

In this paper, the authors perform a bioinformatics homology search analysis of miRNAs in human, mouse, and rat. The pipeline defined uses previously existing tools (Blast and miRDeep2) that are based on homology. This was successfully used to identify novel miRNAs in humans, mouse, and rat. This work was motivated by the finding and experimental validation of mir-155 in the alternate rat genome (Celera), which is missing in the reference assembly and was previously unknown. This miRNA is encoded in a long noncoding RNA (Bic) and had been already discovered in humans and mice but not in rats. The discovery of “hidden” miRNAs opens new perspectives for finding many more nonconserved miRNAs, as well as novel long noncoding RNAs.
Keywords: mir-155, miRNAs prediction, homology search, genome assembly, small RNA sequencing
Abstract
MicroRNAs (miRNAs) are a class of small noncoding RNAs acting as post-transcriptional gene expression regulators in many physiological and pathological conditions. During the last few years, many novel mammalian miRNAs have been predicted experimentally with bioinformatics approaches and validated by next-generation sequencing. Although these strategies have prompted the discovery of several miRNAs, the total number of these genes still seems larger. Here, by exploiting the species conservation of human, mouse, and rat hairpin miRNAs, we discovered a novel rat microRNA, mir-155. We found that mature miR-155 is overexpressed in rat spleen myeloid cells treated with LPS, similarly to humans and mice. Rat mir-155 is annotated only on the alternate genome, suggesting the presence of other “hidden” miRNAs on this assembly. Therefore, we comprehensively extended the homology search also to mice and humans, finally validating 34 novel mammalian miRNAs (two in humans, five in mice, and up to 27 in rats). Surprisingly, 15 of these novel miRNAs (one for mice and 14 for rats) were found only on the alternate and not on the reference genomic assembly. To date, our findings indicate that the choice of genomic assembly, when mapping small RNA reads, is an important option that should be carefully considered, at least for these animal models. Finally, the discovery of these novel mammalian miRNA genes may contribute to a better understanding of already acquired experimental data, thereby paving the way to still unexplored investigations and to unraveling the function of miRNAs in disease models.
INTRODUCTION
Mature microRNAs (miRNAs) are short (∼22 nt) noncoding RNAs that control gene expression post-transcriptionally by base-pairing with 3′ untranslated regions (3′ UTRs) of their regulated transcripts, facilitating mRNA degradation or translation inhibition (Bartel 2004; Bushati and Cohen 2007; Djuranovic et al. 2012). MiRNAs regulate many biological processes and have critical roles in cell proliferation, differentiation, and death (Shivdasani 2006; Gomase and Parundekar 2009). Moreover, miRNA deregulation has been found in different types of human diseases and tumors (Lu et al. 2005; Volinia et al. 2006; Sayed and Abdellatif 2011). Precursor miRNAs (pre-miRNAs) are ∼70-nt-long RNA molecules with a characteristic hairpin structure. They originate in longer primary transcripts (pri-miRNAs) that are cleaved in animals by the Drosha endonuclease in the nucleus (Lee et al. 2003). Following the export of pre-miRNAs to the cytoplasm by Exportin-5, the loop region of the hairpin is removed by the Dicer endonuclease to produce a short, double-stranded RNA (dsRNA) (Cullen 2004). Based on the thermodynamic stability of each end of this duplex (O’Toole et al. 2006), one of the strands is preferentially incorporated in the RNA-induced silencing complex (RISC), producing a biologically active mature miRNA (generally the -5p miR), while the inactive strand (the -3p miR) is degraded (Kim 2005). The RISC complex inhibits translation elongation or triggers mRNA degradation, depending upon the degree of complementarity of the miRNA with its target (Carrington and Ambros 2003; Bartel 2004; Djuranovic et al. 2012).
Since the seminal identification of the first miRNAs in Caenorhabditis elegans (lin-4 and let-7) using genetic approaches (Lee et al. 1993; Reinhart et al. 2000), hundreds of miRNAs have been characterized experimentally in almost 170 species to date. The function of miRNAs in mice and rats has been thoroughly investigated in order to study the pathogenesis of several human diseases. Interestingly, the prevalence of the rat as a model organism in biomedical research is second only to humans (Jacob 2010). It has been widely studied in physiology, pharmacology, toxicology, nutrition, behavior, immunology, and neoplasia for its physiological similarity to humans (Aitman et al. 2008; Huang et al. 2011). Although the importance of the rat model in gene discovery was emphasized recently (Dwinell et al. 2011), it is quite surprising that some miRNAs that are thoroughly investigated elsewhere in humans and mice (i.e., miR-155) have never been discovered, even with the use of next-generation sequencing technologies (NGS or deep-sequencing) in different strains and tissues (Linsen et al. 2010).
In fact, the recent advent of this technique has enabled the simultaneous sequencing of up to millions of DNA or RNA molecules, increasing the discovery of novel miRNAs in an unprecedented way (Creighton et al. 2009). The rapid identification of novel miRNAs has been also accomplished by the coupling of NGS to computational methods. Among the most commonly used computational tools, we recall miRDeep (Bar et al. 2008; Friedlander et al. 2008; Oulas et al. 2009), miRanalyzer (Hackenberg et al. 2011), miRExpress (Wang et al. 2009), deepBase (Yang et al. 2010), miRTRAP (Hendrix et al. 2010), mirTools (Zhu et al. 2010), SSCprofiler (Oulas and Poirazi 2011), mirExplorer (Guan et al. 2011), MIReNA (Mathelier and Carbone 2010), DSAP (Huang et al. 2010), UEA sRNA workbench (Stocks et al. 2012), and miRNAkey (Ronen et al. 2010). Generally, these programs share the same two basic principles: (1) mapping of the reads to the reference genome and (2) checking for the presence of hairpin structures. However, two genomic assemblies are freely available. The Human Genome Sequencing Consortium (HGSC) assembly (the reference) is a composite genome that is derived from haploids of numerous donors (Lander et al. 2001; International Human Genome Sequencing Consortium 2004), whereas the Celera Genomics version of the genome (the alternate) is a consensus sequence, derived from five individuals, obtained from clone-based and random whole genome shotgun sequencing strategies (Venter et al. 2001). Mice (Mouse Genome Sequencing Consortium et al. 2002; Blake et al. 2006; Bult et al. 2010) and rats (Gibbs et al. 2004; Twigger et al. 2007; Dwinell et al. 2009) have alternative assemblies as well. In fact, Celera released these two alternate genomes to help researchers to fill in the gaps and to complete the assembly and validation of mouse and rat genome sequences, provided that Celera sequenced different strains with respect to the reference assembly (Kaiser 2005).
Although not conclusive, various comparative analyses undertaken in the past on HGSC and Celera human genome assemblies and their associated gene sets have revealed discrepancies (Hogenesch et al. 2001; Li et al. 2002, 2003). So far, no data have been obtained about miRNAs localization on alternate assemblies or comparisons between the two assemblies in mice and rats.
Finally, it appears clear that even the last release of miRBase repository (release 18.0 of November 2011) is far from complete, as only a few miRNAs are listed for rats (408 precursors, 680 mature) and mice (741 precursors, 1157 mature) with respect to human (1527 precursors, 1921 mature). It was recognized that some miRNAs are conserved evolutionarily from worms to humans (Lagos-Quintana et al. 2001). So, miRNA genes might have orthologs in other species, suggesting a powerful way to predict the existence of novel ones. Since the differences in their numbers cannot be accounted for merely by species-specific miRNAs alone, we attempted to determine the reasons for such an imbalance. Therefore, we tried to fill this gap by implementing a bioinformatics pipeline integrated with experimental and deep-sequencing validation experiments for the discovery of miRNAs using homology searches on the available genome sequences.
With our approach, we predicted many novel mammalian miRNAs and showed that many of them are “hidden” exclusively in the rat alternative assembly. This suggests that the choice of the genomic assembly in small RNA deep-sequencing experiments can further advance the discovery of miRNAs.
RESULTS
Hairpin mir-155 sequences in mammals
The alignment of the 11 mammalian mir-155 precursor sequences emphasized good conservation of mature miRNA sequences (Fig. 1). Moreover, this information can also be obtained by expanding the alignment of mir-155 beyond the 11 mammalian precursor sequences employing microRNAviewer (http://people.csail.mit.edu/akiezun/microRNAviewer/all_mir-155-align.html) (Supplemental Fig. S1; Kiezun et al. 2012). The length of hairpin mir-155 is variable (mean = 68.3 nt) with the longest sequence (105 nt) belonging to Ornithorhynchus anatinus (oan). The two mature forms, miR-155-5p and miR-155-3p, were outlined in red and in green, respectively. Of note, miR-155-5p is highly conserved in these species, and only two main sequences were found. The first belongs to hsa-miR-155-5p (5′-AAUGCUAAUCGUGAUAGGGGU-3′), and the second belongs to mmu-miR-155-5p (5′-AAUGCUAAUUGUGAUAGGGGU-3′). They differ only in a mismatch at position 10 from the beginning of the miR-5p (Fig. 1). The sequence of miR-155-5p in Sus scrofa (ssc) is not available in miRBase, although it seems identical to that of mmu-miR-155-5p. In the mammals considered, miR-155-3p sequences are more heterogeneous and present many mismatches. On the basis of these alignment results, we hypothesized that miR-155-5p in Rattus norvegicus might be equal to human or mouse sequences.
