

Abstract
TLR ligands are effective vaccine adjuvants because they stimulate robust pro-inflammatory and immune effector responses, and abrogate suppression mediated by regulatory T cells (Tregs)2. Paradoxically, systemic administration of high doses of CpG oligonucleotides (CpGs) that bind to TLR9 ligands stimulated Tregs in mouse spleen to acquire potent suppressor activity dependent on interactions between PD-1 and its ligands. This response to CpG treatment manifested in a few hours, and was mediated by a rare population of plasmacytoid dendritic cells (CD19+ pDCs) induced to express the immunosuppressive enzyme IDO after TLR9 ligation. When IDO was blocked CpG treatment did not activate Tregs, but instead stimulated pDCs to uniformly express the pro-inflammatory cytokine IL-6, which in turn re-programmed Foxp3-lineage Tregs to express IL-17. Thus, CpG-induced IDO activity in pDCs acted as a pivotal molecular switch that induced Tregs to acquire a stable suppressor phenotype, while simultaneously blocking CpG-induced IL-6 expression required to re-program Tregs to become TH17-like effector T cells. These findings support the hypothesis that IDO dominantly controls the functional status of Tregs in response to inflammatory stimuli in physiologic settings.
Introduction
Toll-like receptors (TLRs) are critical in host defense to infectious microorganisms (1, 2). TLR ligands displayed by pathogens stimulate rapid pro-inflammatory responses by host cells of the innate and adaptive immune systems. For this reason pathogen-derived TLR ligands are critical components of vaccines and vaccine adjuvants, and synthetic TLR ligands are under investigation as potential vaccine adjuvants to stimulate more effective immunity in the treatment of cancer, infections and allergies (3). In mice, LPS and oligonucleotides containing un-methylated CpG motifs (CpGs) that bind to TLR4 and TLR9, respectively, trigger pDCs and other cell types to mature, acquire potent T cell stimulatory functions, and produce pro-inflammatory cytokines such as type I IFNs (IFNα) and IL-6. TLR ligands also promote pro-inflammatory outcomes by attenuating suppression mediated by regulatory T cells (Tregs). Thus, TLR ligands block de novo Treg differentiation, promoting differentiation of effector/helper T cells instead, and abrogate suppressor activity of Tregs (4-8). While some TLR ligands may affect Tregs directly, TLR9 ligands block Treg development and functions indirectly by inducing other immune cells to produce pro-inflammatory cytokines such as IL-6 (4). IL-6, in concert with other cytokines such as TGFβ and IL-23 is essential to induce naïve CD4+ T cells to differentiate into effector TH17 T cells expressing the cytokine IL-17 (9-11). Even mature, lineage committed Foxp3+ Tregs are more plastic than previously thought since exposure to IL-6 and other pro-inflammatory cytokines re-programmed mature Tregs to acquire a phenotype resembling pro-inflammatory TH17 helper T cells (12). Thus by binding to TLR9, CpGs possess potent pro-inflammatory properties widely assumed to overcome counter-regulatory mechanisms that may impede beneficial therapeutic outcomes in clinical settings such as cancer and chronic infections where hypo-immunity contributes to disease progression.
Paradoxically, TLR ligands displayed by some pathogens can also stimulate immune counter-regulatory mechanisms that suppress T cell responses, which may stabilize chronic infections in immunocompetent hosts (13). However, the mechanisms by which TLR ligands promote counter-regulatory over pro-inflammatory outcomes in physiologic settings are poorly defined. One potential mechanism involves induced expression of the intracellular enzyme IDO, which catabolizes tryptophan and triggers cellular stress responses that may modify cellular responses to pro-inflammatory stimuli (14). IDO activity correlates with immune counter-regulation in a range of clinically significant syndromes such as chronic infections, autoimmunity, cancer, and allograft resistance to destructive host immunity (14). Phenotypically distinctive subsets of pDCs in mice and humans are competent to express IDO in response to certain inflammatory cues. IDO-expressing pDCs possess potent T cell regulatory properties that block T cell responses to antigenic stimulation, and induce naïve T cells to differentiate into Tregs ex vivo. Constitutive IDO expression by pDCs blocks T cell responses to nominal tumor antigens in draining lymph nodes (dLNs) associated with sites of melanoma growth (15). Moreover, exposing mouse skin to pro-inflammatory the phorbol ester PMA, which also possesses tumor-promoting properties, caused pDCs in inflamed skin dLNs to express IDO, and to acquire potent T cell regulatory properties as a consequence (16). Thus induced IDO activity at sites of inflammation may attenuate the pro-inflammatory effects of TLR ligands.
Previously we showed that administering high doses of CpGs to mice induced rare CD19+ pDCs in spleen to express IDO, and to acquire potent T cell suppressor activity ex vivo (17). This response to TLR9 ligation in vivo manifested rapidly, within 24 hours of administering CpGs, and was dependent on IDO activity in CD19+ pDCs. However, it is unclear how a small population of IDO-expressing pDCs, and the inherently local effects of IDO, could create the dominant suppression that blocked all T cell stimulatory activity in mouse spleen after high dose CpG treatment. We hypothesized that CpG-induced IDO acts on Tregs to activate and stabilize a suppressor phenotype, which is resistant to TLR9-mediated de-activation of Tregs, and mediates dominant counter-regulation in spleen. We report that IDO acts as a molecular switch driving mature, quiescent Tregs into diametrically opposite functional states during CpG-induced innate immune responses, eliciting dominant Treg-mediated suppression when IDO is active, or allowing CpG-induced inflammatory stimuli to drive IL6-dependent Treg conversion into TH17-like T cells when IDO is blocked.
Materials and Methods
Mice
All mice were bred in a specific pathogen-free facility. A1 TCR transgenic, BM3 TCR transgenic, CBK transgenic, IDO-deficient (IDO1-KO), GCN2-deficient (GCN2-KO), CEBP-homologous protein-deficient (CHOP-KO), Act-mOVA, Foxp3GFP, IFNAR-KO, and IFNγR-KO mice were described previously (17-21). IL-6-KO mice were purchased from Jackson Laboratories, (Bar Harbor, ME). All gene-deficient mice were fully backcrossed (>10 generations) onto standard inbred strain backgrounds. The local (MCG) Institutional Animal Care and Use Committee approved all procedures involving mice.
