

Abstract
The Eph–ephrin system, including the EphA2 receptor and the ephrin-A1 ligand, plays a critical role in tumor and vascular functions during carcinogenesis. We previously identified (3α,5β)-3-hydroxycholan-24-oic acid (lithocholic acid) as an Eph-ephrin antagonist able to inhibit EphA2 receptor activation and therefore potentially useful as a novel EphA2 receptor targeting agent. Here, we explore the structure-activity relationships of a focused set of lithocholic acid derivatives, based on molecular modelling investigation and displacement binding assays. Our exploration shows that while the 3-α-hydroxyl group of lithocholic acid has a negligible role in the recognition of the EphA2 receptor, its carboxylate group is critical for disrupting the binding of ephrin-A1 to the EphA2. As a result of our investigation, we identified (5β)-cholan-24-oic acid (cholanic acid) as a novel compound that competitively inhibits EphA2-ephrin-A1 interaction with higher potency than lithocholic acid. Surface plasmon resonance analysis indicates that cholanic acid binds specifically and reversibly to the ligand-binding domain of EphA2, with a steady-state dissociation constant (KD) in the low micromolar range. Furthermore, cholanic acid blocks the phosphorylation of EphA2 and cell retraction and rounding in PC3 prostate cancer cells, two effects that depend on EphA2 activation by the ephrin-A1 ligand. These findings suggest that cholanic acid can be used as a template structure to design effective EphA2 antagonists, with potential impact in the elucidation of the role played by this receptor in pathological conditions.
Keywords: Protein-protein interactions, Structure-activity relationships, Surface plasmon resonance, Steroids, Drug design
Introduction
The 14 erythropoietin-producing hepatocellular carcinoma (Eph) receptors represent the largest family of receptor tyrosine kinases. The Eph receptors and their 8 ephrin ligands are divided into two subclasses (A and B) depending on their affinities for each other and sequence homology. Generally, EphA receptors (EphA1–A8 and EphA10) bind to glycosylphosphatidylinositol-anchored ephrin-A ligands (ephrin-A1–A5), while the EphB receptors (EphB1–B4 and EphB6) interact with transmembrane ephrin-B ligands (ephrin-B1–B3), which have a short cytoplasmic domain.[1]
Eph A and B receptors have a similar modular structure, consisting of a globular N-terminal ephrin-binding domain followed by a cysteine-rich region and two fibronectin type III repeats in the extracellular region. The intracellular region is composed of a juxtamembrane segment, a conserved tyrosine kinase domain, responsible for the signal transduction, a sterile alpha motif (SAM) domain and a PDZ-binding motif, which serves as a docking site for interacting signaling proteins.[2,3]
The result of the membrane localization of both ephrins and Eph receptors is their ability to transduce “reverse” signals into the cells in which the ephrins are expressed, in addition to “forward” signal into Eph receptor-expressing cells. As a consequence, the Eph-ephrin signaling system is responsible for the modulation of several biological activities involving cellular contact both during embryonic development and in adult tissues. In fact, these proteins modulate cell movements in morphogenetic processes, such as gastrulation, segmentation, angiogenesis, axonal pathfinding and neural crest cell migration.[4,5] Moreover, in the adult they are involved in the maintenance of cellular architecture in various ephitelia[6] and play key roles in neural plasticity[7] and in the regeneration of the adult nervous system.[8]
Increasing evidence supports the notion that the Eph–ephrin system, including the EphA2 and EphB4 receptors, plays a critical role in tumor and vascular functions during carcinogenesis. In particular, EphA2 is over-expressed in many types of tumors, such as breast, prostate, urinary bladder, skin, lung, ovary and brain cancers[2] The modulation of EphA2 activity by recombinant proteins such as monoclonal antibodies or soluble EphA receptor-Fc fusion proteins, has been shown to block tumor growth, metastasis and angiogenic processes in animal models.[9] Moreover, genome-wide or kinome screens for somatic mutations in cancer have identified mutations in essentially all Eph receptors, suggesting that mutations that affect Eph receptor function play a role in cancer initiation or progression.[10-14] Therefore, the Eph-ephrin system is emerging as a novel target for the development of anti-tumorigenic and anti-angiogenic therapies.[15]
The development of small molecules capable of blocking the biological activity of EphA2 represents an attractive alternative to antibodies, peptides and recombinant proteins.[16-19] Few examples of EphA2 inhibitors targeting the intracellular kinase-domain have been recently reported in the literature.[20] As these compounds block EphA2 activity by occupying the ATP binding pocket, they suffer from lack of selectivity, which limits their use as pharmacological tools in vivo. Conversely, compounds acting on the extracellular ligand binding domain of the Eph receptors have some advantages with respect to standard tyrosine kinase inhibitors because they can block Eph receptor activity without having to penetrate inside the cell and because they have the potential to be more selective than ATP mimicking agents.[21]
The three-dimensional structure of the EphA2-ephrin-A1 complex has been recently resolved by X-ray crystallography.[22] The interaction between these two proteins is primarily mediated by the amino-terminal ligand-binding domain of EphA2, which forms a large hydrophobic cavity able to accommodate a protruding loop of ephrin-A1 (the G-H loop, Figure 1).[23] The binding interface is dominated by the Van der Waals contacts between two predominantly hydrophobic surfaces and reinforced by a few salt bridges, including the salt bridge between EphA2 Arg103 and ephrin-A1 Glu119 (Figure 1). Despite the large binding interfaces in the EphA2-ephrin-A1 complex, it has been shown that peptides of moderate size (12 amino acids) as well as small molecules, exemplified by salicylic acid derivatives such as compound 76D10 (Figure 2), can prevent Eph receptor-ephrin interaction possibly by occupying the same EphA2 receptor cavity as the G-H loop of the physiological ephrin ligands.[24]
Figure 1.
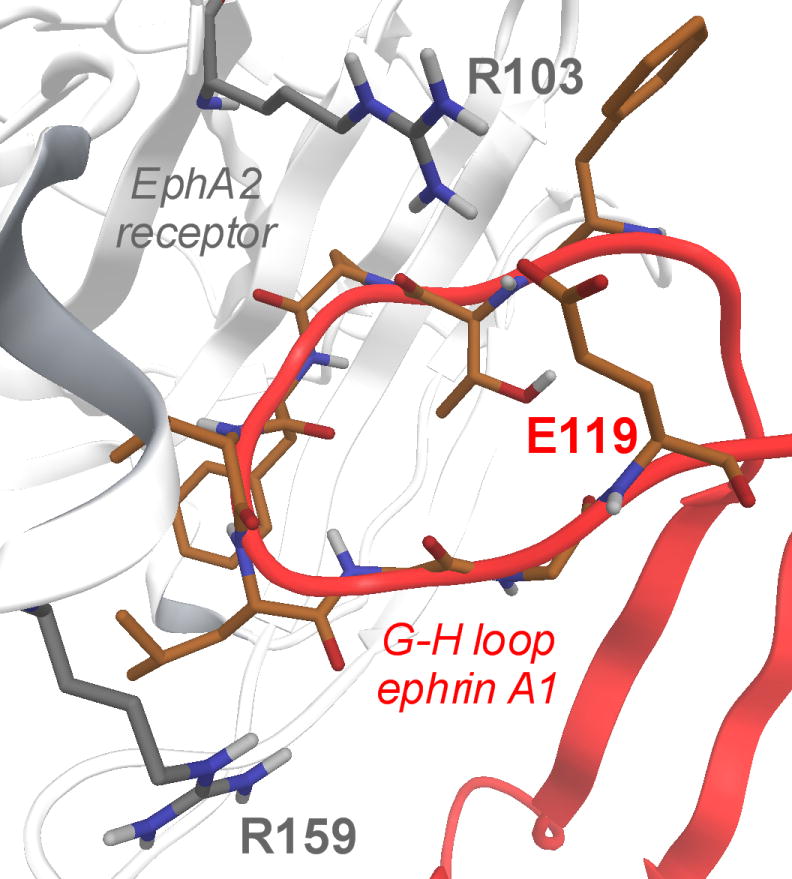
EphA2 ligand binding domain (white cartoon and gray carbon atoms) in complex with ephrin-A1 (red cartoons and orange carbon atoms). In evidence, the crucial salt bridge between R103 (EphA2) and E119 (ephrin-A1).
Figure 2.

Recently identified antagonists of the EphA2 receptor.
In our search for novel EphA2 receptor modulators, we have recently screened an “in house” chemical library of naturally occurring compounds, identifying the secondary bile acid (3α,5β)-3-hydroxycholan-24-oic acid (lithocholic acid, Figure 2) as a non-peptidic ligand of the Eph receptors.[25] Investigation of the mechanism of action of lithocholic acid revealed that this compound acts as a competitive antagonist of the EphA2 receptor (Ki = 49 ± 3 μM). Furthermore, functional experiments showed that lithocholic acid inhibits EphA2 autophosphorylation in a dose-dependent manner and blocks PC3 prostate cancer cell rounding and retraction induced by EphA2 stimulation with ephrin-A1. These results indicate that the lithocholic acid scaffold can be used to design effective EphA2 antagonist.