FIGURE 1.

Multiple sequence alignment (ClustalW 2.0.12, www.ebi.ac.uk) of known hairpin mir-155 sequences. (hsa) Homo sapiens, (ptr) Pan troglodytes, (mml) Macaca mulatta, (ppy) Pongo pygmaeus, (mmu) Mus musculus, (ssc) Sus scrofa, (eca) Equus caballus, (bta) Bos taurus, (cfa) Canis familiaris, (tgu) Taeniopygia guttata, (oan) Ornithorhynchus anatinus. Mature miR-155-5p sequences are represented in red whereas the miR-155-3p forms are represented in green. The black box indicates the mismatch in the two mature miR-155-5p consensus sequences.
Rat total RNA contains the mature miR-155-5p
To assess experimentally the level of expression of miR-155-5p in rats, we conducted real-time qPCR assays on a total RNA sample from rat spleen myeloid cells employing the only two commercially available assays for miR-155: human (hsa-miR-155-5p) and mouse (mmu-miR-155-5p) assays. For amplification to occur, the two assays have the forward primer designed to perfectly hybridize on the mature miRNA-5p sequence, thereby achieving a high degree of selectivity. The use of one or the other of these assays helped us to amplify the template and hypothesize the candidate sequence of rat miR-155-5p. The treatment with LPS was expected to up-regulate the expression of miR-155-5p in keeping with results previously obtained in human monocyte-derived dendritic cells (Ceppi et al. 2009; Del Corno et al. 2009) and in mouse samples (Ruggiero et al. 2009; Worm et al. 2009). The two assays gave different results for product amplification. In fact, only the mmu-miR-155-5p assay gave a detectable product amplification, whereas the hsa-miR-155-5p assay did not (data not shown). Figure 2 reports the quantity of miR-155-5p in spleen myeloid cells that were treated with LPS relative to control (CTRL) cells. Interestingly, we found a statistically significant up-regulation of 5.0 ± 1.2 (P = 0.041) in myeloid cells that had been treated with LPS relative to control cells. All together, these results suggest that mouse and rat mir-155-5p sequence could be identical.
FIGURE 2.
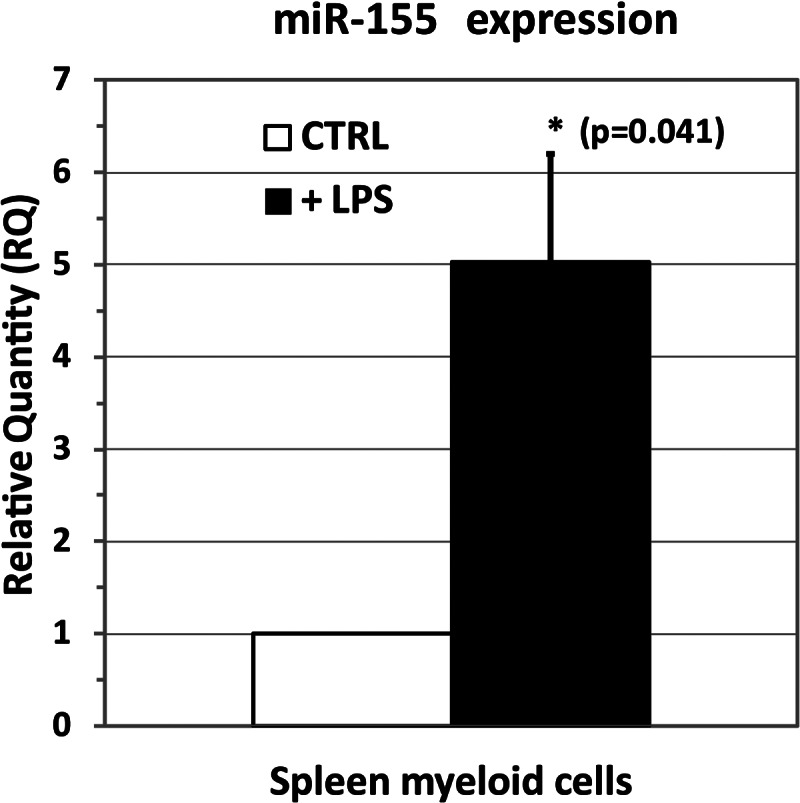
Quantitative PCR (qPCR) assay of miR-155-5p in spleen myeloid cells treated with LPS vs. control (untreated, CTRL) cells showing a statistically significant (P = 0.041) up-regulation of miR-155-5p (fold change = 5.0 ± 1.2).
Only the rat alternate genomic assembly contains the hairpin mir-155 sequence
To amplify the hairpin mir-155 sequence in the rat genome, we first reasoned that, if 86%–94% of rat genes have orthologs in mice (Nilsson et al. 2001; Hancock 2004), the region encompassing rat miRNA-155 could be quite conserved as well. Therefore, we designed a couple of primers (#1 and #2 in Supplemental Table S1) that are able to amplify a region surrounding the mouse mir-155 (Fig. 3). Interestingly, the PCR using these primers gave an amplified DNA fragment of ∼1 kb (994 bp) containing the precursor mir-155 sequence (see Supplemental File 1). To identify the genomic location of the rat hairpin mir-155, we performed a BLAST alignment on the two available genome assemblies (reference and alternate). Surprisingly, a good alignment was found only to the alternate assembly in the locus NW_001084660.1 (RN_Celera; Range: 12060846–12060910) of chromosome 11. The alignment of the whole 994-bp region or part of it (i.e., the putative mir-155 hairpin) on the reference assembly gave no significant results (Fig. 3) even when employing less stringent BLAST algorithms (i.e., blastn or discontiguous megablast instead of megablast). To confirm these preliminary findings, we designed two rat-specific primers (#3 and #4 in Supplemental Table S1) encompassing the mir-155 region that allowed us to amplify and sequence a contiguous DNA region of 917 bp (see Supplemental File 1) that again confirmed the presence of the mir-155 precursor sequence (Fig. 3). These results suggested the presence of a region in the rat genome (alternate assembly) potentially coding for a novel miRNA (namely rno-mir-155) that is orthologous to mmu-mir-155. These unexpected results led us to further investigate this region, exploiting also the information already published for other species.
FIGURE 3.

Genomic localization of rat mir-155 hairpin. Exploiting the homology between mouse and rat, a couple of mouse-specific primers (in green) were employed to amplify a fragment of rat DNA encompassing the mouse ortholog mir-155. This region was sequenced with rat-specific primers (in purple).
Human and mouse mir-155 precursors are encoded in the long noncoding RNA Bic
Human and mouse hairpin mir-155 (hsa-mir-155 and mmu-mir-155) were reported to be encoded and excised from a genomic region known as the B cell integration cluster (Bic) gene. This region encodes for a long noncoding RNA (lncRNA) that was originally identified as a gene transcriptionally activated by promoter insertion at a common retroviral integration site in B cell lymphomas induced by the avian leukosis virus (Tam et al. 1997). The human Bic (hBic) cDNA exhibits 83% identity over 292 nt to the mouse Bic (mBic) cDNA (Tam 2001). In humans and mice, Bic is expressed at a relatively high level in lymphoid organs and cells, implying an evolutionarily conserved function. In his milestone paper, Tam reported that the comparison of Bic genes in humans, mice, and chickens shows 78% identity over 138 nt located in exon III of hBic and mBic, and exon II of chicken (ckBic) (Tam 2001). No evidence of the presence of the lncRNA Bic in rat (rBic) has been reported yet. Therefore, we investigated the existence of the lncRNA Bic in the rat genome. Starting from the available sequence of Bic in humans and mice, we analyzed the similarities on both the reference (Refseq) and alternate (Celera) assemblies in order to predict the putative sequence (and position) of rBic. In fact, owing to the fact that the rat mir-155 hairpin sequence was found only on the alternate, and not on the reference assembly, we deemed it essential to perform comparisons and interspecies homology searches employing both of these assemblies.
Both reference and alternate assemblies encode for human and mouse lncRNA Bic
The human Bic gene (Gene ID: 114614) is contained within a 13-kb region in the locus NC_000021.8 (GRCh37.p5; Range: 26934457–26947480) (Lander et al. 2001) or AC_000153.1 (HuRef; Range: 12337598–12350626) (Levy et al. 2007) on chromosome 21 and has been reported to have three exons (Tam 2001). Both the reference and alternate assemblies encode for the whole 13-kb hBic gene with a 99.0% similarity score. Considering only the three exons, the similarity increased to 100% (Fig. 4A; Supplemental File 2). Therefore, similar results can be obtained by performing the BLAST alignment of hBic on one or the other of the two assemblies, suggesting a high similarity.
FIGURE 4.