CpG Oligonucleotides
CpG-B (#1826, TCCATGACGTTCCTGACGTT), and sequence matched non-CpG B (#2138, TCCATGAGCTTCCTGAGCTT); CpG-C (#2395, TCGTCGTTTTCGGCGCGCGCCG), and sequence matched non-CpG C (#2137, TGCTGCTTTTGTGCTTTTGTGCTT) with fully phosporothioate backbones were a gift of Coley Pharmaceuticals (Ottawa, Canada). Mice were injected with comparatively high doses of ODNs (50μg or 100μg per mouse, i/v) as described (17). The class of CpG used was dependent on the background strain of mice.
1-methyl-[D]-tryptophan (D-1MT)
D-1MT (catalog #45,248-3, Sigma; and clinical grade Newlink Genetics Inc.) was prepared as a 20mM stock solution in 0.1M NaOH, adjusted to pH 7.4, and stored at -20°C protected from light. For in vitro use, D-1MT was added at a final concentration of 100μM. For in vivo treatment, slow-release polymer pellets (5mg/day) containing D-1MT or vehicle alone were inserted under the dorsal skin as described (22) 48 hours before CpG treatment. Alternatively, mice were provided with D-1MT (2mg/ml) in drinking water, with sweetener (Nutrasweet) to enhance palatability, as described (23).
Preparative flow cytometry
Tregs and T cells were isolated from spleens using a Mo-Flo cytometer as described (17, 18). Typically, purity was >92%.
MACS enrichment
MACS enrichment for CD4+, CD8+, CD4+ CD25+ or CD11c+ cell populations was performed according to manufacturer's instructions (Magnetic Cell Separation Technology (MACS) – Miltenyi Biotec Inc., Auburn, CA).
Analytical flow cytometry
Cells were stained with the following antibodies (CD4-FITC [553047], CD8a-PE [553033], CD11c-PE [557401], CD19-PerCP [551001], CD25-PE [553866], B220-FITC [553088], Thy1.1-PerCP [557266] (all from Pharmingen-BD-Biosciences, San Jose, CA) and analyzed using a FACS-Calibur or FACS-Canto flow cytometer (Becton-Dickinson). Intracellular staining to detect IL-6 and IL-17 was performed on erythrocyte-free spleen cell suspensions. Antibodies (1:200 – 1:400) with specificity for cell surface markers were added first for 15 minutes at 4°C in the dark. After washing once in PBS, cells were fixed and permeabilized using Cytofix-Cytoperm (554722 – Pharmingen-BD-Biosciences) for 20 minutes in the dark at room temperature. After washing once in PBS, cells were incubated with (1:50) anti IL-6 (#6672–ABCAM, Cambridge, MA) or anti IL-17 (#17-7177-81–eBioscience Inc., San Diego, CA) antibody overnight at 4°C in the dark. Following another wash in PBS, cells were analyzed as above.
Immunohistochemistry
Sections were stained for IDO using a rabbit polyclonal Ab generated using a synthetic IDO peptide according to previous published methods (24). For IL-6, sections were first exposed to 0.5% Triton-X for 15 minutes at room temperature and washed with PBS prior to addition of the primary anti IL-6 antibody (1:100) [#6672 – ABCAM] and processed as for IDO.
Treg suppression (readout) assays were performed in 96 well V-bottom plates by adding graded numbers (maximum concentration 10 × 103) of Mo-Flo sorted or MACS enriched CD4+CD25+ cells to T cell proliferation (72 hour 3[H] thymidine incorporation) assays containing 1 × 105 or 5 × 104 responders, either (i) H-2Kb-specific T cells (nylon-wool enriched) from BM3 TCR transgenic mice and 2 × 103 CD11c+ APCs (Mo-Flo sorted or MACS enriched) from CBK (H-2Kb transgenic CBA) mice prepared as described (17), or (ii) H-Y-specific T cells (CD4+ MACS enriched) responders from A1 TCR transgenic mice, CD11c+ APCs (MACS enriched) from CBA female spleen and H-Y-Ek cognate peptide as described (19). A cocktail of PD-1, PD-L1 and PD-L2 antibodies was used as previously published (19).
Co-adoptive Transfers
Mo-Flo sorted or MACS enriched Tregs (from 0.4-1.25 × 106/recipient) were isolated from spleens of donor B6 (or CBA) mice treated for 24 hours with CpGs (#1826) or control ODNs (#2138) and mixed with MACS enriched (or nylon-wool purified) OT-1.Thy1.1 (or BM3) T cells (from 1.25-10 × 106/recipient) and co-injected into Act-mOVA transgenic (or CBK) recipients (3 mice/group). OT-1.Thy1.1 T cells were pre-labeled with tracking dye (CFSE) before transfer. Positive control mice received OT-1.Thy1.1 (or BM3) T cells without Tregs; negative control mice were B6 (or CBA) mice that did not express the target antigen for OT-1.Thy1.1 (or BM3) T cells. After 96 hours, mice were sacrificed and spleen cells stained with antibodies to detect donor T cells; anti-CD8a, and anti-Thy1.1 or anti-clonotypic Ab Ti98 (for BM3 - visualized with streptavidin APC (25)).
Statistical Analysis
Analyses were performed using Student's t test to compare data from triplicate wells within a group.
Results
High dose CpGs rapidly activate resting splenic Tregs
Previously, we showed that systemic treatment with high doses (>50μg, i/v) of CpGs induced splenic CD19+ pDCs to express IDO (17, 26). To test the hypothesis that CpG-induced IDO activates splenic Tregs we treated CBA mice with CpGs (#1826) or sequence-matched control (#2138) oligodeoxynucleotides (ODNs), and 24 hours later sorted splenic CD4+CD25+ T cells (Tregs) from treated mice. Graded numbers of sorted Tregs were added to readout assays containing responder H-2Kb-specific (CD8+) T cells from BM3 TCR transgenic mice, and stimulatory APCs from H-2Kb-transgenic (CBK) mice (17). As shown in Fig. 1A, adding only 5000 sorted Tregs from CpG-treated CBA mice abrogated BM3 T cell responses completely (Tregs:T cells = 1:20). In contrast, adding up to 10,000 sorted Tregs from control ODN-treated mice had no significant effect on BM3 T cell proliferation, indicating that TLR9 ligation was essential to stimulate resting Tregs to acquire suppressor activity.
Figure 1. Systemic high dose CpG treatment rapidly activates Tregs.

