukaryotic gene expression. Free Radic. Biol. Med. 1998;24:1520–1534. doi: 10.1016/s0891-5849(97)00461-9. [DOI] [PubMed] [Google Scholar]
- 107.Podda M., Traber M.G., Weber C., Yan L.J., Packer L. UV-irradiation depletes antioxidants and causes oxidative damage in a model of human skin. Free Radic. Biol. Med. 1998;24:55–65. doi: 10.1016/s0891-5849(97)00142-1. [DOI] [PubMed] [Google Scholar]
- 108.Armstrong A.W., Harskamp C.T., Armstrong E.J. Psoriasis and the risk of diabetes mellitus: A systematic review and meta-analysis. JAMA Dermatol. 2013;149:84–91. doi: 10.1001/2013.jamadermatol.406. [DOI] [PubMed] [Google Scholar]
- 109.Azfar R.S., Seminara N.M., Shin D.B., Troxel A.B., Margolis D.J., Gelfand J.M. Increased risk of diabetes mellitus and likelihood of receiving diabetes mellitus treatment in patients with psoriasis. Arch. Dermatol. 2012;148:995–1000. doi: 10.1001/archdermatol.2012.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cheng J., Kuai D., Zhang L., Yang X., Qiu B. Psoriasis increased the risk of diabetes: A meta-analysis. Arch. Dermatol. Res. 2012;304:119–125. doi: 10.1007/s00403-011-1200-6. [DOI] [PubMed] [Google Scholar]
- 111.Maritim A.C., Sanders R.A., Watkins J.B. 3rd Diabetes, oxidative stress, and antioxidants: A review. J. Biochem. Mol. Toxicol. 2003;17:24–38. doi: 10.1002/jbt.10058. [DOI] [PubMed] [Google Scholar]
- 112.Enamandram M., Kimball A.B. Psoriasis epidemiology: The interplay of genes and the environment. J. Investig. Dermatol. 2013;133:287–289. doi: 10.1038/jid.2012.434. [DOI] [PubMed] [Google Scholar]
- 113.Nickoloff B.J., Xin H., Nestle F.O., Qin J.Z. The cytokine and chemokine network in psoriasis. Clin. Dermatol. 2007;25:568–573. doi: 10.1016/j.clindermatol.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 114.Lowes M.A., Bowcock A.M., Krueger J.G. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 115.Zhou Q., Mrowietz U., Rostami-Yazdi M. Oxidative stress in the pathogenesis of psoriasis. Free Radic. Biol. Med. 2009;47:891–905. doi: 10.1016/j.freeradbiomed.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 116.Briganti S., Picardo M. Antioxidant activity, lipid peroxidation and skin diseases. What’s new. J. Eur. Acad. Dermatol. Venereol. 2003;17:663–669. doi: 10.1046/j.1468-3083.2003.00751.x. [DOI] [PubMed] [Google Scholar]
- 117.Racz E., Prens E.P. Molecular pathophysiology of psoriasis and molecular targets of antipsoriatic therapy. Expert Rev. Mol. Med. 2009;11:e38. doi: 10.1017/S146239940900129X. [DOI] [PubMed] [Google Scholar]
- 118.Rocha-Pereira P., Santos-Silva A., Rebelo I., Figueiredo A., Quintanilha A., Teixeira F. Dislipidemia and oxidative stress in mild and in severe psoriasis as a risk for cardiovascular disease. Clin. Chim. Acta. 2001;303:33–39. doi: 10.1016/s0009-8981(00)00358-2. [DOI] [PubMed] [Google Scholar]
- 119.Tekin N.S., Tekin I.O., Barut F., Sipahi E.Y. Accumulation of oxidized low-density lipoprotein in psoriatic skin and changes of plasma lipid levels in psoriatic patients. Mediators Inflamm. 2007;2007:78454. doi: 10.1155/2007/78454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Simonetti O., Ferretti G., Salvi A., Offidani A.M., Bossi G. Plasma lipid changes in psoriatic children. Dermatology. 1992;185:96–100. doi: 10.1159/000247421. [DOI] [PubMed] [Google Scholar]
- 121.Kokcam I., Naziroglu M. Antioxidants and lipid peroxidation status in the blood of patients with psoriasis. Clin. Chim. Acta. 1999;289:23–31. doi: 10.1016/s0009-8981(99)00150-3. [DOI] [PubMed] [Google Scholar]
- 122.Drewa G., Krzyzynska-Malinowska E., Wozniak A., Protas-Drozd F., Mila-Kierzenkowska C., Rozwodowska M., Kowaliszyn B., Czajkowski R. Activity of superoxide dismutase and catalase and the level of lipid peroxidation products reactive with TBA in patients with psoriasis. Med. Sci. Monit. 2002;8:BR338–BR343. [PubMed] [Google Scholar]
- 123.Yildirim M., Inaloz H.S., Baysal V., Delibas N. The role of oxidants and antioxidants in psoriasis. J. Eur. Acad. Dermatol. Venereol. 2003;17:34–36. doi: 10.1046/j.1468-3083.2003.00641.x. [DOI] [PubMed] [Google Scholar]
- 124.Therond P., Gerbaud P., Dimon S., Anderson W.B., Evain-Broin D., Raynaud F. Antioxidant enzymes in psoriatic fibroblasts and erythrocytes. J. Investig. Dermatol. 1996;106:1325–1328. doi: 10.1111/1523-1747.ep12349055. [DOI] [PubMed] [Google Scholar]
- 125.Frank S., Munz B., Werner S. The human homologue of a bovine non-selenium glutathione peroxidase is a novel keratinocyte growth factor-regulated gene. Oncogene. 1997;14:915–921. doi: 10.1038/sj.onc.1200905. [DOI] [PubMed] [Google Scholar]
- 126.Ryu J., Park S.G., Park B.C., Choe M., Lee K.S., Cho J.W. Proteomic analysis of psoriatic skin tissue for identification of differentially expressed proteins: Up-regulation of GSTP1, SFN and PRDX2 in psoriatic skin. Int. J. Mol. Med. 2011;28:785–792. doi: 10.3892/ijmm.2011.757. [DOI] [PubMed] [Google Scholar]
- 127.Armstrong A.W., Voyles S.V., Armstrong E.J., Fuller E.N., Rutledge J.C. Angiogenesis and oxidative stress: Common mechanisms linking psoriasis with atherosclerosis. J. Dermatol. Sci. 2011;63:1–9. doi: 10.1016/j.jdermsci.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 128.Takahashi H., Ibe M., Nakamura S., Ishida-Yamamoto A., Hashimoto Y., Iizuka H. Extracellular regulated kinase and c-Jun N-terminal kinase are activated in psoriatic involved epidermis. J. Dermatol. Sci. 2002;30:94–99. doi: 10.1016/s0923-1811(02)00064-6. [DOI] [PubMed] [Google Scholar]
- 129.Johansen C., Flindt E., Kragballe K., Henningsen J., Westergaard M., Kristiansen K., Iversen L. Inverse regulation of the nuclear factor-kappaB binding to the p53 and interleukin-8 kappaB response elements in lesional psoriatic skin. J. Investig. Dermatol. 2005;124:1284–1292. doi: 10.1111/j.0022-202X.2005.23749.x. [DOI] [PubMed] [Google Scholar]
- 130.Yu X.J., Li C.Y., Dai H.Y., Cai D.X., Wang K.Y., Xu Y.H., Chen L.M., Zhou C.L. Expression and localization of the activated mitogen-activated protein kinase in lesional psoriatic skin. Exp. Mol. Pathol. 2007;83:413–418. doi: 10.1016/j.yexmp.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 131.Abdou A.G., Hanout H.M. Evaluation of survivin and NF-kappaB in psoriasis, an immunohistochemical study. J. Cutan. Pathol. 2008;35:445–451. doi: 10.1111/j.1600-0560.2007.00841.x. [DOI] [PubMed] [Google Scholar]
- 132.Lizzul P.F., Aphale A., Malaviya R., Sun Y., Masud S., Dombrovskiy V., Gottlieb A.B. Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J. Investig. Dermatol. 2005;124:1275–1283. doi: 10.1111/j.0022-202X.2005.23735.x. [DOI] [PubMed] [Google Scholar]
- 133.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009;1 doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Treumer F., Zhu K., Glaser R., Mrowietz U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J. Investig. Dermatol. 2003;121:1383–1388. doi: 10.1111/j.1523-1747.2003.12605.x. [DOI] [PubMed] [Google Scholar]
- 135.Amigo M., Paya M., De Rosa S., Terencio M.C. Antipsoriatic effects of avarol-3′-thiosalicylate are mediated by inhibition of TNF-alpha generation and NF-kappaB activation in mouse skin. Br. J. Pharmacol. 2007;152:353–365. doi: 10.1038/sj.bjp.0707394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Amigo M., Schalkwijk J., Olthuis D., De Rosa S., Paya M., Terencio M.C., Lamme E. Identification of avarol derivatives as potential antipsoriatic drugs using an in vitro model for keratinocyte growth and differentiation. Life Sci. 2006;79:2395–2404. doi: 10.1016/j.lfs.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 137.Teunissen M.B., Koomen C.W., de Waal Malefyt R., Wierenga E.A., Bos J.D. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J. Investig. Dermatol. 1998;111:645–649. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 138.Bito T., Nishigori C. Impact of reactive oxygen species on keratinocyte signaling pathways. J. Dermatol. Sci. 2012;68:3–8. doi: 10.1016/j.jdermsci.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 139.Kao C.C., Garner W.L. Acute burns. Plast. Reconstr. Surg. 2000;101:2482–2493. [PubMed] [Google Scholar]