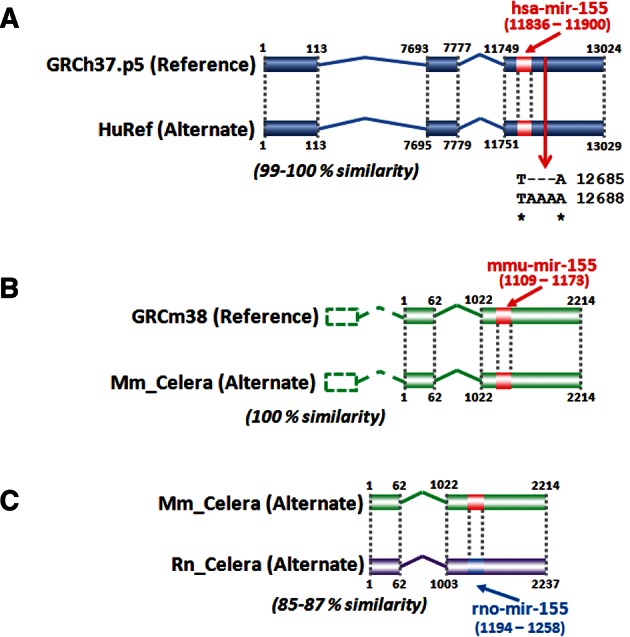
Human (A) and mouse (B) long noncoding RNA Bic have been reported to have three exons (Tam 2001). Since the mouse Bic exon I does not align to the reference or the alternate assembly, it was outlined with dashed lines. The alignment of the human and mouse Bic gene to the human reference and alternate assembly shows a high degree of homology. In (C), the prediction of rat Bic gene has been obtained by alignment of mouse Bic to the rat alternate genome. The hairpin mir-155 for each species is also shown.
The sequence of the mouse Bic gene was reported by Tam (2001) but never formally annotated. The only annotated sequence is the miR-155 stem–loop precursor (Gene ID: 387173) in the locus NC_000082.6 (GRCm38; Range: 84714140–84714204) or AC_000038.1 (Mm_Celera; Range: 84918138–84918202) on chromosome 16. In the original paper, Tam assigned at least three exons to the mBic gene (Tam 2001). The mouse hairpin mir-155 miRNA can be localized within the third exon of the mBic gene (Tam 2001; Lagos-Quintana et al. 2002). Although BLAST analysis failed to align exon I of mBic to the reference or the alternate genome, exons II and III in the two assemblies have a 100% similarity score (Fig. 4B; Supplemental File 3). Therefore, except for exon I, aligning mBic to the reference or the alternate genome gave practically the same result. Due to this imperfect alignment, we decided to omit the short sequence of mBic exon I and to perform all the following analyses employing only the putative sequences of exons II and III.
Prediction of the long noncoding RNA Bic in the rat genome
The presence of the rat ortholog of mouse mir-155 suggested that the genomic coordinates of the whole lncRNA Bic gene in rats could be obtained starting from the alignment of the mBic gene to the rat genome. To carry out this alignment (Fig. 4C), we decided to align mBic to the rat alternate assembly. We had two reasons. The first reason was the identical sequence alignment of mBic (exons II and III) to the reference or the alternate assembly. This allowed us to choose impartially either of these two genomes. The second reason was to compare assemblies obtained primarily by the same technology (whole genome shotgun). The BLAST alignment of the 2.2-kb mBic gene (from exon II) to the alternate rat genome resulted in a similarity score of 85%, whereas mBic exons II and III (∼1.2 kb) showed a similarity score of 87% (see Supplemental File 4). It may be noted that the length of rBic exons II and III are very similar to those of the mBic. This alignment allowed us to predict with good confidence the putative sequence of exon II and III of the rat lncRNA Bic transcript, which is located on chromosome 11 at position 12059653–12061889.
The rat spleen transcriptome contains the lncRNA Bic
To assess the presence of the lncRNA Bic in the rat spleen transcriptome, several PCR reactions were executed on cDNA samples. The PCR reaction with primers #5 and #9 afforded no amplification product, whereas amplification with primers #6 and #9 gave a product of 957 bp that we successfully sequenced (see Supplemental File 1). Aimed at sequencing the full-length rBic transcript and determining the presence of exon I and polyadenylation sites, we employed 5′-RACE and 3′-RACE techniques. Unfortunately, the small amount of total RNA extracted from spleen myeloid cells and the presence of two homopolymeric sequences located near the 5′-end of exon II (A9) and the 3′-end of exon III (T11) prevented a successful gene amplification. These results have two possible explanations. The first is that the rat lncRNA Bic may be shorter than the mBic, lacking exon I. The second is that the reverse-transcriptase employed in the RT reaction is unable to extend the product through homopolymeric sequences, hampering a complete Bic extension. However, additional experiments are needed to exclude definitively the presence of the predicted exon I of rBic.
Finally, having confirmed by several techniques the existence of a novel rat miRNA (mir-155) and also having confirmed its genomic localization only in the alternate assembly (Fig. 5A), we wondered how many rat miRNAs could be “hidden” in the alternate genome and how many novel miRNAs could be found in other mammalian species (i.e., humans and mice). Therefore, we developed a bioinformatics pipeline aimed at identifying other novel miRNAs in human, mouse, and rat genomes (reference and alternate), using a tailored homology search analysis (Fig. 5B).
FIGURE 5.
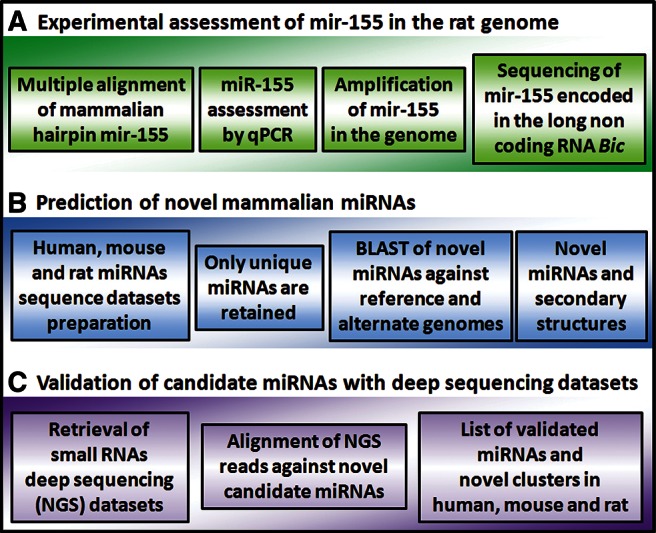
The overall bioinformatics workflow consists of three parts: (A) the experimental assessment of mir-155 hairpin sequence by a wet-lab approach, (B) a complete bioinformatics prediction of novel candidate miRNA sequences, and (C) validation of miRNAs with NGS data.
Homologies and differences of human, mouse, and rat miRNAs
In this step, unique stem–loop miRNA sequences from humans, mice and rats were obtained by proper filtering, since the miRNA name alone cannot be used for sequence discrimination among different species.
After interspecies alignment of miRNA sequences (Supplemental File 5), we identified 12 miRNA hairpin sequences that are identical in humans and mice, 14 between humans and rats, and 47 between mice and rats. Of these, seven hairpins are identical among the three species considered (Table 1; Supplemental Table S2). Furthermore, 34 human and mouse hairpin sequences overlap each other, whereas 23 overlap in humans and rats and 84 in mice and rats (Table 1; Supplemental Table S3). These results are in agreement with the closer evolutionary similarity between rats and mice than the similarity that has been observed between rodents and humans (Mullins and Mullins 2004). Therefore, these hairpins were eliminated from the data sets. We also filtered out hairpin miRNAs having >80% coverage and >90% sequence identity between species (93% for the mouse vs. rat comparison; thresholds were identified by accuracy analysis; see Materials and Methods for details). With these constraints, we obtained a list of 320 and 284 human miRNAs that are similar to mouse and rat miRNAs, respectively. For mice, 315 and 322 miRNAs are similar to those of humans and rats, respectively, while for rats, 303 and 350 miRNAs are similar to those of humans and mice, respectively (Table 1; Supplemental Table S4). After filtering out these hairpin sequences, the final data set containing unique miRNA sequences for each of the three species was retained for further homology searches. In particular, of the unique human miRNA sequences, 1178 miRNAs were absent in mice and 1214 in rats. For mice, 403 and 396 unique miRNAs were absent in humans and rats, respectively. A number of 104 and 57 rat miRNAs were absent in humans and mice, respectively. (Table 1; Supplemental Table S5).
TABLE 1.