A. CBA mice were treated with 50μg (i/v) #1826 CpGs (●) or control #2138 (○) ODNs. After 24 hours graded numbers of FACS-sorted splenic Tregs were added to cultures containing 105 responder T cells (BM3) and APCs from CBK transgenic mice, as described in Methods. Thymidine incorporation was assessed after 72 hrs. B. MACS-enriched Tregs from B6 mice treated with CpGs (100μg, i/v) for the times indicated were added (2000/well) to cultures containing 105 responder T cells (A1), CBA female APCs and cognate (H-Y) peptide. C. Graded numbers of FACS-sorted Tregs from CpG-treated B6 mice (100μg, i/v) cultured with A1 responders and CBA APCs in the absence (●), or presence (○) of a cocktail of anti-PD1, anti-PD-L1 and anti-PD-L2 mAbs. T cell proliferation was assessed after 72 hrs. Data are the mean of triplicate cultures, and are representative of experiments performed on over five (A, C) or two (B) separate occasions. Asterisks highlight significant suppression mediated by Tregs from CpG-treated mice relative to cultures that did not contain sorted Tregs (p <0.001).
Next, we confirmed CpG-induced Treg activation using a second readout assay, in which sorted Tregs from B6 mice were MHC-mismatched with respect to stimulatory APCs from CBA mice used to stimulate H-Y-specific responder T cells from A1 TCR transgenic mice (19). This approach ensures that Treg suppressor activity is not activated during the readout assay itself. When sorted Tregs from B6 mice treated with high dose CpG for 18-48 hours were added to readout assays in the presence of H-Y peptide (Fig. 1B), as few as 2000 Tregs completely suppressed proliferation of A1 readout cells (Tregs:T cells ratio = 1:50), but suppression was not detected 12 hours after CpG treatment (Fig. 1B). Treg suppressor activity in readout assays was abrogated completely by adding a cocktail of mAbs that block the programmed death-1 (PD-1) counter-regulatory pathway, and its ligands PD-L1 and PD-L2 (Fig. 1C). In this regard, suppression mediated by CpG-activated Tregs resembled IDO-activated Tregs from tumor dLNs, which also showed dependence on the PD-1 pathway (19).
IDO is essential for Treg activation following high dose CpG treatment
Next, we tested if IDO was required for CpG-induced Treg activation. IDO-sufficient CBA and background-matched IDO-deficient (IDO1-KO) mice were treated with high-dose CpGs or control ODNs. After 24 hours splenic CD4+CD25+ Tregs were sorted, and added (5000/well) to readout assays containing BM3 responder T cells and stimulatory APCs from CBK mice. As before, Tregs from CpG-treated wild-type (IDO-sufficient) CBA mice were potently suppressive, but Tregs from CpG-treated IDO1-KO mice exhibited no significant suppressor activity relative to mice treated with control ODNs (Fig. 2A). Similarly, Tregs exhibited no CpG-induced suppressor activity when obtained from wild-type mice pre-treated to inhibit IDO pharmacologically using slow-release pellets impregnated with the IDO-inhibitor drug 1-methyl-[D]-tryptophan (D-1MT) implanted 72 hours before CpG treatment (Fig. 2B). Control mice pre-treated with pellets containing vehicle alone showed potent CpG-induced Treg suppressor activity. Naïve splenic CD4+CD25NEG T cells from CpG-treated mice had no effect on BM3 T cell proliferation in readout assays (data not shown), indicating that CpG-induced suppressor activity was induced selectively in the CD4+CD25+ subset. Similar outcomes were obtained in readout assays containing A1 responder T cells; Tregs from CpG-treated IDO1-KO mice (with B6 backgrounds) did not mediate suppression, while Tregs from B6 mice mediated potent PD-1-dependent suppression in this readout assay (data not shown). These findings revealed that functional IDO1 expression was essential to activate the Treg suppressor phenotype following CpG administration in vivo.
Figure 2. IDO activity is essential to activate Tregs after CpG treatment.

Mice were treated with 50μg CpGs (i/v, #1826) or control ODNs (#2138). After 24 hours, FACS-sorted splenic Tregs (5000/well) were added to readout assays containing 105 responder BM3 T cells and APCs from CBK mice. A. CBA or IDO1-KO mice were used as sources of Tregs. B. Tregs were obtained from mice pre-treated for 48 hours with pellets containing vehicle alone or pellets impregnated with IDO inhibitor (D-1MT) before CpG treatment. Thymidine incorporation was assessed after 72 hours. Control cultures containing no sorted Tregs yielded 46+/-1.3 (A) and 31+/-2.5 (b) ×103 CPM respectively. Dotted arrows highlight potent suppression mediated by Tregs from CpG-treated CBA IDO-sufficient mice. Data are the mean of triplicate cultures (+/- 1sd), and are representative of experiments performed on two or more occasions.
IFN type I, and GCN2 signaling is essential for CpG-mediated Treg activation
We have previously shown that signaling via IFN type I receptors (IFNAR) was essential to stimulate splenic CD19+ pDCs to express IDO, while signaling via IFN type II receptors (IFNγR) was not required (17, 27). To elucidate requirements to activate Tregs after TLR ligation we treated IFNAR-deficient (IFNAR-KO) and IFNγR-deficient (IFNγR-KO) mice, and measured Treg suppressor activity. As expected, control Tregs (1200/well) from CpG-treated wild-type (BALB/c) mice mediated potent suppression dependent on PD-1 interactions (Fig. 3A). In contrast, Tregs from CpG-treated IFNAR-KO exhibited no suppressor activity (Fig. 3B). However, Tregs from CpG-treated IFNγR-KO mice mediated potent PD-1 dependent suppression comparable to wild-type (129) controls (Fig. 3C). These outcomes were thus consistent with our previous reports that IDO induction in CD19+ pDCs was dependent on signaling via IFNAR, but not IFNγR signaling (17).
Figure 3. Requirements for TLR9-mediated Treg activation.

Sorted splenic Tregs from CpG-treated (24 hrs) mice were added to A1 T cell suppressor assays with (○) or without (●) PD-1/PD-L blocking mAbs (as in Fig. 1C). Sorted Tregs were obtained from CpG-treated (A) BALB/c, (B) IFNAR-KO, (C) IFNγR-KO, (D) GCN2-KO, and (E) CHOP-KO mice. Data shown are representative of experiments performed on two or more occasions. Sorted Tregs from CpG-treated gene-deficient and WT mice with matched backgrounds were analyzed in parallel (as in panels A & B) as positive controls for CpG-induced suppressor activity within each experiment.