We now report the characterization of the structure-activity relationship (SAR) of (5β)-cholan-24-oic acid derivatives, leading to the identification of compounds with improved binding affinity. We performed molecular modelling studies to identify the putative binding mode of lithocholic acid (compound 1) within the high affinity ephrin binding pocket of the EphA2 receptor. Starting from this theoretical model, a focused set of lithocholic acid derivatives, commercially available or obtained by chemical synthesis, were examined for their ability to disrupt EphA2-ephrin-A1 binding. This led to the discovery of cholanic acid, which resulted more potent and selective than lithocholic acid in both EphA2 binding and inhibition of EphA2 phosphorylation assays.
Results and Discussion
Molecular modelling
The recent resolution of the crystal structure of the ligand-binding domain of the EphA2 receptor in complex with the ephrin-A1 ligand[22] allowed us to investigate the binding mode of lithocholic acid to EphA2 by docking and molecular dynamics simulations. The application of these computational techniques allows to generate working hypotheses on the recognition process involving a ligand and its receptor, helping the design of structural analogues.[26-30]
Figure 3A shows the best solution (in terms of interaction energy, see Experimental Section) obtained by docking lithocholic acid within the high affinity ephrin-binding pocket of the EphA2 receptor. The compound occupies the same space as the ephrin-A1 G–H loop, inserting its cyclopenta[a]perhydro phenanthrene scaffold into a hydrophobic Eph receptor channel. The pentanoic acid fragment, emerging from position 17 of the lithocholic acid core, forms a salt bridge with Arg103, mimicking the interaction of Glu119 from ephrin-A1. Finally, 3-hydroyxl group of lithocholic acid weakly interacts with Arg159 of EphA2, which is usually engaged in a hydrogen bond with Asp86 of ephrin-A1.
Figure 3.

a) Docking of lithocholic acid (LCA, green carbon atoms) in the high-affinity ephrin-binding pocket of the EphA2 receptor (white cartoons, gray carbon atoms). The G-H loop of ephrin-A1 is also displayed (red cartoons) b) EphA2-lithocholic acid complex as obtained at the end of the molecular dynamics simulation. Lithocholic acid (LCA) carbon atoms are in green, hydrogens in white, with the exception of those in position 7α and 12α, highlighted in pink. The alpha and beta faces of LCA are also highlighted. EphA2 carbon atoms are in gray, with white hydrogen atoms.
To evaluate the stability of the proposed binding mode, 30 nanoseconds of molecular dynamics were simulated starting from the structure shown in Figure 3A. After a few nanoseconds of simulation, the lithocholic acid left its initial position and moved deeper inside the EphA2 binding site pointing its alpha face towards the side chain of Phe156 (Figure 3B). Then the lithocholic acid hydrophobic core gave a stable oscillation around this position until the end of the simulation (Figure S1, supporting information), establishing Van der Waals contacts with the hydrophobic surface of the receptor through the beta methyl groups emerging from positions 18 and 19 of lithocholic acid (Figure 3B). These minor rearrangements of the lithocholic acid binding mode did not significantly affect the salt bridge formed by the carboxylic group of lithocholic acid with Arg103 of EphA2, which persisted throughout the simulation (Figure S2, supporting information). On the other hand, the 3-hydroxyl group lost its interaction with Arg159 already in the initial phase of the molecular dynamics simulation, suggesting that the contribution of this interaction to the binding affinity might be negligible.
The simulation also showed that the aromatic ring of Phe156 entered in close contact with the alpha hydrogens at position 7 and 12 of lithocholic acid (Figure 3B), suggesting that there is limited space for introducing bigger substituents at these positions. All together, these analyses suggest that the hydrophobic core of lithocholic acid can mimic the ephrin G-H loop in its interaction with the EphA2 binding site, with the carboxylic group of lithocholic acid being fundamental for EphA2 binding. Furthermore, the computational results also suggest that the alpha-hydroxyl group at position 3 might not be essential for the binding activity.
Structure-activity relationship data for lithocholic acid derivatives
Based on the computational results reported above, fifteen derivatives of lithocholic acid (compound 1) were chosen and tested for their ability to interfere with the EphA2-ephrin-A1 interaction (Table 1). Compounds 2-6 were selected to explore the interaction between the lipophilic scaffold of lithocholic acid and the EphA2 binding site. Compounds 7-12 and 13-16 were selected to examine the role played by the two polar ends of lithocholic acid. The experimental procedures employed to synthesize and characterize these compounds are reported in the experimental section.
Table 1. Structure-activity relationship data for lithocholic acid derivatives.
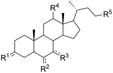
| ||||||
|---|---|---|---|---|---|---|
| Cpd. | R1 | R2 | R3 | R4 | R5 | Ki (μM)[a] |
| 1 |
|
|
|
|
|
49±3.0 |
| 2 |
|
|
|
|
|
>200 |
| 3 |
|
|
|
|
|
>200 |
| 4 |
|
|
|
|
|
>200 |
| 5 |
|
|
|
|
|
114±13 |
| 6 |
|
|
|
|
|
138±20 |
| 7 |
|
|
|
|
|
157±47 |
| 8 |
|
|
|
|
|
114±14 |
| 9 |
|
|
|
|
|
88±11 |
| 10 |
|
|
|
|
|
25±4.0 |
| 11 |
|
|
|
|
|
>200 |
| 12 |
|
|
|
|
|
5.1±1.4 |
| 13 |
|
|
|
|
|
>200 |
| 14 |
|
|
|
|
|
>200 |
| 15 |
|
|
|
|
|
>200 |
| 16 |
|
|
|
|
|
186±27 |
Values are means ± standard error from at least three independent experiments
The potencies for inhibition of EphA2-ephrin-A1 interaction, as indicated by the Ki values reported in Table 1, revealed that the lithocholic acid derivatives are particularly sensitive to the modulation of the cyclopenta[a]perhydro phenanthrene scaffold. Indeed, the introduction of an alpha-hydroxyl group in positions 7 or 12 always produces inactive compounds, as exemplified by the naturally occurring cholic acid (2), deoxycholic acid (3) and chenodeoxycholic acid (4).[25] Similarly, the introduction of a 6- or 7- keto group is detrimental to the binding affinity (compounds 5 and 6).
Modification of the two opposite ends of the hydrophobic core of lithocholic acid gave the following results. Oxidation of the alpha hydroxyl group at position 3 (compounds 7 and 8) and acetylation of this group (compound 9) yielded compounds with lower affinity compared to lithocholic acid (compound 1). By contrast, inversion of the chiral center at position 3 yielded compound 10 ((3β,5β)-3-hydroxycholan-24-oic acid or isolithocholic acid, Ki = 25 ± 4 μM), which inhibits EphA2-ephrin-A1 interaction with a potency similar to compound 1. However, when the beta hydroxyl group of compound 10 was replaced by a sterically hindered substituent, the compound became inactive, as in the case of the sulfonic acid derivative 11. The removal of the alpha hydroxyl group at position 3 yielded compound 12 (cholanic acid). This is the most potent compound of the series (Table 1 and Figure 4) and disrupts EphA2-ephrin-A1 interaction with a Ki of 5.1 ± 1.4 μM. All together, these data indicate that position 3 of lithocholic acid points toward a hydrophobic cavity of limited size, consistent with the binding model shown in Figure 3B.
Figure 4.

Isolithocholic and cholanic acid competitively inhibit EphA2-ephrin-A1 binding. a) Lithocholic (●), isolithocholic (■) and cholanic acid (▲) dose-dependently displace ephrin-A1-Fc from the immobilized EphA2-Fc ectodomain. b) and d), binding of ephrin-A1-Fc to immobilized EphA2-Fc in presence of different concentrations of isolithocholic [(0 μM (■), 12.5 μM (▼), 25 μM (◆), 50 μM (●), 100 μM (□), and 200 μM (×)] or cholanic acid [(0 μM (■), 3 μM (+), 6 μM (○),12.5 μM (▼) and 25 μM (◆)] respectively. c) and e). The dissociation constants (KD) from the displacement experiments shown in b) and d) were used to calculate Log (dose-ratio - 1) and to graph the Schild plots for isolithocholic (slope = 0.96 ± 007) or cholanic acid (slope = 0.98 ± 002). pKi values were estimated by the intersection of the interpolated line with the X-axis. The slope of the interpolated line can be related to the nature of the binding. A slope between 0.8 and 1.2 indicates competitive binding, whereas a higher slope suggests non-specific interactions. (Ki = 25 ± 4 μM for isolithocholic acid; Ki = 5.1 ± 1.4 μM for isolithocholic acid).