Analysis of mammalian miRNA sequences

Prediction of conserved miRNA hairpin sequences
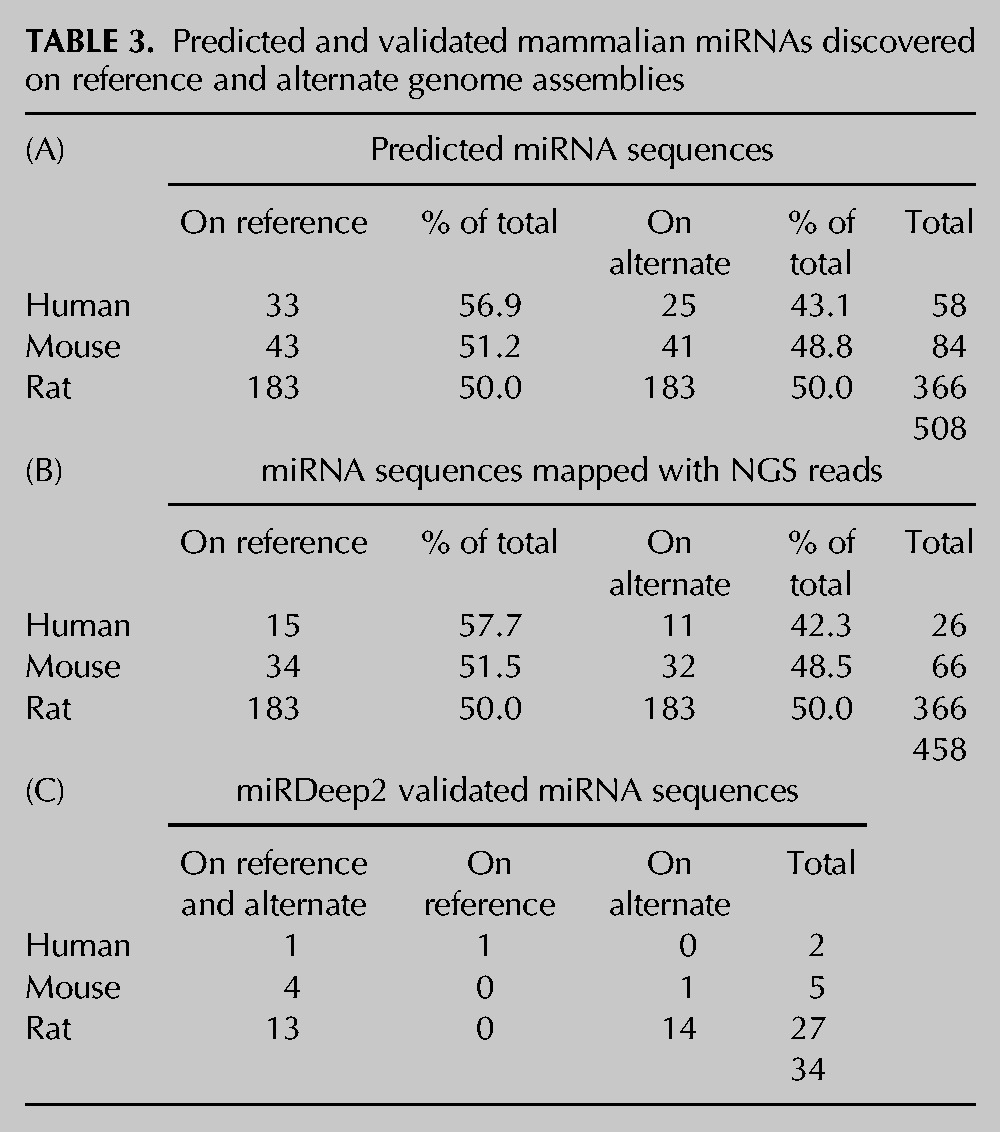
Since the main inclusion criteria that we adopted in our bioinformatics workflow are based on the conservation of the whole hairpin sequence, we expected to retain only those sequences that might have orthologs in the other species. Therefore, the unique miRNA hairpin sequences obtained in the previous filtering step were employed as query sequences and BLASTed against the primary and alternate genome assemblies of the three species (humans, mice, and rats). In particular, we decided to investigate the presence of these novel miRNAs not only on the primary (reference) assembly but also on the alternate (Celera) one, as we found that mir-155 is located only on the rat alternate genome (Fig. 3). The majority of miRNAs found after BLAST search (coverage >80%; identity >90% for human vs. mouse and rat, 93% for mouse vs. rat) aligned to both the reference and the alternate assembly, with a few exceptions for rat. Based on the alignment on the human genome, we predicted the presence of 22 novel putative miRNA genes (the subject sequences, sseq) in both the reference and the alternate assemblies and one putative miRNA (hsa-mir-2182) in the reference assembly only. For mice, 28 putative miRNA hairpins have been found in both assemblies. Two miRNAs (mmu-mir-1973 and mmu-mir-3610) are present only in the reference genome, while mmu-mir-935 is present only in the alternate assembly. As expected from the small number of miRNAs entries annotated so far in miRBase, rats have the greatest number of novel putative conserved hairpin miRNAs. In fact, 110 miRNAs aligned to both assemblies, with the rat mir-2182 present only in the reference genome. Surprisingly, although the majority of miRNAs align on both the reference and alternate genomes, up to 18 novel miRNA hairpins were predicted to align only to the alternate assembly (Table 2). The list of miRNA sequences reported in Table 2 was filtered for already annotated miRNAs. Moreover, since these predicted miRNA sequences may align to multiple positions in the respective genomes, we predicted all their possible alignments to the two assemblies (Supplemental Table S6) reaching a final number of 508 candidate predictions for the three species. The number of predicted miRNA sequences is quite similar in the two assemblies (Table 3A), although a slightly greater number of alignments to the reference relative to the alternate assembly was found.
TABLE 2.
Predicted mammalian miRNAs
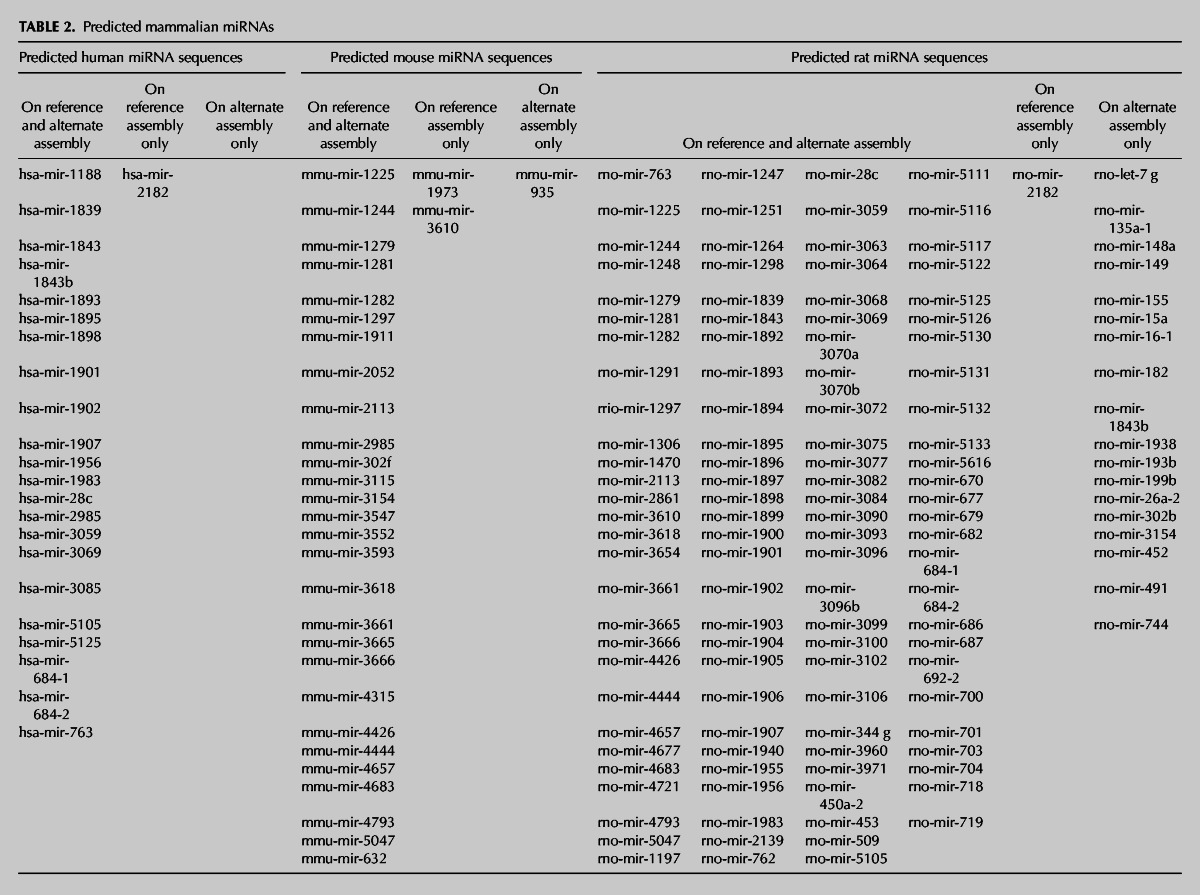
TABLE 3.
Predicted and validated mammalian miRNAs discovered on reference and alternate genome assemblies
Prediction of novel miRNA clusters
Interestingly, many novel putative miRNAs were found to form clusters, that is, groups of miRNAs lying within a genomic region of 10 kb. In particular, we predicted one novel cluster for humans, three for mice, and 14 for rats (Table 4). Once more, these findings outline the miRNA sequence conservation among different species, suggesting also a conserved function for their products (mature miRNAs).
TABLE 4.
Novel predicted mammalian miRNA clusters obtained after BLAST alignment

Many predicted miRNA sequences form stable secondary hairpin structures
To verify that the miRNA predictions obtained after the BLAST search have a secondary structure that is compatible with a miRNA hairpin, we calculated their folding energy by RNAfold. The results indicated that many of these predicted sequences form tight hairpin structures, potentially originating a real miRNA. As an example, Figure 6 shows the secondary structure prediction of hsa-mir-155 (the query sequence), the putative rat ortholog found after BLAST alignment to the alternate genome, and the secondary structure of the known mmu-mir-155 already annotated in the reference mouse genome. The minimum free energy (MFE) calculated for hsa-mir-155 is −29.70 kcal/mol, whereas the putative novel rno-mir-155, whose sequence is identical to mmu-mir-155, has a MFE of −25.62 kcal/mol. The complete list of secondary structure predictions can be found in Supplemental File 6.