- 140.Latha B., Babu M. The involvement of free radicals in burn injury: A review. Burns. 2001;27:309–317. doi: 10.1016/s0305-4179(00)00127-3. [DOI] [PubMed] [Google Scholar]
- 141.Kupper T.S., Deitch E.A., Baker C.C., Wong W.C. The human burn wound as a primary source of interleukin-1 activity. Surgery. 1986;100:409–415. [PubMed] [Google Scholar]
- 142.Ward P.A., Till G.O. Pathophysiologic events related to thermal injury of skin. J. Trauma. 1990;30:S75–S79. doi: 10.1097/00005373-199012001-00018. [DOI] [PubMed] [Google Scholar]
- 143.Till G.O., Beauchamp C., Menapace D., Tourtellotte W., Jr, Kunkel R., Johnson K.J., Ward P.A. Oxygen radical dependent lung damage following thermal injury of rat skin. J. Trauma. 1983;23:269–277. doi: 10.1097/00005373-198304000-00001. [DOI] [PubMed] [Google Scholar]
- 144.Till G.O., Hatherill J.R., Tourtellotte W.W., Lutz M.J., Ward P.A. Lipid peroxidation and acute lung injury after thermal trauma to skin. Evidence of a role for hydroxyl radical. Am. J. Pathol. 1985;119:376–384. [PMC free article] [PubMed] [Google Scholar]
- 145.Gurbuz V., Corak A., Yegen B.C., Kurtel H., Alican I. Oxidative organ damage in a rat model of thermal injury: The effect of cyclosporin A. Burns. 1997;23:37–42. doi: 10.1016/s0305-4179(96)00072-1. [DOI] [PubMed] [Google Scholar]
- 146.Jahovic N., Guzel E., Arbak S., Yegen B.C. The healing-promoting effect of saliva on skin burn is mediated by epidermal growth factor (EGF): Role of the neutrophils. Burns. 2004;30:531–538. doi: 10.1016/j.burns.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 147.Cetinkale O., Konukoglu D., Senel O., Kemerli G.D., Yazar S. Modulating the functions of neutrophils and lipid peroxidation by FK506 in a rat model of thermal injury. Burns. 1999;25:105–112. doi: 10.1016/s0305-4179(98)00147-8. [DOI] [PubMed] [Google Scholar]
- 148.Haycock J.W., Ralston D.R., Morris B., Freedlander E., MacNeil S. Oxidative damage to protein and alterations to antioxidant levels in human cutaneous thermal injury. Burns. 1997;23:533–540. doi: 10.1016/s0305-4179(97)00072-7. [DOI] [PubMed] [Google Scholar]
- 149.Tredget E.E., Yu Y.M. The metabolic effects of thermal injury. World J. Surg. 1992;16:68–79. doi: 10.1007/BF02067117. [DOI] [PubMed] [Google Scholar]
- 150.Bertin-Maghit M., Goudable J., Dalmas E., Steghens J.P., Bouchard C., Gueugniaud P.Y., Petit P., Delafosse B. Time course of oxidative stress after major burns. Intensive Care Med. 2000;26:800–803. doi: 10.1007/s001340051250. [DOI] [PubMed] [Google Scholar]
- 151.Horton J.W. Free radicals and lipid peroxidation mediated injury in burn trauma: The role of antioxidant therapy. Toxicology. 2003;189:75–88. doi: 10.1016/s0300-483x(03)00154-9. [DOI] [PubMed] [Google Scholar]
- 152.Demling R., Ikegami K., Lalonde C. Increased lipid peroxidation and decreased antioxidant activity correspond with death after smoke exposure in the rat. J. Burn Care Rehabil. 1995;16:104–110. doi: 10.1097/00004630-199503000-00003. [DOI] [PubMed] [Google Scholar]
- 153.Hosnuter M., Gurel A., Babuccu O., Armutcu F., Kargi E., Isikdemir A. The effect of CAPE on lipid peroxidation and nitric oxide levels in the plasma of rats following thermal injury. Burns. 2004;30:121–125. doi: 10.1016/j.burns.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 154.Saez J.C., Ward P.H., Gunther B., Vivaldi E. Superoxide radical involvement in the pathogenesis of burn shock. Circ. Shock. 1984;12:229–239. [PubMed] [Google Scholar]
- 155.Oldham K.T., Guice K.S., Till G.O., Ward P.A. Activation of complement by hydroxyl radical in thermal injury. Surgery. 1988;104:272–279. [PubMed] [Google Scholar]
- 156.Sener G., Sehirli A.O., Satiroglu H., Keyer-Uysal M., Yegen B.C. Melatonin improves oxidative organ damage in a rat model of thermal injury. Burns. 2002;28:419–425. doi: 10.1016/s0305-4179(02)00053-0. [DOI] [PubMed] [Google Scholar]
- 157.Saitoh D., Ookawara T., Fukuzuka K., Kawakami M., Sakamoto T., Ohno H., Okada Y. Characteristics of plasma extracellular SOD in burned patients. Burns. 2001;27:577–581. doi: 10.1016/s0305-4179(01)00022-5. [DOI] [PubMed] [Google Scholar]
- 158.Saitoh D., Shirani K.Z., Cioffi W.G., Kizaki T., Ohno H., Okada Y., Mason A.D., Jr, Pruitt B.A., Jr Changes in the tissue and plasma superoxide dismutase (SOD) levels in a burned rat model. Tohoku J. Exp. Med. 2001;193:27–36. doi: 10.1620/tjem.193.27. [DOI] [PubMed] [Google Scholar]
- 159.Cetinkale O., Belce A., Konukoglu D., Senyuva C., Gumustas M.K., Tas T. Evaluation of lipid peroxidation and total antioxidant status in plasma of rats following thermal injury. Burns. 1997;23:114–116. doi: 10.1016/s0305-4179(96)00084-8. [DOI] [PubMed] [Google Scholar]
- 160.Steiling H., Munz B., Werner S., Brauchle M. Different types of ROS-scavenging enzymes are expressed during cutaneous wound repair. Exp. Cell Res. 1999;247:484–494. doi: 10.1006/excr.1998.4366. [DOI] [PubMed] [Google Scholar]
- 161.Sena L.A., Chandel N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Agarwal A., Banerjee A., Banerjee U.C. Xanthine oxidoreductase: A journey from purine metabolism to cardiovascular excitation-contraction coupling. Crit. Rev. Biotechnol. 2011;31:264–280. doi: 10.3109/07388551.2010.527823. [DOI] [PubMed] [Google Scholar]
- 163.Zhang R., Brennan M.L., Shen Z., MacPherson J.C., Schmitt D., Molenda C.E., Hazen S.L. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J. Biol. Chem. 2002;277:46116–46122. doi: 10.1074/jbc.M209124200. [DOI] [PubMed] [Google Scholar]
- 164.Fleming I., Michaelis U.R., Bredenkotter D., Fisslthaler B., Dehghani F., Brandes R.P., Busse R. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ. Res. 2001;88:44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- 165.Tabima D.M., Frizzell S., Gladwin M.T. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic. Biol. Med. 2012;52:1970–1986. doi: 10.1016/j.freeradbiomed.2012.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Andrew P.J., Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999;43:521–531. doi: 10.1016/s0008-6363(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 167.Xia Y., Tsai A.L., Berka V., Zweier J.L. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J. Biol. Chem. 1998;273:25804–25808. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 168.Roe N.D., Ren J. Nitric oxide synthase uncoupling: A therapeutic target in cardiovascular diseases. Vasc. Pharmacol. 2012;57:168–172. doi: 10.1016/j.vph.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 169.Altenhofer S., Kleikers P.W., Radermacher K.A., Scheurer P., Rob Hermans J.J., Schiffers P., Ho H., Wingler K., Schmidt H.H. The NOX toolbox: Validating the role of NADPH oxidases in physiology and disease. Cell. Mol. Life Sci. 2012;69:2327–2343. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Brown D.I., Griendling K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Takac I., Schroder K., Zhang L., Lardy B., Anilkumar N., Lambeth J.D., Shah A.M., Morel F., Brandes R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jiang F., Zhang Y., Dusting G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 173.Lambeth J.D., Kawahara T., Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 175.Chamulitrat W., Stremmel W., Kawahara T., Rokutan K., Fujii H., Wingler K., Schmidt H.H., Schmidt R. A constitutive NADPH oxidase-like system containing gp91phox homologs in human keratinocytes. J. Investig. Dermatol. 2004;122:1000–1009. doi: 10.1111/j.0022-202X.2004.22410.x. [DOI] [PubMed] [Google Scholar]
- 176.Sen C.K., Khanna S., Babior B.M., Hunt T.K., Ellison E.C., Roy S. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. J. Biol. Chem. 2002;277:33284–33290. doi: 10.1074/jbc.M203391200. [DOI] [PubMed] [Google Scholar]