Finally, esterification (compound 13), conjugation with amine derivatives (compounds 14, 15) or reduction to the corresponding alcohol (compound 16) of the lithocholic acid carboxylic group, gave inactive or weakly active compounds, indicating that the presence of a negatively charged group at this position is critical to bind the EphA2 receptor.
Inhibition of EphA2-ephrin-A1 interaction by cholanic and isolithocholic acids
Among the sixteen (5β)-cholan-24-oic acid analogues examined (Table 1), we analyzed further isolithocholic acid (compound 10) and cholanic acid (compound 12), which were found to be more potent inhibitors of EphA2-ephrin-A1 interaction than lithocholic acid (compound 1). These compounds dose-dependently inhibit the binding of the biotinylated ephrin-A1-Fc ectodomain to the immobilized EphA2-Fc-ectodomain (Figure 4A). The IC50 value for isolithocholic acid is 67 μM, while cholanic acid appears to be the most potent derivative of the series, showing an IC50 of 9.6 μM.
In addition to the determination of the IC50 values, we also determined the saturation curves for EphA2-ephrin-A1 binding in the presence of increasing concentrations of isolithocholic or cholanic acid (Figure 4, panels B and D). We calculated the KD or the apparent KD for each curve and drew a Schild plot, where Log [DR-1] is a function of the –Log10 [inhibitor][31] (Figure 4 panels C and E). Both isolithocholic and cholanic acids yielded well-interpolated regression lines (r2 = 0.98 and 0.99, respectively) with slopes of 0.96 and 0.98, respectively, which indicates competitive binding. The intersection of the interpolated line with the X-axis gives a pKi of 4.60 (corresponding to a Ki of 25 μM) for isolithocholic acid and 5.19 (corresponding to a Ki of 5.1 μM) for cholanic acid. We next performed EphA2-ephrin-A1 displacement experiments by incubating the immobilized EphA2 with 100 μM isolithocholic acid or cholanic acid for 1 hour and then washing some wells before adding 50 ng/ml biotinylated ephrin-A1-Fc. Displacement of biotinylated ephrin-A1-Fc binding was observed only in the wells that were not washed, indicating that the binding of isolithocholic and cholanic acids to EphA2 is fully reversible (data not shown)
Selectivity of cholanic and isolithocholic acids for different Eph receptors
We further examined the ability of cholanic and isolithocholic acids to inhibit ephrin binding to all EphA and EphB receptors by using biotinylated ephrin-A1-Fc and biotinylated ephrin-B1-Fc, respectively, at their KD concentration.
Differently from lithocholic acid, which we recently demonstrated to be a promiscuous ligand of all Eph receptors, cholanic and isolithocholic acids demonstrated to be more selective for the EphA receptor subfamily. Particularly, cholanic acid displayed IC50 values for the EphA receptors that were 3 - 30 times lower than those calculated for the EphB receptors (Figure 5).
Figure 5.

Lithocholic acid derivatives partially discriminate between A and B Eph receptor subclasses. a), isolithocholic acid and b), cholanic acid dose-dependently displace the binding of ephrin-A1-Fc and the ephrin-B1-Fc ectodomain from immobilized EphA-Fc or EphB-Fc ectodomains, respectively. Tested concentrations: 3 μM (□), 10 μM (
 ), 30 μM (
), 30 μM (
 ), 100 μM (■). IC50 values are means from at least three independent experiments. The error bars represent standard errors.
), 100 μM (■). IC50 values are means from at least three independent experiments. The error bars represent standard errors.
Notably, cholanic acid was able to inhibit ephrin ligand binding to all members of the EphA receptor subfamily in the low μM range (3.0-7.1). This suggests that cholanic acid interferes with the Eph receptor-ephrin recognition process by occupying a highly conserved region within the EphA receptor ligand binding pocket that is essential for the recruitment of ephrin ligands.
Surface plasmon resonance analysis of the binding of cholanic acid to Eph receptors
To further characterize the mechanism of action of cholanic acid, we investigated the properties of its binding to the EphA2 receptor (and other proteins) by using a surface plasmon resonance (SPR) assay as implemented with Biacore technology.[32]
Dissolved compound was injected over EphA2-Fc immobilized to surfaces attached to an optical biosensor surface, and binding was determined based on the change in mass at the sensor surface.[33] The change in mass depends linearly on the number of molecules bound, making SPR a quantitative technique. After injection, running buffer was flowed over the surface and dissociation of the cholanic acid from the surface was observed. This assay allowed assessment of how cholanic acid associates and dissociates from EphA2 in real time, yielding association/dissociation rate constants (kon, koff) as well as the dissociation equilibrium constant (KD).
As reported by SPR sensorgrams (Figure 6), cholanic acid bound to the immobilized EphA2 receptor in a concentration-dependent manner. The binding was saturable and well fitted by a 1:1 binding interaction model, confirming that the recognition process is specific. The association between EphA2 and cholanic acid was reversible because the protein-compound complex readily dissociated, restoring the baseline signal (Figure 6).
Figure 6.

SPR sensorgrams for the interaction of cholanic acid with EphA2-Fc immobilized on sensor chips. The colored lines denote cholanic acid concentrations: 3 μM (orange line), 6 μM (green line), 12.5 μM (pink line), and 25 μM (maroon line).
Kinetic analysis revealed good binding parameters for cholanic acid. From the sensorgrams, it was possible to determine an association rate (kon) of 4.4 ×104 M −1 s−1 and a dissociation rate (koff) of 3.69 ×10−1 s−1, corresponding to an affinity constant (KD) of 8.45 × 10−6 M. This affinity constant was consistent with the KD of 1.16 × 10−6 M obtained from steady state analysis (i.e. by plotting the binding at equilibrium versus the ligand concentration and considering that the KD equals the concentration yielding 50% of the maximum response,[34] Figure S3, supporting information).
Finally, the SPR analysis was applied to assess the specificity of cholanic acid for the EphA2 receptor as compared to other members of the Eph-ephrin signaling system. This showed that cholanic acid at 6 μM does not bind the EphB1 receptor or the Fc-protein (Figure S4, supporting information), even though it binds the EphA2 receptor (Figure 6).
Cholanic and isolithocholic acids inhibit Eph receptor tyrosine phosphorylation at non-cytotoxic concentrations
To evaluate the functional effects of cholanic and isolithocholic acids on Eph receptors, we performed phosphorylation studies using PC3 human prostate adenocarcinoma cells, which endogenously express the EphA2 receptor, and T47D human mammary carcinoma cells, which endogenously express the EphB4 receptor. Similar to lithocholic acid, the two compounds did not stimulate Eph receptor tyrosine phosphorylation (activation) on their own (data not shown). However, they dose-dependently inhibited EphA2 and EphB4 phosphorylation induced by ephrin-A1-Fc or ephrin-B2-Fc, respectively (Figure 7). The multikinase inhibitor dasatinib (1 μM), used as control, completely blocked EphA2 phosphorylation (data not shown). According to binding data, cholanic acid inhibited Eph receptor activation induced by ephrins more potently than lithocholic acid, showing IC50 values of 12 μM (EphA2) and 38 μM (EphB4) compared to 46 μM and 74 μM for lithocholic acid.
Figure 7.

Lithocholic (●), isolithocholic (■) and cholanic acid (▲) dose-dependently inhibit Eph receptor phosphorylation. a) Inhibition of EphA2 phosphorylation. b) Inhibition of EphB4 phosphorylation. EphA2 phosphorylation was induced by 0.25 μg/ml ephrin-A1-Fc in PC3 cells, whereas EphB4 phosphorylation was induced with 3 μg/ml ephrin-B2-Fc, preclustered with 0.3 μg/ml anti-Fc antibodies in T47D cells. Cells were pretreated for 20 minutes with 1% DMSO, or the indicated concentrations (μM) of compounds and then stimulated for 20 minutes with ephrin-A1/-B2-Fc. Data are the means of at least three independent experiments ± standard error.
Interestingly, despite the similarity between the lithocholic and isolithocholic acid binding profiles, isolithocholic acid more potently inhibited EphA2 and EphB4 phosphorylation in cells, showing IC50 values of 17 μM (EphA2) and 71 μM (EphB4). This suggests an additional inhibitory effect of isolithocholic on the intracellular kinase domain (see next paragraph). Compound concentrations that inhibited receptor tyrosine phosphorylation were not cytotoxic after 2 hours incubation with cells (Figure S5, supporting information).