FIGURE 6.
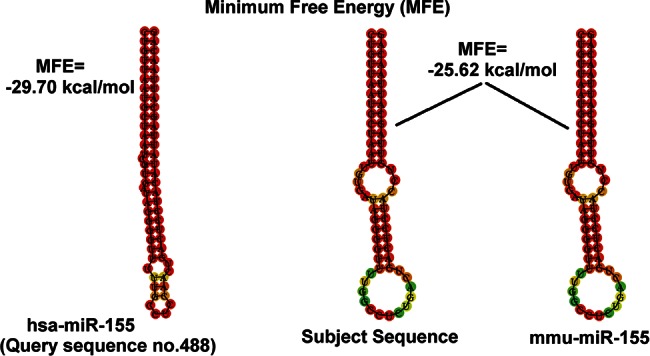
Minimum free energy calculation (MFE) of human hairpin mir-155 compared to the predicted rat mir-155 hairpin and the known mouse mir-155. The predicted rat mir-155 has the same sequence and secondary structure of the already known mouse mir-155.
Deep sequencing confirms the presence of many novel miRNAs and clusters
We reasoned that, if the novel predicted sequences found with our workflow are transcribed into miRNAs, we should have found them on the available small RNA NGS data sets (Supplemental Table S7). Moreover, the reads contained in these data sets have never been aligned to alternate genomes. Overall, the reads aligned to a total number of 458 sequences out of 508 (90.1%) in the three species considered. In particular, a number of 26 putative sequences (15 on the reference and 11 on the alternate assembly) out of 58 in human (44.8%), 66 (34 on the reference and 32 on the alternate assembly) out of 84 in mouse (78.6%), and all the 366 sequences (183 on the reference and 183 on the alternate assembly) in rat (100%) contain at least one read spanning from 19 to 26 nt and containing no or one mismatch (Table 3B; Supplemental Table S8). Sometimes it is difficult to distinguish a real miRNA from other small RNAs, such as endogenous small interfering RNAs (siRNAs), Piwi-interacting RNAs (piRNAs), and mRNA degradation products. Thus, a computational pipeline (miRDeep2) for the systematic identification of miRNAs from deep-sequencing data (Friedlander et al. 2008) was applied to our set of novel putative hairpins. This software is able to identify real hairpin miRNAs and predict the mature (-5p and -3p) sequences. By running miRDeep2 on a total of 185 data sets of NGS experiments, of which 161 are reported in miRBase and an additional 24 for rats (see Supplemental Table S7), the number of miRNAs experimentally supported in the three species decreased to 34. In particular, two miRNAs were found in humans, five in mice, and up to 27 in rats (Table 3C). As an example, the reads distribution for hsa-mir-1843b, mmu-mir-3552, and rno-mir-155 was reported in Figure 7. Interestingly, one human miRNA (hsa-mir-1843b) was found only on the reference assembly, while for mice, only mmu-mir-935 was found on the alternate assembly. For rats, the results are even more surprising. Although 13 novel miRNAs were found on both assemblies, up to 14 novel miRNAs were detected only on the alternate genomic assembly (Table 5; Supplemental Table S9). Interestingly, a number of miRNA clusters have also been validated. In particular, the mouse mir-3154, mir-3552, and mir-3618 form three independent clusters with the already known mir-199b, mir-764, and mir-1306, respectively (Table 4). For rats, we validated the presence of four miRNAs belonging to distinct clusters: mir-3064 (mir-3064/5047), mir-450a (mir-450a/b), mir-5132 (mir-718/5132), and mir-199b (mir-199b/3154). Of note, we validated both the components of the cluster mir-15a/mir-16 that was discovered only on the alternate assembly and not on the reference genome. This latter cluster has been already annotated for humans and mice.
FIGURE 7.

Representation of the number of deep-sequencing reads per base (for the whole hairpin sequence) of validated miRNAs. As indicative examples, the reads for hsa-mir-1843b, mmu-mir-3552, and rno-mir-155 are shown.
TABLE 5.
List of novel miRNAs in human, mouse, and rat

Overall, these data not only validate our bioinformatics prediction but also predict the presence of other novel conserved miRNAs never discovered before.
DISCUSSION
During recent years, we have assisted in a real revolution in the field of small RNAs and miRNAs discovery in eukaryotes by next-generation sequencing (NGS) technologies (Zhou et al. 2011). In fact, direct small RNAs sequencing has several advantages over hybridization-based methodologies (i.e., microarrays). They include (1) an inexpensive increase in throughput that provides a more complete view of the miRNA transcriptome, (2) no requirement for a prior knowledge of candidate regions of the genome, and (3) the opportunity to identify low-abundance miRNAs or those miRNAs with a negligible differential expression between different samples that are otherwise undetectable by conventional hybridization-based methods. However, microarray technology is still employed to validate NGS data, as recently reported for the discovery of miRNAs in the rat kidney (Meng et al. 2012).
To advance the discovery of novel miRNAs and other small RNAs, such as nuclear or nucleolar RNAs (snoRNAs), miRNA-offset RNAs (moRNAs) (Shi et al. 2009), and iso-miRs (Zhou et al. 2012), novel bioinformatics approaches for miRNA prediction coupled to deep-sequencing experiments are emerging continuously (Fasold et al. 2011; Guan et al. 2011; Oulas and Poirazi 2011; Zhang et al. 2012). The predictive strength of these and other tools generally relies on the application of computational constraints derived from the biological knowledge of miRNA biogenesis (i.e., type of Dicer cut, 5′ heterogeneity of -5p miRNA, presence of 3′ overhang between mature miRNAs, stable secondary structure, etc.) that are useful in filtering out many false positive miRNAs.
In our work, we combined a stringent homology search with the miRDeep2 algorithm to obtain a list of novel putative miRNAs. First, we exploited the gene homology among humans, mice, and rats (Nilsson et al. 2001) and the sequence conservation of the whole hairpin miRNA (instead of the mature miRNA only or its seed region) (Lewis et al. 2005). By considering the whole hairpin sequence conservation as a stringent criterion, we sought to reduce the rate of false positive miRNA predictions, although we are aware that our approach could have allowed us to predict many other novel miRNAs by starting the bioinformatics analysis with different criteria. Second, the integration of NGS data sets permitted the identification of many novel rat miRNAs that were “hidden” in the alternative genomic assembly. In the past, these “hidden” miRNAs were never identified in analogous homology search studies (Weber 2005; Artzi et al. 2008), and RefSeq assemblies were always employed as reference genomes (Bentwich et al. 2005; Berezikov et al. 2006; Sheng et al. 2007).
It may be noted that the approach employed permitted the identification of novel miRNA clusters. Not all were completely new, since some had already been found in other species. For example, the human and rat cluster mir-684-1/-2 is already known in mice, while the rat mir-15a/16-1 was already validated in humans and mice.
To the best of our knowledge, this is the first homology study for the discovery of miRNAs performed since the advent of NGS technology. Indeed, our approach allowed us to identify for the first time the existence of the novel rat mir-155 in the alternate assembly and to validate the mature miR-155-5p form, both through a classic molecular biology approach and a NGS approach (on available data sets). In humans and mice, miR-155 is one of the most studied miRNAs for its manifold functions. It is highly expressed in tumors (Eis et al. 2005; Du et al. 2011; Han et al. 2012) and has an important role in tumorigenesis (O’Connell et al. 2008; Jiang et al. 2010; Chang et al. 2011), inflammation (O’Connell et al. 2007, 2010; Tili et al. 2011), immunity (Trotta et al. 2012), autoimmune diseases (Bluml et al. 2011; Murugaiyan et al. 2011), dendritic cell maturation and function (Dunand-Sauthier et al. 2011), infections (Oertli et al. 2011), brain diseases (Junker et al. 2009; Murugaiyan et al. 2011), and transplant complications (Ranganathan et al. 2012). We expect that the discovery of this novel miRNA in rats will pave the way for further model studies focused on specific human diseases. Moreover, our study suggested that the rat reference genome has some differences compared to the alternate genome and that a newer release (the current is almost 10 yr old) is highly desirable and is recommended to fill these gaps. In fact, the availability of a good reference genome and a reliable physical map is fundamental for comparative genomics studies (Lewin et al. 2009). Moreover, the fact that the choice of different genomic assemblies can determine a different final result is not a novel concept. A systematic comparative analysis between the human HGSC and Celera assemblies has been performed by Li et al., who found that most of the unique genes from either genome assembly could be mapped back to the other assembly, suggesting that the gene set discrepancies do not reflect differences in local sequence content but rather in the assemblies and especially the different gene-prediction methodologies (Li et al. 2003). On the other hand, a critical comparison of the Celera and public mouse assemblies has concluded that, although differing in ∼10%, each assembly has advantages over the other (Xuan et al. 2003). Celera has higher accuracy in base pairs and overall higher coverage of the genome, whereas the public assembly has higher sequence quality in some regions. Thus, the authors highly recommended that the two mouse genome assemblies be used in an integrated fashion rather than separately.