We have also shown that susceptibility of T cells and Tregs to the biochemical effects of tryptophan catabolism is critically dependent on intact GCN2-kinase stress response pathways (18, 19). To elucidate whether intact GCN2 was required for CpG-induced splenic Treg activation we assessed suppressor activity of splenic Tregs from CpG-treated GCN2-deficient (GCN2-KO) mice. Tregs from CpG-treated GCN2-KO mice possessed no significant suppressor activity (Fig. 3D). Similarly, Tregs from mice with a defective gene encoding CEBP-homologous protein (CHOP-KO mice), a downstream target of activated GCN2-kinase, did not acquire suppressor activity in response to high dose CpGs (Fig. 3E).
IDO-activated Tregs suppress alloreactive T cell responses elicited in vivo
To evaluate if IDO-activated Tregs inhibited effector T cell responses elicited in vivo we sorted Tregs from donors (Thy1.2), mixed them with CD8+ T cells from OT-1 TCR transgenic (Thy1.1) mice pre-labeled with tracking dye (CFSE), and co-injected Treg/T cell mixtures into Act-mOVA (Thy1.2) transgenic mice (21), which express OVA constitutively as a nominal tissue alloantigen (Fig. 4A). OT-1 T cell responses were assessed 96 hours after Treg/T cell co-adoptive transfers. In this in vivo system, OT-1 T cells injected alone into B6 recipients retained high levels of CFSE, comparable with starting populations before transfer, indicating that they did not divide (Fig. 4B). OT-1 T cells injected alone into Act-mOVA recipients underwent multiple rounds of cell division, as evidenced by extensive dilution of CFSE levels (Fig. 4C). OT-1 T cells also underwent extensive division when co-transferred into Act-mOVA mice with ‘resting’ Tregs from untreated B6 mice (Fig. 4D). In striking contrast, OT-1 T cells did not undergo division, and retained uniformly high levels of CFSE when co-transferred with ‘activated’ Tregs from CpG-treated B6 (WT) mice (Fig. 4E). However, when Tregs originated from CpG-treated IDO1-KO donors CFSE levels on donor OT-1 T cells were extensively diluted (Fig. 4F), indicating that Tregs did not possess a suppressor phenotype. Analyses of the total numbers of OT-1 T cells in recipients revealed that OT-1 T cells underwent clonal expansion (8-10 fold relative to control B6 recipients without target antigen) when no Tregs were co-transferred (data not shown), and when Tregs originated from untreated B6 mice (Fig. 4G), or CpG-treated IDO1-KO mice (Fig. 4H). In contrast, total numbers of OT-1 T cells in spleens of mice that received Tregs from CpG-treated B6 mice were significantly lower, comparable with numbers of OT-1 T cells in spleen of B6 mice that received OT-1 T cells only (Fig. 4G, H). Thus, the ability of splenic Tregs to block in vivo clonal expansion of allo-reactive OT-1 T cells depended on CpG-treatment, and on induced IDO activity in Treg donor mice.
Figure 4. IDO-activated Tregs block alloreactive T cell responses in vivo.

A. Sorted Tregs from donor mice were mixed with MACS enriched CFSE-labeled OT-1 (Thy1.1) CD8+ T cells, and co-injected into Act-mOVA recipients. B-F. CFSE staining profiles of donor OT-1 (gated Thy1.1, CD8+) T cells analyzed 96 hours after co-adoptive transfers with splenic Tregs from the donors indicated. Markers were set to include 95% of OT-1 T cells injected alone into Act-mOVA recipients (as in C). G, H. Total numbers of OT-1 T cells detected in recipient spleens of the mice indicated. Data shown in (B-F) are representative of three mice/group, and (G, H) are the means for each group. Experiments were performed on two or more occasions.
Suppression of in vivo T cell clonal expansion was also observed when CpG-activated Tregs from wild-type (CBA) mice were co-transferred with H-2Kb-specific BM3 T cells into H-2Kb- transgenic (CBK) mice, whereas Tregs from CBA mice treated with control ODNs did not mediate suppression (supplementary Fig. S1). Consistent with these outcomes, spleen histology was normal in CBK mice that received BM3 T cells and IDO-activated Tregs from CpG-treated mice, while spleens from CBK mice that received BM3 T cells and Tregs from control ODN-treated mice were significantly smaller, contained large cohorts of BM3 T cells, and exhibited extensive pathology. Thus, IDO-activated Tregs suppressed clonal expansion and differentiation of allo-reactive effector T cells capable of causing tissue pathology.
Induced IDO activity blocks IL-6 production after high-dose CpG treatment
The preceding studies suggested that IDO induced by high CpG doses antagonized pro-inflammatory responses to CpGs. IL-6 is a classic pro-inflammatory cytokine produced by several cell types following TLR9 ligation (4). We therefore asked whether IDO attenuated IL-6 expression in mice treated with high CpG doses. Consistent with this hypothesis, no cells expressing IL-6 were detected in spleen of wild-type (IDO-sufficient) mice following high-dose CpG treatment (Fig. 5A, lower left). In striking contrast, spleens from CpG-treated IDO1-KO mice showed many IL-6-expressing cells distributed widely in lymphoid follicles, and in non-lymphoid regions of spleen (Fig. 5A, lower right). IDO expression in response to CpG treatment was confirmed by immunohistochemistry (Fig. 5A, upper panels).
Figure 5. IDO blocks IL-6 production following high-dose CpG treatment.