- 177.Chiu C., Maddock D.A., Zhang Q., Souza K.P., Townsend A.R., Wan Y. TGF-beta-induced p38 activation is mediated by Rac1-regulated generation of reactive oxygen species in cultured human keratinocytes. Int. J. Mol. Med. 2001;8:251–255. [PubMed] [Google Scholar]
- 178.Chamulitrat W., Schmidt R., Tomakidi P., Stremmel W., Chunglok W., Kawahara T., Rokutan K. Association of gp91phox homolog Nox1 with anchorage-independent growth and MAP kinase-activation of transformed human keratinocytes. Oncogene. 2003;22:6045–6053. doi: 10.1038/sj.onc.1206654. [DOI] [PubMed] [Google Scholar]
- 179.Steinbeck M.J., Khan A.U., Karnovsky M.J. Extracellular production of singlet oxygen by stimulated macrophages quantified using 9,10-diphenylanthracene and perylene in a polystyrene film. J. Biol. Chem. 1993;268:15649–15654. [PubMed] [Google Scholar]
- 180.Tsiftsoglou A.S., Tsamadou A.I., Papadopoulou L.C. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 2006;111:327–345. doi: 10.1016/j.pharmthera.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 181.Wagener F.A., Volk H.D., Willis D., Abraham N.G., Soares M.P., Adema G.J., Figdor C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003;55:551–571. doi: 10.1124/pr.55.3.5. [DOI] [PubMed] [Google Scholar]
- 182.Ryter S.W., Tyrrell R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000;28:289–309. doi: 10.1016/s0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 183.Halliwell B., Gutteridge J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kumar S., Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005;157:175–188. doi: 10.1016/j.toxlet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 185.Thomas D.D., Espey M.G., Vitek M.P., Miranda K.M., Wink D.A. Protein nitration is mediated by heme and free metals through Fenton-type chemistry: An alternative to the NO/O2– reaction. Proc. Natl. Acad. Sci. USA. 2002;99:12691–12696. doi: 10.1073/pnas.202312699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jeney V., Balla J., Yachie A., Varga Z., Vercellotti G.M., Eaton J.W., Balla G. Pro-oxidant and cytotoxic effects of circulating heme. Blood. 2002;100:879–887. doi: 10.1182/blood.v100.3.879. [DOI] [PubMed] [Google Scholar]
- 187.Balla G., Jacob H.S., Eaton J.W., Belcher J.D., Vercellotti G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. 1991;11:1700–1711. doi: 10.1161/01.atv.11.6.1700. [DOI] [PubMed] [Google Scholar]
- 188.Balla J., Jacob H.S., Balla G., Nath K., Eaton J.W., Vercellotti G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA. 1993;90:9285–9289. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wagener F.A., Abraham N.G., van K.Y., de W.T., Figdor C.G. Heme-induced cell adhesion in the pathogenesis of sickle-cell disease and inflammation. Trends Pharmacol. Sci. 2001;22:52–54. doi: 10.1016/s0165-6147(00)01609-6. [DOI] [PubMed] [Google Scholar]
- 190.Wagener F.A., da Silva J.L., Farley T., de W.T., Kappas A., Abraham N.G. Differential effects of heme oxygenase isoforms on heme mediation of endothelial intracellular adhesion molecule 1 expression. J. Pharmacol. Exp. Ther. 1999;291:416–423. [PubMed] [Google Scholar]
- 191.Wagener F.A., Feldman E., de W.T., Abraham N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med. 1997;216:456–463. doi: 10.3181/00379727-216-44197. [DOI] [PubMed] [Google Scholar]
- 192.Tolosano E., Fagoonee S., Hirsch E., Berger F.G., Baumann H., Silengo L., Altruda F. Enhanced splenomegaly and severe liver inflammation in haptoglobin/hemopexin double-null mice after acute hemolysis. Blood. 2002;100:4201–4208. doi: 10.1182/blood-2002-04-1270. [DOI] [PubMed] [Google Scholar]
- 193.Ma J.L., Yang P.Y., Rui Y.C., Lu L., Kang H., Zhang J. Hemin modulates cytokine expressions in macrophage-derived foam cells via heme oxygenase-1 induction. J. Pharmacol. Sci. 2007;103:261–266. doi: 10.1254/jphs.fp0060270. [DOI] [PubMed] [Google Scholar]
- 194.Cambos M., Bazinet S., Abed E., Sanchez-Dardon J., Bernard C., Moreau R., Olivier M., Scorza T. The IL-12p70/IL-10 interplay is differentially regulated by free heme and hemozoin in murine bone-marrow-derived macrophages. Int J. Parasitol. 2010;40:1003–1012. doi: 10.1016/j.ijpara.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 195.Cambos M., Scorza T. Robust erythrophagocytosis leads to macrophage apoptosis via a hemin-mediated redox imbalance: Role in hemolytic disorders. J. Leukoc. Biol. 2011;89:159–171. doi: 10.1189/jlb.0510249. [DOI] [PubMed] [Google Scholar]
- 196.Wagener F.A., van Beurden H.E., von den Hoff J.W., Adema G.J., Figdor C.G. The heme-heme oxygenase system: A molecular switch in wound healing. Blood. 2003;102:521–528. doi: 10.1182/blood-2002-07-2248. [DOI] [PubMed] [Google Scholar]
- 197.Harris E.D. Regulation of antioxidant enzymes. FASEB J. 1992;6:2675–2683. doi: 10.1096/fasebj.6.9.1612291. [DOI] [PubMed] [Google Scholar]
- 198.Yoshikawa S., Muramoto K., Shinzawa-Itoh K. The O(2) reduction and proton pumping gate mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta. 2011;1807:1279–1286. doi: 10.1016/j.bbabio.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 199.Martinez-Finley E.J., Chakraborty S., Fretham S.J., Aschner M. Cellular transport and homeostasis of essential and nonessential metals. Metallomics. 2012;4:593–605. doi: 10.1039/c2mt00185c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jomova K., Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 201.Ratnam D.V., Ankola D.D., Bhardwaj V., Sahana D.K., Kumar M.N. Role of antioxidants in prophylaxis and therapy: A pharmaceutical perspective. J. Control. Release. 2006;113:189–207. doi: 10.1016/j.jconrel.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 202.Packer L., Valacchi G. Antioxidants and the response of skin to oxidative stress: Vitamin E as a key indicator. Skin Pharmacol. Appl. Skin Physiol. 2002;15:282–290. doi: 10.1159/000064531. [DOI] [PubMed] [Google Scholar]
- 203.Zelko I.N., Mariani T.J., Folz R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002;33:337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 204.Zamocky M., Jakopitsch C., Furtmuller P.G., Dunand C., Obinger C. The peroxidase-cyclooxygenase superfamily: Reconstructed evolution of critical enzymes of the innate immune system. Proteins. 2008;72:589–605. doi: 10.1002/prot.21950. [DOI] [PubMed] [Google Scholar]
- 205.Brigelius-Flohe R. Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem. 2006;387:1329–1335. doi: 10.1515/BC.2006.166. [DOI] [PubMed] [Google Scholar]
- 206.Arthur J.R. The glutathione peroxidases. Cell. Mol. Life Sci. 2000;57:1825–1835. doi: 10.1007/PL00000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.De Haan J.B., Bladier C., Griffiths P., Kelner M., O’Shea R.D., Cheung N.S., Bronson R.T., Silvestro M.J., Wild S., Zheng S.S., et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J. Biol. Chem. 1998;273:22528–22536. doi: 10.1074/jbc.273.35.22528. [DOI] [PubMed] [Google Scholar]
- 208.Auf dem Keller U., Kumin A., Braun S., Werner S. Reactive oxygen species and their detoxification in healing skin wounds. J. Investig. Dermatol. Symp. Proc. 2006;11:106–111. doi: 10.1038/sj.jidsymp.5650001. [DOI] [PubMed] [Google Scholar]
- 209.De Haan J.B., Crack P.J., Flentjar N., Iannello R.C., Hertzog P.J., Kola I. An imbalance in antioxidant defense affects cellular function: The pathophysiological consequences of a reduction in antioxidant defense in the glutathione peroxidase-1 (Gpx1) knockout mouse. Redox Rep. 2003;8:69–79. doi: 10.1179/135100003125001378. [DOI] [PubMed] [Google Scholar]