Finally, future small RNAs sequencing studies will also improve the prediction capability of software such as miRDeep (Friedlander et al. 2008) or miRTRAP (Hendrix et al. 2010), which will perform better when higher genomic coverage is available. Many small RNA molecules (i.e., moR/moR*) of unknown function were recently recognized by deep sequencing experiments and bioinformatics approaches (Hendrix et al. 2010; Bortoluzzi et al. 2011). We expect that, as the deep-sequencing experiments on rat models increase, so will the accuracy of predictions and the discovery of novel small RNA species and of conserved and nonconserved miRNAs.
MATERIALS AND METHODS
Overall workflow
The overall workflow (Fig. 5) consists of three parts. Starting from the experimental assessment of a novel miRNA (mir-155) in the rat genome (Fig. 5A), we extended the prediction of novel candidate miRNAs in humans, mice, and rats by BLAST alignment to reference and alternate genomes and secondary structure analysis (Fig. 5B). Then, we validated the candidate miRNAs by applying miRDeep to an ensemble of 185 small RNA sequencing data sets (Fig. 5C).
Mammalian mir-155 sequences and data set generation
The sequences of hairpin miRNAs were downloaded from miRBase (www.mirbase.org; Release 18.0, Nov 2011) in FASTA format. This miRBase release contains 18,226 hairpin precursor miRNAs, expressing 21,643 mature miRNA products in 168 species. The mir-155 hairpin sequence of 11 mammalian species (Homo sapiens, hsa; Pan troglodytes ptr; Macaca mulatta, mml; Pongo pygmaeus, ppy; Mus musculus, mmu; Sus scrofa, ssc; Equus caballus, eca; Bos taurus, bta; Canis familiaris, cfa; Taeniopygia guttata, tgu; and Ornithorhynchus anatinus, oan) were selected and aligned by ClustalW (2.0.12) (http://www.ebi.ac.uk/Tools/msa/clustalw2/) (Larkin et al. 2007).
Reference and alternate genome assemblies
The available human, mouse, and rat genomes (reference and alternate assemblies) were retrieved from NCBI (http://www.ncbi.nlm.nih.gov/genome/). GRCh37.p5 is the human reference assembly that was released on June 24, 2011 by the Genome Reference Consortium, whereas HuRef is the alternate assembly that represents a composite haploid version of the diploid genome sequence from a single individual (J. Craig Venter). GRCm38 (Dec 2011) is the mouse genome reference assembly that was produced by the Mouse Genome Reference Consortium, and Mm_Celera is the alternate Celera Genomics whole mouse genome shotgun assembly. For rats, RGSC_v3.4 is the reference composite assembly that was made from the WGS data plus finished BAC clone sequences produced by the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) as part of the Rat Genome Sequencing Consortium (RGSC) (November 2004). The whole genome shotgun alternate assembly (Celera Genomics) is represented by Rn_Celera.
Rats and isolation of spleen cells
Female Wistar rats (8–10 wk old) were purchased from Charles River. All animals were manipulated in accordance with the Local Ethical Committee guidelines.
Total spleenocytes were isolated by cutting rat spleens into small fragments. The fragments were then digested with type III collagenase (1 mg/mL, Worthington Biochemical) and DNase I (325 KU/mL, Sigma) in RPMI 1640 containing 10% heat-inactivated FBS (Cambrex) with periodic pipetting, for 25 min at room temperature. At the end of the incubation, EDTA (0.1 M, pH 7.2, Sigma) was added for an additional 5 min, to allow disruption of cell-to-cell complexes. The cells were then washed in PBS without Ca2+ and Mg2+, resuspended in RPMI 1640 containing 10% heat-inactivated FBS, penicillin (100 U/mL), streptomycin (100 µg/mL), 2 mM glutamine, 50 µM 2-mercaptoethanol (all reagents were purchased from Cambrex), and cultured for 2 h at 37°C, 5% CO2, in 6-well-plates (106 cell/mL). Nonadherent cells, represented mostly by lymphocytes, were then removed by washing with PBS without Ca2+ and Mg2+, while the adherent myeloid cell population was maintained in a fresh complete medium containing 200 ng/mL of ultrapure lipopolysaccharide (LPS) from Escherichia coli (serotype EH100, Alexis Biochemicals). Twenty-four hours later, the cells were collected for RNA isolation.
Quantitative PCR analysis of rat miR-155
To assess the presence of the mature form of rat miR-155 (namely, rno-miR-155-5p), total RNA from rat spleen myeloid cells was extracted with the Total RNA Purification Kit (Norgen Biotech Corp.) and reverse-transcribed by use of stem–loop primers that were specifically designed for the analysis of mmu-miR-155-5p (Assay ID: 002571). For comparison purposes, we employed also the human assay for hsa-miR-155-5p (Assay ID: 002623). cDNA was synthesized from 5 ng of total RNA using the TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems) according to the manufacturer’s instructions. Quantitative PCR was performed in triplicate employing the RT product, SensiMix II Probe Kit (Bioline Inc.) and the specific miRNA assay (Applied Biosystems). Amplification and detection were undertaken with a 7900HT Fast Real-Time PCR System (Applied Biosystems). The endogenous control U6 snRNA (Assay ID: 001973) was employed for normalization. The relative quantity (RQ) of miR-155 in LPS-treated cells vs. control (untreated cells) was calculated by the 2−ΔΔCt method (Livak and Schmittgen 2001).
PCR amplification of a rat genomic region containing the mir-155 hairpin sequence
To amplify the region of the rat genome putatively containing the whole mir-155 hairpin sequence, a classic polymerase chain reaction (PCR) was employed. We decided to exploit the gene homology between mice and rats (Nilsson et al. 2001). First, we designed a couple of primers (#1 and #2, see Supplemental Table S1) encompassing the mouse mir-155 hairpin region (Chr16:84714385–84714449) and enclosed in a fragment of ∼1 kb (989 bp), 485 bp upstream of the beginning of mmu-mir-155 (forward primer) and 440 bp downstream from its end (Chr16:84713900-84714889) (reverse primer). PCR was carried out using the BIO-X-ACT Long DNA Polymerase (Bioline USA Inc.), which included also the 10× OptiBuffer, a 5× Hi-Spec Additive, and a 50 mM MgCl2 solution. Each PCR reaction mix contained 1× OptiBuffer (2.5 µL), 100 mM dNTP Mix (0.25 µL), primers 10 µM each (0.25 µL), 50 mM MgCl2 Solution (1 μL), BIO-X-ACT Long polymerase 4 U/µL (0.5 µL), template (rat DNA) (1 µL; ∼20 ng), and water to reach a final volume of 25 µL. The PCR conditions were as follows: a step of 4 min at 94°C, followed by 40 cycles of 30 sec at 94°C, 30 sec at 61°C, 1 min at 72°C, and then finishing with a 7-min incubation at 72°C. The PCR was also repeated using a couple of rat-specific primers (#3 and #4, Supplemental Table S1) with the same PCR program but changing the primer’s annealing temperature (65°C). Product visualization was obtained with agarose gel (2%) electrophoresis that was stained with Gel Red (Biotium Inc.). PCR reactions were purified with the QIAquick Gel Extraction Kit (Qiagen), and sequencing was performed on an 8-capillary 3500 Genetic Analyzer using the BigDye terminator (V3.1) protocol.
Sequencing a fragment of the long noncoding RNA Bic containing mir-155 hairpin
Total RNA was extracted from rat spleen tissue (∼100 mg) with TRIzol Reagent (Life Technologies) according to the manufacturer’s protocol. Total RNA (1 µg) was reverse-transcribed using the QuantiTect Reverse Transcription Kit (Qiagen) that includes an effective reaction step to remove genomic DNA contamination in RNA samples. The RT-PCR reaction was started from 1 µL of cDNA using a couple of primers (#6 and #8 in Supplemental Table S1) that were specifically designed to amplify a 957-bp region in the exon II of the Bic transcript. The RT-PCR conditions were a step of 4 min at 94°C, followed by 45 cycles of 30 sec at 94°C, 30 sec at 62°C, 1 min at 72°C, and then finishing with a 7-min incubation at 72°C. To confirm the presence of the mir-155 hairpin sequence, we employed primers #6, #7, #8, and #9 (see Supplemental Table S1) for sequencing.
Selection of novel candidate miRNAs by homology search
MirBase (ver.18) includes 1527 sequences of hairpin miRNAs for humans, 741 for mice, and 408 for rats. After grouping identical sequences, these data sets were reduced to 1498 unique miRNAs for humans, 718 for mice, and 407 for rats (Table 1). Then, all possible interspecies alignments of hairpin miRNA sequences were generated by BLAST (i.e., human vs. mouse, human vs. rat, and mouse vs. rat) (Altschul et al. 1997). The results were combined in a single file for clarity (Supplemental File 5). Pairs of ortholog hairpin miRNAs were identified by sequence similarity and removed from the data set (coverage, i.e., alignment length/query length, >80% and identity >90% for human vs. mouse and human vs. rat comparison and >93% for mouse vs. rat comparison). Thresholds for coverage and identity were selected by accuracy analysis (Supplemental File 7). Briefly, we constructed 100 sequence sets, including 849 pairs of true miRNA orthologs (i.e., sequence conservation and identical name) and 849 pairs of randomly selected miRNAs. After an all vs. all BLAST search, we computed the accuracy ACC = (True Positives + True Negatives)/(Positives + Negatives) at different values of coverage and identity for each sequence set and for each pair of species independently, as the species have different degrees of divergence (Supplemental File 7). The cut-off values corresponding to the highest accuracy were coverage > 80% and identity > 90% (human vs. mouse and rat) or 93% (mouse vs. rat).