Cholanic acid does not inhibit EphA2 kinase activity
To rule out that the observed inhibition of EphA2 phosphorylation by cholanic acid was due to a direct interaction with the EphA2 kinase domain, the recombinant EphA2 kinase domain was incubated in presence of a peptide substrate, with or without 100 μM cholanic acid. The levels of phosphorylated peptide were detected with a Europium-labeled anti-phosphotyrosine antibody. Cholanic acid did not affect EphA2 kinase activity, confirming that its effect in cells was due to inhibition of Eph-ephrin protein-protein interaction. In contrast, isolithocholic acid significantly reduced EphA2 kinase activity (Figure 8), explaining the unexpectedly high potency of this compound in inhibiting EphA2 phosphorylation induced by ephrin stimulation. On the other hand, the general kinase inhibitor staurosporine fully inhibited the kinase activity of EphA2 at 100 μM (Figure 8).
Figure 8.

Cholanic acid does not inhibit EphA2 kinase activity. The enzymatic activity of the recombinant human EphA2 kinase domain was evaluated with the LANCE® method using ATP and Ulight-TK peptide as substrate. EphA2 was incubated for 30 minutes with the indicated compounds at concentrations of 100 μM, 1 μM staurosporine or 1% DMSO as a control. **, p<0.01 for the comparison to control by one-way ANOVA followed by Tukey's multiple comparison test.
The specificity of lithocholic acid as an Eph receptor antagonist was previously determined by demonstrating lack of effects on other receptor tyrosine kinases, such as the EGF receptor, the VEGF receptor, the insulin receptor and the insulin-like growth factor receptor 1.[25] Similar to lithocholic acid, cholanic and isolithocholic acids at concentrations up to 100 μM also did not interfere with EGF receptor activation induced by EGF (Figure 9).
Figure 9.

Isolithocholic and cholanic acids do not affect EGF receptor activity. PC3 cells were pretreated for 20 minutes with 100 μM lithocholic, isolithocholic or cholanic acid, 10 μM gefitinib, or 1% DMSO as a control, and stimulated for 20 minutes with 30 ng/ml EGF. Phospho-EGF receptor levels are relative to the EGF+DMSO condition. Data are the means of at least three independent experiments ± standard error. **, p<0,01 for the comparison to EGF+DMSO by one-way ANOVA followed by Tukey's multiple comparison test.
Cholanic and isolithocholic acids inhibit EphA2-mediated cell retraction in PC3 cells
Cholanic and isolithocholic acids inhibited EphA2-mediated cell retraction and rounding of PC3 cells stimulated with ephrin-A1 Fc at concentrations as low as 25 μM (Figure 10), suggesting that these compounds can be used to counteract the functional effects mediated by EphA2.[35] As expected, isolithocholic and cholanic acids inhibited cell retraction at concentrations comparable to those needed to inhibit EphA2 phosphorylation. None of the compounds affected cell morphology in the absence of ephrin-A1 stimulation (Figure 10), confirming their lack of toxicity.
Figure 10.

Inhibition of EphA2-dependent retraction and rounding of PC3 prostate cancer cells. a) dose-response curve for isolithocholic (IsoLCA) acid in presence of ephrin-A1 Fc (■), or Fc (
) as a control. b) dose-response curve for cholanic acid in presence of ephrin-A1 Fc (■), or Fc (
) as a control. c) Effects on cell morphology. PC3 cells, pre-treated for 15 min with the indicated concentrations of isolithocholic (IsoLCA) or cholanic acid, were stimulated with 0.5 μg/ml ephrin-A1 Fc (+) or Fc as a control (-) for 20 min in the continued presence of the compounds. The cells were stained with rhodamine-phalloidin to label actin filaments (red) and 4′,6-diamidino-2-phenylindole (DAPI) to label nuclei (blue). DMSO was used as a control. The histograms show the average percentage of retracting cells. Cells having rounded shape and decreased spreading were scored as retracting. The percentages of cell retraction under different conditions were compared with those in the Fc control condition by one-way ANOVA and Dunnett's post test.
Conclusions
The identification of small molecules able to disrupt protein–protein interfaces is a challenging task. Complications include the presence of large protein-protein interacting surfaces, which lack deep cavities where small molecules can bind with good affinity.[21] The ephrin-binding pocket of the EphA2 receptor, however, seems to present favourable features for high-affinity binding of small molecules, as shown here and in other recent papers.[24,35]
In the present work, we report the discovery of a small molecule, cholanic acid ((5β)-cholan-24-oic acid), that binds the ligand binding domain of the EphA2 receptor with an affinity in the low micromolar range. This compound was identified in a focused medicinal chemistry effort aimed at the optimization of lithocholic acid, a weak antagonist of the Eph-ephrin system that was recently discovered by our group.[25]
A computationally-driven exploration of lithocholic acid derivatives allowed us to build a clear structure-activity relationship profile and identify the stereoelectronic requirements for EphA2 binding. In particular, we found that the simultaneous presence of a large hydrophobic region (represented by the cyclopenta[a]perhydro phenanthrene scaffold) and an anionic hydrogen bond acceptor group (represented by a carboxylate functionality) are pivotal to interfere effectively with EphA2-ephrin-A1 binding, consistently with the predicted binding mode for the EphA2-lithocholic acid complex. Notably, surface plasmon resonance experiments indicated that cholanic acid interacts with the ligand-binding domain of the EphA2 receptor, in agreement with our working hypothesis. Surface plasmon resonance was also used to characterize the kinetics for the binding of cholanic acid to EphA2, yielding a steady-state binding constant in the low micromolar range (KD = 1.16 × 10−6 M).
Cholanic acid competitively displaces biotinylated-ephrin-A1 from the EphA2 receptor. Indeed, the shift in the EphA2-ephrin-A1 saturation curves obtained with increasing concentration of cholanic acid produces a Schild plot consistent with competitive antagonism.
The inhibitory activity of cholanic acid towards EphA2 is also confirmed by cell-based assays, where the addition of compound dose-dependently inhibits the ephrin-A1-dependent tyrosine phosphorylation of EphA2 and the retraction of PC3 cells. Cholanic acid is less potent in blocking the ephrin-B1-dependent phosphorylation of EphB4, paralleling the results obtained in the in vitro displacement assay. Furthermore, cholanic acid has no effect on the EphA2 kinase domain, which is instead weakly inhibited by isolithocholic acid.
The substantial binding affinity of cholanic acid together with its ability to block EphA2 activity in cell lines, support the notion that the (5β)-cholan-24-oic acid scaffold can be used as template structure to design an improved generation of EphA2 inhibitors. On the other hand, cholanic acid suffers from high lipophilicity, which might hamper its bioavailability in vivo. However, bile acid derivatives are known to be a good reservoir of biologically active compounds, as in the case of obeticholic acid (INT-747).[36,37] This Farnesoid X receptor (FXR) agonist has been recently advanced to phase III clinical trials for the treatment of chronic liver diseases (clinical trial NCT00570765, study of INT-747 as monotherapy in patients with primary biliary cirrhosis). Thus, a lead optimization program aimed at the improvement of the physicochemical properties of cholanic acid might yield a small molecule that can effectively inhibit the activity of EphA2 in vivo.
Experimental Section
Molecular modelling
Molecular modelling simulations were performed starting from the crystal structure of the EphA2-ephrinA1 complex (3HEI.pdb),[22] using Maestro software [38] and OPLS2005 force field.[39] The EphA2-ephrinA1 complex was submitted to a protein preparation procedure which includes addition of missing side chains and hydrogens, assignment of tautomeric state of histidines to maximize the number of hydrogen bonds, and geometric optimization of the whole system to a root-mean-square displacement (RMSD) value of 0.3 Å. [40] At the end of this procedure, the ephrinA1 ligand was deleted from EphA2 active site. A molecular model of lithocholic acid (1) was also built using Maestro, and its geometry was optimized by energy minimization using OPLS2005 to a gradient of 0.01 kcal/(mol · Å). Docking simulations were then performed using Glide5.5,[41] starting from the minimized structure of lithocholic acid placed in an arbitrary position within a region centered on the surface of channel of EphA2, delimited by Arg103, Phe156 and Arg159, using enclosing and bounding boxes of 20 and 14 Å on each side, respectively. Van der Waals radii of the protein atoms were not scaled, while van der Waals radii of the ligand atoms with partial atomic charges lower than |0.15| were scaled by 0.8. Standard precision mode was applied. The resulting binding poses were ranked according to the G-score, and the best docking solution was selected for MD simulations. The selected EphA2-lithocholic acid docking complex was (i) solvated by approximately 14000 SPC water molecules in a simulation box of 78 Å × 78 Å × 78 Å, (ii) neutralized by adding 5 Na+ ions, and (iii) equilibrated by 30 ns of MD simulations. The simulation was performed in the NPT ensemble under constant pressure of 1 atm and temperature of 300 K. All bond lengths to hydrogen atoms were constrained using M-SHAKE.[42] Short-range electrostatic interactions were cut off at 9 Å whereas long-range electrostatic interactions were computed using the Particle Mesh Ewald method.[43] A RESPA integrator[44] was used with a timestep of 2 fs, and long-range electrostatics were computed every 6 fs. Snapshots were saved every 10 ps, for a total of 3000 structures. The MD simulation was performed with the OPLS2005 force field, using Desmond package v22623.[45]
Analysis of the MD trajectory was performed by evaluating the root mean square deviation (RMSD) of EphA2 receptor and lithocholic acid, using the first frame of the production phase as a reference structure. While for EphA2 the RMSD was measured considering the Cα of the amino acid residue, for lithocholic acid the RMSD was measured only taking into its heavy atoms, after their optimal superposition.