- 210.Hanschmann E.M., Godoy J.R., Berndt C., Hudemann C., Lillig C.H. Thioredoxins, glutaredoxins, and peroxiredoxins-molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013 doi: 10.1089/ars.2012.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Manevich Y., Sweitzer T., Pak J.H., Feinstein S.I., Muzykantov V., Fisher A.B. 1-Cys peroxiredoxin overexpression protects cells against phospholipid peroxidation-mediated membrane damage. Proc. Natl. Acad. Sci. USA. 2002;99:11599–11604. doi: 10.1073/pnas.182384499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wang Y., Manevich Y., Feinstein S.I., Fisher A.B. Adenovirus-mediated transfer of the 1-cys peroxiredoxin gene to mouse lung protects against hyperoxic injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L1188–L1193. doi: 10.1152/ajplung.00288.2003. [DOI] [PubMed] [Google Scholar]
- 213.Kumin A., Huber C., Rulicke T., Wolf E., Werner S. Peroxiredoxin 6 is a potent cytoprotective enzyme in the epidermis. Am. J. Pathol. 2006;169:1194–1205. doi: 10.2353/ajpath.2006.060119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Pak J.H., Manevich Y., Kim H.S., Feinstein S.I., Fisher A.B. An antisense oligonucleotide to 1-cys peroxiredoxin causes lipid peroxidation and apoptosis in lung epithelial cells. J. Biol. Chem. 2002;277:49927–49934. doi: 10.1074/jbc.M204222200. [DOI] [PubMed] [Google Scholar]
- 215.Mo Y., Feinstein S.I., Manevich Y., Zhang Q., Lu L., Ho Y.S., Fisher A.B. 1-Cys peroxiredoxin knock-out mice express mRNA but not protein for a highly related intronless gene. FEBS Lett. 2003;555:192–198. doi: 10.1016/s0014-5793(03)01199-2. [DOI] [PubMed] [Google Scholar]
- 216.Wang X., Phelan S.A., Forsman-Semb K., Taylor E.F., Petros C., Brown A., Lerner C.P., Paigen B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 2003;278:25179–25190. doi: 10.1074/jbc.M302706200. [DOI] [PubMed] [Google Scholar]
- 217.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 218.Wu G., Fang Y.Z., Yang S., Lupton J.R., Turner N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 219.Stanton R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life. 2012;64:362–369. doi: 10.1002/iub.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kletzien R.F., Harris P.K., Foellmi L.A. Glucose-6-phosphate dehydrogenase: A “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J. 1994;8:174–181. doi: 10.1096/fasebj.8.2.8119488. [DOI] [PubMed] [Google Scholar]
- 221.Pandolfi P.P., Sonati F., Rivi R., Mason P., Grosveld F., Luzzatto L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995;14:5209–5215. doi: 10.1002/j.1460-2075.1995.tb00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rosenstraus M., Chasin L.A. Isolation of mammalian cell mutants deficient in glucose-6-phosphate dehydrogenase activity: Linkage to hypoxanthine phosphoribosyl transferase. Proc. Natl. Acad. Sci. USA. 1975;72:493–497. doi: 10.1073/pnas.72.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Serpillon S., Floyd B.C., Gupte R.S., George S., Kozicky M., Neito V., Recchia F., Stanley W., Wolin M.S., Gupte S.A. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am. J. Physiol. Heart .Circ. Physiol. 2009;297:H153–H162. doi: 10.1152/ajpheart.01142.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Haines D.D., Lekli I., Teissier P., Bak I., Tosaki A. Role of haeme oxygenase-1 in resolution of oxidative stress-related pathologies: Focus on cardiovascular, lung, neurological and kidney disorders. Acta Physiol. 2012;204:487–501. doi: 10.1111/j.1748-1716.2011.02387.x. [DOI] [PubMed] [Google Scholar]
- 225.Abraham N.G., Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 226.Maines M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 227.Tenhunen R., Marver H.S., Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wagener F.A., Scharstuhl A., Tyrrell R.M., Von den Hoff J.W., Jozkowicz A., Dulak J., Russel F.G., Kuijpers-Jagtman A.M. The heme-heme oxygenase system in wound healing; Implications for scar formation. Curr. Drug Targets. 2010;11:1571–1585. doi: 10.2174/1389450111009011571. [DOI] [PubMed] [Google Scholar]
- 229.Lundvig D.M., Immenschuh S., Wagener F.A. Heme oxygenase, inflammation, and fibrosis: The good, the bad, and the ugly? Front. Pharmacol. 2012;3:81. doi: 10.3389/fphar.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Grochot-Przeczek A., Dulak J., Jozkowicz A. Haem oxygenase-1: Non-canonical roles in physiology and pathology. Clin. Sci. 2012;122:93–103. doi: 10.1042/CS20110147. [DOI] [PubMed] [Google Scholar]
- 231.Alam J., Shibahara S., Smith A. Transcriptional activation of the heme oxygenase gene by heme and cadmium in mouse hepatoma cells. J. Biol. Chem. 1989;264:6371–6375. [PubMed] [Google Scholar]
- 232.Applegate L.A., Noel A., Vile G., Frenk E., Tyrrell R.M. Two genes contribute to different extents to the heme oxygenase enzyme activity measured in cultured human skin fibroblasts and keratinocytes: Implications for protection against oxidant stress. Photochem. Photobiol. 1995;61:285–291. doi: 10.1111/j.1751-1097.1995.tb03973.x. [DOI] [PubMed] [Google Scholar]
- 233.Cisowski J., Loboda A., Jozkowicz A., Chen S., Agarwal A., Dulak J. Role of heme oxygenase-1 in hydrogen peroxide-induced VEGF synthesis: Effect of HO-1 knockout. Biochem. Biophys. Res. Commun. 2005;326:670–676. doi: 10.1016/j.bbrc.2004.11.083. [DOI] [PubMed] [Google Scholar]
- 234.Tosaki A., Das D.K. The role of heme oxygenase signaling in various disorders. Mol. Cell. Biochem. 2002;232:149–157. doi: 10.1023/a:1014885014600. [DOI] [PubMed] [Google Scholar]
- 235.Tyrrell R.M. Solar ultraviolet A radiation: An oxidizing skin carcinogen that activates heme oxygenase-1. Antioxid. Redox Signal. 2004;6:835–840. doi: 10.1089/ars.2004.6.835. [DOI] [PubMed] [Google Scholar]
- 236.Wagener F.A., Eggert A., Boerman O.C., Oyen W.J., Verhofstad A., Abraham N.G., Adema G., van Kooyk Y., de Witte T., Figdor C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood. 2001;98:1802–1811. doi: 10.1182/blood.v98.6.1802. [DOI] [PubMed] [Google Scholar]
- 237.Wagener F.A., Dankers A.C., van Summeren F., Scharstuhl A., van den Heuvel J.J., Koenderink J.B., Pennings S.W., Russel F.G., Masereeuw R. Heme oxygenase-1 and breast cancer resistance protein protect against heme-induced toxicity. Curr. Pharm. Des. 2013;19:2698–2707. doi: 10.2174/1381612811319150004. [DOI] [PubMed] [Google Scholar]
- 238.Hayashi S., Takamiya R., Yamaguchi T., Matsumoto K., Tojo S.J., Tamatani T., Kitajima M., Makino N., Ishimura Y., Suematsu M. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: Role of bilirubin generated by the enzyme. Circ. Res. 1999;85:663–671. doi: 10.1161/01.res.85.8.663. [DOI] [PubMed] [Google Scholar]
- 239.Vachharajani T.J., Work J., Issekutz A.C., Granger D.N. Heme oxygenase modulates selectin expression in different regional vascular beds. Am. J. Physiol. 2000;278:H1613–H1617. doi: 10.1152/ajpheart.2000.278.5.H1613. [DOI] [PubMed] [Google Scholar]
- 240.Takahashi T., Shimizu H., Morimatsu H., Inoue K., Akagi R., Morita K., Sassa S. Heme oxygenase-1: A fundamental guardian against oxidative tissue injuries in acute inflammation. Mini Rev. Med. Chem. 2007;7:745–753. doi: 10.2174/138955707781024517. [DOI] [PubMed] [Google Scholar]
- 241.Takahashi T., Shimizu H., Morimatsu H., Maeshima K., Inoue K., Akagi R., Matsumi M., Katayama H., Morita K. Heme oxygenase-1 is an essential cytoprotective component in oxidative tissue injury induced by hemorrhagic shock. J. Clin. Biochem. Nutr. 2009;44:28–40. doi: 10.3164/jcbn.08-210-HO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Baranano D.E., Rao M., Ferris C.D., Snyder S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA. 2002;99:16093–16098. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Stocker R., Yamamoto Y., McDonagh A.F., Glazer A.N., Ames B.N. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 244.Kim S.Y., Park S.C. Physiological antioxidative network of the bilirubin system in aging and age-related diseases. Front. Pharmacol. 2012;3:45. doi: 10.3389/fphar.2012.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Dore S., Snyder S.H. Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann. N. Y. Acad. Sci. 1999;890:167–172. doi: 10.1111/j.1749-6632.1999.tb07991.x. [DOI] [PubMed] [Google Scholar]