The final list contained miRNAs with no orthologs in one or two of the other species. Then, we searched for their genomic localization by aligning the miRNAs identified in the previous step to the reference and alternate assemblies using BLAST with the same thresholds as above.
Secondary structure analysis and free energy calculation
To inspect the secondary structure of the predicted miRNA sequences and calculate their minimum free energy, we employed the software RNAfold that is implemented in the Vienna package (Gruber et al. 2008). To facilitate comparisons, we ran RNAfold on the original miRNA sequences (fseq), the query sequences (qseq), and the subject sequences (sseq) identified by BLAST. The MFE for qseq and sseq were reported in Supplemental File 6.
Validation of predicted miRNAs with deep-sequencing data
To validate the miRNAs that were predicted in the previous steps, we retrieved from Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo/) 185 small RNA NGS data sets (161 were already employed to validate human and murine known miRNAs annotated in miRBase, while the remaining 24 represent all the rat small RNA NGS data sets available to date) (Supplemental Table S7). In particular, 79 data sets were obtained from humans, 82 from mice, and 24 from rats. First, NGS reads in base-space were trimmed based on quality values. Then, together with reads in color-space, they were mapped with Bowtie (ver 0.12.7) (Langmead et al. 2009; Langmead and Salzberg 2012) on the novel candidate miRNA sequences using the sequential trimming strategy recently described (Marco and Griffiths-Jones 2012). Only those reads that were 18 to 26 nt in length and aligned to candidate miRNA sequences with zero or one mismatches were considered. Mapped reads were then submitted to the miRDeep2 algorithm for further validation.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. A. Alisi for her preliminary discussions about the role of miRNAs in the rat model, and Dr. S. Barresi and Dr. A. Taranta for providing additional information on amplification protocols. We also thank the Italian Ministry of Health and the Bambino Gesù Children’s Hospital, IRCCS for their financial support (grant RC-201202G002799 to A.M.). A.M. dedicates his contribution to the memory of his uncle Luigi.
Author contributions: P.U. retrieved data, prepared data sets, implemented the bioinformatics pipeline, and wrote part of the manuscript, L.D.S. performed all of the molecular biology experiments, M.D.C. isolated and treated spleen myeloid cells, A.B. set up the PCR and qPCR experiments, P.S. prepared the animals and spleen tissues, M.O. implemented tools for NGS data analysis, A.P. optimized Sanger’s sequencing reactions, S.G. organized and coordinated the experimental procedures of spleen myeloid cell preparation and treatment and wrote part of the manuscript, and A.M. coordinated the work, contributed to data analysis, and wrote and assembled the manuscript.
REFERENCES
- Aitman TJ, Critser JK, Cuppen E, Dominiczak A, Fernandez-Suarez XM, Flint J, Gauguier D, Geurts AM, Gould M, Harris PC, et al. 2008. Progress and prospects in rat genetics: A community view. Nat Genet 40: 516–522 [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artzi S, Kiezun A, Shomron N 2008. miRNAminer: A tool for homologous microRNA gene search. BMC Bioinformatics 9: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, Kroh EM, Bendoraite A, Mitchell PS, Nelson AM, et al. 2008. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells 26: 2496–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP 2004. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. 2005. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 37: 766–770 [DOI] [PubMed] [Google Scholar]
- Berezikov E, van Tetering G, Verheul M, van de Belt J, van Laake L, Vos J, Verloop R, van de Wetering M, Guryev V, Takada S, et al. 2006. Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res 16: 1289–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Eppig JT, Bult CJ, Kadin JA, Richardson JE, Mouse Genome Database Group 2006. The mouse genome database (MGD): Updates and enhancements. Nucleic Acids Res 34: D562–D567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluml S, Bonelli M, Niederreiter B, Puchner A, Mayr G, Hayer S, Koenders MI, van den Berg WB, Smolen J, Redlich K 2011. Essential role of microRNA-155 in the pathogenesis of autoimmune arthritis in mice. Arthritis Rheum 63: 1281–1288 [DOI] [PubMed] [Google Scholar]
- Bortoluzzi S, Biasiolo M, Bisognin A 2011. MicroRNA-offset RNAs (moRNAs): By-product spectators or functional players? Trends Mol Med 17: 473–474 [DOI] [PubMed] [Google Scholar]
- Bult CJ, Kadin JA, Richardson JE, Blake JA, Eppig JT, Mouse Genome Database Group 2010. The mouse genome database: Enhancements and updates. Nucleic Acids Res 38: D586–D592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Cohen SM 2007. microRNA functions. Annu Rev Cell Dev Biol 23: 175–205 [DOI] [PubMed] [Google Scholar]
- Carrington JC, Ambros V 2003. Role of microRNAs in plant and animal development. Science 301: 336–338 [DOI] [PubMed] [Google Scholar]
- Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, Pierre P 2009. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci 106: 2735–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Wang RH, Akagi K, Kim KA, Martin BK, Cavallone L, Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab), Haines DC, Basik M, Mai P, et al. 2011. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med 17: 1275–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Reid JG, Gunaratne PH 2009. Expression profiling of microRNAs by deep sequencing. Brief Bioinform 10: 490–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR 2004. Transcription and processing of human microRNA precursors. Mol Cell 16: 861–865 [DOI] [PubMed] [Google Scholar]
- Del Corno M, Michienzi A, Masotti A, Da Sacco L, Bottazzo GF, Belardelli F, Gessani S 2009. CC chemokine ligand 2 down-modulation by selected Toll-like receptor agonist combinations contributes to T helper 1 polarization in human dendritic cells. Blood 114: 796–806 [DOI] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, Green R 2012. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 336: 237–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du ZM, Hu LF, Wang HY, Yan LX, Zeng YX, Shao JY, Ernberg I 2011. Upregulation of MiR-155 in nasopharyngeal carcinoma is partly driven by LMP1 and LMP2A and downregulates a negative prognostic marker JMJD1A. PLoS One 6: e19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunand-Sauthier I, Santiago-Raber ML, Capponi L, Vejnar CE, Schaad O, Irla M, Seguin-Estevez Q, Descombes P, Zdobnov EM, Acha-Orbea H, et al. 2011. Silencing of c-Fos expression by microRNA-155 is critical for dendritic cell maturation and function. Blood 117: 4490–4500 [DOI] [PubMed] [Google Scholar]
- Dwinell MR, Worthey EA, Shimoyama M, Bakir-Gungor B, DePons J, Laulederkind S, Lowry T, Nigram R, Petri V, Smith J, et al. 2009. The rat genome database 2009: Variation, ontologies and pathways. Nucleic Acids Res 37: D744–D749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwinell MR, Lazar J, Geurts AM 2011. The emerging role for rat models in gene discovery. Mamm Genome 22: 466–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE 2005. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci 102: 3627–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasold M, Langenberger D, Binder H, Stadler PF, Hoffmann S 2011. DARIO: A ncRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res 39: W112–W117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N 2008. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 26: 407–415 [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, et al. 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521 [DOI] [PubMed] [Google Scholar]
- Gomase VS, Parundekar AN 2009. microRNA: Human disease and development. Int J Bioinform Res Appl 5: 479–500 [DOI] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL 2008. The Vienna RNA websuite. Nucleic Acids Res 36: W70–W74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan DG, Liao JY, Qu ZH, Zhang Y, Qu LH 2011. mirExplorer: Detecting microRNAs from genome and next generation sequencing data using the AdaBoost method with transition probability matrix and combined features. RNA Biol 8: 922–934 [DOI] [PubMed] [Google Scholar]
- Hackenberg M, Rodriguez-Ezpeleta N, Aransay AM 2011. miRanalyzer: An update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res 39: W132–W138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han ZB, Chen HY, Fan JW, Wu JY, Tang HM, Peng ZH 2012. Up-regulation of microRNA-155 promotes cancer cell invasion and predicts poor survival of hepatocellular carcinoma following liver transplantation. J Cancer Res Clin Oncol 138: 153–161 [DOI] [PubMed] [Google Scholar]
- Hancock JM 2004. A bigger mouse? The rat genome unveiled. Bioessays 26: 1039–1042 [DOI] [PubMed] [Google Scholar]
- Hendrix D, Levine M, Shi W 2010. miRTRAP, a computational method for the systematic identification of miRNAs from high throughput sequencing data. Genome Biol 11: R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenesch JB, Ching KA, Batalov S, Su AI, Walker JR, Zhou Y, Kay SA, Schultz PG, Cooke MP 2001. A comparison of the Celera and Ensembl predicted gene sets reveals little overlap in novel genes. Cell 106: 413–415 [DOI] [PubMed] [Google Scholar]
- Huang PJ, Liu YC, Lee CC, Lin WC, Gan RR, Lyu PC, Tang P 2010. DSAP: Deep-sequencing small RNA analysis pipeline. Nucleic Acids Res 38: W385–W391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Tong C, Kumbhani DS, Ashton C, Yan H, Ying QL 2011. Beyond knockout rats: New insights into finer genome manipulation in rats. Cell Cycle 10: 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945 [DOI] [PubMed] [Google Scholar]