The interaction between the critical Arg103 of EphA2[22] and lithocholic acid was evaluated by plotting, for each snapshot recorded during the simulation, the shortest of the six possible distances between the three nitrogen atoms of the guanidinium group of Arg103 and the two oxygen atoms of the carboxylic group of lithocholic acid.
Chemistry
Unless otherwise noted, reagents and solvents were purchased from commercial suppliers (Aldrich and Fluka) and were used without purification. Melting points were determined on a Gallenkamp melting point apparatus and were not corrected. The final compounds 1-16, were analyzed on a ThermoQuest (Italia) FlashEA 1112 Elemental Analyzer for C, H and N. The percentages found were within ±0.4% of the theoretical values. The 1H-NMR and 13C-NMR spectra were recorded on a Bruker Avance 400 spectrometer (400MHz); chemical shifts (δ scale) are reported in parts per million (ppm). 1H-NMR spectra are reported in the following order: multiplicity, number of protons and approximate coupling constants (J value) in Hertz (Hz); signals were characterized as s (singlet), d (doublet), t (triplet), m (multiplet), br s (broad signal). Mass spectra were recorded on an Applied Biosystem API-150 EX system spectrometer with ESI interface. The progress of the reaction was monitored by thin-layer chromatography with F254 silica-gel precoated sheets (Merck Darmstadt, Germany). UV light and potassium permanganate solution (10% w/v) were used for detection. Flash chromatography was performed using Merck silica-gel 60 (Si 60, 40-63 μm, 230-400 mesh ASTM). Tetrahydrofurane (THF) was dried by distillation over Na/benzophenone. All reactions were carried out using flame-dried glassware under atmosphere of nitrogen. Compounds 1-8 and 12 were purchased from Sigma and characterized by elemental analysis (see Table S1 supporting information,). Compounds 9-11, and 13-16 were synthesized according to the procedures described below.
Synthesis of (3α5β)-3-acetoxycholan-24-oic acid (9)
A modification of a described procedure[46] was used (Scheme S1, supporting information): lithocholic acid (1) (2.654 mmol) and 4-dimethylaminopyridine.(DMAP) (0.409 mmol) were dissolved in anhydrous pyridine (10 ml). Acetic anhydride (21.58 mmol) was added dropwise to the previous solution; the reaction mixture was stirred at room temperature and kept under nitrogen for 3 hours and then ice and water were added. The mixture was acidified with concentrated hydrochloric acid and the white precipitate was filtered off and washed with water. The solid obtained was purified by flash chromatography [SiO2, CH2Cl2:HCOOH:C2H5OH 89:1:10 (300 ml)]. The crude product was recrystallized from ethanol-water to give 9 (0.730 g, 65%) as white powder. 1H-NMR (400 MHz DMSO-d6) δ = 0.60 (s, 3H, CH3), 0.86 (d, 3H, J = 6.4 CH3); 0.89 (s, 3H, CH3); 0.96-1.67 (m, 26H); 1.72-1.83 (m, 4H); 1.90-1.93 (m, 1H); 1.95 (s,3H, CH3), 2.04-2.12 (m, 1H), 2.18-2.25 (m, 1H), 4.58-4.54 (m, 1H), 11.96 (br s, 1H, OH); 13C-NMR (100 MHz DMSO-d6) δ = 12.30, 18.57, 20.88, 21.48, 23.45, 24.26, 26.40, 26.70, 27.04, 28.16, 31.14, 32.33, 34.60, 34.96, 35.26, 35.75, 41.63, 42.72, 56.02, 56.38, 73.89, 170.14, 175.25. MS (ESI) calcd. for C26H42O4 418.61, found 417 [M−1−]−.
Synthesis of methyl (3α,5β)-3-hydroxycholan-24-oate (13)
A protocol reported in the literature[47] was followed (Scheme S2): to a stirred suspension of lithocholic acid (1) (3.04 mmol) in methanol (15 ml) was added concentrated sulphuric acid (0.5 ml). The reaction mixture was stirred at room temperature (rt) for 3 hours and then diluted with water. The white precipitate obtained was filtered off under vacuum and washed with water. The crude product was recrystallized from ethanol-water to give 13 (1.118 g, 94%) as colorless solid. 1H-NMR (400 MHz CDCl3 δ = 0.60 (s, 3H, CH3); 0.85-0.91 (m, 6H), 1.02-1.98 (m, 28H), 2.17-2.36 (m, 2H), 3.57-3.64 (m, 1H), 3.67 (s, 3H, CH3); 13C-NMR (100 MHz CDCl3) δ = 12.04, 18.26, 20.82, 23.38, 24.20, 26.42, 27.20, 28.19, 30.54, 31.00, 31.06, 34.57, 35.36, 35.84, 36.45, 40.17, 40.43, 42.10, 42.73, 43.73, 51.48, 55.95, 56.49, 71.84,174.79. MS (ESI) calcd for C25H42O3 390.59, found 413 [M+Na+]+.
Synthesis of methyl (3β,5β)-3-benzoyloxycholan-24-oate (10a)
A modification of a described procedure[48] was used (scheme S3): triphenylphosphine (0.648 mmol) was dissolved in anhydrous THF (5 ml) and cooled to 0°C. To the stirred solution under nitrogen was added dropwise a solution of diisopropyl azodicarboxylate (DIAD) (0.637 mmol) in anhydrous THF (1ml), keeping the temperature at 0°C. After the addition was complete, the reaction mixture was warmed to room temperature and then a solution of 13 (0.510 mmol) and benzoic acid (0.510 mmol) in anhydrous THF (5 ml) was added dropwise. After stirring overnight at room temperature, the solvent was removed under reduced pressure and the residue was purified by flash chromatography [SiO2, CH2Cl2: n-hexane from 90:10 (200 ml) to 100% CH2Cl2 (100 ml)]. The crude product was recrystallized from ethanol-water to give 10a (0.245 g, 97%) as white powder. 1H-NMR (400 MHz CDCl3 δ = 0.66 (s, 3H, CH3), 0.92 (d, 3H, J = 6.4 CH3), 1.02-1.21 (m, 9H), 1.27-1.45 (m, 9H), 1.73-2.10 (m, 11H), 2.18-2.26 (m, 1H), 2.32-2.40 (m, 1H), 3.67 (s, 3H, CH3), 5.34 (s, 1H, OH), 7.44 (t, 2H, J = 7.6 Ar), 7.55 (t, 1H, J = 7.4 Ar), 8.05 (d, 2H, J = 8.4 Ar).(s, 3H, CH3), 13C-NMR (100 MHz CDCl3) δ = 12.08, 18.29, 21.14, 24.04, 24.20, 25.22, 26.19, 26.57, 28.19, 30.80, 31.02, 31.07, 31.09, 35.00, 35.38, 35.70, 37.74, 39.96, 40.19, 42.78, 51.48, 55.99, 56.57, 71.40, 121.31 129.51, 131.19, 132.68, 165.92, 174.77. MS (ESI) calcd for C32H46O4 494.70, found 517 [M+Na+]+.
Synthesis of (3β,5β)-3-Hydroxycholan-24-oic acid (10)
A modification of a described procedure[49] was used (scheme S4): to a solution of 10a (1.96 mmol) in ethanol (75 ml) was added a solution of sodium hydroxide 15% w/v (50 ml) and the mixture was refluxed overnight. Ethanol was removed under vacuum and the solution was acidified with concentrated hydrochloric acid until a precipitate was formed. The resulting suspension was extracted with dichloromethane (3 × 100 ml). The organic extracts were washed with water, brine and dried over anhydrous Na2SO4. Evaporation of solvent under reduce pressure yielded a white solid that was purified by flash chromatography [SiO2, CH2Cl2:HCOOH:C2H5OH from 99.37:0.03:0.6 (100 ml) to 97.50:0.5:2 (150 ml)]. The crude product was recrystallized from ethanol-water to give 10 (1.02 g, 83%) as a white solid. 1H-NMR (400 MHz DMSO-d6) δ = 0.60 (s, 3H, CH3); 0.85-0.91 (m, 6H;), 1.02-1.98 (m, 28H), 2.17-2.36 (m, 2H), 3.57-3.64 (m, 1H), 3.67 (s, 3H, CH3); 13C-NMR (100 MHz DMSO-d6) δ = 12.04, 18.26, 20.82, 23.38, 24.20, 26.42, 27.20, 28.19, 30.54, 31.00, 31.06, 34.57, 35.36, 35.84, 36.45, 40.17, 40.43, 42.10, 42.73, 43.73, 51.48, 55.95, 56.49, 71.84 174.79. MS (ESI) calcd for C24H40O3 376.57, found 375 [M−1]−.