- 246.Dore S., Takahashi M., Ferris C.D., Zakhary R., Hester L.D., Guastella D., Snyder S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA. 1999;96:2445–2450. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lanone S., Bloc S., Foresti R., Almolki A., Taille C., Callebert J., Conti M., Goven D., Aubier M., Dureuil B., et al. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. 2005;19:1890–1892. doi: 10.1096/fj.04-2368fje. [DOI] [PubMed] [Google Scholar]
- 248.Jiang F., Roberts S.J., Datla S., Dusting G.J. NO modulates NADPH oxidase function via heme oxygenase-1 in human endothelial cells. Hypertension. 2006;48:950–957. doi: 10.1161/01.HYP.0000242336.58387.1f. [DOI] [PubMed] [Google Scholar]
- 249.Matsumoto H., Ishikawa K., Itabe H., Maruyama Y. Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol. Cell. Biochem. 2006;291:21–28. doi: 10.1007/s11010-006-9190-y. [DOI] [PubMed] [Google Scholar]
- 250.McDonagh A.F. The biliverdin-bilirubin antioxidant cycle of cellular protection: Missing a wheel? Free Radic. Biol. Med. 2010;49:814–820. doi: 10.1016/j.freeradbiomed.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 251.Jomova K., Valko M. Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr. Pharm. Des. 2011;17:3460–3473. doi: 10.2174/138161211798072463. [DOI] [PubMed] [Google Scholar]
- 252.Harrison P.M., Arosio P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- 253.Torti F.M., Torti S.V. Regulation of ferritin genes and protein. Blood. 2002;99:3505–3516. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- 254.Orino K., Watanabe K. Molecular, physiological and clinical aspects of the iron storage protein ferritin. Vet. J. 2008;178:191–201. doi: 10.1016/j.tvjl.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 255.Villacorta L., Azzi A., Zingg J.M. Regulatory role of vitamins E and C on extracellular matrix components of the vascular system. Mol. Asp. Med. 2007;28:507–537. doi: 10.1016/j.mam.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 256.Fan J., Frey R.S., Rahman A., Malik A.B. Role of neutrophil NADPH oxidase in the mechanism of tumor necrosis factor-alpha -induced NF-kappa B activation and intercellular adhesion molecule-1 expression in endothelial cells. J. Biol. Chem. 2002;277:3404–3411. doi: 10.1074/jbc.M110054200. [DOI] [PubMed] [Google Scholar]
- 257.Kono H., Rusyn I., Yin M., Gabele E., Yamashina S., Dikalova A., Kadiiska M.B., Connor H.D., Mason R.P., Segal B.H., et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J. Clin. Investig. 2000;106:867–872. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Sadikot R.T., Zeng H., Yull F.E., Li B., Cheng D.S., Kernodle D.S., Jansen E.D., Contag C.H., Segal B.H., Holland S.M., et al. p47phox deficiency impairs NF-kappa B activation and host defense in Pseudomonas pneumonia. J. Immunol. 2004;172:1801–1808. doi: 10.4049/jimmunol.172.3.1801. [DOI] [PubMed] [Google Scholar]
- 259.Sato K., Kadiiska M.B., Ghio A.J., Corbett J., Fann Y.C., Holland S.M., Thurman R.G., Mason R.P. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: A model for ARDS. FASEB J. 2002;16:1713–1720. doi: 10.1096/fj.02-0331com. [DOI] [PubMed] [Google Scholar]
- 260.Rahman A., Kefer J., Bando M., Niles W.D., Malik A.B. E-selectin expression in human endothelial cells by TNF-alpha-induced oxidant generation and NF-kappaB activation. Am. J. Physiol. 1998;275:L533–L544. doi: 10.1152/ajplung.1998.275.3.L533. [DOI] [PubMed] [Google Scholar]
- 261.Li J.M., Fan L.M., Christie M.R., Shah A.M. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: Role of p47phox phosphorylation and binding to TRAF4. Mol. Cell. Biol. 2005;25:2320–2330. doi: 10.1128/MCB.25.6.2320-2330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Gu Y., Xu Y.C., Wu R.F., Souza R.F., Nwariaku F.E., Terada L.S. TNFalpha activates c-Jun amino terminal kinase through p47(phox) Exp. Cell Res. 2002;272:62–74. doi: 10.1006/excr.2001.5404. [DOI] [PubMed] [Google Scholar]
- 263.Lambeth J.D., Krause K.H., Clark R.A. NOX enzymes as novel targets for drug development. Semin. Immunopathol. 2008;30:339–363. doi: 10.1007/s00281-008-0123-6. [DOI] [PubMed] [Google Scholar]
- 264.Jaquet V., Scapozza L., Clark R.A., Krause K.H., Lambeth J.D. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid. Redox Signal. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 265.Zhang W.J., Wei H., Frei B. Genetic deficiency of NADPH oxidase does not diminish, but rather enhances, LPS-induced acute inflammatory responses in vivo. Free Radic. Biol. Med. 2009;46:791–798. doi: 10.1016/j.freeradbiomed.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Marriott H.M., Jackson L.E., Wilkinson T.S., Simpson A.J., Mitchell T.J., Buttle D.J., Cross S.S., Ince P.G., Hellewell P.G., Whyte M.K., et al. Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 2008;177:887–895. doi: 10.1164/rccm.200707-990OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Liochev S.I., Fridovich I. The role of O2.– in the production of HO.: In vitro and in vivo. Free Radic. Biol. Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 268.Kakhlon O., Cabantchik Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes(1) Free Radic. Biol. Med. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 269.Fleming R.E., Ponka P. Iron overload in human disease. N. Engl. J. Med. 2012;366:348–359. doi: 10.1056/NEJMra1004967. [DOI] [PubMed] [Google Scholar]
- 270.Larsen R., Gouveia Z., Soares M.P., Gozzelino R. Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front. Pharmacol. 2012;3:77. doi: 10.3389/fphar.2012.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Kell D.B. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch. Toxicol. 2010;84:825–889. doi: 10.1007/s00204-010-0577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Perron N.R., Brumaghim J.L. A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem. Biophys. 2009;53:75–100. doi: 10.1007/s12013-009-9043-x. [DOI] [PubMed] [Google Scholar]
- 273.Sestili P., Guidarelli A., Dacha M., Cantoni O. Quercetin prevents DNA single strand breakage and cytotoxicity caused by tert-butylhydroperoxide: Free radical scavenging versus iron chelating mechanism. Free Radic. Biol. Med. 1998;25:196–200. doi: 10.1016/s0891-5849(98)00040-9. [DOI] [PubMed] [Google Scholar]
- 274.Fukai T., Ushio-Fukai M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Marklund S.L. Extracellular superoxide dismutase and other superoxide dismutase isoenzymes in tissues from nine mammalian species. Biochem. J. 1984;222:649–655. doi: 10.1042/bj2220649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Moysan A., Marquis I., Gaboriau F., Santus R., Dubertret L., Morliere P. Ultraviolet A-induced lipid peroxidation and antioxidant defense systems in cultured human skin fibroblasts. J. Investig. Dermatol. 1993;100:692–698. doi: 10.1111/1523-1747.ep12472352. [DOI] [PubMed] [Google Scholar]
- 277.Poswig A., Wenk J., Brenneisen P., Wlaschek M., Hommel C., Quel G., Faisst K., Dissemond J., Briviba K., Krieg T., et al. Adaptive antioxidant response of manganese-superoxide dismutase following repetitive UVA irradiation. J. Investig. Dermatol. 1999;112:13–18. doi: 10.1046/j.1523-1747.1999.00465.x. [DOI] [PubMed] [Google Scholar]
- 278.Choung B.Y., Byun S.J., Suh J.G., Kim T.Y. Extracellular superoxide dismutase tissue distribution and the patterns of superoxide dismutase mRNA expression following ultraviolet irradiation on mouse skin. Exp. Dermatol. 2004;13:691–699. doi: 10.1111/j.0906-6705.2004.00209.x. [DOI] [PubMed] [Google Scholar]