- Jacob HJ 2010. The rat: A model used in biomedical research. Methods Mol Biol 597: 1–11 [DOI] [PubMed] [Google Scholar]
- Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H, Liu MF, Wang ED 2010. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res 70: 3119–3127 [DOI] [PubMed] [Google Scholar]
- Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, Lassmann H, Wekerle H, Hohlfeld R, Meinl E 2009. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain 132: 3342–3352 [DOI] [PubMed] [Google Scholar]
- Kaiser J 2005. Genomics. Celera to end subscriptions and give data to public GenBank. Science 308: 775. [DOI] [PubMed] [Google Scholar]
- Kiezun A, Artzi S, Modai S, Volk N, Isakov O, Shomron N 2012. miRviewer: A multispecies microRNA homologous viewer. BMC Res Notes 5: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN 2005. MicroRNA biogenesis: Coordinated cropping and dicing. Nat Rev Mol Cell Biol 6: 376–385 [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T 2001. Identification of novel genes coding for small expressed RNAs. Science 294: 853–858 [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T 2002. Identification of tissue-specific microRNAs from mouse. Curr Biol 12: 735–739 [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854 [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, et al. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419 [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin HA, Larkin DM, Pontius J, O’Brien SJ 2009. Every genome sequence needs a good map. Genome Res 19: 1925–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20 [DOI] [PubMed] [Google Scholar]
- Li S, Liao J, Cutler G, Hoey T, Hogenesch JB, Cooke MP, Schultz PG, Ling XB 2002. Comparative analysis of human genome assemblies reveals genome-level differences. Genomics 80: 138–139 [DOI] [PubMed] [Google Scholar]
- Li S, Cutler G, Liu JJ, Hoey T, Chen L, Schultz PG, Liao J, Ling XB 2003. A comparative analysis of HGSC and Celera human genome assemblies and gene sets. Bioinformatics 19: 1597–1605 [DOI] [PubMed] [Google Scholar]
- Linsen SE, de Wit E, de Bruijn E, Cuppen E 2010. Small RNA expression and strain specificity in the rat. BMC Genomics 11: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. 2005. MicroRNA expression profiles classify human cancers. Nature 435: 834–838 [DOI] [PubMed] [Google Scholar]
- Marco A, Griffiths-Jones S 2012. Detection of microRNAs in color space. Bioinformatics 28: 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Carbone A 2010. MIReNA: Finding microRNAs with high accuracy and no learning at genome scale and from deep sequencing data. Bioinformatics 26: 2226–2234 [DOI] [PubMed] [Google Scholar]
- Meng F, Hackenberg M, Li Z, Yan J, Chen T 2012. Discovery of novel microRNAs in rat kidney using next generation sequencing and microarray validation. PLoS One 7: e34394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Mullins LJ, Mullins JJ 2004. Insights from the rat genome sequence. Genome Biol 5: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL 2011. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol 187: 2213–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson S, Helou K, Walentinsson A, Szpirer C, Nerman O, Stahl F 2001. Rat-mouse and rat-human comparative maps based on gene homology and high-resolution zoo-FISH. Genomics 74: 287–298 [DOI] [PubMed] [Google Scholar]
- O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D 2007. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci 104: 1604–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, Paquette RL, Baltimore D 2008. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med 205: 585–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D 2010. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 33: 607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertli M, Engler DB, Kohler E, Koch M, Meyer TF, Muller A 2011. MicroRNA-155 is essential for the T cell-mediated control of Helicobacter pylori infection and for the induction of chronic Gastritis and Colitis. J Immunol 187: 3578–3586 [DOI] [PubMed] [Google Scholar]
- O’Toole AS, Miller S, Haines N, Zink MC, Serra MJ 2006. Comprehensive thermodynamic analysis of 3′ double-nucleotide overhangs neighboring Watson-Crick terminal base pairs. Nucleic Acids Res 34: 3338–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulas A, Poirazi P 2011. Utilization of SSCprofiler to predict a new miRNA gene. Methods Mol Biol 676: 243–252 [DOI] [PubMed] [Google Scholar]
- Oulas A, Boutla A, Gkirtzou K, Reczko M, Kalantidis K, Poirazi P 2009. Prediction of novel microRNA genes in cancer-associated genomic regions—a combined computational and experimental approach. Nucleic Acids Res 37: 3276–3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan P, Heaphy CE, Costinean S, Stauffer N, Na C, Hamadani M, Santhanam R, Mao C, Taylor PA, Sandhu S, et al. 2012. Regulation of acute graft-versus-host disease by microRNA-155. Blood 119: 4786–4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G 2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901–906 [DOI] [PubMed] [Google Scholar]
- Ronen R, Gan I, Modai S, Sukacheov A, Dror G, Halperin E, Shomron N 2010. miRNAkey: A software for microRNA deep sequencing analysis. Bioinformatics 26: 2615–2616 [DOI] [PubMed] [Google Scholar]
- Ruggiero T, Trabucchi M, De Santa F, Zupo S, Harfe BD, McManus MT, Rosenfeld MG, Briata P, Gherzi R 2009. LPS induces KH-type splicing regulatory protein-dependent processing of microRNA-155 precursors in macrophages. FASEB J 23: 2898–2908 [DOI] [PubMed] [Google Scholar]
- Sayed D, Abdellatif M 2011. MicroRNAs in development and disease. Physiol Rev 91: 827–887 [DOI] [PubMed] [Google Scholar]
- Sheng Y, Engstrom PG, Lenhard B 2007. Mammalian microRNA prediction through a support vector machine model of sequence and structure. PLoS One 2: e946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Hendrix D, Levine M, Haley B 2009. A distinct class of small RNAs arises from pre-miRNA-proximal regions in a simple chordate. Nat Struct Mol Biol 16: 183–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivdasani RA 2006. MicroRNAs: Regulators of gene expression and cell differentiation. Blood 108: 3646–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks MB, Moxon S, Mapleson D, Woolfenden HC, Mohorianu I, Folkes L, Schwach F, Dalmay T, Moulton V 2012. The UEA sRNA workbench: A suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 28: 2059–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam W 2001. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene 274: 157–167 [DOI] [PubMed] [Google Scholar]
- Tam W, Ben-Yehuda D, Hayward WS 1997. bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol Cell Biol 17: 1490–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tili E, Michaille JJ, Wernicke D, Alder H, Costinean S, Volinia S, Croce CM 2011. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc Natl Acad Sci 108: 4908–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta R, Chen L, Ciarlariello D, Josyula S, Mao C, Costinean S, Yu L, Butchar JP, Tridandapani S, Croce CM, et al. 2012. miR-155 regulates IFN-γ production in natural killer cells. Blood 119: 3478–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigger SN, Shimoyama M, Bromberg S, Kwitek AE, Jacob HJ, RGD Team 2007. The Rat Genome Database, update 2007—Easing the path from disease to data and back again. Nucleic Acids Res 35: D658–D662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. 2001. The sequence of the human genome. Science 291: 1304–1351 [DOI] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. 2006. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci 103: 2257–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WC, Lin FM, Chang WC, Lin KY, Huang HD, Lin NS 2009. miRExpress: Analyzing high-throughput sequencing data for profiling microRNA expression. BMC Bioinformatics 10: 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MJ 2005. New human and mouse microRNA genes found by homology search. FEBS J 272: 59–73 [DOI] [PubMed] [Google Scholar]
- Worm J, Stenvang J, Petri A, Frederiksen KS, Obad S, Elmen J, Hedtjarn M, Straarup EM, Hansen JB, Kauppinen S 2009. Silencing of microRNA-155 in mice during acute inflammatory response leads to derepression of c/ebp Βeta and down-regulation of G-CSF. Nucleic Acids Res 37: 5784–5792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Z, Wang J, Zhang MQ 2003. Computational comparison of two mouse draft genomes and the human golden path. Genome Biol 4: R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Shao P, Zhou H, Chen YQ, Qu LH 2010. deepBase: A database for deeply annotating and mining deep sequencing data. Nucleic Acids Res 38: D123–D130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xu B, Yang Y, Cooke HJ, Ban R, Xue Y, Shi Q 2012. CPSS: A computational platform for the analysis of small RNA deep sequencing data. Bioinformatics 28: 1925–1927 [DOI] [PubMed] [Google Scholar]
- Zhou L, Li X, Liu Q, Zhao F, Wu J 2011. Small RNA transcriptome investigation based on next-generation sequencing technology. J Genet Genomics 38: 505–513 [DOI] [PubMed] [Google Scholar]
- Zhou H, Arcila ML, Li Z, Lee EJ, Henzler C, Liu J, Rana TM, Kosik KS 2012. Deep annotation of mouse iso-miR and iso-moR variation. Nucleic Acids Res 40: 5864–5875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu E, Zhao F, Xu G, Hou H, Zhou L, Li X, Sun Z, Wu J 2010. mirTools: microRNA profiling and discovery based on high-throughput sequencing. Nucleic Acids Res 38: W392–W397 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.