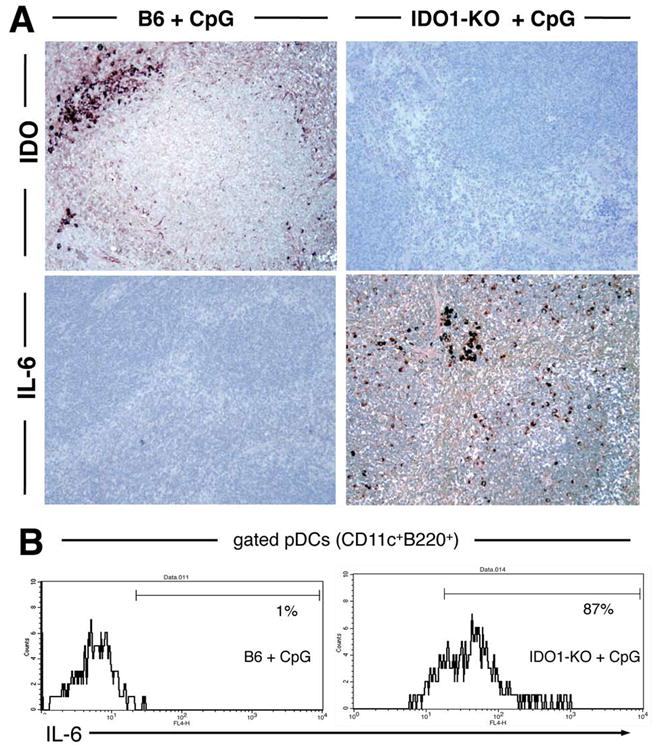
A. Spleen sections from CpG-treated B6 and IDO1-KO mice were stained with anti-IDO and anti-IL-6 Abs 24 hours after injecting CpGs (original magnifications, ×200). B. Splenic pDCs (gated CD11c+B220+ cells) from CpG-treated B6 (left) and IDO1-KO (right) mice were analyzed to detect intracellular IL-6 expression; markers exclude 95% of pDCs stained with isotype control mAbs, and percentages are the proportions of gated pDCs falling within this marker.
Flow cytometric analyses of gated splenic pDCs (CD11c+B220+) to detect intracellular IL-6 showed that pDCs from CpG-treated IDO-sufficient mice were not induced to express IL-6, while pDCs from IDO1-KO mice uniformly expressed IL-6 (Fig. 5B). However, splenic myeloid DCs (CD11c+B220NEG) from CpG-treated mice showed no IL-6 expression, irrespective of whether they originated from IDO-sufficient or IDO1-KO mice (data not shown), indicating that IL-6 production was a selective response to TLR9 ligation by pDCs amongst DCs. Levels of CD80 and PD-L2 surface expression by splenic DCs were unaffected by CpG treatment, while levels of CD86 and PD-L1 were elevated following CpG treatment (Supplementary Fig. S2), indicating that CpG treatment had some effect on expression of positive and negative co-stimulatory markers by DCs. Other splenocyte populations such as subsets of B cells also expressed IL-6 in response to CpG treatment, but only when IDO was not active (data not shown). These data suggested that multiple cell types, including the majority of pDCs were competent to express IL-6 in response to TLR9 ligation, but all these cells were prevented from expressing IL-6 when CpGs also co-induced CD19+ pDCs to express IDO.
CpG treatment reprograms splenic Tregs to express IL-17 in the absence of IDO
In concert with a cocktail of other cytokines (such as IL-1 and IL-23), IL-6 has been reported to reprogram Foxp3-lineage committed Tregs to express the inflammatory cytokine IL-17 in vitro (11, 12, 28). We asked whether CpG-induced IL-6 expression in IDO-deficient mice reprogrammed Tregs to express IL-17. Consistent with this hypothesis, a subset (6-7%) of splenic CD4+ cells expressed intracellular IL-17 in IDO1-KO mice treated with high-dose CpG; in contrast no IL-17-expressing cells were detected in spleen of IDO-sufficient (B6) mice after high-dose CpG treatment (Fig 6A). IL-17 expression in this subset of CD4+ cells was induced rapidly, with maximal IL-17 expression occurring 6-9 hours after CpG administration (Fig 6B). Further analysis revealed that the CD4+CD25+ subset expressed IL-17 uniformly, while the CD4+CD25NEG subset did not express IL-17 (Fig. 6C), suggesting that CpG treatment in the absence of IDO stimulated selective IL-17 expression in Tregs. To test this hypothesis we used Foxp3GFP knock-in mice that express a Foxp3-GFP fusion protein (20). Foxp3GFP mice were treated with oral IDO inhibitor (D-1MT in drinking water) beginning two days before CpG treatment. At baseline, (<6 hours after CpG treatment) no GFP+ Tregs expressed IL-17, but 24 hours after CpG treatment the majority of GFP+ Tregs (>80%) co-expressed IL-17 (Fig. 6D). Moreover, the population of IL-17+ cells corresponded with the GFP+ Treg population. Consistent with this finding CpG treatment induced CD4+CD25+ cells from IDO1-KO mice to express IL-17 uniformly, while CD4+CD25+ cells from CpG-treated B6 mice did not express IL-17 (supplementary Fig. S3); however, induced IL-17 expression correlated with loss of intracellular Foxp3 staining by CD4+CD25+ cells, suggesting that Foxp3 protein may be sequestered, or undergo conformational changes leading to loss of antibody binding sites, despite apparent retention of the Foxp3-GFP fusion protein in Foxp3GFP knock in mice. Because IL-17 expression by Tregs occurred rapidly after CpG treatment, and with no change in the total number of Tregs (Figs. 6, S3), we hypothesized that CpG treatment in the absence of IDO directly re-programmed existing, pre-formed Foxp3+ Tregs to express IL-17 (rather than stimulating de novo generation of IL-17+ Foxp3+ Tregs from naïve CD4+ precursors). To test this hypothesis we sorted splenic GFP+ cells from untreated Foxp3GFP donor mice (pre-formed Foxp3+ Tregs), and injected them into IDO-sufficient (B6) or IDO1-KO recipients. Two days after Treg transfer recipients were treated with high-dose CpG, and IL-17 expression was assessed in the transferred GFP+ cells 24 hours later. Analysis of gated GFP+ cells revealed that donor Tregs uniformly up-regulated IL-17 in CpG-treated IDO1-KO recipients (Fig. 6E), whereas GFP+ donor cells injected into CpG-treated IDO-sufficient recipients did not express IL-17. Collectively, these outcomes support the hypothesis that pre-formed, Foxp3-lineage committed Tregs were directly converted into IL-17-expressing cells by CpG treatment in the absence of IDO, while re-programming was blocked when IDO was active.
Figure 6. TLR ligation stimulates Tregs to express IL-17 when IDO is inactive.

FACS analyses to detect IL-17+ cells after CpG treatment, except where indicated analyses were performed 24 hours after CpG treatment. A, B Analyses of total splenocytes from the mouse strains indicated; percentages show the proportion of CD4+ cells expressing IL-17. C. Analyses of gated CD4+ cells; percentages show the proportion of CD4+ cells co-expressing CD25 and IL-17. D. Analyses of total splenocytes from Foxp3GFP mice treated with CpGs. E. FACS-sorted GFP+ cells from Foxp3GFP donors were injected into B6 and IDO1-KO recipients two days before CpG treatment. Histograms show IL-17 staining profiles for gated GFP+ cells from spleen of CpG-treated recipient mice; the marker excludes 95% of cells stained with isotype control mAb. Data shown are representative of two or more experiments.