- 279.Delanian S., Baillet F., Huart J., Lefaix J.L., Maulard C., Housset M. Successful treatment of radiation-induced fibrosis using liposomal Cu/Zn superoxide dismutase: Clinical trial. Radiother. Oncol. 1994;32:12–20. doi: 10.1016/0167-8140(94)90444-8. [DOI] [PubMed] [Google Scholar]
- 280.Lefaix J.L., Delanian S., Leplat J.J., Tricaud Y., Martin M., Nimrod A., Baillet F., Daburon F. Successful treatment of radiation-induced fibrosis using Cu/Zn-SOD and Mn-SOD: An experimental study. Int. J. Radiat. Oncol. Biol. Phys. 1996;35:305–312. doi: 10.1016/0360-3016(96)00061-2. [DOI] [PubMed] [Google Scholar]
- 281.Luo J.D., Wang Y.Y., Fu W.L., Wu J., Chen A.F. Gene therapy of endothelial nitric oxide synthase and manganese superoxide dismutase restores delayed wound healing in type 1 diabetic mice. Circulation. 2004;110:2484–2493. doi: 10.1161/01.CIR.0000137969.87365.05. [DOI] [PubMed] [Google Scholar]
- 282.Ceradini D.J., Yao D., Grogan R.H., Callaghan M.J., Edelstein D., Brownlee M., Gurtner G.C. Decreasing intracellular superoxide corrects defective ischemia-induced new vessel formation in diabetic mice. J. Biol. Chem. 2008;283:10930–10938. doi: 10.1074/jbc.M707451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Ha H.Y., Kim Y., Ryoo Z.Y., Kim T.Y. Inhibition of the TPA-induced cutaneous inflammation and hyperplasia by EC-SOD. Biochem. Biophys. Res. Commun. 2006;348:450–458. doi: 10.1016/j.bbrc.2006.07.079. [DOI] [PubMed] [Google Scholar]
- 284.Kim Y., Kim B.H., Lee H., Jeon B., Lee Y.S., Kwon M.J., Kim T.Y. Regulation of skin inflammation and angiogenesis by EC-SOD via HIF-1alpha and NF-kappaB pathways. Free Radic. Biol. Med. 2011;51:1985–1995. doi: 10.1016/j.freeradbiomed.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 285.Lee Y.S., Cheon I.S., Kim B.H., Kwon M.J., Lee H.W., Kim T.Y. Loss of extracellular superoxide dismutase induces severe IL-23-mediated skin inflammation in mice. J. Investig. Dermatol. 2013;133:732–741. doi: 10.1038/jid.2012.406. [DOI] [PubMed] [Google Scholar]
- 286.Kwon M.J., Jeon Y.J., Lee K.Y., Kim T.Y. Superoxide dismutase 3 controls adaptive immune responses and contributes to the inhibition of ovalbumin-induced allergic airway inflammation in mice. Antioxid. Redox Signal. 2012;17:1376–1392. doi: 10.1089/ars.2012.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Nozik-Grayck E., Suliman H.B., Piantadosi C.A. Extracellular superoxide dismutase. Int. J. Biochem. Cell Biol. 2005;37:2466–2471. doi: 10.1016/j.biocel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 288.Qin Z., Reszka K.J., Fukai T., Weintraub N.L. Extracellular superoxide dismutase (ecSOD) in vascular biology: An update on exogenous gene transfer and endogenous regulators of ecSOD. Transl. Res. 2008;151:68–78. doi: 10.1016/j.trsl.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Carlsson L.M., Marklund S.L., Edlund T. The rat extracellular superoxide dismutase dimer is converted to a tetramer by the exchange of a single amino acid. Proc. Natl. Acad. Sci. USA. 1996;93:5219–5222. doi: 10.1073/pnas.93.11.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Yamaguchi M., Zhou C., Heistad D.D., Watanabe Y., Zhang J.H. Gene transfer of extracellular superoxide dismutase failed to prevent cerebral vasospasm after experimental subarachnoid hemorrhage. Stroke. 2004;35:2512–2517. doi: 10.1161/01.STR.0000145198.07723.8e. [DOI] [PubMed] [Google Scholar]
- 291.Batinic-Haberle I., Reboucas J.S., Spasojevic I. Superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Signal. 2010;13:877–918. doi: 10.1089/ars.2009.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Day B.J. Catalase and glutathione peroxidase mimics. Biochem. Pharmacol. 2009;77:285–296. doi: 10.1016/j.bcp.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Doctrow S.R., Huffman K., Marcus C.B., Tocco G., Malfroy E., Adinolfi C.A., Kruk H., Baker K., Lazarowych N., Mascarenhas J., et al. Salen-manganese complexes as catalytic scavengers of hydrogen peroxide and cytoprotective agents: Structure-activity relationship studies. J. Med. Chem. 2002;45:4549–4558. doi: 10.1021/jm020207y. [DOI] [PubMed] [Google Scholar]
- 294.Day B.J. Catalytic antioxidants: A radical approach to new therapeutics. Drug Discov. Today. 2004;9:557–566. doi: 10.1016/S1359-6446(04)03139-3. [DOI] [PubMed] [Google Scholar]
- 295.Rong Y., Doctrow S.R., Tocco G., Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc. Natl. Acad. Sci. USA. 1999;96:9897–9902. doi: 10.1073/pnas.96.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Liu R., Liu I.Y., Bi X., Thompson R.F., Doctrow S.R., Malfroy B., Baudry M. Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc. Natl. Acad. Sci. USA. 2003;100:8526–8531. doi: 10.1073/pnas.1332809100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Zhang H.J., Doctrow S.R., Xu L., Oberley L.W., Beecher B., Morrison J., Oberley T.D., Kregel K.C. Redox modulation of the liver with chronic antioxidant enzyme mimetic treatment prevents age-related oxidative damage associated with environmental stress. FASEB J. 2004;18:1547–1549. doi: 10.1096/fj.04-1629fje. [DOI] [PubMed] [Google Scholar]
- 298.Clausen A., Doctrow S., Baudry M. Prevention of cognitive deficits and brain oxidative stress with superoxide dismutase/catalase mimetics in aged mice. Neurobiol. Aging. 2010;31:425–433. doi: 10.1016/j.neurobiolaging.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Liesa M., Luptak I., Qin F., Hyde B.B., Sahin E., Siwik D.A., Zhu Z., Pimentel D.R., Xu X.J., Ruderman N.B., et al. Mitochondrial transporter ATP binding cassette mitochondrial erythroid is a novel gene required for cardiac recovery after ischemia/reperfusion. Circulation. 2011;124:806–813. doi: 10.1161/CIRCULATIONAHA.110.003418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Mahmood J., Jelveh S., Calveley V., Zaidi A., Doctrow S.R., Hill R.P. Mitigation of radiation-induced lung injury by genistein and EUK-207. Int. J. Radiat. Biol. 2011;87:889–901. doi: 10.3109/09553002.2011.583315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Doctrow S.R., Lopez A., Schock A.M., Duncan N.E., Jourdan M.M., Olasz E.B., Moulder J.E., Fish B.L., Mader M., Lazar J., et al. A synthetic superoxide dismutase/catalase mimetic EUK-207 mitigates radiation dermatitis and promotes wound healing in irradiated rat skin. J. Investig. Dermatol. 2013;133:1088–1096. doi: 10.1038/jid.2012.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Soares M.P., Bach F.H. Heme oxygenase-1: From biology to therapeutic potential. Trends Mol. Med. 2009;15:50–58. doi: 10.1016/j.molmed.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 303.Saikawa Y., Kaneda H., Yue L., Shimura S., Toma T., Kasahara Y., Yachie A., Koizumi S. Structural evidence of genomic exon-deletion mediated by Alu-Alu recombination in a human case with heme oxygenase-1 deficiency. Hum. Mutat. 2000;16:178–179. doi: 10.1002/1098-1004(200008)16:2<178::AID-HUMU16>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 304.Radhakrishnan N., Yadav S.P., Sachdeva A., Pruthi P.K., Sawhney S., Piplani T., Wada T., Yachie A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol./Oncol. 2011;33:74–78. doi: 10.1097/MPH.0b013e3181fd2aae. [DOI] [PubMed] [Google Scholar]
- 305.Exner M., Minar E., Wagner O., Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic. Biol. Med. 2004;37:1097–1104. doi: 10.1016/j.freeradbiomed.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 306.Kappas A., Drummond G.S. Control of heme and cytochrome P-450 metabolism by inorganic metals, organometals and synthetic metalloporphyrins. Environ. Health Perspect. 1984;57:301–306. doi: 10.1289/ehp.8457301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Sasaki T., Takahashi T., Maeshima K., Shimizu H., Toda Y., Morimatsu H., Takeuchi M., Yokoyama M., Akagi R., Morita K. Heme arginate pretreatment attenuates pulmonary NF-kappaB and AP-1 activation induced by hemorrhagic shock via heme oxygenase-1 induction. Med. Chem. 2006;2:271–274. doi: 10.2174/157340606776930781. [DOI] [PubMed] [Google Scholar]