Synthesis of methyl (3β,5β)-S-acetyl-3-mercaptocholan-24-oate (11a)
Triphenylphosphine (0.648 mmol) was dissolved in anhydrous THF (5 ml) and cooled to 0 °C (Scheme S5). To the stirred solution under nitrogen was added dropwise a solution of DIAD (0.637 mmol) in anhydrous THF (1 ml), keeping the temperature at 0°C. After the addition was complete, the reaction mixture was warmed to room temperature and then a solution of 13 (0.510 mmol) and thioacetic acid (1.02 mmol) in anhydrous THF (5 ml) was added dropwise. After stirring overnight at room temperature, the solvent was removed under reduced pressure and the residue was purified by flash chromatography [SiO2, CH2Cl2: n-hexane from 50:50 (300 ml) to 90:10 (100 ml)]. The crude product was recrystallized from ethanol-water to give 11a (0.148 g, 65%) as white powder. Mp 128-131 °C; 1H-NMR (400 MHz CDCl3) δ = 0.63 (s, 3H, CH3), 0.89-0.93 (m, 6H); 1.03-1.67 (m, 21H); 1.77-1.96 (m, 5H, CH3); 2.19-2.26 (m, 1H); 2.29 (s, 3H, CH3); 3.65(s, 3H, CH3), 4.09 (s,1H); 13C-NMR (100 MHz CDCl3) δ = 12.05, 18.27, 20.96, 23.90, 24.17, 26.33, 26.38, 26.85, 28.17, 30.97, 31.00, 31.04, 32.01, 32.99, 35.21, 35.36, 35.72, 39.52, 40.13, 40.24, 42.73, 42.79, 51.48, 55.94, 56.49, 174.76, 195.77. MS (ESI) calcd for C27H44O3S 448.70, found 471 [M+Na+]+. Anal. calcd for C27H44O3S: C, 72.27; H, 9.88; found: C, 72.49; H, 9.53.
Synthesis of (3β,5β)-3-mercaptocholan-24-oic acid (11b)
A modification of a described procedure[50] was used (Scheme S6): to a solution of compound 11a (1.55 mmol) in ethanol (60 ml) was added a solution of sodium hydroxide 15% w/v (35 ml) and the mixture under nitrogen was refluxed for 2 hours. Ethanol was removed under vacuum and the solution was acidified with concentrated hydrochloric acid until a precipitate was formed. The resulting suspension was extracted with dichloromethane (3 × 100 ml). The organic extracts were washed with water, brine and dried over anhydrous Na2SO4. Evaporation of solvent under reduce pressure yielded 11b as a white solid (0.583 g, 96%) that was sufficiently pure for the next reaction step.1H-NMR (400 MHz CDCl3 = 0.67 (s, 3H, CH3), 0.95-1.05 (m, 6H), 1.08-1.61 (m, 22H), 1.80-2.00 (m, 6H), 2.22-2.43 (m, 3H), 3.59-3.65 (m,1H);13C-NMR (100 MHz CDCl3) δ = 12.08, 18.25, 20.93, 23.92, 24.17, 26.55, 26.72, 28.17, 28.70, 29.71, 30.24, 30.75, 31.04, 34.31, 35.31, 35.50, 35.71, 36.70, 37.46, 40.20, 40.24, 42.76, 55.96, 56.60,180.66. MS (ESI) calcd. for C24H40O2S 392.64, found 391 [M−1−]−.
Synthesis of (3β,5β)-3-sulfocholan-24-oic acid (11)
To a stirred solution of 11b (1.27 mmol) kept under nitrogen and at 0 °C was added dropwise peracetic acid solution 40% w/w (4.16 mmol, Scheme S7).The mixture under stirring was allowed to warm to rt for 4 hours. Evaporation of the solvent under reduced pressure afforded a white solid that was purified by flash chromatography [SiO2, CH2Cl2:HCOOH:C2H5OH from 83:7:10 (250 ml) to 73:7:20 (200 ml)].The crude product was recrystallized from ethanol-water to give 11 (0.354 g, 63%) as a white amorphous solid. Mp 285-288 °C. 1H-NMR (400 MHz DMSO-d6) δ = 0.60 (S, 3H), 0.84-0.86 (m, 6H), 1.01-1.90 (m, 30H), 2.15-2.20 (m, 1H), 2.25-2.29 (m, 1H), 2.54-2.58 (m, 1H); 13C-NMR (100 MHz DMSO-d6) δ = 12.32, 18.57, 21.03, 21.37, 23.97, 24.28, 26.20, 26.74, 26.85 28.15, 31.06, 31.09, 31.56, 34.54, 35.23, 35.65, 36.36, 40.20, 42.74, 54.35, 55.94, 56.02, 56.56, 60.10, 173.70. MS (ESI) calcd. for C24H40O5S 440.64, found 439 [M-1-]-. Anal. calcd for C24H40O5S*1.4H2O: C, 61.61; H, 9.65, found: C, 61.85; H, 9.62.
Synthesis of (3α,5β)-3-Hydroxycholan-24-hydroxamic acid (14)
A modification of a described procedure[51] was used (Scheme S8): to a solution of hydroxylamine hydrochloride (6.48 mmol) in methanol (4 ml), under nitrogen and cooled at 0°C, was added dropwise potassium hydroxide (12.97 mmol) dissolved in methanol (4 ml). The mixture was stirred for further 20 minutes and then a solution of 13 (0.64 mmol) in methanol (7 ml) was added dropwise keeping the reaction temperature at 0°C. The reaction mixture was then warmed to rt and stirred for 4 hours. Finally, it was diluted with water, cooled and acidified with 6 N hydrochloric acid to afford a white precipitate. The solid was filtered off under vacuum and purified by flash chromatography [SiO2, CH2Cl2:HCOOH:C2H5OH from 94,50:0,5:5 (200 ml) to 82:8:10 (150 ml)]. The crude product was recrystallized from ethanol-water to give 14 (0.140 g, 56%) as reddish amorphous solid. Mp 169-173 °C; 1H-NMR (400 MHz DMSO-d6) δ = 0.59 (s, 3H, CH3), 0.86-0.91 (m, 6H), 1.01-1.92 (m 27H), 3.52-3.59 (m, 1H), 4.42 (s, 1H, OH), 4.42, 8.62 (s, 1H, NH), 10.30 (s, 1H, OH); 13C-NMR (100 MHz DMSO-d6) δ = 12.36, 18.69, 20.88, 23.74, 24.31, 26.63, 27.36, 28.18, 29.67, 30.85, 31.90, 34.67, 35.33, 35.62, 35.85, 36.76, 41.99, 42.73, 56.00, 56.54, 70.32, 169.94. MS (ESI) calcd. for C24H41NO3 391.58, found 390 [M−1−]−.
Synthesis of (3α,5β)-3-Hydroxycholan-24-hydrazide (15)
A modification of a described procedure[52] was used (Scheme S9): to a solution of 13 (0.768 mmol) in methanol (10 ml) wad added dropwise hydrazine monohydrate (103 mmol) and the reaction mixture was stirred for 6 hours at rt. The reaction was diluted with water and the white precipitate obtained was filtered off under vacuum and washed with water. The white solid obtained was recrystallized from ethanol-water to give 15 (0.288 g, 96%). Mp 201-206 °C; 1H-NMR (400 MHz DMSO-d6) δ = 0.59 (s, 3H, CH3), 0.85-0.91 (m, 6H), 1.01-2.04 (m 27H), 3.52-3.59 (m, 1H), 4.11 (s, 2H, NH2), 4.42 (d, 1H, J = 4.4 OH), 8.88(s,1H, NH); 13C-NMR (100 MHz DMSO-d6) δ = 12.37, 18.76, 20.90, 23.71, 24.30, 26.63, 27.40, 28.12, 30.91, 31.99, 34.71, 35.38, 35.68, 35.92, 36.85, 42.09, 42.79, 56.14, 56.61, 70.38, 172.46 MS (ESI) calcd. for C24H42N2O2 390.60, found 389 [M−1−]−.