Treg reprogramming to express IL-17 is dependent on CpG dose and IL-6
Next, we evaluated requirements to reprogram splenic Tregs to express IL-17 following CpG treatment. Consistent with the outcomes in IDO1-KO mice, high dose CpG treatment of IDO-sufficient B6 mice stimulated CD4+CD25+ splenocytes to express IL-17 when IDO was blocked by oral IDO inhibitor (D-1MT, Fig. 7B), but not when IDO was active (Fig. 7A). Administering lower doses (25μg, i/v) of CpGs that fail to induce IDO (17), also stimulated IL-17 upregulation by CD4+CD25+ cells (data not shown). Thus, selecting a lower dose of CpG to avoid inducing IDO stimulated purely pro-inflammatory responses (i.e. Treg conversion) without the need for D-1MT. At higher CpG doses the IDO effect predominated, and Treg conversion occurred only if IDO was blocked by D-1MT. Similarly, IL-17 expression was induced when IFNAR-KO mice were treated with high (Fig. 7C), and low (data not shown) CpG doses, consistent with requirements for IFNAR signaling to induce IDO, and revealing that IFNAR signaling was not required to induce IL-17 expression by Tregs. In contrast, mice lacking IL-6 (IL-6-KO) failed to up-regulate IL-17, irrespective of whether mice were pre-treated with D-1MT to block IDO (Fig. 7D, and data not shown), consistent with the hypothesis that IL-6 was essential for IL-17 induction in Tregs when IDO was inactive. Analyses of gated GFP+ cells from Foxp3GFP mice pre-treated with D-1MT revealed that CpG treatment induced increased expression of intracellular IFNγ, TNFα and IL-2, in addition to IL-17 by GFP+ Tregs, relative to resting GFP+ Tregs from untreated Foxp3GFP mice (Fig. 7E). Thus, when IDO was inactivated Foxp3-lineage Tregs rapidly acquired a polyfunctional pro-inflammatory phenotype after CpG treatment. Taken together, these data support the hypothesis that IDO actively suppresses IL-6-dependent re-programming of Treg functionality, which would otherwise occur in response to TLR9 ligation.
Figure 7. IL-6 is required to re-program Tregs into T cells expressing pro-inflammatory cytokines in the absence of IDO.

A-D. FACS analyses of gated CD4 splenocytes from the CpG-treated mice indicated to detect intracellular IL-17 expression 24 hours after CpG treatment. Where indicated, mice were given oral D-1MT two days before CpG treatment. Percentages indicate the proportions of CD4+ T cells from CpG-treated mice co-expressing CD25 and IL-17. E. FACS analyses to detect intracellular expression of the cytokines indicated by gated GFP+ Tregs from untreated Foxp3GFP mice and CpG-treated Foxp3GFP mice pre-treated with D-1MT. Numbers on histograms indicate mean fluorescence intensities of total gated GFP+ cells. Percentages indicate the proportions of GFP+ cells from treated mice expressing cytokines relative to staining profiles obtained using isotype control mAbs (not shown). Data shown are representative of experiments performed on three or more occasions.
Discussion
In this study we show that IDO acts as a pivotal molecular switch controlling the functional status of Tregs following TLR9 ligation, leading to diametrically opposed counter-regulatory or pro-inflammatory outcomes depending on whether IDO was active or inactive. Counter-regulatory responses manifested only a few hours after CpG administration, and only at high CpG doses that induced pDCs to express IDO. Moreover, IDO-mediated counter-regulation predominated over classic pro-inflammatory and T cell stimulatory responses to TLR9 ligation induced concomitantly. CpG-induced IDO stimulated potent Treg bystander suppressor activity, and simultaneously blocked IL-6 production required to convert Tregs into TH17-like T cells. Conversely, if IDO activity was blocked, CpG treatment elicited purely pro-inflammatory responses, inducing IL-6 expression that drove uniform conversion of mature Foxp3-lineage Tregs into a pro-inflammatory TH17-like phenotype. Thus, high doses of CpG are not intrinsically suppressive; rather, they trigger counter-regulatory responses mediated by IDO that dominantly suppress, or veto their underlying immunostimulatory and inflammatory effects. These findings suggest that under certain circumstances inducible or pre-existing IDO activity at local sites of inflammation may dominantly suppress pro-inflammatory processes, and block effector/helper T cell responses to antigens encountered at such sites. Thus, sufficiently intense inflammation may elicit dominant counter-regulation by IDO-activated Tregs to create local T cell suppression and immune privilege. Conversely, when IDO is absent, even strong pro-inflammatory stimuli do not elicit local Treg suppression, and Tregs are re-programmed to acquire a pro-inflammatory TH17-like phenotype.
Splenic Tregs acquired potent suppressor activity rapidly (12-18 hours) after TLR9 ligation. Activated Tregs blocked T cell proliferation ex vivo, and clonal expansion of allo-specific effector T cells in vivo. Rapid responses to CpG treatment were blocked completely by ablating the IDO1 gene, and by pharmacologic inhibition of IDO prior to CpG administration. Thus, intact IDO2 genes, which are closely related and linked to IDO1 genes (29, 30), did not compensate for loss of IDO1 regulatory functions. Ex vivo, the CpG-induced form of Treg suppressor activity was dependent on intact PD-1 signaling during suppressor assays. PD-1-dependent Treg suppression was also a distinctive feature of IDO-activated Tregs from inflamed LNs draining sites of melanoma growth (19), and skin exposed to the pro-inflammatory tumor promoter phorbol ester (PMA, unpublished data). These models of counter-regulation at sites of localized inflammation share with the CpG model the fact that Treg activation was dependent on IDO expression by pDCs in a physiologic setting. The role of PD-1 in these in vivo systems remains to be elucidated, but the requirement for PD-1 to mediate suppression ex vivo is a characteristic feature of Tregs activated by IDO in all three models. A recent report identified requirements for PD-L1 to generate Tregs from naïve T cells, implying that the PD-1 pathway is critical for Treg differentiation in vivo (31); however, this study did not address if IDO was required to promote Treg differentiation, and to stabilize the Treg suppressor phenotype.