- 308.Tenhunen R., Mustajoki P. Acute porphyria: Treatment with heme. Semin. Liver Dis. 1998;18:53–55. doi: 10.1055/s-2007-1007140. [DOI] [PubMed] [Google Scholar]
- 309.Barbagallo I., Galvano F., Frigiola A., Cappello F., Riccioni G., Murabito P., D’Orazio N., Torella M., Gazzolo D., Li Volti G. Potential therapeutic effects of natural heme oxygenase-1 inducers in cardiovascular diseases. Antioxid. Redox Signal. 2013;18:507–521. doi: 10.1089/ars.2011.4360. [DOI] [PubMed] [Google Scholar]
- 310.Motterlini R., Foresti R., Bassi R., Green C.J. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic. Biol. Med. 2000;28:1303–1312. doi: 10.1016/s0891-5849(00)00294-x. [DOI] [PubMed] [Google Scholar]
- 311.Balogun E., Hoque M., Gong P., Killeen E., Green C.J., Foresti R., Alam J., Motterlini R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003;371:887–895. doi: 10.1042/BJ20021619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.McNally S.J., Harrison E.M., Ross J.A., Garden O.J., Wigmore S.J. Curcumin induces heme oxygenase 1 through generation of reactive oxygen species, p38 activation and phosphatase inhibition. Int. J. Mol. Med. 2007;19:165–172. [PubMed] [Google Scholar]
- 313.Lin H.C., Cheng T.H., Chen Y.C., Juan S.H. Mechanism of heme oxygenase-1 gene induction by quercetin in rat aortic smooth muscle cells. Pharmacology. 2004;71:107–112. doi: 10.1159/000076947. [DOI] [PubMed] [Google Scholar]
- 314.Yao P., Nussler A., Liu L., Hao L., Song F., Schirmeier A., Nussler N. Quercetin protects human hepatocytes from ethanol-derived oxidative stress by inducing heme oxygenase-1 via the MAPK/Nrf2 pathways. J. Hepatol. 2007;47:253–261. doi: 10.1016/j.jhep.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 315.Wannamethee S.G., Lowe G.D., Rumley A., Bruckdorfer K.R., Whincup P.H. Associations of vitamin C status, fruit and vegetable intakes, and markers of inflammation and hemostasis. Am. J. Clin. Nutr. 2006;83:567–574. doi: 10.1093/ajcn.83.3.567. ; quiz 726–567. [DOI] [PubMed] [Google Scholar]
- 316.Homma S., Azuma A., Taniguchi H., Ogura T., Mochiduki Y., Sugiyama Y., Nakata K., Yoshimura K., Takeuchi M., Kudoh S., et al. Efficacy of inhaled N-acetylcysteine monotherapy in patients with early stage idiopathic pulmonary fibrosis. Respirology. 2012;17:467–477. doi: 10.1111/j.1440-1843.2012.02132.x. [DOI] [PubMed] [Google Scholar]
- 317.Demedts M., Behr J., Buhl R., Costabel U., Dekhuijzen R., Jansen H.M., MacNee W., Thomeer M., Wallaert B., Laurent F., et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2005;353:2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- 318.Tomioka H., Kuwata Y., Imanaka K., Hashimoto K., Ohnishi H., Tada K., Sakamoto H., Iwasaki H. A pilot study of aerosolized N-acetylcysteine for idiopathic pulmonary fibrosis. Respirology. 2005;10:449–455. doi: 10.1111/j.1440-1843.2005.00725.x. [DOI] [PubMed] [Google Scholar]
- 319.Behr J., Demedts M., Buhl R., Costabel U., Dekhuijzen R.P., Jansen H.M., MacNee W., Thomeer M., Wallaert B., Laurent F., et al. Lung function in idiopathic pulmonary fibrosis—Extended analyses of the IFIGENIA trial. Respir. Res. 2009;10:101. doi: 10.1186/1465-9921-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Crestani B., Besnard V., Boczkowski J. Signalling pathways from NADPH oxidase-4 to idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2011;43:1086–1089. doi: 10.1016/j.biocel.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 321.Atkuri K.R., Mantovani J.J., Herzenberg L.A., Herzenberg L.A. N-Acetylcysteine—A safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 2007;7:355–359. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Idiopathic Pulmonary Fibrosis Clinical Research, N. Raghu G., Anstrom K.J., King T.E., Jr, Lasky J.A., Martinez F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012;366:1968–1977. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Schagen S.K., Zampeli V.A., Makrantonaki E., Zouboulis C.C. Discovering the link between nutrition and skin aging. Dermato-Endocrinol. 2012;4:298–307. doi: 10.4161/derm.22876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Placzek M., Gaube S., Kerkmann U., Gilbertz K.P., Herzinger T., Haen E., Przybilla B. Ultraviolet B-induced DNA damage in human epidermis is modified by the antioxidants ascorbic acid and D-alpha-tocopherol. J. Investig. Dermatol. 2005;124:304–307. doi: 10.1111/j.0022-202X.2004.23560.x. [DOI] [PubMed] [Google Scholar]
- 325.Morganti P., Bruno C., Guarneri F., Cardillo A., Del Ciotto P., Valenzano F. Role of topical and nutritional supplement to modify the oxidative stress. Int. J. Cosmet. Sci. 2002;24:331–339. doi: 10.1046/j.1467-2494.2002.00159.x. [DOI] [PubMed] [Google Scholar]
- 326.Eberlein-Konig B., Ring J. Relevance of vitamins C and E in cutaneous photoprotection. J. Cosmet. Dermatol. 2005;4:4–9. doi: 10.1111/j.1473-2165.2005.00151.x. [DOI] [PubMed] [Google Scholar]
- 327.Stahl W., Heinrich U., Jungmann H., Sies H., Tronnier H. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am. J. Clin. Nutr. 2000;71:795–798. doi: 10.1093/ajcn/71.3.795. [DOI] [PubMed] [Google Scholar]
- 328.Lee J., Jiang S., Levine N., Watson R.R. Carotenoid supplementation reduces erythema in human skin after simulated solar radiation exposure. Proc. Soc. Exp. Biol. Med. 2000;223:170–174. doi: 10.1046/j.1525-1373.2000.22323.x. [DOI] [PubMed] [Google Scholar]
- 329.Heinrich U., Gartner C., Wiebusch M., Eichler O., Sies H., Tronnier H., Stahl W. Supplementation with beta-carotene or a similar amount of mixed carotenoids protects humans from UV-induced erythema. J. Nutr. 2003;133:98–101. doi: 10.1093/jn/133.1.98. [DOI] [PubMed] [Google Scholar]
- 330.Telfer A., Dhami S., Bishop S.M., Phillips D., Barber J. beta-Carotene quenches singlet oxygen formed by isolated photosystem II reaction centers. Biochemistry. 1994;33:14469–14474. doi: 10.1021/bi00252a013. [DOI] [PubMed] [Google Scholar]
- 331.Bose B., Chatterjee S.N. UVA-induced peroxidation of lipid in the dried film state. J. Photochem. Photobiol. B. 1994;23:119–123. doi: 10.1016/1011-1344(94)06995-6. [DOI] [PubMed] [Google Scholar]
- 332.Eicker J., Kurten V., Wild S., Riss G., Goralczyk R., Krutmann J., Berneburg M. Betacarotene supplementation protects from photoaging-associated mitochondrial DNA mutation. Photochem. Photobiol. Sci. 2003;2:655–659. doi: 10.1039/b300808h. [DOI] [PubMed] [Google Scholar]
- 333.Manach C., Scalbert A., Morand C., Remesy C., Jimenez L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004;79:727–747. doi: 10.1093/ajcn/79.5.727. [DOI] [PubMed] [Google Scholar]
- 334.Nichols J.A., Katiyar S.K. Skin photoprotection by natural polyphenols: Anti-inflammatory, antioxidant and DNA repair mechanisms. Arch. Dermatol. Res. 2010;302:71–83. doi: 10.1007/s00403-009-1001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Agarwal R., Katiyar S.K., Khan S.G., Mukhtar H. Protection against ultraviolet B radiation-induced effects in the skin of SKH-1 hairless mice by a polyphenolic fraction isolated from green tea. Photochem. Photobiol. 1993;58:695–700. doi: 10.1111/j.1751-1097.1993.tb04954.x. [DOI] [PubMed] [Google Scholar]
- 336.Katiyar S.K., Elmets C.A., Agarwal R., Mukhtar H. Protection against ultraviolet-B radiation-induced local and systemic suppression of contact hypersensitivity and edema responses in C3H/HeN mice by green tea polyphenols. Photochem. Photobiol. 1995;62:855–861. doi: 10.1111/j.1751-1097.1995.tb09147.x. [DOI] [PubMed] [Google Scholar]
- 337.Elmets C.A., Singh D., Tubesing K., Matsui M., Katiyar S., Mukhtar H. Cutaneous photoprotection from ultraviolet injury by green tea polyphenols. J. Am. Acad. Dermatol. 2001;44:425–432. doi: 10.1067/mjd.2001.112919. [DOI] [PubMed] [Google Scholar]