Synthesis of (3α,5β)-Cholan-3,24-diol (16)
A modification of a described procedure[53] was used (Scheme S10): a solution of lithocholic acid (1) (2.66 mmol) in anhydrous THF (30 ml) was added dropwise to a suspension of LiAlH4 (10.64 mmol) in anhydrous THF (30 ml), stirred at 0 °C and kept under nitrogen. The reaction mixture was then allowed to warm to room temperature and stirred overnight. The reaction was then chilled to 0 °C and carefully quenched by dropwise addition of a 2N H2SO4 solution (30 ml) and swirled at rt until the reaction mixture became clear. THF was removed under reduced pressure and the mixture was extracted with diethyl ether (3 × 40 ml).The organic phase was washed with water, brine and then dried over anhydrous Na2SO4. Evaporation of the solvent under reduced pressure afforded a white solid which was recrystallized from ethanol-water to give 16 (0.791 g, 81%). 1H-NMR (400 MHz DMSO-d6) δ = 0.62 (s, 3H, CH3), 0.88-0.92 (m, 7H), 0.95-1.93 (m, 31H); 4.32 (t, 1H, J = 6.8 CH2-OH), 4.44 (d,1H, J = 6.4 OH); 13C-NMR (100 MHz DMSO-d6) δ = 12.31, 18.99, 23.71, 24.30, 26.62, 27.37, 28.31, 29.59, 30.82, 32.24, 34.65, 35.57, 35.62, 35.85, 36.73, 42.00, 42.69, 56.27, 56.54, 61.75, 70.31. MS (ESI) calcd for C24H42O2 362.59, found 385 [M+Na+]+
Pharmacology
Reagents
all culture media and supplements were purchased from Lonza. Recombinant proteins and antibodies were from R&D systems. Cells were purchased from ECACC. Leupeptin, aprotinin, NP40, MTT, tween20, BSA and salts for solutions were from Applichem; EDTA and sodium orthovanadate were from Sigma. Human IgG Fc fragment was from Millipore (AG714).
Cell Cultures
PC3 human prostate adenocarcinoma cells were grown in Ham F12 or RPMI 1640 supplemented with 7% fetal bovine serum (FBS) and 1% antibiotic solution. T47D human breast tumor cells were grown in RPMI 1640 with 10% FBS and 1% antibiotic solution. All cell lines were grown in a humidified atmosphere of 95% air, 5% CO2 at 37°C.
ELISA assays and Ki/IC50 determination
ELISA assays were performed as previously described.[54] Briefly, compounds (Table 1) were stocked as 20 mM solutions in dimethyl sulfoxide (DMSO) and tested both in displacing and saturation studies, starting from a concentration of 200 μM. Ninety-six well ELISA high binding plates (Costar #2592) were incubated overnight at 4°C with 100 μl/well of 1 μg/ml EphA2-Fc (R&D 639-A2) diluted in sterile phosphate buffered saline (PBS, 0.2 g/l KCl, 8.0 g/l NaCl, 0.2 KH2PO4, 1.15 g/l Na2HPO4, pH 7.4). The day after wells were washed with washing buffer (PBS +0.05% tween20, pH 7.5) and blocked with blocking solution (PBS +0.5% BSA) for 1 hour at 37°C. Compounds were added to the wells at proper concentration in 1% DMSO and incubated at 37°C for 1 hour. Biotinylated ephrinA1-Fc (R&D Systems BT602) was added at 37°C for 4 hours at its KD in displacement assays or in a range from 1 to 2000 ng/ml in saturation studies. The wells were washed and incubated with 100 μl/well Streptavidin-HRP (Sigma S5512) in blocking solution (0.05 μg/ml in PBS supplemented with 0.5% BSA, pH 7.4) for 20 minutes at room temperature, then washed again and incubated at room temperature with 0.1 mg/ml tetra-methylbenzidine (Sigma T2885) reconstituted in stable peroxide buffer (11.3 g/l citric acid, 9.7 g/l sodium phosphate, pH 5.0) and 0.1% H2O2 (30% m/m in water), added immediately before use. The reaction was stopped with 3N HCl 100 μl/well and the absorbance was measured using an ELISA plate reader (Sunrise, TECAN, Switzerland) at 450 nm.
IC50 values were determined using one-site competition non-linear regression and KD values of the curves with or without antagonists were calculated using one-binding site non-linear regression analysis with Prism software (GraphPad Software Inc.). Ki values were obtained using Schild plot[31] where Log[DR-1] is a function of the negative Log10 of the inhibitor concentration. The Hill's coefficient was calculated using linear fitting to evaluate whether the inhibition was competitive or uncompetitive.
Surface plasmon resonance
EphA2 (3000RU) EphB1 (3000 RU) and Fc fragment (1000RU) were immobilized via NH2 group on dextran matrix of a CM4 sensor chip, respectively on flow cell 2, flow cell 3 and flow cell 4. On flow cell 1 was performed a blank immobilization, in order to be used as reference surface. Cholanic acid was dissolved into a solution of DMSO (final concentration 1mM) and then diluted to 50μM in PBS-0.05% pH7.4. Subsequent dilutions from 25 μM to 3 μM were performed in 5% DMSO-PBS-0.05% pH7.4 that was also used as Running Buffer. Cholanic acid was injected over immobilized EphA2 EphB1 and Fc fragment for 90sec at a flow rate of 30μl/min, followed by a 300 sec dissociation. Kinetics were analyzed with the Biacore T100 evaluation software and were calculated as 1:1 binding model and as steady state affinity.
Cell lysates
PC3 or T47D cells were seeded in 12-well plates at concentration of 105 cells/ml, 1 ml/well, in complete medium until they reached ~70% confluence and serum starved overnight. The day after cells were treated with the compounds under study, vehicle or standard drug, stimulated with the proper agonist (ephrinA1-Fc or ephrinB2-Fc), rinsed with sterile PBS and solubilized in lysis buffer. The lysates were resuspended and rocked at 4°C for 30 minutes and then centrifuged at 14000 × g for 5 minutes. The protein content of supernatant was measured with BCA protein assay kit (Thermo scientific) and standardized to 200 μg/ml.
Phosphorylation of EphA2, EphB4 and EGFR in cells
EphA2-, EphB4- and EGFR-phosphorylation were measured in cell lysates using a DuoSet®IC Sandwich ELISA (R&D Systems, #DYC4056, #DYC4057 and #DYC1095, respectively) following the manufacturer's protocol. Briefly, 96 well ELISA high binding plates (costar 2592) were incubated overnight at room temperature with 100 μl/well of the specific capture antibody diluted in sterile PBS to the proper working concentrations. After blocking, the wells were incubated for 2 hours at room temperature with 100 μl/well of lysates, followed by a 2 hour incubation at room temperature with the detection antibody. Receptor phosphorylation was revealed utilizing a standard HRP format with a colorimetric reaction read at 450 nm.
EphA2 kinase assay
the ability of isolithocholic acid and cholanic acid to interact directly with the intracellular kinase domain of human EphA2 was assessed by measuring the phosphorylation of the substrate Ulight-TK peptide (50 nM), in absence and in presence of 100 μM of the tested compound. The LANCE detection method was applied[55] and the general kinase inhibitor staurosporine was used as reference compound.
LDH assay
cytotoxicity of all compounds was evaluated with CytoTox 96® Non-Radioactive Cytotoxicity Assay, following the manufacturer's protocol (Promega, #1780). Briefly, cells were seeded in 96-well plates at a density of 105cells/ml and the day after treated with compounds or lysis buffer for 2h. After incubation, released LDH in culture supernatants was measured using a 30-minute coupled enzymatic assay, which results in conversion of a tetrazolium salt (INT) into a red formazan product. The amount of colour formed is proportional to the number of lysed cells and quantified by ELISA plate reader (Sunrise, TECAN, Switzerland) at 492 nm. The results were expressed as the ratio between absorbance of the cells treated with the compounds and cells treated with lysis buffer.
Retraction assay
The procedure is similar to that of references 24 and 34. Briefly, PC3 cells (4,000 cells per well) were plated in 96-well plates (Greiner Bio One, Frickenhausen Germany) and grown for 17 hours. The cells were starved for 1 hour in serum-free RPMI, incubated for 15 min with the compounds or DMSO, and stimulated for 10 min with 0.5 μg/ml ephrin-A1 Fc or Fc as a control. The cells were then fixed for 15 min in 4% formaldehyde in PBS, permeabilized for 3 min in 0.5% Triton X-100 in TBS, and stained with rhodamine-conjugated phalloidin (Invitrogen). Nuclei were labeled with 4′,6-diamidino-2- phenylindole (DAPI). Cells were photographed under a fluorescence microscope, and the number of retracted cells was counted in a blinded manner.
Supplementary Material
Acknowledgments
The work was supported by Ministero della Università e della Ricerca, “Futuro in Ricerca” program (project code: RBFR10FXCP), Associazione Italiana per la Ricerca sul Cancro (AIRC), “My first AIRC” grant program (project code: 6181), and NIH grant CA138390.