CpG-induced Treg activation in the physiologic setting of the spleen was also dependent on intact IFNAR-signaling, but not IFNγR signaling. These outcomes are consistent with our previous reports showing an obligatory requirement for IFNAR-signaling to induce CD19+ pDCs to express functional IDO following treatment with soluble CTLA4 (CTLA4-Ig), and CpGs, which ligate B7 and TLR9 respectively (17, 27). We cannot exclude the possibility that IFNAR-signaling may play a direct role in stimulating Treg suppressor activity, but the known role of IFNAR signaling upstream of IDO is sufficient to explain its importance in the high-dose CpG model. However, signaling via IFNAR was not required to re-program Tregs to express IL-17 under IDO-deficient conditions.
IDO mediated inhibition of IL-6 expression was a key novel finding in our study because this provides a plausible explanation for the ability of IDO to prevent Treg re-programming into pro-inflammatory TH17-like cells, which is dependent on a cocktail of cytokines, including IL-6 (8, 11, 12). Uniform expression of IL-6 and IL-17 by pDCs and Tregs respectively, was induced rapidly (between 6-9 hours) after TLR9 ligation in IDO-deficient mice. The mechanism of IDO-mediated blockade of IL-6 expression is not known, but may involve autocrine and paracrine signaling mediated directly by IDO-expressing pDCs as the patterns of IDO and IL-6 expression induced by CpGs under IDO-sufficient and IDO-deficient conditions, respectively were not identical. Metz and colleagues recently reported that induced IDO activity in transfected cell lines induced expression of liver-enriched inhibitory protein (LIP), an inhibitory isoform of the NF-IL-6 transcription factor required to promote IL-6 gene expression (30). Thus, molecular pathways exist by which IDO may directly suppress up-regulation of IL-6 gene expression. Whether by this direct mechanism, or an alternative indirect route, our data unambiguously show that IDO blocked TLR9-induced IL-6 expression. CpG is widely used to induce IL-6 production by B cells and myeloid cells expressing TLR9, but our findings identify CpG dose as a critical factor, presumably because IDO expression by pDCs occurs only above a certain signaling threshold, which causes all splenic IDO-competent pDCs to up-regulate IDO simultaneously. Once this threshold was breached the counter-regulatory effects of induced IDO were dominant, and CpG treatment failed to stimulate IL-6 production, unless IDO was absent.
Our finding that IDO predominates over the immunostimulatory and pro-inflammatory effects of CpG treatment has potentially important implications for understanding the role of IDO in clinically significant inflammatory disease processes, and for treatment of such syndromes. We do not know what the human equivalent would be for the ‘high-dose’ CpG used in our murine model. But many natural infections and inflammatory conditions induce IDO in vivo (14), and CpGs are often administered locally as a vaccine adjuvant (3). If local or systemic levels were high enough to induce IDO, then paradoxical immunosuppression might ensue. The corollary of this however, is that blocking IDO at the time of CpG treatment may allow IL-6 production, leading to local reprogramming of Tregs. A further consideration is that, unlike spleen where IDO has to be co-induced by CpG treatment, IDO is constitutively activated in some settings of chronic inflammation, including lymphoid tissues draining local microenvironments where tumors develop, and draining inflamed skin exposed to phorbol esters that promote tumor development (15, 16). In these settings, constitutive (pre-induced) IDO activity may preclude immunostimulatory and pro-inflammatory responses to a range of insults, including tumors, certain infectious pathogens that activate Tregs (32) and induce local IDO expression, and artificial immunostimulants such as vaccines and vaccine adjuvants (14). Moreover, the use of IDO inhibitors to enhance tumor vaccine efficacy in murine models of tumor growth correlated with loss of suppressor activity by Tregs, and concomitant IL-6-dependent conversion of Tregs into TH17-like T cells in tumor dLNs (33), implying that the potent effects of IDO in spleens of CpG-treated mice are relevant to settings of chronic inflammation created by local tumor growth that induce constitutive IDO activity. IL-6 is known to synergize with other cytokines such as IL-1, and IL-23 to re-program Foxp3-lineage committed Tregs to express IL-17 (11). Cytokine induced functional reprogramming of cultured Foxp3-lineage Tregs to become TH17 T cells has been described, and was observed in mice treated with a novel immune modulator (B7-DC XAb), and in some infectious disease settings (7, 8, 12). However, the physiologic mechanisms that drive Treg to TH17 re-programming remain obscure. The findings we report in this study identify IDO as a critical molecular switch that stimulates potent Treg suppressor functions, and simultaneously blocks IL-6-mediated re-programming of Tregs to generate pro-inflammatory effector T cells expressing IL-17, and other pro-inflammatory cytokines such as IFNγ, TNFα and IL-2. Thus, our findings suggest that manipulating IDO may have profound effects on the balance of effector and counter-regulatory suppressor functions during inflammation. This scenario is also consistent with a recent study showing that absence of functional IDO activity contributed to unregulated local pro-inflammatory responses to a pulmonary infection (34).
In summary, the striking dichotomy of physiologic responses by splenic Tregs to high dose CpG treatment under IDO-sufficient and IDO-deficient conditions suggests that IDO-competent CD19+ pDCs are pivotal regulators of T cell responses at sites of inflammation. Thus, IDO emerges as a key molecular switch controlling the balance between suppressor and effector functions of T cells. Chronic activation of IDO has been reported in a diverse range of clinically relevant syndromes, including persistent infections, and cancer (14). In these syndromes, chronic or excessive IDO activity may create paradoxical local immune suppression and privilege that promotes disease progression by blocking innate T cell immunity to pathogens, and tumor antigens. Conversely, a deficiency in IDO promotes excessive autoimmunity in mice prone to type I diabetes (35, 36), and allows exaggerated pro-inflammatory responses in infected mice genetically predisposed to chronic granulomatous disease (34). Thus, IDO appears to be a pivotal regulator of inflammation in certain settings: suppressing inflammation and maintaining the suppressive phenotype of Tregs when IDO is active, or allowing unchecked inflammation and re-programming of Tregs when IDO is absent. This has significant implications for the pathogenesis of chronic inflammatory disorders, and also has significant practical implications for the use of artificial ligands as vaccine adjuvants. Recently, IDO induced by treatment with the NKT (CD1d) ligand αGalCer was identified as the reason why αGalCer treatment did not stimulate a vaccine adjuvant effect in a murine model of influenza vaccination (37), suggesting that IDO-mediated attenuation of immune responses to vaccine adjuvants may not be restricted to TLR ligands. When it is desirable to achieve the maximal pro-inflammatory effect of immune agonists, it may be crucial to block the dominant counter-regulatory effects of IDO. On the other hand, in settings where it is desirable to reduce excessive inflammation and T cell activation, enhancing IDO activity may be beneficial.