- 338.Katiyar S.K., Elmets C.A. Green tea polyphenolic antioxidants and skin photoprotection (Review) Int. J. Oncol. 2001;18:1307–1313. doi: 10.3892/ijo.18.6.1307. [DOI] [PubMed] [Google Scholar]
- 339.Van Laethem A., Claerhout S., Garmyn M., Agostinis P. The sunburn cell: Regulation of death and survival of the keratinocyte. Int. J. Biochem. Cell Biol. 2005;37:1547–1553. doi: 10.1016/j.biocel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 340.Lambert J.D., Hong J., Yang G.Y., Liao J., Yang C.S. Inhibition of carcinogenesis by polyphenols: Evidence from laboratory investigations. Am. J. Clin. Nutr. 2005;81:284S–291S. doi: 10.1093/ajcn/81.1.284S. [DOI] [PubMed] [Google Scholar]
- 341.Shindo Y., Witt E., Han D., Epstein W., Packer L. Enzymic and non-enzymic antioxidants in epidermis and dermis of human skin. J. Investig. Dermatol. 1994;102:122–124. doi: 10.1111/1523-1747.ep12371744. [DOI] [PubMed] [Google Scholar]
- 342.Kwong L.K., Kamzalov S., Rebrin I., Bayne A.C., Jana C.K., Morris P., Forster M.J., Sohal R.S. Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic. Biol. Med. 2002;33:627–638. doi: 10.1016/s0891-5849(02)00916-4. [DOI] [PubMed] [Google Scholar]
- 343.Sohal R.S., Kamzalov S., Sumien N., Ferguson M., Rebrin I., Heinrich K.R., Forster M.J. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic. Biol. Med. 2006;40:480–487. doi: 10.1016/j.freeradbiomed.2005.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Prahl S., Kueper T., Biernoth T., Wohrmann Y., Munster A., Furstenau M., Schmidt M., Schulze C., Wittern K.P., Wenck H., et al. Aging skin is functionally anaerobic: Importance of coenzyme Q10 for anti aging skin care. BioFactors. 2008;32:245–255. doi: 10.1002/biof.5520320129. [DOI] [PubMed] [Google Scholar]
- 345.Blatt T., Lenz H., Koop U., Jaspers S., Weber T., Mummert C., Wittern K.P., Stab F., Wenck H. Stimulation of skin’s energy metabolism provides multiple benefits for mature human skin. BioFactors. 2005;25:179–185. doi: 10.1002/biof.5520250121. [DOI] [PubMed] [Google Scholar]
- 346.Blatt T., Littarru G.P. Biochemical rationale and experimental data on the antiaging properties of CoQ(10) at skin level. BioFactors. 2011;37:381–385. doi: 10.1002/biof.169. [DOI] [PubMed] [Google Scholar]
- 347.Makrantonaki E., Zouboulis C.C. Skin alterations and diseases in advanced age. Drug Discov. Today Dis. Mech. 2008;5:153–162. [Google Scholar]
- 348.Han R., Liu L., Li J., Du G., Chen J. Functions, applications and production of 2-O-D-glucopyranosyl-L-ascorbic acid. Appl. Microbiol. Biotech. 2012;95:313–320. doi: 10.1007/s00253-012-4150-9. [DOI] [PubMed] [Google Scholar]
- 349.Pawar R.S., Tamta H., Ma J., Krynitsky A.J., Grundel E., Wamer W.G., Rader J.I. Updates on chemical and biological research on botanical ingredients in dietary supplements. Anal. Bioanal. Chem. 2013 doi: 10.1007/s00216-012-6691-2. [DOI] [PubMed] [Google Scholar]
- 350.Ristow M., Zarse K., Oberbach A., Kloting N., Birringer M., Kiehntopf M., Stumvoll M., Kahn C.R., Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Stanner S.A., Hughes J., Kelly C.N., Buttriss J. A review of the epidemiological evidence for the ‘antioxidant hypothesis’. Public Health Nutr. 2004;7:407–422. doi: 10.1079/phn2003543. [DOI] [PubMed] [Google Scholar]
- 352.Marik P.E., Flemmer M. Do dietary supplements have beneficial health effects in industrialized nations: What is the evidence? J. Parenteral Enteral Nutr. 2012;36:159–168. doi: 10.1177/0148607111416485. [DOI] [PubMed] [Google Scholar]
- 353.Mursu J., Robien K., Harnack L.J., Park K., Jacobs D.R., Jr Dietary supplements and mortality rate in older women: The Iowa Women’s Health Study. Arch. Intern. Med. 2011;171:1625–1633. doi: 10.1001/archinternmed.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Sedlak T.W., Saleh M., Higginson D.S., Paul B.D., Juluri K.R., Snyder S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA. 2009;106:5171–5176. doi: 10.1073/pnas.0813132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Dekker D., Dorresteijn M.J., Pijnenburg M., Heemskerk S., Rasing-Hoogveld A., Burger D.M., Wagener F.A., Smits P. The bilirubin-increasing drug atazanavir improves endothelial function in patients with type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2011;31:458–463. doi: 10.1161/ATVBAHA.110.211789. [DOI] [PubMed] [Google Scholar]
- 356.Scheffler I.E. Mitochondria make a come back. Advan. Drug Delivery Rev. 2001;49:3–26. doi: 10.1016/s0169-409x(01)00123-5. [DOI] [PubMed] [Google Scholar]
- 357.Davis R.E., Williams M. Mitochondrial function and dysfunction: An update. J. Pharmacol. Exp. Ther. 2012;342:598–607. doi: 10.1124/jpet.112.192104. [DOI] [PubMed] [Google Scholar]
- 358.Shults C.W., Oakes D., Kieburtz K., Beal M.F., Haas R., Plumb S., Juncos J.L., Nutt J., Shoulson I., Carter J., et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002;59:1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- 359.Shults C.W., Flint Beal M., Song D., Fontaine D. Pilot trial of high dosages of coenzyme Q10 in patients with Parkinson’s disease. Exp. Neurol. 2004;188:491–494. doi: 10.1016/j.expneurol.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 360.Ernster L., Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. 1995;1271:195–204. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 361.Zhang Y., Aberg F., Appelkvist E.L., Dallner G., Ernster L. Uptake of dietary coenzyme Q supplement is limited in rats. J. Nutr. 1995;125:446–453. doi: 10.1093/jn/125.3.446. [DOI] [PubMed] [Google Scholar]
- 362.Matthews R.T., Yang L., Browne S., Baik M., Beal M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA. 1998;95:8892–8897. doi: 10.1073/pnas.95.15.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Cocheme H.M., Kelso G.F., James A.M., Ross M.F., Trnka J., Mahendiran T., Asin-Cayuela J., Blaikie F.H., Manas A.R., Porteous C.M., et al. Mitochondrial targeting of quinones: Therapeutic implications. Mitochondrion. 2007;7:S94–S102. doi: 10.1016/j.mito.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 364.Kelso G.F., Porteous C.M., Coulter C.V., Hughes G., Porteous W.K., Ledgerwood E.C., Smith R.A., Murphy M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 365.Adlam V.J., Harrison J.C., Porteous C.M., James A.M., Smith R.A., Murphy M.P., Sammut I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 366.Murphy M.P., Smith R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Ann. Rev. Pharmacol. Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 367.Skulachev V.P. A biochemical approach to the problem of aging: “Megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry (Mosc.) 2007;72:1385–1396. doi: 10.1134/s0006297907120139. [DOI] [PubMed] [Google Scholar]
- 368.Antonenko Y.N., Roginsky V.A., Pashkovskaya A.A., Rokitskaya T.I., Kotova E.A., Zaspa A.A., Chernyak B.V., Skulachev V.P. Protective effects of mitochondria-targeted antioxidant SkQ in aqueous and lipid membrane environments. J. Membr. Biol. 2008;222:141–149. doi: 10.1007/s00232-008-9108-6. [DOI] [PubMed] [Google Scholar]
- 369.Distelmaier F., Valsecchi F., Forkink M., van Emst-de Vries S., Swarts H.G., Rodenburg R.J., Verwiel E.T., Smeitink J.A., Willems P.H., Koopman W.J. Trolox-sensitive reactive oxygen species regulate mitochondrial morphology, oxidative phosphorylation and cytosolic calcium handling in healthy cells. Antioxid. Redox Signal. 2012;17:1657–1669. doi: 10.1089/ars.2011.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Marrache S., Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc. Natl. Acad. Sci. USA. 2012;109:16288–16293. doi: 10.1073/pnas.1210096109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Bindu S., Pal C., Dey S., Goyal M., Alam A., Iqbal M.S., Dutta S., Sarkar S., Kumar R., Maity P., et al. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J. Biol. Chem. 2011;286:39387–39402. doi: 10.1074/jbc.M111.279893. [DOI] [PMC free article] [PubMed] [Google Scholar]