References
- 1.Gale NW, Yancopoulos GD. Cell Tissue Res. 1997;290:227–241. doi: 10.1007/s004410050927. [DOI] [PubMed] [Google Scholar]
- 2.Wykosky J, Debinski W. Mol Cancer Res. 2008;6:1795–1806. doi: 10.1158/1541-7786.MCR-08-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kullander K, Klein R. Nat Rev Mol Cell Biol. 2002;3:475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 4.Pasquale EB. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Pasquale EB. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- 6.Miao H, Wang B. Int J Biochem Cell Biol. 2009;41:762–770. doi: 10.1016/j.biocel.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein R. Nat Neurosci. 2009;12:15–20. doi: 10.1038/nn.2231. [DOI] [PubMed] [Google Scholar]
- 8.Du J, Fu C, Sretavan DW. Curr Pharm Des. 2007;13:2507–2518. doi: 10.2174/138161207781368594. [DOI] [PubMed] [Google Scholar]
- 9.Mosch B, Reissenweber B, Neuber C, Pietzsch J. J Oncol. 2010;2010:135285. doi: 10.1155/2010/135285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y. Nat Genet. 2009;41:1127–1132. doi: 10.1038/ng.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruhe JE, Streit S, Hart S, Wong CH, Specht K, Knyazev P, Knyazeva T, Tay LS, Loo HL, Foo P, Wong W, Pok S, Lim SJ, Ong H, Luo M, Ho HK, Peng K, Lee TC, Bezler M, Mann C, Gaertner S, Hoefler H, Iacobelli S, Peter S, Tay A, Brenner S, Venkatesh B, Ullrich A. Cancer Res. 2007;67:11368–11376. doi: 10.1158/0008-5472.CAN-07-2703. [DOI] [PubMed] [Google Scholar]
- 13.Pasquale EB. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lisabeth EM, Fernandez C, Pasquale EB. Biochemistry. 2012;51:1464–1675. doi: 10.1021/bi2014079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tandon M, Vemula SV, Mittal SK. Expert Opin Ther Targets. 2011;15:31–51. doi: 10.1517/14728222.2011.538682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lafleur K, Huang D, Zhou T, Caflisch A, Nevado C. J Med Chem. 2009;52:6433–6466. doi: 10.1021/jm9009444. [DOI] [PubMed] [Google Scholar]
- 17.Zhou T, Caflisch A. ChemMedChem. 2010;5:1007–1014. doi: 10.1002/cmdc.201000085. [DOI] [PubMed] [Google Scholar]
- 18.Barlaam B, Ducray R, Lambert-van der Brempt C, Plé P, Bardelle C, Brooks N, Coleman T, Cross D, Kettle JG, Read J. Bioorg Med Chem Lett. 2011;21:2207–2211. doi: 10.1016/j.bmcl.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 19.van Linden OP, Farenc C, Zoutman WH, Hameetman L, Wijtmans M, Leurs R, Tensen CP, Siegal G, de Esch IJ. Eur J Med Chem. 2012;47:493–500. doi: 10.1016/j.ejmech.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 20.Chang Q, Jorgensen C, Pawson T, Hedley DW. Br J Cancer. 2008;99:1074–1082. doi: 10.1038/sj.bjc.6604676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noberini R, Lamberto I, Pasquale EB. Semin Cell Dev Biol. 2012;23:51–57. doi: 10.1016/j.semcdb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Himanen JP, Goldgur Y, Miao H, Myshkin E, Guo H, Buck M, Nguyen M, Rajashankar KR, Wang B, Nikolov DB. EMBO Rep. 2009;10:722–728. doi: 10.1038/embor.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Himanen JP. Semin Cell Dev Biol. 2012;23:35–32. doi: 10.1016/j.semcdb.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 24.Noberini R, De SK, Zhang Z, Wu B, Raveendra-Panickar D, Chen V, Vazquez J, Qin H, Song J, Cosford NDP, Pellecchia M, Pasquale EB. Chem Biol Drug Des. 2011;78:667–678. doi: 10.1111/j.1747-0285.2011.01199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giorgio C, Hassan Mohamed I, Flammini L, Barocelli E, Incerti M, Lodola A, Tognolini M. PLoS One. 2011;6:e18128. doi: 10.1371/journal.pone.0018128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kellogg GE, Fornabaio M, Spyrakis F, Lodola A, Cozzini P, Mozzarelli A, Abraham DJ. J Mol Mod. 2004;22:476–489. doi: 10.1016/j.jmgm.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Leach AR, Shoichet BK, Peishoff CE. J Med Chem. 2006;49:5851–5855. doi: 10.1021/jm060999m. [DOI] [PubMed] [Google Scholar]
- 28.Lodola A, Rivara S, Mor M. Adv Protein Chem Struct Biol. 2011;85:1–26. doi: 10.1016/B978-0-12-386485-7.00001-6. [DOI] [PubMed] [Google Scholar]
- 29.Durrant JD, McCammon JA. BMC Biology. 2011;9:71. doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.King AR, Dotsey EY, Lodola A, Jung KM, Ghomian A, Qiu Y, Fu J, Mor M, Piomelli D. Chem Biol. 2009;16:1045–1052. doi: 10.1016/j.chembiol.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arunlakshana O, Schild HO. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giannetti AM, Koch BD, Browner MF. J Med Chem. 2008;51:574–580. doi: 10.1021/jm700952v. [DOI] [PubMed] [Google Scholar]
- 33.Pini A, Giuliani A, Falciani C, Fabbrini M, Pileri S, Lelli B, Bracci L. J Pept Sci. 2007;13:393–399. doi: 10.1002/psc.858. [DOI] [PubMed] [Google Scholar]
- 34.Núñez S, Venhorst J, Kruse CG. Drug Discov Today. 2012;17:10–22. doi: 10.1016/j.drudis.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 35.Noberini R, Koolpe M, Peddibhotla S, Dahl R, Su Y, Cosford ND, Roth GP, Pasquale EB. J Biol Chem. 2008;283:29461–29472. doi: 10.1074/jbc.M804103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM. J Med Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 37.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Nat Rev Drug Discov. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 38.Maestro, version 9.0. Schrodinger, LLC; New York, NY: 2009. [Google Scholar]
- 39.Jorgensen WL, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 40.Carmi C, Cavazzoni A, Vezzosi S, Bordi F, Vacondio F, Silva C, Rivara S, Lodola A, Alfieri RR, La Monica S, Galetti M, Ardizzoni A, Petronini PG, Mor M. J Med Chem. 2010;53:2038–2050. doi: 10.1021/jm901558p. [DOI] [PubMed] [Google Scholar]
- 41.Glide, version 5.5. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]
- 42.Krautler V, van Gunsteren WF, Hunenberger PH. J Comput Chem. 2001;22:501–508. [Google Scholar]
- 43.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 44.Tuckerman M, Berne BJ, Martyna GJ. J Chem Phys. 1992;97:1990–2001. [Google Scholar]
- 45.Bowers KJ, Chou E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06) Tampa; Florida. November 11-17; 2006. [Google Scholar]
- 46.Kihel LE, Clement M, Bazin MA, Descamps G, Khalid M, Rault S. Bioorg Med Chem. 2008;16:8737–8744. doi: 10.1016/j.bmc.2008.07.046. [DOI] [PubMed] [Google Scholar]
- 47.Katoch R, Trivedi GR, Phadke RS. Steroids. 1999;64:849–855. doi: 10.1016/s0039-128x(99)00072-0. [DOI] [PubMed] [Google Scholar]
- 48.Radomińska-Pyrek A, Huynh T, Lester R, St Pyrek J. J Lipid Res. 1986;27:102–113. [PubMed] [Google Scholar]
- 49.Katona BW, Cummins CL, Ferguson AD, Li T, Schmidt DR, Mangelsdorf DJ, Covey DF. J Med Chem. 2007;50:6048–6058. doi: 10.1021/jm0707931. [DOI] [PubMed] [Google Scholar]
- 50.Steinrauf LK, Cox B, Foster E, Sattar A, Blikenstaff RT. J Pharm Sci. 1978;67:1739–1743. doi: 10.1002/jps.2600671228. [DOI] [PubMed] [Google Scholar]
- 51.Shimizu K, Usui T, Fujioca T, Yamasaki K. J Biochem. 1958;14:13–14. [Google Scholar]
- 52.Siddiqui AU, Siddiqui AH, Ramaiah TS. J Heterocyclic Chem. 1993;30:61–64. [Google Scholar]
- 53.Valkonen A, Sievaenen E, Ikonen S, Lukashev NV, Donez PA, Averin AD, Lahtinen M, Kolehmainen E. J Molec Struct. 2007;846:65–73. [Google Scholar]
- 54.Mohamed IH, Giorgio C, Bruni R, Flammini L, Barocelli E, Rossi D, Domenichini G, Poli F, Tognolini M. Pharmacol Res. 2011;64:464–470. doi: 10.1016/j.phrs.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 55.Olive DM. Expert Rev Proteomics. 2004;1:327–341. doi: 10.1586/14789450.1.3.327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.