Supplementary Material
Acknowledgments
The authors thank Doris McCool, Anita Singh, and Diane Addis for expert technical assistance. We thank David Ron (NYU) for generously providing GCN2-KO and CHOP-KO mice, and Coley Pharmaceuticals and NewLink Genetics Inc. for the generous gifts of CpG oligodeoxynucleotides and D-1MT respectively.
Footnotes
Abbreviations (non-standard): pDC, plasmacytoid dendritic cell; ODN, oligonucleotide; CpG, oligonucleotides containing unmethylated CpG motifs; IFN type 1/2 receptor, IFNAR/IFNγR; PD-1/L, Programmed death-1/ligand; GCN2, general control non-de-repressible-2; CHOP, C/EBP homologous protein; LIP, liver-enriched inhibitory protein.
References
- 1.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annual Review of Immunology. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 3.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5:471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 4.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 5.Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19:39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Dominitzki S, Fantini MC, Neufert C, Nikolaev A, Galle PR, Scheller J, Monteleone G, Rose-John S, Neurath MF, Becker C. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J Immunol. 2007;179:2041–2045. doi: 10.4049/jimmunol.179.4.2041. [DOI] [PubMed] [Google Scholar]
- 7.Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol. 2008;29:429–435. doi: 10.1016/j.it.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Radhakrishnan S, Cabrera R, Schenk EL, Nava-Parada P, Bell MP, Van Keulen VP, Marler RJ, Felts SJ, Pease LR. Reprogrammed FoxP3+ T regulatory cells become IL-17+ antigen-specific autoimmune effectors in vitro and in vivo. J Immunol. 2008;181:3137–3147. doi: 10.4049/jimmunol.181.5.3137. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 10.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Z, O'Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res. 2008;41:87–102. doi: 10.1007/s12026-007-8014-9. [DOI] [PubMed] [Google Scholar]
- 12.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, Feng XH, Jetten AM, Dong C. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, Young L, Forehan SP, Mount A, Steptoe RJ, Shortman KD, de Koning-Ward TF, Belz GT, Carbone FR, Crabb BS, Heath WR, Villadangos JA. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- 14.Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008;8:74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- 15.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller AJ, Sharma MD, Chandler PR, DuHadaway JB, Everhart ME, Johnson BA, Kahler DJ, Pihkala J, Soler AP, Munn DH, Prendergast GC, Mellor AL. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci U S A. 2008;105:17073–17078. doi: 10.1073/pnas.0806173105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mellor AL, Baban B, Chandler PR, Manlapat A, Kahler DJ, Munn DH. Cutting Edge: CpG Oligonucleotides Induce Splenic CD19+ Dendritic Cells to Acquire Potent Indoleamine 2,3-Dioxygenase-Dependent T Cell Regulatory Functions via IFN Type 1 Signaling. J Immunol. 2005;175:5601–5605. doi: 10.4049/jimmunol.175.9.5601. [DOI] [PubMed] [Google Scholar]
- 18.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity. 2005;22:1–10. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Sharma MD, Baban B, Chandler PR, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:1–13. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Ehst BD, Ingulli E, Jenkins MK. Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am J Transplant. 2003;3:1355–1362. doi: 10.1046/j.1600-6135.2003.00246.x. [DOI] [PubMed] [Google Scholar]
- 22.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 23.Hou D, Muller AJ, Sharma M, Mellor AL, Prendergast GC, Munn DH. Stereoisomers of 1-methyl-tryptophan as pharmacologic inhibitors of indoleamine 2,3-dioxygenase. Cancer Research. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 24.Baban B, Chandler P, McCool D, Marshall B, Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J Reprod Immunol. 2004;61:67–77. doi: 10.1016/j.jri.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 25.Tarazona R, Sponaas AM, Mavria G, Zhou M, Schulz R, Tomlinson P, Antoniou J, Mellor AL. Effects of different antigenic microenvironments on the course of CD8+ T cell responses in vivo. International Immunology. 1996;8:351–358. doi: 10.1093/intimm/8.3.351. [DOI] [PubMed] [Google Scholar]
- 26.Manlapat AM, Kahler DJ, Chandler PR, Munn DH, Mellor AL. Cell autonomous control of interferon type I expression by indoleamine 2,3-dioxygenase in regulatory CD19+ dendritic cells. Eur J Immunol. 2007;37:1064–1071. doi: 10.1002/eji.200636690. [DOI] [PubMed] [Google Scholar]
- 27.Baban B, Hansen AM, Chandler PR, Manlapat A, Bingaman A, Kahler DJ, Munn DH, Mellor AL. A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell suppression via type I IFN signaling following B7 ligation. Int Immunol. 2005;17:909–919. doi: 10.1093/intimm/dxh271. [DOI] [PubMed] [Google Scholar]
- 28.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ball HJ, Sanchez-Perez A, Weiser S, Austin CJ, Astelbauer F, Miu J, McQuillan JA, Stocker R, Jermiin LS, Hunt NH. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene. 2007;396:203–213. doi: 10.1016/j.gene.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Metz R, Duhadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007;67:7082–7087. doi: 10.1158/0008-5472.CAN-07-1872. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105:9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 33.Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler PR, Mellor AL, He Y, Munn DH. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009 doi: 10.1182/blood-2008-12-195354. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romani L, Fallarino F, De Luca A, Montagnoli C, D'Angelo C, Zelante T, Vacca C, Bistoni F, Fioretti MC, Grohmann U, Segal BH, Puccetti P. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- 35.Grohmann U, Fallarino F, Bianchi R, Orabona C, Vacca C, Fioretti MC, Puccetti P. A defect in tryptophan catabolism impairs tolerance in nonobese diabetic mice. J Exp Med. 2003;198:153–160. doi: 10.1084/jem.20030633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol. 2007;179:5041–5053. doi: 10.4049/jimmunol.179.8.5041. [DOI] [PubMed] [Google Scholar]
- 37.Guillonneau C, Mintern JD, Hubert FX, Hurt AC, Besra GS, Porcelli S, Barr IG, Doherty PC, Godfrey DI, Turner SJ. Combined NKT cell activation and influenza virus vaccination boosts memory CTL generation and protective immunity. Proc Natl Acad Sci U S A. 2009;106:3330–3335. doi: 10.1073/pnas.0813309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.